[level-membership-for-dermatology-category]
Malignant atrophic papulosis
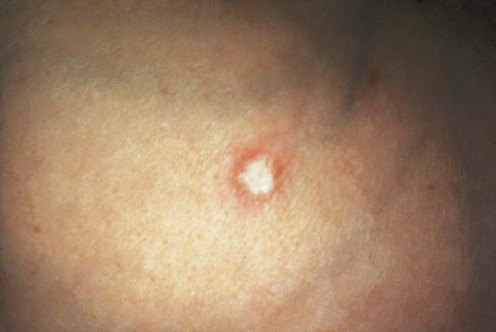
(From Lebwohl MG. The Skin and Systemic Disease: A Color Atlas, 2nd edn. Churchill Livingstone 2003, with permission of Elsevier.)
Specific investigations

 Tissue biopsy (skin primarily, but also tissue from the intestines and other internal organs)
Tissue biopsy (skin primarily, but also tissue from the intestines and other internal organs)



 Protein C, protein S and factor V Lieden, antithrombin III, homocysteine levels
Protein C, protein S and factor V Lieden, antithrombin III, homocysteine levels
 Anticardiolipin antibody titer
Anticardiolipin antibody titer
 Antiphospholipid antibody titer
Antiphospholipid antibody titer
 Endoscopy of the gastrointestinal tract (i.e., stomach, esophagus, duodenum, colon, rectum)
Endoscopy of the gastrointestinal tract (i.e., stomach, esophagus, duodenum, colon, rectum)


 Eculizumab
EculizumabSecond-line therapies
 Coumadin (warfarin)
Coumadin (warfarin) Heparin
Heparin Aspirin
Aspirin Dipyridamole
Dipyridamole Phenformin
Phenformin Ethylestrenol
Ethylestrenol Nicotine patches
Nicotine patches Lansoprazole (for gastrointestinal ulceration)
Lansoprazole (for gastrointestinal ulceration) Cyclosporine
CyclosporineThird-line therapies
 Azathioprine
Azathioprine Cyclophosphamide
Cyclophosphamide Tacrolimus
Tacrolimus Intravenous immunoglobulin
Intravenous immunoglobulin Ultraviolet B therapy
Ultraviolet B therapy
[/level-membership-for-dermatology-category][not-level-membership-for-dermatology-category]
Malignant atrophic papulosis
(From Lebwohl MG. The Skin and Systemic Disease: A Color Atlas, 2nd edn. Churchill Livingstone 2003, with permission of Elsevier.)
