5 Malignant Gliomas in Adults
Introduction
Malignant gliomas (MG) account for more than 75% of the approximately 20,500 newly diagnosed malignant primary brain tumors in the United States each year.1 They represent the most common type of malignant primary brain tumor in adults. More than half of MGs are glioblastomas (GBM), the most aggressive subtype. The majority of the remainder include anaplastic astrocytomas (AA), anaplastic oligodendrogliomas (AO), and anaplastic oligoastrocytomas (AOA).1,2 Other MG subtypes are rare. Glioblastoma is a disease of middle age, with a median age at diagnosis of 64 years. Anaplastic gliomas (AG) affect a younger adult population with a median age of 45 years.1,3 Despite being relatively uncommon, MGs are incurable and are responsible for a disproportionate share of cancer-related morbidity and mortality.4 With optimal treatment, median survival is only 12 to 15 months for GBM and 2 to 5 years for AG. Established adverse prognostic factors in MG include increasing age, GBM histology, poor performance status, and unresectable tumor.5 Recently, there have been important advances in the molecular pathogenesis of MG, some of which may provide additional prognostic information that will help in therapeutic decision-making.6–12 This chapter summarizes the pathology, pathogenesis, diagnosis, and management of adult MGs with a focus on therapy and recent therapeutic advances. This topic has been recently reviewed elsewhere.4,13
Epidemiology
The only established risk factors for MG are exposure to ionizing radiation and rare familial syndromes such as neurofibromatosis types 1 and 2, Li-Fraumeni syndrome, and Turcot syndrome.3,14 Approximately 5% of patients with malignant gliomas have a family history of gliomas. The genetic basis for most of these tumors is unknown. An international consortium, GLIOGENE, has been established to study the genetic basis of familial gliomas.15 Exposures to various carcinogens, infections such as cytomegalovirus, diet, elevated IgE, electromagnetic radiation, and cellular phones have not been conclusively linked to MG risk.3 Malignant glioma epidemiology is reviewed in detail elsewhere in this volume.
Pathology
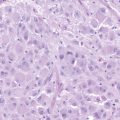
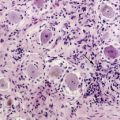
MGs are infiltrative tumors with a presumptive glial cell of origin. The World Health Organization (WHO) classification scheme defines four histologic grades for astrocytomas. These include grade I (pilocytic astrocytoma), grade II (diffuse astrocytoma), grade III (AA), and grade IV (GBM).2 Grade I tumors occur primarily in children. Grade II tumors are considered low-grade gliomas, while grade III and IV tumors are MGs. Typical histologic features of AAs include hypercellularity, nuclear atypia, and mitoses. The presence of microvascular proliferation and/or necrosis often indicates progression to GBM. Oligodendrogliomas are classified as well-differentiated oligodendrogliomas (grade II) or AOs (grade III). Mixed tumors, or oligoastrocytomas, are also classified as well-differentiated (grade II) or AOA (grade III). Tumors with oligodendroglial components may have distinctive features such as perinuclear clearing, giving rise to a “fried-egg” appearance, and a reticular pattern of blood vessel growth.
Molecular Pathogenesis
Recent studies have begun to elucidate the molecular pathogenesis of MGs.10–12,16 Such information may help to improve tumor classification and prognostication.8,9,17–19 As has been observed for many cancers, increasing malignancy in glial tumors is associated with an accumulation of genetic abnormalities that results in impairment of cell cycle control, DNA repair, and signaling.12,20 On the basis of differences in molecular pathogenesis and natural history, GBMs are often classified as primary or secondary tumors (Figure 5-1).12,20 Primary GBMs arise de novo in older patients, and have a characteristic molecular genetic profile that includes epidermal growth factor receptor (EGFR) amplification and mutations, loss of heterozygosity (LOH) of chromosome 10 q, phosphatase and tensin homolog deleted on chromosome 10 (PTEN) mutations, and p16 deletion. Secondary GBMs are much rarer, evolve from lower-grade gliomas, and typically occur in younger patients. Typical genetic abnormalities include TP53 mutations,21 platelet-derived growth factor (PDGFR) overexpression, abnormalities in the p16/retinoblastoma (Rb) pathway, and LOH 10 q.12,22 These tumors may also have mutations of the iso- citrate dehydrogenase gene.23 None of these features allow for definitive GBM classification. Primary and secondary GBMs cannot be distinguished histologically and have not been shown to respond differentially to treatment.
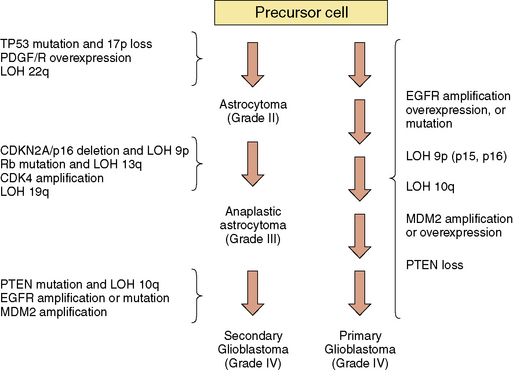
Figure 5-1 Molecular genetic changes associated with glioma progression.
Abbreviations: CDK, cyclin-dependent kinase; EGFR, epidermal growth factor receptor; LOH, loss of heterozygosity; MDM2, murine double minute 2; PDGF/R, platelet-derived growth factor/receptor; PTEN, phosphate and tensin homolog deleted on chromosome 10; Rb, retinoblastoma.
(Adapted from Wen PY, Kesari S, Drappatz J. Expert Rev Anticancer Ther 2006;6:733–54.)
Abnormalities in EGFR signaling are common in MGs.12 EGFR amplification and overexpression are observed in about 40% of primary GBMs. EGFRvIII is a constitutively active EGFR mutant that is expressed by approximately 30% of GBMs, and represents a potentially important therapeutic target because of its exclusive expression by tumor cells.12,18 Activating mutations in the extracellular domain of the EGFR are also present in a subset of GBMs.24 Activation of the PDGFR promotes tumor growth as well, and MGs frequently coexpress PDGF and PDGFR.12 Signaling through these and other growth factor receptors activates fundamental signal transduction pathways such as the Ras/mitogen-activated protein kinase (MAP-kinase) pathway and the phosphoinositide 3′-kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) pathway, both of which promote cell proliferation.12 PTEN is an endogenous inhibitor of PI3K signaling that is mutated or lost in up to 50% of GBMs.12,25 Downstream targets of these growth factor signaling pathways activate transcriptional programs for cell survival, proliferation, and invasion. Additionally, many of these pathways serve to upregulate vascular endothelial growth factor (VEGF) and angiogenesis, which has an emerging pathogenic role in MGs that is discussed in greater detail below.26,27
For cancer in general and MG specifically, there is growing interest in “personalized medicine,” which refers to the effort to individualize therapy based on a particular tumor’s molecular genetic profile.7,9 Examples of molecular predictors with probable therapeutic value are chromosomes 1 p/19 q, the loss of which may confer chemosensitivity in oligodendroglial tumors,28–30 and MGMT promoter methylation, which may predict temozolomide sensitivity in GBM.8,31 Advances in gene sequencing and expression profiling are helping to define molecular subtypes of MG that will allow for optimal selection of targeted therapeutics for individual patients.32,33
Cellular Origins
Despite recent advances, the cell of origin for MGs remains unknown. Emerging evidence suggests that neural stem cells, or related progenitor cells, may undergo malignant transformation and give rise to these tumors.16,34–38 Additionally, glioma stem cells contribute to the treatment resistance of MGs38,39; a recent study found that expression of stem cell markers such as CD133 in MG specimens may predict resistance to both radiation and chemotherapy.40 The radioresistance of stem cells is mediated by robust activation of DNA damage response pathways.41 Mechanisms that contribute to stem cell chemoresistance are different and include increased production of O6-methylguanine-DNA-methyltransferase (MGMT), overexpression of multi-drug resistance genes, and apoptosis inhibition.42–44
Diagnosis
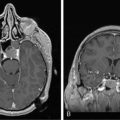
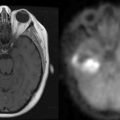
Like other space-occupying lesions, MGs often present with a combination of general- ized and focal symptoms. Generalized symptoms include headaches, mood changes, altered mental status, and psychomotor slowing. Focal symptoms reflect the location of the lesion in the brain and may include seizures, hemiparesis or hemisensory loss, and aphasia. Among the generalized symptoms, headaches are common and are frequently indistinguishable from tension headaches.45 The classical pattern of headaches that are worse in the morning and with recumbency is only occasionally reported. In the face of suspicious neurological symptoms, the possibility of MG is often suggested by findings on contrast-enhanced magnetic resonance imaging (MRI) or computerized tomography (CT). A typical appearance of MG on either MRI or CT is an irregularly enhancing mass with associated edema and mass effect. Although GBMs often have a central necrotic cavity and more peritumoral edema than AGs,46 pathological confirmation of the diagnosis is crucial, as radiographic features are currently inadequate to reliably determine histology and tumor grade.
Medical Management
Common medical and neurological problems in MG patients include seizures, peritumoral edema, venous thromboembolic disease, fatigue, mood disorders, and cognitive dysfunction.47 Patients who experience seizures require treatment with antiepileptic drugs (AED). Many of the older AEDs (e.g., phenytoin, carbamazepine, and phenobarbital) are inducers of hepatic cytochrome P450, the same enzyme system that metabolizes many chemotherapeutic agents. For this reason, newer AEDs that neither induce nor inhibit the cytochrome P450 system (e.g., gabapentin, levetiracetam, and lamotrigine) are recommended. Although the American Academy of Neurology suggests that AEDs not be routinely prescribed to brain tumor patients who have not had seizures,48 many practitioners continue to administer AED prophylaxis.49
Because many MG patients experience symptomatic peritumoral edema, corticosteroids such as dexamethasone are often required.50 Adverse effects such as Cushing syndrome and steroid myopathy frequently develop due to prolonged corticosteroid administration. This treatment also increases the risk of Pneumocystis jirovecii pneumonia. Prophylactic antibiotics are generally prescribed for these patients,47 although the value of this practice is unproven.51 Late complications of corticosteroid use, such as osteoporosis with vertebral body compression fractures, are increasingly observed as new treatments prolong survival. Routine prophylaxis with calcium and vitamin D supplementation and bisphosphonates is an important consideration. Emerging antiangiogenic therapies such as bevacizumab (a humanized monoclonal antibody against VEGF) and VEGFR inhibitors effectively treat peritumoral edema and may permit reduction in corticosteroid doses.47,50,52
Venous thromboembolism from leg and pelvic veins is an important risk for MG patients, with a lifetime incidence of 20% to 30%.47,53 Anticoagulation in MG patients with venous thromboembolism confers a low risk of intratumoral hemorrhage.47,54 Therefore, in most circumstances, anticoagulation is appropriate treatment for venous thromboembolism in a MG patient. When a MG patient has recently had brain surgery or has intracerebral hemorrhage, inferior vena cava filters are a reasonable alternative, although they have high complication rates.55 Results from a large randomized clinical trial performed in cancer patients with venous thromboembolism indicate that treatment with low molecular weight heparin confers a lower risk of recurrent thromboembolic disease than does treatment with warfarin (9% versus 17%), without a difference in bleeding rates or mortality.56 On the basis of these data, which included 27 brain tumor patients, low molecular weight heparin is recommended by many physicians for treatment of venous thromboembolism in MG patients.
Fatigue is a common complaint among MG patients, and may respond to treatment with modafinil or methylphenidate.57 Methylphenidate is also used to treat apathy or abulia. Limited evidence supports the use of donepezil58 and memantine for memory impairment. Depression and anxiety respond to standard psycho-pharmacological interventions59 and are likely underdiagnosed in this population.
Tumor-Directed Therapy (See Table 5-1)
SURGERY
Standard therapy for newly diagnosed MGs begins with a surgical procedure, as a definitive diagnosis cannot be made based on radiographic features alone. Although the infiltrative nature of gliomas precludes surgical cure, maximal surgical resection is recommended in all newly diagnosed patients. Benefits of surgical resection include improvement of symptoms related to mass effect, reduction of tumor volume remaining to be treated with other modalities,60 and removal of the necrotic tumor core which may be poorly accessible to circulating chemotherapy and resistant to radiation therapy (RT). Surgical resection also increases diagnostic accuracy, as biopsy provides a small specimen for pathological review that may not be representative of the entire lesion.61 Mounting evidence of variable quality suggests that surgical resection confers a modest survival benefit as compared to biopsy.62,63 Advances including MRI-guided neuro-navigation, intraoperative MRI, functional MRI, intraoperative mapping,64 and fluorescent-guided surgery65 have reduced surgical complication rates and allowed more complete tumor resections.66 Patients with inoperable tumors located in eloquent areas require stereotactic biopsies for tissue diagnosis.
TABLE 5-1 Summary of Therapeutic Options for Malignant Gliomas
| Setting | Histology | Treatment Options |
|---|---|---|
| Newly Diagnosed Tumor* | Glioblastoma |
Astrocytoma**
Abbreviations: PCV, procarbazine, lomustine (CCNU), and vincristine; RT, radiation therapy; SRS, stereotactic radiosurgery; TMZ, temozolomide.
* Treatment should always begin with maximal surgical resection.
** No standard of care has been defined.
*** Clinical trial enrollment should be offered to recurrent malignant glioma patients whenever possible.
Adapted from: Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med 2008;359:492–507.
Surgery may also be considered in recurrent MG patients with good performance status when the tumor is accessible, symptomatic, and distant from eloquent areas. Surgical resection in the recurrent setting may improve quality of life and allow time for additional therapy.67 The impact of resection on survival in recurrent MG patients is uncertain.
RADIATION THERAPY
Radiation therapy is generally considered to be the most important treatment modality for MGs. The addition of RT to surgery provides a robust survival benefit for GBM patients, increasing median survival from 3 to 4 months to 7 to 12 months.68,69 Standard RT for MG is involved-field external beam irradiation to a total dose of 60 Gy, delivered in 1.8 Gy to 2.0 Gy fractions over approximately 6 weeks. The definition of the involved field varies, but it generally includes the area of T2 hyperintensity on MRI plus a 1 cm to 2 cm margin, often with a boost to the area of contrast enhancement. Conformal planning is used to reduce radiation exposure to surrounding brain. Although MG is an infiltrative disease, 90% of conventionally irradiated tumors recur locally at the original site.70
Many variations on standard RT have been investigated in an attempt to increase efficacy. Randomized studies with higher total radiation doses have confirmed the early impression that doses greater than 60 Gy yield no additional benefit.71 A number of studies using altered fractionation schemes have failed to show any benefit over conventional fractionation.72 Various radiosensitizing agents have been administered to patients with MGs, though none has demonstrated substantial efficacy. Strategies designed to provide additional radiation dose to the resection cavity, such as brachytherapy73 and stereotactic radiosurgery (SRS),74,75 have neither improved local control nor survival. Newer approaches including chemotherapy,13 targeted molecular agents,76 and antiangiogenic agents77 may potentially work synergistically with RT and improve outcomes and are under investigation.
Patients over the age of 70 represent a challenging subgroup of MG patients with an extremely poor prognosis. In this population, the addition of RT to supportive care alone increases median survival from 16.9 weeks to only 29.1 weeks.78 Furthermore, older patients are at increased risk for both acute and delayed radiation toxicity. Common manifestations include disabling fatigue that may last weeks to months,79 nausea, and headache. Treatment options include an abbreviated course of RT (40 Gy in 15 fractions over 3 weeks)80 or chemotherapy alone81; in elderly patients, both approaches produce similar outcomes to conventional RT regimens.
Additional involved-field RT is rarely offered to patients with recurrent MG, as doses higher than 60 Gy offer marginal benefit and increase the risk of radiation necrosis.82 Small nonrandomized studies have demonstrated a survival benefit for MG patients treated with SRS at recurrence.75 However, much of the data is subject to selection bias, and this approach is not routinely utilized. Fractionated stereo-tactic RT has also been evaluated for treatment of recurrent MG.83 Fractionation allows for increased radiation doses to the tumor bed with less risk of injury to surrounding brain, but its efficacy is also unproven.
CHEMOTHERAPY
The importance of chemotherapy in MG treatment has grown in recent years. Early studies of adjuvant nitrosourea therapy for MGs were methodologically flawed and failed to demonstrate a significant survival advantage.68,84 Subsequent meta-analyses confirmed that adjuvant chemotherapy produces a modest survival benefit (6% to 10% increase in 1-year survival).85,86 In 2005, the European Organization for Research and Treatment of Cancer (EORTC) and the National Cancer Institute of Canada (NCIC) reported the results of a large randomized phase III trial in newly diagnosed GBM, comparing RT alone (60 Gy over 6 weeks) with RT and concomitant daily temozolomide (Temodar®, Schering-Plough, Kenilworth, New Jersey; 75 mg/m2/d), followed by adjuvant temozolomide therapy (150 to 200 mg/m2/day for 5 consecutive days out of every 28-day cycle, for 6 cycles).69 Grade 3 or 4 toxicity in the chemotherapy group was modest and included fatigue (13%), nausea (2%), thrombocytopenia (12%), and neutropenia (7%). The addition of temozolomide to RT increased median survival compared to RT alone (14.6 mo vs. 12.1 mo; p< 0.0001). At two years, the proportion of surviving patients in the chemotherapy group was 26.5% as compared to 10.4% in the RT alone group. On the basis of these results, RT with concomitant and adjuvant temozolomide became the standard of care for newly diagnosed GBM (Figure 5-2). Recently, updated results from this study showed that the survival benefit with temozolomide was maintained even at 5 years (9.8% were alive at 5 years with temozolomide, versus 1.9% with radiotherapy alone (hazard ratio 0.6, 95% CI 0.5–0.7; p<0.0001).87
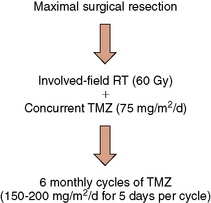
Figure 5-2 Standard treatment algorithm for newly-diagnosed glioblastoma.
Abbreviations: RT, radiation therapy; TMZ, temozolomide
In a companion study, tumor specimens from the EORTC/NCIC study were evaluated for methylation status of the MGMT gene promoter.8 MGMT is an endogenous DNA repair enzyme that removes alkyl groups from DNA and thus confers resistance to temozolomide and other alkylating agents. Methylation is an epigenetic modification that results in decreased gene transcription. Therefore, tumor cells with methylated MGMT promoters have less MGMT enzyme, resulting in less effective DNA repair and increased temozolomide sensitivity. As predicted, the benefit of temozolomide was more dramatic in patients with MGMT promoter methylation. Among GBM patients with MGMT promoter methylation who were treated with temozolomide, median survival was 21.7 months and 2-year survival, 46%. Temozolomide-treated patients with unmethylated MGMT promoters had a significantly shorter median survival of only 12.7 months, and a 2-year survival of 13.8%.8 Because this study was conducted retrospectively in a relatively small sample of patients, temozolomide remains the standard of care for newly diagnosed GBM patients, regardless of MGMT promoter methylation status. A randomized phase III trial sponsored by the Radiation Therapy Oncology Group (RTOG 0525) will definitively evaluate the utility of MGMT promoter methylation in determining temozolomide sensitivity. In the future, patients whose tumors have unmethylated MGMT promoters may be offered alternatives to the standard temozolomide regimen. Investigational approaches to suppress MGMT activity include dose-intense temozolomide regimens (e.g., 21 days on/7 days off,88 7 days on/7days off,89 or continuous dosing90), which may deplete the enzyme,91 and combination therapy with O6-benzylguanine or other MGMT inhibitors.92–94 Inhibitors of other DNA repair enzymes such as poly-(ADP-ribose)-polymerase (PARP) may also improve the efficacy of temozolomide.95
An alternative to systemic chemotherapy involves the implantation of carmustine-containing biodegradable wafers (Gliadel® wafers, MGI Pharma, Bloomington, Minnesota) into the resection cavity following tumor debulking. These wafers gradually release carmustine over several weeks, allowing for a high local chemotherapy concentration with minimal systemic toxicity. In a double-blind, randomized, phase III trial of 240 newly diagnosed MG patients, patients received either conventional therapy (surgery followed by RT) with placement of a carmustine wafer or conventional therapy with placement of an identical placebo wafer. Carmustine wafer treatment increased median survival from 11.6 to 13.9 months (P = 0.03).96 Carmustine wafers were FDA-approved for treatment of newly-diagnosed GBM on the basis of these data.
Although chemotherapy is often considered in treating recurrent MGs, the benefit of standard chemotherapy has been modest in this setting. Phase II trials of temozolomide for recurrent glioblastomas have demonstrated response rates of only 5%, with stable disease in 40% of patients and 6-month progression-free survival (PFS6) in the range of 21%.97–99 Alternative temozolomide dosing regimens may be more effective. Other agents such as carmustine, carboplatin, etoposide, irinotecan, and PCV (procarbazine, lomustine [CCNU], and vincristine) combination therapy produce low response rates and no significant survival benefit.100 In selected patients with recurrent glioblastomas who can undergo resection, carmustine-containing biodegradable wafers (Gliadel® wafers, MGI Pharma, Bloomington, Minnesota) produces a modest survival advantage of approximately 8 weeks.101 In light of limited data, treatment decisions for patients with recurrent glioblastomas must be made on an individual basis. Factors to consider include tumor histology, prior therapy, time to relapse, and performance status. In general, patients with recurrent disease should be enrolled in clinical trials whenever feasible.
THERAPY FOR ANAPLASTIC GLIOMAS
Tumors with oligodendroglial elements, AOs and AOAs, are less common than other subtypes of MG. However, they confer a better prognosis than do pure astrocytic tumors, and they have increased sensitivity to treatment.102 The majority of AOs and 14% to 20% of AOAs have deletions of chromosomes 1 p and 19 q,102 due to an unbalanced translocation of 19 p to 1 q.103 Tumors with 1 p/19 q codeletion are particularly sensitive to PCV chemotherapy.6,104 Codeletion of 1 p/19 q also may confer sensitivity to temozolomide, with an increase in response rate from 34% to 59% in one study.28 The reason for the increased chemosensitivity conferred by 1 p/19 q codeletion is an area of ongoing investigation, but decreased levels of stathmin may be important.105
The value of chemotherapy for newly-diagnosed AO/AOA has recently been evaluated in two large phase III trials.106,107 Although neither study showed an overall survival benefit (2.5 to 4.7 years), patients treated with both RT and PCV chemotherapy had 10 to 12 months of additional time to tumor progression as compared to RT alone. The lack of an overall survival benefit may relate to the fact that most patients who initially received RT alone were subsequently treated with chemotherapy at relapse. In both studies, 1 p/19 q codeletion was associated with marked survival prolongation. Because most published studies in AO/AOA were initiated prior to 2005, the majority of available data involve PCV chemotherapy. Although PCV and temozolomide have not yet been directly compared, temozolomide is likely to have similar activity and less toxicity.102 Several large intergroup trials are underway evaluating the optimal combination of radiation therapy and temozolomide in patients with newly diagnosed AO/AOA.
Experimental Therapies
TARGETED MOLECULAR THERAPIES
As the molecular pathogenesis of MG is elucidated, there is growing interest in targeted molecular therapies against signaling pathways that regulate glioma cell growth, division, and other critical functions.12,76,108,109 Targets of particular importance include receptor tyrosine kinases such as EGFR,110 PDGFR,111,112 and VEGFR.52 Inhibitors of intracellular signaling molecules are also being developed against mTOR,113,114 farnesyltransferase,115 and PI3K, among many others (Figure 5-3).
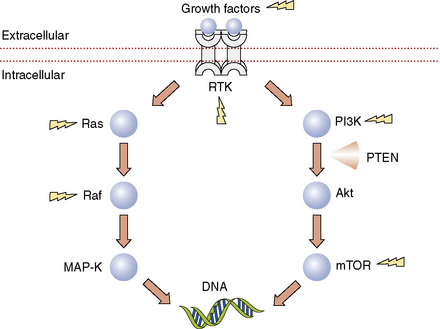
Figure 5-3 Selected signaling pathways in malignant gliomas. By transcriptional regulation, these pathways have important effects on cell growth, division, invasion, apoptosis, and angiogenesis. Lightning bolts indicate targets for which inhibitors are currently available. Some examples of receptor tyrosine kinase inhibitors include the epidermal growth factor receptor (EGFR) inhibitors erlotinib and gefitinib, the platelet-derived growth factor receptor (PDGFR) inhibitor imatinib, and the vascular endothelial growth factor receptor (VEGFR) inhibitor cediranib. An example of an inhibitor directed against a growth factor ligand is bevacizumab, the humanized monoclonal antibody against VEGF. Sorafenib is a multi-targeted tyrosine kinase inhibitor that blocks VEGF, PDGFR, and Raf. PI3K inhibitors such as XL765 (Exelixis, South San Francisco, California) and many others are now entering clinical trials for malignant glioma. A variety of mTOR inhibitors have been evaluated in malignant glioma including rapamycin (sirolimus, Rapamune®, Wyeth, Madison, New Jersey), temsirolimus (CCI-779, Torisel™, Wyeth, Madison, New Jersey), and everolimus (RAD001, Novartis, Basel, Switzerland).
Abbreviations: MAP-K, mitogen-associated protein kinase; mTOR, mammalian target of rapamycin; PI3K, phosphoinositide 3′-kinase; PTEN, phosphatase and tensin homolog deleted on chromosome 10; RTK, receptor tyrosine kinase
The EGFR came to attention as a potential therapeutic target because the gene is amplified in more than 40% of primary GBMs.116 Approximately one quarter to one third of GBMs have a constitutively active EGFR mutant known as EGFRvIII, and all of these EGFRvIII-expressing tumors also exhibit EGFR amplification or overexpression.117 Unchecked EGFR pathway signaling promotes cell proliferation, tumor invasion, angiogenesis, and apoptosis inhibition. Several small-molecule inhibitors of the EGFR such as gefitinib (Iressa®, AstraZeneca, Wilmington, Delaware) and erlotinib (Tarceva®, OSI, Melville, New York) have been evaluated in MGs. Though these agents are generally well-tolerated, reported responses have been limited (0% to 25%) and short-lived. EGFR expression is not predictive of response, but patients with EGFRvIII mutations and intact PTEN may be more likely to benefit from therapy.118
PDGF pathway upregulation is another common feature of MGs that suggests a potential therapeutic target.119 The first clinically useful small-molecule tyrosine kinase inhibitor was imatinib mesylate (Gleevec®, Novartis, East Hanover, New Jersey), an inhibitor of the Bcr-Abl, PDGF, and c-Kit receptor tyrosine kinases, which has activity against chronic myelogenous leukemia and gastrointestinal stromal tumors. Phase II trials of imatinib monotherapy in patients with recurrent MG showed minimal activity.111,112 Preliminary studies of the combination of imatinib with hydroxyurea suggested possible activity in recurrent AG120 and recurrent GBM.121 However, a large multicenter phase III trial failed to confirm any benefit.122
Receptor tyrosine kinase signaling is mediated in part by the MAP-kinase and PI3K pathways. As such, components of these signaling pathways represent potential targets in MG therapy. An early step in activation of the MAP-kinase pathway is localization of Ras to the cell membrane, which depends upon Ras farnesylation by the enzyme farnesyltransferase. Farnesyltransferase inhibitors (FTI) such as tipifarnib123 (R115777, Zarnestr™, Johnson and Johnson, New Brunswick, New Jersey) and lonafarnib (SCH66366, Sarasar®, Schering-Plough, Kenilworth, New Jersey) have shown modest activity as monotherapy in recurrent MG. The mammalian target of rapamycin is a downstream molecule in the PI3K pathway that is an attractive target for therapy.124 The mTOR inhibitor and rapamycin analog temsirolimus (CCI-779, Torisel™, Wyeth, Madison, New Jersey) has been studied in recurrent MG with modest results thus far.125,126 A pilot study of neoadjuvant rapamycin therapy for recurrent PTEN-deficient GBM demonstrated variable mTOR inhibition in tumor tissue and marked reduction in tumor cell proliferation in 7 of 14 (50%) patients, supporting in vitro evidence of increased sensitivity to mTOR inhibitors in this population.127 Inhibitors of PI3K itself are currently in early clinical development for MG.
As these results imply, monotherapy with targeted molecular agents has shown modest activity, with disappointing response rates of 0% to 15% and no improvement in PFS6.76,108,128 These results are not surprising when one considers that most MGs have coactivation of multiple tyrosine kinases129 and highly redundant signaling pathways. In addition, many of the targeted molecular agents are hydrophilic and fail to penetrate tumor tissue sufficiently to inhibit critical targets. In light of these difficulties, efforts are under way to identify biomarkers that may enable prediction of responsiveness to targeted molecular drugs. For example, tumors with EGFRvIII and intact PTEN seem to be more sensitive to EGFR inhibitors in some studies,130 although not in others.131 Similarly, tumors with increased activity of the PI3K/Akt pathway are resistant to these same drugs.132 Approaches that are now being evaluated in clinical trials involve the use of rational combinations of drugs that inhibit multiple targets, drugs that target multiple relevant targets at once, and combinations of drugs with radiotherapy and chemotherapy.12,76,108,128,133 There is also growing interest in clinical trial designs that include posttreatment tissue specimens to verify appropriate target inhibition.134
ANTIANGIOGENIC THERAPIES
It is now well established that angiogenesis is required for the growth of most solid tumors, including MG.27,135,136 The importance of angiogenesis in the pathogenesis of MG was long suspected, because GBMs are highly vascular tumors with abundant endothelial proliferation. Angiogenic factors produced by GBM cells include VEGF,137 basic fibroblast growth factor (bFGF),138 hepatocyte growth factor/scatter factor (HGF/SF).139 The endothelial cells within MG tumor vasculature express VEGFR2 (KDR, Fkl-1), resulting in a paracrine loop in which VEGF secreted locally by the tumor promotes endothelial cell growth and division.140 Higher levels of VEGF expression are observed in more malignant tumors; in one study, VEGF levels were increased more than ten-fold in high-grade gliomas as compared to their low-grade counterparts.139 Other important angiogenic signaling pathways in MGs involve Tie2 and its ligands angiopoietin-1 and -2141 and the Notch-Delta-like ligand (Dll) pathway.142
As described above, glioma stem cells may contribute to treatment resistance41,143 and produce pro-angiogenic molecules such as VEGF.144 Recent data shows that glioma stem cells are found in close proximity to tumor blood vessels,145,146 suggesting that antiangiogenic treatment might preferentially target these treatment-resistant MG constituents. This may also suggest a mechanism by which antiangiogenic treatments and cytotoxic chemotherapy could achieve synergistic antitumor efficacy.147
For these reasons, MGs have become targets of interest for antiangiogenic drug therapy.148 Early and relatively weak antiangiogenic agents such as thalidomide had modest activity,149 but newer agents appear to be more promising (Figure 5-4). In initial studies for recurrent MG, combination therapy with bevacizumab and irinotecan resulted in radiographic response rates of 34% to 66%.150–153 The regimen was well-tolerated with a low incidence of hemorrhage, an adverse effect of major concern for this class of drugs based on initial solid tumor data. Although it has been suggested that the impressive radiographic responses observed in patients treated with bevacizumab may be the result of decreased permeability of the vasculature rather than a true antitumor effect, subsequent clinical trial data have confirmed prolongation of PFS. In the first phase II trial, PFS6 was 43% for GBM patients and 59% for AG patients; these data are substantially better than the PFS6 of 21% reported in recurrent GBM patients treated with temozolomide.99 The radiographic response rate was 57% for GBM patients and 61% for AG patients.154 Treatment was well-tolerated with a single intracranial hemorrhage and four cases of venous thromboembolism, another adverse effect reported in previous solid tumor trials.155,156 A subsequent phase II trial evaluated recurrent GBM patients in first or second relapse who were treated with bevacizumab with or without irinotecan.157 In the monotherapy arm, PFS6 was 42.6% and radiographic response rate 28.2%. In the combination therapy arm, PFS6 was 50.3% and radiographic response rate 37.8%. Median overall survival was approximately 9 months in both groups. The study lacked statistical power to identify any benefit of adding irinotecan to bevacizumab. Most patients were able to reduce their corticosteroid doses by 50% or more. Again, treatment was well-tolerated with rare hemorrhagic complications. More recently, a phase II trial of bevacizumab in heavily pretreated patients with recurrent GBM produced similar results with a response rate of 35% and a PFS6 of 29%.158
There is emerging evidence that inhibitors of angiogenesis may work synergistically with RT.77 Several trials are ongoing in which newly diagnosed GBM patients receive bevacizumab with RT and temozolomide. The regimen appears to be safe, despite a possible increase in wound-healing complications.159 Bevacizumab is being combined in ongoing studies with a variety of targeted molecular agents as well.160
Aside from inhibitors of VEGF like bevacizumab, there are many small-molecule tyrosine kinase inhibitors directed against VEGFR. Cediranib (AZD2171, Recentin; AstraZeneca, London, UK), is an oral pan-VEGFR inhibitor that also has activity against PDGFR and c-Kit. Cediranib was tested in a phase II clinical trial in patients with recurrent GBM. The treatment achieved a promising response rate of 56% and PFS6 of 26%.161,162 As had been noted in the bevacizumab studies, there was a striking steroid-sparing effect. All of the patients who required corticosteroids were able to reduce their doses or stop treatment entirely. Among the 31 patients enrolled, there were no intracranial hemorrhages, although adverse effects such as hypertension, diarrhea, and fatigue were common. The authors used advanced MRI techniques to show that cediranib produced reversible reductions in blood vessel size and permeability, lending support to the concept of vascular normalization, which asserts that antiangiogenic treatment may “normalize” structurally and functionally abnormal tumor blood vessels.27 By virtue of vascular normalization, antiangiogenic therapies may facilitate delivery of chemotherapeutics into the tumor and reduce hypoxia, perhaps improving the efficacy of RT as well.163
Mechanisms of resistance to antiangiogenic therapy are beginning to be elucidated.164,165 Failure of anti-VEGF/VEGFR therapy may correlate with upregulation of alternative pro-angiogenic factors such as bFGF,162 stromal-derived factor-1α (SDF1α),162 Tie2, and placental growth factor (PlGF),166 or with mobilization of circulating endothelial cells or their bone marrow–derived precursor cells.142,162 Some preclinical data suggest that blockade of VEGF-mediated angiogenesis may promote tumor infiltration by cooption of existing cerebral blood vessels.141,167–171 In recurrent MG patients who are treated with bevacizumab, there is an increased risk of infiltrative tumor growth observed on MRI scans, which suggests that bevacizumab may promote tumor infiltration in patients.150,172–174 These findings imply that anti-VEGF/VEGFR therapy may function optimally when combined with agents that target tumor invasion and non–VEGF-mediated angiogenesis.
OTHER THERAPIES
An array of other therapeutic modalities are being explored for MG. Examples include gene therapy,175,176 stem cell targeting,177 synthetic chlorotoxins (TM-601),178 chemotherapeutic agents with enhanced ability to penetrate into tumor tissue, and convection-enhanced delivery of drugs and toxins.179 Antitumor vaccines based on peptide antigens, dendritic cells, or whole tumor cells represent a major avenue of investigation. Among the many promising vaccines are CDX-110,180 a peptide vaccine directed against EGFRvIII, and GVAX,181 which involves administration of irradiated autologous tumor cells mixed with GM-CSF producing cells.
Acknowledgments
We gratefully acknowledge the support of the Melinda Wanatick Brain Tumor Research Fund.
1. CBTRUS. Statistical Report: Primary Brain Tumors in the United States, 2000–2004. Published by the Central Brain Tumor Registry of the United States, 2008.
2. D.N. Louis, H. Ohgaki, O.D. Wiestler, et al. The 2007 WHO classification of tumors of the central nervous system. Lyon, France: IARC Press, 2007.
3. J.L. Fisher, J.A. Schwartzbaum, M. Wrensch, J.L. Wiemels. Epidemiology of brain tumors. Neurol Clin. 2007;25:867-890.
4. P.Y. Wen, S. Kesari. Malignant gliomas in adults. N Engl J Med. 2008;359:492-507.
5. W.J. CurranJr, C.B. Scott, J. Horton, et al. Recursive partitioning analysis of prognostic factors in three Radiation Therapy Oncology Group malignant glioma trials. J Natl Cancer Inst. 1993;85:704-710.
6. Y. Ino, R.A. Betensky, M.C. Zlatescu, et al. Molecular subtypes of anaplastic oligodendroglioma: implications for patient management at diagnosis. Clin Cancer Res. 2001;7:839-845.
7. H. Colman, K. Aldape. Molecular predictors in glioblastoma: toward personalized therapy. Arch Neurol. 2008;65:877-883.
8. M.E. Hegi, A.C. Diserens, T. Gorlia, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997-1003.
9. H.S. Phillips, S. Kharbanda, R. Chen, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9:157-173.
10. R. McLendon, A. Friedman, D. Bigner, et al. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008.
11. D.W. Parsons, S. Jones, X. Zhang, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807-1812.
12. F.B. Furnari, T. Fenton, R.M. Bachoo, et al. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev. 2007;21:2683-2710.
13. R. Stupp, M.E. Hegi, M.R. Gilbert, A. Chakravarti. Chemoradiotherapy in malignant glioma: standard of care and future directions. J Clin Oncol. 2007;25:4127-4136.
14. C.J. Farrell, S.R. Plotkin. Genetic causes of brain tumors: neurofibromatosis, tuberous sclerosis, von Hippel-Lindau, and other syndromes. Neurol Clin. 2007;25:925-946.
15. B. Malmer, P. Adatto, G. Armstrong, et al. GLIOGENE an International Consortium to Understand Familial Glioma. Cancer Epidemiol Biomarkers Prev. 2007;16:1730-1734.
16. D.Y. Lee, D.H. Gutmann. Cancer stem cells and brain tumors: uprooting the bad seeds. Expert Rev Anticancer Ther. 2007;7:1581-1590.
17. C.L. Nutt, D.R. Mani, R.A. Betensky, et al. Gene expression-based classification of malignant gliomas correlates better with survival than histological classification. Cancer Res. 2003;63:1602-1607.
18. C.E. Pelloski, K.V. Ballman, A.F. Furth, et al. Epidermal growth factor receptor variant III status defines clinically distinct subtypes of glioblastoma. J Clin Oncol. 2007;25:2288-2294.
19. V.P. Collins. Mechanisms of disease: genetic predictors of response to treatment in brain tumors. Nat Clin Pract Oncol. 2007;4:362-374.
20. H. Ohgaki, P. Kleihues. Genetic pathways to primary and secondary glioblastoma. Am J Pathol. 2007;170:1445-1453.
21. K. Watanabe, O. Tachibana, K. Sata, Y. Yonekawa, P. Kleihues, H. Ohgaki. Overexpression of the EGF receptor and p53 mutations are mutually exclusive in the evolution of primary and secondary glioblastomas. Brain Pathol. 1996;6:217-223. discussion 23–4
22. K. Ueki, Y. Ono, J.W. Henson, J.T. Efird, A. von Deimling, D.N. Louis. CDKN2/p16 or RB alterations occur in the majority of glioblastomas and are inversely correlated. Cancer Res. 1996;56:150-153.
23. H. Yan, D.W. Parsons, G. Jin, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360:765-773.
24. J.C. Lee, I. Vivanco, R. Beroukhim, et al. Epidermal growth factor receptor activation in glioblastoma through novel missense mutations in the extracellular domain. PLoS Med. 2006;3:e485.
25. P. Steck, M.A. Perhouse, S.A. Jasser, et al. Identification of a candidate tumour suppressor gene, MMAC 1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nature Genet. 1997;15:356-362.
26. P. Guo, B. Hu, W. Gu, et al. Platelet-derived growth factor-B enhances glioma angiogenesis by stimulating vascular endothelial growth factor expression in tumor endothelia and by promoting pericyte recruitment. Am J Pathol. 2003;162:1083-1093.
27. R.K. Jain, E. di Tomaso, D.G. Duda, J.S. Loeffler, A.G. Sorensen, T.T. Batchelor. Angiogenesis in brain tumours. Nat Rev Neurosci. 2007;8:610-622.
28. A.A. Brandes, A. Tosoni, G. Cavallo, et al. Correlations between O6-methylguanine DNA methyltransferase promoter methylation status, 1 p and 19 q deletions, and response to temozolomide in anaplastic and recurrent oligodendroglioma: a prospective GICNO study. J Clin Oncol. 2006;24:4746-4753.
29. Y. Ino, M.C. Zlatescu, H. Sasaki, et al. Long survival and therapeutic responses in patients with histologically disparate high-grade gliomas demonstrating chromosome 1 p loss. J Neurosurg. 2000;92:983-990.
30. J.G. Cairncross, K. Ueki, M.C. Zlatescu, et al. Specific chromosomal losses predict chemotherapeutic response and survival in patients with anaplastic oligodendrogliomas. J Natl Cancer Inst. 1998;90:1473-1479.
31. M.E. Hegi, L. Liu, J.G. Herman, et al. Correlation of O6-methylguanine methyltransferase (MGMT) promoter methylation with clinical outcomes in glioblastoma and clinical strategies to modulate MGMT activity. J Clin Oncol. 2008;26:4189-4199.
32. R. McLendon, A. Friedman, D. Bigner, et al. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008.
33. D.W. Parsons, S. Jones, X. Zhang, et al. An Integrated Genomic Analysis of Human Glioblastoma Multiforme. Science. 2008.
34. M. Assanah, R. Lochhead, A. Ogden, J. Bruce, J. Goldman, P. Canoll. Glial progenitors in adult white matter are driven to form malignant gliomas by platelet-derived growth factor-expressing retroviruses. J Neurosci. 2006;26:6781-6790.
35. N. Sanai, A. Alvarez-Buylla, M.S. Berger. Neural stem cells and the origin of gliomas. N Engl J Med. 2005;353:811-822.
36. M.F. Clarke, M. Fuller. Stem cells and cancer: two faces of eve. Cell. 2006;124:1111-1115.
37. S.K. Singh, C. Hawkins, I.D. Clarke, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396-401.
38. C.D. Stiles, D.H. Rowitch. Glioma stem cells: a midterm exam. Neuron. 2008;58:832-846.
39. P.B. Dirks. Brain tumor stem cells: bringing order to the chaos of brain cancer. J Clin Oncol. 2008;26:2916-2924.
40. A. Murat, E. Migliavacca, T. Gorlia, et al. Stem cell-related “self-renewal” signature and high epidermal growth factor receptor expression associated with resistance to concomitant chemoradiotherapy in glioblastoma. J Clin Oncol. 2008;26:3015-3024.
41. S. Bao, Q. Wu, R.E. McLendon, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756-760.
42. G. Liu, X. Yuan, Z. Zeng, et al. Analysis of gene expression and chemoresistance of CD133 + cancer stem cells in glioblastoma. Mol Cancer. 2006;5:67.
43. A. Salmaggi, A. Boiardi, M. Gelati, et al. Glioblastoma-derived tumorospheres identify a population of tumor stem-like cells with angiogenic potential and enhanced multidrug resistance phenotype. Glia. 2006;54:850-860.
44. M. Dean, T. Fojo, S. Bates. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005;5:275-284.
45. M. Loghin, V.A. Levin. Headache related to brain tumors. Curr Treat Options Neurol. 2006;8:21-32.
46. S. Cha. Update on brain tumor imaging: from anatomy to physiology. AJNR Am J Neuroradiol. 2006;27:475-487.
47. P. Wen, D. Schiff, S. Kesari, J. Drappatz, D. Gigas, L. Doherty. Medical management of patients with brain tumors. J Neurooncol. 2006;80:313-332.
48. M.J. Glantz, B.F. Cole, P.A. Forsyth, et al. Practice parameter: anticonvulsant prophylaxis in patients with newly diagnosed brain tumors. Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2000;54:1886-1893.
49. V. Siomin, L. Angelov, L. Li, M.A. Vogelbaum. Results of a survey of neurosurgical practice patterns regarding the prophylactic use of anti-epilepsy drugs in patients with brain tumors. J Neurooncol. 2005;74:211-215.
50. E.R. Gerstner, D.G. Duda, E. di Tomaso, et al. VEGF inhibitors in the treatment of cerebral edema in patients with brain cancer. Nat Rev Clin Oncol. 2009;6:229-236.
51. H. Green, M. Paul, L. Vidal, L. Leibovici. Prophylaxis of Pneumocystis pneumonia in immunocompromised non-HIV-infected patients: systematic review and meta-analysis of randomized controlled trials. Mayo Clin Proc. 2007;82:1052-1059.
52. T. Batchelor, A. Sorensen, E. di Tomaso, et al. AZD2171, a Pan-VEGF Receptor Tyrosine Kinase Inhibitor, Normalizes Tumor Vasculature and Alleviates Edema in Glioblastoma Patients. Cancer Cell. 2007;11:83-95.
53. D.E. Gerber, S.A. Grossman, M.B. Streiff. Management of venous thromboembolism in patients with primary and metastatic brain tumors. J Clin Oncol. 2006;24:1310-1318.
54. R.L. Ruff, J.B. Posner. Incidence and treatment of peripheral venous thrombosis in patients with glioma. Ann Neurol. 1983;13:334-336.
55. J.M. Levin, D. Schiff, J.S. Loeffler, H.A. Fine, P.M. Black, P.Y. Wen. Complications of therapy for venous thromboembolic disease in patients with brain tumors. Neurology. 1993;43:1111-1114.
56. A.Y. Lee, M.N. Levine, R.I. Baker, et al. Low-molecular-weight heparin versus a coumarin for the prevention of recurrent venous thromboembolism in patients with cancer. N Engl J Med. 2003;349:146-153.
57. C.A. Meyers, M.A. Weitzner, A.D. Valentine, V.A. Levin. Methylphenidate therapy improves cognition, mood, and function of brain tumor patients. J Clin Oncol. 1998;16:2522-2527.
58. E.G. Shaw, R. Rosdhal, R.B. D’AgostinoJr, et al. Phase II study of donepezil in irradiated brain tumor patients: effect on cognitive function, mood, and quality of life. J Clin Oncol. 2006;24:1415-1420.
59. N.S. Litofsky, E. Farace, F. AndersonJr, C.A. Meyers, W. Huang, E.R. LawsJr. Depression in patients with high-grade glioma: results of the Glioma Outcomes Project. Neurosurgery. 2004;54:358-366. discussion 66–7
60. G.E. Keles, K.R. Lamborn, S.M. Chang, M.D. Prados, M.S. Berger. Volume of residual disease as a predictor of outcome in adult patients with recurrent supratentorial glioblastomas multiforme who are undergoing chemotherapy. J Neurosurg. 2004;100:41-46.
61. R.J. Jackson, G.N. Fuller, D. Abi-Said, et al. Limitations of stereotactic biopsy in the initial management of gliomas. Neuro-oncol. 2001;3:193-200.
62. N. Sanai, M.S. Berger. Glioma extent of resection and its impact on patient outcome. Neurosurgery. 2008;62:753-764. discussion 264–6
63. W. Stummer, H.J. Reulen, T. Meinel, et al. Extent of resection and survival in glioblastoma multiforme: identification of and adjustment for bias. Neurosurgery. 2008;62:564-576. discussion -76
64. A.R. Asthagiri, N. Pouratian, J. Sherman, G. Ahmed, M.E. Shaffrey. Advances in brain tumor surgery. Neurol Clin. 2007;25:975-1003.
65. W. Stummer, U. Pichlmeier, T. Meinel, O.D. Wiestler, F. Zanella, H.J. Reulen. Fluorescence-guided surgery with 5-aminolevulinic acid for resection of malignant glioma: a randomised controlled multicentre phase III trial. Lancet Oncol. 2006;7:392-401.
66. A.R. Asthagiri, N. Pouratian, J. Sherman, G. Ahmed, M.E. Shaffrey. Advances in brain tumor surgery. Neurol Clin. 2007;25:975-1003. viii-ix
67. P.D. Brown, M.J. Maurer, T.A. Rummans, et al. A prospective study of quality of life in adults with newly diagnosed high-grade gliomas: the impact of the extent of resection on quality of life and survival. Neurosurgery. 2005;57:495-504. discussion 495–504
68. M.D. Walker, E. AlexanderJr, W.E. Hunt, et al. Evaluation of BCNU and/or radiotherapy in the treatment of anaplastic gliomas. A cooperative clinical trial. J Neurosurg. 1978;49:333-343.
69. R. Stupp, W.P. Mason, M.J. van den Bent, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987-996.
70. F.H. Hochberg, A. Pruitt. Assumptions in the radiotherapy of glioblastoma. Neurology. 1980;30:907-911.
71. S.W. Lee, B.A. Fraass, L.H. Marsh, et al. Patterns of failure following high-dose 3-D conformal radiotherapy for high-grade astrocytomas: a quantitative dosimetric study. Int J Radiat Oncol Biol Phys. 1999;43:79-88.
72. J.B. Fiveash, S.A. Spencer. Role of radiation therapy and radiosurgery in glioblastoma multiforme. Cancer J. 2003;9:222-229.
73. R.G. Selker, W.R. Shapiro, P. Burger, et al. The Brain Tumor Cooperative Group NIH Trial 87–01: a randomized comparison of surgery, external radiotherapy, and carmustine versus surgery, interstitial radiotherapy boost, external radiation therapy, and carmustine. Neurosurgery. 2002;51:343-355. discussion 55–7
74. L. Souhami, W. Seiferheld, D. Brachman, et al. Randomized comparison of stereotactic radiosurgery followed by conventional radiotherapy with carmustine to conventional radiotherapy with carmustine for patients with glioblastoma multiforme: report of Radiation Therapy Oncology Group 93–05 protocol. Int J Radiat Oncol Biol Phys. 2004;60:853-860.
75. M.N. Tsao, M.P. Mehta, T.J. Whelan, et al. The American Society for Therapeutic Radiology and Oncology (ASTRO) evidence-based review of the role of radiosurgery for malignant glioma. Int J Radiat Oncol Biol Phys. 2005;63:47-55.
76. A. Chi, P. Wen. Inhibiting kinases in malignant gliomas. Expert Opin Ther Targets. 2007;11:473-496.
77. D.G. Duda, R.K. Jain, C.G. Willett. Antiangiogenics: the potential role of integrating this novel treatment modality with chemoradiation for solid cancers. J Clin Oncol. 2007;25:4033-4042.
78. F. Keime-Guibert, O. Chinot, L. Taillandier, et al. Radiotherapy for glioblastoma in the elderly. N Engl J Med. 2007;356:1527-1535.
79. E. Davies, C. Clarke, A. Hopkins. Malignant cerebral glioma—I: Survival, disability, and morbidity after radiotherapy. BMJ. 1996;313:1507-1512.
80. W. Roa, P.M. Brasher, G. Bauman, et al. Abbreviated course of radiation therapy in older patients with glioblastoma multiforme: a prospective randomized clinical trial. J Clin Oncol. 2004;22:1583-1588.
81. M. Glantz, M. Chamberlain, Q. Liu, N.S. Litofsky, L.D. Recht. Temozolomide as an alternative to irradiation for elderly patients with newly diagnosed malignant gliomas. Cancer. 2003;97:2262-2266.
82. V.W. Stieber, M.P. Mehta. Advances in radiation therapy for brain tumors. Neurol Clin. 2007;25:1005-1033. ix
83. S.E. Combs, C. Thilmann, L. Edler, J. Debus, D. Schulz-Ertner. Efficacy of fractionated stereotactic reirradiation in recurrent gliomas: long-term results in 172 patients treated in a single institution. J Clin Oncol. 2005;23:8863-8869.
84. Randomized trial of procarbazine, lomustine, and vincristine in the adjuvant treatment of high-grade astrocytoma: a Medical Research Council trial. J Clin Oncol. 2001;19:509-518.
85. H.A. Fine, K.B. Dear, J.S. Loeffler, P.M. Black, G.P. Canellos. Meta-analysis of radiation therapy with and without adjuvant chemotherapy for malignant gliomas in adults. Cancer. 1993;71:2585-2597.
86. L.A. Stewart. Chemotherapy in adult high-grade glioma: a systematic review and meta-analysis of individual patient data from 12 randomised trials. Lancet. 2002;359:1011-1018.
87. R. Stupp, M.E. Hegi, W.P. Mason, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009.
88. A.A. Brandes, A. Tosoni, G. Cavallo, et al. Temozolomide 3 weeks on and 1 week off as first-line therapy for recurrent glioblastoma: phase II study from gruppo italiano cooperativo di neuro-oncologia (GICNO). Br J Cancer. 2006;95:1155-1160.
89. A. Wick, J. Felsberg, J.P. Steinbach, et al. Efficacy and tolerability of temozolomide in an alternating weekly regimen in patients with recurrent glioma. J Clin Oncol. 2007;25:3357-3361.
90. J.R. Perry, W.P. Mason, K. Belanger, et al. The temozolomide RESCUE study: A phase II trial of continuous (28/28) dose-intense temozolomide (TMZ) after progression on conventional 5/28 day TMZ in patients with recurrent malignant glioma. J Clin Oncol. 2010;2008:26.
91. A.W. Tolcher, S.L. Gerson, L. Denis, et al. Marked inactivation of O6-alkylguanine-DNA alkyltransferase activity with protracted temozolomide schedules. Br J Cancer. 2003;88:1004-1011.
92. A. Broniscer, S. Gururangan, T.J. MacDonald, et al. Phase I trial of single-dose temozolomide and continuous administration of o6-benzylguanine in children with brain tumors: a pediatric brain tumor consortium report. Clin Cancer Res. 2007;13:6712-6718.
93. J.A. Quinn, S.X. Jiang, D.A. Reardon, et al. Phase 1 trial of temozolomide plus irinotecan plus O(6)-benzylguanine in adults with recurrent malignant glioma. Cancer. 2009.
94. J.A. Quinn, S.X. Jiang, D.A. Reardon, et al. Phase II trial of temozolomide plus o6-benzylguanine in adults with recurrent, temozolomide-resistant malignant glioma. J Clin Oncol. 2009;27:1262-1267.
95. T. Zaremba, N.J. Curtin. PARP inhibitor development for systemic cancer targeting. Anticancer Agents Med Chem. 2007;7:515-523.
96. M. Westphal, D. Hilt, E. Bortey, et al. A phase 3 trial of local chemotherapy with biodegradable carmustine (BCNU) wafers (Gliadel wafers) in patients with primary malignant glioma. Neurooncol. 2003;5:79-88.
97. M. Brada, K. Hoang-Xuan, R. Rampling, et al. Multicenter phase II trial of temozolomide in patients with glioblastoma multiforme at first relapse. Ann Oncol. 2001;12:259-266.
98. A.A. Brandes, M. Ermani, U. Basso, et al. Temozolomide as a second-line systemic regimen in recurrent high-grade glioma: a phase II study. Ann Oncol. 2001;12:255-257.
99. W.K. Yung, R.E. Albright, J. Olson, et al. A phase II study of temozolomide vs. Procarbazine in patients with glioblastoma multiforme at first relapse. Br J Cancer. 2000;83:588-593.
100. J. Laterra, S. Grossman, K. Carson, G. Lesser, F. Hochberg, M. Gilbert. Suramin and radiotherapy in newly diagnosed glioblastoma: phase 2 NABTT CNS Consortium study. Neuro-oncol. 2004;6:15-20.
101. H. Brem, S. Piantadosi, P.C. Burger, et al. Placebo-controlled trial of safety and efficacy of intraoperative controlled delivery by biodegradable polymers of chemotherapy for recurrent malignant gliomas: The Polymer-brain Tumor Treatment Group. Lancet. 1999;345:1008-1012.
102. M.J. van den Bent. Anaplastic oligodendroglioma and oligoastrocytoma. Neurol Clin. 2007;25:1089-1109.
103. R.B. Jenkins, H. Blair, K.V. Ballman, et al. A t(1;19)(q10;p10) mediates the combined deletions of 1 p and 19 q and predicts a better prognosis of patients with oligodendroglioma. Cancer Res. 2006;66:9852-9861.
104. J.G. Cairncross, K. Ueki, M.C. Zlatescu, et al. Specific genetic predictors of chemotherapeutic response and survival in patients with anaplastic oligodendrogliomas. J Natl Cancer Inst. 1998;90:1473-1479.
105. T. Ngo, T. Peng, X. Liang, et al. The 1 p-encoded protein stathmin and resistance of malignant gliomas to nitrosoureas. J Natl Cancer Inst. 2007;99:639-652.
106. G. Cairncross, B. Berkey, E. Shaw, et al. Phase III trial of chemotherapy plus radiotherapy compared with radiotherapy alone for pure and mixed anaplastic oligodendroglioma: Intergroup Radiation Therapy Oncology Group Trial 9402. J Clin Oncol. 2006;24:2707-2714.
107. M.J. van den Bent, A.F. Carpentier, A.A. Brandes, et al. Adjuvant procarbazine, lomustine, and vincristine improves progression-free survival but not overall survival in newly diagnosed anaplastic oligodendrogliomas and oligoastrocytomas: a randomized European Organisation for Research and Treatment of Cancer phase III trial. J Clin Oncol. 2006;24:2715-2722.
108. S. Sathornsumetee, D.A. Reardon, A. Desjardins, J.A. Quinn, J.J. Vredenburgh, J.N. Rich. Molecularly targeted therapy for malignant glioma. Cancer. 2007;110:13-24.
109. A. Idbaih, F. Ducray, M. Sierra Del Rio, K. Hoang-Xuan, J.Y. Delattre. Therapeutic application of noncytotoxic molecular targeted therapy in gliomas: growth factor receptors and angiogenesis inhibitors. Oncologist. 2008;13:978-992.
110. J. Rich, D. Reardon, T. Peery, et al. Phase II trial of gefitinib in recurrent glioblastoma. J Clin Oncol. 2004;22:133-142.
111. P.Y. Wen, W.K. Yung, K.R. Lamborn, et al. Phase I/II study of imatinib mesylate for recurrent malignant gliomas: North American Brain Tumor Consortium Study 99–08. Clin Cancer Res. 2006;12:4899-4907.
112. E. Raymond, A.A. Brandes, C. Dittrich, et al. Phase II study of imatinib in patients with recurrent gliomas of various histologies: a European Organisation for Research and Treatment of Cancer Brain Tumor Group Study. J Clin Oncol. 2008;26:4659-4665.
113. E. Galanis, J. Buckner, M. Maurer, et al. Phase II trial of temsirolimus (CCI-779) in recurrent glioblastoma multiforme: a North Central Cancer Treatment Group Study. J Clin Oncol. 2005;23:5294-5304.
114. S. Chang, P. Wen, T. Cloughesy, et al. Phase II study of CCI-779 in patients with recurrent glioblastoma multiforme. Invest New Drugs. 2005;23:357-361.
115. T. Cloughesy, P. Wen, H. Robins, et al. Phase II trial of tipifarnib in patients with recurrent malignant glioma either receiving or not receiving enzyme-inducing antiepileptic drugs: a North American Brain Tumor Consortium Study. J Clin Oncol. 2006;24:3651-3656.
116. E.A. Maher, F.B. Furnari, R.M. Bachoo, et al. Malignant glioma: genetics and biology of a grave matter. Genes Dev. 2001;15:1311-1333.
117. K.D. Aldape, K. Ballman, A. Furth, et al. Immunohistochemical detection of egfrviii in high malignancy grade astrocytomas and evaluation of prognostic significance. J Neuropathol Exp Neurol. 2004;63:700-707.
118. I. Mellinghoff, M. Wang, I. Vivanco, et al. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med. 2005;353:2012-2024.
119. A.H. Shih, E.C. Holland. Platelet-derived growth factor (PDGF) and glial tumorigenesis. Cancer Lett. 2006;232:139-147.
120. A. Desjardins, J.A. Quinn, J.J. Vredenburgh, et al. Phase II study of imatinib mesylate and hydroxyurea for recurrent grade III malignant gliomas. J Neurooncol. 2007;83:53-60.
121. D.A. Reardon, M.J. Egorin, J.A. Quinn, et al. Phase II study of imatinib mesylate plus hydroxyurea in adults with recurrent glioblastoma multiforme. J Clin Oncol. 2005;23:9359-9368.
122. G. Dresemann, M. Rosenthal, K. Höffken, et al. Imatinib plus hydroxyurea versus hydroxyurea monotherapy in progressive glioblastoma (GBM)−an international open label randomised phase III study. Neuro Oncol. 2007;9:519.
123. T.F. Cloughesy, P.Y. Wen, H.I. Robins, et al. Phase II trial of tipifarnib in patients with recurrent malignant glioma either receiving or not receiving enzyme-inducing antiepileptic drugs: a North American Brain Tumor Consortium Study. J Clin Oncol. 2006;24:3651-3656.
124. G.G. Chiang, R.T. Abraham. Targeting the mtor signaling network in cancer. Trends Mol Med. 2007;13:433-442.
125. S.M. Chang, P. Wen, T. Cloughesy, et al. Phase II study of CCI-779 in patients with recurrent glioblastoma multiforme. Invest New Drugs. 2005;23:357-361.
126. E. Galanis, J.C. Buckner, M.J. Maurer, et al. Phase II trial of temsirolimus (CCI-779) in recurrent glioblastoma multiforme: a North Central Cancer Treatment Group Study. J Clin Oncol. 2005;23:5294-5304.
127. T.F. Cloughesy, K. Yoshimoto, P. Nghiemphu, et al. Antitumor activity of rapamycin in a Phase I trial for patients with recurrent PTEN-deficient glioblastoma. PLoS Med. 2008;5:e8.
128. S. Sathornsumetee, J.N. Rich, D.A. Reardon. Diagnosis and Treatment of High-Grade Astrocytoma. Neurol Clin. 2007;25:1111-1139.
129. J.M. Stommel, A.C. Kimmelman, H. Ying, et al. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science. 2007;318:287-290.
130. I.K. Mellinghoff, M.Y. Wang, I. Vivanco, et al. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med. 2005;353:2012-2024.
131. P.D. Brown, S. Krishnan, J.N. Sarkaria, et al. Phase I/II trial of erlotinib and temozolomide with radiation therapy in the treatment of newly diagnosed glioblastoma multiforme: North Central Cancer Treatment Group Study N0177. J Clin Oncol. 2008;26:5603-5609.
132. D. Haas-Kogan, M. Prados, K. Lamborn, T. Tihan, M. Berger, D. Stokoe. Biomarkers to predict response to epidermal growth factor receptor inhibitors. Cell Cycle. 2005;4:1369-1372.
133. P.Y. Wen. New developments in targeted molecular therapies for glioblastoma. Expert Rev Anticancer Ther. 2009;9:7-10.
134. S.M. Chang, K.R. Lamborn, J.G. Kuhn, et al. Neurooncology clinical trial design for targeted therapies: lessons learned from the North American Brain Tumor Consortium. Neuro Oncol. 2008;10:631-642.
135. J. Folkman. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285:1182-1186.
136. J. Folkman. Angiogenesis. Annu Rev Med. 2006;57:1-18.
137. K. Plate, G. Breier, H. Weich, W. Risau. Vascular endothelial growth factor is a potential tumour angiogenesis factor in human gliomas in vivo. Nature. 1992;359:845-848.
138. D.F. Stefanik, L.R. Rizkalla, A. Soi, S.A. Goldblatt, W.M. Rizkalla. Acidic and basic fibroblast growth factors are present in glioblastoma multiforme. Cancer Res. 1991;51:5760-5765.
139. N.O. Schmidt, M. Westphal, C. Hagel, et al. Levels of vascular endothelial growth factor, hepatocyte growth factor/scatter factor and basic fibroblast growth factor in human gliomas and their relation to angiogenesis. Int J Cancer. 1999;84:10-18.
140. B. Millauer, L.K. Shawver, K.H. Plate, W. Risau, A. Ullrich. Glioblastoma growth inhibited in vivo by a dominant-negative Flk-1 mutant. Nature. 1994;367:576-579.
141. J. Holash, P.C. Maisonpierre, D. Compton, et al. Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF. Science. 1999;284:1994-1998.
142. R.S. Kerbel. Tumor angiogenesis. N Engl J Med. 2008;358:2039-2049.
143. J.N. Rich. Cancer stem cells in radiation resistance. Cancer Res. 2007;67:8980-8984.
144. S. Bao, Q. Wu, S. Sathornsumetee, et al. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res. 2006;66:7843-7848.
145. C. Calabrese, H. Poppleton, M. Kocak, et al. A perivascular niche for brain tumor stem cells. Cancer Cell. 2007;11:69-82.
146. R.J. Gilbertson, J.N. Rich. Making a tumour’s bed: glioblastoma stem cells and the vascular niche. Nat Rev Cancer. 2007;7:733-736.
147. C. Folkins, S. Man, P. Xu, Y. Shaked, D.J. Hicklin, R.S. Kerbel. Anticancer therapies combining antiangiogenic and tumor cell cytotoxic effects reduce the tumor stem-like cell fraction in glioma xenograft tumors. Cancer Res. 2007;67:3560-3564.
148. A.D. Norden, J. Drappatz, P.Y. Wen. Novel anti-angiogenic therapies for malignant gliomas. Lancet Neurol. 2008;7:1152-1160.
149. H.A. Fine, P.Y. Wen, E.A. Maher, et al. Phase II trial of thalidomide and carmustine for patients with recurrent high-grade gliomas. J Clin Oncol. 2003;21:2299-2304.
150. A.D. Norden, G.S. Young, K. Setayesh, et al. Bevacizumab for recurrent malignant gliomas: efficacy, toxicity, and patterns of recurrence. Neurology. 2008;70:779-787.
151. W.B. Pope, A. Lai, P. Nghiemphu, P. Mischel, T.F. Cloughesy. MRI in patients with high-grade gliomas treated with bevacizumab and chemotherapy. Neurology. 2006;66:1258-1260.
152. V. Stark Vance. Bevacizumab (Avastin®) and CPT-11 (Camptosar®) in the Treatment of Relapsed Malignant Glioma [abstract]. Neuro-oncol. 2005;7:369.
153. P.L. Nghiemphu, W. Liu, Y. Lee, et al. Bevacizumab and chemotherapy for recurrent glioblastoma: a single-institution experience. Neurology. 2009;72:1217-1222.
154. S.A. Wagner, A. Desjardins, D.A. Reardon, et al. Update on survival from the original phase II trial of bevacizumab and irinotecan in recurrent malignant gliomas [abstract]. J Clin Oncol. 2008;26:2021.
155. J.J. Vredenburgh, A. Desjardins, J.E. Herndon2nd, et al. Phase II trial of bevacizumab and irinotecan in recurrent malignant glioma. Clin Cancer Res. 2007;13:1253-1259.
156. J.J. Vredenburgh, A. Desjardins, J.E. Herndon2nd, et al. Bevacizumab plus irinotecan in recurrent glioblastoma multiforme. J Clin Oncol. 2007;25:4722-4729.
157. T.F. Cloughesy, M.D. Prados, T. Mikkelsen, et al. A phase II, randomized, non-comparative clinical trial of the effect of bevacizumab (BV) alone or in combination with irinotecan (CPT) on 6-month progression free survival (PFS6) in recurrent, treatment-refractory glioblastoma (GBM) [abstract]. J Clin Oncol. 2008;26:2010b.
158. T.N. Kreisl, L. Kim, K. Moore, et al. Phase II trial of single-agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. J Clin Oncol. 2009;27:740-745.
159. A. Lai, E. Filka, B. McGibbon, et al. Phase II Pilot Study of Bevacizumab in Combination With Temozolomide and Regional Radiation Therapy for Up-Front Treatment of Patients With Newly Diagnosed Glioblastoma Multiforme: Interim Analysis of Safety and Tolerability. Int J Radiat Oncol Biol Phys. 2008.
160. A.D. Norden, J. Drappatz, P.Y. Wen. Antiangiogenic therapy in malignant gliomas. Curr Opin Oncol. 2008;20:652-661.
161. T. Batchelor, A.G. Sorensen, M. Ancukiewicz, et al. A phase II trial of AZD2171 (cediranib), an oral pan-VEGF receptor tyrosine kinase inhibitor, in patients with recurrent glioblastoma [abstract]. J Clin Oncol. 2007;25:2001.
162. T.T. Batchelor, A.G. Sorensen, E. di Tomaso, et al. AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell. 2007;11:83-95.
163. R.K. Jain. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307:58-62.
164. G. Bergers, D. Hanahan. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer. 2008;8:592-603.
165. L.M. Ellis, D.J. Hicklin. Pathways mediating resistance to vascular endothelial growth factor-targeted therapy. Clin Cancer Res. 2008;14:6371-6375.
166. C.G. Willett, Y. Boucher, D.G. Duda, et al. Surrogate markers for antiangiogenic therapy and dose-limiting toxicities for bevacizumab with radiation and chemotherapy: continued experience of a phase I trial in rectal cancer patients. J Clin Oncol. 2005;23:8136-8139.
167. A. Chi, A.D. Norden, P.Y. Wen. Inhibition of angiogenesis and invasion in malignant gliomas. Expert Rev Anticancer Ther. 2007;7:1537-1560.
168. P. Kunkel, U. Ulbricht, P. Bohlen, et al. Inhibition of glioma angiogenesis and growth in vivo by systemic treatment with a monoclonal antibody against vascular endothelial growth factor receptor-2. Cancer Res. 2001;61:6624-6628.
169. K. Lamszus, P. Kunkel, M. Westphal. Invasion as limitation to anti-angiogenic glioma therapy. Acta Neurochir Suppl. 2003;88:169-177.
170. J.L. Rubenstein, J. Kim, T. Ozawa, et al. Anti-VEGF antibody treatment of glioblastoma prolongs survival but results in increased vascular cooption. Neoplasia. 2000;2:306-314.
171. M. Paez-Ribes, E. Allen, J. Hudock, et al. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell. 2009;15:220-231.
172. A.B. Lassman, F.M. Iwamoto, P.H. Gutin, L.E. Abrey. Patterns of relapse and prognosis after bevacizumab (BEV) failure in recurrent glioblastoma (GBM) [abstract]. J Clin Oncol. 2008;26:2028.
173. R.M. Zuniga, R. Torcuator, T. Doyle, et al. Retrospective analysis of patterns of recurrence seen on MRI in patients with recurrent glioblastoma multiforme treated with bevacizumab plus irinotecan [abstract]. J Clin Oncol. 2008;26:13013.
174. A. Narayana, S. Raza, J.G. Golfinos, et al. Bevacizumab therapy in recurrent high grade glioma: Impact on local control and survival [abstract]. J Clin Oncol. 2008;26:13000.
175. G. Fulci, E.A. Chiocca. The status of gene therapy for brain tumors. Expert Opin Biol Ther. 2007;7:197-208.
176. M. Aghi, S. Rabkin, R.L. Martuza. Effect of chemotherapy-induced DNA repair on oncolytic herpes simplex viral replication. J Natl Cancer Inst. 2006;98:38-50.
177. A. Nakamizo, F. Marini, T. Amano, et al. Human bone marrow-derived mesenchymal stem cells in the treatment of gliomas. Cancer Res. 2005;65:3307-3318.
178. A. Mamelak, S. Rosenfeld, R. Bucholz, et al. Phase I single-dose study of intracavitary-administered iodine-131-TM-601 in adults with recurrent high-grade glioma. J Clin Oncol. 2006;24:3644-3650.
179. S. Ferguson, M.S. Lesniak. Convection enhanced drug delivery of novel therapeutic agents to malignant brain tumors. Curr Drug Deliv. 2007;4:169-180.
180. J.H. Sampson, G.E. Archer, D.D. Bigner, et al. Effect of egfrviii-targeted vaccine (CDX-110) on immune response and TTP when given with simultaneous standard and continuous temozolomide in patients with GBM. J Clin Oncol. 2008;26:2011.
181. J. Nemunaitis, T. Jahan, H. Ross, et al. Phase 1/2 trial of autologous tumor mixed with an allogeneic GVAX vaccine in advanced-stage non-small-cell lung cancer. Cancer Gene Ther. 2006;13:555-562.