[level-membership-for-hematology-oncology-and-palliative-medicine-category]
B- and T-Lymphocyte Development
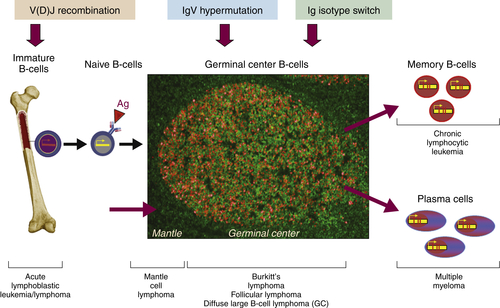
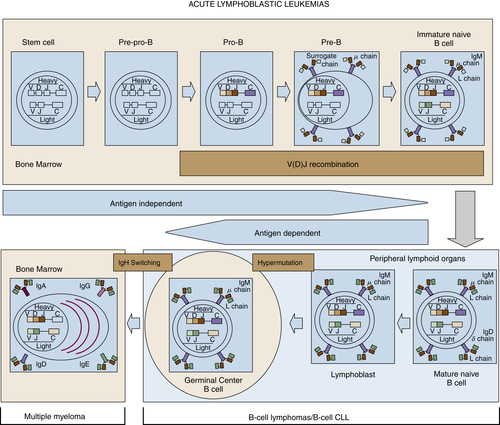
The ontogeny of B cells is complex and proceeds through a series of developmental and genetic steps, leading to the creation of unique B cells carrying unique B-cell receptors, capable of identifying unique antigen (
Figure 29-3 ). These developmental and genetic steps are defined by changes in the genome at the antibody loci. B lymphocytes play a fundamental role in the humoral branch of the adaptive immune system and can be distinguished from other types of lymphocytes by the panoply of cell surface markers (CD19, CD20, CD22, CD23) and the presence of the B-cell receptor (BCR), which itself is a surface immunoglobulin. Undifferentiated and immature B cells are produced in the intramedullary compartment of the bone. The antibodies produced by individual B cells are composed of two identical light chains (κ or λ), composed of two segments called the
V and J segments, and one heavy chain (M, D, A, G) composed of three segments called the
V, D, and J segments, where V denotes the variable region that comprises the terminal Fab portion of the antibody.
1 At the genetic level, early lymphocytes in the bone marrow undergo a process referred to as
VDJ recombination (see
Figure 29-2), which is the mechanism by which B cells produce immunoglobulin diversity. Once B lymphocytes undergo VDJ recombination, they exit the bone marrow, where they then hone to secondary lymphoid organs such as the lymph node. There they undergo highly controlled mutagenesis, in a process referred to as
somatic hypermutation (SMH). SMH plays a crucial role in the affinity maturation of the humoral response, by mediating the introduction of discrete random mutations in the loci of the genes encoding the variable (V) regions of the immunoglobulin.
2 Once engaged by antigen, the subsequent proliferation of the B cell undergoes an incredibly high rate of somatic mutation that is at least 100,000- to 1 millionfold higher than the normal rate of mutation across the genome. This highly regulated mutagenesis, mediated initially by activation-induced cytidine deaminase (AID), introduces single base substitutions predominantly at hotspots in the DNA called
hypervariable regions.
2 Paradoxically, it is this directed hypermutation of the BCR variable regions that allows for the selection of B cells with an enhanced capability to identify and bind specific foreign antigens, which, when dysregulated or uncontrolled, creates an opportunity for aberrant somatic hypermutation, leading to lymphomagenesis. This potential for any error, or error-prone mutagenesis of B lymphocytes, can create the background for malignant transformation.
In contrast, T lymphocytes play a central role in cell-mediated immunity and can be distinguished from other lymphocytes by the expression of their own unique set of surface proteins (e.g., CD3, CD4, CD8) and the T-cell receptor (TCR). T lymphocytes typically mature in the thymus, in contrast to the germinal center for B lymphocytes. In the thymus, T lymphocytes expand through multiple series of cell division to produce a large population of more immature cells. Initially, these cells do not express CD4 or CD8 and are referred to as double-negative thymocytes.
3 As they differentiate into a specific subset of T lymphocytes, they eventually become positive for both CD4 and CD8 (that is, double positive). In their final step of thymic maturation they emerge as single positive T lymphocytes, including CD4-positive/CD8-negative T-helper cells or CD4-negative/CD8-positive cytotoxic T cells. The TCR recognizes antigen bound to major histocompatibility complex (MHC). Engagement of the TCR by antigen incites a number of downstream events important to the
lymphocytes role in mediating cellular immunity. The generation of the TCR is quite similar to that discussed earlier for the BCR. Composed of four chains and existing as a dimeric structure (alpha pairing with the beta chain or the gamma chain pairing with the delta chain), unique TCR is produced through VJ (the alpha and gamma chains) or VDJ (the beta and delta chains) recombination. Like the heavy and light chains comprising the BCR, it is the unique combination of these specific regions that accounts for the diversity of the TCR.
3
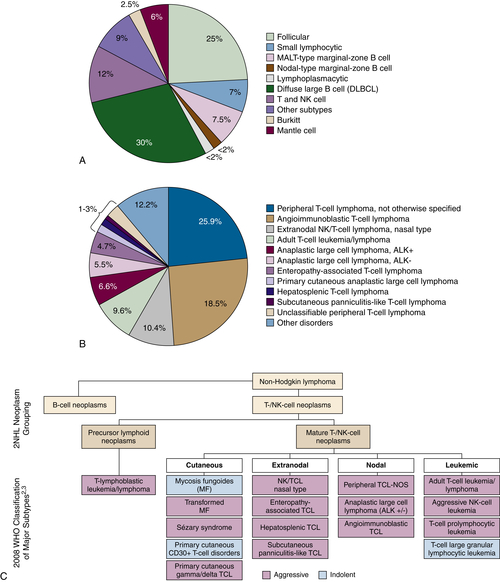
Figure 29-1 (A) Relative incidence of select B- and T-cell lymphomas. (B) Relative incidence of select T-cell lymphomas. A survey of 1314 peripheral T-cell and natural killer/T-cell leukemia cases from 22 sites around the world evaluated the histopathology of previously untreated patients consecutively diagnosed between 1990 and 2002. PTCL-NOS was the most common subtype at 25.9%. Anaplastic large cell lymphoma (ALCL) ALK+/−, angioimmunoblastic lymphoma, adult T-cell leukemia/lymphoma, and natural killer/T-cell lymphoma were also common subtypes. (C) Classification of the mature T-cell lymphoma (WHO). PTCL refers to a heterogeneous group of aggressive T- and NK-cell lymphomas, characterized by the involvement of mature (thymic) T or NK cells. The term peripheral in PTCL does not refer to anatomic sites. T- and NK-cell lymphomas may be characterized as cutaneous, nodal, extranodal, or leukemic disseminated. Data from Armitage JO et al. Ann Oncol. 2004;15:1447-1449; Rodriguez J et al. Crit Rev Oncol Hematol. 2008; Swerdlow SH et al., WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; 2008. Data in B from Armitage J et al. J Clin Oncol. 2008;26:4124-4130.
Figure 29-2 Schematic representation of the ontogeny of B-lymphocytes and select lymphoma relative to their sites of origin.
Although complex, the ontogeny of B and T lymphocytes provides the basis for thinking about the diversity of different forms of non-Hodgkin and Hodgkin lymphoma, and in particular the cell of origin (see
Figure 29-3). In the case of the lymphoid malignancies, identifying the cell of origin goes well beyond the identification of the lymphocyte and is ascribed to the discrete step in development at which the clonal expansion is thought to occur. Increasingly, the understanding of immunohistochemical staining and cytogenetics has allowed for a description of the natural ontogeny of lymphocytes and, by extension, the more accurate identification of the cell of origin, leading to a more biologically relevant classification of different subtypes of lymphomas (
Table 29-2 ).
Pathogenesis of Diffuse Large B-Cell Lymphoma
Diffuse large B-cell lymphoma (DLBCL) comprises a group of aggressive lymphomas with very heterogeneous clinicopathologic and molecular genetic features. The current 2008 World Health Organization (WHO) classification
4 defines DLBCL as a diffuse growth of neoplastic large B-lymphoid cells with a nuclear size equal to or exceeding normal macrophage nuclei or more than twice the size of a normal lymphocyte. DLBCL is the most common subtype of lymphoma and accounts for 30% to 40% of adult NHL.
5 The 2008 WHO classification recognizes a large number of DLBCL subgroups primarily based on their distinct morphologic, biologic, immunophenotypic, or clinical features. Despite revisions to the classification, a significant proportion of large-cell lymphomas remain biologically heterogeneous, not fitting into any specific DLBCL subgroup. These are typically defined as DLBCL, not otherwise specified (DLBCL-NOS). DLBCL-NOS is a diagnosis of exclusion, not corresponding to one of the specific subtypes. It is still the most common type of lymphoma, accounting for 25% to 30% of NHLs.
5
Figure 29-3 Stages of B-cell development and correlation with different lymphoid diseases. CLL, Chronic lymphocytic leukemia.
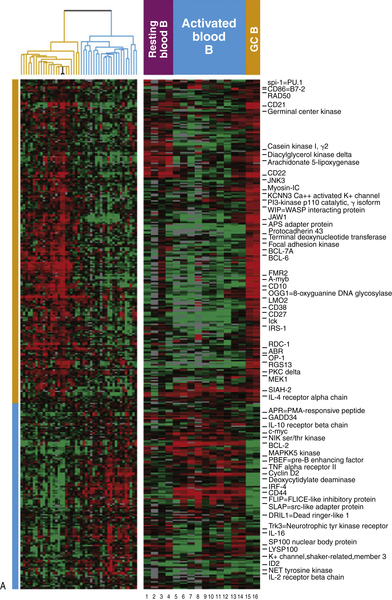
The focus in recent years has shifted toward the identification of molecular alterations and specific pathogenetic pathways leading to transformation in DLBCL. Gene expression profiling (GEP) of DLBCLs has identified at least two major molecular subtypes, which correlate with prognosis and may have relevance for treatment based on the dominant signaling pathways. DLBCL could be classified by GEP as resembling either germinal center B cells (GCBs) or activated B cells (ABCs), establishing a putative “cell of origin”
5 (
Figure 29-4 ). Because GEP is technically difficult and therefore cannot be performed in every case for diagnostic purposes, various algorithms based on immunohistochemical profiles have been proposed as surrogates of the GEP.
5,6 Although the correspondence is not precise, similar prognostic correlations can be drawn with immunohistochemically defined subgroups. These immunohistochemical algorithms have facilitated risk stratification of DLBCL patients and DLBCL research using archival materials.
5 The ABC and GCB DLBCL subtypes, originally formulated based on a cell-of-origin model, have more recently been shown to be characterized by the different pathways of cellular transformation and oncogenesis. Although substantial progress has been made toward molecular subclassification of DLBCLs, the translation to effective treatment strategies is only now beginning to be explored.
5 Next-generation sequencing (NGS) platforms have evolved to provide an accurate and comprehensive means for the detection of molecular mutations and will likely contribute to a more sophisticated understanding of DLBCL biology and, it can be hoped, more biologically relevant treatment.
7
Table 29-2
Characteristic Immunophenotypic and Genetic Features of Selected Lymphoma Subtypes
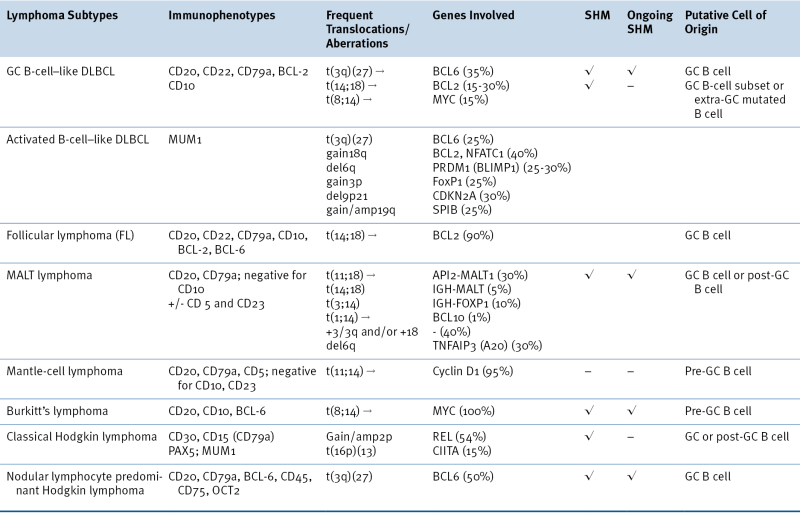
DLBCL, Diffuse large B-cell lymphoma; GC, germinal center; MALT, extranodal marginal zone B-cell lymphoma of mucosa-associated lymphoid tissues; SHM, somatic hypermutation.
Although increased understanding of DLBCL based on cell of origin is emerging, it is clear that the dysregulated pathways across this entity are often overlapping, providing redundancy to promote growth and prevent cell death signals. These signaling pathways are often necessary for the development and differentiation of normal memory B cells and plasma cells and are coopted during malignant transformation.
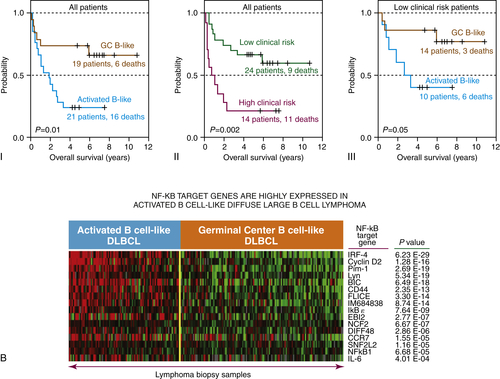
For example, in ABC-derived DLBCL, most of the deranged signaling pathways converge to aberrantly activate the NFκB pathway, leading to evasion of cell death and resistance to chemotherapy (see
Figure 29-4,
B). Activation of this molecular pathway translates into poor outcomes for patients with ABC-DLBCL compared to GC-DLBCL, with 5-year overall survival of 40% versus 60%, respectively. Constitutive activation of NFκB is complex, involving both “classical” and “alternative” pathways, and can be regulated by many different mechanisms. Derangements leading to activation of NFκB can be attributed to a number of different molecular lesions, including tonic signaling of the BCR. BCR signaling is essential to B-cell development, antigen selection, and humoral immunity. Aberrations in BCR signaling, including tonic signaling that occurs in the absence of antigenic stimulation, can lead to uncontrolled B-cell growth and survival.
BCR signaling, whether it be in a normal or malignant B-cells, leads to the formation of the CBM complex consisting of
CARD11,
BCL10, and
MALT1. This complex leads to activation of NFκB through direct activation of NFκB and inhibition of negative NFκB regulators. Gain-of-function somatic mutations of the coiled-coil domain of CARD11 are found in nearly 10% of ABC-DLCBL and 16% of primary CNS lymphoma (an ABC variant). This mutant CARD11 leads to initiation of the NFκB cascade via IKKβ signaling in cooperation with casein kinase 1α (CK1α) and MALT1. MALT1 can also augment NFκB signaling through cleavage of two negative NFκB regulators, namely, A20 and CYLD.
TNFAIP3, the gene encoding A20, can also be inactivated through mutations, deletions, or epigenetic silencing in ABC-DLCBL. Mutations of other negative regulators of NFκB, such as
IKBKA encoding the inhibitor IkBα, lead to uninhibited translocation of NFκB to the nucleus and increased transcription of NFκB-dependent genes. Interestingly, linkage of this intrinsic ABC biology to B-cell receptor signaling may offer an interesting opportunity to affect the adverse prognostic features of this DLBCL phenotype.
Figure 29-4 (A) Classification of diffuse large B-cell lymphoma based on cell of origin. The left panel represents hierarchical clustering of the genes selectively expressed in GC-derived DLBCL (yellow) and ABC-derived DLBCL (blue). The right panel compares normal lymphocytes from blood and lymph nodes in various states of B-cell activation.
(B) Gene expression profile for the GC and ABC subtypes of DLBCL reveal that the ABC phenotype is enriched for NFκB-regulated genes, while the GC phenotype is enriched for Bcl-6-regulated genes. Gene expression profiling has revealed three subtypes of DLBCL, two of which include the germinal center subtype (GC) and the activated B-cell subtype (ABC). These molecular phenotypes override the prognostic impact of the International Prognostic Index (IPI). Courtesy Wyndham Wilson. Data in A from Alizadeh et al. Nature 2000;403:503-511.
In addition to the BCR-CBM effects on NFκB regulation, the BCR can activate NFκB through a number of alternative mechanisms. On antigenic stimulation or tonic dysregulation, the BCR heavy chains are coupled to cell surface markers CD79a and CD79b. This leads to phosphorylation of immunoreceptor tyrosine-based activation motifs (ITAM) by LYN, FYN, and BLK, which then recruit SYK, engaging the NFκB, PI3K, NFAT MAP kinase and RAS signaling pathways. Currently, an inhibitor of SYK is under investigation as a therapeutic target in those DLBCLs known to be addicted to BCR signaling. Substitutions and deletions of the CD79a and CD79b ITAMs are present in more than 20% of ABC-DLBCL and lead to a gain of function of the BCR signaling pathway. These mutants evade negative feedback by LYN and also allow for increased cell surface localization of the BCR.
Tonic BCR signaling also recruits Bruton’s tyrosine kinase (BTK) via the PI3K pathway. This tends to occur in ABC-DLBCL that do not possess CARD11 mutations. BTK is required for the survival of wild-type CARD11 ABC-DLBCL and knockdown of its components or effectors SYK, BLNK, PLCγ2, PI3Kδ, PKCβ abrogates NFκB signaling. BTK is activated by LYN or SYK, and on activation initiates signaling of the classical NFκB pathway. In addition, BTK forms a complex with BLNK, which, through a series of events that activates CARD11, contributes further to activation of the NFκB pathway. BTK inhibitors have demonstrated promising activity in patients with ABC-DLBCL and other lymphomas with aberrant BCR signaling, such as CLL.
All known ABC-DLBCL cell lines and 30% of tissue samples from patients with ABC DLBCL harbor MYD88
gain-of-function mutations in the toll-interleukin receptor (TIR) domain. MYD88 is an adaptor protein that interacts with IRAK4 and IRAK1 to activate both the NFκB and interferon pathways through TRAF6. Mutant MYD88 promotes secretion of the cytokines IL6 and IL10 activating the JAK/STAT pathway. This potentiates NFκB through a positive feedback loop.
Many of the mutations just discussed are expressed in tandem and are restricted to the ABC-DLBCL variant over GCB-derived DLBCL. As testament to the redundancy in these signaling pathways, knockdown of parallel pathways in cell lines or mouse models has demonstrated synergistic cytotoxicity in ABC-DLBCL, compared to the knockdown of singular elements in a pathway. The clarification of this underlying biology has led to the identification of several new drugs potentially affecting these specific pathways, including inhibitors of Syk, BTK, and CD79a and b, and proteasome inhibitors that inhibit degradation of IkB, leading to inhibition of NFκB.
Although there is overlap in the pathogenesis of GC- and ABC-derived DLBCL, there are unique features of both that may create opportunities to affect the discrete natural history of these subtypes rationally. For example, transcription factors, oncogenes, and epigenetic alterations are often responsible for driving proliferation and survival in GC-DLBCL. BCL6, an oncogene, is a transcriptional repressor considered the hallmark of GC-DLBCL, and acts as a master regulator of the germinal center reaction. Although physiologically active in normal germinal center B cells promoting somatic hypermutation (SMH) and class switch recombination, this pathway is co-opted in malignant lymphomas to enforce the “damage phenotype” of GC-DLBCL. Dysregulation through point mutations and translocations of the 3q27 locus lead to constitutive activation of this transcription factor in over 50% of DLBCL lymphomas. Specifically, mutations in the BCL6 autoregulatory domains upstream from the promoter regions via somatic hypermutation and AID result in lost feedback loop inhibition from Bcl6, IRF4, and STAT5. This phenomenon leads to the GC-DLBCL subtype. In addition, the Bcl6 BTB domain recruits the co-repressors NCOR, BCOR, and SMRT, along with HDACs to repress genes involved in the DNA damage response such as ATR, p53, CHK1, p21, and p27. This repression leads to unchecked cell cycle progression in the face of inhibited apoptosis and permissive DNA damage. Furthermore, the Bcl6 RD2 domain recruits MTA3 to repress PRDM1, which encodes Blimp-1, the master regulator of plasma cell development, trapping centroblasts within the germinal center. Interestingly, Bcl6 inhibits both MYC and BCL2, but in double-hit lymphomas, where these two oncogenes are overexpressed by translocations, they overcome repression by Bcl6, leading to even more aggressive variants of DLBCL.
Another target gene of Bcl6 is IRF4, a transcription factor that facilitates exit from the germinal center, leading to plasmacytic differentiation. Bcl6 silencing of IRF4 further inhibits transcription of Blimp-1, which would retain cells in the germinal center. IRF4 may also be inhibited by translocations in GC-DLBCL (predominantly pediatric cases). In these cases, IRF4 may serve as an oncogene. Nonstructural aberrations not interfering with IRF4 function have been found to be required for survival of ABC-DLBCL. The effects of IRF4 in ABC and GC-DLBCL are clearly different, and this likely contributes to their classification by cell of origin. One mechanism proposed is that IRF4 works as a positive feedback loop involving CARD11.
Although Bcl6 translocations occur in GC-DLBCL (10%), translocations of the 3q27 locus involving the BCL6 gene that lead to arrest of cells in the plasmablastic stage of development can be seen in 30% of ABC-DLBCL. These translocations have promiscuous partners and ultimately lead to silencing of PRDM1. The tumor suppressor functions of PRDM1 may be silenced by truncation, deletion, mutation, or epigenetic modifications as well.
Transcriptional control has also been influenced by mutations in epigenetic regulators. CREBBP and EP300 encode histone acetyl-transferases (HATs) that have classically been known to modify chromatin condensation and, by extension, transcription. It is now known that these enzymes also influence the activity of key modulators of DLBCL such as p53, NFκB, and Bcl6. Acetylation can act either as an activating posttranslational modification (p53), or as an inhibitory posttranslational modification (Bcl6). Nonoverlapping mutations of CREBBP (40%) and EP300 (10%) are found in GC-DLBCL. The inactivating mutations in these two genes lead to the activation of Bcl6 and inhibition of p53 by impaired acetylation. Reversal of this effect may be accomplished by therapeutic alteration of these posttranslational modifications pharmacologically with pan-Class1-2 histone deacetylase (HDAC) inhibitors.
GC-DLBCL has also been found to have mutations in the EZH2 component of the polycomb repression complex-2 (PRC2). This complex has histone methyltransferase activity and primarily trimethylates histone H3 on lysine 27 (i.e., H3K27me3), a mark of transcriptionally silent chromatin. In order to produce the pathologic effect, mutations are required to be heterozygous, allowing the wild-type allele to methylate H3K27, followed by the uncontrolled methylation mediated by the enzyme encoded by the mutant allele. This action leads to hypermethylated CpG islands, which in turn attracts methyl-CpG binding domain proteins (MBDP: MBD1, MBD2, MBD3, MeCP2 i Kaiso) to recruit HDACs, leading chromatin condensation and transcriptional silencing. Hypomethylating drugs have been shown in preclinical models to have therapeutic potential
and synergize with HDAC inhibitors to restore normal methylation and acetylation patterns on histones.
Although only recently described, the role of microRNAs in DLBCL lymphomagenesis has begun to emerge. MiRNAs can have both oncogenic and tumor suppressor properties. The miR-17-92 family, also known as oncomir-1, has been found to be amplified specifically in GC-DLBCL, which leads to increased expression of MYC and downregulation of BIM and p21. miR-17-92 also has an inverse relationship with p53, each inhibiting the other.
Molecular Pathogenesis of Follicular Lymphoma
Follicular lymphoma is a B-cell lymphoma derived from germinal center B cells with rearranged heavy- and light-chain genes. This derivation is depicted by the immunoarchitectural features of follicular lymphoma, particularly reflected in the expression of a germinal center immunohistochemical profile as well as a follicular growth pattern. Cytogenetically, about 90% of follicular lymphoma cases harbor the t(14;18) or variant B-cell CLL/lymphoma 2 (BCL2) rearrangements with immunoglobulin kappa or lambda on chromosome 2 and 22, respectively. Although centrocytes and centroblasts are regarded as the particular cell of origin, the lymphomatous counterpart demonstrates ongoing somatic hypermutations, which is of particular interest as it decreases the detection rate of follicular lymphoma by standard polymerase chain reaction (PCR) testing.
The most prominent cytogenetic abnormality in follicular lymphoma involves t(14;18) with rearrangement of BCL2. Normal BCL2 protein has an antiapoptotic effect by affecting mitochondrial permeability and release of cytochrome c. During the translocation of the IgH regulatory apparatus on chromosome 14 and the bcl-2 gene on chromosome 18, BCL2 is juxtaposed to immunoglobulin heavy-chain gene (IgH) sequences, including enhancer sequences, which brings BCL2 under the influence of the immunoglobulin promoter region. This leads to deregulated expression/overexpression of a functional protein, promoting increased cell survival and neoplastic transformation.
The rearrangement can occur in different breakpoint regions as noted:
• 65% to 70% occur in the major breakpoint region (MBR) located in an untranslated region of the last exon (exon 3).
• 20% occur at the 3′ end of the BCL2 gene or 5′ to the minor cluster region (mcr).
• 10% occur in the mcr, which is about 30 kb outside of the BCL2 gene.
Although additional breakpoints are being studied, efforts to associate these different breakpoints with different clinical characteristics or clinical outcome have been unrevealing.
8 Testing for the
BCL2 rearrangement can be determined by conventional cytogenetic karyotyping, fluorescence in situ hybridization (FISH), or PCR. Although conventional cytogenetic karyotyping detects the majority of cases with
BCL2 rearrangement, independent of the partner gene, the limiting factor with this approach remains the availability of viable tissue. In comparison, FISH can be performed on paraffin-embedded tissue and detects the translocation in up to 90% of cases. Lower detection rates have been reported depending on the ethnic group studied—such as in Japanese patients, where the frequency of the t(14:18) has been shown less frequently compared to patients from Western countries.
9 Of interest, PCR testing has been proven to be less valuable, as most PCR assays contain primers targeting the MBR and mcr regions, which decreases the detection rate to 60%.
10 Although increased detection rates can be achieved by adding more primers to the 5′ mcr and 3′ MBR breakpoint regions, targeting incomplete DH-JH rearrangements or mutations of the primer-binding sites, false-negative PCR results are common and still occur with alternate breakpoints as mentioned earlier.
11 Therefore, up-to-date FISH analysis remains the preferred testing modality.
11 The absence of
BCL2 translocation using the routinely available methods is described in a subset of follicular lymphomas, particularly primary cutaneous and pediatric follicular lymphoma. Further complicating the matter is the fact that multiple follicular lymphoma cases have been shown to increase their
BCL2 expression through alternate mechanisms, as shown in cases with an addition of 18q or trisomy 18.
12 Furthermore, the frequency of
BCL2 rearrangement is not only variable in different subsets of follicular lymphomas, but also differs as a function of the grade of the follicular lymphoma. More than 80% of low-grade follicular lymphoma cases possess the translocation, whereas higher grade follicular lymphoma cases harbor the translocation with a frequency of 70% and less than 15% in grades 3A and 3B, respectively.
13 However, a positive result for
BCL2 rearrangement demands additional studies, as the rearrangement is not specific and has been identified in other subtypes of lymphomas as well as in healthy individuals and in benign follicular hyperplasia.
14 An additional random locus of genetic alteration in follicular lymphoma includes alterations of BCL6 at 3q27. Grade 3B follicular lymphomas frequently have rearrangements of this locus similar to diffuse large B-cell lymphoma.
13,15 The Bcl6 protein is a zinc-finger protein that acts as a repressor of IRF-4/MUM-1 transactivating ability, as well as influencing cell cycle control and proapoptotic
genes. The major breakpoint region of this translocation is within the 5′ first exon and a portion of the first intron of the Bcl-6 gene. In addition to translocation events, the coding region of the Bcl6 gene is also altered by somatic mutation.
Because t(14;18) can be identified in healthy individuals, the translocation is thought to be an early event in B-cell development, neither necessary nor sufficient for lymphomagenesis in B cells. The
BCL2 rearrangement is thought to enhance the life span of the neoplastic cell, which in return retains the capability to develop additional genetic defects. So far a myriad of additional abnormalities have been described, including gains of 1q, 2p, 7, 8q, 12q, 18q, and X and losses of 1p, 6q, 10q, 13q, and 17p.
16 After detection of these additional abnormalities, prognostic factors have been identified, including gains of 1q, 12, and X, as well as losses of 1p, 17p, and 17q, all considered late events with a poor prognosis or associated with histologic transformation.
16 Transformation to diffuse large B-cell lymphoma is a relatively common event, occurring in 25% to 35% of follicular lymphoma cases.
16 To date, large-cell transformation has been linked to gains of 7, 12q, and X; losses of 4q, 13q, and 17p;
MYC deregulation; and inactivation of
TP53 and
CDKN2A. Although
MYC deregulation has only been occasionally described in follicular lymphoma, it harbors a particularly poor diagnosis.
17 Newer diagnostic modalities such as comparative genomic hybridization (CGH) have firmly established that additional chromosomal abnormalities, such as gains in 12p and 18p in more than 10% of cases and deletions of 3q, 9p, and 11q in less than 10% of cases, are associated with particularly poor prognostic factors. Although these deletions involve the
CDKN2A and
CDKN2B loci as well as the LIM domain and have been partially associated with decreased survival, the overall significance of these findings has yet to be determined.
18 Next-generation sequencing has been used to elucidate the mutational landscape of follicular lymphoma. With this modality it was discovered that 89% of cases harbored MLL2 mutations, 33% of cases showed CREBBP mutations, and 13% of cases depicted MEF2B mutations. E300 mutations were identified in 9% of cases, EZH2 mutations in 7% of cases, and the TBL1XR1/TP63 fusion gene was identified at a very low incidence. So far mainly the presence of EZH2 is of interest, as this particular mutation supports the hypothesis that the polycomb repressor complex-2 orchestrates the pathogenesis in a subset of germinal center–derived lymphomas, but in general the implications of these findings are still under investigation.
19 Additional factors thought to be involved in the pathogenesis of follicular lymphoma include the nature of the microenvironment. In particular, the role of T cells and dendritic cells appears to be of substantial importance. GEP of the microenvironment has identified two distinct signatures. These signatures have been categorized as immune response 1 (IR1) and immune response 2 (IR2). Whereas immune response 1 (IR1) shows increased expression of T-cell genes and the macrophage genes
TNFSF13B and
ACTN1, immune response 2 (IR2) shows increased expression of follicular dendritic cell genes and different macrophage genes. Immune response 1 has been linked to a more favorable prognosis, but immune response 2 has been linked to an unfavorable prognosis.
16 To date, these genetic changes have not yet led to the tailoring of specific therapies for specific genetic subsets of the disease.
Molecular Pathogenesis of Marginal Zone Lymphoma and Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma
Marginal zone lymphoma (MZL) arises from memory B cells that normally reside in the marginal zone of secondary lymphoid follicles. MZL is the third most common type of non-Hodgkin lymphoma, representing about 10% of all cases of NHL. At least three distinct entities of MZL have been included in the WHO classification, including extranodal MZL of mucosa-associated lymphatic tissue (MALT) lymphoma, which represents more than two thirds of all MZL cases; splenic MZL with circulating villous lymphocytes; and nodal MZL. Clinically, MZL typically presents as early-stage disease in most patients and exhibits an indolent course in most patients. However, MZL can become disseminated or transform into aggressive large-cell lymphoma. Over the past two decades we have witnessed unprecedented progress in our understanding of the biology of MZL at the molecular level, and some of that knowledge is being translated into the development of rational treatment paradigms.
Because MALT lymphoma develops in tissues that are not necessarily abundantly enriched with lymphocytes, the first step in the pathogenesis is the abnormal aggregation of lymphoid tissues in extranodal sites and organs. The etiologic factor in this process can be infectious and/or inflammatory. Gastric MALT lymphoma represents more than 50% of all MALT lymphoma and has been shown to be intricately associated with colonization of the stomach by the bacterium
Helicobacter pylori.
20 Although aggregation of lymphocytes stimulated by
H. pylori is a prerequisite for MALT lymphoma,
H. pylori infection alone is not sufficient to cause the malignant transformation of lymphocytes into lymphoma cells.
H. pylori is found in more than 90% of patients with MALT lymphoma, yet most patients with
H. pylori infection do not develop lymphoma.
H. pylori also produces inflammation and reactive oxygen species (ROS)
that further increase genomic instability, which is essential for lymphomagenesis. In support of the theory of genomic instability in gastric MALT lymphoma, a balanced translocation t(11;18)(q21;q21) was reported in up to 50% of all cases of the disease.
21 The translocation results in the production of fusion protein API/BIRC3-MALT1, which interacts with CARD11/CARMA1 to activate the critical pro-survival transcription factor NFκB.
22 The translocation t(11;18) has been found to be mutually exclusive of other cytogenetic abnormalities, further suggesting its role as an important, if not the singular, driver for the development of MALT lymphoma.
23 Importantly, the emergence of t(11;18) is associated with unfavorable clinical outcomes and resistance to antibiotics and chemotherapy in the treatment of gastric MALT lymphoma.
24 In addition to its occurrence in the gastrointestinal tract, MALT lymphoma has been reported in the lung, ocular adnexa, salivary gland, and less commonly the skin and thyroid. MALT lymphoma in these sites often involves a different etiology. For example,
Chlamydophila psittaci or
Borrelia burgdorferi is thought to be responsible for MALT lymphoma of the ocular adnexa and skin, respectively, whereas inflammation caused by autoimmune disease is responsible for MALT lymphoma in the thyroid and salivary gland. Cytogenetic abnormalities are well described for MALT lymphoma in these sites and include t(14;18) and t(1;14), which involve MALT1 and BCL10, respectively. Other genetic abnormalities in MALT lymphoma include t(3;14) involving IGH-FOP1, +3/3q, +18/18q, and
−6q23, many of which target TNFAIP3 (A20), a negative regulator of NFkB, which is also mutated and silenced by methylation in subsets of MZL.
25 The pathogenetic role of many of these genes and loci are poorly understood at present.
Splenic MZL is a less common entity of MZL and has been shown to be associated with hepatitis C infection (HCV) in some cases.
26 In these cases, HCV glycoprotein E2 interacts with CD81 on B cells and causes activation of the BCR and its downstream signals.
27,28 Similar to other etiologic factors in MZL, HCV is required for the initiation and maintenance of the malignant phenotype of MZL, but not sufficient to cause malignant transformation of memory B cells in the marginal zone. This is evident from the fact that most patients with HCV do not develop splenic MZL. Furthermore, the majority of patients with splenic MZL are not associated with HCV. Clearly, genetic and epigenetic alterations are required for the development of splenic MZL. Although t(11;18) is not associated with splenic MZL, cytogenetic abnormalities are very common in splenic MZL, including partial trisomy of 3q, gain of 12q, and deletion or translocation of 7q32.
21,29 Furthermore, recent studies have demonstrated that deregulation of the NFκB pathway is frequently observed in splenic MZL. For example, mutations and copy number abnormalities of NFκB genes have been shown to occur in 36 (36%) of 101 splenic MZLs in one series; in another series, inactivating mutations were found in the NFκB negative regulator A20 (6/46 = 13%), and activating mutations were found in MYD88 (6/46 = 13%) and CARD11 (3/34 = 8.8%). Interestingly, these mutations are largely mutually exclusive of each other, suggesting that they may play vital roles in the pathogenesis of splenic MZL. Mutations in NOTCH2, a gene required for marginal-zone B-cell development, is a recurrent genetic lesion in splenic MZL, accounting for approximately 20% to 25% of cases.
30 Importantly, NOTCH2 mutations are restricted to splenic MZL but not other indolent B-cell lymphoproliferative disorders such as MALT lymphoma.
31 These mutations are predicted to cause impaired degradation of the NOTCH2 protein or may result in a gain of function of the truncated NOTCH2 in splenic MZL. The 7q deletion mentioned earlier was investigated recently using a combination of high-resolution array comparative genomic hybridization and integrated transcriptomic analysis. No single gene from these analyses could be identified as a potential driving event for splenic MZL with del7q.
32 Rather, a number of genes and microRNA species were found to be downregulated, some attributed to promoter hypermethylation. Another study of the 7q deletion arrived at a similar conclusion and was able to find nucleotide changes in IRF5 in two of the patients, resulting in fourfold downregulation of the IRF5 gene in lymphomas with 7q32 deletion versus deleted tumors.
33 Importantly, ectopic expression of IRF5 in marginal-zone lymphoma cells decreased proliferation and increased apoptosis in vitro, and impaired lymphoma development in vivo, suggesting that IRF5 may be a haploinsufficient tumor suppressor in this lymphoma entity.
Another layer of complexity in the pathogenesis of splenic MZL was revealed by the discovery of frequent microRNA dysregulation.
34 When compared to reactive lymphoid hyperplasia and other lymphoproliferative diseases, splenic MZL is found to have distinct expression of miRNAs, including highly overexpressed miR-155 and miR-34a, and repressed expression of miR-377 and miR-145, which are candidate diagnostic tools for splenic MZL.
Nodal MZL is a rare entity of MZL and is also associated with HCV infection, though probably to a lesser degree than splenic MZL. No characteristic translocations or chromosome imbalances, such as the t(11;18) in MALT lymphoma and del7q in splenic MZL, have been described in nodal MZL. Only a few cytogenetic alterations of NMZL have been reported, including trisomy 3 in 50% to 70% of cases, none with a clearly established role in either the diagnosis or pathogenesis of nodal MZL. Recent studies by Arribas and colleagues demonstrated that nodal MZL is characterized by upregulation of the IL6 and IL2 cytokine pathways and CD40 signaling, which are involved in B-cell
survival.
35 Some of the highly expressed genes include SYK, which is involved in BCR signaling, and TAC1, which is involved in activating NFκB. In the molecular signature of nodal MZL, a large number of overexpressed genes were associated with the NFκB signaling pathway, such as CD74, CD81, CD82, RELA, and TRAF4, reaffirming the central role of NFκB as a converging point for the proliferation and survival of MZL cells. Interestingly, some microRNA species known to be involved in memory B-cell development, including, for example, miR-221, miR-223, and let-7f, were overexpressed, suggesting their potential utility in the diagnosis of nodal MZL.
Our increased understanding of the biology of MZL in the past decade has already changed how we manage these patients. Because the infectious agents have been found to be required to initiate and maintain the malignant phenotype of some MZL subtypes, antimicrobial treatment has become a highly successful strategy in many patients. The majority of patients with
H. pylori–associated MALT lymphoma can be cured with antibiotics that eliminate the bacterium, thus sparing a large number of patients more aggressive and expensive treatments.
36 Treatment of HCV and
C. psittaci are effective strategies to manage patients with splenic MZL and MALT lymphoma of the ocular adnexa. The discovery of antigen stimulation and the central role of the BCR and NFκB signaling in MZL certainly supports the rationale to explore inhibitors of these pathways with Syk and BTK inhibitors, and even proteasome inhibitors. Targeting NOTCH2, which has been studied predominantly in ALL, could also be an effective strategy in the treatment of splenic MZL.
In contrast to MZL, CLL/SLL are diseases with clearly identified cytogenetic aberrations that have been shown to correlate with prognosis and survival. The clonal population of B lymphocytes that characterize CLL express CD19, CD5, and CD23, with often dim expression of CD20. In addition, these cells typically exhibit reduced levels of surface immunoglobulin. Collectively, this features are the hallmark of mature and activated B lymphocytes.
37 As with many of the other lymphoid malignancies, as our understanding of the pathogenesis of these diseases evolves, our appreciation of their heterogeneity has broadened. This heterogeneity seen in CLL can be characterized at numerous levels, including the status of mutation of the V genes, the expression of CD38, ZAP-70, and other discrete cytogenetic lesions.
For example, CLL cases can be subdivided on the basis of mutations in the V genes, based on the direct comparison of the DNA sequence of germline V genes with the V genes in the CLL cells.
38 CLL cases can then be classified as mutated (that is, they exhibit a more than 2% difference from the germline sequence) or unmutated. Expression of ZAP-70, a protein that is normally expressed near the surface of T cells known to play a role in signaling through the TCR, has also been shown to be an independent prognostic factor.
39–41 In CLL it has been found to be profoundly prognostic. Patients who are ZAP 70
− have been shown to have a median survival of 8 years, compared to patients who are ZAP-70
+, who have a median survival of approximately 25 years. Collectively, these factors have been used to risk stratify patients with CLL, such that those patients with B-CLL clones having few to no V-gene mutations, or with CD38
+ and ZAP-70
+ B-CLL, have been found to have comparatively more aggressive, usually fatal disease. In contrast, those patients with mutated V-genes and/or little to no CD38 or ZAP-70 are typically considered to have a very indolent natural history.
42 Recurring cytogenetic lesions have also been described and may be more valuable in risk stratifying patients with CLL, and ultimately in developing better tailored therapies for the disease. One of the most common is deletion of 13q14.3, which occurs in more than 50% of cases over time.
43 This region of the chromosome is thought to encode two miRNA genes.
44 However, some of the highest risk cytogenetic lesions include deletions of 11q22-23, 17p13, and 6q21.
45 Although the specific genes involved in these deletions remain to be precisely identified, loss of 17p13 is thought to be associated with loss of the tumor suppressor p53 function, and deletion of 11q22-23 is thought to be associated with loss of the ataxia-telangiectasia mutated (ATM) gene. Specific treatment regimens for these discrete genetic subcategories of CLL are in development, and some principles may be emerging. First, patients with loss of 17p are thought to have a very poor prognosis and are typically very refractory to chemotherapy. Some anecdotes suggest that these patients may respond well to anti-CD52–based therapy (alemtuzumab), and it is thought that BTK inhibitors might override this adverse prognostic feature. Similarly, patients who have loss of the ATM function through deletion of 11q22-23 may benefit from the introduction of alkylating agents into the treatment program.
Pathogenesis of Mantle-Cell Lymphoma
Mantle-cell lymphoma (MCL) accounts for approximately 6% of all cases of non-Hodgkin lymphoma, or about 3000 cases per year in the United States.
1 Only a decade ago, it was primarily thought to be one of the most challenging subtypes of lymphoma to treat, carrying the incurable characteristics of indolent lymphoma, and the unfavorable features of aggressive lymphoma. Over the past decade, treatment paradigms for the disease have changed rapidly, and so too has the natural history. These changes are attributed to a
number of observations: (1) the recognition that MCL is not one disease and, like DLBCL, is a very heterogeneous disease composed of more indolent and aggressive variants; (2) the finding that intensive induction chemotherapy regimens consolidated by ASCT used in the front line can consistently produce a response rate of 90% to 100% and produce prolonged progression-free survival compared to traditional R-CHOP–based chemotherapy regimens; and (3) the emergence of novel drugs affecting unique targets, such as proteasome inhibitors and immunomodulatory drugs, which have led to new opportunities to either complement existing treatment paradigms or manage relapsed or refractory disease without specific cytotoxic therapy. What has become painfully clear over the past several years, however, is that given the heterogeneity of the disease, certainly at a biologic level, failure to characterize future populations of patients with MCL on clinical study in this regard will lead to confounding of conclusions about future treatments.
The molecular hallmark of MCL is overexpression of cyclin D1.
46 Although dysregulation of cyclin D1 has been recognized for some time, it has now become evident that MCL is a disease, possibly the prototypical disease, defined by gross cell cycle dysregulation. Cyclin D1 dysregulation is the pathognomonic chromosomal translocation t(11;14)(q13;q32) of the disease, which places cyclin D1 downstream of the highly active IgH enhancer.
47 Essential to the pathogenesis of the disease, the mRNA of cyclin D1 undergoes alternative splicing to produce two unique transcripts: cyclin D1a, which has been clearly shown to drive much of the cell cycle dysregulation; and cyclin D1b, whose expression is more variable and whose role is less well defined.
48 Deletion or mutation of the cyclin D1a mRNA tail region produces a truncated version of cyclin D1a mRNA, which has been shown to be 6 to 10 times more stable than the wild-type full-length cyclin D1a mRNA.
49 In addition to the pivotal role the splice variants of cyclin D1 mRNA play, microRNAs, specifically miRNA61, downregulate cyclin D1a, potentially modulating the cell cycle kinetics. The influence of miR61 can also be influenced by mutations in the tail region of the cyclin D1a mRNA in MCL, which can abrogate the miR61 binding site, allowing for the marked overproduction of cyclin D1 protein.
50 In addition, a number of important translational events have been shown to regulate cyclin D1 level. Phosphorylation of cyclin D1 by GSK3β leads to polyubiquitination by the E3 ligase FBX4, rendering it a substrate for the proteasome and proteolytic degradation.
51 Inactivation of GSK3β would preempt cyclin D1 phosphorylation, which has been shown to occur in the setting where AKT is aberrantly activated,
52 whereas impaired proteolytic degradation of cyclin D1 can be prevented by mutation of the E3 ligase FBX4,
53 resulting in reduced ubiquitination of the D1 protein. Collectively, these overlapping mechanisms of cyclin D1 regulation ensure high levels of cyclin D1 protein in MCL and, in select settings, can lead to marked impact on the cell cycle kinetics.
Although cyclin D1 levels are central in the pathogenesis of MCL, it is by no means the only corrupted cell cycle pathway.
54 Cyclin D1 normally interacts with CDK4/CDK6 to facilitate cell cycle progression through the G1-S checkpoint.
55 CDK4 is frequently overexpressed or amplified in MCL,
56 which would further limit the checkpoint control. Conversely, impaired inhibition of cell cycle regulation can play a major role in the disease, as evidenced by the fact that patients with MCL often have little to no accumulation of the CDK inhibitors p16 and p27.
57 Loss of p16 influence has been shown in patients with MCL—this tumor suppressor is deleted in about half of all MCL cases, and either mutated or silenced by promoter hypermethylation.
58 Impaired cell cycle control due to loss of p27 has been attributed to a number of mechanisms. The protein level of p27 in MCL is regulated by the ubiquitin proteasome. The p27-specific F-box protein Skp2 is inducible and been shown to be overexpressed in some patients with MCL, in particular those with disease that is known to be more aggressive. Increased levels of Skp2 lead to more active ubiquitin ligases, which leads to more prompt and efficient ubiquitination of p27 and thus a theoretically shorter half-life for the protein. This loss of cell cycle inhibition has been noted in patients with more aggressive forms of the disease.
59 This convergence of markedly dysregulated cell cycle control processes, involving both drivers and inhibitors of cell cycle kinetics, conspires to affect the proliferative rate of the disease and, as we now know, the risk stratification of select patient subpopulations.
The notion that the proliferative rate of the disease can be prognostic has now been confirmed at a number of levels. Patients with the truncated version of the mRNA of cyclin D1a, the stabler form, are known to have disease with a higher proliferative index
49 and more aggressive histologies, such as blastoid MCL.
60 Remarkably, the median overall survival (OS) of MCL patients with the truncated cyclin D1a mRNA is only 1.38 years, compared to 3.28 years for patients with the full-length and unstable mRNA.
49 The levels of the full-length mRNA transcript of cyclin D1a have been shown to positively correlate with the proliferation index as assayed by positive immunostaining of Ki-67. Ki-67 is a nuclear protein found in all states of the cell cycle (G
1, S, G
2, and M), but is absent from resting cells (G
0). Many cell cycle regulatory factors have been shown to correlate with the protein level of cyclin D1 on immunohistochemical staining, including the percentage of MCL cells with positive nuclear staining of the protein or the amount of nuclear staining of cyclin D1.
60 Conversely, the expression of p27 is inversely associated with Ki-67, where high levels of p27 appear to be
associated with lower proliferative index and improved overall survival.
57 These lines of data strongly support the contention that MCL is a disease grossly characterized by cell cycle dysregulation, and that these features of the underlying disease biology can be highly prognostic.
Further confirming the significance of the proliferative index, several studies have shown that nuclear staining of Ki-67, and using a cutoff of 30%, predicted OS, where patients below and above the 30% cutoff experienced a median OS of 13 months and 45 months, respectively.
61 Using yet another approach, a group at the National Cancer Institute (NCI) employed GEP to characterize patients with MCL (
Figure 29-5 ). Their profiling experience firmly established that heterogeneity of the disease, which they used to define the proliferation signature of MCL. They discovered 48 genes whose expression levels were highly correlated with survival in a statistically significant manner. Among those 48 genes were a group of 20 genes that were variably expressed, though strongly correlated with cellular proliferation. They called the pattern of gene expression the “proliferation signature,” which could essentially stratify patients along a continuum from highly proliferative to less proliferative disease.
62 Calculation of the average expression levels of these 20 genes in the proliferation signature allowed for the separation of the population into quartiles. The median OS of patients with the lowest expression of proliferation signature genes (quartile 1) was 6.71 years, whereas those with the highest expression (quartile 4) exhibited an OS of only 0.83 years. Surprisingly, cyclin D1 was not among the proliferation signature genes identified using the data generated from DNA microarrays, because the chip-based technology detected only the full-length mRNA of cyclin D1, which is unstable. Employing a reverse transcriptase (RT)-PCR assay to differentiate the two species of the cyclin D1 mRNA showed that patients with the truncated mRNA of cyclin D1 (3′ UTR low) demonstrated a higher expression level of cyclin D1, a higher expression of the 20 “proliferation signature” genes, and a substantially shorter OS, compared to patients with the full-length mRNA of cyclin D1 (i.e., 3′ UTR high).
62 When RT-PCR was designed to detect only
the coding region of cyclin D1 mRNA, which accounted for both the full-length and truncated mRNA, the levels of cyclin D1 were again shown to correlate with patient survival.
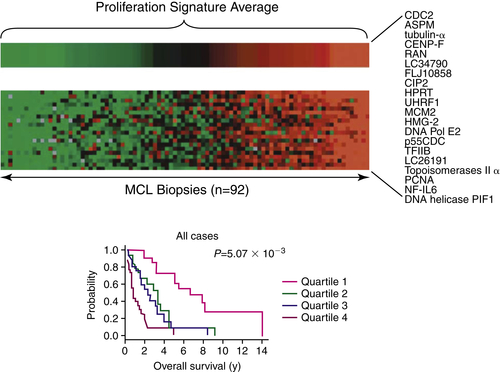
Figure 29-5 Gene expression profiling of mantle-cell lymphoma cases reveals prognostic significance of the proliferation index Gene expression profiling of primary mantle-cell lymphoma cases has demonstrated that the most aggressive forms of the disease are associated with an enrichment of genes involved in cell proliferation, a higher proliferative index, and a poorer prognosis. Cases with a lower proliferative index have a better prognosis. Proliferation gene expression signature may predict survival in MCL. These authors established a molecular diagnosis of MCL based on gene expression that can distinguish the disease from other lymphoma types. Using this diagnostic tool, they were able to investigate gene expression patterns in patient samples and evaluate whether there was a correlation with survival. In addition, they were able to investigate the genetic aberrations that are the basis of the pathobiology of MCL. This figure shows the proliferation signature average (PSA) for 92 samples from patients with MCL. The PSA is the average expression of the top 20 genes that were expressed most variably in MCL (i.e., the top third). To visualize the power of this as a predictive model, patients were ranked according to the expression of these “signature” genes in their tumors and then divided into four equal quartiles as shown. This analysis allows a suggested quantitative relationship between proliferation and survival. The Kaplan-Meier plots of overall survival of patients in each of these quartiles indicate that the proliferation signature can identify subgroups of patients with different survival times. Median survival times for each quartile were 6.71 years (1), 3.28 years (2), 2.31 years (3), and 0.83 years (4). Data from Rosenwald A et al. Cancer Cell. 2003;3:185-197.
There is no question that the proliferative index of individual patients with MCL is prognostic. There is also no doubt that failing to characterize study populations with regard to this biologic parameter will lead to confounding influences in our interpretation of clinical trial data. The prognostic impact of the proliferation index remains relatively significant irrespective of the therapy, as had been shown with rituximab,
63 high-dose chemotherapy and stem cell transplant,
64 and the proteasome inhibitor bortezomib.
65 Moving forward, it will be critical to determine if the proliferation signature will prove useful in predicting responses to specific treatments, or even if it could be used to tailor treatment to a particular disease context. Although prognosticating patients with MCL using DNA microarray-based gene expression profiling remains an experimental approach, RT-PCR–based measurement of the expression of proliferation signature genes
62 may be validated and practical in the near future.
Pathogenesis of Lymphomas Derived from Thymic B cells: Primary Mediastinal Large B-Cell Lymphoma, Hodgkin Lymphoma, and Gray Zone Lymphomas
Lymphomas arising in the anterior mediastinum have posed significant diagnostic and management challenges over the past several decades, especially given the overlapping biologic and clinical features of these diseases. Emerging molecular data have begun to demonstrate important differences in the molecular pathogenesis of these diseases, especially the cell of origin. Both bone marrow stroma and thymic stromal cells express ligands and cytokines required for B-cell differentiation. Despite the shared roles marrow and thymic stroma play in B-cell differentiation, B-cell development in the thymus is very restricted. B-cell precursors are present in the thymus and are felt to be distinct from B-cell precursors in the bone marrow. Although the thymic microenvironment is the source of lymphopoietic factors that include interleukin-7 (IL-7), pro-B cells exposed to the thymic microenvironment are hyporesponsive to IL-7, whereas pro-B cells derived from the bone marrow are typically responsive. Thus, pro-B cells from the thymus accumulate at an early pro–B-cell stage of development, cycle less than their bone marrow counterparts, and fail to differentiate efficiently.
66 This difference in the microenvironment and its effects on the maturation of B cells may account for the unique features for lymphomas that arise from thymic B cells compared to lymphomas arising from the bone marrow and other lymphoid tissue.
Based merely on histopathologic features, including the expression of CD20, primary mediastinal large B-cell lymphoma (PMBL) had always been considered a diffuse large B-cell lymphoma (DLBCL), at least until it was recognized as a discrete entity in the REAL Classification in 1994.
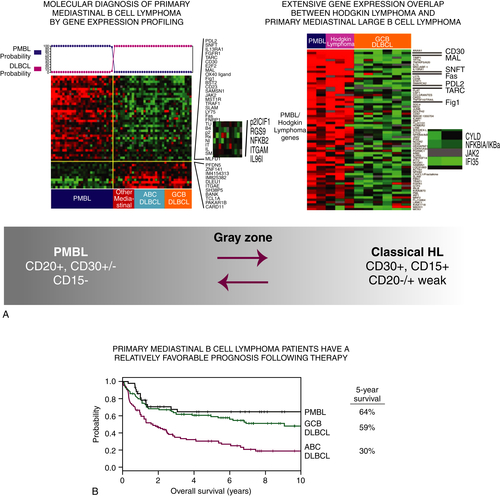
67 Despite the histopathologic similarities with DLBCL, it was becoming evident in the early 1980s that the clinical presentation of PMBL and DLBCL, as well as the prognosis, was significantly different. Differences in the molecular pathogenesis of these diseases became evident when differences in gene expression profiling demonstrated extensive overlap between PMBL and classical Hodgkin lymphoma–nodular sclerosing (cHL-NS)
68 (
Figure 29-6 ). Though unclear for years, it is now widely recognized that the Hodgkin–Reed Sternberg (HRS) cells are characterized by suppression of many components of the B-cell program, and they are incapable of immunoglobulin secretion.
69 Efforts to identify the differences between PMBL and other subtypes of B-cell lymphoma using gene expression profiling led to the surprising observation that PMBL was strikingly similar to the profiles seen for cHL rather than for other subtypes of DLBCL. In a study by Rosenwald and colleagues, more than one third of the genes that were more highly expressed in PMBL compared to DLBCLs were also characteristically expressed in Hodgkin lymphoma cells.
68 In this study, the PDL2 gene, which encodes a regulator of T-cell activation, was the gene that best discriminated PMBL from other DLBCLs. PDL2 was more highly expressed in PMBL and cHL and either not expressed or expressed to minor levels in DLBCL. The genomic loci for PDL2 and several neighboring genes were amplified in more than half of cases of PMBL and Hodgkin lymphoma and expressed to much lower levels in DLBCL. Collectively, these data gave credence to the observation that B-cell lymphomas derived from the thymus were a unique entity based on the shared pathogenetic features and likely clinical history. It also established that they were markedly different compared to those B-cell lymphomas derived from the bone marrow compartment.
Genetic studies of PMBL frequently demonstrate amplification of recurring regions on chromosome 9p and 2p, which, interestingly, have also been described in cHL, and rarely in other subtypes of DLBCL.
70 The 9p region encodes JAK2, a tyrosine kinase that phosphorylates and activates the transcription factor STAT6. SOCS1, which suppresses JAK signaling, is regularly deleted in both PMBL and cHL, contributing to further dysregulation of the JAK-STAT pathway.
71 Other genes thought to be involved at 9p include PDL1 and PDL2, whereas c-Rel may be involved at 2p. Like cHL, PMBL also has constitutively activated NFκB and loss of CIITA (MHC 2), which may help explain
the ability of these cells to evade immune surveillance.
72 Although PMBCL and cHL share a cell of origin in the form of the thymic B-cell, as well as molecular characteristics, there are still important morphologic and immunophenotypic distinguishing features between these two diseases with significant clinical and treatment implications.
Figure 29-6 (A) Gene expression profiling of primary mediastinal B-cell and Hodgkin lymphoma reveal marked similarities with each other, and marked dissimilarity with DLBCL. (B) Patients with PMBL have a prognosis similar to that for Hodgkin lymphoma, and superior to that for patients with DLBCL.
Another example of a thymic B-cell lymphoma, recognized only in 2008 by the WHO Classification of Tumors of Hematopoietic and Lymphoid Tissues, is the entity known as
gray zone lymphomas (GZL), now more widely referred to as
B-cell lymphoma with features intermediate between diffuse large B-cell lymphoma and classical Hodgkin lymphoma.
4 GZL represents a mediastinal B-cell lymphoma that does not fulfill the morphologic and/or phenotypic criteria for PMBL or CHL, but instead represents a spectrum between these two entities.
Interphase FISH studies of GZL have demonstrated a number of recurring molecule lesions, including amplification of the REL/BCL11A locus, alterations affecting the JAK2/PDL2 locus, and rearrangement of the CIITA (MHC2)
locus, as well as gains of MYC.
73 Although the molecular signature of GZL has not been elucidated, a recent large-scale methylation analysis of PMBL, CHL, and GZL has demonstrated a number of similarities in the methylation profile of these diseases, although GZL did exhibit its own unique profile distinct from PMBL and cHL.
74 Importantly, GZL could be distinguished from cHL-NS and PMBL by differential methylation of selected CpG islands, and a class prediction model could be established to segregate the various entities. Thus, the global methylation profile of these entities may be useful not only to establish new diagnostic tools and clarify the pathogenesis of these lymphomas, but also to identify possible targets for future therapies.
While lymphomas of the thymic B-cell origin (cHL-NS, GZL, and PMBL) are closely related diseases that share clinical and biologic characteristics, there remains a spectrum of heterogeneity.
As the field of medical oncology enters the molecular and targeted therapy era, the biology that connects this disease and sets some lymphomas apart from DLBCL can pave the way for new therapeutic platforms that optimize efficacy and limit toxicity. The elucidation of pathways and molecular targets that are unique to the thymic B-cell lymphoma group of disease gives credence to therapies targeting NFκB, Janus kinases (JAK), programmed death-1 (PD-1), and epigenetic-based therapies.
Molecular Pathogenesis of the Peripheral T-Cell Lymphomas
The mature T-cell lymphomas, also referred to as
peripheral T-cell lymphomas (PTCL), arise from thymic T cells or NK cells. These diseases are associated with often diverse presentation, arising in either the nodal or extranodal compartments.
4 Molecular and genetic characterization of PTCLs has lagged well behind that of B-cell non-Hodgkin lymphomas—in part because of their rarity and the relatively specific morphologic and immunophenotypic features of these neoplasms, which has precluded reliable identification of the cell of origin and classification into distinct disease subtypes.
Studies using conventional cytogenetic analysis over the past two decades have revealed only limited, disease-specific, recurrent karyotypic abnormalities.
75 CGH studies have helped delineate genetic differences, as well as similarities, between different subtypes of PTCL.
76 Gene expression profile analyses are providing insights into cell-intrinsic and microenvironment-related pathogenetic features in disease subsets,
77 as well as genetic signatures associated with clinical outcomes.
77 Higher resolution genetic analyses, specifically exome and genome sequencing using next-generation techniques, might help refine diagnostic categories and provide prognostic and therapeutically actionable information.
Recent advances in immunophenotypic analysis and systematic clinical characterization of large series of PTCL have led to the recognition of more than 22 different defined or provisional clinicopathologic subtypes of T-cell lymphoma that are distributed among four different subcategories in the present WHO classification (2008).
4 Molecular and genetic aspects of disease pathogenesis of some of the more common and better defined subtypes of PTCL are described here.
Anaplastic Large-Cell Lymphoma (ALCL)
Two different types of ALCLs are presently recognized, including ALK
+ ALCL and ALK
− ALCL, the former associated with a superior outcome relative to ALK
− ALCL. Although these entities share similar morphologic and immunophenotypic features, there are a number of key differences in their biologic and clinical characteristics. ALK
+ ALCL is characterized by the chromosome translocation t(2;5)(p23;q35) in 55% to 85% of systemic ALCL, or variant translocations involving ALK and other partner genes in a minority of cases.
78,79 The translocation t(2;5)(p23;q35) results in a fusion protein, nucleophosmin (NPM)/ALK (NPM-ALK), which leads to the constitutive activation of the ALK tyrosine kinase and alterations in multiple signaling pathways, including the Janus kinase 3 (JAK3)/signal transducer and activator of transcription (STAT3), phosphatidylinositol 3-kinase (PI3-kinase)/protein kinase (AKT)/mammalian target of rapamycin (mTOR), and the phospholipase C-γ (PLC-γ) mediated RAS-extracellular signal–regulated kinase (ERK) pathways.
78 Activation of Notch1 signaling by its ligand Jagged1, which is expressed on neoplastic and neoplastic cells, has also been proposed to play a role in the pathogenesis of ALK
+ ALCL.
80 Secondary MYC translocations have been reported in some cases with an aggressive behavior.
81 It is unclear at present whether ALK
+ ALCL originates from cytotoxic T cells or acquires a cytotoxic phenotype, but studies have reported activation of a Th17 differentiation program in this disease.
77 Molecular alterations in signaling pathways as a consequence of the variant translocations are not adequately understood, but similarities and differences in gene expression profiles with NPM-ALK have been described.
82 Differences in gene expression profiles between certain morphologic subtypes of ALK
+ ALCL have also been reported,
83 but array CGH analysis of NPM-ALK and variant ALK translocations associated with ALCL have revealed similar recurrent secondary genetic abnormalities.
76 The cell of origin of ALK
− ALCL is also not known at present. Array CGH analysis of ALK
+ and ALK
− ALCL has highlighted differences in secondary genetic aberrations between the two subtypes,
76 and differential expression of microRNAs has also been reported.
84 Comparative gene expression analysis of ALK
+ and ALK
− ALCL has revealed deregulation of common kinase signaling cascades and regulators of apoptosis, as well as differences between these subtypes, such as alterations in cell cycle regulators, signal transduction proteins, and various transcription factors.
85 ALK
+ ALCL shows overexpression of genes implicated in immune or inflammatory responses, regulation of NFκB signaling, and lymphocyte migration and adhesion, whereas ALK
− ALCL exhibits overexpression of genes involved in certain cytokine signaling pathways.
83 Analysis of ALK
− ALCL using next-generation massively parallel sequencing has revealed a recurrent balanced translocation t(6;7)(p25.3;q32.3), leading to the juxtaposition of the DUSP22 phosphatase gene with the fragile site FRA7H, which results in the downregulation of DUSP22 and upregulation of MIR29 microRNAs located on 7q32.3.
86 This translocation was observed in 45% of ALK
− ALCLs with 6p25.3 rearrangements and in both systemic and cutaneous forms of ALK
− ALCL. The biologic consequence of this translocation awaits further investigation.
Peripheral T-Cell Lymphoma Not Otherwise Specified
PTCL-NOS is the most common subtype of PTCL, accounting for up to 25% of all PTCL occurring worldwide. It is a clinically heterogeneous entity, potentially comprising different PTCL subtypes at different stages of disease evolution. Comparison of PTCL-NOS expression profiles with those of purified T-cell subsets has suggested a relationship between PTCL-NOS subtypes with either activated CD4
+ or CD8
+ T cells,
87 and cases exhibiting a gene expression profile similar to cytotoxic T cells have been shown to have inferior survival.
77 PTCL-NOS lacks specific, recurrent cytogenetic abnormalities, but complex cytogenetic aberrations have been associated with a poor prognosis in PTCL-NOS.
75 Recently, the translocation t(5:9)(q33:32) was reported in 17% of PTCL-NOS, which results in the fusion of the
IL-2 inducible T-cell kinase (ITK) gene with the
spleen tyrosine kinase (SYK) gene.
88 Transgenic mice expressing the ITK-SYK fusion transcript develop T-cell lymphomas mimicking the human disease.
89,90 Overexpression of Syk tyrosine kinase and Syk phosphorylation, in the absence of
SYK translocations, has also been observed in PTCL.
86 Based on the expression levels of the NFκB pathway genes, PTCL-NOS can be segregated into two groups showing differences in survival.
91 The differential expression of a set of proliferation and cell cycle–associated genes, including CCNA, CCNB, TOP2A, and PCNA, has been shown to predict prognosis.
92 PTCL-NOS shows deregulation of pathways controlling apoptosis, cell proliferation, adhesion, matrix remodeling, and chemoresistance, and upregulation of platelet-derived growth factor receptor alpha has been observed in many cases.
87 Distinguishing PTCL-NOS from ALK
− ALCL can be difficult using current cytomorphologic and immunophenotypic criteria. This is mirrored at the chromosomal and genetic level. Early gene expression profiling studies were unable to distinguish PTCL-NOS from other PTCL subtypes.
91 However, analysis restricted to nodal PTCL allowed discrimination between PTCL-NOS and other PTCL subtypes and further subclassification based on alterations of different biologic processes or signaling pathways.
9 Refined, supervised gene expression profile analysis can provide a signature allowing distinction between PTCL-NOS and ALK
− ALCL; however, the pathogenetic significance of the involved genes and pathways is not yet clear.
94 Recently a model comprising three genes, TNFRSF8, BATF3, and TMOD1, obtained from a meta-analysis of the transcriptional profiles of a large series of PTCL, was shown to distinguish ALK
− ALCL from PTCL-NOS.
95 Future studies might illuminate the biologic basis of this observation. PTCL-NOS and ALK
− ALCL also share karyotypic abnormalities, including gains of chromosomes 1q and 3p and losses on chromosome 6q, although the loci on 6q have been shown to differ.
75 CGH analysis has shown overlapping aberrations, including 6q and 13q losses, as well as subtype-specific abnormalities.
96 Recurrent chromosome gains of 7q that target cyclin-dependent kinase 6
97 and 8q involving the
MYC locus
98 have been reported in PTCL-NOS. A recent genome-wide NGS analysis of PTCL led to the identification of recurrent translocations involving p53-related genes, including rearrangements of the
TP63 gene with TBL1XR1 and ATXN1 genes.
99 These gene fusions encode proteins that inhibit the p53 pathway and are associated with adverse clinical outcomes. Screening a large series of cases for
TP63 rearrangements by FISH showed similar frequencies in PTCL-NOS (9.4%), ALK
− ALCL (12.5%), and primary cutaneous ALCL (10.5%).
Angioimmunoblastic T-Cell Lymphoma
AITL is thought to derive from T-follicular helper (TFH) cells based on phenotypic features and overexpression of genes characteristic of normal TFH cells (CXCL13, BCL6, PD-1, CD40LG, and NFATC1).
100,101 Microenvironmental factors and signals orchestrating tumor-stroma cross talk are
thought to play a role in AITL pathogenesis. Recent studies, however, have also demonstrated a TFH phenotype for a subset of PTCL-NOS.
102 This might be one of the explanations for the inability of gene expression profiling to segregate AITL from subsets of PTCL-NOS in some instances.
87 Gains of chromosomes 3q, 5q, and 21 have been described as recurrent alterations in AITL, although the genes affected by these changes are not known.
75 Studies from transgenic mouse models and genome sequencing studies have begun to provide insights into AITL pathogenesis and its relationship with other types of PTCL. Mice engineered to disrupt the function of the
Tet2 gene showed an increase in T-cell progenitors and developed T-cell lymphomas. On sequencing human lymphoma specimens, heterozygous insertions and deletions of
Tet2 were detected in 33% of AITL and smaller subsets of other PTCLs.
103 Analysis of a large cohort of PTCL revealed a higher frequency of
Tet2 mutations in AITL (47%) and PTCL-NOS (38%) and an association with adverse clinical features.
104 Of interest, PTCL-NOS expressing TFH markers, despite showing or lacking a histopathologic resemblance to AITL, demonstrated a significantly higher frequency of
Tet2 mutations. Furthermore,
DNMT3A mutations occur in a high frequency (73%) of cases harboring
TET2, including PTCL-NOS and AITL, suggesting oncogenic cooperation between pathways regulated by
TET2 and
DNMT3A such as cytosine methylation and demethylation processes in PTCL.
105 Recently, heterozygous IDH2 mutations, mostly resulting in an R172 substitution, have been described in 20% to 45% of AITL.
106 The prognostic implications of this mutation, if any, are unclear at present. It also remains to be seen whether this mutation is specific for AITL or other subtypes of PTCL derived from TFH cells. Overall, these studies indicate an important contribution of epigenetic alterations in the pathogenesis of AITL and at least a subset of PTCL-NOS.
Translating Molecular Pathogenesis into Novel Treatment Platforms
One of the many objectives for improving our understanding of the molecular basis of any disease is ultimately to use that information to treat disease in a more biologically rational manner. The explosion of detailed mechanistic studies into the pathogenesis of lymphoma has begun to create new opportunities for therapeutic intervention. Although we could point to many studies in various stages of development, the focus here is on one in ABC-DLBCL.
As discussed earlier, the emergence of new small molecules targeting BTK has provided an immediate opportunity to treat B-cell lymphomas at a fundamental level. Enrichment for dysregulated BCR signaling in ABC-DLBCL offers an opportunity to improve the outcome of a subset of DLBCL associated with an inferior prognosis. The recent development of the BTK inhibitor ibrutinib has been shown to completely block BCR signaling and induce apoptosis. Ibrutinib forms a bond with cysteine-148 in BTK, inhibiting the kinase with an IC50 of 0.5 nM, and with a relatively high degree of specificity. In a recent study reported by the NCI, 49 patients with relapsed or refractory DLBCL derived from the GC (n = 20) or ABC origin (n = 29) were treated with ibrutinib at the maximum tolerated dose. These patients were overall very heavily treated, with a median of three to four prior therapies, with a substantial number having refractory disease. With the caveats of a small unrandomized study, the overall response rates were 41% among those patients with ABC DLBCL (12 of 29 responding), compared to 5% (1 of 20) among those patients with GC-DLBCL. In addition, although no complete remissions were seen in the GC group, 17% of the responses seen in the ABC subtype were complete remissions. When the response in the ABC group was characterized as a function of CD70b and MYD88 mutation, the response rates seen among patients with mutant CD79b, wild-type CD79b, and mutant 79b/MYD88 were 71% (5 of 7), 34% (10 of 29), and 80% (4 of 5), further supporting the idea that drugs targeting specific pathways known to be dysregulated in a particular biologic subset of disease may be able to help overcome some of the adverse prognostic features of that disease.
Future Directions
Our rapidly evolving understanding of lymphoma at the molecular level has afforded new opportunities to better risk stratify patients, which will likely lead to more reasonable tailoring of treatment, and has firmly created the opportunity to more precisely treat these diseases with respect to their underlying biologic basis. At present, small molecules targeting BCR signaling mechanisms, PI3-kinase, Bcl-2 family members, and a host of monoclonal antibodies and antibody drug conjugates offer the potential to tailor the use of these small molecules with complementing current treatment paradigms, or creating new ones with less of an emphasis on specific cytotoxic therapy.
1. Li A. et al. Utilization of Ig heavy chain variable, diversity, and joining gene segments in children with B-lineage acute lymphoblastic leukemia: implications for the mechanisms of VDJ recombination and for pathogenesis . Blood . 2004 ; 103 : 4602 – 4609 .
2. Peled J.U. et al. The biochemistry of somatic hypermutation . Annu Rev Immunol . 2008 ; 26 : 481 – 511 .
3. Takahama Y. Journey through the thymus: stromal guides for T-cell development and selection . Nat Rev Immunol . 2006 ; 6 : 127 – 135 .
4. Swerdlow S.H. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues . Lyon, France : International Agency for Research on Cancer ; 2008 .
5. Menon M.P. , Pittaluga S. , Jaffe E.S. The histological and biological spectrum of diffuse large B-cell lymphoma in the World Health Organization classification . Cancer J . 2012 ; 18 : 411 – 420 .
6. Choi W.W. et al. A new immunostain algorithm classifies diffuse large B-cell lymphoma into molecular subtypes with high accuracy . Clin Cancer Res . 2009 ; 15 : 5494 – 5502 .
7. Kohlmann A. et al. Next-generation sequencing—feasibility and practicality in haematology . Br J Haematol . 2013 ; 160 : 736 – 753 .
8. Buchonnet G. et al. Distribution of BCL2 breakpoints in follicular lymphoma and correlation with clinical features: specific subtypes or same disease? Leukemia . 2002 ; 16 : 1852 – 1856 .
9. Sekiguchi N. et al. Follicular lymphoma subgrouping by fluorescence in situ hybridization analysis . Cancer Sci . 2005 ; 96 : 77 – 82 .
10. van Dongen J.J. et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: report of the BIOMED-2 Concerted Action BMH4-CT98-3936 . Leukemia . 2003 ; 17 : 2257 – 2317 .
11. Gu K. , Chan W.C. , Hawley R.C. Practical detection of t(14;18)(IgH/BCL2) in follicular lymphoma . Arch Pathol Lab Med . 2008 ; 132 : 1355 – 1361 .
12. Leich E. et al. Follicular lymphomas with and without translocation t(14;18) differ in gene expression profiles and genetic alterations . Blood . 2009 ; 114 : 826 – 834 .
13. Ott G. et al. Cytomorphologic, immunohistochemical, and cytogenetic profiles of follicular lymphoma: 2 types of follicular lymphoma grade 3 . Blood . 2002 ; 99 : 3806 – 3812 .
14. Schmitt C. et al. The bcl-2/IgH rearrangement in a population of 204 healthy individuals: occurrence, age and gender distribution, breakpoints, and detection method validity . Leuk Res . 2006 ; 30 : 745 – 750 .
15. Katzenberger T. et al. Cytogenetic alterations affecting BCL6 are predominantly found in follicular lymphomas grade 3B with a diffuse large B-cell component . Am J Pathol . 2004 ; 165 : 481 – 490 .
16. Hartmann E.M. , Ott G. , Rosenwald A. Molecular biology and genetics of lymphomas . Hematol Oncol Clin North Am . 2008 ; 22 : 807 – 823 vii .
17. Voorhees P.M. et al. Follicular lymphoma with a Burkitt translocation—predictor of an aggressive clinical course: a case report and review of the literature . Arch Pathol Lab Med . 2004 ; 128 : 210 – 213 .
18. Schwaenen C. et al. Microarray-based genomic profiling reveals novel genomic aberrations in follicular lymphoma which associate with patient survival and gene expression status . Genes Chromosomes Cancer . 2009 ; 48 : 39 – 54 .
19. Slack G.W. , Gascoyne R.D. Next-generation sequencing discoveries in lymphoma . Adv Anat Pathol . 2013 ; 20 : 110 – 116 .
20. Parsonnet J. et al. Helicobacter pylori infection and gastric lymphoma . N Engl J Med . 1994 ; 330 : 1267 – 1271 .
21. Dierlamm J. et al. Characteristic pattern of chromosomal gains and losses in marginal zone B cell lymphoma detected by comparative genomic hybridization . Leukemia . 1997 ; 11 : 747 – 758 .
22. Thome M. CARMA1, BCL-10 and MALT1 in lymphocyte development and activation . Nat Rev Immunol . 2004 ; 4 : 348 – 359 .
23. Kwee I. et al. Genomic profiles of MALT lymphomas: variability across anatomical sites . Haematologica . 2011 ; 96 : 1064 – 1066 .
24. Levy M. et al. Prognostic value of translocation t(11;18) in tumoral response of low-grade gastric lymphoma of mucosa-associated lymphoid tissue type to oral chemotherapy . J Clin Oncol . 2005 ; 23 : 5061 – 5066 .
25. Novak U. et al. The NFκB negative regulator TNFAIP3 (A20) is inactivated by somatic mutations and genomic deletions in marginal zone lymphomas . Blood . 2009 ; 113 : 4918 – 4921 .
26. Udomsakdi-Auewarakul C. et al. Hepatitis C virus infection in patients with Hodgkin lymphoma in Thailand . Blood . 2000 ; 95 : 3640 – 3641 .
27. Pileri P. et al. Binding of hepatitis C virus to CD81 . Science . 1998 ; 282 : 938 – 941 .
28. Cocquerel L. et al. CD81-dependent binding of hepatitis C virus E1E2 heterodimers . J Virol . 2003 ; 77 : 10677 – 10683 .
29. Salido M. et al. Cytogenetic aberrations and their prognostic value in a series of 330 splenic marginal zone B-cell lymphomas: a multicenter study of the Splenic B-Cell Lymphoma Group . Blood . 2010 ; 116 : 1479 – 1488 .
30. Rossi D. et al. The coding genome of splenic marginal zone lymphoma: activation of NOTCH2 and other pathways regulating marginal zone development . J Exp Med . 2012 ; 209 : 1537 – 1551 .
31. Mensah A.A. et al. Absence of NOTCH1 gene mutations in MALT lymphomas . Br J Haematol . 2012 ; 157 : 382 – 384 .
32. Watkins A.J. et al. An integrated genomic and expression analysis of 7q deletion in splenic marginal zone lymphoma . PLoS One . 2012 ; 7 e44997 .
33. Fresquet V. et al. High-throughput sequencing analysis of the chromosome 7q32 deletion reveals IRF5 as a potential tumour suppressor in splenic marginal-zone lymphoma . Br J Haematol . 2012 ; 158 : 712 – 726 .
34. Arribas A.J. et al. Splenic marginal zone lymphoma: comprehensive analysis of gene expression and miRNA profiling . Mod Pathol . 2013 ; 26 : 889 – 901 .
35. Arribas A.J. et al. Nodal marginal zone lymphoma: gene expression and miRNA profiling identify diagnostic markers and potential therapeutic targets . Blood . 2012 ; 119 : e9 – e21 .
36. Nakamura S. et al. Long-term clinical outcome of gastric MALT lymphoma after eradication of Helicobacter pylori: a multicentre cohort follow-up study of 420 patients in Japan . Gut . 2012 ; 61 : 507 – 513 .
37. Ternynck T. et al. Comparison of normal and CLL lymphocyte surface Ig determinants using peroxidase-labeled antibodies. I. Detection and quantitation of light chain determinants . Blood . 1974 ; 43 : 789 – 795 .
38. Damle R.N. et al. Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia . Blood . 1999 ; 94 : 1840 – 1847 .
39. Crespo M. et al. ZAP-70 expression as a surrogate for immunoglobulin-variable-region mutations in chronic lymphocytic leukemia . N Engl J Med . 2003 ; 348 : 1764 – 1775 .
40. Chen L. et al. Expression of ZAP-70 is associated with increased B-cell receptor signaling in chronic lymphocytic leukemia . Blood . 2002 ; 100 : 4609 – 4614 .
41. Wiestner A. et al. ZAP-70 expression identifies a chronic lymphocytic leukemia subtype with unmutated immunoglobulin genes, inferior clinical outcome, and distinct gene expression profile . Blood . 2003 ; 101 : 4944 – 4951 .
42. Oscier D.G. et al. Multivariate analysis of prognostic factors in CLL: clinical stage, IGVH gene mutational status, and loss or mutation of the p53 gene are independent prognostic factors . Blood . 2002 ; 100 : 1177 – 1184 .
43. Migliazza A. et al. Nucleotide sequence, transcription map, and mutation analysis of the 13q14 chromosomal region deleted in B-cell chronic lymphocytic leukemia . Blood . 2001 ; 97 : 2098 – 2104 .
44. Calin G.A. et al. Frequent deletions and down-regulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia . Proc Natl Acad Sci U S A . 2002 ; 99 : 15524 – 15529 .
45. Dohner H. et al. Genomic aberrations and survival in chronic lymphocytic leukemia . N Engl J Med . 2000 ; 343 : 1910 – 1916 .
46. Perez-Galan P. , Dreyling M. , Wiestner A. Mantle cell lymphoma: biology, pathogenesis, and the molecular basis of treatment in the genomic era . Blood . 2011 ; 117 : 26 – 38 .
47. Bosch F. et al. PRAD-1/cyclin D1 gene overexpression in chronic lymphoproliferative disorders: a highly specific marker of mantle cell lymphoma . Blood . 1994 ; 84 : 2726 – 2732 .
48. Marzec M. et al. Mantle cell lymphoma cells express predominantly cyclin D1a isoform and are highly sensitive to selective inhibition of CDK4 kinase activity . Blood . 2006 ; 108 : 1744 – 1750 .
49. Wiestner A. et al. Point mutations and genomic deletions in CCND1 create stable truncated cyclin D1 mRNAs that are associated with increased proliferation rate and shorter survival . Blood . 2007 ; 109 : 4599 – 4606 .
50. Chen R.W. et al. Truncation in CCND1 mRNA alters miR-16-1 regulation in mantle cell lymphoma . Blood . 2008 ; 112 : 822 – 829 .
51. Alt J.R. et al. Phosphorylation-dependent regulation of cyclin D1 nuclear export and cyclin D1-dependent cellular transformation . Genes Dev . 2000 ; 14 : 3102 – 3114 .
52. Dal Col J. et al. Distinct functional significance of Akt and mTOR constitutive activation in mantle cell lymphoma . Blood . 2008 ; 111 : 5142 – 5151 .
53. Barbash O. et al. Mutations in Fbx4 inhibit dimerization of the SCF(Fbx4) ligase and contribute to cyclin D1 overexpression in human cancer . Cancer Cell . 2008 ; 14 : 68 – 78 .
54. Quelle D.E. et al. Overexpression of mouse D-type cyclins accelerates G1 phase in rodent fibroblasts . Genes Dev . 1993 ; 7 : 1559 – 1571 .
55. Sherr C.J. Cancer cell cycles . Science . 1996 ; 274 : 1672 – 1677 .
56. Hofmann W.K. et al. Altered apoptosis pathways in mantle cell lymphoma detected by oligonucleotide microarray . Blood . 2001 ; 98 : 787 – 794 .
57. Chiarle R. et al. Increased proteasome degradation of cyclin-dependent kinase inhibitor p27 is associated with a decreased overall survival in mantle cell lymphoma . Blood . 2000 ; 95 : 619 – 626 .
58. Dreyling M.H. et al. Alterations of the cyclin D1/p16-pRB pathway in mantle cell lymphoma . Cancer Res . 1997 ; 57 : 4608 – 4614 .
59. Lwin T. et al. Cell adhesion induces p27Kip1-associated cell-cycle arrest through down-regulation of the SCFSkp2 ubiquitin ligase pathway in mantle-cell and other non-Hodgkin B-cell lymphomas . Blood . 2007 ; 110 : 1631 – 1638 .
60. Shakir R. , Ngo N. , Naresh K.N. Correlation of cyclin D1 transcript levels, transcript type and protein expression with proliferation and histology among mantle cell lymphoma . J Clin Pathol . 2008 ; 61 : 920 – 927 .
61. Raty R. et al. Ki-67 expression level, histological subtype, and the International Prognostic Index as outcome predictors in mantle cell lymphoma . Eur J Haematol . 2002 ; 69 : 11 – 20 .
62. Rosenwald A. et al. The proliferation gene expression signature is a quantitative integrator of oncogenic events that predicts survival in mantle cell lymphoma . Cancer Cell . 2003 ; 3 : 185 – 197 .
63. Determann O. et al. Ki-67 predicts outcome in advanced-stage mantle cell lymphoma patients treated with anti-CD20 immunochemotherapy: results from randomized trials of the European MCL Network and the German Low Grade Lymphoma Study Group . Blood . 2008 ; 111 : 2385 – 2387 .
64. Garcia M. et al. Proliferation predicts failure-free survival in mantle cell lymphoma patients treated with rituximab plus hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone alternating with rituximab plus high-dose methotrexate and cytarabine . Cancer . 2009 ; 115 : 1041 – 1048 .
65. Goy A. et al. Potential biomarkers of bortezomib activity in mantle cell lymphoma from the phase 2 PINNACLE trial . Leuk Lymphoma . 2010 ; 51 : 1269 – 1277 .
66. Hashimoto Y. et al. B-cell development in the thymus is limited by inhibitory signals from the thymic microenvironment . Blood . 2002 ; 100 : 3504 – 3511 .
67. Barth T.F. et al. Mediastinal (thymic) large B-cell lymphoma: where do we stand? Lancet Oncol . 2002 ; 3 : 229 – 334 .
68. Rosenwald A. et al. Molecular diagnosis of primary mediastinal B cell lymphoma identifies a clinically favorable subgroup of diffuse large B cell lymphoma related to Hodgkin lymphoma . J Exp Med . 2003 ; 198 : 851 – 862 .
69. Schmitz R. et al. Pathogenesis of classical and lymphocyte-predominant Hodgkin lymphoma . Annu Rev Pathol . 2009 ; 4 : 151 – 174 .
70. Green M.R. et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma . Blood . 2010 ; 116 : 3268 – 3277 .
71. Melzner I. et al. Biallelic mutation of SOCS-1 impairs JAK2 degradation and sustains phospho-JAK2 action in the MedB-1 mediastinal lymphoma line . Blood . 2005 ; 105 : 2535 – 2542 .
72. Steidl C. et al. MHC class II transactivator CIITA is a recurrent gene fusion partner in lymphoid cancers . Nature . 2011 ; 471 : 377 – 381 .
73. Eberle F.C. et al. Gray zone lymphoma: chromosomal aberrations with immunophenotypic and clinical correlations . Mod Pathol . 2011 ; 24 : 1586 – 1597 .
74. Eberle F.C. et al. Methylation profiling of mediastinal gray zone lymphoma reveals a distinctive signature with elements shared by classical Hodgkin’s lymphoma and primary mediastinal large B-cell lymphoma . Haematologica . 2011 ; 96 : 558 – 566 .
75. Nelson M. et al. Cytogenetic abnormalities and clinical correlations in peripheral T-cell lymphoma . Br J Haematol . 2008 ; 141 : 461 – 469 .
76. Salaverria I. et al. Genomic profiling reveals different genetic aberrations in systemic ALK-positive and ALK-negative anaplastic large cell lymphomas . Br J Haematol . 2008 ; 140 : 516 – 526 .
77. Iqbal J. et al. Molecular signatures to improve diagnosis in peripheral T-cell lymphoma and prognostication in angioimmunoblastic T-cell lymphoma . Blood . 2010 ; 115 : 1026 – 1036 .
78. Amin H.M. , Lai R. Pathobiology of ALK+ anaplastic large-cell lymphoma . Blood . 2007 ; 110 : 2259 – 2267 .
79. Stein H. et al. CD30+ anaplastic large cell lymphoma: a review of its histopathologic, genetic, and clinical features . Blood . 2000 ; 96 : 3681 – 3695 .
80. Jundt F. et al. Activated Notch1 signaling promotes tumor cell proliferation and survival in Hodgkin and anaplastic large cell lymphoma . Blood . 2002 ; 99 : 3398 – 3403 .
81. Moritake H. et al. C-MYC rearrangement may induce an aggressive phenotype in anaplastic lymphoma kinase positive anaplastic large cell lymphoma: identification of a novel fusion gene ALO17/C-MYC . Am J Hematol . 2011 ; 86 : 75 – 78 .
82. Bohling S.D. et al. Analysis of gene expression profile of TPM3-ALK positive anaplastic large cell lymphoma reveals overlapping and unique patterns with that of NPM-ALK positive anaplastic large cell lymphoma . Leuk Res . 2008 ; 32 : 383 – 393 .
83. Lamant L. et al. Gene-expression profiling of systemic anaplastic large-cell lymphoma reveals differences based on ALK status and two distinct morphologic ALK+ subtypes . Blood . 2007 ; 109 : 2156 – 2164 .
84. Merkel O. et al. Identification of differential and functionally active miRNAs in both anaplastic lymphoma kinase (ALK)+ and ALK- anaplastic large-cell lymphoma . Proc Natl Acad Sci U S A . 2010 ; 107 : 16228 – 16233 .
85. Thompson M.A. et al. Differential gene expression in anaplastic lymphoma kinase-positive and anaplastic lymphoma kinase-negative anaplastic large cell lymphomas . Hum Pathol . 2005 ; 36 : 494 – 504 .
86. Feldman A.L. et al. Overexpression of Syk tyrosine kinase in peripheral T-cell lymphomas . Leukemia . 2008 ; 22 : 1139 – 1143 .
87. Piccaluga P.P. et al. Gene expression analysis of peripheral T cell lymphoma, unspecified, reveals distinct profiles and new potential therapeutic targets . J Clin Invest . 2007 ; 117 : 823 – 834 .
88. Streubel B. et al. Novel t(5;9)(q33;q22) fuses ITK to SYK in unspecified peripheral T-cell lymphoma . Leukemia . 2006 ; 20 : 313 – 318 .
89. Pechloff K. et al. The fusion kinase ITK-SYK mimics a T cell receptor signal and drives oncogenesis in conditional mouse models of peripheral T cell lymphoma . J Exp Med . 2010 ; 207 : 1031 – 1044 .
90. Dierks C. et al. The ITK-SYK fusion oncogene induces a T-cell lymphoproliferative disease in mice mimicking human disease . Cancer Res . 2010 ; 70 : 6193 – 6204 .
91. Martinez-Delgado B. et al. Expression profiling of T-cell lymphomas differentiates peripheral and lymphoblastic lymphomas and defines survival related genes . Clin Cancer Res . 2004 ; 10 : 4971 – 4982 .
92. Cuadros M. et al. Identification of a proliferation signature related to survival in nodal peripheral T-cell lymphomas . J Clin Oncol . 2007 ; 25 : 3321 – 3329 .
93. Ballester B. et al. Gene expression profiling identifies molecular subgroups among nodal peripheral T-cell lymphomas . Oncogene . 2006 ; 25 : 1560 – 1570 .
94. Piva R. et al. Gene expression profiling uncovers molecular classifiers for the recognition of anaplastic large-cell lymphoma within peripheral T-cell neoplasms . J Clin Oncol . 2010 ; 28 : 1583 – 1590 .
95. Agnelli L. et al. Identification of a 3-gene model as a powerful diagnostic tool for the recognition of ALK-negative anaplastic large-cell lymphoma . Blood . 2012 ; 120 : 1274 – 1281 .
96. Zettl A. et al. Genomic profiling of peripheral T-cell lymphoma, unspecified, and anaplastic large T-cell lymphoma delineates novel recurrent chromosomal alterations . Am J Pathol . 2004 ; 164 : 1837 – 1848 .
97. Nagel S. et al. Amplification at 7q22 targets cyclin-dependent kinase 6 in T-cell lymphoma . Leukemia . 2008 ; 22 : 387 – 392 .
98. Thorns C. et al. Chromosomal aberrations in angioimmunoblastic T-cell lymphoma and peripheral T-cell lymphoma unspecified: a matrix-based CGH approach . Genes Chromosomes Cancer . 2007 ; 46 : 37 – 44 .
99. Vasmatzis G. et al. Genome-wide analysis reveals recurrent structural abnormalities of TP63 and other p53-related genes in peripheral T-cell lymphomas . Blood . 2012 ; 120 : 2280 – 2289 .
100. de Leval L. , Gisselbrecht C. , Gaulard P. Advances in the understanding and management of angioimmunoblastic T-cell lymphoma . Br J Haematol . 2010 ; 148 : 673 – 689 .
101. de Leval L. et al. The gene expression profile of nodal peripheral T-cell lymphoma demonstrates a molecular link between angioimmunoblastic T-cell lymphoma (AITL) and follicular helper T (TFH) cells . Blood . 2007 ; 109 : 4952 – 4963 .
102. Rodriguez-Pinilla S.M. et al. Peripheral T-cell lymphoma with follicular T-cell markers . Am J Surg Pathol . 2008 ; 32 : 1787 – 1799 .
103. Quivoron C. et al. TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis . Cancer Cell . 2011 ; 20 : 25 – 38 .
104. Lemonnier F. et al. Recurrent TET2 mutations in peripheral T-cell lymphomas correlate with TFH-like features and adverse clinical parameters . Blood . 2012 ; 120 : 1466 – 1469 .
105. Couronne L. , Bastard C. , Bernard O.A. TET2 and DNMT3A mutations in human T-cell lymphoma . N Engl J Med . 2012 ; 366 : 95 – 96 .
106. Cairns R.A. et al. IDH2 mutations are frequent in angioimmunoblastic T-cell lymphoma . Blood . 2012 ; 119 : 1901 – 1903 .
[/level-membership-for-hematology-oncology-and-palliative-medicine-category][not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
B- and T-Lymphocyte Development
The ontogeny of B cells is complex and proceeds through a series of developmental and genetic steps, leading to the creation of unique B cells carrying unique B-cell receptors, capable of identifying unique antigen (
Figure 29-3 ). These developmental and genetic steps are defined by changes in the genome at the antibody loci. B lymphocytes play a fundamental role in the humoral branch of the adaptive immune system and can be distinguished from other types of lymphocytes by the panoply of cell surface markers (CD19, CD20, CD22, CD23) and the presence of the B-cell receptor (BCR), which itself is a surface immunoglobulin. Undifferentiated and immature B cells are produced in the intramedullary compartment of the bone. The antibodies produced by individual B cells are composed of two identical light chains (κ or λ), composed of two segments called the
V and J segments, and one heavy chain (M, D, A, G) composed of three segments called the
V, D, and J segments, where V denotes the variable region that comprises the terminal Fab portion of the antibody.
1 At the genetic level, early lymphocytes in the bone marrow undergo a process referred to as
VDJ recombination (see
Figure 29-2), which is the mechanism by which B cells produce immunoglobulin diversity. Once B lymphocytes undergo VDJ recombination, they exit the bone marrow, where they then hone to secondary lymphoid organs such as the lymph node. There they undergo highly controlled mutagenesis, in a process referred to as
somatic hypermutation (SMH). SMH plays a crucial role in the affinity maturation of the humoral response, by mediating the introduction of discrete random mutations in the loci of the genes encoding the variable (V) regions of the immunoglobulin.
2 Once engaged by antigen, the subsequent proliferation of the B cell undergoes an incredibly high rate of somatic mutation that is at least 100,000- to 1 millionfold higher than the normal rate of mutation across the genome. This highly regulated mutagenesis, mediated initially by activation-induced cytidine deaminase (AID), introduces single base substitutions predominantly at hotspots in the DNA called
hypervariable regions.
2 Paradoxically, it is this directed hypermutation of the BCR variable regions that allows for the selection of B cells with an enhanced capability to identify and bind specific foreign antigens, which, when dysregulated or uncontrolled, creates an opportunity for aberrant somatic hypermutation, leading to lymphomagenesis. This potential for any error, or error-prone mutagenesis of B lymphocytes, can create the background for malignant transformation.
In contrast, T lymphocytes play a central role in cell-mediated immunity and can be distinguished from other lymphocytes by the expression of their own unique set of surface proteins (e.g., CD3, CD4, CD8) and the T-cell receptor (TCR). T lymphocytes typically mature in the thymus, in contrast to the germinal center for B lymphocytes. In the thymus, T lymphocytes expand through multiple series of cell division to produce a large population of more immature cells. Initially, these cells do not express CD4 or CD8 and are referred to as double-negative thymocytes.
3 As they differentiate into a specific subset of T lymphocytes, they eventually become positive for both CD4 and CD8 (that is, double positive). In their final step of thymic maturation they emerge as single positive T lymphocytes, including CD4-positive/CD8-negative T-helper cells or CD4-negative/CD8-positive cytotoxic T cells. The TCR recognizes antigen bound to major histocompatibility complex (MHC). Engagement of the TCR by antigen incites a number of downstream events important to the
lymphocytes role in mediating cellular immunity. The generation of the TCR is quite similar to that discussed earlier for the BCR. Composed of four chains and existing as a dimeric structure (alpha pairing with the beta chain or the gamma chain pairing with the delta chain), unique TCR is produced through VJ (the alpha and gamma chains) or VDJ (the beta and delta chains) recombination. Like the heavy and light chains comprising the BCR, it is the unique combination of these specific regions that accounts for the diversity of the TCR.
3
Figure 29-1 (A) Relative incidence of select B- and T-cell lymphomas. (B) Relative incidence of select T-cell lymphomas. A survey of 1314 peripheral T-cell and natural killer/T-cell leukemia cases from 22 sites around the world evaluated the histopathology of previously untreated patients consecutively diagnosed between 1990 and 2002. PTCL-NOS was the most common subtype at 25.9%. Anaplastic large cell lymphoma (ALCL) ALK+/−, angioimmunoblastic lymphoma, adult T-cell leukemia/lymphoma, and natural killer/T-cell lymphoma were also common subtypes. (C) Classification of the mature T-cell lymphoma (WHO). PTCL refers to a heterogeneous group of aggressive T- and NK-cell lymphomas, characterized by the involvement of mature (thymic) T or NK cells. The term peripheral in PTCL does not refer to anatomic sites. T- and NK-cell lymphomas may be characterized as cutaneous, nodal, extranodal, or leukemic disseminated. Data from Armitage JO et al. Ann Oncol. 2004;15:1447-1449; Rodriguez J et al. Crit Rev Oncol Hematol. 2008; Swerdlow SH et al., WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; 2008. Data in B from Armitage J et al. J Clin Oncol. 2008;26:4124-4130.
Figure 29-2 Schematic representation of the ontogeny of B-lymphocytes and select lymphoma relative to their sites of origin.
Although complex, the ontogeny of B and T lymphocytes provides the basis for thinking about the diversity of different forms of non-Hodgkin and Hodgkin lymphoma, and in particular the cell of origin (see
Figure 29-3). In the case of the lymphoid malignancies, identifying the cell of origin goes well beyond the identification of the lymphocyte and is ascribed to the discrete step in development at which the clonal expansion is thought to occur. Increasingly, the understanding of immunohistochemical staining and cytogenetics has allowed for a description of the natural ontogeny of lymphocytes and, by extension, the more accurate identification of the cell of origin, leading to a more biologically relevant classification of different subtypes of lymphomas (
Table 29-2 ).
Pathogenesis of Diffuse Large B-Cell Lymphoma
Diffuse large B-cell lymphoma (DLBCL) comprises a group of aggressive lymphomas with very heterogeneous clinicopathologic and molecular genetic features. The current 2008 World Health Organization (WHO) classification
4 defines DLBCL as a diffuse growth of neoplastic large B-lymphoid cells with a nuclear size equal to or exceeding normal macrophage nuclei or more than twice the size of a normal lymphocyte. DLBCL is the most common subtype of lymphoma and accounts for 30% to 40% of adult NHL.
5 The 2008 WHO classification recognizes a large number of DLBCL subgroups primarily based on their distinct morphologic, biologic, immunophenotypic, or clinical features. Despite revisions to the classification, a significant proportion of large-cell lymphomas remain biologically heterogeneous, not fitting into any specific DLBCL subgroup. These are typically defined as DLBCL, not otherwise specified (DLBCL-NOS). DLBCL-NOS is a diagnosis of exclusion, not corresponding to one of the specific subtypes. It is still the most common type of lymphoma, accounting for 25% to 30% of NHLs.
5
Figure 29-3 Stages of B-cell development and correlation with different lymphoid diseases. CLL, Chronic lymphocytic leukemia.
The focus in recent years has shifted toward the identification of molecular alterations and specific pathogenetic pathways leading to transformation in DLBCL. Gene expression profiling (GEP) of DLBCLs has identified at least two major molecular subtypes, which correlate with prognosis and may have relevance for treatment based on the dominant signaling pathways. DLBCL could be classified by GEP as resembling either germinal center B cells (GCBs) or activated B cells (ABCs), establishing a putative “cell of origin”
5 (
Figure 29-4 ). Because GEP is technically difficult and therefore cannot be performed in every case for diagnostic purposes, various algorithms based on immunohistochemical profiles have been proposed as surrogates of the GEP.
5,6 Although the correspondence is not precise, similar prognostic correlations can be drawn with immunohistochemically defined subgroups. These immunohistochemical algorithms have facilitated risk stratification of DLBCL patients and DLBCL research using archival materials.
5 The ABC and GCB DLBCL subtypes, originally formulated based on a cell-of-origin model, have more recently been shown to be characterized by the different pathways of cellular transformation and oncogenesis. Although substantial progress has been made toward molecular subclassification of DLBCLs, the translation to effective treatment strategies is only now beginning to be explored.
5 Next-generation sequencing (NGS) platforms have evolved to provide an accurate and comprehensive means for the detection of molecular mutations and will likely contribute to a more sophisticated understanding of DLBCL biology and, it can be hoped, more biologically relevant treatment.
7
Table 29-2
Characteristic Immunophenotypic and Genetic Features of Selected Lymphoma Subtypes
DLBCL, Diffuse large B-cell lymphoma; GC, germinal center; MALT, extranodal marginal zone B-cell lymphoma of mucosa-associated lymphoid tissues; SHM, somatic hypermutation.
Although increased understanding of DLBCL based on cell of origin is emerging, it is clear that the dysregulated pathways across this entity are often overlapping, providing redundancy to promote growth and prevent cell death signals. These signaling pathways are often necessary for the development and differentiation of normal memory B cells and plasma cells and are coopted during malignant transformation.
For example, in ABC-derived DLBCL, most of the deranged signaling pathways converge to aberrantly activate the NFκB pathway, leading to evasion of cell death and resistance to chemotherapy (see
Figure 29-4,
B). Activation of this molecular pathway translates into poor outcomes for patients with ABC-DLBCL compared to GC-DLBCL, with 5-year overall survival of 40% versus 60%, respectively. Constitutive activation of NFκB is complex, involving both “classical” and “alternative” pathways, and can be regulated by many different mechanisms. Derangements leading to activation of NFκB can be attributed to a number of different molecular lesions, including tonic signaling of the BCR. BCR signaling is essential to B-cell development, antigen selection, and humoral immunity. Aberrations in BCR signaling, including tonic signaling that occurs in the absence of antigenic stimulation, can lead to uncontrolled B-cell growth and survival.
BCR signaling, whether it be in a normal or malignant B-cells, leads to the formation of the CBM complex consisting of
CARD11,
BCL10, and
MALT1. This complex leads to activation of NFκB through direct activation of NFκB and inhibition of negative NFκB regulators. Gain-of-function somatic mutations of the coiled-coil domain of CARD11 are found in nearly 10% of ABC-DLCBL and 16% of primary CNS lymphoma (an ABC variant). This mutant CARD11 leads to initiation of the NFκB cascade via IKKβ signaling in cooperation with casein kinase 1α (CK1α) and MALT1. MALT1 can also augment NFκB signaling through cleavage of two negative NFκB regulators, namely, A20 and CYLD.
TNFAIP3, the gene encoding A20, can also be inactivated through mutations, deletions, or epigenetic silencing in ABC-DLCBL. Mutations of other negative regulators of NFκB, such as
IKBKA encoding the inhibitor IkBα, lead to uninhibited translocation of NFκB to the nucleus and increased transcription of NFκB-dependent genes. Interestingly, linkage of this intrinsic ABC biology to B-cell receptor signaling may offer an interesting opportunity to affect the adverse prognostic features of this DLBCL phenotype.
Figure 29-4 (A) Classification of diffuse large B-cell lymphoma based on cell of origin. The left panel represents hierarchical clustering of the genes selectively expressed in GC-derived DLBCL (yellow) and ABC-derived DLBCL (blue). The right panel compares normal lymphocytes from blood and lymph nodes in various states of B-cell activation.
(B) Gene expression profile for the GC and ABC subtypes of DLBCL reveal that the ABC phenotype is enriched for NFκB-regulated genes, while the GC phenotype is enriched for Bcl-6-regulated genes. Gene expression profiling has revealed three subtypes of DLBCL, two of which include the germinal center subtype (GC) and the activated B-cell subtype (ABC). These molecular phenotypes override the prognostic impact of the International Prognostic Index (IPI). Courtesy Wyndham Wilson. Data in A from Alizadeh et al. Nature 2000;403:503-511.
In addition to the BCR-CBM effects on NFκB regulation, the BCR can activate NFκB through a number of alternative mechanisms. On antigenic stimulation or tonic dysregulation, the BCR heavy chains are coupled to cell surface markers CD79a and CD79b. This leads to phosphorylation of immunoreceptor tyrosine-based activation motifs (ITAM) by LYN, FYN, and BLK, which then recruit SYK, engaging the NFκB, PI3K, NFAT MAP kinase and RAS signaling pathways. Currently, an inhibitor of SYK is under investigation as a therapeutic target in those DLBCLs known to be addicted to BCR signaling. Substitutions and deletions of the CD79a and CD79b ITAMs are present in more than 20% of ABC-DLBCL and lead to a gain of function of the BCR signaling pathway. These mutants evade negative feedback by LYN and also allow for increased cell surface localization of the BCR.
Tonic BCR signaling also recruits Bruton’s tyrosine kinase (BTK) via the PI3K pathway. This tends to occur in ABC-DLBCL that do not possess CARD11 mutations. BTK is required for the survival of wild-type CARD11 ABC-DLBCL and knockdown of its components or effectors SYK, BLNK, PLCγ2, PI3Kδ, PKCβ abrogates NFκB signaling. BTK is activated by LYN or SYK, and on activation initiates signaling of the classical NFκB pathway. In addition, BTK forms a complex with BLNK, which, through a series of events that activates CARD11, contributes further to activation of the NFκB pathway. BTK inhibitors have demonstrated promising activity in patients with ABC-DLBCL and other lymphomas with aberrant BCR signaling, such as CLL.
All known ABC-DLBCL cell lines and 30% of tissue samples from patients with ABC DLBCL harbor MYD88
gain-of-function mutations in the toll-interleukin receptor (TIR) domain. MYD88 is an adaptor protein that interacts with IRAK4 and IRAK1 to activate both the NFκB and interferon pathways through TRAF6. Mutant MYD88 promotes secretion of the cytokines IL6 and IL10 activating the JAK/STAT pathway. This potentiates NFκB through a positive feedback loop.
Many of the mutations just discussed are expressed in tandem and are restricted to the ABC-DLBCL variant over GCB-derived DLBCL. As testament to the redundancy in these signaling pathways, knockdown of parallel pathways in cell lines or mouse models has demonstrated synergistic cytotoxicity in ABC-DLBCL, compared to the knockdown of singular elements in a pathway. The clarification of this underlying biology has led to the identification of several new drugs potentially affecting these specific pathways, including inhibitors of Syk, BTK, and CD79a and b, and proteasome inhibitors that inhibit degradation of IkB, leading to inhibition of NFκB.
Although there is overlap in the pathogenesis of GC- and ABC-derived DLBCL, there are unique features of both that may create opportunities to affect the discrete natural history of these subtypes rationally. For example, transcription factors, oncogenes, and epigenetic alterations are often responsible for driving proliferation and survival in GC-DLBCL. BCL6, an oncogene, is a transcriptional repressor considered the hallmark of GC-DLBCL, and acts as a master regulator of the germinal center reaction. Although physiologically active in normal germinal center B cells promoting somatic hypermutation (SMH) and class switch recombination, this pathway is co-opted in malignant lymphomas to enforce the “damage phenotype” of GC-DLBCL. Dysregulation through point mutations and translocations of the 3q27 locus lead to constitutive activation of this transcription factor in over 50% of DLBCL lymphomas. Specifically, mutations in the BCL6 autoregulatory domains upstream from the promoter regions via somatic hypermutation and AID result in lost feedback loop inhibition from Bcl6, IRF4, and STAT5. This phenomenon leads to the GC-DLBCL subtype. In addition, the Bcl6 BTB domain recruits the co-repressors NCOR, BCOR, and SMRT, along with HDACs to repress genes involved in the DNA damage response such as ATR, p53, CHK1, p21, and p27. This repression leads to unchecked cell cycle progression in the face of inhibited apoptosis and permissive DNA damage. Furthermore, the Bcl6 RD2 domain recruits MTA3 to repress PRDM1, which encodes Blimp-1, the master regulator of plasma cell development, trapping centroblasts within the germinal center. Interestingly, Bcl6 inhibits both MYC and BCL2, but in double-hit lymphomas, where these two oncogenes are overexpressed by translocations, they overcome repression by Bcl6, leading to even more aggressive variants of DLBCL.
Another target gene of Bcl6 is IRF4, a transcription factor that facilitates exit from the germinal center, leading to plasmacytic differentiation. Bcl6 silencing of IRF4 further inhibits transcription of Blimp-1, which would retain cells in the germinal center. IRF4 may also be inhibited by translocations in GC-DLBCL (predominantly pediatric cases). In these cases, IRF4 may serve as an oncogene. Nonstructural aberrations not interfering with IRF4 function have been found to be required for survival of ABC-DLBCL. The effects of IRF4 in ABC and GC-DLBCL are clearly different, and this likely contributes to their classification by cell of origin. One mechanism proposed is that IRF4 works as a positive feedback loop involving CARD11.
Although Bcl6 translocations occur in GC-DLBCL (10%), translocations of the 3q27 locus involving the BCL6 gene that lead to arrest of cells in the plasmablastic stage of development can be seen in 30% of ABC-DLBCL. These translocations have promiscuous partners and ultimately lead to silencing of PRDM1. The tumor suppressor functions of PRDM1 may be silenced by truncation, deletion, mutation, or epigenetic modifications as well.
Transcriptional control has also been influenced by mutations in epigenetic regulators. CREBBP and EP300 encode histone acetyl-transferases (HATs) that have classically been known to modify chromatin condensation and, by extension, transcription. It is now known that these enzymes also influence the activity of key modulators of DLBCL such as p53, NFκB, and Bcl6. Acetylation can act either as an activating posttranslational modification (p53), or as an inhibitory posttranslational modification (Bcl6). Nonoverlapping mutations of CREBBP (40%) and EP300 (10%) are found in GC-DLBCL. The inactivating mutations in these two genes lead to the activation of Bcl6 and inhibition of p53 by impaired acetylation. Reversal of this effect may be accomplished by therapeutic alteration of these posttranslational modifications pharmacologically with pan-Class1-2 histone deacetylase (HDAC) inhibitors.
GC-DLBCL has also been found to have mutations in the EZH2 component of the polycomb repression complex-2 (PRC2). This complex has histone methyltransferase activity and primarily trimethylates histone H3 on lysine 27 (i.e., H3K27me3), a mark of transcriptionally silent chromatin. In order to produce the pathologic effect, mutations are required to be heterozygous, allowing the wild-type allele to methylate H3K27, followed by the uncontrolled methylation mediated by the enzyme encoded by the mutant allele. This action leads to hypermethylated CpG islands, which in turn attracts methyl-CpG binding domain proteins (MBDP: MBD1, MBD2, MBD3, MeCP2 i Kaiso) to recruit HDACs, leading chromatin condensation and transcriptional silencing. Hypomethylating drugs have been shown in preclinical models to have therapeutic potential
and synergize with HDAC inhibitors to restore normal methylation and acetylation patterns on histones.
Although only recently described, the role of microRNAs in DLBCL lymphomagenesis has begun to emerge. MiRNAs can have both oncogenic and tumor suppressor properties. The miR-17-92 family, also known as oncomir-1, has been found to be amplified specifically in GC-DLBCL, which leads to increased expression of MYC and downregulation of BIM and p21. miR-17-92 also has an inverse relationship with p53, each inhibiting the other.
Molecular Pathogenesis of Follicular Lymphoma
Follicular lymphoma is a B-cell lymphoma derived from germinal center B cells with rearranged heavy- and light-chain genes. This derivation is depicted by the immunoarchitectural features of follicular lymphoma, particularly reflected in the expression of a germinal center immunohistochemical profile as well as a follicular growth pattern. Cytogenetically, about 90% of follicular lymphoma cases harbor the t(14;18) or variant B-cell CLL/lymphoma 2 (BCL2) rearrangements with immunoglobulin kappa or lambda on chromosome 2 and 22, respectively. Although centrocytes and centroblasts are regarded as the particular cell of origin, the lymphomatous counterpart demonstrates ongoing somatic hypermutations, which is of particular interest as it decreases the detection rate of follicular lymphoma by standard polymerase chain reaction (PCR) testing.
The most prominent cytogenetic abnormality in follicular lymphoma involves t(14;18) with rearrangement of BCL2. Normal BCL2 protein has an antiapoptotic effect by affecting mitochondrial permeability and release of cytochrome c. During the translocation of the IgH regulatory apparatus on chromosome 14 and the bcl-2 gene on chromosome 18, BCL2 is juxtaposed to immunoglobulin heavy-chain gene (IgH) sequences, including enhancer sequences, which brings BCL2 under the influence of the immunoglobulin promoter region. This leads to deregulated expression/overexpression of a functional protein, promoting increased cell survival and neoplastic transformation.
The rearrangement can occur in different breakpoint regions as noted:
• 65% to 70% occur in the major breakpoint region (MBR) located in an untranslated region of the last exon (exon 3).
• 20% occur at the 3′ end of the BCL2 gene or 5′ to the minor cluster region (mcr).
• 10% occur in the mcr, which is about 30 kb outside of the BCL2 gene.
Although additional breakpoints are being studied, efforts to associate these different breakpoints with different clinical characteristics or clinical outcome have been unrevealing.
8 Testing for the
BCL2 rearrangement can be determined by conventional cytogenetic karyotyping, fluorescence in situ hybridization (FISH), or PCR. Although conventional cytogenetic karyotyping detects the majority of cases with
BCL2 rearrangement, independent of the partner gene, the limiting factor with this approach remains the availability of viable tissue. In comparison, FISH can be performed on paraffin-embedded tissue and detects the translocation in up to 90% of cases. Lower detection rates have been reported depending on the ethnic group studied—such as in Japanese patients, where the frequency of the t(14:18) has been shown less frequently compared to patients from Western countries.
9 Of interest, PCR testing has been proven to be less valuable, as most PCR assays contain primers targeting the MBR and mcr regions, which decreases the detection rate to 60%.
10 Although increased detection rates can be achieved by adding more primers to the 5′ mcr and 3′ MBR breakpoint regions, targeting incomplete DH-JH rearrangements or mutations of the primer-binding sites, false-negative PCR results are common and still occur with alternate breakpoints as mentioned earlier.
11 Therefore, up-to-date FISH analysis remains the preferred testing modality.
11 The absence of
BCL2 translocation using the routinely available methods is described in a subset of follicular lymphomas, particularly primary cutaneous and pediatric follicular lymphoma. Further complicating the matter is the fact that multiple follicular lymphoma cases have been shown to increase their
BCL2 expression through alternate mechanisms, as shown in cases with an addition of 18q or trisomy 18.
12 Furthermore, the frequency of
BCL2 rearrangement is not only variable in different subsets of follicular lymphomas, but also differs as a function of the grade of the follicular lymphoma. More than 80% of low-grade follicular lymphoma cases possess the translocation, whereas higher grade follicular lymphoma cases harbor the translocation with a frequency of 70% and less than 15% in grades 3A and 3B, respectively.
13 However, a positive result for
BCL2 rearrangement demands additional studies, as the rearrangement is not specific and has been identified in other subtypes of lymphomas as well as in healthy individuals and in benign follicular hyperplasia.
14 An additional random locus of genetic alteration in follicular lymphoma includes alterations of BCL6 at 3q27. Grade 3B follicular lymphomas frequently have rearrangements of this locus similar to diffuse large B-cell lymphoma.
13,15 The Bcl6 protein is a zinc-finger protein that acts as a repressor of IRF-4/MUM-1 transactivating ability, as well as influencing cell cycle control and proapoptotic
genes. The major breakpoint region of this translocation is within the 5′ first exon and a portion of the first intron of the Bcl-6 gene. In addition to translocation events, the coding region of the Bcl6 gene is also altered by somatic mutation.
Because t(14;18) can be identified in healthy individuals, the translocation is thought to be an early event in B-cell development, neither necessary nor sufficient for lymphomagenesis in B cells. The
BCL2 rearrangement is thought to enhance the life span of the neoplastic cell, which in return retains the capability to develop additional genetic defects. So far a myriad of additional abnormalities have been described, including gains of 1q, 2p, 7, 8q, 12q, 18q, and X and losses of 1p, 6q, 10q, 13q, and 17p.
16 After detection of these additional abnormalities, prognostic factors have been identified, including gains of 1q, 12, and X, as well as losses of 1p, 17p, and 17q, all considered late events with a poor prognosis or associated with histologic transformation.
16 Transformation to diffuse large B-cell lymphoma is a relatively common event, occurring in 25% to 35% of follicular lymphoma cases.
16 To date, large-cell transformation has been linked to gains of 7, 12q, and X; losses of 4q, 13q, and 17p;
MYC deregulation; and inactivation of
TP53 and
CDKN2A. Although
MYC deregulation has only been occasionally described in follicular lymphoma, it harbors a particularly poor diagnosis.
17 Newer diagnostic modalities such as comparative genomic hybridization (CGH) have firmly established that additional chromosomal abnormalities, such as gains in 12p and 18p in more than 10% of cases and deletions of 3q, 9p, and 11q in less than 10% of cases, are associated with particularly poor prognostic factors. Although these deletions involve the
CDKN2A and
CDKN2B loci as well as the LIM domain and have been partially associated with decreased survival, the overall significance of these findings has yet to be determined.
18 Next-generation sequencing has been used to elucidate the mutational landscape of follicular lymphoma. With this modality it was discovered that 89% of cases harbored MLL2 mutations, 33% of cases showed CREBBP mutations, and 13% of cases depicted MEF2B mutations. E300 mutations were identified in 9% of cases, EZH2 mutations in 7% of cases, and the TBL1XR1/TP63 fusion gene was identified at a very low incidence. So far mainly the presence of EZH2 is of interest, as this particular mutation supports the hypothesis that the polycomb repressor complex-2 orchestrates the pathogenesis in a subset of germinal center–derived lymphomas, but in general the implications of these findings are still under investigation.
19 Additional factors thought to be involved in the pathogenesis of follicular lymphoma include the nature of the microenvironment. In particular, the role of T cells and dendritic cells appears to be of substantial importance. GEP of the microenvironment has identified two distinct signatures. These signatures have been categorized as immune response 1 (IR1) and immune response 2 (IR2). Whereas immune response 1 (IR1) shows increased expression of T-cell genes and the macrophage genes
TNFSF13B and
ACTN1, immune response 2 (IR2) shows increased expression of follicular dendritic cell genes and different macrophage genes. Immune response 1 has been linked to a more favorable prognosis, but immune response 2 has been linked to an unfavorable prognosis.
16 To date, these genetic changes have not yet led to the tailoring of specific therapies for specific genetic subsets of the disease.
Molecular Pathogenesis of Marginal Zone Lymphoma and Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma
Marginal zone lymphoma (MZL) arises from memory B cells that normally reside in the marginal zone of secondary lymphoid follicles. MZL is the third most common type of non-Hodgkin lymphoma, representing about 10% of all cases of NHL. At least three distinct entities of MZL have been included in the WHO classification, including extranodal MZL of mucosa-associated lymphatic tissue (MALT) lymphoma, which represents more than two thirds of all MZL cases; splenic MZL with circulating villous lymphocytes; and nodal MZL. Clinically, MZL typically presents as early-stage disease in most patients and exhibits an indolent course in most patients. However, MZL can become disseminated or transform into aggressive large-cell lymphoma. Over the past two decades we have witnessed unprecedented progress in our understanding of the biology of MZL at the molecular level, and some of that knowledge is being translated into the development of rational treatment paradigms.
Because MALT lymphoma develops in tissues that are not necessarily abundantly enriched with lymphocytes, the first step in the pathogenesis is the abnormal aggregation of lymphoid tissues in extranodal sites and organs. The etiologic factor in this process can be infectious and/or inflammatory. Gastric MALT lymphoma represents more than 50% of all MALT lymphoma and has been shown to be intricately associated with colonization of the stomach by the bacterium
Helicobacter pylori.
20 Although aggregation of lymphocytes stimulated by
H. pylori is a prerequisite for MALT lymphoma,
H. pylori infection alone is not sufficient to cause the malignant transformation of lymphocytes into lymphoma cells.
H. pylori is found in more than 90% of patients with MALT lymphoma, yet most patients with
H. pylori infection do not develop lymphoma.
H. pylori also produces inflammation and reactive oxygen species (ROS)
that further increase genomic instability, which is essential for lymphomagenesis. In support of the theory of genomic instability in gastric MALT lymphoma, a balanced translocation t(11;18)(q21;q21) was reported in up to 50% of all cases of the disease.
21 The translocation results in the production of fusion protein API/BIRC3-MALT1, which interacts with CARD11/CARMA1 to activate the critical pro-survival transcription factor NFκB.
22 The translocation t(11;18) has been found to be mutually exclusive of other cytogenetic abnormalities, further suggesting its role as an important, if not the singular, driver for the development of MALT lymphoma.
23 Importantly, the emergence of t(11;18) is associated with unfavorable clinical outcomes and resistance to antibiotics and chemotherapy in the treatment of gastric MALT lymphoma.
24 In addition to its occurrence in the gastrointestinal tract, MALT lymphoma has been reported in the lung, ocular adnexa, salivary gland, and less commonly the skin and thyroid. MALT lymphoma in these sites often involves a different etiology. For example,
Chlamydophila psittaci or
Borrelia burgdorferi is thought to be responsible for MALT lymphoma of the ocular adnexa and skin, respectively, whereas inflammation caused by autoimmune disease is responsible for MALT lymphoma in the thyroid and salivary gland. Cytogenetic abnormalities are well described for MALT lymphoma in these sites and include t(14;18) and t(1;14), which involve MALT1 and BCL10, respectively. Other genetic abnormalities in MALT lymphoma include t(3;14) involving IGH-FOP1, +3/3q, +18/18q, and
−6q23, many of which target TNFAIP3 (A20), a negative regulator of NFkB, which is also mutated and silenced by methylation in subsets of MZL.
25 The pathogenetic role of many of these genes and loci are poorly understood at present.
Splenic MZL is a less common entity of MZL and has been shown to be associated with hepatitis C infection (HCV) in some cases.
26 In these cases, HCV glycoprotein E2 interacts with CD81 on B cells and causes activation of the BCR and its downstream signals.
27,28 Similar to other etiologic factors in MZL, HCV is required for the initiation and maintenance of the malignant phenotype of MZL, but not sufficient to cause malignant transformation of memory B cells in the marginal zone. This is evident from the fact that most patients with HCV do not develop splenic MZL. Furthermore, the majority of patients with splenic MZL are not associated with HCV. Clearly, genetic and epigenetic alterations are required for the development of splenic MZL. Although t(11;18) is not associated with splenic MZL, cytogenetic abnormalities are very common in splenic MZL, including partial trisomy of 3q, gain of 12q, and deletion or translocation of 7q32.
21,29 Furthermore, recent studies have demonstrated that deregulation of the NFκB pathway is frequently observed in splenic MZL. For example, mutations and copy number abnormalities of NFκB genes have been shown to occur in 36 (36%) of 101 splenic MZLs in one series; in another series, inactivating mutations were found in the NFκB negative regulator A20 (6/46 = 13%), and activating mutations were found in MYD88 (6/46 = 13%) and CARD11 (3/34 = 8.8%). Interestingly, these mutations are largely mutually exclusive of each other, suggesting that they may play vital roles in the pathogenesis of splenic MZL. Mutations in NOTCH2, a gene required for marginal-zone B-cell development, is a recurrent genetic lesion in splenic MZL, accounting for approximately 20% to 25% of cases.
30 Importantly, NOTCH2 mutations are restricted to splenic MZL but not other indolent B-cell lymphoproliferative disorders such as MALT lymphoma.
31 These mutations are predicted to cause impaired degradation of the NOTCH2 protein or may result in a gain of function of the truncated NOTCH2 in splenic MZL. The 7q deletion mentioned earlier was investigated recently using a combination of high-resolution array comparative genomic hybridization and integrated transcriptomic analysis. No single gene from these analyses could be identified as a potential driving event for splenic MZL with del7q.
32 Rather, a number of genes and microRNA species were found to be downregulated, some attributed to promoter hypermethylation. Another study of the 7q deletion arrived at a similar conclusion and was able to find nucleotide changes in IRF5 in two of the patients, resulting in fourfold downregulation of the IRF5 gene in lymphomas with 7q32 deletion versus deleted tumors.
33 Importantly, ectopic expression of IRF5 in marginal-zone lymphoma cells decreased proliferation and increased apoptosis in vitro, and impaired lymphoma development in vivo, suggesting that IRF5 may be a haploinsufficient tumor suppressor in this lymphoma entity.
Another layer of complexity in the pathogenesis of splenic MZL was revealed by the discovery of frequent microRNA dysregulation.
34 When compared to reactive lymphoid hyperplasia and other lymphoproliferative diseases, splenic MZL is found to have distinct expression of miRNAs, including highly overexpressed miR-155 and miR-34a, and repressed expression of miR-377 and miR-145, which are candidate diagnostic tools for splenic MZL.
Nodal MZL is a rare entity of MZL and is also associated with HCV infection, though probably to a lesser degree than splenic MZL. No characteristic translocations or chromosome imbalances, such as the t(11;18) in MALT lymphoma and del7q in splenic MZL, have been described in nodal MZL. Only a few cytogenetic alterations of NMZL have been reported, including trisomy 3 in 50% to 70% of cases, none with a clearly established role in either the diagnosis or pathogenesis of nodal MZL. Recent studies by Arribas and colleagues demonstrated that nodal MZL is characterized by upregulation of the IL6 and IL2 cytokine pathways and CD40 signaling, which are involved in B-cell
survival.
35 Some of the highly expressed genes include SYK, which is involved in BCR signaling, and TAC1, which is involved in activating NFκB. In the molecular signature of nodal MZL, a large number of overexpressed genes were associated with the NFκB signaling pathway, such as CD74, CD81, CD82, RELA, and TRAF4, reaffirming the central role of NFκB as a converging point for the proliferation and survival of MZL cells. Interestingly, some microRNA species known to be involved in memory B-cell development, including, for example, miR-221, miR-223, and let-7f, were overexpressed, suggesting their potential utility in the diagnosis of nodal MZL.
Our increased understanding of the biology of MZL in the past decade has already changed how we manage these patients. Because the infectious agents have been found to be required to initiate and maintain the malignant phenotype of some MZL subtypes, antimicrobial treatment has become a highly successful strategy in many patients. The majority of patients with
H. pylori–associated MALT lymphoma can be cured with antibiotics that eliminate the bacterium, thus sparing a large number of patients more aggressive and expensive treatments.
36 Treatment of HCV and
C. psittaci are effective strategies to manage patients with splenic MZL and MALT lymphoma of the ocular adnexa. The discovery of antigen stimulation and the central role of the BCR and NFκB signaling in MZL certainly supports the rationale to explore inhibitors of these pathways with Syk and BTK inhibitors, and even proteasome inhibitors. Targeting NOTCH2, which has been studied predominantly in ALL, could also be an effective strategy in the treatment of splenic MZL.
In contrast to MZL, CLL/SLL are diseases with clearly identified cytogenetic aberrations that have been shown to correlate with prognosis and survival. The clonal population of B lymphocytes that characterize CLL express CD19, CD5, and CD23, with often dim expression of CD20. In addition, these cells typically exhibit reduced levels of surface immunoglobulin. Collectively, this features are the hallmark of mature and activated B lymphocytes.
37 As with many of the other lymphoid malignancies, as our understanding of the pathogenesis of these diseases evolves, our appreciation of their heterogeneity has broadened. This heterogeneity seen in CLL can be characterized at numerous levels, including the status of mutation of the V genes, the expression of CD38, ZAP-70, and other discrete cytogenetic lesions.
For example, CLL cases can be subdivided on the basis of mutations in the V genes, based on the direct comparison of the DNA sequence of germline V genes with the V genes in the CLL cells.
38 CLL cases can then be classified as mutated (that is, they exhibit a more than 2% difference from the germline sequence) or unmutated. Expression of ZAP-70, a protein that is normally expressed near the surface of T cells known to play a role in signaling through the TCR, has also been shown to be an independent prognostic factor.
39–41 In CLL it has been found to be profoundly prognostic. Patients who are ZAP 70
− have been shown to have a median survival of 8 years, compared to patients who are ZAP-70
+, who have a median survival of approximately 25 years. Collectively, these factors have been used to risk stratify patients with CLL, such that those patients with B-CLL clones having few to no V-gene mutations, or with CD38
+ and ZAP-70
+ B-CLL, have been found to have comparatively more aggressive, usually fatal disease. In contrast, those patients with mutated V-genes and/or little to no CD38 or ZAP-70 are typically considered to have a very indolent natural history.
42 Recurring cytogenetic lesions have also been described and may be more valuable in risk stratifying patients with CLL, and ultimately in developing better tailored therapies for the disease. One of the most common is deletion of 13q14.3, which occurs in more than 50% of cases over time.
43 This region of the chromosome is thought to encode two miRNA genes.
44 However, some of the highest risk cytogenetic lesions include deletions of 11q22-23, 17p13, and 6q21.
45 Although the specific genes involved in these deletions remain to be precisely identified, loss of 17p13 is thought to be associated with loss of the tumor suppressor p53 function, and deletion of 11q22-23 is thought to be associated with loss of the ataxia-telangiectasia mutated (ATM) gene. Specific treatment regimens for these discrete genetic subcategories of CLL are in development, and some principles may be emerging. First, patients with loss of 17p are thought to have a very poor prognosis and are typically very refractory to chemotherapy. Some anecdotes suggest that these patients may respond well to anti-CD52–based therapy (alemtuzumab), and it is thought that BTK inhibitors might override this adverse prognostic feature. Similarly, patients who have loss of the ATM function through deletion of 11q22-23 may benefit from the introduction of alkylating agents into the treatment program.
Pathogenesis of Mantle-Cell Lymphoma
Mantle-cell lymphoma (MCL) accounts for approximately 6% of all cases of non-Hodgkin lymphoma, or about 3000 cases per year in the United States.
1 Only a decade ago, it was primarily thought to be one of the most challenging subtypes of lymphoma to treat, carrying the incurable characteristics of indolent lymphoma, and the unfavorable features of aggressive lymphoma. Over the past decade, treatment paradigms for the disease have changed rapidly, and so too has the natural history. These changes are attributed to a
number of observations: (1) the recognition that MCL is not one disease and, like DLBCL, is a very heterogeneous disease composed of more indolent and aggressive variants; (2) the finding that intensive induction chemotherapy regimens consolidated by ASCT used in the front line can consistently produce a response rate of 90% to 100% and produce prolonged progression-free survival compared to traditional R-CHOP–based chemotherapy regimens; and (3) the emergence of novel drugs affecting unique targets, such as proteasome inhibitors and immunomodulatory drugs, which have led to new opportunities to either complement existing treatment paradigms or manage relapsed or refractory disease without specific cytotoxic therapy. What has become painfully clear over the past several years, however, is that given the heterogeneity of the disease, certainly at a biologic level, failure to characterize future populations of patients with MCL on clinical study in this regard will lead to confounding of conclusions about future treatments.
The molecular hallmark of MCL is overexpression of cyclin D1.
46 Although dysregulation of cyclin D1 has been recognized for some time, it has now become evident that MCL is a disease, possibly the prototypical disease, defined by gross cell cycle dysregulation. Cyclin D1 dysregulation is the pathognomonic chromosomal translocation t(11;14)(q13;q32) of the disease, which places cyclin D1 downstream of the highly active IgH enhancer.
47 Essential to the pathogenesis of the disease, the mRNA of cyclin D1 undergoes alternative splicing to produce two unique transcripts: cyclin D1a, which has been clearly shown to drive much of the cell cycle dysregulation; and cyclin D1b, whose expression is more variable and whose role is less well defined.
48 Deletion or mutation of the cyclin D1a mRNA tail region produces a truncated version of cyclin D1a mRNA, which has been shown to be 6 to 10 times more stable than the wild-type full-length cyclin D1a mRNA.
49 In addition to the pivotal role the splice variants of cyclin D1 mRNA play, microRNAs, specifically miRNA61, downregulate cyclin D1a, potentially modulating the cell cycle kinetics. The influence of miR61 can also be influenced by mutations in the tail region of the cyclin D1a mRNA in MCL, which can abrogate the miR61 binding site, allowing for the marked overproduction of cyclin D1 protein.
50 In addition, a number of important translational events have been shown to regulate cyclin D1 level. Phosphorylation of cyclin D1 by GSK3β leads to polyubiquitination by the E3 ligase FBX4, rendering it a substrate for the proteasome and proteolytic degradation.
51 Inactivation of GSK3β would preempt cyclin D1 phosphorylation, which has been shown to occur in the setting where AKT is aberrantly activated,
52 whereas impaired proteolytic degradation of cyclin D1 can be prevented by mutation of the E3 ligase FBX4,
53 resulting in reduced ubiquitination of the D1 protein. Collectively, these overlapping mechanisms of cyclin D1 regulation ensure high levels of cyclin D1 protein in MCL and, in select settings, can lead to marked impact on the cell cycle kinetics.
Although cyclin D1 levels are central in the pathogenesis of MCL, it is by no means the only corrupted cell cycle pathway.
54 Cyclin D1 normally interacts with CDK4/CDK6 to facilitate cell cycle progression through the G1-S checkpoint.
55 CDK4 is frequently overexpressed or amplified in MCL,
56 which would further limit the checkpoint control. Conversely, impaired inhibition of cell cycle regulation can play a major role in the disease, as evidenced by the fact that patients with MCL often have little to no accumulation of the CDK inhibitors p16 and p27.
57 Loss of p16 influence has been shown in patients with MCL—this tumor suppressor is deleted in about half of all MCL cases, and either mutated or silenced by promoter hypermethylation.
58 Impaired cell cycle control due to loss of p27 has been attributed to a number of mechanisms. The protein level of p27 in MCL is regulated by the ubiquitin proteasome. The p27-specific F-box protein Skp2 is inducible and been shown to be overexpressed in some patients with MCL, in particular those with disease that is known to be more aggressive. Increased levels of Skp2 lead to more active ubiquitin ligases, which leads to more prompt and efficient ubiquitination of p27 and thus a theoretically shorter half-life for the protein. This loss of cell cycle inhibition has been noted in patients with more aggressive forms of the disease.
59 This convergence of markedly dysregulated cell cycle control processes, involving both drivers and inhibitors of cell cycle kinetics, conspires to affect the proliferative rate of the disease and, as we now know, the risk stratification of select patient subpopulations.
The notion that the proliferative rate of the disease can be prognostic has now been confirmed at a number of levels. Patients with the truncated version of the mRNA of cyclin D1a, the stabler form, are known to have disease with a higher proliferative index
49 and more aggressive histologies, such as blastoid MCL.
60 Remarkably, the median overall survival (OS) of MCL patients with the truncated cyclin D1a mRNA is only 1.38 years, compared to 3.28 years for patients with the full-length and unstable mRNA.
49 The levels of the full-length mRNA transcript of cyclin D1a have been shown to positively correlate with the proliferation index as assayed by positive immunostaining of Ki-67. Ki-67 is a nuclear protein found in all states of the cell cycle (G
1, S, G
2, and M), but is absent from resting cells (G
0). Many cell cycle regulatory factors have been shown to correlate with the protein level of cyclin D1 on immunohistochemical staining, including the percentage of MCL cells with positive nuclear staining of the protein or the amount of nuclear staining of cyclin D1.
60 Conversely, the expression of p27 is inversely associated with Ki-67, where high levels of p27 appear to be
associated with lower proliferative index and improved overall survival.
57 These lines of data strongly support the contention that MCL is a disease grossly characterized by cell cycle dysregulation, and that these features of the underlying disease biology can be highly prognostic.
Further confirming the significance of the proliferative index, several studies have shown that nuclear staining of Ki-67, and using a cutoff of 30%, predicted OS, where patients below and above the 30% cutoff experienced a median OS of 13 months and 45 months, respectively.
61 Using yet another approach, a group at the National Cancer Institute (NCI) employed GEP to characterize patients with MCL (
Figure 29-5 ). Their profiling experience firmly established that heterogeneity of the disease, which they used to define the proliferation signature of MCL. They discovered 48 genes whose expression levels were highly correlated with survival in a statistically significant manner. Among those 48 genes were a group of 20 genes that were variably expressed, though strongly correlated with cellular proliferation. They called the pattern of gene expression the “proliferation signature,” which could essentially stratify patients along a continuum from highly proliferative to less proliferative disease.
62 Calculation of the average expression levels of these 20 genes in the proliferation signature allowed for the separation of the population into quartiles. The median OS of patients with the lowest expression of proliferation signature genes (quartile 1) was 6.71 years, whereas those with the highest expression (quartile 4) exhibited an OS of only 0.83 years. Surprisingly, cyclin D1 was not among the proliferation signature genes identified using the data generated from DNA microarrays, because the chip-based technology detected only the full-length mRNA of cyclin D1, which is unstable. Employing a reverse transcriptase (RT)-PCR assay to differentiate the two species of the cyclin D1 mRNA showed that patients with the truncated mRNA of cyclin D1 (3′ UTR low) demonstrated a higher expression level of cyclin D1, a higher expression of the 20 “proliferation signature” genes, and a substantially shorter OS, compared to patients with the full-length mRNA of cyclin D1 (i.e., 3′ UTR high).
62 When RT-PCR was designed to detect only
Buy Membership for Hematology, Oncology and Palliative Medicine Category to continue reading.
Learn more here
[/not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
The Molecular Basis of Cancer Expert Consult - Online