[level-membership-for-neurology-category]
Chapter 204 Management of Cranial Nerve Injuries
The incidence of traumatic cranial nerve injury is approximately 5% to 23%.1 Nontraumatic and iatrogenic injuries are not uncommon. Cranial nerve injuries are caused by direct impact or indirect transference of energy from the skull base and/or bony elements surrounding the cranial nerves. Neuroimaging studies are crucial for diagnosis; however, the clinician must rely on accurate history and the neurologic examination to confirm the appropriate diagnosis of cranial nerve injury. The neurologic examination can be limited by mechanical ventilation and sedation, especially in patients with significant multisystem trauma. The knowledge of the pathologic anatomy of cranial nerve injuries is essential. With this knowledge, the clinician can develop appropriate suspicion for potential cranial nerve injury, make the appropriate diagnosis, and treat patients based on current standards of care. Delay in diagnosis and treatment leads to significant morbidity.
Traumatic Olfactory Nerve Injury
The literature reports a wide range of percentages for traumatic olfactory nerve dysfunction. Injury to the olfactory nerve varies from 4% to 60%.2 Clinical diagnosis of traumatic olfactory nerve dysfunction depends on patient age and their recognition of the dysfunction. The likelihood of traumatic olfactory dysfunction increases with age.
Self-assessment may under- or over-estimate the incidence of olfactory nerve dysfunction because of the lack of awareness of olfactory function. Only 40% of patients with olfactory dysfunction were aware of their deficit.2 Most dysfunction is present immediately after injury, but delayed onset has been observed. It is thought that delay is secondary to excessive fibrous tissue around a fractured cribriform plate; there is little radiographic or histologic evidence, however, to prove this.
The probability of traumatic olfactory nerve dysfunction does not depend on the severity of the traumatic head injury, as recorded by the Glasgow coma scale (GCS).2 The severity of dysfunction, however, is related to the severity of head injury. Traumatic olfactory nerve dysfunction is also related to whether the anterior skull base and/or frontal lobe are involved. Radiographic evidence of anterior skull base or frontal lobe injuries is, however, absent in 24% of cases.2 Patients with skull base fractures present not only with olfactory nerve dysfunction, but can also present with rhinorrhea.3 Coup and contracoup injuries that occur with frontal and occipital trauma are the most common type of injuries producing olfactory nerve dysfunction.4 There are literature reports of minor head injury associated with olfactory nerve dysfunction.
Traumatic olfactory nerve dysfunction involves damage to the olfactory epithelium containing the receptor neurons in the nasal mucosa and/or mechanical stretching/shearing of unmyelinated nerve rootlets passing through the cribriform plate to form the olfactory bulb. Olfactory information is processed by the olfactory tract via the olfactory bulb to the entorhinal cortex (primary sensory areas) and the orbitofrontal cortex (secondary sensory areas). Therefore, olfactory dysfunction after traumatic head injury may not only be a result of shearing of the olfactory nerve filaments, but also by damage to frontotemporal cortical structures. Experimental animal models and human histopathologic studies in mild traumatic brain injury patients have shown axonal damage, mainly located in the frontal lobes and olfactory bulbs.5 Impairment of olfactory stimuli processing leads to distortion of olfactory sensation known as “dysosmia.” Patients complaining of dysosmia have abnormal regeneration of injured olfactory pathways. Recent animal studies demonstrate that recovery of the olfactory system varies with the severity of injury and that dexamethasone treatment may have therapeutic value by reducing injury-associated edema.6
There are a variety of tests used for olfactory nerve dysfunction. These include the Brief Smell Identification Test, testing using Sniffin’ Sticks, and chemosensory-evoked potentials. Another widely available test is the 40-odor University of Pennsylvania Smell Identification Test (UPSIT), which has been administered to over 180,000 people in Europe and North America.7 Hypometabolism in the orbitofrontal cortex and the medial prefrontal cortex were found in positron emission tomography (PET) studies in patients with post-traumatic anosmia.8 In head injury patients with anosmia or hyposmia, coexistence of olfactory bulb and tract damage and frontal encephalomalacia on magnetic resonance imaging (MRI) has also been found.9
Treatment of traumatic olfactory nerve dysfunction is usually conservative. Approximately one third of patients have significant recovery with conservative management.10 Unlike the other cranial nerves, the olfactory nerve contains olfactory ensheathing cells rather than Schwann cells. The olfactory pathway continuously rebuilds itself throughout life. More invasive treatment, including removal of the olfactory bulbs and tracts, is reserved for patients with severe dysosmia. Case reports demonstrated patients with severe dysosmia causing significant anorexia.
Traumatic Optic Nerve Injury
Traumatic optic nerve injuries occur in 0.5% to 5% of head injuries.11 More recent surveys of craniofacial trauma data suggest an incidence of traumatic optic nerve injury in 2% to 5%.12
Optic nerve injury can be caused by primary and secondary mechanisms. Primary injury is by permanent axonal injury at the moment of impact from mechanical shearing, contusion, and ischemic necrosis of nerve axons. Secondary mechanisms are due to apoptosis, edema, and cell death, incorporating a variety of mechanisms leading to further axonal damage after the initial impact.13–16
Traumatic optic nerve injury can also be divided into direct and indirect injuries. Direct injuries occur with penetrating objects into the orbit causing direct damage the optic nerve. Indirect traumatic optic nerve injury is a closed injury resulting from the transmitted impact to the optic nerve.17
Direct traumatic optic nerve injuries typically have a worse prognosis in comparison to indirect injuries.18 Direct optic nerve injury anterior to the central retinal artery entry point disturbs the retinal circulation due to the associated vascular injury. Injuries posterior to the entry point maintain normal circulation. Partial or complete optic nerve avulsion can result in a partial or complete hemorrhagic ring at the optic nerve head.19–20 Computer modeling of direct optic nerve trauma has shown that the main site of stress is at the insertion of the nerve to the sclera and opposite to the side of impact.21 Nerve damage at this point leads to optic disc swelling.22 Rotation of the nerve at its insertion point to the sclera contributes the most damage in direct traumatic injury.21 A sudden increase in intraocular pressure is more likely to injure the optic nerve head.21 The intracranial segment of the optic nerve can also be injured against the falciform ligament.23
Indirect optic nerve injury can be intracanalicular, within the optic canal, or intracranial affecting the optic nerve or chiasm. Studies utilizing laser interferometry demonstrated forces applied to the frontal bone are transferred to the optic canal.24 Deceleration forces on the facial bones deform the sphenoid bone, with energy being transferred to the optic nerve.25,26 Indirect iatrogenic injury has been reported from heat transmission during electric cautery of the posterior ethmoidal artery during treatment of epistasis, and mechanical or heat transmission injuries during transnasal endoscopic surgery. Orbital hemorrhage can cause an intraorbital compartment syndrome, a potentially reversible cause of traumatic optic neuropathy.
Injury to the optic nerve can cause significant disability. Patients typically present initially with an afferent papillary defect and visual field defect. Significant visual loss is almost always accompanied by an afferent papillary defect.27 Diagnosis of traumatic optic nerve injury can be challenging and can be delayed weeks to months. This is attributed to multiple factors including severe craniofacial fractures, traumatic brain injury, and multi-system trauma requiring mechanical ventilation and sedation. Retinal examination is usually not helpful in diagnosing optic nerve injuries, as optic atrophy does not become apparent for 3 to 4 weeks.28 A retinal exam is useful in the diagnosis of retinal hemorrhages and other ocular trauma.
The initial clinical assessment should document the time interval between injury and the visual examination. Delayed visual loss was reported in 10% of patients included in the International Optic Nerve Trauma Study.29
Patients with direct disruption of the optic nerve will have minimal, if any, recovery. Indirect or secondary injury can be treated and prevented. Treatment includes observation, medical treatment with steroids and optic canal decompression or nerve sheath fenestration. Conservative management (i.e., observation) can achieve a 40% to 60% recovery rate.28 Prognostic factors have been studied. Initial visual acuity is the most influential indicator of good outcome.29 Poor prognostic factors include blood in the posterior ethmoidal cells, age over 40 years, loss of consciousness, and absence of recovery within 48 hours of steroid treatment.30 Blood in the posterior ethmoidal cells reflects the severity of injury, as the frontal bones transfer energy to the optic canal region.24
Steroid treatment is based on the National Acute Spinal Cord Injury studies. High-dose corticosteroids were translated from acute spinal cord injury to optic nerve injury. The International Optic Nerve Trauma Study (IONTS) revealed no clear benefit for either high-dose corticosteroid therapy or surgical decompression when compared to observation alone,31 reporting 54% improvement after 3 months with steroid therapy, and 57% improvement with observation. Rat models have not shown histologic evidence of either improved axonal regeneration or prevention of further degeneration.32 The IONTS study reported that mega-doses of steroids within the first few hours of injury most likely provides the maximum benefit out of all treatment modalities studied. Adverse effects of steroids should be considered in light of other injuries.30,31
Surgery should be considered with any obvious nerve compression or continual visual loss despite steroid therapy.28,33,34 Timing of surgery is controversial. Surgical decompression has similar outcomes up to 4 months after the injury in patients who are not completely blind.35
Evaluation of optic canal fractures can be visualized with high resolution computed tomography (CT) scan. Fracture of the optic canal potentially results in direct impingement on the nerve by bony fragments or associated hematoma and/or optic nerve edema (Fig. 204-1). Intrasheath hematoma should be treated with optic nerve fenestration. Perisheath hematoma can, however, be confused with an intrasheath hematoma. Optic nerve decompression has been performed by a variety of approaches, including transnasal trans-sphenoidal/transethmoidal, transpalpebral, and supraorbital transcranial. There is no statistical difference between steroids only or steroids plus surgical decompression.36
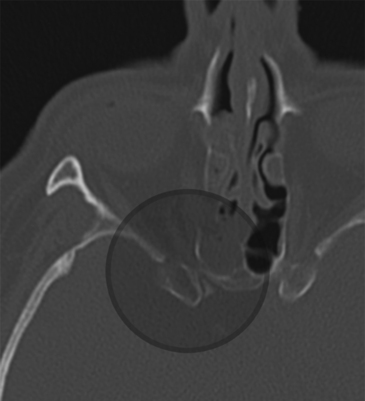
FIGURE 204-1 Axial computed tomography demonstrating optic canal fracture with compression of the canal by bony elements.
Endoscopic decompressive approaches have proven to maintain or improve vision in 50% of cases, and have shown to have fewer adverse outcomes.36 Endoscopic transnasal approaches are recently been favored due to the anatomic proximity of the optic canal to the sphenoid sinus, lack of external scars, preservation of olfaction, decreased morbidity, and faster recovery time. Endoscopic decompression can be considered earlier than conventional surgery, especially in patients with poor prognosis.36
Oculomotor Nerve
Oculomotor nerve palsy due to closed head injury is uncommon (5% to 15%).37–39 Injury to multiple ocular cranial nerves including oculomotor, trochlear, and abducens, can occur with post-traumatic, carotid-cavernous fistula. The oculomotor nerve exits the midbrain into the interpeduncular fossa after its rootlets traverse the red nucleus. The nerve separates into two divisions upon entering the orbit. The superior division innervates the superior rectus and levator palpebrae superioris. The inferior division innervates the medial and inferior recti and the inferior oblique muscles. Preganglionic parasympathetic innervation of the eye runs off of a branch of the nerve to the inferior oblique muscle. The complex anatomy of the oculomotor nerve results in a spectrum of presentations. In the midbrain, subnuclei within the oculomotor nuclei specifically innervate individual ocular muscles. The superior rectus muscle receives input from the contralateral subnuclei. Both levator palpebrae superioris muscles receive bilateral input.
Oculomotor nerve injury may result from distraction of the nerve, avulsion, and compression or displacement of the oculomotor nerve by traumatic intracerebral hematoma or hemorrhagic contusion.40,41 Oculomotor nerve injury can be associated with severe traumatic head injury.39 Ophthalmoplegia may result from downward displacement of the brain stem at the time of impact, and direct injury to the pupillomotor fibers on the ventromedial surface of the third nerve at the posterior petroclinoid ligament.42 Disturbance of the oculomotor nerve’s blood supply and biochemical changes from head injury can also contribute to the mechanism of injury.
Diagnosis of an oculomotor nerve injury can be challenging, especially in patients with orbital edema and/or ecchymosis. An adequate exam must be made, excluding any extraocular influences on ocular movement. Waiting for edema to resolve is usually required. Localization of oculomotor nerve injuries is based on the anatomic specifics of the nerve and nuclei. Involvement of the cerebral peduncle will not only present with an oculomotor nerve palsy, but also a contralateral hemiplegia and tremor. Involvement of the nuclei itself will present with not only ipsilateral oculomotor weakness (except superior rectus, which would be contralateral), but also bilateral ptosis and contralateral superior rectus weakness.
Oculomotor nerve injury is associated with a lower GCS when compared to other traumatic cranial neuropathies.43 Patients were involved mainly in motor vehicle accidents and had higher number of temporal lobe abnormalities (43%) on CT and MRI. Patients with bilateral traumatic oculomotor nerve palsies or unilateral oculomotor paresis were associated with severe injuries.3 The prognosis of traumatic oculomotor palsy is poor and full recovery is uncommon. A prolonged period (up to years) of healing is usually anticipated.44
Trochlear Nerve
The trochlear nerve is the smallest and longest of the ocular motor nerves. It runs at the free tentorial edge around the midbrain after decussating around the dorsal midbrain. Contracoup injury can occur when the nerve is compressed against the tentorium. Unilateral trochlear nerve injury can occur after frontolateral impact whereas bilateral trochlear nerve injury can occur from midfrontal impact.45 The presence of a Horner syndrome or an afferent pupillary defect should alert the clinician to a nuclear cause of the trochlear nerve palsy. The trochlear nucleus is in close proximity to the sympathetic fibers and the pupillomotor fibers. Trochlear nerve palsies can also be challenging to diagnosis in the comatose patient. There is large variation in the amount of head trauma that translates into trochlear nerve injury.4
Trigeminal Nerve
Trigeminal nerve injuries not only causes significant neurosensory deficits and facial pain, but can cause significant comorbidities due to changes in eating habits from muscular denervation of masticator muscles or altered sensation of the oral mucosa. Approximately 70% of patients with trigeminal nerve injuries complain about paresthesias.46 Ten to fifteen percent present with neuropathic pain confirmed with nerve block testing. The development of neuropathic pain is similar in susceptibility as peripheral nerves.47
Risk factors include direct maxillofacial trauma with facial bone fractures, injury secondary to local anesthetic injections, and during surgical intervention for facial trauma repair. There is an association between trigeminal nerve injury and facial bone fractures. Facial fractures affect the peripheral branches of the trigeminal nerve. With penetrating injuries including gunshot wounds, any portion of the trigeminal nerve can be affected. Traumatic trigeminal nerve injury associated with fractures of the upper third of the face and temporal bone is rare.48 Fractures extending from the temporal bone to the clivus can cause injury to the trigeminal root or ganglion. Forceful impact to the posterior third of the skull may crush the petrous apex against the dorsal sella leading to injury of the trigeminal ganglion. Facial fractures directly involving the trigeminal nerve and dislocated fractures have a higher incidence of traumatic trigeminal nerve injury than nondisplaced fractures. Traumatic trigeminal nerve injury was found in 88.2% of dislocated fractures, 54.4% of nondislocated fractures, and 100% of fractures with direct nerve involvement.49 Nondisplaced midfacial fractures had the highest incidence of trigeminal nerve impairment, but had the best prognosis out of all trigeminal nerve impairment associated with facial fractures.
Trigeminal nerve injuries are also a well known risk after oral and dental procedures, especially the inferior alveolar nerve during mandibular procedures.50 The second most common nerve injured in dental procedures is the lingual nerve. Injuries to the long buccal nerve, greater palantine, and nasopalantine are usually clinically insignificant. Tay and Zuniga reported a higher incidence of females presenting with trigeminal nerve injuries. Lower third molar surgery was the procedure most commonly associated with trigeminal nerve injury. Patients typically presented 3 to 9 months with subjective functional problems, but minimal signs of self-injury to the hypesthetic or hyperpathic oral mucosa.
Initial evaluation begins with the neurologic exam when there are suspicions of trigeminal nerve trauma. Patients with neurosensory dysfunction of the trigeminal nerve should be evaluated serially for up to 3 months.51 Patients with unacceptable recovery at 3 months should be offered exploration and/or repair of the nerve.
If trigeminal nerve trauma is due to a fracture, open reduction with decompression is indicated unless there are other contraindications. Closed reductions of fractures continue to show deformity of the nerve and attenuation of nerve at the site of the fracture despite gentle reduction of fractured elements.51 This is due to reactive osseous proliferation causing subsequent compression of the nerve. Decompression by enlarging surrounding bone and foramina prevents nerve compression secondary to post-traumatic nerve edema and ossification of surrounding bone. Nerve repair produces significant improvement or complete recovery in 86% of patients.51 Similar results are achieved when patients undergo surgical repair up to 6 to 9 months after the initial injury.
Abducens Nerve
Abducens nerve palsy is uncommon following traumatic brain injury, with an incidence of 1% to 2.7%.52 Postmortem examination in severe head trauma revealed abducens nerve injury at the dural entry point (Dorello’s canal), petrous apex and the lateral wall of the ICA.53 The petrous bone is the most common of the three causing stretching of the nerve. Other theories include neck hyperextension causing stretch of the nerve as it passes through Dorello’s canal. Isolated abducens nerve palsies without traumatic brain injury have been described in the literature.54,55 There was an association with fractures of the cervical vertebrae.
Abducens nerve injury has been reported after lumbar puncture.56 The mechanism is thought to be downward sagging of the brain after intracranial hypotension resulting in traction of the nerve. Patients usually recover from this.
The recovery rate after conservative management is high, 71%, with palsy or paresis.57 Predictors of nonrecovery include the failure to recover by 6 months from onset, complete paresis or bilateral involvement.58 Botulinum treatment does show significantly improved outcomes with 73% showing recovery when given within 3 months of injury.57
Facial Nerve
Facial nerve injury usually results from blunt or penetrating trauma to the petrous portion of the temporal bone. Approximately 5% of patients who suffered head injuries have temporal bone fractures.61 Facial nerve weakness can be partial or complete; it can manifest immediately or in a delayed fashion. Late presentation is secondary to pressure effect from hemorrhage, edema, or granulation tissue.
Fractures of the temporal bone are historically divided into two types based on the orientation of the fracture line to the long axis of the petrous temporal bone. Longitudinal fractures are more common and are usually from parietal impacts (Fig. 204-2). The fracture line is along the long axis of the petrous apex. These fractures spare the middle ear, but course through the external auditory canal to the foramen lacerum. Transverse fractures run from the foramen magnum through the otic capsule and then toward the foramen lacerum and are oriented perpendicular to the long axis of the petrous apex (Fig. 204-3). Longitudinal fractures comprise 72% of temporal bone fractures and transverse fractures comprise 20%.59 Mixed fractures containing elements of both fractures are also common.
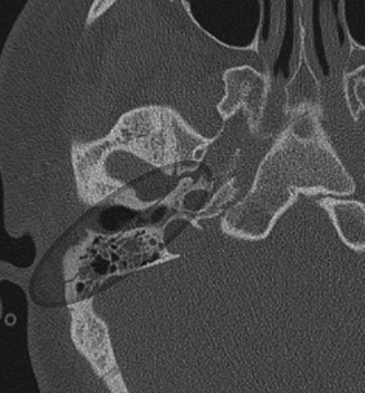
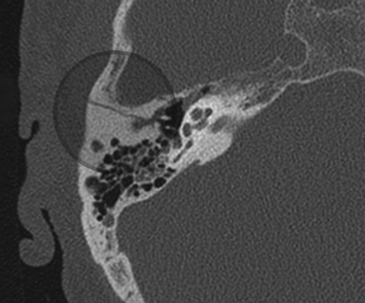
Facial paralysis occurs after transverse fractures of the temporal bone in 50% of cases. However, paralysis after longitudinal fractures occurs in 25% of cases. Traumatic facial nerve injuries result from transection, bony impingement on the nerve, intraneural hematoma, and edema. Chang and Cass reviewed 67 longitudinal fractures and revealed that 76% of fractures had bony impingement or intraneural hematoma, and 15% had nerve transection.60 The remainder had no visible pathology except nerve edema. Meanwhile, in 11 transverse fractures reviewed, 92% had nerve transection and 8% had bony impingement.
Evaluation can be challenging, especially in the acute trauma setting. Battle’s sign, cerebral spinal fluid otorrhea, air or fluid in the mastoid air cells, or fracture of the temporal bone should alert the clinician to further pursue any facial nerve injury. The House-Brackmann scale is used to grade the severity of facial paralysis. Fracture of the temporal bone is almost always associated with facial nerve injury.4
High-resolution CT with 1-mm axial and coronal cuts should be used to localize the site of injury. High-resolution, gradient echo sequences on magnetic resonance imaging have also been used to evaluate the facial nerve (Fig. 204-4). Audiologic testing, when possible, should also be performed not only for diagnostic purposes, but can also help to guide surgical approaches.
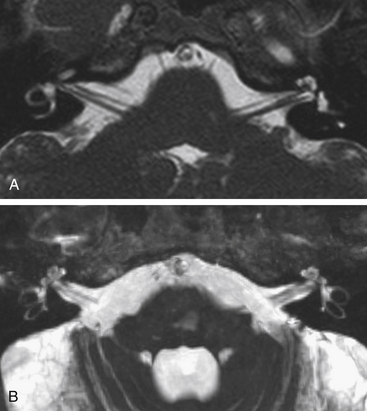
Sites of cerebral spinal fluid otorrhea usually close spontaneously within 1 week and are managed conservatively.61 After 1 week, surgery or spinal fluid diversion should be considered. The risk of meningitis increases up to 23% after 1 week.61
Various electrodiagnostic testing is utilized in determining the extent of injury. Electroneuronography (ENOG) determines the extent of degeneration via the amount of abnormal electrical activity. As long as muscle fibers are not degenerated, the test is useful.62 In the acute setting, ENOG is useful in the first 3 weeks of facial paralysis. Performed 3 to 4 days after paralysis, surgical intervention should be considered if there is less than 10% function diagnosed by ENOG.63 Wallerian degeneration takes 3 to 4 days to become apparent; the ENOG would not be helpful in the first several days in the evaluation of injury. Hindrances to the test include muscle artifact, skin impedance, poor electrode placement, and equipment variability. Factors determining good prognosis include early maximal denervation with consecutive testing (80% to 100%) and early voluntary activity within the first 2 to 3 weeks.
Cutaneous nerve testing using the Hilger nerve stimulator is inexpensive and easy to use. It uses visual output and hence, is limited by its subjectivity.64 The test can be painful. A 2- to 3.5-mA difference between sides of stimulation is considered significant.62
Electromyelography (EMG) measures the postsynaptic membrane potentials. Polyphasic potentials are a good prognostic finding. Fibrillations signify continued denervation. Nerve growth is approximately 1 mm/day. With this knowledge, 6-month reevaluation typically provides enough time for adequate evaluation of maximal nerve regrowth.62 After 15 days, Grosheva et al. report a positive predictive value and negative predictive value of 100% and 96% for EMG, respectively.65 Values before that are lower, as it takes up that amount of time for lesions to be detectable.
Patients with paresis can be managed with observation. Medical therapy is in the form of steroids. Patients presenting House-Brackmann grade V facial function should be evaluated with EMG testing if there is concern that the physical exam is confounded by muscles not innervated by the facial nerve.62
Chen and Arriaga62 have developed treatment algorithms regarding facial paralysis associated with closed head injury utilizing CT evaluation of nerve and surrounding bony structures and tissue and electrical testing. Continued observation is suggested at 3 weeks from injury in patients without CT evidence of more than 1-mm diastasis along the facial nerve or bony spicule in the nerve itself. Observation is indicated for those with negative electrical testing. Criteria for positive electrical testing is less than 10% residual function on ENOG. Surgery is recommended for those with positive CT findings and positive electrical testing. At 6 months, patients with evidence of recovering facial nerve function are encouraged to continue observation.
Patients presenting with paralysis with penetrating temporal bone trauma are managed in a similar fashion. An important consideration, however, involves the associated heat and force transmitted with penetrating missile injury. Because of associated heat injury, those without CT findings and positive electrical testing are also offered surgery.62 In fact, those with positive electrical testing, regardless of CT findings are offered surgery in patients with penetrating trauma.
Radiographic findings are important to guide surgical approaches to repair traumatic facial nerve injuries.62,66 Most of the traumatic facial nerve injuries are in proximity to the geniculate ganglion, proximal tympanic segments, or labyrinthine segments.67 Surgical exploration of the facial nerve after a temporal bone fracture is initially performed through a transmastoid approach. The facial nerve is decompressed from the proximal tympanic segment to the distal vertical segment (mastoid). Exploration of the geniculate ganglion, labyrinthine and canalicular segments can be performed via middle cranial fossa craniotomy and exposure of the middle cranial fossa floor. If the patient suffers total sensorineural hearing loss, a translabyrinthine approach is preferred to avoid temporal lobe retraction.
If the facial nerve is anatomically intact, it has to be widely decompressed around the fracture site and the nerve sheath should be opened. Partial transection of the facial nerve poses a decisional dilemma regarding the use of an interpositional graft. Most surgeons agree if more than 50% of the nerve is transected, it should be grafted. However, obtaining an accurate estimate of transection can be difficult due to challenging anatomic visualization and post-traumatic granulation tissue. Large diastasis of the facial nerve requires an interpositional graft. The greater auricular nerve is an excellent source of grafting material, with the sural nerve as an alternative.
Surgical exploration of patients with traumatic facial nerve injuries remains controversial. Nonpenetrating trauma to the facial nerve has an overall outcome of 50% improvement to House-Brackmann grade 1 or 2 in patients who undergo decompression.68 However, there is data suggesting a similar percentage of facial nerve recovery in nonoperative conservative management. Darrouzet et al. showed 94% recovery to House-Brackmann grades 1 to 3 after facial nerve decompression in patients without nerve transection in a 2-year follow-up.69 Complete facial nerve transection caries an unfavorable prognosis, with the highest recovery being House-Brackmann grade 3.69
Vestibulocochclear Nerve
Traumatic injury to the vestibulocochlear nerve is common after transverse fractures of the temporal bone.70 Ninety-six percent of patients suffering from traumatic conductive hearing loss have an associated temporal bone fracture.71 Conductive hearing loss is due to bleeding into the middle ear, ossicular injury and, rarely, tympanic membrane perforation. Fractures to the otic capsule can lead to complete degeneration of the cochlea and vestibular organs. Patients suffering from vestibulocochlear dysfunction without damage to the cochlea, vestibule, or temporal bone have petechial hemorrhage in the brain stem from the shearing effect of trauma.72
The literature reports the incidence of hearing loss in traumatic brain injury ranges from 7% to 50%.73 It is very rare for hearing loss after traumatic injury to be localized to the cortex in conscious individuals. Injury would have to be severe and bilateral; patients are usually unconscious with such a severe injury complex. Bilateral midbrain injury in awake and alert patients can cause deafness, but isolated lesions are very rare.74
Patients suffering from a cochlear concussion have an injury to the cochlea itself. Mechanisms of injury include disturbance of cochlear microcirculation, hemorrhage into the fluid spaces of the cochlea, or disruption of the cochlea’s membranous portion by pressure waves transmitted from increases in intracranial pressure.73 Damage to the cochlea is from damage to hair cells, traumatic Ménière’s syndrome, or perilymph fistula.73 Hair cell damage contributing to cochlear concussion is greatest in the 4000- to 8000-Hz area of the cochlea. Hearing loss is immediate after injury and gradually recovers over 6 months. Evoked otoacoustic emissions can distinguish between cochlear pathology and retrocochlear pathology.75 These emissions are produced in the outer hair cells of the cochlea by contractile elements. This test allows for indirect measurement of the status of the cochlea via these outer hair cells. In contrast to patients with hair cell damage, patients presenting with fluctuating or delayed onset of symptoms may be suffering from traumatic Ménière’s syndrome. Hearing loss, however, is less frequent than vestibular symptoms. An endolymphatic shunt is effective in 50% of patients for up to 5 years.73 Patients with severe, traumatic sensorineural hearing loss can be treated with cochlear implantation and intense rehabilitation.
Patients with perilymph fistula typically present with fluctuating/progressive sensorineural hearing loss and vertigo. Diagnosis can be challenging as patients presenting with traumatic perforation with or without fistula development have similar symptoms.76 Two mechanisms cause perilymph fistulas. The explosive fistula is caused by increased cerebrospinal fluid (CSF) pressure transmitted to the perilymph resulting in rupture of the round window. Implosive fistulas are a result of pressure through the tympanic membrane. Patients are managed conservatively. Exploratory tympanotomy is indicated with persistence of symptoms. The goal of surgical treatment is prevention of further hearing loss. Surgical treatment improves symptoms in 40% of patients.77
Hearing loss from blast-related injuries is increasing in incidence.78 Blast injuries are caused by explosives that transmit shock waves. These shock waves are distinct in that they can affect the middle and inner ear because they affect both gas- and fluid-filled structures. This can lead to increased sensorineural hearing loss. Lew et al.78 reported an increased incidence of hearing loss and tinnitus in U.S. veterans suffering from blast-related traumatic brain injury. Sixty-two percent of U.S. veterans suffering from blast-related traumatic brain injury complained of hearing loss and 38% complained of tinnitus. This is significantly higher than the 44% and 18% complaining of hearing loss and tinnitus, respectively, in patients who suffered non–blast-related traumatic brain injury. Pure sensorineural hearing loss was diagnosed in 58% of the blast-related traumatic brain injuries compared to 47% in non–blast-related traumatic brain injuries. Only 20% of patients suffering from clinically significant, refractory tinnitus were treated with cognitive behavioral therapy. There is a potentially high incidence of other neurologic injuries in blast-injury patients who suffer tympanic membrane perforations.79
Vertigo and ataxia are common clinical presentations after traumatic brain injury. Complaints of vertigo with rapid head movement are common. Most of these patients do not have CT scan evidence of injury.85 For patients with chronic vertigo after traumatic brain injury with negative neuroimaging evidence for dysfunction, there is little information that provides predictive value for outcomes.80 Balance testing does not predict return of functional status. Symptoms of vestibular dysfunction are due to complete or partial unilateral interruption of the vestibular information with attempted compensation by the central nervous system using information from the contralateral healthy side. Benign paroxysmal positional vertigo is a frequent diagnosis after trauma.73 Patients experience severe vertigo when rolling in one particular direction or dizziness with quick head motion. Post-traumatic, benign, paroxysmal positional vertigo is usually self-limiting.
Patients suffering from vertigo can also have a labyrinthine concussion, injury to the labyrinthine end organ. Patients present with severe, debilitating vertigo and unsteadiness which worsens in darkness and with fatigue. These patients typically have vigorous horizontal nystagmus.85 Direct microvascular injury to the vestibule has been demonstrated by clots in the vestibular microcirculation in animal models.81,82 Local hypoxia and fibrosis with reactive ossification has also been shown to play a role.83,84 Patients with traumatic injury to the labyrinthine end organ or to central vestibular nuclei present in a similar fashion.85 Localization to either the vestibule or brain stem is by demonstration of labyrinthine end organ pathology by magnetic resonance imaging showing hemorrhage in a semicircular canal. Labyrinthine concussion is usually self-limited.85 Treatment is conservative during the acute denervation phase. Once the acute denervation phase has resolved, physical therapy assists the patient in compensating for the loss.
Patients suffering from peripheral vestibular end-organ damage are offered labyrinthectomy (hearing sacrificing) or selective vestibular nerve section (hearing preserving) if conservative treatment has failed. Labyrinthectomy and selective vestibular nerve section disconnect the injured organ’s vestibular input from the central nervous system with the hopes that the central nervous system will compensate over time by utilizing other senses, including vision and proprioception. These are offered 6 to 8 months from injury to allow for maximal recovery with conservative treatment. Patient selection is important when deciding to offer labyrinthectomy or vestibular nerve section.73 Central adaptation to unilateral vestibular information depends on a healthy vestibular apparatus on the nonoperative side and central nervous system plasticity as patients older than 60 years may not adapt appropriately. Surgical candidacy also depends on the ability of the central nervous system to rely more on visual and proprioceptive input after removal of unilateral vestibular input. Hence, poor surgical candidates would be patients aged over 60 years and patients with diabetic neuropathy and retinopathy. Labyrinthectomy and selective vestibular nerve section are not offered to patients with injury to the brain stem, as these lesions are too proximal for labyrinthectomy and selective vestibular nerve section to be helpful. Patients start to show recovery within 6 weeks after surgery.73
Vagus Nerve
The recurrent laryngeal nerve is at risk for injury during thyroid surgery. The incidence of recurrent laryngeal nerve injury used to be 21%.86 Modern surgical techniques allowing routine identification of the recurrent laryngeal nerve and intraoperative neuromonitoring reduced the temporary injury rate to 6% and permanent injury rate to 1%.87 Causes of injury include transection, stretching or ischemia by compression from ligature entrapment or clamping, and electrothermal injury.87 The duration of injury has a mean of approximately 30 days, but can range from 3 days to 4 months. Recovery from vocal cord palsy diminishes after 2 months.
Injury to the vagus nerve during posterior atrial ablation for atrial fibrillation is rare; only case reports have been produced. Patients can present with gastroparesis and pancreatic dysfunction.88 Injury is usually noted early after the ablation, but diagnosis can take up to 1 year from the ablation.89 Diagnostic tests include gastric emptying studies, pancreatic polypeptide secretion with sham feeding, or endoscopic studies. Treatment is primarily symptomatic. Patients can be instructed to restrict oral intake to smaller meals and eliminate foods containing fiber, and use gastric motility agents like metoclopramide. Botulinum toxin can be used for severe cases of pyloric spasm. Early diagnosis is key in preventing chronic complications from gastrointestinal symptoms, including excessive weight loss and metabolic deficiencies. Fifty-seven percent of patients have favorable outcomes.88
Spinal Accessory Nerve
Injury to the spinal accessory nerve not only results in debilitating trapezius dysfunction, but can also result in chronic pain and debilitation with prolonged shoulder girdle dysfunction.90 It usually presents after diagnostic lymph node biopsies of the posterior triangle of the neck, with an incidence of 3% to 8%.91 Spinal accessory nerve injury can also result from minor trauma, including wrestling and whiplash. Stretch or traction compromises the microvascular network inside the nerve, which leads to ischemia and axonal degeneration. Chandawarkar et al. question the diagnostic utility of such biopsies, as most report a benign pathology. Although microsurgical repair of the nerve has excellent results, patients with significant deficit do not benefit because of delays in diagnosis.90
Patients present initially not only with depressed motor function, but also heaviness of the arm and pain over the muscle.92 Patients with chronic presentations exhibit muscle atrophy, paresthesias, and symptoms of frozen shoulder (e.g., adhesive capsulitis and sternoclavicular joint hypertrophy). Diagnosis can be confounded by presence of trapezius muscle innervation by the cervical plexus.93
Treatment groups are divided into observation and surgical exploration.94 Surgical candidates are those who do not improve clinically, have severe symptoms, known transection, and show denervation indicated by electromyographic studies. Patients who present with very poor trapezius function will most likely need operative exploration. High-resolution ultrasonography allows the clinician to see changes in the accessory nerve, atrophic changes in the surrounding musculature, granuloma, or scar formation. Transection, however, is not visible on high-resolution ultrasound.95
Surgical exploration includes primary repair of diastatic nerve endings that are not under tension, interpositional nerve graft using the sural or great auricular nerve for nerve endings under tension, or resection of injured nerve segments with subsequent interpositional nerve graft. Neuromas are also excised at the time of exploration and subsequent repair is made primarily or using grafts. Patients unable to undergo nerve reconstruction or repair can undergo muscle transposition to return muscular function of the shoulder joint. Surgical exploration is most effective if done within 3 months of injury.90
Response to surgical treatment must be monitored on a frequent basis with serial examinations and/or electromyographic testing. Recovery of function is noticeable at 6 weeks postsurgery. Aggressive physical therapy is important after surgery to maximize potential functional improvement and to limit further fibrosis of the shoulder capsule. Preoperative EMG studies can be used to guide physical therapy and to monitor trapezius function.96 Chandawarkar et al.90 report complete resolution of shoulder pain. Five out of the six study patients had complete recovery of the trapezius muscle, with the remaining patient attaining partial recovery. The average time to maximal recovery was 6 months, with a range of 4 to 12 months. Early surgical exploration yields better results, especially in those with severe accessory nerve dysfunction.
Vernet’s Syndrome: Glossopharyngeal, Vagus, and Spinal Accessory Nerve
Lower cranial nerves IX, X, and XI can be injured at the jugular foramen. Figure 204-5 demonstrates fracture of the jugular foramen. Vernet’s syndrome, also known as the syndrome of the jugular foramen, typically presents with jugular foramen tumors.
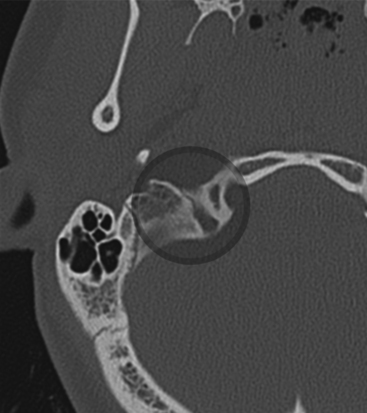
Case reports of Vernet’s syndrome have been associated with traumatic injury to the jugular foramen.97 Patients suffer from fractures of the jugular foramen, which can cause hematoma, and/or thrombosis of the inferior petrosal and sigmoid sinus. The trio of presenting symptoms includes loss of taste and sensation in the posterior third of the tongue, vocal cord paralysis, decreases sensation of the pharynx and larynx and weakness of the trapezius muscles and sternocleidomastoid.
Glossopharyngeal Nerve
Glossopharyngeal nerve injury has been reported in cases of occipital condyle fractures.98 Injury of the glossopharyngeal nerve can be isolated or in combination with the other lower cranial nerves. The anatomic relationship of the occipital condyle to the jugular foramen explains the logical anatomy of the injury. This is due to injury to the nerve by fractured bone or compression by associated hematoma or tissue edema.
Hypoglossal Nerve
Injury to the hypoglossal nerve is typically associated with occipital condyle fracture. Despite the anatomic proximity of the occipital condyle to the brain stem and cerebellum, most patients present with isolated lower cranial injuries.98,99 The incidence of brain stem involvement may be higher as some patients do not present to emergency departments because of a higher fatality rate. There are case reports in the literature reporting minor head trauma causing unilateral traumatic hypoglossal palsy without fracture.100 Patients with tongue deviation, dysphagia, or dysarthria should be evaluated for hypoglossal nerve injury.
Collet-Sicard Syndrome: Glossopharyngeal, Vagus, Spinal Accessory, and Hypoglossal Nerve
Originally described in the traumatic setting in the early 1900s, this is similar to Vernet’s syndrome, but includes the hypoglossal nerve. Occipital condyle fracture is typically associated with this syndrome.101,102 High-resolution, 1-mm axial and coronal CT imaging typically shows the injury. Gunshot wounds to the craniocervical junction have also been described103 in causing a Collet-Sicard syndrome. Metastastic disease to the occipital condyle region can be associated with this syndrome.104,105
Neck Surgery and Lower Cranial Nerve Injuries
Swallowing can be divided into three phases: oral, pharyngeal, and esophageal.106 The oral phase is controlled by the hypoglossal nerve. Muscles of mastication and the tongue manipulate the food into a broken-down bolus. The facial and glossopharyngeal nerves also are involved in this part. The pharyngeal phase is controlled by the superior laryngeal and the recurrent laryngeal nerve. The broken-down bolus is passed into the pharynx. During this phase, respiration ceases with the closure of the true and false vocal cords. At the same time, the larynx elevates and the epiglottis inverts. This prevents the food bolus from entering the airway. The esophageal phase is controlled by the vagus nerve. The myenteric plexus of Auerbach controls the coordination of peristalsis, with the longitudinal and circular muscles moving the food bolus in coordination through the lower esophageal sphincter. Videofluoroscopic studies evaluating dysphagia after anterior cervical spine surgery demonstrated a variety of dysphagia patterns involving any of the three stages.107 Lower cranial nerves can be vulnerable to injury according to the level of surgery. Surgery at the C3 level or above can affect the glossopharyngeal and hypoglossal nerves. The superior laryngeal nerve can be injured at the C3–C4 level. Surgery at the C6 level or below can affect the recurrent laryngeal nerve. The vagus nerve is anatomically located within the carotid sheath and can be injured during retraction at any level.
The hypoglossal nerve can be injured during exposure for carotid endarterectomy. Recent literature showed an average incidence of 9% injury of the hypoglossal, vagus, or facial nerve.108
Albert T.J., Lee J., Lim M. Cervical Spine Surgery Challenges: Diagnosis and Management. Stuttgart: Thieme; 2008.
Bagheri S.C., Meyer R.A., Kahn H.A., Steed M.B. Microsurgical repair of peripheral trigeminal nerve injuries. J Oral Maxillofac Surg. 2009;67:1791-1799.
Brodie H.A., Thompson T.C. Management of complications from 820 temporal bone fractures. Am J Otol. 1997;18:188-197.
Carta A., Ferrigno L., Salvo M., et al. Visual prognosis after indirect traumatic optic neuropathy. J Neurol Neurosurg Psychiatry. 2003;74:246-248.
Chandawarkar R.Y., Cervino A.L., Pennington G.A. Management of iatrogenic injury to the spinal accessory nerve. Plast Reconstr Surg. 2003;111:611-617.
Chang C.Y., Cass S.P. Management of facial nerve injury due to temporal bone trauma. Am J Otol. 1999;20(1):96-114.
Chen D.A., Arriaga M.A. Acute facial paralysis. In: Pensak M.L., editor. Controversies in Otolaryngology. New York: Thieme; 2001:227-231.
Chung S.M., Fenton G.A., Schmidt J.G., Selhorst J.B. Trauma to the cranial nerves and brainstem. In: Narayan R.K., Wilberger J.E., Povlishock J.T. Neurotrauma. New York: McGraw-Hill, Health Professions Division; 1996:621-637.
Cirovic S., Bhola R.M., Hose D.R., et al. Computer modelling study of the mechanism of optic nerve injury in blunt trauma. Br J Ophthalmol. 2006;90:778-783.
Darrouzet V., Duclos J.Y., Liguoro D., et al. Management of facial paralysis resulting from temporal bone fractures: our experience in 115 cases. Otolaryngol Head Neck Surg. 2001;125(1):77-84.
Fisch U. Prognostic value of electrical tests in acute facial paralysis. Am J Otol. 1984;5(6):494-498.
Fitzgerald D.C. Head Trauma: hearing loss and dizziness. J Trauma Inj Infect Crit Care. 1996;40(3):488-496.
Haxel B.R., Grant L., Mackay-Sim A. Olfactory dysfunction after head injury. J Head Trauma Rehabil. 2008;23:207-413.
Holmes J.M., Beck R.W., Kip K.E., et al. Predictors of nonrecovery in acute traumatic sixth nerve palse and paresis. Ophthalmology. 2001;108:1457-1460.
Kline L.B., Morawetz R.B., Swaid S.N. Indirect injury of the optic nerve. Neurosurgery. 1984;14:756-764.
Lagreze W.A. Neuro-ophthalmology of trauma. Curr Opin Ophthalmol. 1998;9:33-39.
Lew H.L., Jerger J.F., Guillory S.B., Henry J.A. Auditory dysfunction in traumatic brain injury. J Rehab Res Dev. 2007;44(7):921-928.
Li H., Zhou B., Shi J., et al. Treatment of traumatic optic neuropathy: our experience of endoscopic optic nerve decompression. J Laryngol Otol. 2008;122:1325-1329.
May M. Total Facial nerve exploration: transmastoid, extralabyrinthine, and subtemporal indications and results. Laryngoscope. 1979;89:906-917.
Sharma B.S., Mahajan R.K., Bhatia S., Khosla V.K. Collet-Sicard syndrome after closed head injury. Clin Neurol Neurosurg. 1994;96:197-198.
Tay A.B.G., Zuniga J.R. Clinical characteristics of trigeminal nerve injury referrals to a university centre. Int J Oral Maxilofac Surg. 2007;36:922-927.
Wang B.H., Robertson B.C., Girotto J.A., et al. Traumatic optic neuropathy: a review of 61 patients. Plast Reconstr Surg. 2001;107:1655-1664.
Yang W.G., Chen C.T., Tsay P.K., et al. Outcome for traumatic optic neuropathy—surgical versus nonsurgical treatment. Ann Plast Surg. 2004;52:36-42.
Yu-Wai-Man P., Griffiths P.G. Steroids for traumatic optic neuropathy. Cochrane Database Syst Rev. 2007;4:CD006032.
1. Keane J.R., Baloh R.W. Post-traumatic cranial neuropathies. In: Evans R.W., editor. The Neurology of Trauma. Philadelphia: Saunders; 1992:849-868.
2. Haxel B.R., Grant L., Mackay-Sim A. Olfactory dysfunction after head injury. J Head Trauma Rehab. 2008;23:407-413.
3. Patel P., Kalyanaraman S., Reginald J., et al. Post-traumatic cranial nerve injury. Indian J Neurotrauma. 2005;2:27-32.
4. Chung S.M., Fenton G.A., Schmidt J.G., Selhorst J.B. Trauma to the cranial nerves and brainstem. In: Narayan R.K., Wilberger J.E., Povlishock J.T. Neurotrauma. New York: McGraw-Hill, Health Professions Division; 1996:621-637.
5. Kern R., Quinn B., Rosseau G., et al. Post-traumatic olfactory dysfunction. Laryngoscope. 2000;110:2106-2109.
6. Kobayashi M., Costanzo R.M. Chem senses. 2009;34(7):573-580.
7. Doty R. Olfaction. Ann Rev Psychol. 2001;54:423-452.
8. Varney N., Pinkston J., Wu J. Quantitative PET findings in patients with post-traumatic anosmia. J Head Trauma Rehab. 2001;16:253-259.
9. Yousem D., Geckle R., Bilker W., et al. Posttraumatic smell loss: relationship of psychophysical tests and volumes of the olfactory bulbs and tracts and the temporal lobes. Acad Radiol. 1999;6:264-272.
10. Kern R.C., Quinn B., Rosseau G., Farbman A.I. Post-traumatic olfactory dysfunction. Laryngoscope. 2000;110:2106-2109.
11. Steinsapir K.D., Goldberg R.A. Traumatic optic neuropathy. Surv Ophthalmol. 1994;38(6):487-518.
12. Al-Qurainy A., Stassen L.F.A., Dutton G.N., et al. The characteristics of midfacial fractures and the association with ocular injury: a prospective study. Br J Oral Maxillofac Surg. 1991;29:291-301.
13. Walsh F.B., Hoyt W.F., 3rd ed, Clinical Neuro-Ophthalmology, Vol. 3;Baltimore, Williams & Wilkins, 1969
14. Anderson D.R. Ascending and descending optic atrophy produced experimentally in squirrel monkeys. Am J Ophthalmol. 1973;76:693-711.
15. Klöcker N., Zerfowski M., Gellrich N.C., Bähr M. Morphological and functional analysis of an incomplete CNS fiber tract lesion: graded crush of the rat optic nerve. J Neurosci Methods. 2001;110:147-153.
16. Vorwerk C.K., Zurakowski D., McDermott L.M., et al. Effects of axonal injury on ganglion cell survival and glutamate homeostasis. Brain Res Bull. 2004;62:485-490.
17. Kline L.B., Morawetz R.B., Swaid S.N. Indirect injury of the optic nerve. Neurosurgery. 1984;14:756-764.
18. Wang B.H., Robertson B.C., Girotto J.A., et al. Traumatic optic neuropathy: a review of 61 patients. Plast Reconstr Surg. 2001;107:1655-1664.
19. Hedges T.R., Gragoudas E.S. Traumatic anterior ischemic optic neuropathy. Ann Ophthalmol. 1981;13:625-628.
20. Park J.H., Frenkel M., Dobbie J.G., Choromokos E. Avulsion of the optic nerve. Am J Ophthalmol. 1971;72:969-971.
21. Cirovic S., Bhola R.M., Hose D.R., et al. Computer modelling study of the mechanism of optic nerve injury in blunt trauma. Br J Ophthalmol. 2006;90:778-783.
22. Lagreze W.A. Neuro-ophthalmology of trauma. Curr Opin Ophthalmol. 1998;9:33-39.
23. Crompton M.R. Visual lesions in closed head injury. Brain. 1970;93:785-792.
24. Gross C.E., DeKock J.R., Panje W.R., et al. Evidence for orbital deformation that may contribute to monocular blindness following minor frontal head trauma. J Neurosurg. 1981;55:963-966.
26. Spetzler R.F., Spetzler H. Holographic interferometry applied to the study of the human skull. J Neurosurg. 1980;52:825-828.
27. Mavrakanas N.A., Schutz J.S. Feigned visual loss misdiagnosed as occult traumatic optic neuropathy: diagnostic guidelines and medical-legal issues. Surv Ophthalmol. 2009;54:412-416.
28. Yu-Wai-Man P., Griffiths P.G. Steroids for traumatic optic neuropathy. Cochrane Database Syst Rev.. 2007;4:CD006032.
29. Yang W.G., Chen C.T., Tsay P.K., et al. Outcome for traumatic optic neuropathy—surgical versus nonsurgical treatment. Ann Plast Surg. 2004;52:36-42.
30. Carta A., Ferrigno L., Salvo M., et al. Visual prognosis after indirect traumatic optic neuropathy. J Neurol Neurosurg Psychiatry. 2003;74:246-248.
31. Levin L.A., Beck R.W., Joseph M.P., et al. The treatment of traumatic optic neuropathy: the international optic nerve trauma study. Ophthalmology. 1999;106:1268-1277.
32. Ohlsson M., Westerlund U.l.f., Langmoen I.A., Svensson M. Methylprednisolone treatment does not influence axonal regeneration or degeneration following optic nerve injury in the adult rat. J Neuroophthalmol. 2004;24:11-18.
33. Wang B.H., Robertson B.C., Girotto J.A., et al. Traumatic optic neuropathy: a review of 61 patients. Plast Reconstr Surg. 2001;107:1655-1664.
34. Rajiniganth M.G., Gupta A.K., Gupta A., Bapuraj J.R. Traumatic optic neuropathy: visual outcome following combined therapy protocol. Arch Otolaryngol Head Neck Surg. 2003;129:203-206.
35. Thakar A., Mahapatra A.K., Tandon D.A. Delayed optic nerve decompression for indirect optic nerve injury. Laryngoscope. 2003;113:112-119.
36. Li H., Zhou B., Shi J., et al. Treatment of traumatic optic neuropathy: our experience of endoscopic optic nerve decompression. J Laryngol Otol. 2008;122:1325-1329.
37. Memon M.Y., Paine K.W.E. Direct injury of the oculomotor nerve in craniocerebral trauma. Neurosurgery. 35, 1971. 464–464
38. Eyster E.F., Hoyt W.F., Wilson C.B. Oculomotor palsy from minor head trauma. JAMA. 1972;220:1083-1086.
39. Elston J.S. Traumatic third nerve palsy. Br J Ophthalmol. 1984;68:538-543.
40. Muthu P., Pritty P. Mild head injury with isolated third nerve palsy. Emerg Med J. 2001;18:310-311.
41. Chen C.C., Pai Y.M., Wang R.F., et al. Isolated Oculomotor nerve palsy from minor head trauma. Br J Sports Med. 2005;39:e34.
42. Nagaseki Y., Shimizu T., Kakizawa T., et al. Primary internal ophthalmoplegia due to head injury. Acta Neurochir. 1989;97:117-122.
43. Dhaliwal A., West A.L., Trobe J.D., Musch D.C. Third, fourth, and sixth cranial nerve palsies following closed head injury. J Neuro-Ophthalmol. 2006;26(1):4-10.
44. Heinz J. Cranial nerve avulsion and other neural injuries. Med J Aust. 1969;2:1246-1249.
45. Buter L.J., Kalvin N.H., Smith J.L. Acquired lesions of the fourth cranial nerve. Brain. 1970;93:567-574.
46. Sandstedt P., Sorensen S. Neurosensory disturbances of the trigeminal nerve: a long-term follow-up of traumatic injuries. J Oral Maxillofac Surg. 1995;53(5):498-505.
47. Jääskeläinen S.K., Teerijoki-Oksa T., Virtanen A., et al. Sensory regeneration following intraoperatively verified trigeminal nerve injury. Neurology. 2004;62:1951-1957.
48. Ghorayeb B.Y., Yeakley J.W., Hall J.W., Jones B.E. Unusual complications of temporal bone fractures. Arch Otolaryngol Head Neck Surg. 1987;113:749-753.
49. Renzi G., Carboni A., Perugini M., et al. Posttraumatic trigeminal nerve impairment: a prospective analysis of recovery patterns in a series of 103 consecutive facial fractures. J Oral Maxillofac Surg. 2004;62:1341-1346.
50. Tay A.B.G., Zuniga J.R. Clinical characteristics of trigeminal nerve injury referrals to a university centre. Int J Oral Maxillofac Surg. 2007;36:922-927.
51. Bagheri S.C., Meyer R.A., Kahn H.A., Steed M.B. Microsurgical repair of peripheral trigeminal nerve injuries. J Oral Maxillofac Surg. 2009;67:1791-1799.
52. Turner J.W.A. Indirect Injuries of the optic nerve. Brain. 1943;66:140-151.
53. Sama B., Ozverenb M.F., Akdemirb I., et al. The mechanism of injury of the abducens nerve in severe head trauma: a postmortem study. Forensic Sci Int. 2004;140:25-32.
54. Lindenberg R., Freytag E. Brainstem lesions characteristic of traumatic hyperextension of the head. Arch Pathol. 1970;90:509-515.
55. Rosa L., Carol M., Bellegarrique R., et al. Multiple cranial nerve palsies due to a hyperextension injury to the cervical spine. Case report. J Neurosurg. 1984;61:172-173.
56. Niedermuller U., Trinka E., Bauer G. Abducens palsy after lumbar puncture. Clin Neurol Neurosurg. 2002;104:61-63.
57. Holmes J.M., Beck R.W., Kip K.E., Droste P.J., Leske D.A., et al. Botulinum toxin treatment versus conservative management in acute traumatic sixth nerve palsy or paresis. J AAPOS. 2000;4:145-149.
58. Holmes J.M., Beck R.W., Kip K.E., et al. Predictors of nonrecovery in acute traumatic sixth nerve palse and paresis. Ophthalmology. 2001;108:1457-1460.
59. Nosan D.K., Benecke J.E.Jr., Murr A.H. Current perspective on temporal bone trauma. Otolaryngol Head Neck Surg. 1997;117(1):67-71.
60. Chang C.Y., Cass S.P. Management of facial nerve injury due to temporal bone trauma. Am J Otol. 1999;20:96-114.
61. Brodie H.A., Thompson T.C. Management of complications from 820 temporal bone fractures. Am J Otol. 1997;18:188-197.
62. Chen D.A., Arriaga M.A. Acute facial paralysis. In: Pensak M.L., editor. Controversies in Otolaryngology. New York: Thieme; 2001:227-231.
63. Fisch U. Prognostic Value of electrical tests in acute facial paralysis. Am J Otol. 1984;5:494-498.
64. Lewis B.I., Adour K.K., Kahn J.M., et alLewis A.J. Hilger Facial nerve stimulator: a 25-year update. Laryngoscope. 1991;101:71-74.
65. Grosheva M., Wittekindt C., GuntinasLichius O., et al. Electroneurography and electromyography in facial palsy. Laryngoscope. 2008;118:394-397.
66. May M. Total facial nerve exploration: transmastoid, extralabyrinthine, and subtemporal indications and results. Laryngoscope. 1979;89:906-917.
67. Green D.J., Shelton C., Brackmann D.E. Iatrogenic facial nerve injury during otologic surgery. Otololaryngol Head Neck Surg. 1986;96:47-55.
68. McKennan K.X., Chole R.A. Facial paralysis in temporal bone trauma. Am J Otol. 1992;13(2):167-172.
69. Darrouzet V., Duclos J.Y., Liguoro D., et al. Management of facial paralysis resulting from temporal bone fractures: our experience in 115 cases. Otolaryngol Head Neck Surg. 2001;125:77-84.
70. Katzen J.T., Jarrahy R., Eby J.B., et al. Craniofacial and skull base trauma. J Trauma. 2003;54(5):1026-1034.
71. Griffiths M.V. The Incidence of auditory and vestibular concussion following minor head injury. J Laryngol Otol. 1979;93:253.
72. Strick S.J. Shearing of nerve fibers as a cause of brain damage due to head injury. Lancet. 1961;11:443.
73. Fitzgerald D.C. Head trauma: hearing loss and dizziness. J Trauma Inj Infect Crit Care. 1996;40(3):488-496.
74. Jane N.N., Laureno R., Mark A.S., et al. Deafness after bilateral midbrain contusion: a correlation of magnetic resonance imaging with auditory brain stem evoked responses. Neurosurgery. 1991;29:106.
75. Kemp D.T. Stimulated acoustic emissions from within the human auditory system. J Acoust Soc Am. 1978;64:1386.
76. Emmett J.R., Shea J.J. Traumatic perilymph fistula. Laryngoscope. 1980;90:1513-1520.
77. Black F.O., Pesznecker S., Norton T., et al. Surgical management of perilymphatic fistulas: a portland experience. Am J Otol. 1992;13:254.
78. Lew H.L., Jerger J.F., Guillory S.B., Henry J.A. Auditory dysfunction in traumatic brain injury. J Rehab Res Dev.. 2007;44:921-928.
79. Scott S.G., Vanderploeg R.D., Belanger H.G. Centers for Disease Control and Prevention. CDC Injury Prevention. Explosions and Blast Injuries: a primer for clinicians. Atlanta, GA: CDC; 2003.
80. Marzo S.J., Leonetti J.P., Raffin M.J., Letarte P. Diagnosis and management of post-traumatic vertigo. Laryngoscope. 2004;114:1720-1723.
81. Brunner H. Commotio auris internae. In: Alexander H.V., Marburg O. Handbuch Neurologie des Ohres. Munich: Urban & Schwarzenberg; 1928:37.
82. Axelsson A., Hallen C. The healing of the external cochlea wall in the guinea pig after mechanical injury. Arch Otolaryngol. 1973;76:136.
83. Lindsay J.R., Zajtchuk J. Concussion of the inner ear. Ann Otol Rhinol Laryngol. 1970;79:699.
84. Ilberg C. Die Innenohrschwerhorigkeit nach strumpfen Schaltrauma. Laryngorhinootologie. 1977;56:323.
85. Kelly K.E., Tami T.A. Temporal bone and skull base trauma. In: Jackler R.K., Brackmann D.E. Neurotology. St. Louis: Mosby; 1994:1127-1147.
86. Chiang F.Y., Wang L.F., Huang Y.F., et al. Recurrent laryngeal nerve palsy after thyroidectomy with routine identification of the recurrent laryngeal nerve. Surgery. 2005;137(3):342-347.
87. Chiang F.Y., Lu I.C., Kuo W.R., et al. The mechanism of recurrent laryngeal nerve injury during thyroid surgery—the application of intraoperative neuromonitoring. Surgery. 2008;143(6):743-749.
88. Bunch T.J., Ellenbogen K.A., Packer D.L., Asirvatham S.J. Vagal nerve injury after posterior atrial radiofrequency ablation. Heart Rhythm. 2008;5(9):1327-1330.
89. Bauer A., Deisenhofer I., Schneider R., et al. Effects of circumferential or segmental pulmonary vein ablation for paroxysmal atrial fibrillation on cardiac autonomic function. Heart Rhythm. 2006;3:1428-1435.
90. Chandawarkar R.Y., Cervino A.L., Pennington G.A. Management of iatrogenic injury to the spinal accessory nerve. Plast Reconstr Surg. 2003;111:611-617.
91. Valtonen E.J., Lilius H.G. Late Sequelae of iatrogenic spinal accessory nerve injury. Acta Chir Scand. 1974;140:453.
92. Wright T.A. Accessory spinal nerve injury. Clin Orthop. 1975;108:15.
93. Strauss W.L.Jr., Howell A.B. The spinal accessory nerve and its musculature. Q Rev Biol. 1936;11:387.
94. Donner T.R., Kline D.G. Extracranial spinal accessory nerve injury. Neurosurgery. 1993;32(6):907-910.
95. Bodner G., Harpf C., Gardetto A., et al. Ultrasonography of the Accessory Nerve: normal and pathologic findings in cadavers and patients with iatrogenic accessory nerve palsy. J Ultrasound Med. 2002;21:1159-1163.
96. Cappiello J., Piazza C., Giudice M., et al. Shoulder disability after different selective neck dissections (levels II–IV versus levels II–V): a comparative study. Laryngoscope. 2005;115(2):259-263.
97. Kim H.S., Ko K. Penetrating trauma of the posterior fossa resulting in vernet’s syndrome and internuclear ophthalmoplegia. J Trauma Inj Infect Crit Care. 1996;40:647-649.
98. Urculo E., Arrazola M., Arrazola M.Jr., et al. Delayed glossopharyngeal and vagus nerve paralysis following occipital condyle fracture case report. J Neurosurg. 1996;84:522-525.
99. Bozboga M., Unal F., Hepgul K. Fracture of the occipital condyle. case report. Spine. 1992;17:1119-1121.
100. Loro W.A., Owens B. Unilateral hypoglossal nerve injury in a collegiate wrestler: a case report. J Athletic Training. 2009;44(5):534-537.
101. Sharma B.S., Mahajan R.K., Bhatia S., Khosla V.K. Collet-Sicard syndrome after closed head injury. Clin Neurol Neurosurg. 1994;96:197-198.
102. Hashimoto T., Watanabe O., Takase M., et al. Collet-Sicard Syndrom after minor head injury. Neurosurgery. 1988;23(3):367-370.
103. Lee J.S. Craniofacial gunshot injury resulting in pseudoaneurysm of the left internal maxillary artery and Collet-Sicard syndrome. J Craniofac Surg. 2009;20(2):568-571.
104. Prashant R., Franks A. Collet-Sicard syndrome—a report and review. Lancet Oncol. 2003;4:376-377.
105. Tappin J.A., Satchi G., Corless J.A., Ashword F. Multiple myeloma presenting as the Collet-Sicard syndrome. J Neurol Neurosurg Psychiatry. 1996;60:14.
106. Albert T.J., Lee J., Lim M. Cervical Spine Surgery Challenges: diagnosis and management. Stuttgart: Thieme; 2008.
107. Martin R.E., Neary M.A., Diamant N.E. Dysphagia following anterior cervical spine surgery. Dysphagia. 1997;12:2-8. discussion 9-10
108. Sajid M.S., Vijaynagar B., Singh P., Hamilton G. Literature review of cranial nerve injuries during carotid endarterectomy. Acta Chir Belg. 2007;107:25-28.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Chapter 204 Management of Cranial Nerve Injuries
The incidence of traumatic cranial nerve injury is approximately 5% to 23%.1 Nontraumatic and iatrogenic injuries are not uncommon. Cranial nerve injuries are caused by direct impact or indirect transference of energy from the skull base and/or bony elements surrounding the cranial nerves. Neuroimaging studies are crucial for diagnosis; however, the clinician must rely on accurate history and the neurologic examination to confirm the appropriate diagnosis of cranial nerve injury. The neurologic examination can be limited by mechanical ventilation and sedation, especially in patients with significant multisystem trauma. The knowledge of the pathologic anatomy of cranial nerve injuries is essential. With this knowledge, the clinician can develop appropriate suspicion for potential cranial nerve injury, make the appropriate diagnosis, and treat patients based on current standards of care. Delay in diagnosis and treatment leads to significant morbidity.
Traumatic Olfactory Nerve Injury
The literature reports a wide range of percentages for traumatic olfactory nerve dysfunction. Injury to the olfactory nerve varies from 4% to 60%.2 Clinical diagnosis of traumatic olfactory nerve dysfunction depends on patient age and their recognition of the dysfunction. The likelihood of traumatic olfactory dysfunction increases with age.
Self-assessment may under- or over-estimate the incidence of olfactory nerve dysfunction because of the lack of awareness of olfactory function. Only 40% of patients with olfactory dysfunction were aware of their deficit.2 Most dysfunction is present immediately after injury, but delayed onset has been observed. It is thought that delay is secondary to excessive fibrous tissue around a fractured cribriform plate; there is little radiographic or histologic evidence, however, to prove this.
The probability of traumatic olfactory nerve dysfunction does not depend on the severity of the traumatic head injury, as recorded by the Glasgow coma scale (GCS).2 The severity of dysfunction, however, is related to the severity of head injury. Traumatic olfactory nerve dysfunction is also related to whether the anterior skull base and/or frontal lobe are involved. Radiographic evidence of anterior skull base or frontal lobe injuries is, however, absent in 24% of cases.2 Patients with skull base fractures present not only with olfactory nerve dysfunction, but can also present with rhinorrhea.3 Coup and contracoup injuries that occur with frontal and occipital trauma are the most common type of injuries producing olfactory nerve dysfunction.4 There are literature reports of minor head injury associated with olfactory nerve dysfunction.
Traumatic olfactory nerve dysfunction involves damage to the olfactory epithelium containing the receptor neurons in the nasal mucosa and/or mechanical stretching/shearing of unmyelinated nerve rootlets passing through the cribriform plate to form the olfactory bulb. Olfactory information is processed by the olfactory tract via the olfactory bulb to the entorhinal cortex (primary sensory areas) and the orbitofrontal cortex (secondary sensory areas). Therefore, olfactory dysfunction after traumatic head injury may not only be a result of shearing of the olfactory nerve filaments, but also by damage to frontotemporal cortical structures. Experimental animal models and human histopathologic studies in mild traumatic brain injury patients have shown axonal damage, mainly located in the frontal lobes and olfactory bulbs.5 Impairment of olfactory stimuli processing leads to distortion of olfactory sensation known as “dysosmia.” Patients complaining of dysosmia have abnormal regeneration of injured olfactory pathways. Recent animal studies demonstrate that recovery of the olfactory system varies with the severity of injury and that dexamethasone treatment may have therapeutic value by reducing injury-associated edema.6
There are a variety of tests used for olfactory nerve dysfunction. These include the Brief Smell Identification Test, testing using Sniffin’ Sticks, and chemosensory-evoked potentials. Another widely available test is the 40-odor University of Pennsylvania Smell Identification Test (UPSIT), which has been administered to over 180,000 people in Europe and North America.7 Hypometabolism in the orbitofrontal cortex and the medial prefrontal cortex were found in positron emission tomography (PET) studies in patients with post-traumatic anosmia.8 In head injury patients with anosmia or hyposmia, coexistence of olfactory bulb and tract damage and frontal encephalomalacia on magnetic resonance imaging (MRI) has also been found.9
Treatment of traumatic olfactory nerve dysfunction is usually conservative. Approximately one third of patients have significant recovery with conservative management.10 Unlike the other cranial nerves, the olfactory nerve contains olfactory ensheathing cells rather than Schwann cells. The olfactory pathway continuously rebuilds itself throughout life. More invasive treatment, including removal of the olfactory bulbs and tracts, is reserved for patients with severe dysosmia. Case reports demonstrated patients with severe dysosmia causing significant anorexia.
Traumatic Optic Nerve Injury
Traumatic optic nerve injuries occur in 0.5% to 5% of head injuries.11 More recent surveys of craniofacial trauma data suggest an incidence of traumatic optic nerve injury in 2% to 5%.12
Optic nerve injury can be caused by primary and secondary mechanisms. Primary injury is by permanent axonal injury at the moment of impact from mechanical shearing, contusion, and ischemic necrosis of nerve axons. Secondary mechanisms are due to apoptosis, edema, and cell death, incorporating a variety of mechanisms leading to further axonal damage after the initial impact.13–16
Traumatic optic nerve injury can also be divided into direct and indirect injuries. Direct injuries occur with penetrating objects into the orbit causing direct damage the optic nerve. Indirect traumatic optic nerve injury is a closed injury resulting from the transmitted impact to the optic nerve.17
Direct traumatic optic nerve injuries typically have a worse prognosis in comparison to indirect injuries.18 Direct optic nerve injury anterior to the central retinal artery entry point disturbs the retinal circulation due to the associated vascular injury. Injuries posterior to the entry point maintain normal circulation. Partial or complete optic nerve avulsion can result in a partial or complete hemorrhagic ring at the optic nerve head.19–20 Computer modeling of direct optic nerve trauma has shown that the main site of stress is at the insertion of the nerve to the sclera and opposite to the side of impact.21 Nerve damage at this point leads to optic disc swelling.22 Rotation of the nerve at its insertion point to the sclera contributes the most damage in direct traumatic injury.21 A sudden increase in intraocular pressure is more likely to injure the optic nerve head.21 The intracranial segment of the optic nerve can also be injured against the falciform ligament.23
Indirect optic nerve injury can be intracanalicular, within the optic canal, or intracranial affecting the optic nerve or chiasm. Studies utilizing laser interferometry demonstrated forces applied to the frontal bone are transferred to the optic canal.24 Deceleration forces on the facial bones deform the sphenoid bone, with energy being transferred to the optic nerve.25,26 Indirect iatrogenic injury has been reported from heat transmission during electric cautery of the posterior ethmoidal artery during treatment of epistasis, and mechanical or heat transmission injuries during transnasal endoscopic surgery. Orbital hemorrhage can cause an intraorbital compartment syndrome, a potentially reversible cause of traumatic optic neuropathy.
Injury to the optic nerve can cause significant disability. Patients typically present initially with an afferent papillary defect and visual field defect. Significant visual loss is almost always accompanied by an afferent papillary defect.27 Diagnosis of traumatic optic nerve injury can be challenging and can be delayed weeks to months. This is attributed to multiple factors including severe craniofacial fractures, traumatic brain injury, and multi-system trauma requiring mechanical ventilation and sedation. Retinal examination is usually not helpful in diagnosing optic nerve injuries, as optic atrophy does not become apparent for 3 to 4 weeks.28 A retinal exam is useful in the diagnosis of retinal hemorrhages and other ocular trauma.
The initial clinical assessment should document the time interval between injury and the visual examination. Delayed visual loss was reported in 10% of patients included in the International Optic Nerve Trauma Study.29
Patients with direct disruption of the optic nerve will have minimal, if any, recovery. Indirect or secondary injury can be treated and prevented. Treatment includes observation, medical treatment with steroids and optic canal decompression or nerve sheath fenestration. Conservative management (i.e., observation) can achieve a 40% to 60% recovery rate.28 Prognostic factors have been studied. Initial visual acuity is the most influential indicator of good outcome.29 Poor prognostic factors include blood in the posterior ethmoidal cells, age over 40 years, loss of consciousness, and absence of recovery within 48 hours of steroid treatment.30 Blood in the posterior ethmoidal cells reflects the severity of injury, as the frontal bones transfer energy to the optic canal region.24
Steroid treatment is based on the National Acute Spinal Cord Injury studies. High-dose corticosteroids were translated from acute spinal cord injury to optic nerve injury. The International Optic Nerve Trauma Study (IONTS) revealed no clear benefit for either high-dose corticosteroid therapy or surgical decompression when compared to observation alone,31 reporting 54% improvement after 3 months with steroid therapy, and 57% improvement with observation. Rat models have not shown histologic evidence of either improved axonal regeneration or prevention of further degeneration.32 The IONTS study reported that mega-doses of steroids within the first few hours of injury most likely provides the maximum benefit out of all treatment modalities studied. Adverse effects of steroids should be considered in light of other injuries.30,31
Surgery should be considered with any obvious nerve compression or continual visual loss despite steroid therapy.28,33,34 Timing of surgery is controversial. Surgical decompression has similar outcomes up to 4 months after the injury in patients who are not completely blind.35
Evaluation of optic canal fractures can be visualized with high resolution computed tomography (CT) scan. Fracture of the optic canal potentially results in direct impingement on the nerve by bony fragments or associated hematoma and/or optic nerve edema (Fig. 204-1). Intrasheath hematoma should be treated with optic nerve fenestration. Perisheath hematoma can, however, be confused with an intrasheath hematoma. Optic nerve decompression has been performed by a variety of approaches, including transnasal trans-sphenoidal/transethmoidal, transpalpebral, and supraorbital transcranial. There is no statistical difference between steroids only or steroids plus surgical decompression.36
FIGURE 204-1 Axial computed tomography demonstrating optic canal fracture with compression of the canal by bony elements.
Endoscopic decompressive approaches have proven to maintain or improve vision in 50% of cases, and have shown to have fewer adverse outcomes.36 Endoscopic transnasal approaches are recently been favored due to the anatomic proximity of the optic canal to the sphenoid sinus, lack of external scars, preservation of olfaction, decreased morbidity, and faster recovery time. Endoscopic decompression can be considered earlier than conventional surgery, especially in patients with poor prognosis.36
Oculomotor Nerve
Oculomotor nerve palsy due to closed head injury is uncommon (5% to 15%).37–39 Injury to multiple ocular cranial nerves including oculomotor, trochlear, and abducens, can occur with post-traumatic, carotid-cavernous fistula. The oculomotor nerve exits the midbrain into the interpeduncular fossa after its rootlets traverse the red nucleus. The nerve separates into two divisions upon entering the orbit. The superior division innervates the superior rectus and levator palpebrae superioris. The inferior division innervates the medial and inferior recti and the inferior oblique muscles. Preganglionic parasympathetic innervation of the eye runs off of a branch of the nerve to the inferior oblique muscle. The complex anatomy of the oculomotor nerve results in a spectrum of presentations. In the midbrain, subnuclei within the oculomotor nuclei specifically innervate individual ocular muscles. The superior rectus muscle receives input from the contralateral subnuclei. Both levator palpebrae superioris muscles receive bilateral input.
Oculomotor nerve injury may result from distraction of the nerve, avulsion, and compression or displacement of the oculomotor nerve by traumatic intracerebral hematoma or hemorrhagic contusion.40,41 Oculomotor nerve injury can be associated with severe traumatic head injury.39 Ophthalmoplegia may result from downward displacement of the brain stem at the time of impact, and direct injury to the pupillomotor fibers on the ventromedial surface of the third nerve at the posterior petroclinoid ligament.42 Disturbance of the oculomotor nerve’s blood supply and biochemical changes from head injury can also contribute to the mechanism of injury.
Diagnosis of an oculomotor nerve injury can be challenging, especially in patients with orbital edema and/or ecchymosis. An adequate exam must be made, excluding any extraocular influences on ocular movement. Waiting for edema to resolve is usually required. Localization of oculomotor nerve injuries is based on the anatomic specifics of the nerve and nuclei. Involvement of the cerebral peduncle will not only present with an oculomotor nerve palsy, but also a contralateral hemiplegia and tremor. Involvement of the nuclei itself will present with not only ipsilateral oculomotor weakness (except superior rectus, which would be contralateral), but also bilateral ptosis and contralateral superior rectus weakness.
Oculomotor nerve injury is associated with a lower GCS when compared to other traumatic cranial neuropathies.43 Patients were involved mainly in motor vehicle accidents and had higher number of temporal lobe abnormalities (43%) on CT and MRI. Patients with bilateral traumatic oculomotor nerve palsies or unilateral oculomotor paresis were associated with severe injuries.3 The prognosis of traumatic oculomotor palsy is poor and full recovery is uncommon. A prolonged period (up to years) of healing is usually anticipated.44
Trochlear Nerve
The trochlear nerve is the smallest and longest of the ocular motor nerves. It runs at the free tentorial edge around the midbrain after decussating around the dorsal midbrain. Contracoup injury can occur when the nerve is compressed against the tentorium. Unilateral trochlear nerve injury can occur after frontolateral impact whereas bilateral trochlear nerve injury can occur from midfrontal impact.45 The presence of a Horner syndrome or an afferent pupillary defect should alert the clinician to a nuclear cause of the trochlear nerve palsy. The trochlear nucleus is in close proximity to the sympathetic fibers and the pupillomotor fibers. Trochlear nerve palsies can also be challenging to diagnosis in the comatose patient. There is large variation in the amount of head trauma that translates into trochlear nerve injury.4
Trigeminal Nerve
Trigeminal nerve injuries not only causes significant neurosensory deficits and facial pain, but can cause significant comorbidities due to changes in eating habits from muscular denervation of masticator muscles or altered sensation of the oral mucosa. Approximately 70% of patients with trigeminal nerve injuries complain about paresthesias.46 Ten to fifteen percent present with neuropathic pain confirmed with nerve block testing. The development of neuropathic pain is similar in susceptibility as peripheral nerves.47
Risk factors include direct maxillofacial trauma with facial bone fractures, injury secondary to local anesthetic injections, and during surgical intervention for facial trauma repair. There is an association between trigeminal nerve injury and facial bone fractures. Facial fractures affect the peripheral branches of the trigeminal nerve. With penetrating injuries including gunshot wounds, any portion of the trigeminal nerve can be affected. Traumatic trigeminal nerve injury associated with fractures of the upper third of the face and temporal bone is rare.48 Fractures extending from the temporal bone to the clivus can cause injury to the trigeminal root or ganglion. Forceful impact to the posterior third of the skull may crush the petrous apex against the dorsal sella leading to injury of the trigeminal ganglion. Facial fractures directly involving the trigeminal nerve and dislocated fractures have a higher incidence of traumatic trigeminal nerve injury than nondisplaced fractures. Traumatic trigeminal nerve injury was found in 88.2% of dislocated fractures, 54.4% of nondislocated fractures, and 100% of fractures with direct nerve involvement.49 Nondisplaced midfacial fractures had the highest incidence of trigeminal nerve impairment, but had the best prognosis out of all trigeminal nerve impairment associated with facial fractures.
Trigeminal nerve injuries are also a well known risk after oral and dental procedures, especially the inferior alveolar nerve during mandibular procedures.50 The second most common nerve injured in dental procedures is the lingual nerve. Injuries to the long buccal nerve, greater palantine, and nasopalantine are usually clinically insignificant. Tay and Zuniga reported a higher incidence of females presenting with trigeminal nerve injuries. Lower third molar surgery was the procedure most commonly associated with trigeminal nerve injury. Patients typically presented 3 to 9 months with subjective functional problems, but minimal signs of self-injury to the hypesthetic or hyperpathic oral mucosa.
Initial evaluation begins with the neurologic exam when there are suspicions of trigeminal nerve trauma. Patients with neurosensory dysfunction of the trigeminal nerve should be evaluated serially for up to 3 months.51 Patients with unacceptable recovery at 3 months should be offered exploration and/or repair of the nerve.
If trigeminal nerve trauma is due to a fracture, open reduction with decompression is indicated unless there are other contraindications. Closed reductions of fractures continue to show deformity of the nerve and attenuation of nerve at the site of the fracture despite gentle reduction of fractured elements.51 This is due to reactive osseous proliferation causing subsequent compression of the nerve. Decompression by enlarging surrounding bone and foramina prevents nerve compression secondary to post-traumatic nerve edema and ossification of surrounding bone. Nerve repair produces significant improvement or complete recovery in 86% of patients.51 Similar results are achieved when patients undergo surgical repair up to 6 to 9 months after the initial injury.
Abducens Nerve
Abducens nerve palsy is uncommon following traumatic brain injury, with an incidence of 1% to 2.7%.52 Postmortem examination in severe head trauma revealed abducens nerve injury at the dural entry point (Dorello’s canal), petrous apex and the lateral wall of the ICA.53 The petrous bone is the most common of the three causing stretching of the nerve. Other theories include neck hyperextension causing stretch of the nerve as it passes through Dorello’s canal. Isolated abducens nerve palsies without traumatic brain injury have been described in the literature.54,55 There was an association with fractures of the cervical vertebrae.
Abducens nerve injury has been reported after lumbar puncture.56 The mechanism is thought to be downward sagging of the brain after intracranial hypotension resulting in traction of the nerve. Patients usually recover from this.
The recovery rate after conservative management is high, 71%, with palsy or paresis.57 Predictors of nonrecovery include the failure to recover by 6 months from onset, complete paresis or bilateral involvement.58 Botulinum treatment does show significantly improved outcomes with 73% showing recovery when given within 3 months of injury.57
Facial Nerve
Facial nerve injury usually results from blunt or penetrating trauma to the petrous portion of the temporal bone. Approximately 5% of patients who suffered head injuries have temporal bone fractures.61 Facial nerve weakness can be partial or complete; it can manifest immediately or in a delayed fashion. Late presentation is secondary to pressure effect from hemorrhage, edema, or granulation tissue.
[/not-level-membership-for-neurology-category]
