Chapter 42 Macular Dystrophies
 For additional online content visit http://www.expertconsult.com
For additional online content visit http://www.expertconsult.com
Introduction
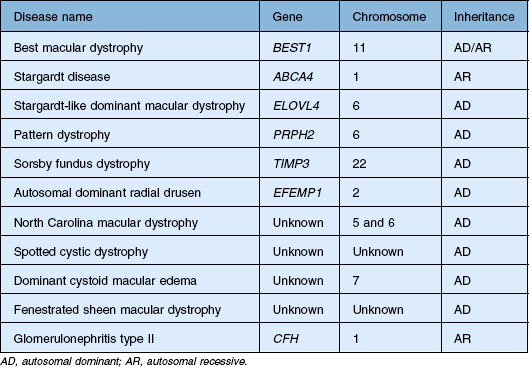
The genes that cause most of the disorders in this chapter have been identified and it is tempting to try to eliminate the inconsistencies in the historical clinical nomenclature by abandoning it in favor of a scheme that is solely based on the causative genes. However, there are several serious disadvantages to such an approach. First, patients present with symptoms and signs – not with genetic test results. In addition, the near-term disease course in a given patient is usually more tightly correlated to their current clinical appearance and visual function than it is to their genotype. Thus, even in the molecular era, clinicians need to think first in terms of clinical patterns and second about the molecular mechanisms underlying these patterns. Prior to 1985 the clinical diagnosis was for all practical purposes the final diagnosis. Today the initial clinical diagnosis is really a hypothesis that has some prognostic weight by itself, and also serves to focus the search for a more precise and definitive molecular understanding of the patient’s disease through genetic testing. Clinicians are often bothered by the fact that there is an imperfect correlation between mutations in specific genes and the resulting clinical outcomes. That is, mutations in a single gene (e.g., PRPH2 and ABCA4) can cause quite different clinical appearances in different patients (e.g., retinitis pigmentosa and macular dystrophy) while a single clinical appearance (e.g., retinitis pigmentosa) can be caused by mutations in many different genes. The best way to cope with this imperfect correlation is to simply accept that the clinical diagnosis has greater validity in certain contexts while the molecular diagnosis has greater validity in others – and that the one can often strengthen the other. For example, prior to the discovery of the causative genes, characteristic clinical features were used to select genetically similar patients from heterogeneous clinic populations for the purpose of gene discovery. Now that many of the disease genes are known, genetic testing can be used to select genetically similar cohorts from clinically heterogeneous populations for the purpose of identifying the range of clinical features that can be associated with mutations in each gene. In this chapter we have taken advantage of molecular diagnosis in our patients to illustrate some unusual clinical presentations of these macular dystrophies that would have been difficult to include in such a chapter in the pre-molecular era. For the most part, we have retained the historical names associated with specific phenotypes and have grouped these whenever they are known to be caused by mutations in a single gene. We have also organized the disorders in the approximate order of their prevalence in the population with the more common diseases first. Table 42.1 summarizes the genetic characterization for all of the macular dystrophies discussed in this chapter.
The initial approach to a patient with macular dystrophy
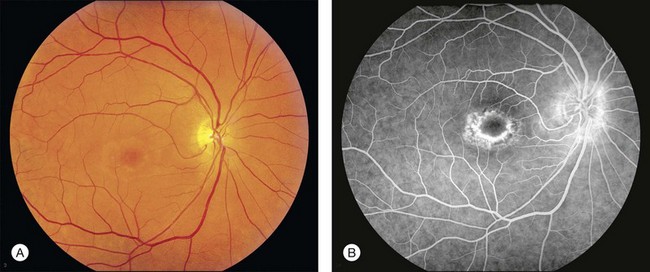
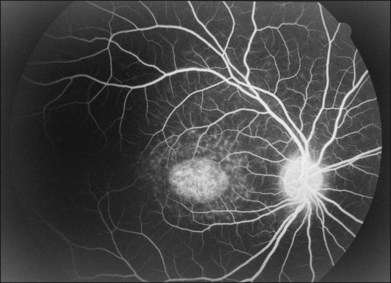
Perhaps the most important step in managing a patient with a macular dystrophy is to convince oneself that the patient truly has a Mendelian condition and not one of the many toxic, infectious, autoimmune, and multigenic disorders that can mimic them. Except in extraordinary circumstances, macular dystrophies are bilateral, and in the early years of the disease are usually extremely symmetrical in their fundus appearance. Autoimmune disorders like the presumed ocular histoplasmosis syndrome and multifocal choroiditis are often bilateral but much less symmetrical in their appearance (Fig. 42.1). Most of the macular dystrophies are inherited in an autosomal dominant fashion and thus one or more living affected relatives often exist. Identification of such a relative is one of the most powerful and most underutilized diagnostic maneuvers the clinician has at his or her disposal. One should always take a careful family history from patients suspected to have a macular dystrophy, realizing that affected individuals over the age of 50 will often carry the diagnosis of age-related macular degeneration (AMD). One should also remember that dominant macular dystrophies often exhibit variable expressivity and incomplete penetrance, such that a patient’s parents can be normal by history while more distant relatives can exhibit macular disease. Thus, when taking the history one should be very suspicious of any relative reported to have a macular disease of any kind. One should also examine any first-degree relatives who accompany the patient to the clinic and request fundus photographs and other ophthalmic records from all relatives with a history of macular disease. Not infrequently an affected relative will exhibit the “classic” features of a specific macular dystrophy but carry the diagnosis of AMD while the patient before you will exhibit a more puzzling fundus appearance and/or set of symptoms.
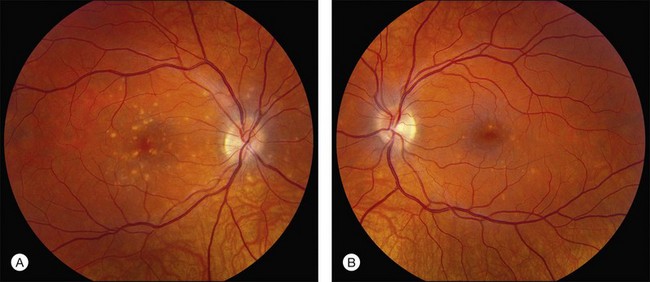
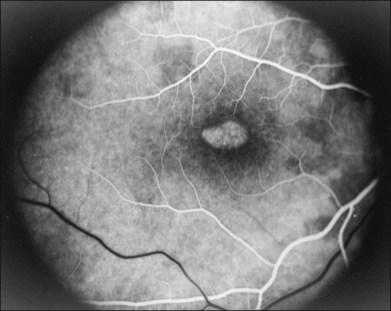
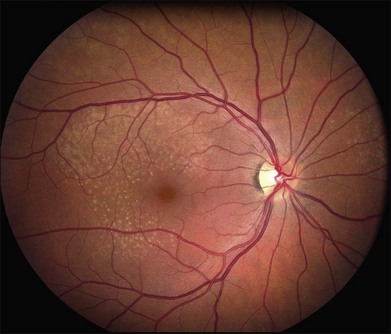
There are two classes of disease one should be especially careful to think about when initially considering a diagnosis of macular dystrophy: neuronal ceroid lipofuscinosis (NCL) and drug toxicity. In children between the ages of six and eight years, NCL (a fatal systemic disease) can present with a Stargardt-like appearance and no systemic features of any kind (Fig. 42.2). The electroretinogram is usually markedly abnormal in visually symptomatic patients with NCL but is much less likely to be abnormal in a Stargardt patient of that age. The visual dysfunction associated with NCL (both visual field and visual acuity) also tends to progress much more rapidly than Stargardt disease – over months instead of years. The other class of disease to explicitly consider and exclude in every patient suspected to have a macular dystrophy is drug toxicity. One should ask patients whether they are taking or have ever taken any medications over an extended period of time, especially medications for arthritis or skin disease (Fig. 42.3). Table 42.2 provides a list of medications that can cause macular lesions that mimic a macular dystrophy.
| Agent | Reference(s) |
|---|---|
| Chloroquine | Hobbs et al., 1959; Marmor et al., 2011 |
| Hydroxychloroquine | Shearer et al., 1965; Marmor et al., 2011; |
| Thioridazine (mellaril) | Weekley et al., 1960 |
| Chloropromazine (thorazine) | Delong et al., 1965 |
| Clofazimine | Craythorn et al., 1986 |
| Tamoxifen | Kaiser-Kupfer and Lippman, 1978 |
| Oxalosis/methoxyfluorane | Bullock and Albert, 1975; Albert et al., 1975 |
| Canthaxanthine | Boudreault et al., 1983; Ros et al., 1985 |
| Nitrofurantoin | Ibanez et al., 1994 |
| Talc | AtLee, 1972 |
| Deferoxamine | Haimovici et al., 2002; Gonzales et al., 2004 |
Albert DM, Bullock JD, Lahav M, et al. Flecked retina secondary to oxalate crystals from methoxyflurane anesthesia: clinical and experimental studies. Trans Sect Ophthalmol Am Acad Ophthalmol Otolaryngol 1975;79(6):OP817–26.
AtLee WE, Jr. Talc and cornstarch emboli in eyes of drug abusers. JAMA 1972;219(1):49–51.
Boudreault G, Cortin P, Corriveau LA, et al. [Canthaxanthine retinopathy: 1. Clinical study in 51 consumers.] Can J Ophthalmol 1983;18(7):325–8.
Bullock JD, Albert DM. Flecked retina. Appearance secondary to oxalate crystals from methoxyflurane anesthesia. Arch Ophthalmol 1975;93(1):26–31.
Craythorn JM, Swartz M, Creel DJ. Clofazimine-induced bull’s-eye retinopathy. Retina 1986;6(1):50–2.
Delong SL, Poley BJ, McFarlane JR, Jr. Ocular changes associated with long-term chlorpromazine therapy. Arch Ophthalmol 1965;73:611–17.
Gonzales CR, Lin AP, Engstrom RE, et al. Bilateral vitelliform maculopathy and deferoxamine toxicity. Retina 2004;24(3):464–7.
Haimovici R, D’Amico DJ, Gragoudas ES, et al. The expanded clinical spectrum of deferoxamine retinopathy. Ophthalmology 2002;109(1):164–71.
Hobbs HE, Sorsby A, Freedman A. Retinopathy following chloroquine therapy. Lancet 1959;2(7101):478–80.
Ibanez HE, Williams DF, Boniuk I. Crystalline retinopathy associated with long-term nitrofurantoin therapy. Arch Ophthalmol 1994;112(3):304–5.
Kaiser-Kupfer MI, Lippman ME. Tamoxifen retinopathy. Cancer Treat Rep 1978;62(3):315–20.
Marmor MF, Kellner U, Lai TY, et al. Revised recommendations on screening for chloroquine and hydroxychloroquine retinopathy. Ophthalmology 2011;118(2):415–22.
Ros AM, Leyon H, Wennersten G. Crystalline retinopathy in patients taking an oral drug containing canthaxanthine. Photodermatol 1985;2(3):183–5.
Shearer RV, Dubois EL. Ocular changes induced by long-term hydroxychloroquine (plaquenil) therapy. Am J Ophthalmol 1967;64(2):245–52.
Weekley RD, Potts AM, Reboton J, et al.. Pigmentary retinopathy in patients receiving high doses of a new phenothiazine. Arch Ophthalmol 1960;64:65–76.
Most macular dystrophies are inherited in an autosomal dominant fashion and the identification of affected individuals in multiple generations make ABCA4-associated disease less likely. As a general rule, patients with any of the autosomal dominant macular dystrophies often have visual acuities that are better than one might expect given the striking nature of their fundus findings (e.g., Fig. 42.59) while patients with autosomal recessive Stargardt disease caused by mutations in ABCA4 often have acuities that are poorer than one might expect given the relatively mild abnormality of their fundus (e.g., Fig. 42.29).
Best macular dystrophy
Best macular dystrophy (BMD), or Best disease, is an autosomal dominant condition caused by mutations in the BEST1 gene (OMIM #607854, formerly known as VMD2).1,2 The first family with this dystrophy was described by Friedrich Best in 1905.3 Other designations for this disease have since been used, including vitelline dystrophy,4 vitelliruptive degeneration,5 and vitelliform dystrophy.6 It is one of the most common Mendelian macular dystrophies, occurring in about 1 in 10 000 individuals. BMD refers to the “classic” form of a single, symmetric egg-yolk-like lesion centered on the fovea of each eye (Figs 42.4–42.7). However it is important to realize that BEST1 mutations are also associated with multiple other phenotypes (multifocal Best dystrophy (Fig. 42.8), autosomal dominant vitreoretinochoroidopathy (Fig. 42.9), and autosomal recessive bestrophinopathy (Fig. 42.10), which share only a few clinical features, detailed below.
Clinical features of BMD
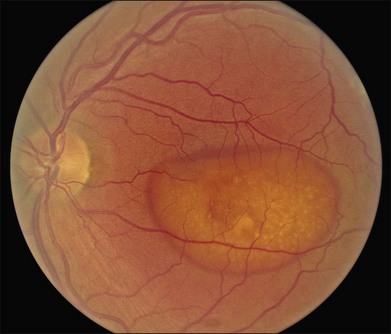
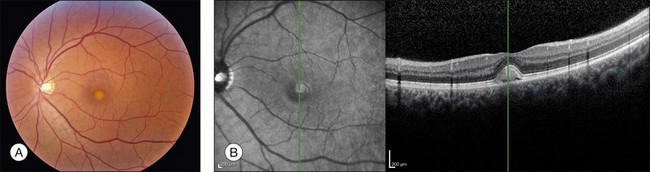
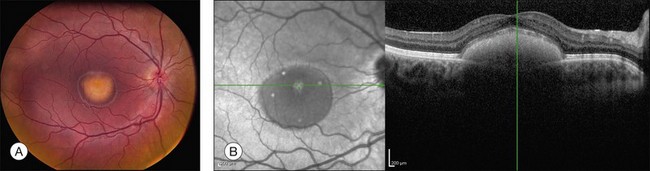
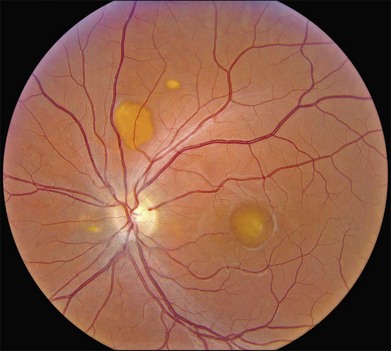
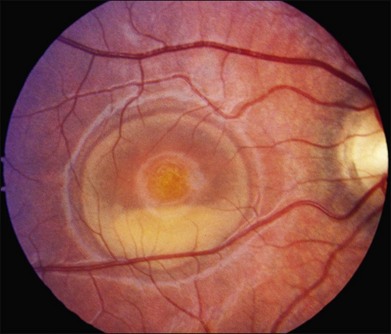
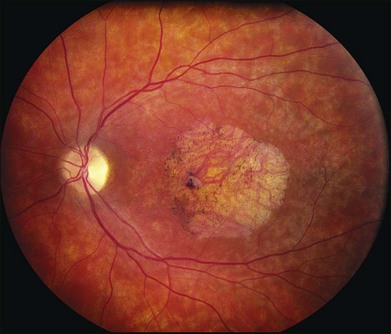
The fundus findings associated with BMD are quite varied. The macular lesions that are most characteristic of the disease are known as “vitelliform” because of their egg-yolk-like appearance (Figs 42.4–42.7). These lesions are typically solitary, round or horizontally oval, yellow, slightly elevated, and are centered on the fovea. Vitelliform lesions in patients with BMD can range in size from a few hundred microns (Fig. 42.5) to a few millimeters in diameter (Fig. 42.4). The larger lesions can be seen within the first few years of life, while the smaller lesions typically develop after age 20 and sometimes as late as age 60. As a result, the smaller lesions are sometimes referred to as “adult vitelliform” lesions (Fig. 42.5, Fig. 42.11), a phenotype that also occurs in patients with mutations in the PRPH2 gene (discussed more fully below). Some individuals who harbor disease-causing mutations in BEST1 never develop significant macular lesions.
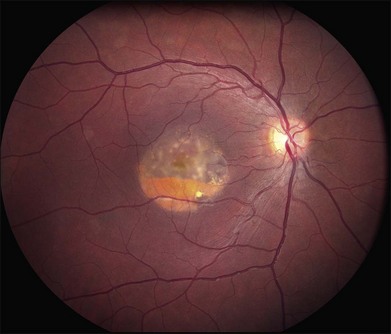
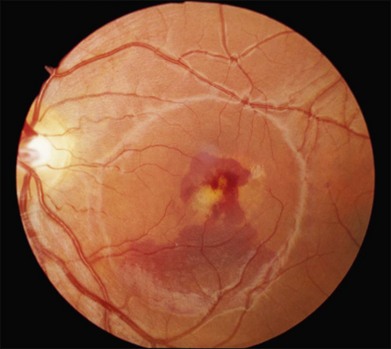
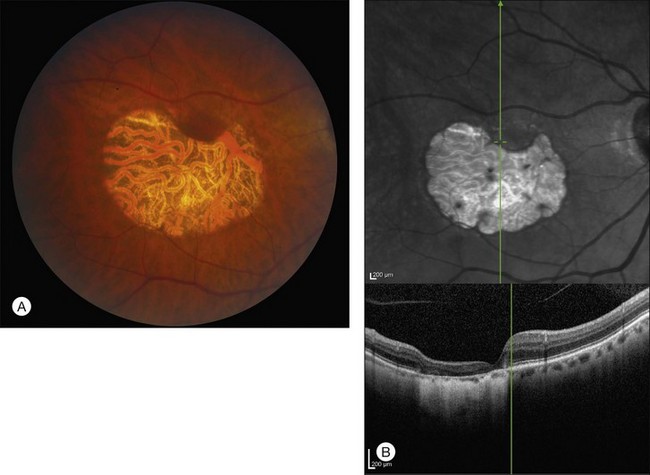
Over time, many vitelliform lesions develop a “pseudohypopyon” appearance in which the yellow material gravitates inferiorly in the subretinal space (Figs 42.12, 42.13). Other lesions develop varying amounts of subretinal and sub-RPE fibrosis, RPE atrophy in addition to hyperpigmentation, and atrophy of the RPE. This is sometimes known as a “scrambled-egg lesion” (Fig. 42.14). Many patients develop a single nodule of sub-RPE fibrosis centered very near the fovea (Fig. 42.15). Geographic atrophy is also fairly common after age 60 (Figs 42.16, 42.17), but can occur earlier in some patients. As with all diseases that disturb the RPE, true choroidal neovascularization can develop in a few percent of cases (Fig. 42.18).7 In addition, a few patients with vitelliform lesions develop subretinal hemorrhage following fairly modest blunt trauma to the head or eye (Fig. 42.19). Fortunately, these hemorrhages usually resolve and good vision returns without treatment. While these many clinical patterns have been described as “stages” by some authors, there is not always a predictable progression of these fundus changes from one to the other in a given patient. Although the maculas of patients with BMD are usually quite symmetric in the early years of the disease, the two eyes can become quite strikingly different in function and appearance as the disease progresses.
Although most patients with Best disease exhibit a single lesion centered on the fovea, there have also been numerous reports of patients with multiple vitelliform lesions who also manifest abnormalities on electro-oculography (EOG)8,9 and mutations in the BEST1 gene.10–14 In the most striking form of this multifocal phenotype, sometimes called multifocal Best dystrophy, there are multiple vitelliform lesions scattered throughout the posterior pole of both eyes (see Fig. 42.8). As with typical Best disease, these patients are asymptomatic unless the vitelliform lesions develop fibrotic scarring affecting the center of the macula. Autofluorescence and optical coherence tomography (OCT) imaging of multifocal lesions demonstrate characteristics similar to the solitary lesions seen in typical Best disease. This condition is distinguished from acute exudative polymorphous vitelliform maculopathy15,16 as patients with the latter disorder have a normal EOG and lack variations in BEST1. Notably, the canine model for BMD, which is due to a recessive mutation in the canine BEST1 gene, is associated with multifocal vitelliform lesions17,18 and some human patients with mutations in both alleles of BEST1 also exhibit multifocal vitelliform lesions.10,11,13 However, a few individuals with dominantly inherited disease and solitary lesions as children or young adults will also develop additional extramacular lesions later in life (Figs 42.20, 42.21).
Visual function
Visual acuity sufficient to drive is usually preserved in at least one eye throughout the first six decades of life, with more substantial visual loss occurring when BMD is complicated by nodular fibrosis, choroidal neovascularization19–21 or central geographic atrophy.22 Visual acuity is often 20/20 or better in eyes with undisturbed vitelliform lesions, which is surprising considering the substantial physical separation of the photoreceptor outer segments and the RPE (see Fig. 42.6) that exists for decades in some individuals (see Fig. 42.7). This suggests that the fluid within vitelliform lesions has an ionic composition relatively similar to that of the normal interphotoreceptor matrix and quite distinct from the composition of the subretinal fluid associated with rhegmatogenous retinal detachments. Some vitelliform lesions gradually flatten over time with persistence of good acuity, while others develop nodular sub-RPE scars or RPE atrophy which are associated with poorer visual acuities that are somewhat proportional to the size of the scar. Peripheral visual fields are usually completely normal in BMD, although patients with other BEST1 phenotypes do exhibit abnormal visual fields in some cases (see below).
Refractive error
It is not uncommon for patients with BMD to have hyperopia,22,23 which is likely due to shortened axial length.24 These findings are sometimes associated with narrow angles and/or angle closure glaucoma22,24,25 requiring peripheral iridotomy. Hyperopia and angle closure have also been demonstrated in other BEST1 phenotypes, including autosomal dominant vitreoretinochoroidopathy (ADVIRC)26,27 and autosomal recessive bestrophinopathy (ARB).13 Examination of the anterior segment with particular attention to the intraocular pressure and angle is warranted in patients with BMD and other phenotypes associated with BEST1 mutations.
Optical coherence tomography (OCT)
Although there have been no histopathologic studies of eyes with undisturbed vitelliform lesions, spectral domain OCT allows in vivo determination of the anatomy of macular lesions at near histopathologic resolution. The yellow material of a classic vitelliform lesion lies in the subretinal space and appears fairly homogeneous on OCT (see Figs 42.5B, 42.6B).28–32 In some patients, over time, some of the yellow pigment disappears and is replaced by clear fluid. The yellow pigment is denser than the clear fluid and settles gravitationally to the bottom of the vitelliform lesion with a fairly sharp horizontal line demarcating the pigment–fluid interface (see Figs 42.12, 42.13). This configuration is known as a “pseudohypopyon” and on OCT the yellow pigment appears hyperreflective while the clear subretinal fluid appears hyporeflective (black). Another lesion that is quite common in patients with Best disease and that has a very dramatic appearance on OCT is a fibrotic pillar that develops in the sub-RPE space, usually within 100 µm of the foveal center. These lesions appear hyperreflective on spectral domain OCT and seem to elevate the retina like a circus tent such that they are usually flanked by clear (hyporeflective on OCT) subretinal fluid (see Fig. 42.15B).
Fluorescein angiography and autofluorescence
The primary clinical use of fluorescein angiography (FA) in patients with Best disease is to help differentiate non-neovascular alterations in lesion anatomy (e.g., irregular resorption of yellow pigment) from active choroidal neovascularization in patients with a recent decrease in visual acuity. Although the vitelliform lesions of Best disease resemble the pigment epithelial detachments that occur in AMD, their anatomy and composition, however, are quite different and this causes some difficulty when interpreting fluorescein angiograms in patients with Best disease. For example, the yellow pigment filling some vitelliform lesions, especially in very young patients, is extremely hydrophobic and completely excludes fluorescein from the lesion. In such patients the vitelliform lesion blocks the underlying choroidal fluorescence almost as completely as blood would in a patient with AMD (see Fig. 42.7). Over time, the contents of the subretinal vitelliform cavities in some patients become more hydrophilic and in such cases the lesions briskly and completely fill with dye during an angiogram, much as a serous pigment epithelial detachment would in a patient with AMD (Fig. 42.22). In patients with the pseudohypopyon configuration the serous component of the lesion will fill with dye while the yellow pigment in the inferior portion of the lesion will both exclude the fluorescein from the vitelliform cavity and block the underlying choroidal fluorescence. This results in an angiogram that closely resembles a “notched” pigment epithelial detachment, which in AMD patients would suggest the presence of a choroidal neovascular membrane. In patients with Best disease this appearance is much less ominous. Autofluorescence imaging is not typically needed to make the diagnosis of Best disease or to make treatment decisions. From a research perspective, however, it is interesting that undisturbed vitelliform lesions usually exhibit uniform hyperfluorescence while those that have some amount of sub-RPE fibrosis or atrophy usually exhibit hypofluorescence.22,33,34 The increased autofluorescence in the undisturbed vitelliform lesions is likely a reflection of the increased amounts of lipofuscin in eyes with Best disease.35–38
Electrophysiology
Before the discovery of the gene responsible for Best disease, electrophysiologic testing played a very prominent role in the diagnosis. The electro-oculogram is usually markedly abnormal in molecularly affected individuals, even when macular lesions are not evident. In normal individuals the cornea positive standing potential of the eye is nearly twofold higher in bright light than it is in darkness, while in patients with Best disease the ratio of the light peak to dark trough (the Arden ratio) is typically less than 1.5. Full-field electroretinography (ERG) cone and rod a- and b-wave amplitudes are usually normal. The advantage of EOG over genetic testing is that diagnostic information can be obtained within an hour or two, while the patient is in the clinic. The disadvantage is that it is usually more expensive and less sensitive than molecular testing. That is, a normal EOG does not exclude the possibility of Best disease,39–43 as 37.5% of patients in one series had a BEST1 mutation despite normal a EOG.39
Genetics
Best disease was mapped to chromosome 11q13 in 199244 and the causative gene was identified six years later.1,45,46 Now known as BEST1, this gene encodes a 585 amino acid protein known as bestrophin that localizes to the basolateral membrane of the RPE.47 To date, more than 100 different mutations in BEST1 have been associated with Best disease.1,2,22,23,34,41,42,48–62 Most disease-causing mutations are missense variants, and a substantial fraction (about 25%) occur in exon 8, suggesting that the portion of the protein encoded by this exon may be critical to its function.22 More than 90% of families that have two or more individuals with the clinical diagnosis of Best disease will have a detectable mutation in the coding sequence of BEST1. As a result, a negative molecular result is clinically meaningful and suggests that the patient’s disease is caused by another gene or that it is a non-Mendelian phenocopy. The gene that most commonly causes macular lesions that are clinically similar to those of Best disease is PRPH2 (formerly RDS), while the most common non-Mendelian phenocopy is a pigment epithelial detachment associated with age-related macular degeneration.
Pathophysiology and histopathology
Although Best disease is rare, there have been a number of histopathologic reports describing the structural changes that accompany this condition. Histopathologic findings include increased RPE lipofuscin,37,38,63 loss of photoreceptors38 (often seen over a relatively intact RPE layer35,58), sub-RPE drusenoid material,35,36 and accumulation of cells and material in the subretinal space (Fig. 42.23). Following the discovery of the Best disease gene, efforts have also been made to identify the relationship between anatomical findings and specific genotypes. For example, eyes have been characterized from patients with Tyr227Asn,58 Thr6Arg,63 and a homozygous Trp93Cys donor.38 From these studies it has been suggested that the eyes of patients with Tyr227Asn mutations are notable for extramacular flecks.58 A donor eye with this mutation was also found to mislocalize bestrophin.58
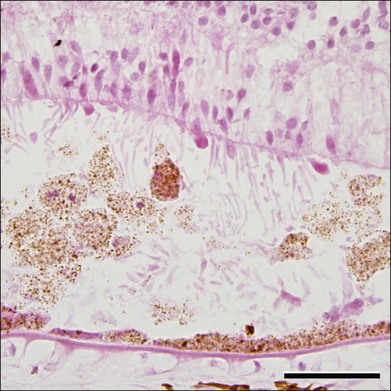
The molecular pathophysiology of Best disease is somewhat controversial – owing in part to the difficulty of comparing the behavior of different types of cultured cells with the RPE in vivo – and a thorough discussion of the evidence for the possible functions of the bestrophin-1 protein is beyond the scope of this chapter; it has been well reviewed elsewhere.64–66 Briefly, the normal function of bestrophin-1 appears to include the regulation of the ionic milieu in the RPE and/or the subretinal space. When overexpressed in some cell types, bestrophin-1 appears to function as a calcium-sensitive chloride channel,67,68 whereas mice lacking bestrophin-169 or harboring a mutant allele (Tryp93Cys)70 show altered uptake of calcium by the RPE. It has been suggested that impaired ionic flow across the RPE could result in alterations in the adhesiveness between the interphotoreceptor matrix and the RPE or a diminution of outer segment phagocytosis, both of which are sensitive to levels of calcium.71,72
Bestrophin-1 is expressed in all RPE cells, not just those underlying the macula. Moreover, the large response of the EOG to changes in light (the Arden ratio) suggests that the entire retina participates in this response, not just the macula. Why then are the most typical Best lesions found in the macula, centered on fixation? The answer may be in part due to differences in bestrophin-1 expression in different regions of the retina.63 However, it also seems likely that there are regional differences in the adhesion of the photoreceptors to the retinal pigment epithelium. The patient shown in Fig. 42.24 is supportive of this hypothesis. She developed an aggressive epiretinal membrane that created localized traction detachments in her left eye and she developed yellow vitelliform material in each of these locations. After successful surgery to remove the membrane and relieve the traction, the extramacular vitelliform deposits persisted for years.
Additional phenotypes associated with mutations in BEST1
Autosomal dominant vitreoretinochoroidopathy (ADVIRC)
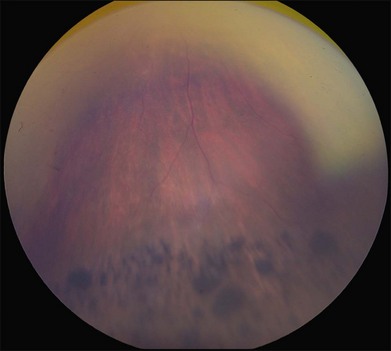
ADVIRC was first described by Kaufman et al. in 198273 as a condition with: (1) an autosomal dominant inheritance pattern; (2) peripheral pigmentary retinopathy for 360 degrees, with a discrete posterior boundary near the equator (see Fig. 42.9); (3) punctate whitish opacities in the retina; (4) vitreous cells and fibrillar condensation; (5) blood–retinal barrier breakdown; (6) retinal arteriolar narrowing and occlusion; (7) retinal neovascularization; (8) choroidal atrophy; and (9) presenile cataracts (see Fig. 42.9).74 The EOG is usually abnormal with a relatively normal ERG,75 but the first electrophysiologic studies of ADVIRC patients27,75,76 occurred in the pre-molecular era when genetic testing was not available. It has since been discovered that ADVIRC is caused by splice-altering mutations in BEST1 and that these patients can also have concomitant developmental abnormalities, including microcornea, hyperopia, and shortened axial length.26,77,78 Some patients have a severe form of ADVIRC in which both the ERG and EOG are abnormal, thus resembling retinitis pigmentosa.79,80
Autosomal recessive bestrophinopathy (ARB)
The first description of compound heterozygous BEST1 mutations causing multifocal yellowish changes in the macula and cystoid macular edema was published in 2006.11 Subsequent descriptions of patients with mutation in both BEST1 alleles have included both compound heterozygous and homozygous mutations and demonstrate a wide spectrum of fundus findings that are not present in their carrier parents.10,13,81–83 Burgess et al.13 coined the term autosomal recessive bestrophinopathy (ARB) to refer to this unusual presentation of BEST1-associated retinal disease. Hyperopia and an abnormal EOG are common. The visual acuity in ARB can be normal but tends to be worse than in autosomal dominant Best disease. Some patients exhibit cystoid macular edema or shallow subretinal fluid that can extend throughout the macula and beyond the arcades. Both solitary and mutifocal vitelliform lesions can occur, sometimes associated with flecks within and outside the macula (see Fig. 42.10). OCT can be helpful in detecting the low-lying subretinal fluid, especially in young patients who lack vitelliform lesions. Subretinal fibrosis is more common in ARB than in autosomal dominant disease.
Treatment
Treatment for BEST1 disease consists primarily of recognizing choroidal neovascularization and hastening its regression with anti-VEGF therapy. Though the natural history of subretinal hemorrhage in BMD is relatively good,19 preservation of visual function for choroidal neovascularization (CNV) has been reported in retrospective studies using intravitreal bevacizumab and ranibizumab.81,84–88 In our experience, it is not usually possible to completely eradicate subretinal fluid that exists adjacent to the nodular fibrotic pillars that occur beneath the RPE (see Fig. 42.15B). Thus, when this configuration is seen following treatment of suspected CNV, we would recommend elongating the intervals between anti-VEGF injections, and then discontinuing them altogether, once the visual acuity is stabilized and all subretinal blood has been resorbed. Even in the absence of CNV, subretinal hemorrhage can occur in patients with Best disease following relatively modest head or eye trauma (see Fig. 42.19).19,89,90 As a result, we usually caution patients against playing sports in which frequent blows to the head are to be expected. Protective eyewear is recommended for all sports. Many patients with Best disease have poor vision in one eye but retain vision sufficient in the fellow eye to drive for many years. In such individuals we recommend wearing spectacles with safety plastic lenses at all times.
Stargardt disease
Variations in ABCA4 (OMIM #601691) are the most common cause of autosomal recessive retinal disease in humans. ABCA4 mutations were first found in patients with autosomal recessive Stargardt disease91 and were later shown to also cause cone dystrophy, cone–rod dystrophy, and retinitis pigmentosa.92–97 As discussed more fully below, a patient’s position within this disease spectrum is determined largely by the total amount of residual ABCA4 function.97 First described in 1909,98 Stargardt disease is the mildest of the ABCA4 phenotypes while a form of retinitis pigmentosa is the most severe.
Clinical features of Stargardt disease
The clinical presentation of Stargardt disease is quite variable in age of onset, presenting symptoms, and fundus appearance, and this variability is often daunting to ophthalmologists who see the condition infrequently. Most of the differences in clinical findings in patients with ABCA4 disease can be explained by the interplay of three factors that vary among patients: (1) the severity of their ABCA4 genotype (and hence the rate at which toxic bisretinoids form in the photoreceptors); (2) the relative sensitivity of the foveal cones to the genotype; and (3) the relative sensitivity of the retinal pigment epithelium to the genotype (Fig. 42.25).97 The first of these variables can be directly assessed by molecular testing of the ABCA4 gene while the molecular nature of the latter two remains to be determined.
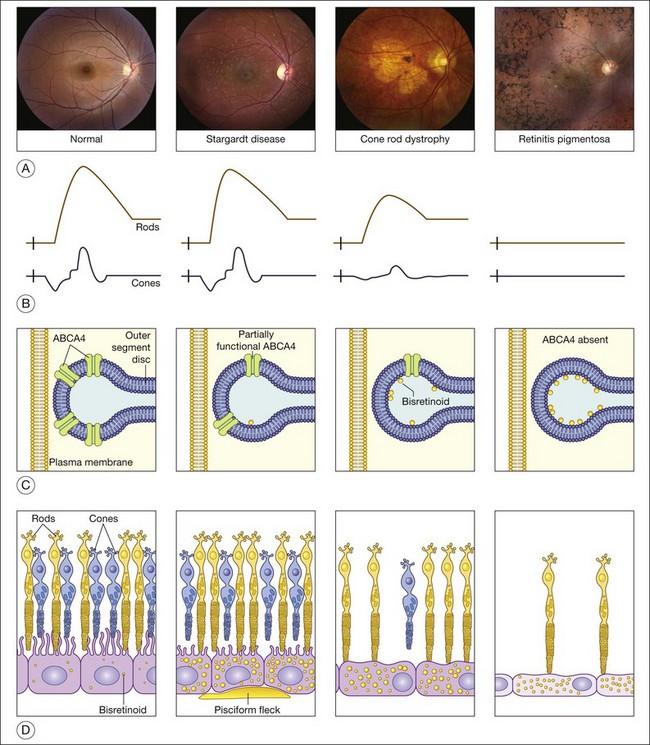
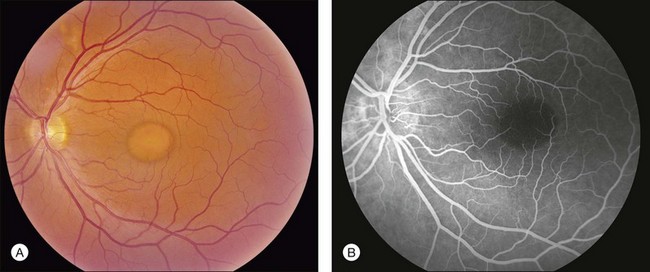
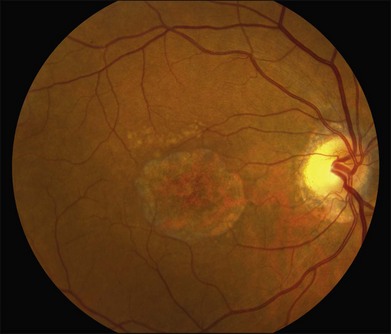
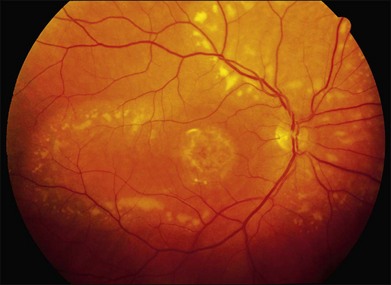
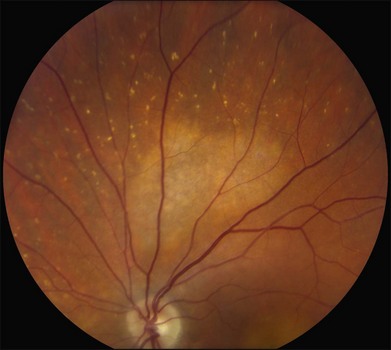
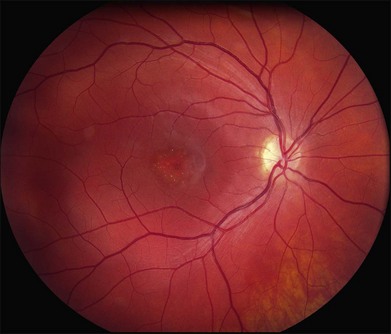
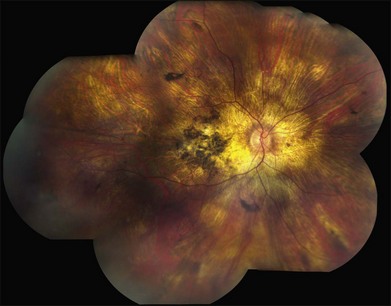
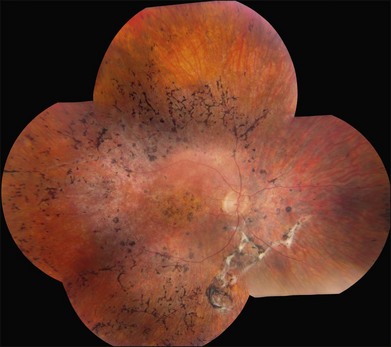
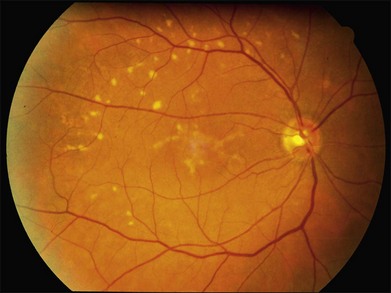
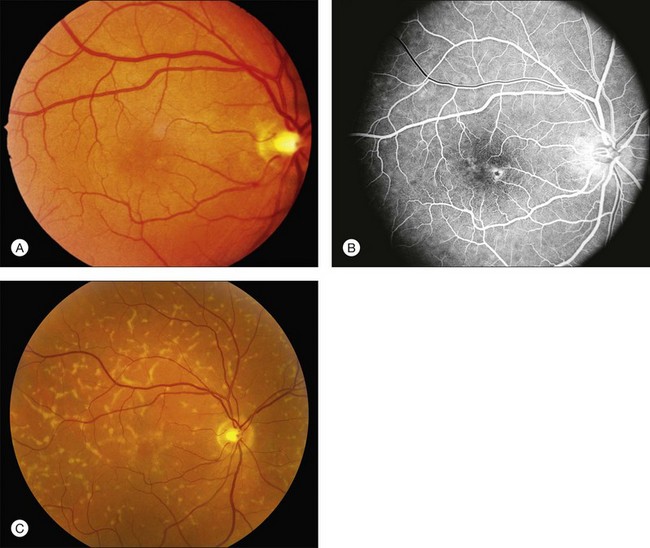
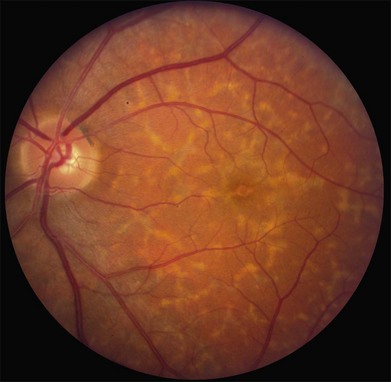
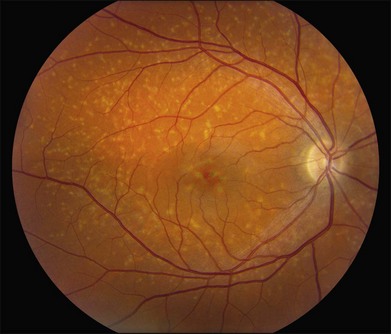
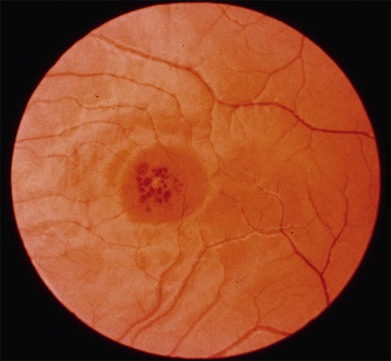
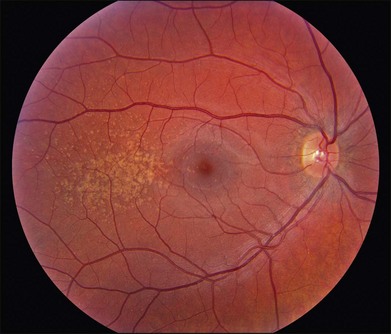
The most characteristic fundus findings in Stargardt disease are light-colored flecks at the level of the retinal pigment epithelium (Fig. 42.26). These flecks differ from drusen in that they are usually more elongated than round and they often contact each other at angles that create a branching or net-like appearance. Occasionally, two adjacent flecks form an obtuse angle that Franceschetti called “pisciform” because of its resemblance to a fish tail.99 In some patients a cluster of flecks is entirely contained within a one-by-two disc diameter horizontal ellipse centered on fixation (Fig. 42.27), while in other patients the flecks extend well beyond the temporal vascular arcades (Fig. 42.28), almost reaching the equator.
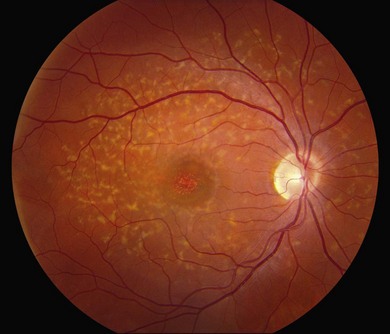
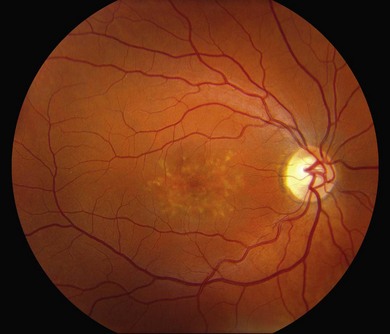
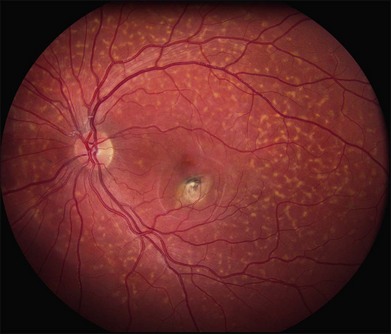
In addition to the difference in distribution, Stargardt flecks also differ widely in number, size, color, aspect ratio, and edge definition among different patients. Some patients have no flecks at all (Fig. 42.29) while others have hundreds (see Fig. 42.28). Flecks are most commonly yellow but can range from dirty-white to orange. Some flecks have pigmented edges and in a few cases this RPE hyperpigmentation can be quite dramatic. Some patients have very small deposits near the fovea that have a crystalline character on biomicroscopy (Fig. 42.30). Flecks outside the central 2 mm of the macula tend to be a bit larger than those nearer the fovea. Some patients have flecks that are almost round while others have flecks that are several times longer than they are wide. In some individuals with severe ABCA4 genotypes, the flecks are small and white and are admixed with small patches of subretinal fibrosis that resemble confetti (Fig. 42.31). This is presumably because the widespread photoreceptor injury associated with such genotypes reduces the production of bisretinoids. Some flecks are stable in position, size and number for many years, while others grow in number and/or progress to RPE atrophy (Fig. 42.32).
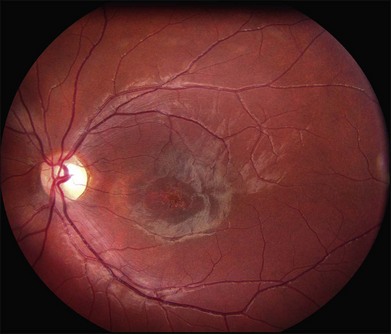
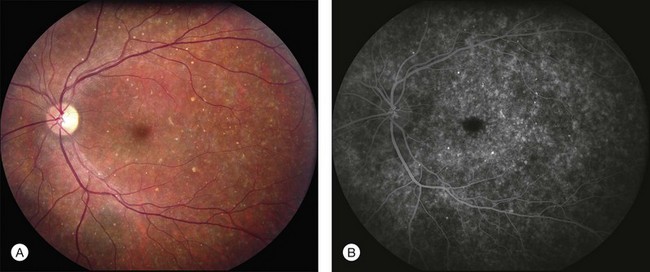
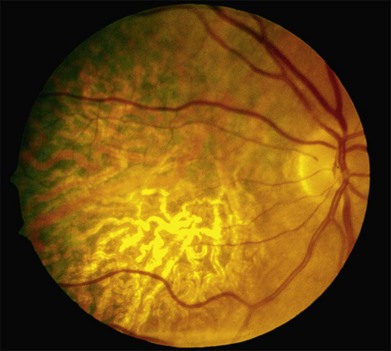
In addition to the many different fleck configurations, the RPE itself responds to ABCA4 mutations quite differently in different patients depending in part on the severity of the ABCA4 genotype and in part on the sensitivity of the RPE to the accumulation of bisretinoids. In some patients with relatively mild genotypes, there is little photoreceptor injury and thus there is a steady supply of bisretinoid to the RPE. In some patients the RPE retains this material intracellularly and as a result becomes somewhat opaque to visible light. On ophthalmoscopy this is recognized as a very uniform vermillion or light-brown color to the fundus with complete obscuration of the underlying choroidal details (see Fig. 42.27, Figs 42.33, 42.34). On fluorescein angiography, this is seen as a complete masking of the choroidal circulation. As a result, the dye-filled retinal vessels lie upon a completely hypofluorescent background (see Fig. 42.43). In other patients with a very similar bisretinoid load emanating from the photoreceptors, the RPE gradually thins, presumably from apoptotic death of some RPE cells and compensatory stretching of those that survive (Fig. 42.35). Frank RPE atrophy is commonly seen in the center of the macula and the bases of these atrophic lesions have a metallic sheen to them (Fig. 42.32) that is distinctly different from the geographic atrophy that occurs in age-related macular degeneration. In some patients these atrophic lesions become dusted with dark pigment over time (Fig. 42.35). In the later stages of disease patients with severe genotypes can develop nummular atrophy of the extramacular RPE and choriocapillaris that somewhat resembles choroideremia (Fig. 42.36).
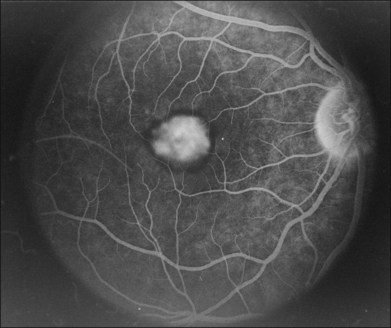
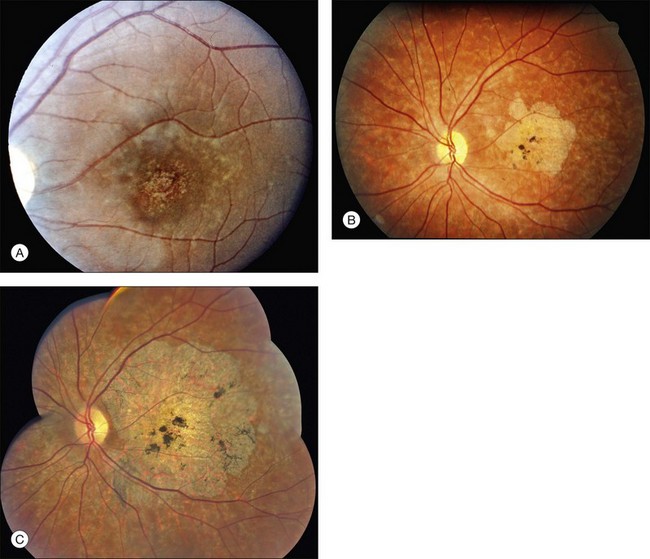
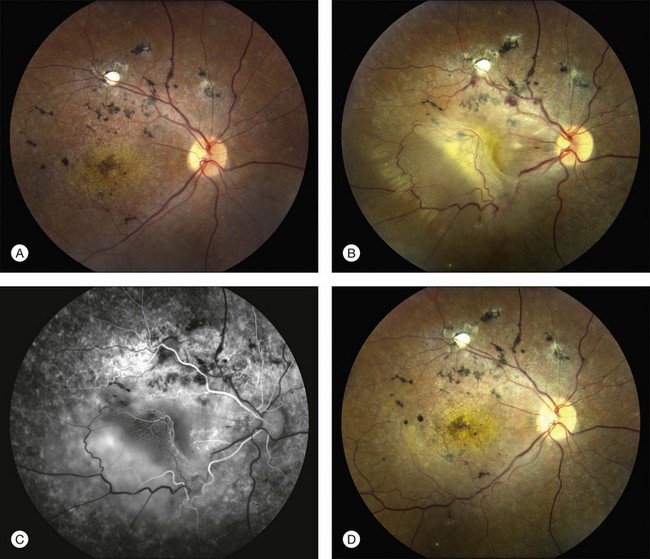
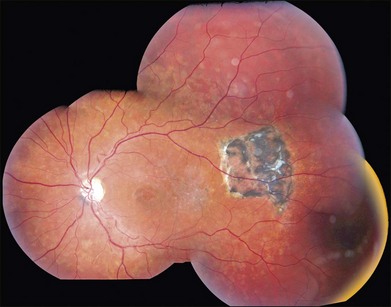
A fairly reliable diagnostic sign of ABCA4-associated retinal disease is a relative sparing of the peripapillary RPE. This sign is often more visible on fluorescein angiography100 (see Fig. 42.31) or autofluorescence;101,102 however, it can also be easily observed on ophthalmoscopy alone (Fig. 42.37). The mechanism of this sparing is currently unknown. Patients with ABCA4-associated retinal disease are not immune to inflammatory or traumatic retinal injuries and the coexistence of these disorders can make it difficult to establish the correct diagnoses. Although the frequency with which epiretinal membranes (Fig. 42.38A-D), subretinal fibrosis (Figs 42.39, 42.40) and inflammatory nodules (see Fig. 42.38A) occur in Stargardt patients may not be greater than would be expected by chance, the exuberant nature of some of these lesions suggests an adjuvant effect of either the bisretinoids themselves or the low-grade inflammation that is commonly present in degenerative retinal disease.
Visual function
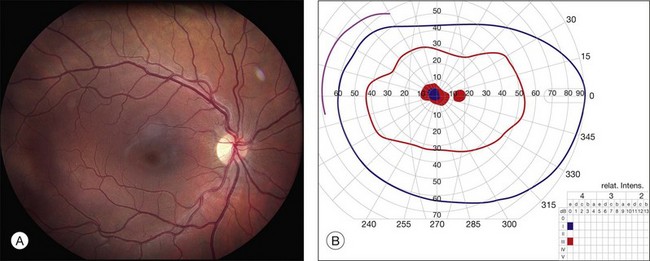
It is important to recognize that the effect of ABCA4 mutations on visual acuity (VA) is frequently at odds with their effect on the visual field. As a result, many patients will have quite full visual fields for many years after their acuity has fallen below the threshold of legal blindness (Fig. 42.41), while a few patients will retain acuity of better than 20/40 despite extensive field loss (Fig. 42.42). Fishman studied 95 patients with Stargardt disease and found that the probability of maintaining a VA of 20/40 in at least one eye was 52% by age 19, 32% by age 29, and 22% by age 39.103 In a larger cohort of Stargardt disease patients, cross-sectional analysis showed that almost a quarter had VA of 20/40 or better, whereas 4% had VA worse than 20/400.104 In general, patients with extensive extramacular flecks have a poorer long-term visual prognosis than patients with flecks and/or atrophy that are limited to the macula.105,106 Similarly, patients with significant loss of the I2e isopter on Goldmann perimetry have more severe genotypes than those with a more normal I2e response.97
Fluorescein angiography and autofluorescence
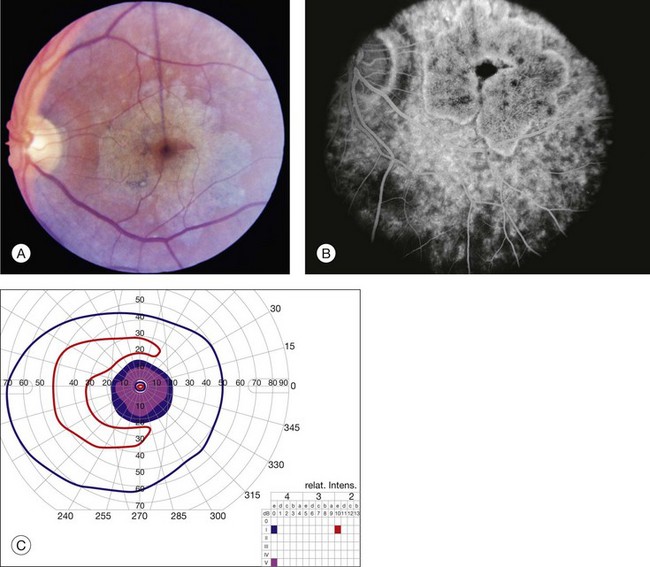
The accumulation of A2E within the retinal pigment epithelium results in a detectable abnormality on both fluorescein angiography and autofluorescence imaging. With angiography, the A2E blocks the exciting blue light from reaching the dye in the choroidal circulation, resulting in a finding that is variously known as a dark, silent, or masked choroid.107,108 In these angiograms, the retinal vessels stand out in sharp contrast to the dark background (Fig. 42.43). Flecks and any areas of atrophy are hyperfluorescent, although an annulus of a few hundred microns around the optic disc usually remains hypofluorescent even in the presence of very extensive disease.100 Although quite helpful in the past, we do not routinely employ fluorescein angiography in the workup of patients suspected of ABCA4 disease for several reasons. First, there is some evidence that exposure to visible light is a cofactor in the disease (knockout mice reared in darkness do not accumulate A2E109 and there is certainly a lot of light exposure associated with a fluorescein angiogram). Second, molecular testing is becoming more sensitive and more widely available (https://www.carverlab.org/; cited February 2012) and has the added utility of providing some prognostic information when well-characterized mutations are present.97 Third, many patients with molecularly proven ABCA4 disease have a readily visible choroidal circulation on angiography (Fig. 42.44). Thus, as with current molecular tests, the absence of a positive result is not helpful. Fourth, there is a small risk of serious complications from the intravenous dye, and finally, the dynamic range of digital cameras is noticeably less than film, tending to accentuate the contrast between the retinal circulation and the normal choroidal circulation, resulting in a false-positive interpretation of the test in some cases. In patients with Stargardt disease, autofluorescence (AF) imaging can also show areas of RPE atrophy, bull’s-eye changes in the macula (see Fig. 42.33B), flecks and peripapillary sparing.42,101,102,110–123 However, AF uses high-intensity illumination at the low end of the visible light spectrum and thus we do not routinely employ this testing modality in patients with suspected ABCA4 disease.
Optical coherence tomography
OCT can reveal the extent of outer retinal loss and RPE atrophy (see Fig. 42.33C), and it can also distinguish the anatomic level of flecks with accuracy.112,116,124–126 The test is very sensitive to early changes; one study revealed three patients with photoreceptor abnormalities on OCT without an equivalent abnormality on AF.116 Peripapillary nerve fiber layer thickness can also be altered on OCT but the significance of this is not yet known.127
Electrophysiology
Somewhat by definition, the full-field ERG is typically normal in patients with Stargardt disease while cone and cone–rod dysfunction are seen in more severe forms of ABCA4 disease.111,128–133 With mild ABCA4 genotypes, bisretinoids do not seem to accumulate rapidly enough to injure the photoreceptors directly, except for cones in and near the fovea, which seem to be the most sensitive to ABCA4 dysfunction in most patients. Loss of the latter cells can cause quite a bit of acuity loss without any detectable effect on the full-field ERG. With moderate ABCA4 genotypes cones throughout the fundus are directly affected, leading to cone-selective abnormalities in the ERG. With the most severe ABCA4 genotypes even rods experience direct injury from A2E accumulation, resulting in effects on all components of the ERG and a fundus appearance that can reasonably be called retinitis pigmentosa (Fig. 42.39).97,134 It is important to realize that patients with the more severe ABCA4 genotypes can progress from a clinical pattern consistent with the label “Stargardt disease” to one consistent with “retinitis pigmentosa” over the course of their disease and it can be very distressing to patients to have their diagnosis change from one doctor to the next. A few minutes spent explaining that these descriptive labels are used to describe different aspects (e.g., ophthalmoscopic and electrophysiologic) and stages (early and late) of a single disease, and that these descriptions can change over time in an individual patient, can reduce this distress significantly. One of the most valuable uses of the full-field ERG in patients suspected to have ABCA4-related retinal disease is in differentiating the earliest onset forms of this condition from juvenile NCL (see Fig. 42.2). In patients with NCL, the ERG is usually severely reduced or extinguished before the age of 10 years. If it is recordable at all, the rod ERG is more severely affected than the cone ERG, and the maximum stimulus intensity scotopic response typically shows an electronegative configuration (greater loss of b-wave than a-wave).135 In contrast, profound reduction in the full-field scotopic ERG response to the standard maximum stimulus intensity is rare in the first decade of life in patients with ABCA4 disease, and any reductions that do occur are noticeably cone-selective. Finally, electronegative waveforms have not been reported in ABCA4-associated retinal disease. The pattern ERG can be abnormal in patients with Stargardt disease even when the fundus looks relatively normal.111,126 This led to the proposal of three groups of SD based on electrophysiology: in group 1 there is a severe pattern ERG abnormality with normal scotopic and full-field ERGs; in group 2 there is additional loss of photopic function; and in group 3 there is loss of both photopic and scotopic function.111 The multifocal ERG is less useful in Stargardt patients than the full-field ERG. Its test–retest reliability and interocular symmetry in SD is significantly lower than in controls.136
Genetics
Since the discovery in 1997 that a recessively inherited ABCA4 mutation causes Stargardt disease,91 there have been more than 250 different disease-causing alleles identified in ABCA4 and many non-disease-causing polymorphisms as well. Individuals who counsel patients about their ABCA4 genotype need to have a thorough understanding of the differences between these two classes of genetic variation. Some laboratories provide a pathogenicity score for the mutations they identify that can be helpful in this regard.137,138 Another challenge in counseling patients with ABCA4-associated retinal disease is that a significant number of disease-causing mutations lie outside the coding and promoter sequences of the gene, making them difficult to identify. Thus, many patients will have only one of their two disease alleles identified by current testing methods. Mutations in ABCA4 are thought to be responsible for more than 95% of cases of clinical Stargardt disease, 30–50% of cases of cone–rod dystrophy96 and 8% of autosomal recessive retinitis pigmentosa.97 Most of the remaining 5% of cases with a Stargardt disease phenotype are caused by mutations in ELOVL4, PRPH2, or BEST1.
The wide range of phenotypes seen in ABCA4-associated disease can be attributed to: (1) the variable severity of the many disease alleles in the population; (2) the interaction of these alleles with genetic and environmental modifiers (e.g. light exposure, smoking, and diet); and (3) the complex interaction between rods, cones, and RPE cells. Systematic investigation of the clinical findings of large cohorts of patients with known ABCA4 genotypes has the potential to deduce the specific pathogenic contribution of individual alleles. For example, multiple regression analysis based on an additive model of ABCA4 function recently enabled the quantification of specific disease-causing power of some of the more common disease-causing ABCA4 alleles.97 As additional studies of this kind are performed and combined, it will allow clinicians to better inform patients of their prognosis, help balance enrollment in clinical trials of novel treatments, and facilitate the identification of disease-modifying factors that may form the basis of such treatments.
Pathophysiology and histopathology
Histological studies of eyes with Stargardt disease reveal remarkable lipofuscin accumulation in the RPE. For example, an eye from a 9-year-old child with Stargardt, enucleated for retinoblastoma at 16 months of age, showed striking RPE autofluorescence when compared to normal aging.139 Another unusual finding observed in autopsy tissue from a 62-year-old donor is the presence of significant lipofuscin accumulation within photoreceptor cell inner segments, suggesting significantly altered processing of retinoids in the outer retina.140 Attenuation of photoreceptor inner and outer segments over areas of still-organized RPE cells was also noted, consistent with a primary photoreceptor cell defect. In addition, end-stage central atrophy of the outer retina, gliosis, and RPE hypertrophy or loss have been seen.140,141
The pathophysiology of ABCA4-associated disease has been recently reviewed134,142,143 and was elucidated in large part through elegant biochemical studies of the Abca4-/- mouse.144 The normal role of ABCA4 is the clearance of a retinoid intermediate of the visual cycle (N-retinylidene phosphatidylethanolamine) from the intradiscal lumen of the outer segments of rods and cones. Condensation of this retinoid with a second vitamin A moiety, which may occur in the photoreceptor cell or in the RPE following outer segment phagocytosis, results in the formation of A2E, a toxic detergent-like compound that can trigger death of RPE cells145,146, complement activation,147 and both direct and indirect loss of photoreceptors (see Fig. 42.25).
Treatment
Since a primary defect in ABCA4-associated retinal disease is an accumulation of toxic bisretinoids in the RPE and photoreceptors, drugs that modulate the visual cycle (e.g. isoretinoin and fenretinide), have been investigated for their potential to slow the formation of these toxic products in Abca4 knockout mice.148–150 Similarly, gene replacement therapy has been proposed for ABCA4 disease and efficacy has been demonstrated in the mouse model.151–153 Proof of concept, safety, and efficacy for ocular gene therapy in humans has already been shown for RPE65-associated Leber congenital amaurosis154,155 and thus gene therapy for ABCA4-associated disease seems promising for patients who still have substantial visual function. Patients who have already experienced extensive loss of RPE and photoreceptors will likely need some type of cell replacement therapy, and one human clinical trial of RPE cell replacement is already underway (http://clinicaltrials.gov/ct2/show/NCT01345006?term=advanced+cell+technology&rank=2; cited February 2012).
Stargardt-like dominant macular dystrophy (SLDMD)
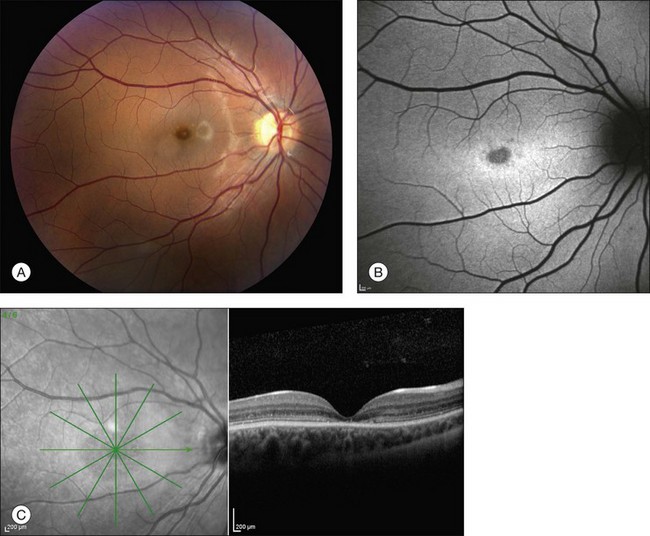
In 1994 Stone and coworkers described a large family with a Stargardt-like phenotype and mapped the gene to chromosome 6.156 In contrast to typical Stargardt disease, this family displayed a clear autosomal dominant pattern of inheritance with high penetrance. Zhang et al. later identified a 5 base pair deletion in the gene ELOVL4 in the affected members of five families affected with this disease.157 Although additional disease-causing mutations in ELOVL4 have been identified,158,159 the 5 bp deletion is responsible for more than 90% of cases in North America. This condition is characterized by progressive central vision loss; some patients develop symptoms in the first decade of life and the majority have a visual acuity of 20/200 or worse by 30 years of age.156 The range of fundus findings is almost identical to that of autosomal recessive Stargardt disease and includes pisciform flecks (Figs 42.45, 42.46), peripapillary sparing (Fig. 42.47) and macular atrophy. The bases of the atrophic macular lesions are less likely to have a distinct reflective sheen than those of autosomal recessive Stargardt disease and some patients display a round or cuneiform clump of dark pigment very near fixation (Fig. 42.47). As in ABCA4-associated Stargardt disease, a few patients have a severe loss of foveal cones with an otherwise normal fundus (Fig. 42.48). The lipofuscin deposits in SLDMD tend to be a bit larger than those of recessive Stargardt disease and often take on a “butterfly” appearance at some point in the evolution of the macular lesions (Fig. 42.46). For a given degree of fundus abnormality, however, the acuity is usually much more affected in patients with SLDMD than it is in patients with PRPH2-associated pattern dystrophy. The ERG is usually normal. These patients are less likely than patients with autosomal recessive Stargardt disease to have a dark choroid on fluorescein angiography but the flecks are similarly hyperfluorescent in both conditions.
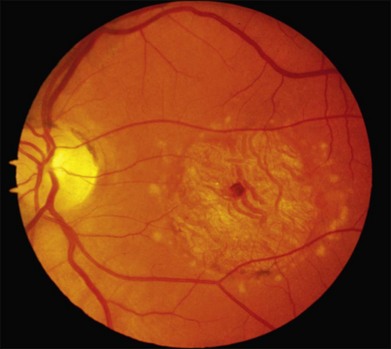
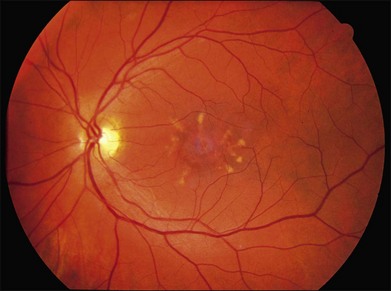
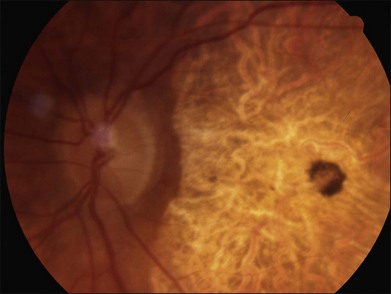
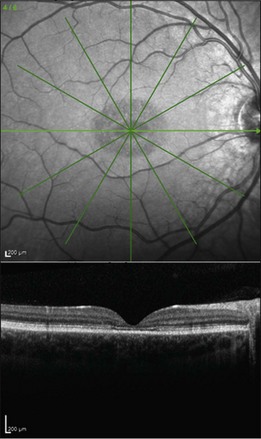
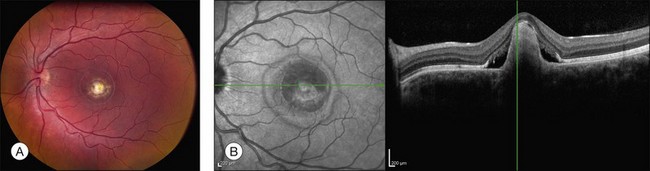
Fig. 42.48 Infrared fundus photograph and spectral domain optical coherence tomogram of the right eye of a 14-year-old male with the common Leu263 del5tttCTTAA mutation in ELOVL4 causing Stargardt-like dominant macular dystrophy. The visual acuity in this eye is 20/125. Ophthalmoscopically, the fundus is near normal, but SD-OCT reveals loss of foveal photoreceptors. This selective loss of foveal cones is also seen in a subset of patients with ABCA4-associated recessive Stargardt disease (see for example Fig. 42.33).
Pathophysiology
The ELOVL4 gene encodes the protein “elongation of very long chain fatty acids-4,” a biosynthetic enzyme in the endoplasmic reticulum responsible for the synthesis of fatty acids with more than 26 carbons.160,161 This gene is expressed in brain and retina, where expression is restricted to photoreceptor cells.157 Mutations associated with Stargardt-like macular degeneration cause mistrafficking of the mutant protein in vitro,162 which results in cell death.163 Mice harboring a single mutant allele of Elovl4 display mistrafficking of the ELOVL4 protein, show increased lipofuscin formation, and develop peripheral photoreceptor loss.164,165
Pattern dystrophy
Pattern dystrophy (PD) refers to a group of inherited retinal dystrophies characterized by pigment changes at the level of the RPE.166–168 PD encompasses a broad spectrum of clinical appearances that were originally given names based on the pattern of pigment distribution; such as, butterfly-shaped pigment dystrophy (Fig. 42.49),166 adult-onset vitelliform pattern dystrophy169,170 (Fig. 42.50) peculiar foveomacular dystrophy171 (Fig. 42.51A, B), Sjögren reticular dystrophy of the RPE,172–174 (Figs 42.51C, 42.52), and fundus pulverulentus.175,176 The most common causes of all of these different patterns have proven to be mutations in a single gene, PRPH2 (originally RDS – OMIM #179605). Mutations in this gene also cause some cases of central areolar choroidal dystrophy (Fig. 42.53),177 retinitis pigmentosa,178–181 and some cases that are nearly identical to the fundus flavimaculatus variant of Stargardt disease (Fig. 42.54). Patients with extensive RPE atrophy will sometimes exhibit peripapillary sparing very similar to that seen in ABCA4 disease (Fig. 42.55). PRPH2 mutations usually cause disease in the heterozygous state and the disorders are thus inherited in an autosomal dominant fashion. Most patients with a PRPH2-associated PD will experience macular photostress in their daily life; that is, their central acuity will be slow to recover following exposure to bright light. Thus, a patient who can read 20/30 in a dim clinic lane may be incapable of reading their mail for tens of minutes after walking down a sunlit driveway to retrieve it. Similarly, a waitress with excellent acuity under optimal circumstances may be unable to make change for her customers after walking through a bright kitchen and returning to a dark restaurant. All of the PRPH2-associated pattern dystrophies also share an 18% lifetime risk of choroidal neovascularization.182
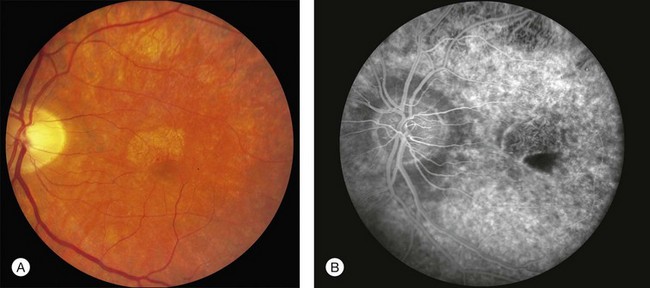
Like most autosomal dominant disorders, the genetic background plays an important role in the specific clinical manifestations that result from PRPH2 mutations and the same point mutation can cause vitelliform lesions in one patient, peculiar foveomacular dystrophy in another, and butterfly-shaped pigment dystrophy in others.179,183 Gutman described a patient with a vitelliform lesion in one eye and a butterfly-shaped lesion in the other.184
Clinical features and history of specific pattern dystrophies
Butterfly-shaped pigment dystrophy
Deutman described a family with autosomal dominant inheritance that had butterfly-shaped pigmentation located in the RPE.166 The pigmentation can be yellow, white, or black and accumulate in an unusual configuration of three to five “arms” or “wings” that resemble the wings of a butterfly (see Fig. 42.49). An area of depigmentation often occurs around the pigment. Additional pigment deposits that look like drusen or flecks can be seen peripheral to the central lesion. These changes can occasionally be seen in the teens but symptoms usually do not occur until patients are in their 20s–30s. Several mutations in PRPH2 have been shown to be causative of this macular pattern.183,185–188
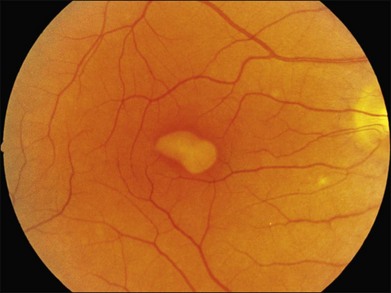
Adult-onset foveomacular vitelliform pattern dystrophy
Patients with adult-onset foveomacular vitelliform dystrophy (described by Gass as a “peculiar foveomacular dystrophy”171) present asymptomatically or with mild blurring. Fundus examination reveals symmetric, solitary, autofluorescent vitelliform lesions in the macula (usually centered on or just adjacent to the fovea) that are much smaller in size (less than one-third disc diameter) than the vitelliform lesions of Best disease. These lesions often have a central pigmented spot (see Fig. 42.51B) that is best seen when a narrow beam of a slit lamp is placed just adjacent to the lesion. Partial or complete resorption of the vitelliform material is common and is usually accompanied by outer retinal loss and decreased visual acuity.189
Arnold et al. performed histopathologic evaluation of eyes from patients with adult vitelliform lesions. Although genotypes were not determined, the phenotype was consistent with a mutation in PRPH2 or BEST1. These authors noted significant material in the subretinal space beneath the fovea, consisting of outer-segment material and pigment-laden cells. Atrophic changes appeared to be secondary to the accumulation of subretinal material.190
Sjögren reticular dystrophy of the RPE
In reticular dystrophy of the RPE, first reported by Sjögren,172 the fundus is characterized by a clearly defined network of black-pigmented lines at the level of the RPE that resemble a fishnet with knots or chicken wire. Early cases may have only pigment granules in the fovea but still give the impression of a pigmented network in the process of disintegration. The pigment alterations may be more visible with fluorescein angiography. The network usually starts in the fovea and can extend into the periphery. Some patients in the same family as those with reticular dystrophy also have vitelliform and/or butterfly-shaped pigment changes.174,191–195 The genotypes of Sjögren original patients are unknown but individuals with PRPH2 mutations can exhibit a pattern very similar to the one he described (see Figs 42.51C, 42.52).
Central areolar choroidal dystrophy (central areolar retinochoroidal dystrophy)
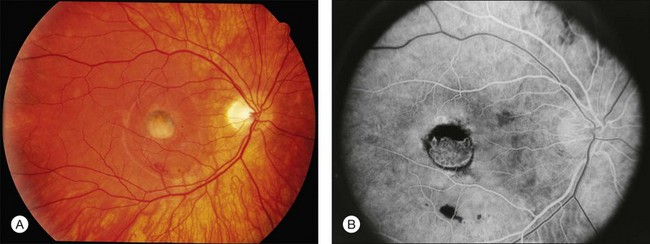
Central areolar choroidal dystrophy was, as its name implies, originally thought to be a primarily choroidal disease. However, numerous reports also link this macular pattern to mutations in PRPH2.177,196–204 Thus, the choroidal changes are secondary to a genetic abnormality expressed at the level of the photoreceptors, and a more accurate term might be “central areolar retinochoroidal dystrophy (CARCD).” Visual fields are normal except for central scotomas corresponding to the macular lesions. The earliest change is a fine, mottled depigmentation in the macula of both eyes that appears between the second and fourth decades and gradually evolves into symmetric, sharply outlined, bull’s-eye oval or round areas of geographic atrophy of the RPE (see Fig. 42.53). Within the area of RPE atrophy, the reddish-orange color of the large choroidal vessels is replaced by a yellow–white color, sometimes called “choroidal sclerosis” in older literature. Visual acuity deteriorates somewhat in the fourth and fifth decades but can remain as good as 20/100 to 20/200 into the seventh and eighth decades.205–210
Electrophysiology
Most patients with pattern dystrophy show normal cone and rod amplitudes and implicit times on the full-field ERG211 but some reduction can be seen when there are more extensive changes. EOG light-peak to dark-trough ratios are most frequently normal or only modestly subnormal.211 Genetic testing for variations in PRPH2 is less expensive than electrophysiology in most institutions and thus we reserve electrophysiologic testing for patients who lack mutations in PRPH2 and/or who have ophthalmoscopic or perimetric findings suggestive of more widespread photoreceptor disease.
Pathophysiology
PRPH2 encodes a structural protein (peripherin) that plays a critical role in establishing and maintaining the morphology of photoreceptor outer-segment discs.212,213 Zhang et al.187 studied eyes from a donor with butterfly dystrophy due to a Cys213Tyr mutation in PRPH2 and noted an abrupt transition between healthy and degenerated retina and RPE, with massively lipofuscin-laden RPE cells at the transitions. In another report, surgical samples of retina were collected from a living patient with an Arg172Trp mutation in PRPH2 and manifested abnormal localization of mutant peripherin protein, as well as ultrastructural alterations in outer-segment disc structure.214 resembling the whorls observed in mice heterozygous for the rds mutation.215 Thus, the primary defect is both genetically and structurally present in photoreceptor cells, with presumed subsequent injury to the RPE and choriocapillaris. The pathophysiologic interplay between the photoreceptors, the RPE, and the choriocapillaris plays a very important role in PRPH2-associated diseases, just as it does in the diseases caused by mutations in ABCA4. The most severe mutations in both genes cause direct photoreceptor death and this in turn leads to bone-spicule-like pigmentation, narrowed arterioles, and clinical symptoms and signs characteristic of retinitis pigmentosa. In such patients, there is very little yellow pigment deposition because the source of the pigment (the photoreceptor outer segments) is lost. In contrast, the milder mutations of both genes, although expressed in the photoreceptors, have their most visible pathologic effects at the level of the retinal pigment epithelium because the normal phagocytic activity of the latter tissue imports the toxic bisretinoid (in the case of ABCA4) or misshaped discs (in the case of PRPH2). Apoptotic death of the RPE in response to this long-term stress causes death of the underlying choriocapillaris just as it does in age-related macular degeneration.
Treatment
A significant risk for vision loss exists for patients with PD; Francis et al. reported 50% eventually developed poor central acuity due to either geographic atrophy or subretinal neovascularization.182 However, as in Best disease, many patients can retain driving vision in at least one eye well into their seventh decade of life. Although CNV is slightly less frequent than in age-related macular degeneration, it can be just as devastating to vision and anti-VEGF injections may result in regression and limitation of vision loss.216 It is also helpful to discuss the practical aspects of delayed recovery from exposure to bright light with PD patients because wearing dark glasses and a hat when outside can allow them to adapt more readily when coming inside. Similarly, adjusting the lighting in the patient’s home or office to minimize large changes in illumination from one room to the next (e.g., bright kitchen, dim family room) can make a big difference in their activities of daily living.
Sorsby fundus dystrophy
In 1949 Sorsby described five families with an autosomal dominantly inherited “fundus dystrophy with unusual features” characterized by macular hemorrhages that usually begin early in the fifth decade of life and pigmentary changes in both maculae.217 Progressive atrophy of the peripheral choroid and RPE is common and can severely limit ambulatory vision later in life. Now known as Sorsby fundus dystrophy (SFD), this disease is caused by mutations in TIMP-3 (OMIM #188826).218
Clinical features of SFD
One of the earliest symptoms of the disease is night blindness,219,220 although this is not typically severe enough to bring a patient to medical attention. When individuals are seen at this early stage, it is often because they are concerned about the severe macular disease in one of their parents. At this point in the disease course ophthalmoscopically visible yellow-to-gray material is present at the level of Bruch’s membrane. In some parts of the fundus this material can take the form of drusen (Fig. 42.56) while in other areas the material coalesces into a fairly uniform yellowish-gray sheet that becomes more prominent with increasing age (Fig. 42.57A). Patients most commonly present to an ophthalmologist in the fourth or fifth decade of life,217 when they develop bilateral subfoveal neovascular membranes (Figs 42.57A, 42.58) and their visual acuity worsens suddenly. Untreated CNV often results in extensive disciform scarring that severely reduces visual acuity.221 Marked pigment alterations consisting of atrophy admixed with angular pigment proliferation commonly develop in the macula with or without CNV. Unlike age-related macular degeneration, which rarely extends beyond the temporal vascular arcades, SFD relentlessly extends peripherally and commonly reduces a patient’s acuity to hand motions late in life.
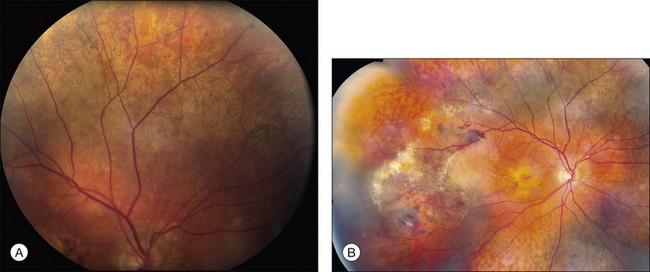
Genetics
Since its discovery in 1994, at least 14 mutations in TIMP3 have been identified.218,222–229 Most people with SFD in the United Kingdom share a common ancestor and thus harbor the same mutation, Ser181Cys.230,231 A very unusual feature of SFD is that almost all of the reported mutations create a new cysteine residue in the mutant protein, suggesting that a perturbation of tertiary structure through altered disulfide bonding is required for the development of the disease.
Pathophysiology
The TIMP3 gene encodes a protein, tissue inhibitor of metalloproteinases-3 (TIMP3), which belongs to a family of negative regulators of matrix metalloproteinase activity. TIMP3 physically interacts with several metalloproteinases232 and regulates their activity in normal tissue homeostasis. Mutations in this gene are likely to cause altered turnover of extracellular matrix. Given that pathologic angiogenesis is a major clinical feature of SFD, it is notable that TIMP3 suppresses angiogenesis by blocking the interaction of VEGF with its receptor KDR.233
The most striking characteristic of eyes with SD is the accumulation of lipidic and proteinaceous material between Bruch’s membrane and the RPE up to 30 µm in thickness.234 Histopathologic analyses of eyes from a donor with SFD due to a Ser181Cys mutation in TIMP3 revealed CNV and thick deposits beneath the RPE that contained TIMP3 and other extracellular matrix components. The presence of basement membrane proteins in the SFD lesions is consistent with the notion that TIMP3 gain-of-function mutations are pathogenic by their dysregulation of matrix turnover. Ultrastructurally, this material contains banded proteinaceous aggregates with a periodicity suggestive of type VI collagen.235 Type VI collagen has also been shown to be present at the margins of the lesions by immunohistochemistry.236
The TIMP3 protein is a normal component of Bruch’s membrane237 as well as a component of drusen238 and the abnormal deposits in SFD.236,239 The accumulation of TIMP3 in SFD may result from the relatively poor turnover of the mutant protein caused by missense mutations.240
Dominant mutations in TIMP3 have been recapitulated in mice. Mice harboring a mutant allele of TIMP3 (Ser156Cys) show abnormalities in the RPE basal infoldings and increased extracellular material between the RPE plasma membrane and its basal lamina.241 Thus, animal studies and biochemical findings strongly suggest that SFD-causing mutations in TIMP3 cause impaired regulation of metalloproteinases and reduced remodeling of the matrix. Normal synthesis, reduced remodeling of extracellular matrix, and impaired control of angiogenesis would appear sufficient to describe the pathology of SFD. However, when Fogarasi et al. assayed the proteinase inhibitory activity of a TIMP3 protein carrying a common SFD mutation, they found no impairment of proteinase inhibition.242 Thus, some significant questions about the molecular basis of SFD remain to be answered.
Treatment
Treatment of patients with SFD is aimed at CNV control. Argon laser therapy and PDT are not effective in treating SFD-associated CNVs, possibly due to the thickening of Bruch’s membrane.221,243 However, intravenous bevacizumab has been used in one patient and resulted in CNV regression and improvement in visual acuity.244
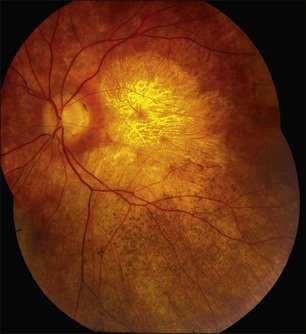
Autosomal dominant radial drusen (doyne honeycomb retinal dystrophy, malattia leventinese)
A familial macular condition with a honeycomb appearance was described by Doyne in the UK in 1899245 and a similar condition (malattia leventinese, ML) was recognized by Vogt in patients residing in the Leventine Valley of Switzerland in 1925.246 Surprisingly, it was not until the late 1980s that Gass drew specific attention to one of the most striking clinical features of the condition, the radial distribution of drusen at the temporal periphery of the macular lesions (Fig. 42.59),247 although at that time he did not explicitly connect this observation to the entities described by Doyne and Vogt. In 1996 Héon and colleagues mapped the disease-causing gene for malattia leventinese to chromosome 2,248 and in 1999 Stone and coworkers showed that Doyne’s honeycomb retinal dystrophy and ML were in fact one disorder caused by a single point mutation (Arg345Trp) in the EFEMP1 gene.249 In this chapter, we will use the descriptive term “autosomal dominant radial drusen” (ADRD) to refer to this disease.
Clinical features of ADRD
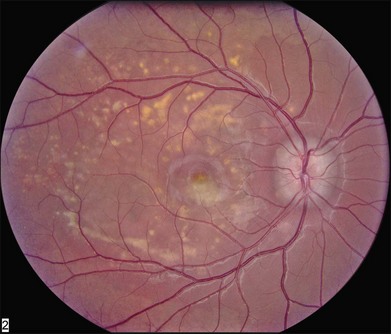
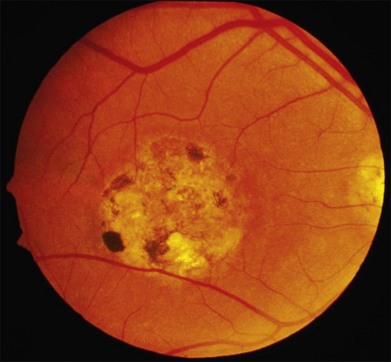
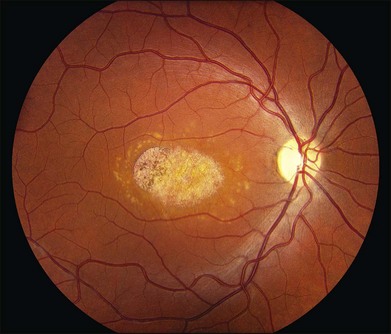
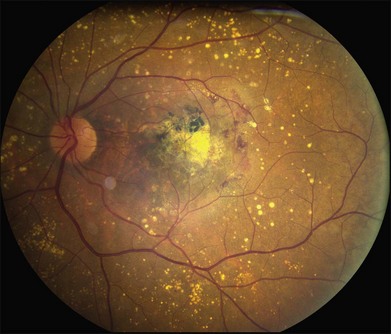
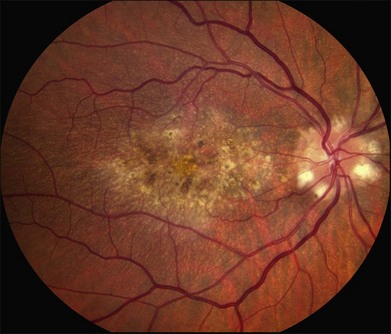
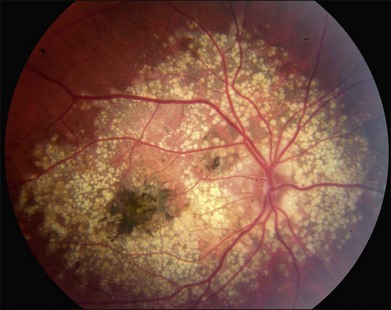
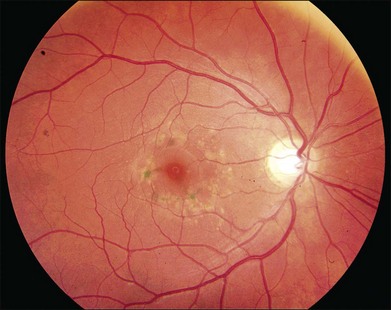
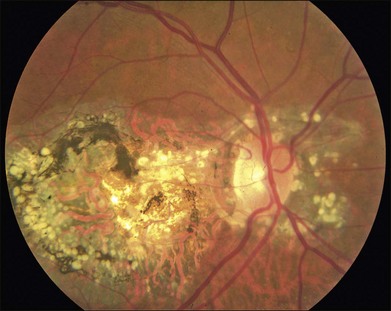
As with most autosomal dominant conditions, ADRD has a wide range of clinical appearances, presumably due to the variable effect of other genes and environmental factors in different affected individuals. Drusen can be seen in the second decade in some patients while others have barely detectable drusen in the seventh decade. The drusen in the center of the macula and on the nasal edge of the optic disc tend to be large and round (Fig. 42.60), while those at the temporal margin of the macula tend to be smaller, elongated, and radial (see Fig. 42.59). In some patients the small radial drusen can be nearly invisible and reticular pigment changes in the macula can simulate a pattern dystrophy (Fig. 42.61). Most eyes have a group of drusen abutting the nasal aspect of the nerve, even when macular drusen are few or absent.250 Over time, many eyes develop central atrophy, scarring, and pigment proliferation that can look similar to SFD (Fig. 42.62). However, for a given degree of macular abnormality, the visual acuity in ADRD is typically much better than SFD. Choroidal neovascularization can also complicate ADRD but this occurs much less frequently than it does in SFD.
Visual function and electrophysiology
Even in the presence of extensive drusen, the visual acuity is usually excellent until central atrophy, pigment proliferation, or CNV develop later in life.250,251 Scotopic sensitivity is reduced and dark-adaptation kinetics are prolonged over confluent macular deposits but are normal elsewhere. The full-field ERG is usually normal but the pattern ERG is abnormal in most eyes.252
Imaging
In contrast to drusen in AMD that tend to be hypoautofluorescent, the drusen in ADRD are hyperautofluorescent.250,253 Autofluorescence can also be useful to distinguish areas of atrophy that can be challenging to see on clinical examination when there is extensive pathology present. Fluorescein angiography is useful for the detection of active choroidal neovascularization, while OCT can demonstrate retinal edema, loss of foveal contour, pigment epithelial detachments, and intense sub-RPE reflectivity.251
Pathophysiology and histopathology
The EFEMP1 gene encodes an extracellular matrix protein known as fibulin-3. Marmorstein et al. studied eyes from an 86-year-old donor with ADRD and found large sub-RPE deposits containing fibulin-3, localized along the apical (i.e., sub-RPE) surface of the deposit, but generally not throughout the druse.254 Ultrastructural studies have also shown deposition of membranous material in Bruch’s membrane.255 Histologically, drusen in ADRD have a layered appearance that is distinct from age-related drusen (see Fig. 42.63). The drusen tend to be large and are often fused together. Cellular infiltration may be present.
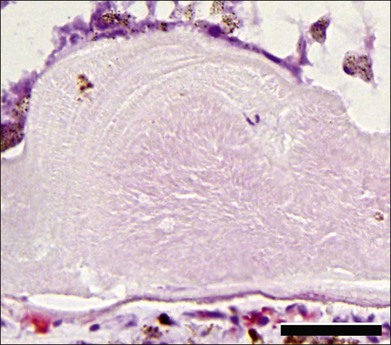
Physiological studies have provided some insight into the pathogenesis of ADRD. Fibulin-3 bearing the Arg345Trp mutation is poorly secreted by RPE cells in vitro and its accumulation in the endoplasmic reticulum activates the unfolded protein response.256 Knockin mice harboring one or more Arg345Trp alleles develop sub-RPE deposits that, while modest in size compared to the drusen observed in ADRD, share molecular components such as fibulin-3 and TIMP3.257,258 EFEMP1 is expressed in both neural retina and RPE/choroid layers259 and fibulin-3 has been detected in photoreceptor cells, inner retinal neurons, and Bruch’s membrane.254
North carolina macular dystrophy
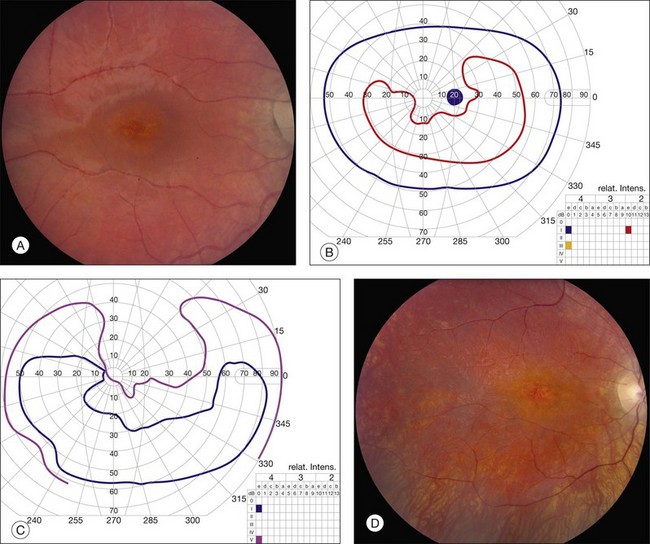
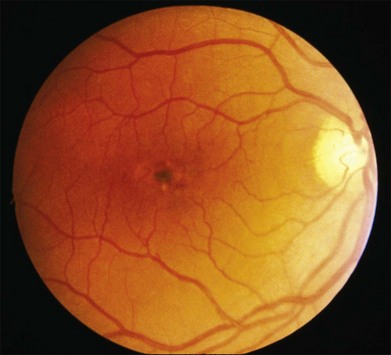
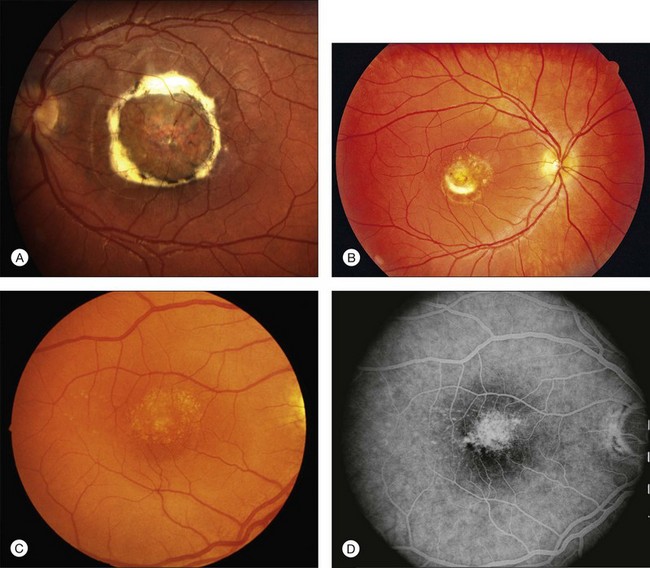
North Carolina Macular Dystrophy (NCMD) was first described as “dominant macular degeneration and aminoaciduria” by Lefler, Wadsworth, and Sidbury,260 who studied a kindred originating from two Irish brothers who settled in North Carolina in the 1830s.261 The original North Carolina pedigree now consists of more than 5000 individuals,262 but families with similar phenotypes have been identified throughout the world.263–267 It has also been called central areolar pigment epithelial dystrophy268 and dominant progressive foveal dystrophy.261 The latter term is a particularly unfortunate misnomer because Small and colleagues clearly demonstrated that NCMD is a developmental abnormality that is almost completely stationary in most individuals.269 Large lesions are large at birth and do not progress from smaller ones. This complete lack of progression is one of the most reliable diagnostic features of the disease, and accounts to some degree for the amazingly good visual acuity in some patients with very large lesions. That is, because the lesions are present well before maturation of the visual system, patients learn to fixate on the edges of the lesions and develop their visual pathways accordingly. The fundus findings in NCMD tend to be bilateral and quite symmetric. The most characteristic lesion is a circular coloboma centered on fixation with a shiny concave base surrounded by a thick, white fibrotic rim (Fig. 42.64A). Much more difficult to diagnose correctly in the absence of other family members is the milder manifestation of the disease that can appear as nothing more than a patch of drusen in a young adult with excellent acuity (Fig. 42.64B,C). The visual acuity in NCMD correlates fairly well with the size of the fundus lesions as follows: 20/20 to 20/30 for small (less than 50 µm) lesions; 20/25 to 20/60 for confluent yellow specks in the central macula; and 20/40 to 20/200 when large (500–1000 µm) colobomatous lesions are present (Fig. 42.64A,D).269,270
NCMD was mapped by Small and coworkers to chromosome 6271–273 but the causative gene has not yet been identified. Another dominant macular dystrophy with clinical features very similar to that of NCMD has recently been mapped to chromosome 5274,275 but the causative gene at this locus has also not yet been identified. In a histopathologic study of one eye from a donor with NCMD, lipofuscin and choriocapillaris atrophy were noted.276 Currently little is known about the pathophysiology of this disorder or the site of expression of the mutant gene.
Spotted cystic dystrophy
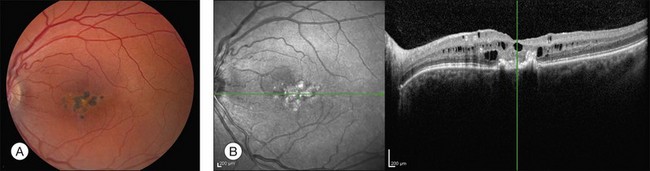
Mahajan et al.277 recently described seven members of a three-generation family with autosomal dominant inheritance of a new dystrophy limited to the macula and characterized by round, flat pigmented spots with or without surrounding hypopigmentation (Fig. 42.65A), cysts in multiple retinal layers on OCT (Fig. 42.65B), and neovascularization. Amblyopia and strabismus were frequently present in affected individuals. Visual acuity ranged from 20/20 to 20/200. The pathophysiology and genetic mutation responsible for this condition have not been identified. When active macular neovascularization occurs in affected individuals, it has been responsive to either focal laser or a single injection of bevacizumab.277
Dominant cystoid macular dystrophy
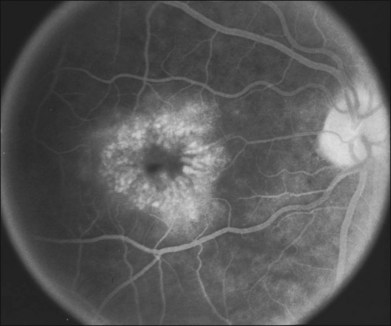
Dominant cystoid macular dystrophy (DCMD) was described in 1976 by Deutman278 as an autosomal dominant condition characterized by leaking perimacular capillaries, whitish punctate deposits in the vitreous, a normal ERG, a subnormal EOG, and hyperopia (Fig. 42.66). In the late stages of the disease, an atrophic central “beaten-bronze” macula was common. A second family was identified a few years later279 and linkage analysis mapped the chromosomal location of the disease-causing gene to the short arm of chromosome 7.280 The disease-causing gene for DCMD has not yet been identified. Hogewind and coworkers evaluated intramuscular injections of a somatostatin analog (octreotide acetate) in four patients with DCMD and seven of the eight eyes showed improvement on fluorescein angiography, with stabilization of visual acuity.281
Fenestrated sheen macular dystrophy (FSMD)
Several families have been described with an autosomal dominant macular disorder characterized by central macular sheen with small red fenestrations, occurring as early as the first decade of life and seen as late as the fifth decade (Fig. 42.67). Some middle-aged family members develop a bull’s-eye pattern of stippled hypopigmentation in the central macula. Mild functional abnormalities roughly correlate with more advanced age but patients with the red fenestrations have 20/20 visual acuity. Normal or mildly abnormal ERG findings have been reported.282–285 The chromosomal location of the disease-causing gene is currently unknown.
Glomerulonephritis type II and drusen
The majority of patients with membranproliferative glomerulonephritis (MPGN) type II (also known as dense-deposit disease) develop subretinal deposits with the clinical appearance of basal laminar drusen (Fig. 42.68)286 D’Souza and coworkers followed four MPGN patients with such drusen for 10 years and observed no progression and no vision loss during this interval.287 In general, visual acuity tends to be preserved unless CNV, exudative drusen, or serous detachment complicate the disease.288–290 An abnormal EOG with a relatively normal ERG can be seen in some patients, suggesting a more global retinal dysfunction than the visible drusen would suggest.291,292 Histopathologic studies of the Bruch’s membrane deposits found in MPGN II demonstrate that they are morphologically293 and compositionally294 similar to the drusen found in AMD.295 Abnormal urinalysis with this phenotype in young adults should prompt a referral for work-up of kidney disease.
 For online acknowledgments visit http://www.expertconsult.com
For online acknowledgments visit http://www.expertconsult.com
1 Petrukhin K, Koisti MJ, Bakall B, et al. Identification of the gene responsible for Best macular dystrophy. Nat Genet. 1998;19(3):241–247.
2 Marquardt A, Stohr H, Passmore LA, et al. Mutations in a novel gene, VMD2, encoding a protein of unknown properties cause juvenile-onset vitelliform macular dystrophy (Best’s disease). Hum Mol Genet. 1998;7(9):1517–1525.
3 Best F. Ueber eine hereditaere Makulaaffection. Zschr Augenheilk. 1905:199–212.
4 Braley AE, Spivey BE. Hereditary Vitelline macular degeneration: a clinical and functional evaluation of a new pedigree with variable expressivity and dominant inheritance. Trans Am Ophthalmol Soc. 1963;61:339–371.
5 Krill AE, Morse PA, Potts AM, et al. Hereditary vitelliruptive macular degeneration. Am J Ophthalmol. 1966;61(6):1405–1415.
6 Zanen J, Rausin G. Kyste vitelliforme congenital de la macula. Bull Soc Belge Ophtalmol. 1950;96:544.
7 Miller SA, Bresnick GH, Chandra SR. Choroidal neovascular membrane in Best’s vitelliform macular dystrophy. Am J Ophthalmol. 1976;82(2):252–255.
8 Miller SA. Multifocal Best’s vitelliform dystrophy. Arch Ophthalmol. 1977;95(6):984–990.
9 Ciulla TA, Frederick AR, Jr. Acute progressive multifocal Best’s disease in a 61-year-old man. Am J Ophthalmol. 1997;123(1):129–131.
10 Kinnick TR, Mullins RF, Dev S, et al. Autosomal recessive vitelliform macular dystrophy in a large cohort of vitelliform macular dystrophy patients. Retina. 2011;31(3):581–595.
11 Schatz P, Klar J, Andreasson S, et al. Variant phenotype of Best vitelliform macular dystrophy associated with compound heterozygous mutations in VMD2. Ophthalmic Genet. 2006;27(2):51–56.
12 Wittstrom E, Ekvall S, Schatz P, et al. Morphological and functional changes in multifocal vitelliform retinopathy and biallelic mutations in BEST1. Ophthalmic Genet. 2011;32(2):83–96.
13 Burgess R, Millar ID, Leroy BP, et al. Biallelic mutation of BEST1 causes a distinct retinopathy in humans. Am J Hum Genet. 2008;82(1):19–31.
14 Querques G, Zerbib J, Santacroce R, et al. Functional and clinical data of Best vitelliform macular dystrophy patients with mutations in the BEST1 gene. Mol Vis. 2009;15:2960–2972.
15 Chan CK, Gass JD, Lin SG. Acute exudative polymorphous vitelliform maculopathy syndrome. Retina. 2003;23(4):453–462.
16 Gass JD, Chuang EL, Granek H. Acute exudative polymorphous vitelliform maculopathy. Trans Am Ophthalmol Soc. 1988;86:354–366.
17 Guziewicz KE, Zangerl B, Lindauer SJ, et al. Bestrophin gene mutations cause canine multifocal retinopathy: a novel animal model for best disease. Invest Ophthalmol Vis Sci. 2007;48(5):1959–1967.
18 Guziewicz KE, Slavik J, Lindauer SJ, et al. Molecular consequences of BEST1 gene mutations in canine multifocal retinopathy predict functional implications for human bestrophinopathies. Invest Ophthalmol Vis Sci. 2011.
19 Chung MM, Oh KT, Streb LM, et al. Visual outcome following subretinal hemorrhage in Best disease. Retina. 2001;21(6):575–580.
20 Mohler CW, Fine SL. Long-term evaluation of patients with Best’s vitelliform dystrophy. Ophthalmology. 1981;88(7):688–692.
21 Fishman GA, Baca W, Alexander KR, et al. Visual acuity in patients with best vitelliform macular dystrophy. Ophthalmology. 1993;100(11):1665–1670.
22 Sohn EH, Francis PJ, Duncan JL, et al. Phenotypic variability due to a novel Glu292Lys variation in exon 8 of the BEST1 gene causing best macular dystrophy. Arch Ophthalmol. 2009;127(7):913–920.
23 Renner AB, Tillack H, Kraus H, et al. Late onset is common in Best macular dystrophy associated with VMD2 gene mutations. Ophthalmology. 2005;112(4):586–592.
24 Wittstrom E, Ponjavic V, Bondeson ML, et al. Anterior Segment abnormalities and angle-closure glaucoma in a family with a mutation in the Best1 gene and Best vitelliform macular dystrophy. Ophthalmic Genet. 2011;32(4):217–227.
25 Wabbels B, Preising MN, Kretschmann U, et al. Genotype-phenotype correlation and longitudinal course in ten families with Best vitelliform macular dystrophy. Graefes Arch Clin Exp Ophthalmol. 2006;244(11):1453–1466.
26 Burgess R, MacLaren RE, Davidson AE, et al. ADVIRC is caused by distinct mutations in BEST1 that alter pre-mRNA splicing. J Med Genet. 2009;46(9):620–625.
27 Lafaut BA, Loeys B, Leroy BP, et al. Clinical and electrophysiological findings in autosomal dominant vitreoretinochoroidopathy: report of a new pedigree. Graefes Arch Clin Exp Ophthalmol. 2001;239(8):575–582.
28 Kay CN, Abramoff MD, Mullins RF, et al. Three dimensional distribution of the vitelliform lesion, photoreceptors, and retinal pigment epithelium in the macula of patients with Best vitelliform macular dystrophy. Retina (submitted).
29 Querques G, Regenbogen M, Quijano C, et al. High-definition optical coherence tomography features in vitelliform macular dystrophy. Am J Ophthalmol. 2008;146(4):501–507.
30 Ferrara DC, Costa RA, Tsang S, et al. Multimodal fundus imaging in Best vitelliform macular dystrophy. Graefes Arch Clin Exp Ophthalmol. 2010;248(10):1377–1386.
31 Spaide RF, Noble K, Morgan A, et al. Vitelliform macular dystrophy. Ophthalmology. 2006;113(8):1392–1400.
32 Pianta MJ, Aleman TS, Cideciyan AV, et al. In vivo micropathology of Best macular dystrophy with optical coherence tomography. Exp Eye Res. 2003;76(2):203–211.
33 von Ruckmann A, Fitzke FW, Bird AC. In vivo fundus autofluorescence in macular dystrophies. Arch Ophthalmol. 1997;115(5):609–615.
34 Boon CJ, Theelen T, Hoefsloot EH, et al. Clinical and molecular genetic analysis of best vitelliform macular dystrophy. Retina. 2009;29(6):835–847.
35 Frangieh GT, Green WR, Fine SL. A histopathologic study of Best’s macular dystrophy. Arch Ophthalmol. 1982;100(7):1115–1121.
36 O’Gorman S, Flaherty WA, Fishman GA, et al. Histopathologic findings in Best’s vitelliform macular dystrophy. Arch Ophthalmol. 1988;106(9):1261–1268.
37 Weingeist TA, Kobrin JL, Watzke RC. Histopathology of Best’s macular dystrophy. Arch Ophthalmol. 1982;100(7):1108–1114.
38 Bakall B, Radu RA, Stanton JB, et al. Enhanced accumulation of A2E in individuals homozygous or heterozygous for mutations in BEST1 (VMD2). Exp Eye Res. 2007;85(1):34–43.
39 Meunier I, Senechal A, Dhaenens CM, et al. Systematic screening of BEST1 and PRPH2 in juvenile and adult vitelliform macular dystrophies: a rationale for molecular analysis. Ophthalmology. 2011;118(6):1130–1136.
40 Pollack K, Kreuz FR, Pillunat LE. [Best’s disease with normal EOG. Case report of familial macular dystrophy.]. Ophthalmologe. 2005;102(9):891–894.
41 Testa F, Rossi S, Passerini I, et al. A normal electro-oculography in a family affected by Best disease with a novel spontaneous mutation of the BEST1 gene. Br J Ophthalmol. 2008;92(11):1467–1470.
42 Wabbels B, Demmler A, Paunescu K, et al. Fundus autofluorescence in children and teenagers with hereditary retinal diseases. Graefes Arch Clin Exp Ophthalmol. 2006;244(1):36–45.
43 Yu K, Qu Z, Cui Y, et al. Chloride channel activity of bestrophin mutants associated with mild or late-onset macular degeneration. Invest Ophthalmol Vis Sci. 2007;48(10):4694–4705.
44 Stone EM, Nichols BE, Streb LM, et al. Genetic linkage of vitelliform macular degeneration (Best’s disease) to chromosome 11q13. Nat Genet. 1992;1(4):246–250.
45 Graff C, Forsman K, Larsson C, et al. Fine mapping of Best’s macular dystrophy localizes the gene in close proximity to but distinct from the D11S480/ROM1 loci. Genomics. 1994;24(3):425–434.
46 Nichols BE, Bascom R, Litt M, et al. Refining the locus for Best vitelliform macular dystrophy and mutation analysis of the candidate gene ROM1. Am J Hum Genet. 1994;54(1):95–103.
47 Marmorstein AD, Marmorstein LY, Rayborn M, et al. Bestrophin, the product of the Best vitelliform macular dystrophy gene (VMD2), localizes to the basolateral plasma membrane of the retinal pigment epithelium. Proc Natl Acad Sci U S A. 2000;97(23):12758–12763.
48 Allikmets R, Seddon JM, Bernstein PS, et al. Evaluation of the Best disease gene in patients with age-related macular degeneration and other maculopathies. Hum Genet. 1999;104(6):449–453.
49 Bakall B, Marknell T, Ingvast S, et al. The mutation spectrum of the bestrophin protein – functional implications. Hum Genet. 1999;104(5):383–389.
50 Ponjavic V, Eksandh L, Andreasson S, et al. Clinical expression of Best’s vitelliform macular dystrophy in Swedish families with mutations in the bestrophin gene. Ophthalmic Genet. 1999;20(4):251–257.
51 Lotery AJ, Munier FL, Fishman GA, et al. Allelic variation in the VMD2 gene in Best disease and age-related macular degeneration. Invest Ophthalmol Vis Sci. 2000;41(6):1291–1296.
52 White K, Marquardt A, Weber BH. VMD2 mutations in vitelliform macular dystrophy (Best disease) and other maculopathies. Hum Mutat. 2000;15(4):301–308.
53 Kramer F, White K, Pauleikhoff D, et al. Mutations in the VMD2 gene are associated with juvenile-onset vitelliform macular dystrophy (Best disease) and adult vitelliform macular dystrophy but not age-related macular degeneration. Eur J Hum Genet. 2000;8(4):286–292.
54 Eksandh L, Bakall B, Bauer B, et al. Best’s vitelliform macular dystrophy caused by a new mutation (Val89Ala) in the VMD2 gene. Ophthalmic Genet. 2001;22(2):107–115.
55 Seddon JM, Afshari MA, Sharma S, et al. Assessment of mutations in the Best macular dystrophy (VMD2) gene in patients with adult-onset foveomacular vitelliform dystrophy, age-related maculopathy, and bull’s-eye maculopathy. Ophthalmology. 2001;108(11):2060–2067.
56 Stohr H, Marquardt A, Nanda I, et al. Three novel human VMD2-like genes are members of the evolutionary highly conserved RFP-TM family. Eur J Hum Genet. 2002;10(4):281–284.
57 Seddon JM, Sharma S, Chong S, et al. Phenotype and genotype correlations in two Best families. Ophthalmology. 2003;110(9):1724–1731.
58 Mullins RF, Oh KT, Heffron E, et al. Late development of vitelliform lesions and flecks in a patient with Best disease: clinicopathologic correlation. Arch Ophthalmol. 2005;123(11):1588–1594.
59 Apushkin MA, Fishman GA, Taylor CM, et al. Novel de novo mutation in a patient with Best macular dystrophy. Arch Ophthalmol. 2006;124(6):887–889.
60 Marchant D, Yu K, Bigot K, et al. New VMD2 gene mutations identified in patients affected by Best vitelliform macular dystrophy. J Med Genet. 2007;44(3):e70.
61 Boon CJ, Klevering BJ, Leroy BP, et al. The spectrum of ocular phenotypes caused by mutations in the BEST1 gene. Prog Retin Eye Res. 2009;28(3):187–205.
62 Schatz P, Bitner H, Sander B, et al. Evaluation of macular structure and function by OCT and electrophysiology in patients with vitelliform macular dystrophy due to mutations in BEST1. Invest Ophthalmol Vis Sci. 2010;51(9):4754–4765.
63 Mullins RF, Kuehn MH, Faidley EA, et al. Differential macular and peripheral expression of bestrophin in human eyes and its implication for Best disease. Invest Ophthalmol Vis Sci. 2007;48(7):3372–3380.
64 Marmorstein AD, Cross HE, Peachey NS. Functional roles of bestrophins in ocular epithelia. Prog Retin Eye Res. 2009;28(3):206–226.
65 Xiao Q, Hartzell HC, Yu K. Bestrophins and retinopathies. Pflugers Arch. 2010;460(2):559–569.
66 Hartzell C, Qu Z, Putzier I, et al. Looking chloride channels straight in the eye: bestrophins, lipofuscinosis, and retinal degeneration. Physiology (Bethesda). 2005;20:292–302.
67 Sun H, Tsunenari T, Yau KW, et al. The vitelliform macular dystrophy protein defines a new family of chloride channels. Proc Natl Acad Sci U S A. 2002;99(6):4008–4013.
68 Fischmeister R, Hartzell HC. Volume sensitivity of the bestrophin family of chloride channels. J Physiol. 2005;562(Pt 2):477–491.
69 Marmorstein LY, Wu J, McLaughlin P, et al. The light peak of the electroretinogram is dependent on voltage-gated calcium channels and antagonized by bestrophin (best-1). J Gen Physiol. 2006;127(5):577–589.
70 Zhang Y, Stanton JB, Wu J, et al. Suppression of Ca2+ signaling in a mouse model of Best disease. Hum Mol Genet. 2010;19(6):1108–1118.
71 Marmor MF, Yao XY, Hageman GS. Retinal adhesiveness in surgically enucleated human eyes. Retina. 1994;14(2):181–186.
72 Hall MO, Abrams TA, Mittag TW. ROS ingestion by RPE cells is turned off by increased protein kinase C activity and by increased calcium. Exp Eye Res. 1991;52(5):591–598.
73 Kaufman SJ, Goldberg MF, Orth DH, et al. Autosomal dominant vitreoretinochoroidopathy. Arch Ophthalmol. 1982;100(2):272–278.
74 Blair NP, Goldberg MF, Fishman GA, et al. Autosomal dominant vitreoretinochoroidopathy (ADVIRC). Br J Ophthalmol. 1984;68(1):2–9.
75 Han DP, Lewandowski MF. Electro-oculography in autosomal dominant vitreoretinochoroidopathy. Arch Ophthalmol. 1992;110(11):1563–1567.
76 Kellner U, Jandeck C, Kraus H, et al. Autosomal dominant vitreoretinochoroidopathy with normal electrooculogram in a German family. Graefes Arch Clin Exp Ophthalmol. 1998;236(2):109–114.
77 Vincent A, McAlister C, Vandenhoven C, et al. BEST1-related autosomal dominant vitreoretinochoroidopathy: a degenerative disease with a range of developmental ocular anomalies. Eye (Lond). 2011;25(1):113–118.
78 Yardley J, Leroy BP, Hart-Holden N, et al. Mutations of VMD2 splicing regulators cause nanophthalmos and autosomal dominant vitreoretinochoroidopathy (ADVIRC). Invest Ophthalmol Vis Sci. 2004;45(10):3683–3689.
79 Oh KT, Vallar C. Central cone dysfunction in autosomal dominant vitreoretinochoroidopathy (ADVIRC). Am J Ophthalmol. 2006;141(5):940–943.
80 Davidson AE, Millar ID, Urquhart JE, et al. Missense mutations in a retinal pigment epithelium protein, bestrophin-1, cause retinitis pigmentosa. Am J Hum Genet. 2009;85(5):581–592.
81 Iannaccone A, Kerr NC, Kinnick TR, et al. Autosomal recessive best vitelliform macular dystrophy: report of a family and management of early-onset neovascular complications. Arch Ophthalmol. 2011;129(2):211–217.
82 Davidson AE, Sergouniotis PI, Burgess-Mullan R, et al. A synonymous codon variant in two patients with autosomal recessive bestrophinopathy alters in vitro splicing of BEST1. Mol Vis. 2010;16:2916–2922.
83 Gerth C, Zawadzki RJ, Werner JS, et al. Detailed analysis of retinal function and morphology in a patient with autosomal recessive bestrophinopathy (AR(B). Doc Ophthalmol. 2009;118(3):239–246.
84 Leu J, Schrage NF, Degenring RF. Choroidal neovascularisation secondary to Best’s disease in a 13-year-old boy treated by intravitreal bevacizumab. Graefes Arch Clin Exp Ophthalmol. 2007;245(11):1723–1725.
85 Montero JA, Ruiz-Moreno JM, De La Vega C. Intravitreal bevacizumab for adult-onset vitelliform dystrophy: a case report. Eur J Ophthalmol. 2007;17(6):983–986.
86 Querques G, Bocco MC, Soubrane G, et al. Intravitreal ranibizumab (Lucentis) for choroidal neovascularization associated with vitelliform macular dystrophy. Acta Ophthalmol. 2008;86(6):694–695.
87 Rishi E, Rishi P, Mahajan S. Intravitreal bevacizumab for choroidal neovascular membrane associated with Best’s vitelliform dystrophy. Indian J Ophthalmol. 2010;58(2):160–162.
88 Heidary F, Hitam WH, Ngah NF, et al. Intravitreal ranibizumab for choroidal novascularization in Best’s vitelliform macular dystrophy in a 6-year-old boy. J Pediatr Ophthalmol Strabismus. 2011;48:e19–e22. Online
89 Boon CJ, den Hollander AI, Hoyng CB, et al. The spectrum of retinal dystrophies caused by mutations in the peripherin/RDS gene. Prog Retin Eye Res. 2008;27(2):213–235.
90 Chowers I, Zamir E, Banin E, et al. Blunt trauma in Best’s vitelliform macular dystrophy. Br J Ophthalmol. 2000;84(11):1330–1331.
91 Allikmets R. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet. 1997;17(1):122.
92 Cremers FP, van de Pol DJ, van Driel M, et al. Autosomal recessive retinitis pigmentosa and cone–rod dystrophy caused by splice site mutations in the Stargardt’s disease gene ABCR. Hum Mol Genet. 1998;7(3):355–362.
93 Martinez-Mir A, Paloma E, Allikmets R, et al. Retinitis pigmentosa caused by a homozygous mutation in the Stargardt disease gene ABCR. Nat Genet. 1998;18(1):11–12.
94 Fishman GA, Stone EM, Grover S, et al. Variation of clinical expression in patients with Stargardt dystrophy and sequence variations in the ABCR gene. Arch Ophthalmol. 1999;117(4):504–510.
95 Maugeri A, Klevering BJ, Rohrschneider K, et al. Mutations in the ABCA4 (ABCR) gene are the major cause of autosomal recessive cone–rod dystrophy. Am J Hum Genet. 2000;67(4):960–966.
96 Fishman GA, Stone EM, Eliason DA, et al. ABCA4 gene sequence variations in patients with autosomal recessive cone–rod dystrophy. Arch Ophthalmol. 2003;121(6):851–855.
97 Schindler EI, Nylen EL, Ko AC, et al. Deducing the pathogenic contribution of recessive ABCA4 alleles in an outbred population. Hum Mol Genet. 2010;19(19):3693–3701.
98 Stargardt K. Uber familiare, progressive degeneration in der maculagegend des auges. Albrecht von Graefes Arch Ophthalmol. 1909;71:534–550.
99 Franceschetti A. La Retinopathie ponctuée albescente. In: Bulletins et memoires de la Société Française d’Ophthalmologie. Paris: Masson&Cie; 1963:14–19.
100 Jayasundera T, Rhoades W, Branham K, et al. Peripapillary dark choroid ring as a helpful diagnostic sign in advanced Stargardt disease. Am J Ophthalmol. 2010;149(4):656–660.
101 Cideciyan AV, Swider M, Aleman TS, et al. ABCA4-associated retinal degenerations spare structure and function of the human parapapillary retina. Invest Ophthalmol Vis Sci. 2005;46(12):4739–4746.
102 Lois N, Halfyard AS, Bird AC, et al. Fundus autofluorescence in Stargardt macular dystrophy–fundus flavimaculatus. Am J Ophthalmol. 2004;138(1):55–63.
103 Fishman GA, Farber M, Patel BS, et al. Visual acuity loss in patients with Stargardt’s macular dystrophy. Ophthalmology. 1987;94(7):809–814.
104 Rotenstreich Y, Fishman GA, Anderson RJ. Visual acuity loss and clinical observations in a large series of patients with Stargardt disease. Ophthalmology. 2003;110(6):1151–1158.
105 Oh KT, Weleber RG, Oh DM, et al. Clinical phenotype as a prognostic factor in Stargardt disease. Retina. 2004;24(2):254–262.
106 Hadden OB, Gass JD. Fundus flavimaculatus and Stargardt’s disease. Am J Ophthalmol. 1976;82(4):527–539.
107 Anmarkrud N. Fundus fluorescein angiography in fundus flavimaculatus and Stargardt’s disease. Acta Ophthalmol (Copenh). 1979;57(2):172–182.
108 Ernest JT, Krill AE. Fluorescein studies in fundus flavimaculatus and drusen. Am J Ophthalmol. 1966;62(1):1–6.
109 Radu RA, Mata NL, Bagla A, et al. Light exposure stimulates formation of A2E oxiranes in a mouse model of Stargardt’s macular degeneration. Proc Natl Acad Sci U S A. 2004;101(16):5928–5933.
110 Lois N, Holder GE, Fitzke FW, et al. Intrafamilial variation of phenotype in Stargardt macular dystrophy–fundus flavimaculatus. Invest Ophthalmol Vis Sci. 1999;40(11):2668–2675.
111 Lois N, Holder GE, Bunce C, et al. Phenotypic subtypes of Stargardt macular dystrophy–fundus flavimaculatus. Arch Ophthalmol. 2001;119(3):359–369.
112 Ergun E, Hermann B, Wirtitsch M, et al. Assessment of central visual function in Stargardt’s disease/fundus flavimaculatus with ultrahigh-resolution optical coherence tomography. Invest Ophthalmol Vis Sci. 2005;46(1):310–316.
113 Sunness JS, Ziegler MD, Applegate CA. Issues in quantifying atrophic macular disease using retinal autofluorescence. Retina. 2006;26(6):666–672.
114 Boon CJ, Jeroen Klevering B, Keunen JE, et al. Fundus autofluorescence imaging of retinal dystrophies. Vision Res. 2008;48(26):2569–2577.
115 Sunness JS, Steiner JN. Retinal function and loss of autofluorescence in stargardt disease. Retina. 2008;28(6):794–800.
116 Gomes NL, Greenstein VC, Carlson JN, et al. A comparison of fundus autofluorescence and retinal structure in patients with Stargardt disease. Invest Ophthalmol Vis Sci. 2009;50(8):3953–3959.
117 Smith RT, Gomes NL, Barile G, et al. Lipofuscin and autofluorescence metrics in progressive STGD. Invest Ophthalmol Vis Sci. 2009;50(8):3907–3914.
118 Shah SN, Koozekanani DD, Kim JE. Phenotypic heterogeneity and lesion size measurements in Stargardt macular dystrophy. Ophthalmic Surg Lasers Imaging. 2009;40(5):506–512.
119 Sodi A, Bini A, Passerini I, et al. Different patterns of fundus autofluorescence related to ABCA4 gene mutations in Stargardt disease. Ophthalmic Surg Lasers Imaging. 2010;41(1):48–53.
120 Burke TR, Allikmets R, Smith RT, et al. Loss of peripapillary sparing in non-group I Stargardt disease. Exp Eye Res. 2010;91(5):592–600.
121 Anastasakis A, Fishman GA, Lindeman M, et al. Infrared scanning laser ophthalmoscope imaging of the macula and its correlation with functional loss and structural changes in patients with Stargardt disease. Retina. 2011;31(5):949–958.
122 Chen Y, Ratnam K, Sundquist SM, et al. Cone photoreceptor abnormalities correlate with vision loss in patients with Stargardt disease. Invest Ophthalmol Vis Sci. 2011;52(6):3281–3292.
123 Chen B, Tosha C, Gorin MB, et al. Analysis of autofluorescent retinal images and measurement of atrophic lesion growth in Stargardt disease. Exp Eye Res. 2010;91(2):143–152.
124 Cella W, Greenstein VC, Zernant-Rajang J, et al. G1961E mutant allele in the Stargardt disease gene ABCA4 causes bull’s eye maculopathy. Exp Eye Res. 2009;89(1):16–24.
125 Lim JI, Tan O, Fawzi AA, et al. A pilot study of Fourier-domain optical coherence tomography of retinal dystrophy patients. Am J Ophthalmol. 2008;146(3):417–426.
126 Lenassi E, Jarc-Vidmar M, Glavac D, et al. Pattern electroretinography of larger stimulus field size and spectral-domain optical coherence tomography in patients with Stargardt disease. Br J Ophthalmol. 2009;93(12):1600–1605.
127 Genead MA, Fishman GA, Anastasakis A. Spectral-domain OCT peripapillary retinal nerve fibre layer thickness measurements in patients with Stargardt disease. Br J Ophthalmol. 2011;95(5):689–693.
128 Klevering BJ, Deutman AF, Maugeri A, et al. The spectrum of retinal phenotypes caused by mutations in the ABCA4 gene. Graefes Arch Clin Exp Ophthalmol. 2005;243(2):90–100.
129 Oh KT, Weleber RG, Stone EM, et al. Electroretinographic findings in patients with Stargardt disease and fundus flavimaculatus. Retina. 2004;24(6):920–928.
130 Fukui T, Yamamoto S, Nakano K, et al. ABCA4 gene mutations in Japanese patients with Stargardt disease and retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2002;43(9):2819–2824.
131 Gerth C, Andrassi-Darida M, Bock M, et al. Phenotypes of 16 Stargardt macular dystrophy/fundus flavimaculatus patients with known ABCA4 mutations and evaluation of genotype-phenotype correlation. Graefes Arch Clin Exp Ophthalmol. 2002;240(8):628–638.
132 Scholl HP, Besch D, Vonthein R, et al. Alterations of slow and fast rod ERG signals in patients with molecularly confirmed Stargardt disease type 1. Invest Ophthalmol Vis Sci. 2002;43(4):1248–1256.
133 Birch DG, Peters AY, Locke KL, et al. Visual function in patients with cone–rod dystrophy (CRD) associated with mutations in the ABCA4(ABCR) gene. Exp Eye Res. 2001;73(6):877–886.
134 Sheffield VC, Stone EM. Genomics and the eye. N Engl J Med. 2011;364(20):1932–1942.
135 Weleber RG. The dystrophic retina in multisystem disorders: the electroretinogram in neuronal ceroid lipofuscinoses. Eye (Lond). 1998;12(Pt 3b):580–590.
136 Tosha C, Gorin MB, Nusinowitz S. Test–retest reliability and inter-ocular symmetry of multi-focal electroretinography in Stargardt disease. Curr Eye Res 2010;35(1):63–72.
137 Stone EM. Finding and interpreting genetic variations that are important to ophthalmologists. Trans Am Ophthalmol Soc. 2003;101:437–484.
138 Philp AR, Jin M, Li S, et al. Predicting the pathogenicity of RPE65 mutations. Hum Mutat. 2009;30(8):1183–1188.
139 Steinmetz RL, Garner A, Maguire JI, et al. Histopathology of incipient fundus flavimaculatus. Ophthalmology. 1991;98(6):953–956.
140 Birnbach CD, Jarvelainen M, Possin DE, et al. Histopathology and immunocytochemistry of the neurosensory retina in fundus flavimaculatus. Ophthalmology. 1994;101(7):1211–1219.
141 Klien BA, Krill AE. Fundus flavimaculatus. Clinical, functional and histopathologic observations. Am J Ophthalmol. 1967;64(1):3–23.
142 Tsybovsky Y, Molday RS, Palczewski K. The ATP-binding cassette transporter ABCA4: structural and functional properties and role in retinal disease. Adv Exp Med Biol. 2010;703:105–125.
143 Molday RS, Zhang K. Defective lipid transport and biosynthesis in recessive and dominant Stargardt macular degeneration. Prog Lipid Res. 2010;49(4):476–492.
144 Weng J, Mata NL, Azarian SM, et al. Insights into the function of Rim protein in photoreceptors and etiology of Stargardt’s disease from the phenotype in abcr knockout mice. Cell. 1999;98(1):13–23.
145 Sparrow JR, Nakanishi K, Parish CA. The lipofuscin fluorophore A2E mediates blue light-induced damage to retinal pigmented epithelial cells. Invest Ophthalmol Vis Sci. 2000;41(7):1981–1989.
146 Suter M, Remé C, Grimm C, et al. Age-related macular degeneration. The lipofusion component N-retinyl-N-retinylidene ethanolamine detaches proapoptotic proteins from mitochondria and induces apoptosis in mammalian retinal pigment epithelial cells. J Biol Chem. 2000;275:39625–39630.
147 Zhou J, Kim SR, Westlund BS, et al. Complement activation by bisretinoid constituents of RPE lipofuscin. Invest Ophthalmol Vis Sci. 2009;50(3):1392–1399.
148 Radu RA, Mata NL, Nusinowitz S, et al. Treatment with isotretinoin inhibits lipofuscin accumulation in a mouse model of recessive Stargardt’s macular degeneration. Proc Natl Acad Sci U S A. 2003;100(8):4742–4747.
149 Radu RA, Han Y, Bui TV, et al. Reductions in serum vitamin A arrest accumulation of toxic retinal fluorophores: a potential therapy for treatment of lipofuscin-based retinal diseases. Invest Ophthalmol Vis Sci. 2005;46(12):4393–4401.
150 Ma L, Kaufman Y, Zhang J, et al. C20-D3-vitamin A slows lipofuscin accumulation and electrophysiological retinal degeneration in a mouse model of Stargardt disease. J Biol Chem. 2011;286(10):7966–7974.
151 Allocca M, Doria M, Petrillo M, et al. Serotype-dependent packaging of large genes in adeno-associated viral vectors results in effective gene delivery in mice. J Clin Invest. 2008;118(5):1955–1964.
152 Cideciyan AV, Swider M, Aleman TS, et al. ABCA4 disease progression and a proposed strategy for gene therapy. Hum Mol Genet. 2009;18(5):931–941.
153 Kong J, Kim SR, Binley K, et al. Correction of the disease phenotype in the mouse model of Stargardt disease by lentiviral gene therapy. Gene Ther. 2008;15(19):1311–1320.
154 Bainbridge JW, Smith AJ, Barker SS, et al. Effect of gene therapy on visual function in Leber’s congenital amaurosis. N Engl J Med. 2008;358(21):2231–2239.
155 Maguire AM, Simonelli F, Pierce EA, et al. Safety and efficacy of gene transfer for Leber’s congenital amaurosis. N Engl J Med. 2008;358(21):2240–2248.
156 Stone EM, Nichols BE, Kimura AE, et al. Clinical features of a Stargardt-like dominant progressive macular dystrophy with genetic linkage to chromosome 6q. Arch Ophthalmol. 1994;112(6):765–772.
157 Zhang K, Kniazeva M, Han M, et al. A 5-bp deletion in ELOVL4 is associated with two related forms of autosomal dominant macular dystrophy. Nat Genet. 2001;27(1):89–93.
158 Bernstein PS, Tammur J, Singh N, et al. Diverse macular dystrophy phenotype caused by a novel complex mutation in the ELOVL4 gene. Invest Ophthalmol Vis Sci. 2001;42(13):3331–3336.
159 Maugeri A, Meire F, Hoyng CB, et al. A novel mutation in the ELOVL4 gene causes autosomal dominant Stargardt-like macular dystrophy. Invest Ophthalmol Vis Sci. 2004;45(12):4263–4267.
160 Vasireddy V, Wong P, Ayyagari R. Genetics and molecular pathology of Stargardt-like macular degeneration. Prog Retin Eye Res. 2010;29(3):191–207.
161 Agbaga MP, Brush RS, Mandal MN, et al. Role of Stargardt-3 macular dystrophy protein (ELOVL4) in the biosynthesis of very long chain fatty acids. Proc Natl Acad Sci U S A. 2008;105:12843–12848.
162 Ambasudhan R, Wang X, Jablonski MM, et al. Atrophic macular degeneration mutations in ELOVL4 result in the intracellular misrouting of the protein. Genomics. 2004;83(4):615–625.
163 Karan G, Yang Z, Zhang K. Expression of wild type and mutant ELOVL4 in cell culture: subcellular localization and cell viability. Mol Vis. 2004;31(10):248–253.
164 Vasireddy V, Jablonski MM, Khan NW, et al. Elovl4 5-bp deletion knock-in mouse model for Stargardt-like macular degeneration demonstrates accumulation of ELOVL4 and lipofuscin. Exp Eye Res. 2009;89(6):905–912.
165 Vasireddy V, Jablonski MM, Mandal MN, et al. Elovl4 5-bp-deletion knock-in mice develop progressive photoreceptor degeneration. Invest Ophthalmol Vis Sci. 2006;47(10):4558–4568.
166 Deutman AF, van Blommestein JD, Henkes HE, et al. Butterfly-shaped pigment dystrophy of the fovea. Arch Ophthalmol. 1970;83(5):558–569.
167 Hsieh RC, Fine BS, Lyons JS. Patterned dystrophies of the retinal pigment epithelium. Arch Ophthalmol. 1977;95(3):429–435.
168 Marmor MF, Byers B. Pattern dystrophy of the pigment epithelium. Am J Ophthalmol. 1977;84(1):32–44.
169 Epstein GA, Rabb MF. Adult vitelliform macular degeneration: diagnosis and natural history. Br J Ophthalmol. 1980;64(10):733–740.
170 Bloom LH, Swanson DE, Bird AC. Adult vitelliform macular degeneration. Br J Ophthalmol. 1981;65(11):800–801.
171 Gass JD. A clinicopathologic study of a peculiar foveomacular dystrophy. Trans Am Ophthalmol Soc. 1974;72:139–156.
172 Sjogren H. Dystrophia reticularis laminae pigmentosae retinae, an earlier not described hereditary eye disease. Acta Ophthalmol (Copenh). 1950;28(3):279–295.
173 Kingham JD, Fenzl RE, Willerson D, et al. Reticular dystrophy of the retinal pigment epithelium. A clinical and electrophysiologic study of three generations. Arch Ophthalmol. 1978;96(7):1177–1184.
174 Deutman AF, Rumke AM. Reticular dystrophy of the retinal pigment epithelium. Dystrophia reticularis laminae pigmentosa retinae of H. Sjogren. Arch Ophthalmol. 1969;82(1):4–9.
175 Slezak H, Hommer K. [Fundus pulverulentus.] Albrecht Von Graefes Arch Klin Exp Ophthalmol. 1969;178(2):176–82.
176 de Jong PT, Delleman JW. Pigment epithelial pattern dystrophy. Four different manifestations in a family. Arch Ophthalmol. 1982;100(9):1416–1421.
177 Hoyng CB, Heutink P, Testers L, et al. Autosomal dominant central areolar choroidal dystrophy caused by a mutation in codon 142 in the peripherin/RDS gene. Am J Ophthalmol. 1996;121(6):623–629.
178 Kajiwara K, Hahn LB, Mukai S, et al. Mutations in the human retinal degeneration slow gene in autosomal dominant retinitis pigmentosa. Nature. 1991;354(6353):480–483.
179 Weleber RG, Carr RE, Murphey WH, et al. Phenotypic variation including retinitis pigmentosa, pattern dystrophy, and fundus flavimaculatus in a single family with a deletion of codon 153 or 154 of the peripherin/RDS gene. Arch Ophthalmol. 1993;111(11):1531–1542.
180 Farrar GJ, Kenna P, Jordan SA, et al. A three-base-pair deletion in the peripherin-RDS gene in one form of retinitis pigmentosa. Nature. 1991;354(6353):478–480.
181 Farrar GJ, Jordan SA, Kenna P, et al. Autosomal dominant retinitis pigmentosa: localization of a disease gene (RP6) to the short arm of chromosome 6. Genomics. 1991;11(4):870–874.
182 Francis PJ, Schultz DW, Gregory AM, et al. Genetic and phenotypic heterogeneity in pattern dystrophy. Br J Ophthalmol. 2005;89(9):1115–1119.
183 Nichols BE, Sheffield VC, Vandenburgh K, et al. Butterfly-shaped pigment dystrophy of the fovea caused by a point mutation in codon 167 of the RDS gene. Nat Genet. 1993;3(3):202–207.
184 Gutman I, Walsh JB, Henkind P. Vitelliform macular dystrophy and butterfly-shaped epithelial dystrophy: a continuum? Br J Ophthalmol. 1982;66(3):170–173.
185 Nichols BE, Drack AV, Vandenburgh K, et al. A 2 base pair deletion in the RDS gene associated with butterfly-shaped pigment dystrophy of the fovea. Hum Mol Genet. 1993;2(8):1347.
186 Fossarello M, Bertini C, Galantuomo M, et al. Deletion in the peripherin/RDS gene in two unrelated Sardinian families with autosomal dominant butterfly-shaped macular dystrophy. Arch Ophthalmol. 1996;114:448–456.
187 Zhang K, Garibaldi DC, Li Y, et al. Butterfly-shaped pattern dystrophy: a genetic, clinical, and histopathological report. Arch Ophthalmol. 2002;120(4):485–490.
188 Yang Z, Li Y, Jiang L, et al. A novel RDS/peripherin gene mutation associated with diverse macular phenotypes. Ophthalmic Genet. 2004;25(2):133–145.
189 Querques G, Forte R, Querques L, et al. Natural course of adult-onset foveomacular vitelliform dystrophy: a spectral-domain optical coherence tomography analysis. Am J Ophthalmol. 2011;152(2):304–313.
190 Arnold JJ, Sarks JP, Killingsworth MC, et al. Adult vitelliform macular degeneration: a clinicopathological study. Eye (Lond). 2003;17(6):717–726.
191 Fishman GA, Woolf MB, Goldberg MF, et al. Reticular tapeto-retinal dystrophy. As a possible late stage of Sjogren’s reticular dystrophy. Br J Ophthalmol. 1976;60(1):35–40.
192 Chopdar A. Reticular dystrophy of retina. Br J Ophthalmol. 1976;60(5):342–344.
193 Kempeneers HP, Dewachter A, Kempeneers GM. Pattern dystrophies of the retinal pigment epithelium. The study of three generations in a family. Doc Ophthalmol. 1990;76(3):261–272.
194 Cardillo Piccolino F, Zingirian M. Pattern dystrophy of the retinal pigment epithelium with vitelliform macular lesion: evolution in ten years. Int Ophthalmol. 1988;11(4):207–217.
195 Giuffre G, Lodato G. Vitelliform dystrophy and pattern dystrophy of the retinal pigment epithelium: concomitant presence in a family. Br J Ophthalmol. 1986;70(7):526–532.
196 Boon CJ, Klevering BJ, Cremers FP, et al. Central areolar choroidal dystrophy. Ophthalmology. 2009;116(4):771–782. 82 e1
197 Yanagihashi S, Nakazawa M, Kurotaki J, et al. Autosomal dominant central areolar choroidal dystrophy and a novel Arg195Leu mutation in the peripherin/RDS gene. Arch Ophthalmol. 2003;121(10):1458–1461.
198 Reig C, Serra A, Gean E, Vidal M, et al. A point mutation in the RDS-peripherin gene in a Spanish family with central areolar choroidal dystrophy. Ophthalmic Genet. 1995;16(2):39–44.
199 Klevering BJ, van Driel M, van Hogerwou AJ, et al. Central areolar choroidal dystrophy associated with dominantly inherited drusen. Br J Ophthalmol. 2002;86(1):91–96.
200 Keilhauer CN, Meigen T, Weber BH. Clinical findings in a multigeneration family with autosomal dominant central areolar choroidal dystrophy associated with an Arg195Leu mutation in the peripherin/RDS gene. Arch Ophthalmol. 2006;124(7):1020–1027.
201 Keilhauer CN, Meigen T, Stohr H, et al. Late-onset central areolar choroidal dystrophy caused by a heterozygous frame-shift mutation affecting codon 307 of the peripherin/RDS gene. Ophthalmic Genet. 2006;27(4):139–144.
202 Hoyng CB, van Rijn PM, Deutman AF. Central areolar choroidal dystrophy and slowly progressive sensorineural hearing loss. Acta Ophthalmol Scand. 1996;74(6):639–641.
203 Renner AB, Fiebig BS, Weber BH, et al. Phenotypic variability and long-term follow-up of patients with known and novel PRPH2/RDS gene mutations. Am J Ophthalmol. 2009;147(3):518–530. e1
204 Coco RM, Telleria JJ, Sanabria MR, et al. PRPH2 (Peripherin/RDS) mutations associated with different macular dystrophies in a Spanish population: a new mutation. Eur J Ophthalmol. 2010;20(4):724–732.
205 Nettleship E. Central senile areolar choroidal atrophy. Trans Ophthalmol Soc UK. 1884;4:165–166.
206 Hoyng CB, Pinckers AJ, Deutman AF. Early findings in central areolar choroidal dystrophy. Acta Ophthalmol (Copenh). 1990;68(3):356–360.
207 Carr RE. Central areolar choroidal dystrophy. Arch Ophthalmol. 1965;73:32–35.
208 Mansour AM. Central areolar choroidal dystrophy in a family with pseudoachondroplastic spondyloepiphyseal dysplasia. Ophthalmic Paediatr Genet. 1988;9(1):57–65.
209 Noble KG. Central areolar choroidal dystrophy. Am J Ophthalmol. 1977;84(3):310–318.
210 Gass J. Stereoscopic atlas of macular diseases, 4th ed. St Louis: Mosby; 2001.
211 Fishman GA, Birch DG, Holder GE, et al. Electrophysiologic testing in disorders of the retina, optic nerve, and visual pathway. (Ophthalmology Monograph 2.). 2nd ed. San Francisco: Foundation of the American Academy of Ophthalmology; 2001.
212 Farjo R, Naash MI. The role of Rds in outer segment morphogenesis and human retinal disease. Ophthalmic Genet. 2006;27(4):117–122.
213 Bok D. Cellular mechanisms of retinal degenerations: RPE65, ABCA4, RDS, and bicarbonate transporter genes as examples. Retina. 2005;8 Suppl:S18–S20.
214 Wickham L, Chen FK, Lewis GP, et al. Clinicopathological case series of four patients with inherited macular disease. Invest Ophthalmol Vis Sci. 2009;50(8):3553–3561.
215 Kedzierski W, Lloyd M, Birch DG, et al. Generation and analysis of transgenic mice expressing P216L-substituted rds/peripherin in rod photoreceptors. Invest Ophthalmol Vis Sci. 1997;38(2):498–509.
216 Parodi MB, Iacono P, Cascavilla M, et al. Intravitreal bevacizumab for subfoveal choroidal neovascularization associated with pattern dystrophy. Invest Ophthalmol Vis Sci. 2010;51(9):4358–4361.
217 Sorsby A, Mason ME. A fundus dystrophy with unusual features. Br J Ophthalmol. 1949;33(2):67–97.
218 Weber BH, Vogt G, Pruett RC, et al. Mutations in the tissue inhibitor of metalloproteinases-3 (TIMP3) in patients with Sorsby’s fundus dystrophy. Nat Genet. 1994;8(4):352–356.
219 Lin RJ, Blumenkranz MS, Binkley J, et al. A novel His158Arg mutation in TIMP3 causes a late-onset form of Sorsby fundus dystrophy. Am J Ophthalmol. 2006;142(5):839–848.
220 Lip PL, Good PA, Gibson JM. Sorsby’s fundus dystrophy: a case report of 24 years follow-up with electrodiagnostic tests and indocyanine green angiography. Eye (Lond). 1999;13(Pt 1):16–25.
221 Sivaprasad S, Webster AR, Egan CA, et al. Clinical course and treatment outcomes of Sorsby fundus dystrophy. Am J Ophthalmol. 2008;146(2):228–234.
222 Tabata Y, Isashiki Y, Kamimura K, et al. A novel splice site mutation in the tissue inhibitor of the metalloproteinases-3 gene in Sorsby’s fundus dystrophy with unusual clinical features. Hum Genet. 1998;103(2):179–182.
223 Langton KP, McKie N, Curtis A, et al. A novel tissue inhibitor of metalloproteinases-3 mutation reveals a common molecular phenotype in Sorsby’s fundus dystrophy. J Biol Chem. 2000;275(35):27027–27031.
224 Barbazetto IA, Hayashi M, Klais CM, et al. A novel TIMP3 mutation associated with Sorsby fundus dystrophy. Arch Ophthalmol. 2005;123(4):542–543.
225 Jacobson SG, Cideciyan AV, Bennett J, et al. Novel mutation in the TIMP3 gene causes Sorsby fundus dystrophy. Arch Ophthalmol. 2002;120(3):376–379.
226 Jacobson SG, Cideciyan AV, Regunath G, et al. Night blindness in Sorsby’s fundus dystrophy reversed by vitamin A. Nat Genet. 1995;11(1):27–32.
227 Felbor U, Stohr H, Amann T, et al. A novel Ser156Cys mutation in the tissue inhibitor of metalloproteinases-3 (TIMP3) in Sorsby’s fundus dystrophy with unusual clinical features. Hum Mol Genet. 1995;4(12):2415–2416.
228 Felbor U, Benkwitz C, Klein ML, et al. Sorsby fundus dystrophy: reevaluation of variable expressivity in patients carrying a TIMP3 founder mutation. Arch Ophthalmol. 1997;115(12):1569–1571.
229 Saihan Z, Li Z, Rice J, et al. Clinical and biochemical effects of the E139K missense mutation in the TIMP3 gene, associated with Sorsby fundus dystrophy. Mol Vis. 2009;15:1218–1230.
230 Wijesuriya SD, Evans K, Jay MR, et al. Sorsby’s fundus dystrophy in the British Isles: demonstration of a striking founder effect by microsatellite-generated haplotypes. Genome Res. 1996;6(2):92–101.
231 Gregory-Evans K. What is Sorsby’s fundus dystrophy? Br J Ophthalmol. 2000;84(7):679–680.
232 Apte SS, Olsen BR, Murphy G. The gene structure of tissue inhibitor of metalloproteinases (TIMP)-3 and its inhibitory activities define the distinct TIMP gene family. J Biol Chem. 1995;270(24):14313–14318.
233 Qi JH, Ebrahem Q, Moore N, et al. A novel function for tissue inhibitor of metalloproteinases-3 (TIMP3): inhibition of angiogenesis by blockage of VEGF binding to VEGF receptor-2. Nat Med. 2003;9(4):407–415.
234 Capon MR, Marshall J, Krafft JI, et al. Sorsby’s fundus dystrophy. A light and electron microscopic study. Ophthalmology. 1989;96(12):1769–1777.
235 Knupp C, Chong NH, Munro PM, et al. Analysis of the collagen VI assemblies associated with Sorsby’s fundus dystrophy. J Struct Biol. 2002;137(1–2):31–40.
236 Chong NH, Alexander RA, Gin T, et al. TIMP-3, collagen, and elastin immunohistochemistry and histopathology of Sorsby’s fundus dystrophy. Invest Ophthalmol Vis Sci. 2000;41(3):898–902.
237 Fariss RN, Apte SS, Olsen BR, et al. Tissue inhibitor of metalloproteinases-3 is a component of Bruch’s membrane of the eye. Am J Pathol. 1997;150(1):323–328.
238 Kamei M, Apte SS, Rayborn ME, et al. TIMP-3 accumulation in Bruch’s membrane and drusen in eyes from normal and age-related macular degeneration donors. In: LaVail MM, Anderson RE, Hollyfield JG. Degenerative retinal diseases. New York: Plenum Press; 1997:11–15.
239 Fariss RN, Apte SS, Luthert PJ, et al. Accumulation of tissue inhibitor of metalloproteinases-3 in human eyes with Sorsby’s fundus dystrophy or retinitis pigmentosa. Br J Ophthalmol. 1998;82(11):1329–1334.
240 Langton KP, McKie N, Smith BM, et al. Sorsby’s fundus dystrophy mutations impair turnover of TIMP-3 by retinal pigment epithelial cells. Hum Mol Genet. 2005;14(23):3579–3586.
241 Weber BH, Lin B, White K, et al. A mouse model for Sorsby fundus dystrophy. Invest Ophthalmol Vis Sci. 2002;43(8):2732–2740.
242 Fogarasi M, Janssen A, Weber BH, et al. Molecular dissection of TIMP3 mutation S156C associated with Sorsby fundus dystrophy. Matrix Biol. 2008;27(5):381–392.
243 Holz FG, Haimovici R, Wagner DG, et al. Recurrent choroidal neovascularization after laser photocoagulation in Sorsby’s fundus dystrophy. Retina. 1994;14(4):329–334.
244 Prager F, Michels S, Geitzenauer W, et al. Choroidal neovascularization secondary to Sorsby fundus dystrophy treated with systemic bevacizumab (Avastin). Acta Ophthalmol Scand. 2007;85(8):904–906.
245 Doyne R. A peculiar condition of choroiditis occurring in several members of the same family. Trans Ophthalmol Soc UK. 1899;19:71.
246 Vogt A. Handbuch der gesammten Augenheilkunde. Untersuchungsmethoden, 3rd ed. Berlin: Springer Verlag; 1925.
247 Gass J. Diseases causing choroidal exudative and hemorrhagic localized (disciform) detachment of the retina and pigment epithelium. In: Stereoscopic atlas of macular diseases. St Louis: Mosby; 1987:96–97.
248 Heon E, Piguet B, Munier F, et al. Linkage of autosomal dominant radial drusen (malattia leventinese) to chromosome 2p16–21. Arch Ophthalmol. 1996;114(2):193–198.
249 Stone EM, Lotery AJ, Munier FL, et al. A single EFEMP1 mutation associated with both Malattia Leventinese and Doyne honeycomb retinal dystrophy. Nat Genet. 1999;22(2):199–202.
250 Michaelides M, Jenkins SA, Brantley MA, Jr., et al. Maculopathy due to the R345W substitution in fibulin-3: distinct clinical features, disease variability, and extent of retinal dysfunction. Invest Ophthalmol Vis Sci. 2006;47(7):3085–3097.
251 Sohn EH Patel PJ, MacLaren RE, et al. Responsiveness of choroidal neovascular membranes in patients with R345W mutation in fibulin-3 (Doyne honeycomb retinal dystrophy) to anti-VEGF therapy. Arch Ophthalmol. 2011;129(12):1626–1628.
252 Haimovici R, Wroblewski J, Piguet B, et al. Symptomatic abnormalities of dark adaptation in patients with EFEMP1 retinal dystrophy (Malattia Leventinese/Doyne honeycomb retinal dystrophy). Eye (Lond). 2002;16(1):7–15.
253 Evans K, Gregory CY, Wijesuriya SD, et al. Assessment of the phenotypic range seen in Doyne honeycomb retinal dystrophy. Arch Ophthalmol. 1997;115(7):904–910.
254 Marmorstein LY, Munier FL, Arsenijevic Y, et al. Aberrant accumulation of EFEMP1 underlies drusen formation in Malattia Leventinese and age-related macular degeneration. Proc Natl Acad Sci U S A. 2002;99(20):13067–13072.
255 Holz FG, Owens SL, Marks J, et al. Ultrastructural findings in autosomal dominant drusen. Arch Ophthalmol. 1997;115(6):788–792.
256 Roybal CN, Marmorstein LY, Vander Jagt DL, et al. Aberrant accumulation of fibulin-3 in the endoplasmic reticulum leads to activation of the unfolded protein response and VEGF expression. Invest Ophthalmol Vis Sci. 2005;46(11):3973–3979.
257 Fu L, Garland D, Yang Z, et al. The R345W mutation in EFEMP1 is pathogenic and causes AMD-like deposits in mice. Hum Mol Genet. 2007;16(20):2411–2422.
258 Marmorstein LY, McLaughlin PJ, Peachey NS, et al. Formation and progression of sub-retinal pigment epithelium deposits in Efemp1 mutation knock-in mice: a model for the early pathogenic course of macular degeneration. Hum Mol Genet. 2007;16(20):2423–2432.
259 Blackburn J, Tarttelin EE, Gregory-Evans CY, et al. Transcriptional regulation and expression of the dominant drusen gene FBLN3 (EFEMP1) in mammalian retina. Invest Ophthalmol Vis Sci. 2003;44(11):4613–4621.
260 Lefler WH, Wadsworth JA, Sidbury JB, Jr. Hereditary macular degeneration and amino-aciduria. Am J Ophthalmol. 1971;71(1 Pt 2):224–230.
261 Frank HR, Landers MB, 3rd., Williams RJ, et al. A new dominant progressive foveal dystrophy. Am J Ophthalmol. 1974;78(6):903–916.
262 Small KW. North Carolina macular dystrophy: clinical features, genealogy, and genetic linkage analysis. Trans Am Ophthalmol Soc. 1998;96:925–961.
263 Reichel MB, Kelsell RE, Fan J, et al. Phenotype of a British North Carolina macular dystrophy family linked to chromosome 6q. Br J Ophthalmol. 1998;82(10):1162–1168.
264 Pauleikhoff D, Sauer CG, Muller CR, et al. Clinical and genetic evidence for autosomal dominant North Carolina macular dystrophy in a German family. Am J Ophthalmol. 1997;124(3):412–415.
265 Small KW, Puech B, Mullen L, et al. North Carolina macular dystrophy phenotype in France maps to the MCDR1 locus. Mol Vis. 1997;3:1.
266 Rabb MF, Mullen L, Yelchits S, et al. A North Carolina macular dystrophy phenotype in a Belizean family maps to the MCDR1 locus. Am J Ophthalmol. 1998;125(4):502–508.
267 Kim SJ, Woo SJ, Yu HG. A Korean family with an early-onset autosomal dominant macular dystrophy resembling North Carolina macular dystrophy. Korean J Ophthalmol. 2006;20(4):220–224.
268 Fetkenhour CL, Gurney N, Dobbie JG, et al. Central areolar pigment epithelial dystrophy. Am J Ophthalmol. 1976;81(6):745–753.
269 Small KW, Killian J, McLean WC. North Carolina’s dominant progressive foveal dystrophy: how progressive is it? Br J Ophthalmol. 1991;75(7):401–406.
270 Small KW. North Carolina macular dystrophy, revisited. Ophthalmology. 1989;96(12):1747–1754.
271 Small KW, Weber JL, Hung WY, et al. North Carolina macular dystrophy: exclusion map using RFLPs and microsatellites. Genomics. 1991;11(3):763–766.
272 Small KW, Weber JL, Roses A, et al. North Carolina macular dystrophy is assigned to chromosome 6. Genomics. 1992;13(3):681–685.
273 Small KW, Udar N, Yelchits S, et al. North Carolina macular dystrophy (MCDR1) locus: a fine resolution genetic map and haplotype analysis. Mol Vis. 1999;5:38.
274 Michaelides M, Johnson S, Tekriwal AK, et al. An early-onset autosomal dominant macular dystrophy (MCDR3) resembling North Carolina macular dystrophy maps to chromosome 5. Invest Ophthalmol Vis Sci. 2003;44(5):2178–2183.
275 Rosenberg T, Roos B, Johnsen T, et al. Clinical and genetic characterization of a Danish family with North Carolina macular dystrophy. Mol Vis. 2010;16:2659–2668.
276 Small KW, Voo I, Flannery J, et al. North Carolina macular dystrophy: clinicopathologic correlation. Trans Am Ophthalmol Soc. 2001;99:233–237. discussion 7–8
277 Mahajan VB, Russell SR, Stone EM. A new macular dystrophy with anomalous vascular development, pigment spots, cystic spaces, and neovascularization. Arch Ophthalmol. 2009;127(11):1449–1457.
278 Deutman AF, Pinckers AJ, Aan de Kerk AL. Dominantly inherited cystoid macular edema. Am J Ophthalmol. 1976;82(4):540–548.
279 Fishman GA, Goldberg MF, Trautmann JC. Dominantly inherited cystoid macular edema. Ann Ophthalmol. 1979;11(1):21–27.
280 Kremer H, Pinckers A, van den Helm B, et al. Localization of the gene for dominant cystoid macular dystrophy on chromosome 7p. Hum Mol Genet. 1994;3(2):299–302.
281 Hogewind BF, Pieters G, Hoyng CB. Octreotide acetate in dominant cystoid macular dystrophy. Eur J Ophthalmol. 2008;18(1):99–103.
282 Sneed SR, Sieving PA. Fenestrated sheen macular dystrophy. Am J Ophthalmol. 1991;112(1):1–7.
283 Daily MJ, Mets MB. Fenestrated sheen macular dystrophy. Arch Ophthalmol. 1984;102(6):855–856.
284 Slagsvold JE. Fenestrated sheen macular dystrophy. A new autosomal dominant maculopathy. Acta Ophthalmol (Copenh). 1981;59(5):683–688.
285 O’Donnell FE, Jr., Welch RB. Fenestrated sheen macular dystrophy. A new autosomal dominant maculopathy. Arch Ophthalmol. 1979;97(7):1292–1296.
286 McAvoy CE, Silvestri G. Retinal changes associated with type 2 glomerulonephritis. Eye (Lond). 2005;19(9):985–989.
287 D’Souza Y, Short CD, McLeod D, et al. Long-term follow-up of drusen-like lesions in patients with type II mesangiocapillary glomerulonephritis. Br J Ophthalmol. 2008;92(7):950–953.
288 Leys A, Vanrenterghem Y, Van Damme B, et al. Sequential observation of fundus changes in patients with long standing membranoproliferative glomerulonephritis type II (MPGN type II). Eur J Ophthalmol. 1991;1(1):17–22.
289 Leys A, Vanrenterghem Y, Van Damme B, et al. Fundus changes in membranoproliferative glomerulonephritis type II. A fluorescein angiographic study of 23 patients. Graefes Arch Clin Exp Ophthalmol. 1991;229(5):406–410.
290 Ulbig MR, Riordan-Eva P, Holz FG, et al. Membranoproliferative glomerulonephritis type II associated with central serous retinopathy. Am J Ophthalmol. 1993;116(4):410–413.
291 Kim RY, Faktorovich EG, Kuo CY, et al. Retinal function abnormalities in membranoproliferative glomerulonephritis type II. Am J Ophthalmol. 1997;123(5):619–628.
292 O’Brien C, Duvall-Young J, Brown M, et al. Electrophysiology of type II mesangiocapillary glomerulonephritis with associated fundus abnormalities. Br J Ophthalmol. 1993;77(12):778–780.
293 Duvall-Young J, MacDonald MK, McKechnie NM. Fundus changes in (type II) mesangiocapillary glomerulonephritis simulating drusen: a histopathological report. Br J Ophthalmol. 1989;73(4):297–302.
294 Mullins RF, Aptsiauri N, Hageman GS. Structure and composition of drusen associated with glomerulonephritis: implications for the role of complement activation in drusen biogenesis. Eye (Lond). 2001;15(Pt 3):390–395.
295 Mullins RF, Russell SR, Anderson DH, et al. Drusen associated with aging and age-related macular degeneration contain proteins common to extracellular deposits associated with atherosclerosis, elastosis, amyloidosis, and dense deposit disease. FASEB J. 2000;14(7):835–846.