CHAPTER 51 Malformations of Cortical Development
Malformations of cortical development (MCDs) are a heterogeneous group of disorders characterized by abnormal cerebral cortical cytoarchitecture. MCDs represent a spectrum of disorders from subtle microdysgenesis affecting a small area of the cortex to devastating lissencephaly in which the regional, gyral, and laminar patterning of the cortex is lost. A unifying feature of MCDs is their high association with intractable epilepsy and in many cases with cognitive disabilities, autism, or pervasive developmental disorders. With the routine use of magnetic resonance imaging (MRI), many subtle MCDs are being identified with greater frequency and with higher resolution,1–7 although so-called minimal MCDs (mMCDs) often remain radiographically undetectable.
Classification of Malformations of Cortical Development
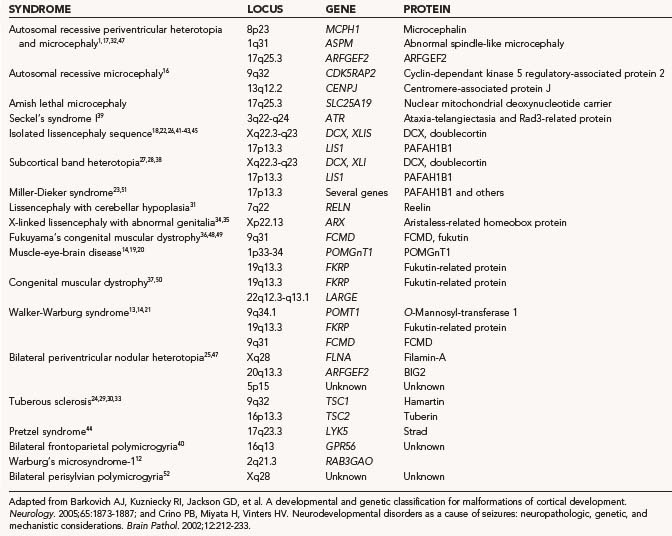
The majority of patients with MCDs do not have a known specific genetic syndrome. Development of the normal six-layered human cortex results from a complex series of events, in large part controlled by known and as yet unknown genes.8–11 Disruptions in these developmentally regulated genes can result in cortical malformations. A number of new genes have been discovered that cause certain forms of MCD (Table 51-1).12–52 The specific malformation resulting from each mutation is related to the normal temporal and anatomic expression of the gene product that has been lost.
The dynamic process of cortical development can be disrupted at many time points by gene mutation or environmental events. Formation of the normal cerebral cortex extends from weeks 8 to 26 of human gestation and is orchestrated by a complex array of genes5–11 that ultimately result in the correct cortical laminar organization. The cortex forms from neuroglial progenitor (stem) cells born in the ventricular and subventricular zones that undergo successive rounds of mitosis. Once exiting the cell cycle, neurons migrate via both radial and tangential pathways in response to various trophic and repulsive cues to form the cortical plate (the nascent cortex). Excitatory neurons follow a radial inside-out migratory gradient along radial glial cells from layer VI to layer II. Layer I is an early embryonic layer that predates the deeper layers. Inhibitory neurons are born in the ganglionic eminences and migrate by tangential pathways into the cortical plate. Cells arriving at their appropriate cortical laminar destination cease migration, begin to extend axons and dendrites, and form early functional synapses. Conceptually, normal corticogenesis can be broadly divided into three stages: proliferation, migration, and organization.
Based on the proposed steps for normal cortical development and the recent identification of single genes responsible for many MCDs, a classification scheme for abnormal cortical development (i.e., MCDs) was defined in 1996 and later modified in 2001 and 2004.53–55 The MCD taxonomy is organized according to the putative cellular mechanisms that are disrupted in development. These groups include (1) disorders of cellular proliferation, (2) disorders of neuronal migration, (3) disorders of cortical organization, and (4) disorders with unknown mechanisms (Table 51-2). Functional in vitro studies have been used to mechanistically support parcellation of particular MCDs into each category.
TABLE 51-2 Classification of Malformations of Cortical Development
| GROUP | SUBGROUP | CONDITION |
|---|---|---|
| Disorders of cellular proliferation | Abnormal proliferation | Microcephaly |
| Macrocephaly | ||
| Cortical tubers (TSC) | ||
| FCD with balloon cells | ||
| Hemimegalencephaly | ||
| Abnormal proliferation | Dysembryoplastic neuroepithelial tumor | |
| Neoplastic process | Ganglioglioma | |
| Gangliocytoma | ||
| Disorders of neuronal migration | Lissencephaly | |
| Heterotopia | ||
| Muscular dystrophy | Congenital muscular dystrophy | |
| Muscle-eye-brain disease | ||
| Walker-Warburg syndrome | ||
| Disorders of cortical organization | Polymicrogyria | |
| Schizencephaly |
FCD, focal cortical dysplasia; TSC, tuberous sclerosis complex.
Adapted from Barkovich AJ, Kuzniecky RI, Jackson GD, et al. A developmental and genetic classification for malformations of cortical development. Neurology. 2005;65:1873-1887.
Disorders of Cellular Proliferation
Malformations in this group include disorders that result in either increased or decreased growth or in increased or decreased apoptosis. These disorders are often manifested as abnormal brain size or localized areas of enlargement. Abnormalities in brain size can be manifested as microcephaly with a normal to thin cortex, microlissencephaly, microcephaly with polymicrogyria, or macrocephaly.15,17,32,47
Microcephaly is defined as a head circumference that is more than 2 SD smaller than normal.56 Patients with this condition have a brain that may be up to 70% smaller than normal, but the brain has essentially normal cortical laminar architecture. In some cases of microcephaly, a simplified gyral folding pattern is observed on MRI. Because any process that interferes with brain growth can lead to reduced head/brain size, microcephaly is associated with a highly heterogeneous group of disorders. It can result from a number of prenatal insults, including hypoxia-ischemia and fetal alcohol exposure. Additionally, a number of genes (ASPM, MCPH1, CENPJ, and CDK5RAP2) have been shown to result in autosomal recessive forms of microcephaly.15–17,32,57,58 Rare syndromic forms include Amish lethal microcephaly and Seckel’s syndrome.16,46 The ASPM (abnormal spindle-like microcephaly associated) gene is of particular interest because it is the single most common genetic mutation associated with microcephaly and has provided new insight into evolutionary mechanisms governing brain size across species. ASPM is an orthologue of a Drosophila gene that has been shown to be essential for normal mitotic spindle function in embryonic neuroblasts. When absent, neuroblasts are arrested in the cell cycle and fail to proliferate, which results in a reduction in the number of cells in the brain and consequently a smaller brain size. When the human ASPM gene sequence was compared with that of primates, there was a correlation between sequence and brain size.59 Thus, ASPM appears to have played a central role in the genetic component of the evolutionary expansion in brain size from primates to Homo sapiens.
Macrocephaly, defined as measurable enlargement of the head (>2 SD), can occur in conjunction with a large number of syndromes and conditions that are not associated with MCDs, including hydrocephalus, autism, leukodystrophies, and organic acidurias.60 Hemimegalencephaly (HME) is a malformation characterized by the abnormal enlargement of one entire hemisphere (Fig. 51-1); a variant of HME is lobar dysplasia, in which an entire brain lobe is enlarged. HME may be seen in isolation or in the setting of a known syndrome such as linear sebaceous nevus (of Jadassohn), hypomelanosis of Ito, tuberous sclerosis complex (TSC), or Proteus syndrome. HME is highly associated with intractable seizures and, frequently, infantile spasms. Pathologic analysis of HME brain demonstrates loss of cortical lamination, dysmorphic neurons, and cytomegalic cells similar to those seen in focal cortical dysplasia (FCD) and TSC (see later).61–63 As yet, no specific gene has been associated with HME, although reports suggest an association with PTEN mutations in Proteus syndrome.64–67
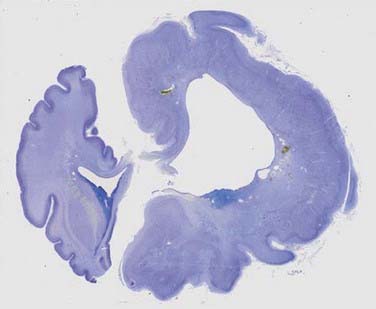
In addition to changes across broad cortical areas, disorders of cellular proliferation can result in focal malformations or dysplasias. Such lesions include FCDs, in particular, cortical dysplasias containing balloon cells, tubers in TSC, and low-grade neoplastic lesions such as dysembryoplastic neuroepithelial tumor, ganglioglioma (Fig. 51-2), and gangliocytoma.24,29,30,33,44,61–63,68 TSC results from mutations in TSC1, which encodes TSC1 or hamartin, or from mutations in TSC2, which encodes TSC2 or tuberin.24,29,30,33 TSC1 and TSC2 are known modulators of the mammalian target of rapamycin (mTOR) pathway, which governs cell size and proliferation. Neurological manifestations of TSC include epilepsy, infantile spasms, cognitive disabilities, and autism.69 Cortical tubers are focal areas of disorganized cortical lamination characterized by the presence of cytomegalic cells known as giant cells.69,70 The number of tubers across patients is variable and does not seem to vary with mutation type, patient age, or necessarily, neurological phenotype.
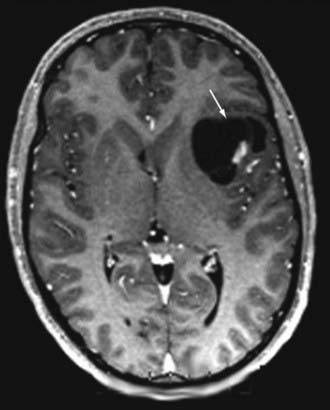
FCD with balloon cells (so-called Palmini type 2B dysplasia; Fig. 51-3) is a sporadic condition for which no specific gene mutation has been found.68,71 Like the tubers in TSC, FCD 2B consists of focal areas of cortical laminar disorganization characterized histopathologically by neuronal dysmorphism and pathognomonic enlarged cells, known as balloon cells, that are similar to the giant cells found in TSC. In addition to appearance, balloon cells and giant cells share many immunohistochemical features.62,70,72–74
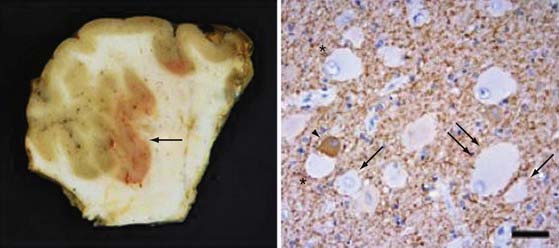
Disorders of Neuronal Migration
Disorders of migration are a heterogeneous set of syndromes characterized by altered gyral patterning and cortical laminar organization or the presence of ectopic neurons in the subcortical white matter or periventricular region. Such disorders include lissencephaly, subependymal/periventricular heterotopia, marginal glioneuronal heterotopia, subcortical band heterotopia, and heterotopia syndromes.75,76 Disorders of neuronal migration can exhibit various clinicopathologic phenotypes, depending on the timing and location of the migratory disruption.
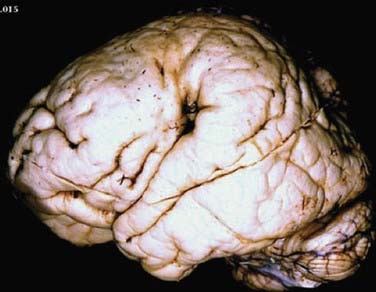
Lissencephaly (“smooth brain”) refers to a heterogeneous group of disorders characterized by loss of the normal gyral pattern and significant disorganization of cortical laminar cytoarchitecture (Fig. 51-4). Lissencephaly is separated into two pathologic subtypes: type 1 or classic and type II or cobblestone lissencephaly. In type I lissencephaly, the gyral patterning across the entire cortex is altered. Because of abnormal neuronal migration, neuronal progenitor cells fail to move to their correct laminar location and the normal six-layer cortex is reduced to a four-layered pattern.
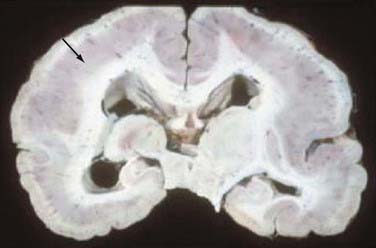
FIGURE 51-4 Unfixed specimen of lissencephaly. Note the thickened cortex (arrow) and absence of cortical gyri.
Several autosomal recessive and X-linked forms of type I lissencephaly exist and result from mutations in the LIS1, DCX, ARX, or RELN genes.* These genes are believed to be involved in important cell processes, such as movement of the neuronal nucleus or dynamic changes in the neuronal cytoskeleton, that are required for neuronal progenitor cells to migrate properly from the ventricular zone into the cortical plate.77–79 LIS1 mutations are seen in isolated lissencephaly, as well as in association with Miller-Dieker lissencephaly syndrome.
Syndromic forms of type II or cobblestone lissencephaly are associated with rare congenital forms of muscular dystrophy, muscle-eye-brain disease, and Walker-Warburg syndrome.12–14,19–21,36,37,48–50 In type II lissencephaly, the altered gyral patterning is more heterogeneously distributed across the cortical mantle. The combination of a muscular dystrophy and lissencephaly should prompt a search for one of these syndromes.
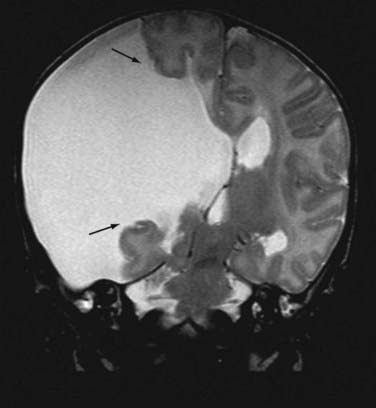
Subcortical band heterotopia (Fig. 51-5) and periventricular nodular heterotopia (Fig. 51-6) syndromes are X-linked disorders that are manifested as sexually dimorphic phenotypes. For example, in females, mutations in the DCX (doublecortin) gene are associated with a type of subcortical band heterotopia in which a broad focal band of cortical neurons is trapped within the subcortical white matter bilaterally.26,41,79,80 The band consists of neurons of both excitatory and inhibitory phenotypes and makes connections with the overlying cortices. In males, the DCX mutation is associated with lissencephaly. Periventricular nodular heterotopia is a disorder resulting from mutations in the filamin-1 (FLN1) gene in which focal nodules of cortical neurons are observed within the walls of the lateral ventricles. These collections are also composed of both excitatory and inhibitory neurons, and recent tensor tract imaging techniques have revealed that these cells make connections with the overlying cortex. FLN1 mutations in males lead to embryonic lethality. It has been proposed that the differential mutational features in males and females reflect the effects of the mutation expressed on a single X chromosome in males versus the presence of both mutated and normal X chromosomes in females. The focal pathology in both disorders is hypothesized to be variable X chromosome inactivation in developing neural stem cells, which occurs in the affected female brain.22,43
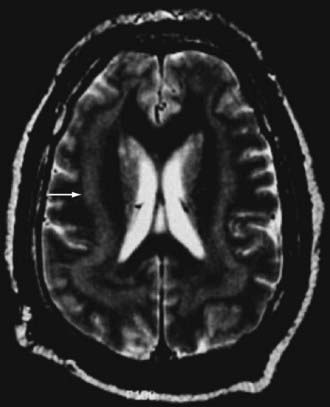
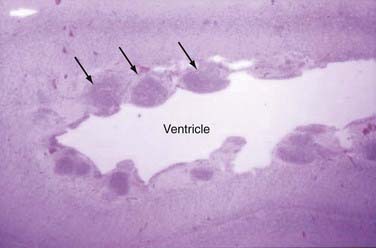
Disorders of Cortical Organization
Polymicrogyria is characterized by an excessive number of abnormally small gyri (Fig. 51-7). It can occur secondary to genetic mutations and has been hypothesized to result from prenatal infections or abnormal twinning.40,52,81 There are rare case reports in which abnormal twin pregnancies resulted in cortical disruption and polymicrogyria.82,83 Bilateral perisylvian polymicrogyria (BPP) is the most common type of polymicrogyria and is characterized clinicopathologically by bilateral polymicrogyria in the perisylvian region, pseudobulbar paresis, mental retardation, and epilepsy. BPP has been mapped to Xq27-28. Additionally, there are a number of congenital conditions in which polymicrogyria is part of the syndrome, including Joubert’s, Delleman’s, Adams-Oliver, and Galloway-Mowat syndromes.
Schizencephaly is characterized by a cerebral cleft that, unlike a porencephalic cyst, is lined by gray matter (Fig. 51-8).84–88 The schizencephalic cleft extends from the surface of the cortex to the ventricle. The cleft can be filled with cerebrospinal fluid (the open-lipped form) or the two surfaces can be fused (the closed-lipped form). Schizencephaly is almost always associated with localized polymicrogyria around the cleft.
Surgery for Malformations of Cortical Development
MCDs are the most common cause of intractable pediatric epilepsy.89,90 For example, 70% to 90% of patients with TSC, HME, or lissencephaly suffer from seizures. In studies on epilepsy surgical samples, MCDs may be seen in 30% of cortical resections, and therefore these lesions and the conditions associated with them are particularly important for epilepsy surgeons. Untreated epilepsy in infants, such as infantile spasms, can result in profound developmental problems for the child.91 Although surgery can be difficult in very young patients, surgical treatment of MCDs associated with intractable seizures can lead to cure and ameliorate some of the developmental problems.92–97
The surgical treatment of epilepsy associated with these lesions includes simple lesionectomy, lesionectomy and resection of the surrounding epileptic focus, resection of the lesion and the mesial temporal structures, and hemispherectomy. Each lesion and the treatment strategy are approached individually based on the extent of the malformation and the age of the patient. In addition, in the setting of long-standing seizures, the mesial temporal structures may be involved and must be resected along with the seizure focus.98
The outcome of surgical treatment of these disorders varies according to the condition. Even with significant advances in localization and surgical technique, surgical treatment of FCD results in cure in only 42% to 55% of patients, with around 70% having a good surgical outcome (Engel classes I and II).99–101 Resection of glioneuronal tumors such as gangliogliomas results in freedom from seizures in 58% to 86% of patients.102–106 Better surgical outcome was associated with a shorter duration of seizures, absence of generalized seizures, younger patient age, and total resection.102
Epilepsy surgery for TSC lesions results in freedom from seizures in 53% to 68% of patients and good seizure outcome in 64% to 92%.107–109 Presurgical evaluation of patients with TSC is particularly important and is primarily directed at identifying the specific seizure-causing lesion. In the setting of a patient with multiple tubers, treatment may require invasive mapping to identify the seizure focus or may require multiple resections.109,110 A worse outcome is associated with younger age at seizure onset, history of infantile spasms, and bilateral focal interictal discharges.108
HME is often treated by hemispherectomy, and both functional and anatomic methods have been described. HME may be associated with a worse outcome than other MCDs with regard to freedom from seizures or developmental outcome; however, the number of patients in most studies is small, and it is difficult to draw major conclusions from such studies.111–114 In a study of 10 patients with HME, 60% were seizure free and 80% had a good outcome.111 A study involving 9 infants with HME demonstrated freedom from seizures in 57% after hemispherectomy.115 Anatomic resection may be preferable to disconnection procedures in the management of HME because disconnection procedures for this condition can be technically difficult with an increased risk for incomplete hemispheric disconnection.
In the limited number of series on schizencephalic lesions, surgery has been shown to be particularly effective, especially when the resection was tailored to the seizure focus and not just the schizencephalic region. However, studies on schizencephaly are limited to case reports and small case series, and it is difficult to draw major conclusions on outcomes after seizure surgery for this condition.116–118
In addition to surgical resection, other options for the treatment of epilepsy associated with MCDs include corpus callosotomy and vagal nerve stimulation. These options are used in patients in whom the seizure focus is in eloquent areas, is bilateral, or cannot be well defined or in patients who are poor surgical candidates for focal resection. Even though these treatments rarely result in cure, they can lead to a significant reduction in seizure frequency in patients with MCDs.119–124
Andermann F. Cortical dysplasias and epilepsy: a review of the architectonic, clinical, and seizure patterns. Adv Neurol. 2000;84:479-496.
Barkovich AJ, Kuzniecky RI, Jackson GD, et al. A developmental and genetic classification for malformations of cortical development. Neurology. 2005;65:1873-1887.
Bond J, Roberts E, Mochida GH, et al. ASPM is a major determinant of cerebral cortical size. Nat Genet. 2002;32:316-320.
Crino PB. Molecular pathogenesis of focal cortical dysplasia and hemimegalencephaly. J Child Neurol. 2005;20:330-336.
Cross JH. Functional neuroimaging of malformations of cortical development. Epileptic Disord. 2003;5(suppl 2):S73-S80.
Granata T, Freri E, Caccia C, et al. Schizencephaly: clinical spectrum, epilepsy, and pathogenesis. J Child Neurol. 2005;20:313-318.
Mathern GW, Andres M, Salamon N, et al. A hypothesis regarding the pathogenesis and epileptogenesis of pediatric cortical dysplasia and hemimegalencephaly based on MRI cerebral volumes and NeuN cortical cell densities. Epilepsia. 2007;48(suppl 5):74-78.
Olney AH. Macrocephaly syndromes. Semin Pediatr Neurol. 2007;14:128-135.
Palmini A, Najm I, Avanzini G, et al. Terminology and classification of the cortical dysplasias. Neurology. 2004;62:S2-S8.
Puffenberger EG, Strauss KA, Ramsey KE, et al. Polyhydramnios, megalencephaly and symptomatic epilepsy caused by a homozygous 7-kilobase deletion in LYK5. Brain. 2007;130:1929-1941.
Weiner HL, Carlson C, Ridgway EB, et al. Epilepsy surgery in young children with tuberous sclerosis: results of a novel approach. Pediatrics. 2006;117:1494-1502.
Zhang J. Evolution of the human ASPM gene, a major determinant of brain size. Genetics. 2003;165:2063-2070.
1 Barkovich AJ, Kuzniecky RI. Neuroimaging of focal malformations of cortical development. J Clin Neurophysiol. 1996;13:481-494.
2 Burneo JG, Kuzniecky RI, Bebin M, et al. Cortical reorganization in malformations of cortical development: a magnetoencephalographic study. Neurology. 2004;63:1818-1824.
3 Colombo N, Tassi L, Galli C, et al. Focal cortical dysplasias: MR imaging, histopathologic, and clinical correlations in surgically treated patients with epilepsy. AJNR Am J Neuroradiol. 2003;24:724-733.
4 Cross JH. Functional neuroimaging of malformations of cortical development. Epileptic Disord. 2003;5(suppl 2):S73-S80.
5 Detre JA, Crino PB. A multilayered approach to studying cortical malformations: EEG-fMRI. Neurology. 2005;64:1108-1110.
6 Mackay MT, Becker LE, Chuang SH, et al. Malformations of cortical development with balloon cells: clinical and radiologic correlates. Neurology. 2003;60:580-587.
7 Widjaja E, Wilkinson ID, Griffiths PD. Magnetic resonance perfusion imaging in malformations of cortical development. Acta Radiol. 2007;48:907-917.
8 Golden JA. Cell migration and cerebral cortical development. Neuropathol Appl Neurobiol. 2001;27:22-28.
9 McConnell SK. Constructing the cerebral cortex: neurogenesis and fate determination. Neuron. 1995;15:761-768.
10 Rakic P. Principles of neural cell migration. Experientia. 1990;46:882-891.
11 Walsh CA. Genetic malformations of the human cerebral cortex. Neuron. 1999;23:19-29.
12 Aligianis IA, Johnson CA, Gissen P, et al. Mutations of the catalytic subunit of RAB3GAP cause Warburg Micro syndrome. Nat Genet. 2005;37:221-223.
13 Beltran-Valero de Bernabe D, Currier S, Steinbrecher A, et al. Mutations in the O-mannosyltransferase gene POMT1 give rise to the severe neuronal migration disorder Walker-Warburg syndrome. Am J Hum Genet. 2002;71:1033-1043.
14 Beltran-Valero de Bernabe D, Voit T, Longman C, et al. Mutations in the FKRP gene can cause muscle-eye-brain disease and Walker-Warburg syndrome. J Med Genet. 2004;41:e61.
15 Bond J, Roberts E, Mochida GH, et al. ASPM is a major determinant of cerebral cortical size. Nat Genet. 2002;32:316-320.
16 Bond J, Roberts E, Springell K, et al. A centrosomal mechanism involving CDK5RAP2 and CENPJ controls brain size (erratum appears in Nat Genet. 2005 May;37(5):555). Nat Genet. 2005;37:353-555.
17 Bond J, Scott S, Hampshire DJ, et al. Protein-truncating mutations in ASPM cause variable reduction in brain size. Am J Hum Genet. 2003;73:1170-1177.
18 Cardoso C, Leventer RJ, Matsumoto N, et al. The location and type of mutation predict malformation severity in isolated lissencephaly caused by abnormalities within the LIS1 gene. Hum Mol Genet. 2000;9:3019-3028.
19 Cormand B, Avela K, Pihko H, et al. Assignment of the muscle-eye-brain disease gene to 1p32-p34 by linkage analysis and homozygosity mapping. Am J Hum Genet. 1999;64:126-135.
20 Cormand B, Pihko H, Bayes M, et al. Clinical and genetic distinction between Walker-Warburg syndrome and muscle-eye-brain disease. Neurology. 2001;56:1059-1069.
21 de Bernabe DB-V, van Bokhoven H, van Beusekom E, et al. A homozygous nonsense mutation in the fukutin gene causes a Walker-Warburg syndrome phenotype. J Med Genet. 2003;40:845-848.
22 des Portes V, Francis F, Pinard JM, et al. Doublecortin is the major gene causing X-linked subcortical laminar heterotopia (SCLH). Hum Mol Genet. 1998;7:1063-1070.
23 Dobyns WB, Curry CJ, Hoyme HE, et al. Clinical and molecular diagnosis of Miller-Dieker syndrome. Am J Hum Genet. 1991;48:584-594.
24 European Chromosome 16 Tuberous Sclerosis Consortium. Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell. 1993;75:1305-1315.
25 Fox JW, Lamperti ED, Eksioglu YZ, et al. Mutations in filamin 1 prevent migration of cerebral cortical neurons in human periventricular heterotopia. Neuron. 1998;21:1315-1325.
26 Gleeson JG, Allen KM, Fox JW, et al. Doublecortin, a brain-specific gene mutated in human X-linked lissencephaly and double cortex syndrome, encodes a putative signaling protein. Cell. 1998;92:63-72.
27 Gleeson JG, Luo RF, Grant PE, et al. Genetic and neuroradiological heterogeneity of double cortex syndrome. Ann Neurol. 2000;47:265-269.
28 Gleeson JG, Minnerath SR, Fox JW, et al. Characterization of mutations in the gene doublecortin in patients with double cortex syndrome. Ann Neurol. 1999;45:146-153.
29 Haines JL, Short MP, Kwiatkowski DJ, et al. Localization of one gene for tuberous sclerosis within 9q32-9q34, and further evidence for heterogeneity. Am J Hum Genet. 1991;49:764-772.
30 Harris RM, Carter NP, Griffiths B, et al. Physical mapping within the tuberous sclerosis linkage group in region 9q32-q34. Genomics. 1993;15:265-274.
31 Hong SE, Shugart YY, Huang DT, et al. Autosomal recessive lissencephaly with cerebellar hypoplasia is associated with human RELN mutations (erratum appears in Nat Genet 2001 Feb;27(2):225). Nat Genet. 2000;26:93-96.
32 Jackson AP, Eastwood H, Bell SM, et al. Identification of microcephalin, a protein implicated in determining the size of the human brain. Am J Hum Genet. 2002;71:136-142.
33 Kandt RS, Haines JL, Smith M, et al. Linkage of an important gene locus for tuberous sclerosis to a chromosome 16 marker for polycystic kidney disease. Nat Genet. 1992;2:37-41.
34 Kato M, Das S, Petras K, et al. Mutations of ARX are associated with striking pleiotropy and consistent genotype-phenotype correlation. Hum Mutat. 2004;23:147-159.
35 Kitamura K, Yanazawa M, Sugiyama N, et al. Mutation of ARX causes abnormal development of forebrain and testes in mice and X-linked lissencephaly with abnormal genitalia in humans. Nat Genet. 2002;32:359-369.
36 Kobayashi K, Nakahori Y, Miyake M, et al. An ancient retrotransposal insertion causes Fukuyama-type congenital muscular dystrophy. Nature. 1998;394:388-392.
37 Longman C, Brockington M, Torelli S, et al. Mutations in the human LARGE gene cause MDC1D, a novel form of congenital muscular dystrophy with severe mental retardation and abnormal glycosylation of alpha-dystroglycan. Hum Mol Genet. 2003;12:2853-2861.
38 Matsumoto N, Leventer RJ, Kuc JA, et al. Mutation analysis of the DCX gene and genotype/phenotype correlation in subcortical band heterotopia. Eur J Hum Genet. 2001;9:5-12.
39 O’Driscoll M, Ruiz-Perez VL, Woods CG, et al. A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat Genet. 2003;33:497-501.
40 Piao X, Hill RS, Bodell A, et al. G protein–coupled receptor–dependent development of human frontal cortex. Science. 2004;303:2033-2036.
41 Pilz DT, Kuc J, Matsumoto N, et al. Subcortical band heterotopia in rare affected males can be caused by missense mutations in DCX (XLIS) or LIS1. Hum Mol Genet. 1999;8:1757-1760.
42 Pilz DT, Macha ME, Precht KS, et al. Fluorescence in situ hybridization analysis with LIS1 specific probes reveals a high deletion mutation rate in isolated lissencephaly sequence. Genet Med. 1998;1:29-33.
43 Pilz DT, Matsumoto N, Minnerath S, et al. LIS1 and XLIS (DCX) mutations cause most classical lissencephaly, but different patterns of malformation. Hum Mol Genet. 1998;7:2029-2037.
44 Puffenberger EG, Strauss KA, Ramsey KE, et al. Polyhydramnios, megalencephaly and symptomatic epilepsy caused by a homozygous 7-kilobase deletion in LYK5. Brain. 2007;130:1929-1941.
45 Reiner O, Carrozzo R, Shen Y, et al. Isolation of a Miller-Dieker lissencephaly gene containing G protein beta-subunit–like repeats. Nature. 1993;364:717-721.
46 Rosenberg MJ, Agarwala R, Bouffard G, et al. Mutant deoxynucleotide carrier is associated with congenital microcephaly. Nat Genet. 2002;32:175-179.
47 Sheen VL, Ganesh VS, Topcu M, et al. Mutations in ARFGEF2 implicate vesicle trafficking in neural progenitor proliferation and migration in the human cerebral cortex. Nat Genet. 2004;36:69-76.
48 Toda T, Kobayashi K. Fukuyama-type congenital muscular dystrophy: the first human disease to be caused by an ancient retrotransposal integration. J Mol Med. 1999;77:816-823.
49 Toda T, Segawa M, Nomura Y, et al. Localization of a gene for Fukuyama type congenital muscular dystrophy to chromosome 9q31-33 (erratum appears in Nat Genet 1994 May;7(1):113). Nat Genet. 1993;5:283-286.
50 Topaloglu H, Brockington M, Yuva Y, et al. FKRP gene mutations cause congenital muscular dystrophy, mental retardation, and cerebellar cysts. Neurology. 2003;60:988-992.
51 Toyo-oka K, Shionoya A, Gambello MJ, et al. 14-3-3epsilon is important for neuronal migration by binding to NUDEL: a molecular explanation for Miller-Dieker syndrome. Nat Genet. 2003;34:274-285.
52 Villard L, Nguyen K, Cardoso C, et al. A locus for bilateral perisylvian polymicrogyria maps to Xq28. Am J Hum Genet. 2002;70:1003-1008.
53 Barkovich AJ, Kuzniecky RI, Dobyns WB, et al. A classification scheme for malformations of cortical development. Neuropediatrics. 1996;27:59-63.
54 Barkovich AJ, Kuzniecky RI, Jackson GD, et al. Classification system for malformations of cortical development: update 2001. Neurology. 2001;57:2168-2178.
55 Barkovich AJ, Kuzniecky RI, Jackson GD, et al. A developmental and genetic classification for malformations of cortical development. Neurology. 2005;65:1873-1887.
56 Mochida GH, Walsh CA. Molecular genetics of human microcephaly. Curr Opin Neurol. 2001;14:151-156.
57 Kumar A, Markandaya M, Girimaji SC. Primary microcephaly: microcephalin and ASPM determine the size of the human brain. J Biosci. 2002;27:629-632.
58 Pattison L, Crow YJ, Deeble VJ, et al. A fifth locus for primary autosomal recessive microcephaly maps to chromosome 1q31. Am J Hum Genet. 2000;67:1578-1580.
59 Zhang J. Evolution of the human ASPM gene, a major determinant of brain size. Genetics. 2003;165:2063-2070.
60 Olney AH. Macrocephaly syndromes. Semin Pediatr Neurol. 2007;14:128-135.
61 Aronica E, Boer K, Baybis M, et al. Co-expression of cyclin D1 and phosphorylated ribosomal S6 proteins in hemimegalencephaly. Acta Neuropathol. 2007;114:287-293.
62 Crino PB. Molecular pathogenesis of focal cortical dysplasia and hemimegalencephaly. J Child Neurol. 2005;20:330-336.
63 Mathern GW, Andres M, Salamon N, et al. A hypothesis regarding the pathogenesis and epileptogenesis of pediatric cortical dysplasia and hemimegalencephaly based on MRI cerebral volumes and NeuN cortical cell densities. Epilepsia. 2007;48(suppl 5):74-78.
64 Butler MG, Dasouki MJ, Zhou XP, et al. Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J Med Genet. 2005;42:318-321.
65 Buxbaum JD, Cai G, Chaste P, et al. Mutation screening of the PTEN gene in patients with autism spectrum disorders and macrocephaly. Am J Med Genet B Neuropsychiatr Genet. 2007;144:484-491.
66 Reardon W, Zhou XP, Eng C. A novel germline mutation of the PTEN gene in a patient with macrocephaly, ventricular dilatation, and features of VATER association. J Med Genet. 2001;38:820-823.
67 Zhou XP, Marsh DJ, Hampel H, et al. Germline and germline mosaic PTEN mutations associated with a Proteus-like syndrome of hemihypertrophy, lower limb asymmetry, arteriovenous malformations and lipomatosis. Hum Mol Genet. 2000;9:765-768.
68 Taylor DC, Falconer MA, Bruton CJ, et al. Focal dysplasia of the cerebral cortex in epilepsy. J Neurol Neurosurg Psychiatry. 1971;34:369-387.
69 Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med. 2006;355:1345-1356.
70 Crino PB. Molecular pathogenesis of tuber formation in tuberous sclerosis complex. J Child Neurol. 2004;19:716-725.
71 Palmini A, Najm I, Avanzini G, et al. Terminology and classification of the cortical dysplasias. Neurology. 2004;62:S2-S8.
72 Baybis M, Yu J, Lee A, et al. mTOR cascade activation distinguishes tubers from focal cortical dysplasia. Ann Neurol. 2004;56:478-487.
73 Lee A, Maldonado M, Baybis M, et al. Markers of cellular proliferation are expressed in cortical tubers. Ann Neurol. 2003;53:668-673.
74 Taylor JP, Sater R, French J, et al. Transcription of intermediate filament genes is enhanced in focal cortical dysplasia. Acta Neuropathol. 2001;102:141-148.
75 Barkovich AJ. Morphologic characteristics of subcortical heterotopia: MR imaging study. AJNR Am J Neuroradiol. 2000;21:290-295.
76 Barkovich AJ. Subcortical heterotopia: a distinct clinicoradiologic entity. AJNR Am J Neuroradiol. 1996;17:1315-1322.
77 Feng Y, Olson EC, Stukenberg PT, et al. LIS1 regulates CNS lamination by interacting with mNudE, a central component of the centrosome. Neuron. 2000;28:665-679.
78 Gleeson JG, Lin PT, Flanagan LA, et al. Doublecortin is a microtubule-associated protein and is expressed widely by migrating neurons. Neuron. 1999;23:257-271.
79 Sapir T, Elbaum M, Reiner O. Reduction of microtubule catastrophe events by LIS1, platelet-activating factor acetylhydrolase subunit. EMBO J. 1997;16:6977-6984.
80 des Portes V, Pinard JM, Billuart P, et al. A novel CNS gene required for neuronal migration and involved in X-linked subcortical laminar heterotopia and lissencephaly syndrome. Cell. 1998;92:51-61.
81 Barkovich AJ, Lindan CE. Congenital cytomegalovirus infection of the brain: imaging analysis and embryologic considerations. AJNR Am J Neuroradiol. 1994;15:703-715.
82 Barth PG, van der Harten JJ. Parabiotic twin syndrome with topical isocortical disruption and gastroschisis. Acta Neuropathol. 1985;67:345-349.
83 Sugama S, Kusano K. Monozygous twin with polymicrogyria and normal co-twin. Pediatr Neurol. 1994;11:62-63.
84 Barkovich AJ, Kjos BO. Schizencephaly: correlation of clinical findings with MR characteristics. AJNR Am J Neuroradiol. 1992;13:85-94.
85 Denis D, Chateil JF, Brun M, et al. Schizencephaly: clinical and imaging features in 30 infantile cases. Brain Dev. 2000;22:475-483.
86 Granata T, Farina L, Faiella A, et al. Familial schizencephaly associated with EMX2 mutation. Neurology. 1997;48:1403-1406.
87 Granata T, Freri E, Caccia C, et al. Schizencephaly: clinical spectrum, epilepsy, and pathogenesis. J Child Neurol. 2005;20:313-318.
88 Lopes CF, Cendes F, Piovesana AM, et al. Epileptic features of patients with unilateral and bilateral schizencephaly. J Child Neurol. 2006;21:757-760.
89 Andermann F. Cortical dysplasias and epilepsy: a review of the architectonic, clinical, and seizure patterns. Adv Neurol. 2000;84:479-496.
90 Crino PB, Chou K. Epilepsy and cortical dysplasias. Curr Treat Options Neurol. 2000;2:543-552.
91 Asarnow RF, LoPresti C, Guthrie D, et al. Developmental outcomes in children receiving resection surgery for medically intractable infantile spasms. Dev Med Child Neurol. 1997;39:430-440.
92 Daniel RT, Meagher-Villemure K, Roulet E, et al. Surgical treatment of temporoparietooccipital cortical dysplasia in infants: report of two cases. Epilepsia. 2004;45:872-876.
93 Duchowny M, Jayakar P, Resnick T, et al. Epilepsy surgery in the first three years of life. Epilepsia. 1998;39:737-743.
94 Duchowny MS, Resnick TJ, Alvarez LA, et al. Focal resection for malignant partial seizures in infancy. Neurology. 1990;40:980-984.
95 Kang H-C, Jung DE, Kim KM, et al. Surgical treatment of two patients with infantile spasms in early infancy. Brain Dev. 2006;28:453-457.
96 Olavarria G, Petronio JA. Epilepsy surgery in infancy. A review of four cases. Pediatr Neurosurg. 2003;39:44-49.
97 Sugimoto T, Otsubo H, Hwang PA, et al. Outcome of epilepsy surgery in the first three years of life. Epilepsia. 1999;40:560-565.
98 Kobayashi E, Bagshaw AP, Grova C, et al. Grey matter heterotopia: what EEG-fMRI can tell us about epileptogenicity of neuronal migration disorders. Brain. 2006;129:366-374.
99 Hader WJ, Mackay M, Otsubo H, et al. Cortical dysplastic lesions in children with intractable epilepsy: role of complete resection. J Neurosurg. 2004;100:110-117.
100 Hong SC, Kang KS, Seo DW, et al. Surgical treatment of intractable epilepsy accompanying cortical dysplasia. J Neurosurg. 2000;93:766-773.
101 Sisodiya SM. Surgery for malformations of cortical development causing epilepsy. Brain. 2000;123:1075-1091.
102 Aronica E, Leenstra S, van Veelen CW, et al. Glioneuronal tumors and medically intractable epilepsy: a clinical study with long-term follow-up of seizure outcome after surgery. Epilepsy Res. 2001;43:179-191.
103 Giulioni M, Galassi E, Zucchelli M, et al. Seizure outcome of lesionectomy in glioneuronal tumors associated with epilepsy in children. J Neurosurg. 2005;102:288-293.
104 Giulioni M, Gardella E, Rubboli G, et al. Lesionectomy in epileptogenic gangliogliomas: seizure outcome and surgical results. J Clinical Neurosci. 2006;13:529-535.
105 Im SH, Chung CK, Cho BK, et al. Supratentorial ganglioglioma and epilepsy: postoperative seizure outcome. J Neurooncol. 2002;57:59-66.
106 Radhakrishnan A, Abraham M, Radhakrishnan VV, et al. Medically refractory epilepsy associated with temporal lobe ganglioglioma: characteristics and postoperative outcome. Clin Neurol Neurosurg. 2006;108:648-654.
107 Jansen FE, Van Huffelen AC, Van Rijen PC, et al. Epilepsy surgery in tuberous sclerosis: the Dutch experience. Seizure. 2007;16:445-453.
108 Madhavan D, Schaffer S, Yankovsky A, et al. Surgical outcome in tuberous sclerosis complex: a multicenter survey. Epilepsia. 2007;48:1625-1628.
109 Weiner HL, Carlson C, Ridgway EB, et al. Epilepsy surgery in young children with tuberous sclerosis: results of a novel approach. Pediatrics. 2006;117:1494-1502.
110 Weiner HL, Ferraris N, LaJoie J, et al. Epilepsy surgery for children with tuberous sclerosis complex. J Child Neurol. 2004;19:687-689.
111 Battaglia D, Di Rocco C, Iuvone L, et al. Neuro-cognitive development and epilepsy outcome in children with surgically treated hemimegalencephaly. Neuropediatrics. 1999;30:307-313.
112 Di Rocco C, Iannelli A. Hemimegalencephaly and intractable epilepsy: complications of hemispherectomy and their correlations with the surgical technique. A report on 15 cases. Pediatr Neurosurg. 2000;33:198-207.
113 Edwards JC, Wyllie E, Ruggeri PM, et al. Seizure outcome after surgery for epilepsy due to malformation of cortical development. Neurology. 2000;55:1110-1114.
114 Jonas R, Nguyen S, Hu B, et al. Cerebral hemispherectomy: hospital course, seizure, developmental, language, and motor outcomes. Neurology. 2004;62:1712-1721.
115 Gonzalez-Martinez JA, Gupta A, Kotagal P, et al. Hemispherectomy for catastrophic epilepsy in infants. Epilepsia. 2005;46:1518-1525.
116 Leblanc R, Tampieri D, Robitaille Y, et al. Surgical treatment of intractable epilepsy associated with schizencephaly. Neurosurgery. 1991;29:421-429.
117 Maehara T, Shimizu H, Nakayama H, et al. Surgical treatment of epilepsy from schizencephaly with fused lips. Surg Neurol. 1997;48:507-510.
118 Silbergeld DL, Miller JW. Resective surgery for medically intractable epilepsy associated with schizencephaly. J Neurosurg. 1994;80:820-825.
119 Alpman A, Serdaroglu G, Cogulu O, et al. Ring chromosome 20 syndrome with intractable epilepsy. Dev Med Child Neurol. 2005;47:343-346.
120 Landy HJ, Curless RG, Ramsay RE, et al. Corpus callosotomy for seizures associated with band heterotopia. Epilepsia. 1993;34:79-83.
121 Nei M, O’Connor M, Liporace J, et al. Refractory generalized seizures: response to corpus callosotomy and vagal nerve stimulation. Epilepsia. 2006;47:115-122.
122 Pallini R, Aglioti S, Tassinari G, et al. Callosotomy for intractable epilepsy from bihemispheric cortical dysplasias. Acta Neurochir (Wein). 1995;132:79-86.
123 Parain D, Penniello MJ, Berquen P, et al. Vagal nerve stimulation in tuberous sclerosis complex patients. Pediatr Neurol. 2001;25:213-216.
124 Ray A, Wyllie E. Treatment options and paradigms in childhood temporal lobe epilepsy. Expert Rev Neurother. 2005;5:785-801.