[level-membership-for-dermatology-category]
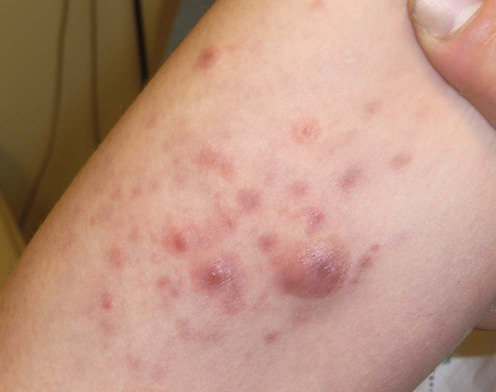
Lymphomatoid papulosis
Rachel S. Klein, Elisha Singer, Jacqueline M. Junkins-Hopkins, Carmela C. Vittorio, Alain H. Rook and Ellen J. Kim
Specific investigations



CD30+ cutaneous lymphoproliferative disorders: the Stanford experience in lymphomatoid papulosis and primary cutaneous anaplastic large cell lymphoma.
Liu HL, Hoppe RT, Kohler S, Harvell JD, Reddy S, Kim YH. J Am Acad Dermatol 2003; 49: 1049–58.
The higher association with malignancy may represent selection bias.
First-line therapies
 Therapy not required
Therapy not required PUVA
PUVA Low-dose methotrexate
Low-dose methotrexate Topical corticosteroids
Topical corticosteroidsEORTC, ISCL, and USCLC consensus recommendations for the treatment of primary cutaneous CD30-positive lymphoproliferative disorders: lymphomatoid papulosis and primary cutaneous anaplastic large-cell lymphoma.
Kempf W, Pfaltz K, Vermeer M, Cozzio A, Ortiz-Romero P, Bagot M, et al. Blood 2011; 118: 4024–35.
 Topical mechlorethamine (nitrogen mustard)
Topical mechlorethamine (nitrogen mustard) Topical carmustine
Topical carmustine Topical bexarotene
Topical bexaroteneThird-line therapies
 Oral bexarotene
Oral bexarotene Recombinant interferon
Recombinant interferon Excimer laser
Excimer laser Radiotherapy
Radiotherapy Topical methotrexate
Topical methotrexate Imiquimod cream
Imiquimod cream SGN-30
SGN-30
[/level-membership-for-dermatology-category][not-level-membership-for-dermatology-category]
Lymphomatoid papulosis
Rachel S. Klein, Elisha Singer, Jacqueline M. Junkins-Hopkins, Carmela C. Vittorio, Alain H. Rook and Ellen J. Kim
Specific investigations
CD30+ cutaneous lymphoproliferative disorders: the Stanford experience in lymphomatoid papulosis and primary cutaneous anaplastic large cell lymphoma.
Liu HL, Hoppe RT, Kohler S, Harvell JD, Reddy S, Kim YH. J Am Acad Dermatol 2003; 49: 1049–58.
[/not-level-membership-for-dermatology-category]
