[level-membership-for-surgery-category]
Chapter 74 Management of ascites in cirrhosis and portal hypertension
Historic Overview
One of the most visible manifestations of liver disease is that of ascites, broadly defined as the pathologic accumulation of fluid within the peritoneal cavity (Runyon, 2009). The almost reflexive association of ascites with liver disease notwithstanding, ascitic fluid volume and composition demonstrate a variability that reflects the range of disease processes, from benign to sinister, from whence it originates.
Descriptions of ascites exist in both human history and prehistory. Indian medical treatises from 1500 bce, Mayan figurines with protuberant abdomens and everted umbilici, and the writings of Hippocrates all testify to the experience of varied cultures with this problem. Even the term ascites—derived from the Greek askos, a bag made of leather or sheepskin used to contain liquids—reflects its ancient origins (Reuben, 2004).
High-volume ascites is readily evident on physical examination; however, smaller volumes may evade detection, particularly in the obese. Ultrasound is the best imaging modality for detection of ascites. It is inexpensive, avoids ionizing radiation, and has the added benefit of providing information on liver architecture and portal vein patency (see Chapter 13).
All clinicians should maintain a low threshold for performing paracentesis. The procedure is necessary in the diagnostic evaluation of newly developed ascites, or when there is a change in the clinical condition in an individual with cirrhosis and ascites. A percutaneous approach that avoids the epigastric vessels offers an avascular plane that is safe even in coagulopathic and thrombocytopenic patients (Wong et al, 2008).
Portal Hypertension and Mechanisms of Ascites Formation
Ascites is the most common complication of portal hypertension arising from cirrhosis, occurring at an annual incidence of 1% (see Chapter 70A, Chapter 70B ; Ginès et al, 1987). Its development heralds a significant change in clinical condition, with a median survival of 50% over 2 years (D’Amico et al, 1986). Portal hypertension can arise from cirrhotic and noncirrhotic causes, although as a manifestation of portal hypertension, ascites is most common in disorders that increase pressures within the hepatic sinusoids, either from sinusoidal processes (cirrhosis) or postsinusoidal processes (heart failure, hepatic venous obstruction). By comparison, ascites is uncommon in presinusoidal portal hypertension.
The normal liver architecture consists of sinusoids that convey blood from the portal vein to the central vein. The sinusoids are separated from cords of hepatocytes by a fenestrated endothelium through which oxygen, cells, and plasma components are permitted to diffuse. The principal collagen-producing cells of the liver, the stellate cells, reside within the space of Disse, and activation of these cells leads to collagen deposition (Friedman et al, 1992). Other cells that contribute to the extracellular matrix within the liver are bone marrow–derived myofibroblasts and fibroblasts derived from epithelial-mesenchymal transition (Forbes et al, 2004; Kalluri & Neilson, 2003).
In cirrhosis, extracellular basement membrane deposition of collagen fibers within the space of Disse results in capillarization of the hepatic sinusoids (Huet et al, 1982). The resulting architectural changes cause a static increase in pressure within the splanchnic circulation (see Chapter 6).
The endothelial cells of the liver also play an important role in controlling dynamic changes to the hepatic microcirculation through the elaboration of nitric oxide (Mittal et al, 1994; Shah et al, 1997) and expression of endothelin-1 receptors (Bauer et al, 2000). Decreased endothelial nitric oxide synthase (eNOS) activity (Gupta et al, 1998; Shah et al, 1999), increased eNOS inactivator activity (Liu et al, 2005), and endothelin-1 overexpression in cirrhosis (Pinzani et al, 1996) have all been proposed as mechanisms that promote endothelial cell dysfunction in cirrhosis (see Chapter 6).
In the splanchnic circulation outside of the hepatic environment, different but equally important changes occur that contribute to portal hypertension. The most prominent of these is splanchnic arterial vasodilation. In experimental models of cirrhosis, vasodilation is mediated by NO-dependent (Sieber & Groszmann, 1992; Sieber et al, 1993) and NO-independent processes. The NO-independent processes include those related to endogenous vasodilatory cannabanoids (Garcia et al, 2001) and overexpression of vascular endothelial growth factor (VEGF) and VEGF receptor-2 to promote splanchnic angiogenesis, thereby augmenting blood flow in the splanchnic circulation (Fernandez et al, 2007). Vasodilation in the splanchnic circulation would decrease the effective arterial circulation if not for compensatory increases in cardiac output (Iwakiri & Groszmann, 2006).
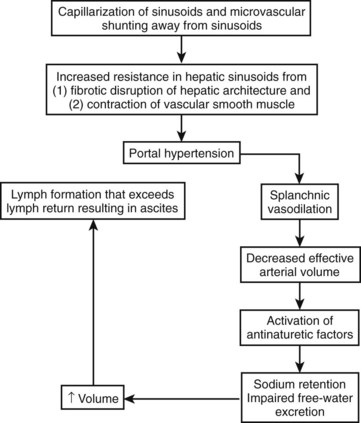
With progression of portal hypertension, other compensatory mechanisms are activated to maintain the arterial circulation in the face of even greater increases in vasodilation and declines in cardiac output. These mechanisms include activation of the renin-angiotensin system and sympathetic nervous system to stimulate sodium retention by the kidneys (Arroyo et al, 1983). The nonosmotic release of arginine vasopressors is an additional compensatory mechanism to increase the effective arterial volume, even at the expense of tonicity; this is reflected in the development of hyponatremia (Arroyo et al, 1994). The cumulative effect of increased hydrostatic pressure in the hepatic microcirculation in the setting of increased splanchnic volume is that of hepatic lymph formation in excess of its removal; the resulting excess fluid weeps into the peritoneal cavity and is recognized as ascites (Fig. 74.1).
Characteristics and Evaluation of Ascites
Ascitic fluid in cirrhotics is transparent but will generally take on a yellow or amber color. The fluid generally has a low leukocyte (<100 µL/mm) and red blood cell content. The fluid protein is typically less than 2.5 mg/dL, and the protein content varies inversely with the severity of the portal hypertension (Hoefs, 1983). Measurement of the serum albumin ascites gradient (SAAG) is both a highly accurate and clinically facile technique for assessing the origins of ascites. The SAAG is calculated by subtracting the concentration of albumin in the ascites from that in the plasma (Pare et al, 1983). With an approximately 97% accuracy, a gradient greater than 1.1 g/dL indicates the presence of portal or sinusoidal hypertension (Runyon et al, 1992).
Opacification of ascitic fluid can arise from a number of disparate processes. Bloody ascites (hematocrit >0.5) can be seen in traumatic paracentesis or in the spontaneous rupture of hepatocellular cancer. Chylous ascites is milky in appearance from increased concentration of chylomicron-rich triglyceride (Aalami et al, 2000). It arises from processes that disrupt lymphatic flow, most commonly lymphangiectasia and lymphoma, but it can also occur with abdominal trauma and surgical disruption of the cysterna chyli. Cirrhotic ascites can also take on a chylous appearance, owing to rupture of abdominal lymphatics from portal hypertension (Rector, 1984). In these cases, known as pseudochylous ascites, the triglyceride concentration is generally less than the threshold value of 110 mg/dL found in pure cases of chylous ascites.
Both malignancy and tuberculosis peritonitis can result in ascites, and in both circumstances the SAAG is less than 1.1 g/dL; however, confusion may arise when liver disease coexists, as in the case of tuberculosis and alcohol-related liver disease (Aguado et al, 1990; Shakil et al, 1996). The diagnosis of malignant ascites is established by the finding of cancer cells within the peritoneal cavity. This can be done by conventional cytology with a diagnostic sensitivity of 40% to 60% (Siddiqui et al, 1992). The accuracy of cytology can be improved when combined with immunohistologic staining (Aslam & Marino, 2001).
The peritoneum is a common site of involvement of tuberculosis (TB), and in the United States, the peritoneum is the sixth most common extrapulmonary site. Peritoneal cell counts typically vary between 500 and 1500 cells/mm3 with a lymphocyte predominance in 68% (Sanai & Bzeizi, 2005), although the absence of a lymphocyte predominance does not exclude TB, particularly in patients with underlying renal failure, in whom the cells are mostly neutrophils (Lui et al, 2001). Mycobacterial culture of the fluid has a diagnostic sensitivity of 34% and requires several weeks of intubation. Measurement of adenosine deaminase activity in the peritoneal fluid has been proposed as another diagnostic test with high sensitivity and specificity, although the positive predictive value has been reported to be low in the setting of concomitant cirrhosis (Hillebrand et al, 1996). Of all diagnostic strategies, laparoscopy with peritoneal biopsy affords the highest sensitivity and specificity and permits exclusion of other granulomatous and nongranulomatous processes that can produce a low-SAAG ascites. The ascites concentration of lactate dehydrogenase (LDH) tends to be higher than that of serum LDH in malignant ascites and less than half that of serum in tuberculous peritonitis.
Management
Dietary Sodium Restriction
Avid renal sodium retention is the initial response to splanchnic arterial vasodilation; thus initial treatment strategies involve tipping the balance in favor of a net loss of sodium; this is most simply accomplished with dietary sodium restriction. The estimated mean sodium consumption among nonhypertensive adults in the United States is 3600 mg/day (Ajani et al, 2005). Those with ascites are commonly advised to restrict dietary salt intake to 1.5 to 2 g per day (67 to 87 mmoL/day), the lower value of which is considered adequate for daily needs. In patients with mild degrees of ascites, sodium restriction may be singularly effective. This group usually has baseline rates of sodium excretion of at least 40 mEq/L per day and normal plasma sodium concentrations (Arroyo et al, 1981). Although seemingly a simple intervention, success with dietary salt restriction requires counseling and vigilance. Most of the sodium present is added during food processing, and the patient’s actual consumption may not be apparent to them unless food labeling is read and understood (Cook, 2008).
Medical Management
Diuretics
Most patients will at some point require diuretics. Between 500 and 750 mL per day of ascites can be mobilized without intravascular depletion, and greater amounts of ascitic fluid losses may be tolerable in the presence of edema, which tends to act as a buffer (Pockros & Reynolds, 1986). The avoidance of rapid fluid loss is critical because precipitous and excessive volume contraction can give rise to hepatorenal syndrome.
Aldosterone Antagonists
The aldosterone antagonists spironolactone and amiloride (Table 74.1) can be used as either monotherapy or in combination with loop diuretics. These agents prevent sodium reabsorption in the distal tubule and cortical collecting duct. Although aldosterone antagonists are weak natriuretics, they are effective in patients with cirrhosis (Perez-Ayuso et al, 1983).
Table 74.1 Diuretics and Dosages Commonly Used in the Management of Ascites
| Medication | Dose | Comments |
|---|---|---|
| Aldosterone Antagnosists | ||
| Spironolactone | 50-400 mg daily | |
| Amiloride | 5-10 mg daily | Suitable substitute when spironolactone use is associated with painful gynecomastia |
| Loop Diuretics | ||
| Furosemide | 20-160 mg daily | Avoid intravenous use |
Urine sodium excretion and plasma aldosterone concentration are hyperbolically and inversely related in patients with cirrhosis, and a greater sensitivity to the dose-response curve is observed in those with ascites (Bernardi et al, 1983). One explanation for the effectiveness of these agents in cirrhotic ascites may be that they target the functional hyperaldosteronism that would otherwise permit sodium reabsorption in the cortical collecting tube of fluid filtered in the loop of Henle. In addition, unlike other diuretics that require access to the tubular lumen, spironolactone enters the principal cell of the collecting duct from the plasma compartment and thereby circumvents decreases in renal blood flow and hypoalbuminemia, commonly encountered in patients with cirrhosis, that might otherwise impair its activity (Horisberger & Giebisch, 1987).
Loop Diuretics
As monotherapy for ascites, loop diuretics are often unsuccessful. The reasons for this are unclear but may relate to a decreased rate of drug entry into the tubular lumen or to a compensatory increase in distal tubular sodium resorption mediated by aldosterone. By comparison, the combination of loop diuretics and aldosterone antagonists is the most commonly used combination for moderate to severe ascites and can achieve reductions in ascites beyond that of aldosterone antagonists alone. The most commonly used loop diuretic is furosemide (see Table 74.1), beginning at doses of 20 to 40 mg daily. The dose is doubled in a coordinated fashion with increases in spironolactone, until therapeutic effect is achieved. Like spironolactone, a fourfold doubling of the dose (160 mg daily) is considered a maximal dose. An added benefit of loop diuretics is that they counteract the hyperkalemia associated with aldosterone antagonists, and they may also benefit those who develop dependent edema in addition to ascites.
Albumin
Hypoalbuminemia is a frequent finding in patients with cirrhosis, and it may influence the development of edema and ascites through alterations in plasma oncotic pressure and the activity of loop diuretics in the lumen of the collecting tubule. Limited evidence supports a role for albumin administration as an adjunct to diuretic therapy in ascites that is difficult to control (Gentilini et al, 1999). However, this strategy may not be broadly applicable with the diuretics currently available or when there is recourse to other treatments (Blendis & Wong, 1999). Intravenous albumin administration does have a secure role in the prevention of renal dysfunction, which occurs in the one third of patients who develop spontaneous bacterial peritonitis (Sort et al, 1999). In addition, vasoconstrictor agents used in the treatment of hepatorenal syndrome are more effective when coadministered with albumin than when administered with saline or other crystalloids (Ortega et al, 2002). Thus albumin appears to achieve a successful volume expansion in patients with cirrhosis, greater than that that of crystalloid, which is translatable into improvement in solid clinical end points.
Arginine Vasopression Receptor Antagonists
The nonosmotic release of antidiuretic hormone (ADH) is commonly observed in patients with cirrhosis as a compensatory mechanism for the decrease in effective arterial volume that occurs with splanchnic vasodilation. ADH exerts its effects through arginine vasopressin receptor-2 (AVPR2), which is predominantly expressed in the distal convoluted tubule and collecting ducts of the kidney (van den Ouweland et al, 1992). Satavaptan is a selective AVPR2 receptor antagonist that has been tested in clinical studies of both the syndrome of inappropriate ADH release (Soupart et al, 2006) and hyponatremia occurring with cirrhosis (Ginès et al, 2008). The study in patients with cirrhosis was a multicenter, double-blind, randomized controlled study of 110 subjects with ascites and hyponatremia. Treatment with satavaptan and spironolactone was associated with improvement in ascites volume concomitant with improvements in hyponatremia more often than the placebo. The use of satavaptan and other aquaretic agents represents a promising area for pharmacologic management of ascites if these early successes are confirmed in other clinical trials.
Refractory Ascites
Ascites that persists despite dietary sodium restriction and high-dose diuretics (spironolactone 400 mg/day and furosemide 160 mg/day) is referred to as refractory ascites (Arroyo et al, 1996). Diuretic-intolerant ascites describes diuretic failure because of intolerant side effects—such as azotemia, hyponatremia, and encephalopathy—that prevent the attainment of a dose sufficient to effect an adequate ascites loss. The clinical significance of refractory ascites should not be overlooked; the survival curves of individuals with refractory ascites approximate those with type II hepatorenal syndrome (Ginès et al, 2004).
Before labeling a patient as having refractory ascites, it is important to exclude excessive sodium intake and other medications that may influence the diuretic response. In particular, nonsteroidal antiinflammatory agents, including aspirin (Planas et al, 1983), may both decrease the response of diuretics and contribute to azotemia.
Treatment options for refractory ascites includes therapeutic paracentesis, transjugular intrahepatic portosystemic shunting (TIPS) (see Chapter 76E), peritoneal shunting, and liver transplantation (see Chapter 97A).
Paracentesis
High-volume paracentesis, also known as therapeutic paracentesis, was a technique known to and practiced by physicians as far back as ancient Greece. It was also the most effective treatment in practice before the development of modern diuretics, after which the practice fell out of favor until it was reintroduced in 1987 as safe and effective (Ginès et al, 1987).
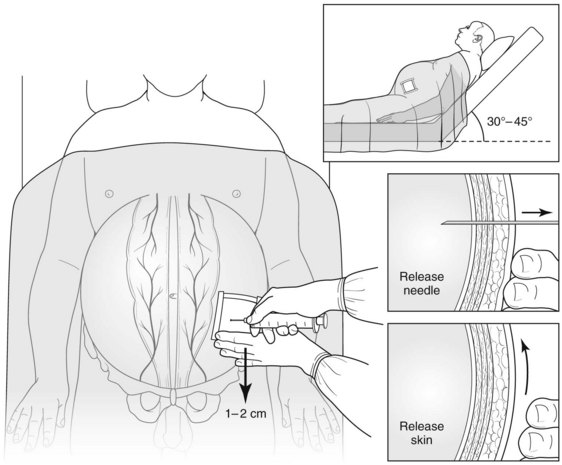
A number of large-bore needles (15 gauge or smaller) of various configurations and features have been manufactured for paracentesis. The procedure can be done with or without ultrasound guidance; commonly chosen sites for needle placement are over the right or left lower quadrant. A variety of techniques have been described for fluid removal, includng drainage by gravity, peristaltic pumps, and vacuum suction, but no single technique is superior to the others (Fig. 74.2).
There are two central debates in paracentesis. The first centers on the maximal volume of fluid that can be removed at one time, and the second centers on whether circulatory volume expanders are required. Because ascites is extravascular fluid, paracentesis can be safely done until fluid withdrawal is complete. Immediately after paracentesis, cardiac stroke volume and output increase, and the renin-angiotensin-aldosterone (RAA) axis is suppressed. More than 12 hours after paracentesis, a rebound increase in the RAA and increased arterial vasodilation are seen. These changes are reflected in a reduction in systemic vascular resistance and form the basis of postparacentesis circulatory dysfunction (Ruiz-del-Arbol et al, 1997), a condition associated with renal dysfunction, a higher incidence of hyponatremia, and decreased survival rate.
The administration of volume expanders at the time of paracentesis is principally done to ameliorate these changes. Studies comparing albumin to other colloidal agents have shown it to be more effective in preventing circulatory dysfunction in paracentesis when the volume removed is greater than 5 L (Ginès et al, 1996). Similar findings have also been described in studies that have compared albumin to saline after total paracentesis (Sola-Vera et al, 2003). In practice, 12.5 g of 25% albumin can be infused for every 2 L of ascites removed. Other groups recommend an infusion of 8 g of 20% albumin for every 1 L of ascites removed (Cardenas et al, 2009). The timing of administration has not been rigorously studied, but because of the long half-life of albumin in the circulation, its administration after completion of paracentesis is likely to be sufficient. The benefits of albumin notwithstanding, no compelling data are available to suggest that albumin administered with paracentesis improves patient survival; however, the sample size of the studies to date may make detecting any survival advantage difficult (Cardenas et al, 2009).
Potential immediate adverse events resulting from paracentesis include hematoma and hemorrhage into the peritoneal cavity. Leakage of ascites can be reduced by using the Z-tract technique (see Fig. 74.2); modest leaks are best controlled by covering the site with an ostomy appliance, but larger volume leaks may require suture closure of the site. No compelling data are available to suggest that repeated high-volume paracentesis predisposes to bacterial peritonitis. There is also no evidence to suggest that the typical patient with high-volume cirrhotic ascites is at risk for abdominal compartment syndrome; thus high-volume paracentesis should not be expected to improve renal function simply by decreased peritoneal ascites volume (Lott, 2010).
Transjugular Intrahepatic Portosystemic Shunts
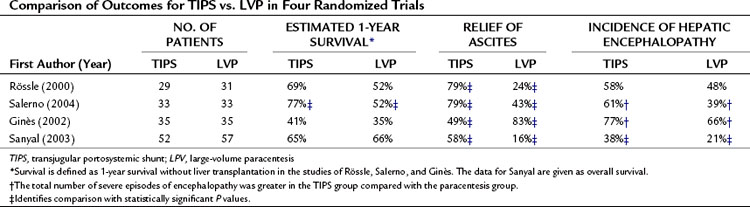
TIPS (see Chapter 76E) was first introduced experimentally in 1971 (Rosch et al, 1971); its purpose is to create a pathway to bypass sinusoidal hypertension and thereby relieve portal hypertension. To date, four randomized controlled trials have compared TIPS with repeated high-volume paracentesis in the management of ascites. Three of the trials are from sites in Europe (Italy, Salerno et al, 2004; Spain, Ginès et al, 2002; and Germany, Rössle et al, 2000); the fourth is from North America (Sanyal et al, 2003). In all the studies, TIPS was associated with a greater sustained relief of ascites compared with paracentesis; between 49% and 79% of those who received TIPS for refractory ascites were able to achieve sustained relief. The effectiveness of TIPS comes at the cost of an increased risk for episodes of severe encephalopathy. Some studies have shown a survival benefit, but others have not. In one of the multicenter trials, quality of life changes were similar in the two groups (Campbell et al, 2005). A comparison between TIPS and the paracentesis groups for the outcomes of survival, relief of ascites, and incidence of encephalopathy is summarized in Table 74.2.
The magnitude of portal decompression necessary to affect ascites formation is not known. The most common indirect measure of portal hypertension is the hepatic venous pressure gradient (HVPG), which is measured by hepatic vein catheterization with a balloon-tipped, pressure-transducing catheter. However, the HVPG necessary for control of refractory cirrhotic ascites is unknown (Boyer & Haskal, 2009).
The risk of dying after TIPS is based both on the mortality immediately related to the procedure and mortality after 1 to 3 months, likely reflecting a deterioration of hepatic function from the shunt. The procedure-related risk of death ranges between 1.7% and 3% and may show a dependence on the volume of TIPS done at any particular center (Haskal et al, 2003).
Unlike procedure-related mortality, which cannot be predicted, a number of systems have been offered to predict the short-term mortality. Scoring systems that have been studied include serum bilirubin alone (Rajan et al, 2002), the Acute Physiology and Chronic Health Evaluation II (APACHE II) (Rubin et al, 1995), Child-Turcotte-Pugh (CTP) class, and the Model for End-Stage Liver Disease (MELD) (Malinchoc et al, 2000). The simplest of these is serum bilirubin as a predictor of 30-day mortality, with a 40% increased risk of death for each 1 mg/dL increase greater than 3 mg/dL (Rajan et al, 2002). Although MELD is now most commonly used for organ allocation for transplantation in the United States, it was originally developed to predict the 3-month mortality after TIPS (see Chapter 70B). The MELD score is calculated from the serum bilirubin (Tbil), international normalized ratio (INR), and serum creatinine (Scr) from the equation below, given a maximum value for any variable of 1, a serum creatinine value of 4 or less, and a MELD score no higher than 40.
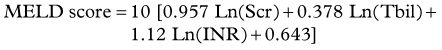
Decreased survival after TIPS has been reported with MELD scores of 15 and greater (Ferral et al, 2004; Pan et al, 2008). In patients with high MELD scores, the risks and benefits of TIPS should be carefully considered, as should the patient’s candidacy for liver transplantation as an alternative to TIPS or as a backup if TIPS results in deterioration of liver function.
Peritoneovenous Shunt
Peritoneovenous shunting (PVS) was introduced by Leveen in 1974 for the management of refractory ascites. The shunt is a plastic cannula subcutaneously tunneled with one end in the peritoneal cavity and the other in the central venous circulation. One-way flow is established by a pressure-sensitive valve positioned between the two ends. These surgically placed shunts go by a variety of names—including Leveen, Minnesota, and Denver shunts—that reflect proprietary differences in their technical details. The placement of shunts results in increases in plasma volume, glomerular filtration rate, and urinary sodium excretion. All these effects reflect favorable physiologic changes to decrease ascites formation; in fact, shunts have been effective in improving ascites.
One of the largest trials to investigate PVS for cirrhotic ascites was the Veterans Administration Cooperative Study on Patients with Alcoholic Cirrhosis with Ascites (Stanley et al, 1989). In this trial, PVS was compared with medical therapy in 299 men with alcoholic cirrhosis. Because the trial predated the discovery of hepatitis C, it is impossible to know how many of the men had cirrhosis from this condition. Regardless, PVS resulted in a slightly faster time to resolution of ascites and a greater time free of ascites. No difference in survival was reported among medically treated subjects and those undergoing PVS. Despite these and other positive clinical trials, PVS is uncommon in current practice. One reason for this is that 40% of shunts stop working within 1 year of placement. This most commonly reflects obstruction, either of the shunt, its valve, or the central vein to which it drains. Adverse events also reported with PVS include pulmonary edema, variceal bleeding, disseminated intravascular coagulation (DIC), and peritonitis (Moskovitz, 1990; Scholz et al, 1989). PVS has also been overshadowed by the widespread availability of both TIPS and liver transplantation; in their own way, each is effective for refractory ascites. Nonetheless, PVS continues to have a role in the management of ascites in children, in the management of chylous ascites, and in patients for whom neither TIPS nor liver transplantation is feasible, such as those with extensive hepatic metastasis.
Liver Transplantation
Liver transplantation is effective in both relieving ascites and improving survival (see Chapter 97A). Although cases of persistent ascites have been reported after liver transplantation (Urbani et al, 2003), these cases are the exception. In all patients with refractory cirrhotic ascites, consideration should be given to liver transplantation as a treatment option.
Complications
Hepatic Hydrothorax
Hepatic hydrothorax is defined as a recurrent pleural effusion in patients with end-stage liver disease and portal hypertension in the absence of comorbid cardiac or pulmonary disease (Strauss & Boyer, 1997). Theories proposed to explain the development of hydrothorax include transdiaphragmatic leakage of fluid from lymphatic channels and azygos vein hypertension (Roussos et al, 2007). However, since 1955, it has been known that those who have a hydrothorax develop frequently have defects in the diaphragm that permit egress of ascites into the negative-pressure space of the pleural cavity (Emerson & Davies, 1955). It has been proposed that intrabdominal pressure from ascites leads to herniation of the peritoneum through gaps in the diaphragmatic muscle that eventually rupture into the pleural space. This communication has been further shown by following the passage of various tracers—air, dyes, and radiolabeled substances—from the peritoneal to pleural compartments and by direct thorascopic visualization of the defects (Benet et al, 1992; Datta et al, 1984; Huang et al, 2005). Hydrothorax is uncommon; its reported incidence is 5% to 12%, which is remarkably similar to the incidence of hydrothorax with continuous ambulatory peritoneal dialysis (Gur et al, 2004; Lew, 2010). The effusions are mostly right sided, but they can be left sided or bilateral and can be found in patients with minimal or no abdominal ascites (Rubinstein et al, 1985).
Hepatic hydrothorax should be suspected in any patient with cirrhosis and portal hypertension with a pleural effusion. Diagnostic thoracentesis should be performed as a starting point in the evaluation. The hepatic hydrothorax is transudative by traditional criteria of Light and colleagues (1972). Tracking the migration of technetium-99m–labeled albumin or sulfur colloid has been proposed and used to establish the diagnosis of hydrothorax (Holt et al, 1999), although in practice, such tests are rarely used. Rather, diagnosis is reasonable in the setting of a rapidly recurring transudative effusion in a patient with cirrhosis in whom other diseases that would predispose to transudative effusion have been excluded. If doubt persists, measurement of the hepatic venous pressure gradient can help support the diagnosis and exclude right-sided heart failure.
Therapeutic thoracentesis is safe and can provide immediate relief from dyspnea (Castellote et al, 2001; Xiol et al, 2001). By comparison, tube thoracostomy, also known as chest tube placement, should be avoided if at all possible. Prominent complications associated with chest tube placement for hydrothorax include infection and acute renal failure, the latter likely reflecting large volume losses. Chest tube placement is also associated with poor outcomes in small, retrospective studies (Orman & Lok, 2009).
The principles of management of a hydrothorax are similar to those of ascites. Dietary salt restriction followed by diuretics is the first line of treatment. Even if these treatments are modestly successful, they may still be inadequate to relieve symptoms given the small capacitance of the pleural space. In these situations, TIPS should be considered early. Although the data supporting a role for TIPS in hepatic hydrothorax are not as certain as those for ascites, in one study of 73 patients undergoing TIPS for hepatic hydrothorax, a favorable clinical response was found in 79% and 75% at 1 and 6 months, respectively (Dhanasekaran et al, 2009). Not surprisingly, liver transplantation is also an effective treatment of hydrothorax.
Surgical repair of the diaphragmatic defects has been described in small series of patients (Mouroux et al, 1996). Obvious limitations to the procedure are the operative morbidity and mortality in patients with end-stage liver disease. Management of hydrothorax is particularly challenging in individuals for whom TIPS is contraindicated and in those whose hydrothorax is refractory to TIPS. One therapy used for refractory hydrothorax is tunneled pleural catheters (Park et al, 1997), which are used for the management of refractory malignant effusions. The risk of infection is theoretically reduced because the catheters are tunneled and it is a closed system. Furthermore, because they are designed for periodic use, the rate of fluid loss can be controlled. Catheters have also been proposed as useful in the palliative care of patients with end-stage liver disease (Sanchez & Talwalkar, 2006), but further studies are needed to determine whether catheters have any role beyond that of palliation.
Spontaneous Bacterial Peritonitis
Spontaneous bacterial infection of cirrhotic ascites in the absence of suppurative infection or bowel perforation was first described by Conn and Fessel in 1971. Since then, this condition, termed spontaneous bacterial peritonitis (SBP), has emerged as one of the most common and feared complications of cirrhotic ascites. The prevalence and consequences of SBP are substantial: between 10% and 27% of patients with cirrhosis who have ascites will have SBP at the time of hospitalization (Guevara et al, 1998). It is also both a potentially lethal complication of cirrhosis and a marker of decreased survival. Twenty years ago, studies of patients presenting with their first episode of SBP reported a mortality rate of 47% (Tito et al, 1988). Those who survived the immediate infection still faced a high risk of dying, and in a majority of these late deaths, renal failure was judged to have played a significant role.
The forces that promote infection of ascites remain incompletely understood. The most common organisms to cause SBP are enteric gram-negative aerobic bacteria, and studies of cirrhosis in animal models show increased bacterial translocation (Casafont et al, 1997). The cirrhotic host may also be uniquely susceptible to infection as a combined effect of decreased reticuloendothelial and leukocyte function (Rimola et al, 1984) and diminished opsonic activity of ascites fluid (Runyon, 1988). The opsonic activity correlates with ascites protein levels, and patients with ascites protein less than 10 g/L have a greater probability of developing peritonitis than do those with higher ascites protein levels (Runyon, 1986).
Paracentesis to exclude SBP should be performed in all subjects with new-onset ascites and in those with a change in clinical condition. The diagnosis of SBP rests on the demonstration of an abnormally high neutrophil count in the ascites fluid or culture of an organism from the fluid. The combination of the two is properly termed spontaneous bacterial peritonitis. By comparison, culture-negative neutrascites (CNNA) is the appropriate term when neutrascites alone is present. These terms notwithstanding, the natural history of SBP and CNNA are indistinguishable and should be treated in a similar fashion (Runyon, 1990). Bacterascites is the term applied to a positive ascites culture in the absence of neutrascites. Limited information on this subset of patients suggests that the infection may be transient (Pelletier et al, 1991).
No fewer than 747 citations have been reported to test the diagnostic accuracy of neutrophil counts in the diagnosis of SBP (Wong et al, 2008). The threshold that is commonly accepted is a neutrophil count greater than 250 polymorphonuclear cells (PMNs)/µL. If the ascites is bloody or the paracentesis traumatic, the neutrophil counts in the fluid should be adjusted for the number of red blood cells present. Inoculating ascites directly into blood culture media at the bedside increases the likelihood that organism will be recovered (Runyon et al, 1990). It has been estimated that for every five patients whose ascites is immediately inoculated into culture media rather than delayed by inoculation in the microbiology lab, one additional bacterial infection will be detected (Wong et al, 2008).
Particularly high neutrophil counts or the recovery of multiple microbiologic organisms should raise concern for secondary peritonitis that would reflect either a suppurative abdominal infection or bowel perforation. Measurement of ascites fluid protein, LDH, and glucose can help distinguish between spontaneous and secondary peritonitis. Secondary peritonitis should be strongly suspected when any two of the three following conditions are met: 1) glucose levels are less than 50 mg/dL, 2) ascites protein concentration is greater than 1 g/dL, and 3) LDH is greater than the upper limit of the reference range of serum LDH (Akriviadis & Runyon, 1990).
Antimicrobial therapy should be promptly started after SBP is diagnosed. The initial treatment should be parenteral and cover the typical organisms associated with SBP. In common practice, the antibiotics of choice are third-generation cephalosporins, particularly cefotaxime (2 g/12 h) (Rimola et al, 1995) and ceftriaxone (1 to 2 g/24 h). Other antibiotic choices may be appropriate when chosen because of local microbial resistance patterns, medication allergies, or the patient’s prior pattern of infection. Five days of therapy are as efficacious as 10 days of therapy (Runyon et al, 1991). Antifungal therapy is generally not required as empiric, first-line therapy. This treatment may, however, have a role in surgical patients who develop SBP while receiving antibacterial therapy, patients with peritonitis despite adequate antibacterial therapy, or in patients with secondary peritonitis.
The administration of intravenous albumin has emerged as a critical adjunct to antibiotics for the treatment of SBP. The major benefit of albumin is in the prevention of renal dysfunction, which occurs in as many as 30% of those with SBP despite effective antibiotic therapy. In a trial comparing a control group that received antibiotics alone to a group that received albumin and antibiotics, the rates of renal impairment were reduced from 33% to 10%, and the hospital mortality rate was reduced from 29% to 10%, respectively (Sort et al, 1999). The dose of albumin used was 1.5 g/kg body weight at diagnosis and 1 g/kg body weight on day 3. Subgroup analysis suggests that patients with preexisting renal impairment or serum bilirubin level greater than 4 mg/dL are most likely to benefit from this intervention.
Antibiotic prophylaxis has also been proposed for subjects with prior episodes of SBP or in those with an ascites protein content less than 10 g/L. A Cochrane review of the subject, limited to those without GI bleeding, found that antibiotic prophylaxis was associated with reductions in both episodes of SBP and death, with very few reported adverse events; however, the study also raised concern about the methodology of many of the studies used to reach the conclusion (Cohen et al, 2009).
Aalami OO, et al. Chylous ascites: a collective review. Surgery. 2000;128(5):761-778.
Aguado JM, et al. Tuberculous peritonitis: a study comparing cirrhotic and noncirrhotic patients. J Clin Gastroenterol. 1990;12(5):550-554.
Ajani UA, et al. Sodium intake among people with normal and high blood pressure. Am J Prev Med. 2005;29(5 Suppl 1):63-67.
Akriviadis EA, Runyon BA. Utility of an algorithm in differentiating spontaneous from secondary bacterial peritonitis. Gastroenterology. 1990;98(1):127-133.
Arroyo V, et al. Plasma renin activity and urinary sodium excretion as prognostic indicators in nonazotemic cirrhosis with ascites. Ann Intern Med. 1981;94(2):198-201.
Arroyo V, et al. Sympathetic nervous activity, renin–angiotensin system and renal excretion of prostaglandin E2 in cirrhosis: relationship to functional renal failure and sodium and water excretion. Eur J Clin Invest. 1983;13(3):271-278.
Arroyo V, et al. Antidiuretic hormone and the pathogenesis of water retention in cirrhosis with ascites. Semin Liver Dis. 1994;14(1):44-58.
Arroyo V, et al. Definition and diagnostic criteria of refractory ascites and hepatorenal syndrome in cirrhosis. International Ascites Club. Hepatology. 1996;23(1):164-176.
Aslam N, Marino CR. Malignant ascites: new concepts in pathophysiology, diagnosis, and management. Arch Intern Med. 2001;161(22):2733-2737.
Bauer M, et al. Functional significance of endothelin B receptors in mediating sinusoidal and extrasinusoidal effects of endothelins in the intact rat liver. Hepatology. 2000;31(4):937-947.
Benet A, et al. Diagnosis of hepatic hydrothorax in the absence of ascites by intraperitoneal injection of 99 m-Tc-Fluor colloid. Postgrad Med J. 1992;68(796):153.
Bernardi M, et al. Aldosterone related blood volume expansion in cirrhosis before and during the early phase of ascites formation. Gut. 1983;24(8):761-766.
Blendis L, Wong F. Intravenous albumin with diuretics: protean lessons to be learnt? J Hepatol. 1999;30(4):727-730.
Boyer TD, Haskal ZJ. The role of transjugular intrahepatic portosystemic shunt (TIPS) in the management of portal hypertension: update 2009. Hepatology. 2009;51(1):306.
Campbell MS, et al. Quality of life in refractory ascites: transjugular intrahepatic portal-systemic shunting versus medical therapy. Hepatology. 2005;42(3):635-640.
Cardenas A, Ginès P, Runyon BA. Is albumin infusion necessary after large volume paracentesis? Liver Int. 2009;29(5):636-640.
Casafont F, et al. Influence of malnutrition on the prevalence of bacterial translocation and spontaneous bacterial peritonitis in experimental cirrhosis in rats. Hepatology. 1997;25(6):1334-1337.
Castellote J, et al. Complications of thoracentesis in cirrhotic patients with pleural effusion. Rev Esp Enferm Dig. 2001;93(9):566-575.
Cohen MJ, et al. Antibiotic prophylaxis for spontaneous bacterial peritonitis in cirrhotic patients with ascites, without gastro-intestinal bleeding. Cochrane Database Syst Rev. 2, 2009. CD004791
Conn HO, Fessel JM. Spontaneous bacterial peritonitis in cirrhosis: variations on a theme. Medicine (Baltimore). 1971;50(3):161-197.
Cook NR. Salt intake, blood pressure and clinical outcomes. Curr Opin Nephrol Hypertens. 2008;17(3):310-314.
D’Amico G, et al. Survival and prognostic indicators in compensated and decompensated cirrhosis. Dig Dis Sci. 1986;31(5):468-475.
Datta N, et al. Radionuclide demonstration of peritoneal–pleural communication as a cause for pleural fluid. JAMA. 1984;252(2):210.
Dhanasekaran R, et al. Transjugular intrahepatic portosystemic shunt for symptomatic refractory hepatic hydrothorax in patients with cirrhosis. Am J Gastroenterol. 2010;105(3):635-641.
Emerson PA, Davies JH. Hydrothorax complicating ascites. Lancet. 1955;268(6862):487-488.
Fernandez M, et al. Reversal of portal hypertension and hyperdynamic splanchnic circulation by combined vascular endothelial growth factor and platelet-derived growth factor blockade in rats. Hepatology. 2007;46(4):1208-1217.
Ferral H, et al. Survival after elective transjugular intrahepatic portosystemic shunt creation: prediction with model for end-stage liver disease score. Radiology. 2004;231(1):231-236.
Forbes SJ, et al. A significant proportion of myofibroblasts are of bone marrow origin in human liver fibrosis. Gastroenterology. 2004;126(4):955-963.
Friedman SL, et al. Isolated hepatic lipocytes and Kupffer cells from normal human liver: morphological and functional characteristics in primary culture. Hepatology. 1992;15(2):234-243.
Garcia NJr, et al. Systemic and portal hemodynamic effects of anandamide. Am J Physiol Gastrointest Liver Physiol. 2001;280(1):G14-G20.
Gentilini P, et al. Albumin improves the response to diuretics in patients with cirrhosis and ascites: results of a randomized, controlled trial. J Hepatol. 1999;30(4):639-645.
Ginès A, et al. Randomized trial comparing albumin, dextran 70, and polygeline in cirrhotic patients with ascites treated by paracentesis. Gastroenterology. 1996;111(4):1002-1010.
Ginès P, et al. Comparison of paracentesis and diuretics in the treatment of cirrhotics with tense ascites: results of a randomized study. Gastroenterology. 1987;93(2):234-241.
Ginès P, et al. Compensated cirrhosis: natural history and prognostic factors. Hepatology. 1987;7(1):122-128.
Ginès P, et al. Transjugular intrahepatic portosystemic shunting versus paracentesis plus albumin for refractory ascites in cirrhosis. Gastroenterology. 2002;123(6):1839-1847.
Ginès P, et al. Management of cirrhosis and ascites. N Engl J Med. 2004;350(16):1646-1654.
Ginès P, et al. Effects of satavaptan, a selective vasopressin V(2) receptor antagonist, on ascites and serum sodium in cirrhosis with hyponatremia: a randomized trial. Hepatology. 2008;48(1):204-213.
Guevara M, et al. Transjugular intrahepatic portosystemic shunt in hepatorenal syndrome: effects on renal function and vasoactive systems. Hepatology. 1998;28(2):416-422.
Gupta TK, et al. Endothelial dysfunction and decreased production of nitric oxide in the intrahepatic microcirculation of cirrhotic rats. Hepatology. 1998;28(4):926-931.
Gur C, Ilan Y, Shibolet O. Hepatic hydrothorax: pathophysiology, diagnosis and treatment: review of the literature. Liver Int. 2004;24(4):281-284.
Haskal ZJ, et al. Quality improvement guidelines for transjugular intrahepatic portosystemic shunts. J Vasc Interv Radiol. 2003;14(9 Pt 2):S265-S270.
Hillebrand DJ, et al. Ascitic fluid adenosine deaminase insensitivity in detecting tuberculous peritonitis in the United States. Hepatology. 1996;24(6):1408-1412.
Hoefs JC. Serum protein concentration and portal pressure determine the ascitic fluid protein concentration in patients with chronic liver disease. J Lab Clin Med. 1983;102(2):260-273.
Holt KA, Oliviera E, Rohatgi PK. Hepatic hydrothorax demonstration by Tc-99 m sulfur colloid ascites scan. Clin Nucl Med. 1999;24(8):609.
Horisberger JD, Giebisch G. Potassium-sparing diuretics. Ren Physiol. 1987;10(3-4):198-220.
Huang P-M, et al. The morphology of diaphragmatic defects in hepatic hydrothorax: thoracoscopic finding. J Thorac Cardiovasc Surg. 2005;130(1):141-145.
Huet PM, et al. Assessment of liver microcirculation in human cirrhosis. J Clin Invest. 1982;70(6):1234-1244.
Iwakiri Y, Groszmann RJ. The hyperdynamic circulation of chronic liver diseases: from the patient to the molecule. Hepatology. 2006;43(2 Suppl 1):S121-S131.
Kalluri R, Neilson EG. Epithelial–mesenchymal transition and its implications for fibrosis. J Clin Invest. 2003;112(12):1776-1784.
Leveen HH, et al. Peritoneo-venous shunting for ascites. Ann Surg. 1974;180(4):580-591.
Lew SQ. Hydrothorax: pleural effusion associated with peritoneal dialysis. Perit Dial Int. 2010;30(1):13-18.
Light RW, et al. Pleural effusions: the diagnostic separation of transudates and exudates. Ann Intern Med. 1972;77(4):507-513.
Liu S, et al. A crucial role for GRK2 in regulation of endothelial cell nitric oxide synthase function in portal hypertension. Nat Med. 2005;11(9):952-958.
Lott JP. Renal failure in cirrhosis. N Engl J Med. 2010;362(1):79. author reply 80-71
Lui SL, et al. Tuberculosis infection in Chinese patients undergoing continuous ambulatory peritoneal dialysis. Am J Kidney Dis. 2001;38(5):1055-1060.
Malinchoc M, et al. A model to predict poor survival in patients undergoing transjugular intrahepatic portosystemic shunts. Hepatology. 2000;31(4):864-871.
Mittal MK, et al. Nitric oxide modulates hepatic vascular tone in normal rat liver. Am J Physiol. 1994;267(3 Pt 1):G416-422.
Moskovitz M. The peritoneovenous shunt: expectations and reality. Am J Gastroenterol. 1990;85(8):917-929.
Mouroux J, et al. Management of pleural effusion of cirrhotic origin. Chest. 1996;109(4):1093-1096.
Orman ES, Lok AS. Outcomes of patients with chest tube insertion for hepatic hydrothorax. Hepatol Int. 2009;3(4):582-586.
Ortega R, et al. Terlipressin therapy with and without albumin for patients with hepatorenal syndrome: results of a prospective, nonrandomized study. Hepatology. 2002;36(4 Pt 1):941-948.
Pan JJ, et al. Factors predicting survival after transjugular intrahepatic portosystemic shunt creation: 15 years’ experience from a single tertiary medical center. J Vasc Interv Radiol. 2008;19(11):1576-1581.
Pare P, Talbot J, Hoefs JC. Serum-ascites albumin concentration gradient: a physiologic approach to the differential diagnosis of ascites. Gastroenterology. 1983;85(2):240-244.
Park MDSZ, et al. Treatment of refractory, nonmalignant hydrothorax with a pleurovenous shunt. Ann Thorac Surg. 1997;63(6):1777-1779.
Pelletier G, et al. Asymptomatic bacterascites: is it spontaneous bacterial peritonitis? Hepatology. 1991;14(1):112-115.
Perez-Ayuso RM, et al. Randomized comparative study of efficacy of furosemide versus spironolactone in nonazotemic cirrhosis with ascites: relationship between the diuretic response and the activity of the renin–aldosterone system. Gastroenterology. 1983;84(5 Pt 1):961-968.
Pinzani M, et al. Endothelin 1 is overexpressed in human cirrhotic liver and exerts multiple effects on activated hepatic stellate cells. Gastroenterology. 1996;110(2):534-548.
Planas R, et al. Acetylsalicylic acid suppresses the renal hemodynamic effect and reduces the diuretic action of furosemide in cirrhosis with ascites. Gastroenterology. 1983;84(2):247-252.
Pockros PJ, Reynolds TB. Rapid diuresis in patients with ascites from chronic liver disease: the importance of peripheral edema. Gastroenterology. 1986;90(6):1827-1833.
Rajan DK, Haskal ZJ, Clark TW. Serum bilirubin and early mortality after transjugular intrahepatic portosystemic shunts: results of a multivariate analysis. J Vasc Interv Radiol. 2002;13(2 Pt 1):155-161.
Rector WGJr. Spontaneous chylous ascites of cirrhosis. J Clin Gastroenterol. 1984;6(4):369-372.
Reuben A. My cup runneth over. Hepatology. 2004;40(2):503-507.
Rimola A, et al. Reticuloendothelial system phagocytic activity in cirrhosis and its relation to bacterial infections and prognosis. Hepatology. 1984;4(1):53-58.
Rimola A, et al. Two different dosages of cefotaxime in the treatment of spontaneous bacterial peritonitis in cirrhosis: results of a prospective, randomized, multicenter study. Hepatology. 1995;21(3):674-679.
Rosch J, et al. Transjugular intrahepatic portacaval shunt: an experimental work. Am J Surg. 1971;121(5):588-592.
Rössle M, et al. A comparison of paracentesis and transjugular intrahepatic portosystemic shunting in patients with ascites. N Engl J Med. 2000;342(23):1701-1707.
Roussos A, et al. Hepatic hydrothorax: pathophysiology diagnosis and management. J Gastroenterol Hepatol. 2007;22(9):1388-1393.
Rubin RA, et al. Transjugular intrahepatic portosystemic shunting: decreased survival for patients with high APACHE II scores. Am J Gastroenterol. 1995;90(4):556-563.
Rubinstein D, McInnes IE, Dudley FJ. Hepatic hydrothorax in the absence of clinical ascites: diagnosis and management. Gastroenterology. 1985;88(1 Pt 1):188-191.
Ruiz-del-Arbol L, et al. Paracentesis-induced circulatory dysfunction: mechanism and effect on hepatic hemodynamics in cirrhosis. Gastroenterology. 1997;113(2):579-586.
Runyon BA. Low-protein-concentration ascitic fluid is predisposed to spontaneous bacterial peritonitis. Gastroenterology. 1986;91(6):1343-1346.
Runyon BA. Patients with deficient ascitic fluid opsonic activity are predisposed to spontaneous bacterial peritonitis. Hepatology. 1988;8(3):632-635.
Runyon BA. Monomicrobial nonneutrocytic bacterascites: a variant of spontaneous bacterial peritonitis. Hepatology. 1990;12(4 Pt 1):710-715.
Runyon BA. Management of adult patients with ascites due to cirrhosis: an update. Hepatology. 2009;49(6):2087-2107.
Runyon BA, et al. Bedside inoculation of blood culture bottles with ascitic fluid is superior to delayed inoculation in the detection of spontaneous bacterial peritonitis. J Clin Microbiol. 1990;28(12):2811-2812.
Runyon BA, et al. Short-course versus long-course antibiotic treatment of spontaneous bacterial peritonitis: a randomized controlled study of 100 patients. Gastroenterology. 1991;100(6):1737-1742.
Runyon BA, et al. The serum-ascites albumin gradient is superior to the exudate-transudate concept in the differential diagnosis of ascites. Ann Intern Med. 1992;117(3):215-220.
Salerno F, et al. Randomized controlled study of TIPS versus paracentesis plus albumin in cirrhosis with severe ascites. Hepatology. 2004;40(3):629-635.
Sanai FM, Bzeizi KI. Systematic review: tuberculous peritonitis—presenting features, diagnostic strategies and treatment. Aliment Pharmacol Ther. 2005;22(8):685-700.
Sanchez W, Talwalkar JA. Palliative care for patients with end-stage liver disease ineligible for liver transplantation. Gastroenterol Clin North Am. 2006;35(1):201-219.
Sanyal AJ, et al. The North American Study for the Treatment of Refractory Ascite Group. Gastroenterology. 2003;124(3):634-641.
Scholz DG, Nagorney DM, Lindor KD. Poor outcome from peritoneovenous shunts for refractory ascites. Am J Gastroenterol. 1989;84(5):540-543.
Shah V, et al. Liver sinusoidal endothelial cells are responsible for nitric oxide modulation of resistance in the hepatic sinusoids. J Clin Invest. 1997;100(11):2923-2930.
Shah V, et al. Impaired endothelial nitric oxide synthase activity associated with enhanced caveolin binding in experimental cirrhosis in the rat. Gastroenterology. 1999;117(5):1222-1228.
Shakil AO, et al. Diagnostic features of tuberculous peritonitis in the absence and presence of chronic liver disease: a case control study. Am J Med. 1996;100(2):179-185.
Siddiqui RA, et al. Evaluation of fibronectin as a marker of malignant ascites. J Gastroenterol Hepatol. 1992;7(2):161-164.
Sieber CC, Groszmann RJ. Nitric oxide mediates hyporeactivity to vasopressors in mesenteric vessels of portal hypertensive rats. Gastroenterology. 1992;103(1):235-239.
Sieber CC, Lopez-Talavera JC, Groszmann RJ. Role of nitric oxide in the in vitro splanchnic vascular hyporeactivity in ascitic cirrhotic rats. Gastroenterology. 1993;104(6):1750-1754.
Sola-Vera J, et al. Randomized trial comparing albumin and saline in the prevention of paracentesis-induced circulatory dysfunction in cirrhotic patients with ascites. Hepatology. 2003;37(5):1147-1153.
Sort P, et al. Effect of intravenous albumin on renal impairment and mortality in patients with cirrhosis and spontaneous bacterial peritonitis. N Engl J Med. 1999;341(6):403-409.
Soupart A, et al. Successful long-term treatment of hyponatremia in syndrome of inappropriate antidiuretic hormone secretion with satavaptan (SR121463B), an orally active nonpeptide vasopressin V2-receptor antagonist. Clin J Am Soc Nephrol. 2006;1(6):1154-1160.
Stanley MM, et al. Peritoneovenous shunting as compared with medical treatment in patients with alcoholic cirrhosis and massive ascites. Veterans Administration Cooperative Study on Treatment of Alcoholic Cirrhosis with Ascites. N Engl J Med. 1989;321(24):1632-1638.
Strauss RM, Boyer TD. Hepatic hydrothorax. Semin Liver Dis. 1997;17(3):227-232.
Tito L, et al. Recurrence of spontaneous bacterial peritonitis in cirrhosis: frequency and predictive factors. Hepatology. 1988;8(1):27-31.
Urbani L, et al. Management of massive and persistent ascites and/or hydrothorax after liver transplantation. Transplant Proc. 2003;35(4):1473-1475.
van den Ouweland AM, et al. Colocalization of the gene for nephrogenic diabetes insipidus (DIR) and the vasopressin type 2 receptor gene (AVPR2) in the Xq28 region. Genomics. 1992;13(4):1350-1352.
Wong CL, et al. Does this patient have bacterial peritonitis or portal hypertension? How do I perform a paracentesis and analyze the results? JAMA. 2008;299(10):1166-1178.
Xiol X, et al. Usefulness and complications of thoracentesis in cirrhotic patients. Am J Med. 2001;111(1):67-69.
[/level-membership-for-surgery-category][not-level-membership-for-surgery-category]
Chapter 74 Management of ascites in cirrhosis and portal hypertension
Historic Overview
One of the most visible manifestations of liver disease is that of ascites, broadly defined as the pathologic accumulation of fluid within the peritoneal cavity (Runyon, 2009). The almost reflexive association of ascites with liver disease notwithstanding, ascitic fluid volume and composition demonstrate a variability that reflects the range of disease processes, from benign to sinister, from whence it originates.
Descriptions of ascites exist in both human history and prehistory. Indian medical treatises from 1500 bce, Mayan figurines with protuberant abdomens and everted umbilici, and the writings of Hippocrates all testify to the experience of varied cultures with this problem. Even the term ascites—derived from the Greek askos, a bag made of leather or sheepskin used to contain liquids—reflects its ancient origins (Reuben, 2004).
High-volume ascites is readily evident on physical examination; however, smaller volumes may evade detection, particularly in the obese. Ultrasound is the best imaging modality for detection of ascites. It is inexpensive, avoids ionizing radiation, and has the added benefit of providing information on liver architecture and portal vein patency (see Chapter 13).
All clinicians should maintain a low threshold for performing paracentesis. The procedure is necessary in the diagnostic evaluation of newly developed ascites, or when there is a change in the clinical condition in an individual with cirrhosis and ascites. A percutaneous approach that avoids the epigastric vessels offers an avascular plane that is safe even in coagulopathic and thrombocytopenic patients (Wong et al, 2008).
Portal Hypertension and Mechanisms of Ascites Formation
Ascites is the most common complication of portal hypertension arising from cirrhosis, occurring at an annual incidence of 1% (see Chapter 70A, Chapter 70B ; Ginès et al, 1987). Its development heralds a significant change in clinical condition, with a median survival of 50% over 2 years (D’Amico et al, 1986). Portal hypertension can arise from cirrhotic and noncirrhotic causes, although as a manifestation of portal hypertension, ascites is most common in disorders that increase pressures within the hepatic sinusoids, either from sinusoidal processes (cirrhosis) or postsinusoidal processes (heart failure, hepatic venous obstruction). By comparison, ascites is uncommon in presinusoidal portal hypertension.
The normal liver architecture consists of sinusoids that convey blood from the portal vein to the central vein. The sinusoids are separated from cords of hepatocytes by a fenestrated endothelium through which oxygen, cells, and plasma components are permitted to diffuse. The principal collagen-producing cells of the liver, the stellate cells, reside within the space of Disse, and activation of these cells leads to collagen deposition (Friedman et al, 1992). Other cells that contribute to the extracellular matrix within the liver are bone marrow–derived myofibroblasts and fibroblasts derived from epithelial-mesenchymal transition (Forbes et al, 2004; Kalluri & Neilson, 2003).
In cirrhosis, extracellular basement membrane deposition of collagen fibers within the space of Disse results in capillarization of the hepatic sinusoids (Huet et al, 1982). The resulting architectural changes cause a static increase in pressure within the splanchnic circulation (see Chapter 6).
The endothelial cells of the liver also play an important role in controlling dynamic changes to the hepatic microcirculation through the elaboration of nitric oxide (Mittal et al, 1994; Shah et al, 1997) and expression of endothelin-1 receptors (Bauer et al, 2000). Decreased endothelial nitric oxide synthase (eNOS) activity (Gupta et al, 1998; Shah et al, 1999), increased eNOS inactivator activity (Liu et al, 2005), and endothelin-1 overexpression in cirrhosis (Pinzani et al, 1996) have all been proposed as mechanisms that promote endothelial cell dysfunction in cirrhosis (see Chapter 6).
In the splanchnic circulation outside of the hepatic environment, different but equally important changes occur that contribute to portal hypertension. The most prominent of these is splanchnic arterial vasodilation. In experimental models of cirrhosis, vasodilation is mediated by NO-dependent (Sieber & Groszmann, 1992; Sieber et al, 1993) and NO-independent processes. The NO-independent processes include those related to endogenous vasodilatory cannabanoids (Garcia et al, 2001) and overexpression of vascular endothelial growth factor (VEGF) and VEGF receptor-2 to promote splanchnic angiogenesis, thereby augmenting blood flow in the splanchnic circulation (Fernandez et al, 2007). Vasodilation in the splanchnic circulation would decrease the effective arterial circulation if not for compensatory increases in cardiac output (Iwakiri & Groszmann, 2006).
With progression of portal hypertension, other compensatory mechanisms are activated to maintain the arterial circulation in the face of even greater increases in vasodilation and declines in cardiac output. These mechanisms include activation of the renin-angiotensin system and sympathetic nervous system to stimulate sodium retention by the kidneys (Arroyo et al, 1983). The nonosmotic release of arginine vasopressors is an additional compensatory mechanism to increase the effective arterial volume, even at the expense of tonicity; this is reflected in the development of hyponatremia (Arroyo et al, 1994). The cumulative effect of increased hydrostatic pressure in the hepatic microcirculation in the setting of increased splanchnic volume is that of hepatic lymph formation in excess of its removal; the resulting excess fluid weeps into the peritoneal cavity and is recognized as ascites (Fig. 74.1).
Characteristics and Evaluation of Ascites
Ascitic fluid in cirrhotics is transparent but will generally take on a yellow or amber color. The fluid generally has a low leukocyte (<100 µL/mm) and red blood cell content. The fluid protein is typically less than 2.5 mg/dL, and the protein content varies inversely with the severity of the portal hypertension (Hoefs, 1983). Measurement of the serum albumin ascites gradient (SAAG) is both a highly accurate and clinically facile technique for assessing the origins of ascites. The SAAG is calculated by subtracting the concentration of albumin in the ascites from that in the plasma (Pare et al, 1983). With an approximately 97% accuracy, a gradient greater than 1.1 g/dL indicates the presence of portal or sinusoidal hypertension (Runyon et al, 1992).
Opacification of ascitic fluid can arise from a number of disparate processes. Bloody ascites (hematocrit >0.5) can be seen in traumatic paracentesis or in the spontaneous rupture of hepatocellular cancer. Chylous ascites is milky in appearance from increased concentration of chylomicron-rich triglyceride (Aalami et al, 2000). It arises from processes that disrupt lymphatic flow, most commonly lymphangiectasia and lymphoma, but it can also occur with abdominal trauma and surgical disruption of the cysterna chyli. Cirrhotic ascites can also take on a chylous appearance, owing to rupture of abdominal lymphatics from portal hypertension (Rector, 1984). In these cases, known as pseudochylous ascites, the triglyceride concentration is generally less than the threshold value of 110 mg/dL found in pure cases of chylous ascites.
Both malignancy and tuberculosis peritonitis can result in ascites, and in both circumstances the SAAG is less than 1.1 g/dL; however, confusion may arise when liver disease coexists, as in the case of tuberculosis and alcohol-related liver disease (Aguado et al, 1990; Shakil et al, 1996). The diagnosis of malignant ascites is established by the finding of cancer cells within the peritoneal cavity. This can be done by conventional cytology with a diagnostic sensitivity of 40% to 60% (Siddiqui et al, 1992). The accuracy of cytology can be improved when combined with immunohistologic staining (Aslam & Marino, 2001).
The peritoneum is a common site of involvement of tuberculosis (TB), and in the United States, the peritoneum is the sixth most common extrapulmonary site. Peritoneal cell counts typically vary between 500 and 1500 cells/mm3 with a lymphocyte predominance in 68% (Sanai & Bzeizi, 2005), although the absence of a lymphocyte predominance does not exclude TB, particularly in patients with underlying renal failure, in whom the cells are mostly neutrophils (Lui et al, 2001). Mycobacterial culture of the fluid has a diagnostic sensitivity of 34% and requires several weeks of intubation. Measurement of adenosine deaminase activity in the peritoneal fluid has been proposed as another diagnostic test with high sensitivity and specificity, although the positive predictive value has been reported to be low in the setting of concomitant cirrhosis (Hillebrand et al, 1996). Of all diagnostic strategies, laparoscopy with peritoneal biopsy affords the highest sensitivity and specificity and permits exclusion of other granulomatous and nongranulomatous processes that can produce a low-SAAG ascites. The ascites concentration of lactate dehydrogenase (LDH) tends to be higher than that of serum LDH in malignant ascites and less than half that of serum in tuberculous peritonitis.
Management
Dietary Sodium Restriction
Avid renal sodium retention is the initial response to splanchnic arterial vasodilation; thus initial treatment strategies involve tipping the balance in favor of a net loss of sodium; this is most simply accomplished with dietary sodium restriction. The estimated mean sodium consumption among nonhypertensive adults in the United States is 3600 mg/day (Ajani et al, 2005). Those with ascites are commonly advised to restrict dietary salt intake to 1.5 to 2 g per day (67 to 87 mmoL/day), the lower value of which is considered adequate for daily needs. In patients with mild degrees of ascites, sodium restriction may be singularly effective. This group usually has baseline rates of sodium excretion of at least 40 mEq/L per day and normal plasma sodium concentrations (Arroyo et al, 1981). Although seemingly a simple intervention, success with dietary salt restriction requires counseling and vigilance. Most of the sodium present is added during food processing, and the patient’s actual consumption may not be apparent to them unless food labeling is read and understood (Cook, 2008).
Medical Management
Diuretics
Most patients will at some point require diuretics. Between 500 and 750 mL per day of ascites can be mobilized without intravascular depletion, and greater amounts of ascitic fluid losses may be tolerable in the presence of edema, which tends to act as a buffer (Pockros & Reynolds, 1986). The avoidance of rapid fluid loss is critical because precipitous and excessive volume contraction can give rise to hepatorenal syndrome.
Aldosterone Antagonists
The aldosterone antagonists spironolactone and amiloride (Table 74.1) can be used as either monotherapy or in combination with loop diuretics. These agents prevent sodium reabsorption in the distal tubule and cortical collecting duct. Although aldosterone antagonists are weak natriuretics, they are effective in patients with cirrhosis (Perez-Ayuso et al, 1983).
Table 74.1 Diuretics and Dosages Commonly Used in the Management of Ascites
| Medication | Dose | Comments |
|---|---|---|
| Aldosterone Antagnosists | ||
| Spironolactone | 50-400 mg daily | |
| Amiloride | 5-10 mg daily | Suitable substitute when spironolactone use is associated with painful gynecomastia |
| Loop Diuretics | ||
| Furosemide | 20-160 mg daily | Avoid intravenous use |
Urine sodium excretion and plasma aldosterone concentration are hyperbolically and inversely related in patients with cirrhosis, and a greater sensitivity to the dose-response curve is observed in those with ascites (Bernardi et al, 1983). One explanation for the effectiveness of these agents in cirrhotic ascites may be that they target the functional hyperaldosteronism that would otherwise permit sodium reabsorption in the cortical collecting tube of fluid filtered in the loop of Henle. In addition, unlike other diuretics that require access to the tubular lumen, spironolactone enters the principal cell of the collecting duct from the plasma compartment and thereby circumvents decreases in renal blood flow and hypoalbuminemia, commonly encountered in patients with cirrhosis, that might otherwise impair its activity (Horisberger & Giebisch, 1987).
Loop Diuretics
As monotherapy for ascites, loop diuretics are often unsuccessful. The reasons for this are unclear but may relate to a decreased rate of drug entry into the tubular lumen or to a compensatory increase in distal tubular sodium resorption mediated by aldosterone. By comparison, the combination of loop diuretics and aldosterone antagonists is the most commonly used combination for moderate to severe ascites and can achieve reductions in ascites beyond that of aldosterone antagonists alone. The most commonly used loop diuretic is furosemide (see Table 74.1), beginning at doses of 20 to 40 mg daily. The dose is doubled in a coordinated fashion with increases in spironolactone, until therapeutic effect is achieved. Like spironolactone, a fourfold doubling of the dose (160 mg daily) is considered a maximal dose. An added benefit of loop diuretics is that they counteract the hyperkalemia associated with aldosterone antagonists, and they may also benefit those who develop dependent edema in addition to ascites.
Albumin
Hypoalbuminemia is a frequent finding in patients with cirrhosis, and it may influence the development of edema and ascites through alterations in plasma oncotic pressure and the activity of loop diuretics in the lumen of the collecting tubule. Limited evidence supports a role for albumin administration as an adjunct to diuretic therapy in ascites that is difficult to control (Gentilini et al, 1999). However, this strategy may not be broadly applicable with the diuretics currently available or when there is recourse to other treatments (Blendis & Wong, 1999). Intravenous albumin administration does have a secure role in the prevention of renal dysfunction, which occurs in the one third of patients who develop spontaneous bacterial peritonitis (Sort et al, 1999). In addition, vasoconstrictor agents used in the treatment of hepatorenal syndrome are more effective when coadministered with albumin than when administered with saline or other crystalloids (Ortega et al, 2002). Thus albumin appears to achieve a successful volume expansion in patients with cirrhosis, greater than that that of crystalloid, which is translatable into improvement in solid clinical end points.
Arginine Vasopression Receptor Antagonists
The nonosmotic release of antidiuretic hormone (ADH) is commonly observed in patients with cirrhosis as a compensatory mechanism for the decrease in effective arterial volume that occurs with splanchnic vasodilation. ADH exerts its effects through arginine vasopressin receptor-2 (AVPR2), which is predominantly expressed in the distal convoluted tubule and collecting ducts of the kidney (van den Ouweland et al, 1992). Satavaptan is a selective AVPR2 receptor antagonist that has been tested in clinical studies of both the syndrome of inappropriate ADH release (Soupart et al, 2006) and hyponatremia occurring with cirrhosis (Ginès et al, 2008). The study in patients with cirrhosis was a multicenter, double-blind, randomized controlled study of 110 subjects with ascites and hyponatremia. Treatment with satavaptan and spironolactone was associated with improvement in ascites volume concomitant with improvements in hyponatremia more often than the placebo. The use of satavaptan and other aquaretic agents represents a promising area for pharmacologic management of ascites if these early successes are confirmed in other clinical trials.
Refractory Ascites
Ascites that persists despite dietary sodium restriction and high-dose diuretics (spironolactone 400 mg/day and furosemide 160 mg/day) is referred to as refractory ascites (Arroyo et al, 1996). Diuretic-intolerant ascites describes diuretic failure because of intolerant side effects—such as azotemia, hyponatremia, and encephalopathy—that prevent the attainment of a dose sufficient to effect an adequate ascites loss. The clinical significance of refractory ascites should not be overlooked; the survival curves of individuals with refractory ascites approximate those with type II hepatorenal syndrome (Ginès et al, 2004).
Before labeling a patient as having refractory ascites, it is important to exclude excessive sodium intake and other medications that may influence the diuretic response. In particular, nonsteroidal antiinflammatory agents, including aspirin (Planas et al, 1983), may both decrease the response of diuretics and contribute to azotemia.
Treatment options for refractory ascites includes therapeutic paracentesis, transjugular intrahepatic portosystemic shunting (TIPS) (see Chapter 76E), peritoneal shunting, and liver transplantation (see Chapter 97A).
Paracentesis
High-volume paracentesis, also known as therapeutic paracentesis, was a technique known to and practiced by physicians as far back as ancient Greece. It was also the most effective treatment in practice before the development of modern diuretics, after which the practice fell out of favor until it was reintroduced in 1987 as safe and effective (Ginès et al, 1987).
[/not-level-membership-for-surgery-category]
