[level-membership-for-neurology-category]
Chapter 4 Long-Term Effects of Seizures on Brain Structure and Function
Perhaps one of the most common questions asked by patients with epilepsy or by parents of children with epilepsy is, “What are the seizures doing to the brain?” Implicit in the question is a concern that the seizures are having a negative effect. The number of questions that derive from this one are almost endless: “Are the seizures causing brain damage?” “Will my child’s cognitive development be impaired by having seizures?” “Will the seizures cause more seizures?” The answers to these and other related questions are as varied as the causes of epilepsy, the types of seizure, and the ages of the patient when the seizures occur. In short, there is no single, simple answer. The physiology associated with seizures varies from a short run of rhythmic spike and wave activity to prolonged tonic discharges. Some types of epilepsy are seemingly benign and self-limited, whereas others are inexorably progressive. Seizures can occur in a neonatal brain with years of development to come or in aging brains that have suffered the ravages of time and are hanging by their fingertips over the abyss of oblivion. In approaching these questions, we therefore have to consider, among other things, which kind of seizure, what cause, and what type of brain. Many of the answers are often colored by the physician’s interpretation of imperfect data, an interpretation that can be further clouded by attempts to extrapolate data across seizure types, developmental state, and species. In this chapter, we will provide an overview of some of the relevant data. However, we cannot offer clarity where none exists. Thus, readers will find answers that contain enough caveats to cast doubt on any attempt at certainty. Our goal is to provide a perspective on the complexities involved in answering your patients’ and their families’ legitimate questions.
Before we enter the discussion regarding the effect of seizures on the brain, it is essential that we differentiate among the different types of epilepsy and seizures because it is clear that the pathophysiology of the many syndromes and situations are quite varied in both cause and consequence. These are important distinctions to make, as one of the problems that we encounter in determining the effect of seizures on the brain is that many of the causes involve preexisting abnormalities or destructive injuries. Thus, claiming any change found as a potential consequence of seizure activity cannot be supported. The primary distinction to be made between seizure types is between status epilepticus (SE) and intermittent spontaneous seizures. Even though SE is often caused by events such as stroke, infection, or trauma, there is ample evidence, especially from the laboratory, that after a duration of as short as 30 minutes, the seizure activity itself can induce neuronal loss.1,2 However, it is likely that even in the general area of SE, there are types that cause injury and types that do not. Convulsive SE or SE associated with high-frequency discharges on electroencephalogram (EEG) clearly can cause damage, even when the high-intensity physical convulsions are controlled.3,4 Absence SE or spontaneous episodes of prolonged partial SE associated with lower frequency discharges (3 c/s spike and wave or rhythmic delta/theta activity) may not be, as there appear to be no long-term consequences to these episodes.
Neonatal Seizures
The developing brain is predisposed to seizures,5,6 and seizures are common in the neonate with an estimated incidence of 1 to 6 per 1000 live births.7–11 The term neonatal seizures encompasses seizures of diverse etiologies ranging from acute symptomatic causes (e.g., hypoxia-ischemia, intracranial hemorrhage, infections, and metabolic derangements such as hypocalcemia) to remote symptomatic causes (e.g., cerebral dysgenesis and inborn errors of metabolism) to idiopathic, genetically determined epilepsies (e.g., early myoclonic encephalopathy of infancy and familial neonatal convulsions).
As a group, the outcome for children with neonatal seizures, although improving, is poor.12 The mortality rate for neonates with seizures is high, with values ranging from 15 to 30%. In survivors, approximately 30% will develop neurological sequelae that include epilepsy, mental retardation, and cerebral palsy. Those most at risk for mortality and neurological morbidity are those neonates with persistent, difficult-to-treat seizures.13–17 It has been proposed that these seizures produce neurological injury as the result of energy depletion and cerebral metabolism dysfunction.18 However, because these prolonged, persistent seizures tend to be observed in those neonates with the most severe CNS involvement and injury (e.g., severe hypoxic ischemic encephalopathy), it is difficult to distinguish between the secondary damage, if any, imposed by the seizures and the primary injury imposed on the brain by the underlying etiology.
There are good examples in which neonatal seizures tend to be associated with a more favorable outcome. One example is the genetically determined benign familial neonatal convulsions, an autosomal dominantly inherited epilepsy syndrome resulting from mutations in voltage-gated potassium channel. These convulsions are brief events commencing as tonic posturing with apnea and other autonomic features, which can evolve to clonic movements in otherwise healthy neonates. These convulsions typically commence during the first week of life, can be observed multiple times per day, and may be difficult to treat. In most cases, the seizures will remit by 16 months of age. Although there is a increased risk of febrile and afebrile seizures later in life, psychomotor development tends to be normal.19 For those with psychomotor delay, it is likely that this outcome is the result of genetic factors and not the seizures.20
In summary, caveats abound when attempting to answer the question of whether seizures damage the neonatal brain. Although all recognize that the answer to this question is important, clinical data are limited.21 It is assumed that few would argue against the concept that in some circumstances seizures in the neonatal brain impose additional injury, but not all would agree on the circumstances or the extent of the injury, especially as some neonatal seizures have a benign outcome.
The Childhood Epilepsies: Good versus Bad
For many children with epilepsy, the outcome is good. The seizures remit and may not recur, even after medication is discontinued. Although cognitive and behavioral disorders may be observed transiently during the time period when the epilepsy was most active and required treatment with an antiepileptic medication, these children have no apparent evidence of a significant, long-term disability resulting from their epilepsy. In contrast, a group of children with epilepsy will have an associated psychomotor slowing or regression that persists even in the absence of seizures. In some of these children, data suggest that the seizures or the interictal EEG abnormalities contributed to the regression. Most childhood epilepsies fall somewhere between these two extremes (Table 4-1).
| Benign Epilepsies |
|---|
| Benign familial neonatal convulsions |
| Benign myoclonic epilepsy of infancy |
| Childhood absence epilepsy |
| Juvenile absence epilepsy |
| Benign rolandic epilepsy |
| Panayiotopoulos syndrome |
| Juvenile myoclonic epilepsy |
| Malignant Epilepsies |
|---|
| Ohtahara syndrome |
| Early myoclonic encephalopathy |
| West syndrome |
| Dravet syndrome |
| Lennox-Gastaut syndrome |
| Landau-Kleffner syndrome |
BENIGN ROLANDIC EPILEPSY
Given that the seizure frequency associated with this syndrome is low,22 and that the seizures most commonly occur during sleep, the majority of children with this syndrome do not require antiepileptic therapy.23 In addition, these children outgrow the epilepsy, and the interictal EEG abnormality resolves during their teenage years.24 That the seizures remit and the rate of epilepsy in these children in adulthood is no greater than that in the general population24 is in contrast to the adage that seizures beget seizures.
Although BRE is considered a benign epilepsy, cognitive and behavioral difficulties may be observed.25–28 However, for the most part, these difficulties are transient and appear to be related to the severity of the interictal discharges and not the result of a permanent injury to the brain.29 Indeed, as a group, the developmental outcome for these children, including those with cognitive and behavioral difficulties, whether treated or not, is good. A potential alternative explanation is that an underlying genetic predisposition underlies both the epilepsy and cognitive dysfunction.
WEST SYNDROME
West syndrome is a cryptogenic or symptomatic generalized epilepsy syndrome composed of infantile spasms, a hypsarrhythmic EEG, and mental retardation. The spasms, which tend to occur in clusters, are characterized by a brief, massive flexion, extension, or mixed contraction of the axial musculature coincident with an electrodecrement on the EEG. The incidence for this syndrome ranges from 3 to 4.5 per 10,000 live births, and the majority of children present during the first year of life.30–32 Causes of West syndrome are multiple and include the neurocutaneous syndromes (e.g., tuberous sclerosis complex), brain malformation and tumors, inborn disorders of metabolism, and genetic disorders, as well as acquired injuries from fetal infections, trauma, and hypoxic-ischemic injury.
As a group, the long-term prognosis is poor and includes medically refractory epilepsy, cognitive impairments ranging from learning disorders to mental retardation, and autism. To a large extent, like neonatal seizures, the prognosis is dependent on the etiology.33 However, medical and surgical reports demonstrating improved developmental outcome in those children with the shortest epilepsy duration and prompt response to therapy suggest that the seizure activity itself may be detrimental. But once again, caution in interpretation is required, as those with a short treatment lag or who responded quickly to therapy or potentially those most amenable to surgical resection may be those who would have had a better developmental outcome, even in the absence of effective treatment.34
Febrile Seizures and Mesial Temporal Lobe Epilepsy
On the other hand, a number of epidemiological studies indicate that the risk for developing epilepsy (or any other negative consequence) following febrile seizures is not greater than for children who never experienced them. However, the studies have also revealed that febrile seizures are not a single entity. Some are classified as simple and last less than 5 minutes, and these do not carry any additional risk for a poor long-term outcome. Complex febrile seizures (focal ictal behavior or seizures lasting more than 30 minutes), especially those that are prolonged, have a much higher risk for poor long-term outcome and the development of chronic epilepsy.
Mesial Temporal Lobe Epilepsy
This syndrome has been the source of the greatest debates regarding the potential negative impact that seizures have on the brain, as it goes straight to the heart of the issue of whether uncontrolled (often single) seizures cause neuronal loss. This is the single area in which the thoughts of the epilepsy community are most divided. The opinions range from the conclusion that each single seizure causes a measurable neuronal loss to the position that no evidence exists for progressive neuronal loss as the consequence of any number of recurrent, spaced seizures, even thousands. The mesial temporal lobe epilepsy (MTLE) or chronic limbic (CLE) syndrome is associated with specific patterns of neuronal loss in the hippocampus and adjacent mesial temporal regions known as hippocampal or mesial temporal sclerosis.35 This particular syndrome has a well-established pattern of neuronal loss that can be quantified in vivo through magnetic resonance imaging (MRI) volume measurements36 or spectroscopy for N-acetyl-aspartase (NAA), a compound that is predominantly neuron specific.37 Anatomical studies have been variably interpreted to show evidence of significant progressive neuronal loss or to show minimal to no progressive loss over prolonged periods of time.38,39
In view of the strong opinions on both sides of the issue (progressive vs. no loss), what do the data really show? A number of clinical correlational studies use surgical or postmortem specimens to correlate the duration of epilepsy or, less commonly, total numbers of seizures (always difficult to estimate) in comparison to the severity of neuronal loss. Although there is not an absolute consensus, the most recent studies have suggested that there is not tremendous neuronal loss as a cumulative consequence of recurrent seizures.38,40 One study that examined tissue obtained at the time of surgery found a slight reduction of neuronal density in patients with a 30-year epilepsy history. However all patients had significant neuronal loss, even patients with 5-year histories and relatively few seizures. This finding suggested that the overwhelming majority of neuronal loss preceded the onset of seizures.38
Laboratory Insights
Animal studies may avoid the issues of cause versus consequence, as one is able to control the inducing lesion and evaluate the tissue at specified times after the induction of the causative lesion and after specified numbers of seizures. Although there is a lack of unanimity,41 the data tend to support the conclusion that much of the neuronal loss precedes the onset of epilepsy and is not a consequence of recurrent seizures.42–45 In animals in which seizures were induced, the method used was direct electrical stimulation in the limbic system (the kindling model).46 As in the human studies there is some disagreement about the interpretation of the data, but overall the studies suggest that recurrent seizures do not cause progressive neuronal loss, even after 1000 induced seizures over a number of months.42,43 This observation does not mean that the seizures have no effect on the brain. They clearly activate glial cells and cause them to hypertrophy,45,47 It is just that, at least in this one model of intermittent seizures, neuronal loss is not a consequence of seizures.
Is there a human parallel of induced seizures in animals with normal brains so that we might extrapolate from the models to the clinic regarding the potential effect of seizures on the brain? There is in one circumstance: electroconvulsive therapy (ECT). Concern has been raised that this therapy causes brain damage, but in recent follow-up studies using MRI volumetrics, no evidence was found for detectable brain injury as a consequence of ECT. Although most courses of ECT involve a small number of induced seizures and most patients undergo the therapy once, there is a report of a patient who had approximately 1200 stimulated seizures through ECT over the course of his life. On autopsy his brain showed no evidence of damage.48
Whether seizures are harmful to the developing brain has also been addressed in the laboratory in which seizures are induced in young animals through the use of chemoconvulsants such as the excitotoxin kainate acid, the cholinergic agonist pilocarpine, and the general anesthetic flurothyl. Some studies have demonstrated that repetitive seizures over several days starting soon after49–53 or even a single episode of repetitive seizures over an approximately 3-hour period in rat pups can result in long-term changes in seizure susceptibility and cognitive function. Yet, in another study, a single prolonged seizure induced in young rats had no effect on later seizure susceptibility or performance on tests of visuospatial learning, memory, responses to novel environment, and emotionality.54
One of the primary lessons learned from laboratory studies is that the developing hippocampus, unlike the adult hippocampus, is resistant to gross neuronal injury secondary to seizures.55–57 However, recurrent seizures in rat pups can induce synaptic rearrangement,58 a reduction in neurogenesis, and altered expression of neurotransmitter receptors. In addition, neuronal loss can be observed in other parts of the brain. Following severe prolonged seizures induced with a combination of lithium and pilocarpine, neuronal loss was observed in the central and lateral segments of the mediodorsal nucleus,59 and repetitive prolonged seizures may result in a change in the distribution of interneurons in the cortex. Although these studies are confounded by the use of excitotoxins, and the nature of the seizures varies by method of induction and the age of the animal, it is reasonable to conclude that the developing brain is less susceptible to seizure-induced injury, but not immune to it. The basis for the difference is unclear, but it likely includes differences in intrinsic cell properties as well as receptors, channels, and type of connectivity.
Other Effects of Seizures
If seizures don’t cause neuronal loss, what effects do they have? There is no question that seizures activate astrocytes.45,47 The activated glia gradually subside if seizures do not occur over several months.45 In addition to the activation of glia, clear changes can be observed in the physiology of neurons following seizures, but the nature of the change varies from region to region. Although there are clear changes in a multitude of channels and receptors in the neurons from epileptic animals and people, the limited available data suggest that many of these changes precede the onset of epilepsy, although one cannot exclude the possibility that some of the changes are a consequence of recurrent seizures. Many of the ion channels have not been studied to the same degree in kindled animals as they have in the poststatus epilepticus spontaneous epilepsy models. However, where they have been studied, very little overlap is seen in terms of changes in ion channel expression in epileptic and kindled animals, suggesting that the changes in epilepsy are part of the disorder as opposed to a consequence of the seizures. There are reports about A-type potassium channels and T-type calcium channels from kindled animals,60,61 as well as other calcium currents that do exhibit short- and long-term alterations.62–65 Changes in the GABA receptor after just a few induced seizures have also been reported.66 A number of reports have demonstrated that the changes associated with kindling are quite different than the changes found in epilepsy. Although there are still many areas for study, the data to date suggest that the consequences of intermittent seizures are quite different and likely of less consequence than the changes that cause the epilepsy (Table 4-2).
| Activation of astrocytes |
| Altered expression of voltage-gated ion channels (sodium, potassium, calcium) |
| Altered expression of receptors (GABA, NMDA) |
| Synaptic rearrangement (minimal mossy fiber spouting in dentate gyrus) |
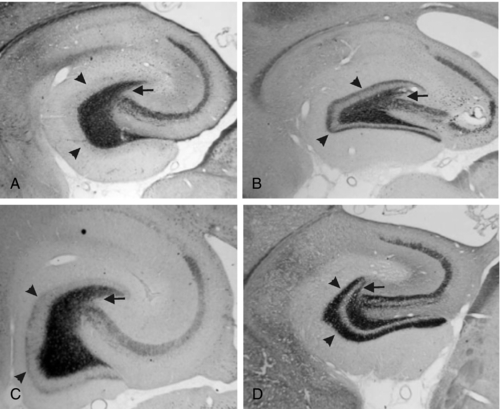
Sprouting of mossy fibers in the dentate gyrus has been observed in MTLE as well as in the kindling models. These aberrant axons, which originate in the granule cells of the dentate gyrus and normally project to the pyramidal cells of CA3 in the hippocampus, turn back and move into the inner molecular layer in MTLE, where they form excitatory synapses.67,68 Although they have been described in kindling, the degree of mossy fiber sprouting in kindling is far less than in models of MTLE44 (Figure 4-1). This observation suggests that although seizures may cause some synaptic reorganization, it is a relatively minor change compared to the changes that occur in MTLE. Changes in the physiology of the dentate gyrus have been described in kindling, so that these changes, along with others, are likely having an effect on the local system physiology. The net effect of these changes with regard to changes in the larger limbic system excitability is still unknown.
Seizures Beget Seizures?
This phrase has been around for over a century.69 It implies that each seizure induces changes that increase the probability that another seizure will occur. If true, “Seizures beget seizures” also implies that there is some urgency in suppressing seizures to improve long-term outcomes. The observation that seizures can induce changes in channels and receptors could support that hypothesis, but what is the clinical experience regarding the natural history of the epilepsies? As noted earlier, there is a significant variability in the ultimate outcomes of the different disorders. Some, such as the Lennox-Gastaut syndrome, are difficult to control and are ultimately associated with a long history of intractable seizures. Others, such as rolandic or benign epilepsy of centrotemporal spikes, enter into spontaneous remission regardless of whether the seizures were controlled or not. Absence epilepsy has a high rate of spontaneous remission (although not complete). These observations would suggest that early treatment may not have an effect on outcome, as it appears that the outcome regarding ultimate remission is independent of treatment. So how did the concept arise? Likely from the observation that some patients who had more than a few seizures were likely to have more, and if they had more seizures, they might be more difficult to control. Although the data have never really been analyzed in this way, it is very possible that the patients who are having more seizures before they start treatment may be having them in a shorter period of time and that their frequent seizures are a sign of a less-benign syndrome.
Animal studies do not completely clarify the issue, unfortunately. Kindling, during which seizures are induced repeatedly over days and weeks (and sometimes months) in rats by direct brain stimulation, results in longer and more behaviorally severe seizures (that ultimately plateau), but do not consistently result in spontaneous seizures. Although some investigators do report the development of spontaneous seizures after many (150 to 300 stimulated seizures),70,71 it is not a universal finding across labs. Primate kindling shows some evidence that genetics may play a role, as there are some primate species that neither kindle well nor ever develop spontaneous seizures.72,73 As a consequence, we are left with the conclusion that although in some situations after many seizures the seizure activity itself may cause changes that result in spontaneous seizures, little evidence supports the notion of seizures begetting seizures. It is far more likely that the natural history of the particular epilepsy syndrome reflects the natural history of the underlying cause.
The issue of kindling to spontaneous seizures is relevant for the theoretical question of secondary epileptogenesis in people. It is a hypothetical process in which seizure activity that initiates in one region of the brain spreads to other regions. Over time the repeated seizures could potentially cause changes in other regions that eventually result in another seizure focus that could act independently. One of the primary reasons for developing this hypothesis was the gradual appearance of independent spikes away from the presumed original (or primary) spike focus. It was also used to explain the presence of a second focus and seizure type in patients who had a single seizure type for many years before the second became active. The first focus was often associated with an obvious abnormality, whereas the second was not. This phenomenon has been called into question because of several developments, not the least of which was improved neuroimaging. This allowed the visualization of subtle cortical changes that indicate the existence of multiple abnormalities that could be independent foci for seizures. In addition, unless there is a well-documented second seizure type that is independent of the primary seizure, the existence of multifocal spikes does not prove the existence of multiple seizure foci, as spikes are not always good indicators of a seizure focus. Rather, they are a sign of a potential to have seizures. Thus, in humans, although we cannot completely exclude the eventual kindling of a secondary focus, at this time there is no convincing evidence that the original primary seizures kindle a secondary and eventually independent seizure focus.
1. Lothman E. The biochemical basis and pathophysiology of status epilepticus. Neurology. 1990;40:13.
2. Fountain NB, Lothman EW. Pathophysiology of status epilepticus. J Clin Neurophysiol. 1995;12:326.
3. Blennow G, Brierley JB, Meldrum BS, et al. Epileptic brain damage: the role of systemic factors that modify cerebral energy metabolism. Brain. 1978;101:687.
4. Nevander G., Ingvar M., Auer R, et al. Status epilepticus in well-oxygenated rats causes neuronal necrosis. Ann Neurol. 1985;18:281-290.
5. Wong M. Advances in the pathophysiology of developmental epilepsies. Semin Pediatr Neurol. 2005;12:72.
6. Ben-Ari Y, Holmes GL. Effects of seizures on developmental processes in the immature brain. Lancet Neurol. 2006;5:1055.
7. Hauser WA, Kurland LT. The epidemiology of epilepsy in Rochester, Minnesota, 1935 through 1967. Epilepsia. 1975;16:1.
8. Ellenberg JH, Hirtz DG, Nelson KB. Age at onset of seizures in young children. Ann Neurol. 1984;15:127.
9. Lanska MJ, Lanska DJ, Baumann RJ, Kryscio RJ. A population-based study of neonatal seizures in Fayette County, Kentucky. Neurology. 1995;45:724.
10. Ronen GM, Penney S, Andrews W. The epidemiology of clinical neonatal seizures in Newfoundland: a population-based study. J Pediatr. 1999;134:71.
11. Cowan LD. The epidemiology of the epilepsies in children. Mental Retard Dev Disabil Res Rev. 2002;8:171.
12. Ronen GM, Buckley D, Penney S, et al. Long-term prognosis in children with neonatal seizures: a population-based study. Neurology. 2007;69:1816.
13. Mellits ED, Holden KR, Freeman JM. Neonatal seizures. II. A multivariate analysis of factors associated with outcome. Pediatrics. 1982;70:177.
14. Legido A, Clancy RR, Berman PH. Neurologic outcome after electroencephalographically proven neonatal seizures. Pediatrics. 1991;88:583.
15. Ortibus EL, Sum JM, Hahn JS. Predictive value of EEG for outcome and epilepsy following neonatal seizures. Electroencephalogr Clin Neurophysiol. 1996;98:175.
16. Bye AM, Cunningham CA, Chee KY, Flanagan D. Outcome of neonates with electrographically identified seizures, or at risk of seizures. Pediatr Neurol. 1997;16:225.
17. Pisani F, Cerminara C, Fusco C, Sisti L. Neonatal status epilepticus vs recurrent neonatal seizures: clinical findings and outcome. Neurology. 2007;69:2177.
18. Miller SP, Weiss J, Barnwell A, et al. Seizure-associated brain injury in term newborns with perinatal asphyxia. Neurology. 2002;58:542.
19. Zonana J, Silvey K, Strimling B. Familial neonatal and infantile seizures: an autosomal-dominant disorder. Am J Med Genet. 1984;18:455.
20. Steinlein OK, Conrad C, Weidner B. Benign familial neonatal convulsions: always benign? Epilepsy Res. 2007;73:245.
21. Clancy RR. Summary proceedings from the neurology group on neonatal seizures. Pediatrics. 2006;117:S23.
22. Kramer U, Zelnik N, Lerman-Sagie T, Shahar E. Benign childhood epilepsy with centrotemporal spikes: clinical characteristics and identification of patients at risk for multiple seizures. J Child Neurol. 2002;17:17.
23. Peters JM, Camfield CS, Camfield PR. Population study of benign rolandic epilepsy: is treatment needed? Neurology. 2001;57:537.
24. Bouma PA, Bovenkerk AC, Westendorp RG, Brouwer OF. The course of benign partial epilepsy of childhood with centrotemporal spikes: a meta-analysis. Neurology. 1997;48:430.
25. Staden U, Isaacs E, Boyd SG, Brandl U, Neville BG. Language dysfunction in children with rolandic epilepsy. Neuropediatrics. 1998;29:242.
26. Ong HT, Wyllie E. Benign childhood epilepsy with centrotemporal spikes: is it always benign? Neurology. 2000;54:1182.
27. Baglietto MG, Battaglia FM, Nobili L, et al. Neuropsychological disorders related to interictal epileptic discharges during sleep in benign epilepsy of childhood with centrotemporal or rolandic spikes. Dev Med Child Neurol. 2001;43:407.
28. Massa R, de Saint-Martin A, Carcangiu R, et al. EEG criteria predictive of complicated evolution in idiopathic rolandic epilepsy. Neurology. 2001;57:1071.
29. Boxerman JL, Hawash K, Bali B, et al. Is rolandic epilepsy associated with abnormal findings on cranial MRI? Epilepsy Res. 2007;75:180.
30. Brna PM, Gordon KE, Dooley JM, et al. The epidemiology of infantile spasms. Can J Neurol Sci. 2002;28:309.
31. Luthvigsson P, Olafsson E, Sigurthardotir S, et al. Epidemiologic features of infantile spasms in Iceland. Epilepsia. 1994;35:802.
32. Sidenvall R, Eeg-Olofssn O. Epidemiology of infantile spasms in Sweden. Epilepsia. 1995;36:572.
33. Koo B, Hwang PA, Logan WJ. Infantile spasms: outcome and prognostic factors of cryptogenic and symptomatic groups. Neurology. 1993;43:2322.
34. Hrachovy RA, Glaze DG, Frost JDJr. A retrospective study of spontaneous remission and long-term outcome in patients with infantile spasms. Epilepsia. 1991;32:212.
35. Margerison JH, Corsellis JA. Epilepsy and the temporal lobes. A clinical, electroencephalographic and neuropathological study of the brain in epilepsy, with particular reference to the temporal lobes. Brain. 1966;89:499.
36. Quigg MS, Bertram EH, Jackson T, et al. Evidence for bilateral atrophy in unilateral mesial temporal lobe epilepsy. Epilepsia. 1997;38:588.
37. Suhy J, Laxer KD, Capizzano AA, et al. 1H MRSI predicts surgical outcome in MRI-negative temporal lobe epilepsy. Neurology. 2002;58:8213.
38. Mathern GW, Adelson PD, Cahan LD, et al. Hippocampal neuron damage in human epilepsy: Meyer’s hypothesis revisited. Prog in Brain Res. 2002;135:237.
39. Cendes F, Andermann F, Gloor P, et al. Atrophy of mesial structures in patients with temporal lobe epilepsy: cause or consequence of repeated seizures? Ann of Neurol. 1993;34:795.
40. Thom M, Zhou J, Martinian L, et al. Quantitative post-mortem study of the hippocampus in chronic epilepsy: seizures do not inevitably cause neuronal loss. Brain. 2005;128:1344.
41. Cavazos JE, Das I, Sutula TP. Neuronal loss induced in limbic pathways by kindling: evidence for induction of hippocampal sclerosis by repeated brief seizures. J Neurosci. 14:3106.
42. Bertram EH, Lothman EW, Lenn NJ. The hippocampus in experimental chronic epilepsy: a morphometric analysis. Ann Neurol. 1990;27:43.
43. Bertram EH, Lothman EW. Morphometric effects of intermittent kindled seizures and limbic status epilepticus in the dentate gyrus of the rat. Brain Res. 1993;603:25.
44. Mathern GW, Bertram EHIII, Babb TL, et al. In contrast to kindled seizures, the frequency of spontaneous epilepsy in the limbic status model correlates with greater aberrant fascia dentata excitatory and inhibitory axon sprouting, and increased staining for N-methyl-D-aspartate, AMPA and GABAA receptors. Neurosci. 1997;77:1003-1019.
45. Adams B, Von Ling E, Vaccarella L, et al. Time course for kindling-induced changes in the hilar area of the dentate gyrus: reactive gliosis as a potential mechanism. Brain Res. 1998;804:331.
46. Goddard GV, McIntyre DC, Leech CK. A permanent change in brain function resulting from daily electrical stimulation. Experimental Neurol. 1969;25:295.
47. Torre ER, Lothman E, Steward O. Glial response to neuronal activity: GFAP-mRNA and protein levels are transiently increased in the hippocampus after seizures. Brain Res. 1993;631:256.
48. Lippman S, Manshadi M, Wehry M, et al. 1250 electroconvulsive treatments without evidence of brain injury. Brit Jour of Psychiat. 1985;147:203.
49. Holmes GL, Gairsa JL, Chevassus-Au-Louis N, et al. Consequences of neonatal seizures in the rat: morphological and behavioral effects. Ann Neurol. 1989;44:845-857.
50. Huang L, Cilio MR, Silveira DC, et al. Long-term effects of neonatal seizures: a behavioral, electrophysiological, and histological study. Brain Res Dev Brain Res. 1999;118:99.
51. de Rogalski Landrot I, Minokoshi M, Silveira DC, et al. Recurrent neonatal seizures: relationship of pathology to the electroencephalogram and cognition. Brain Res Dev Brain Res. 2001;129:27.
52. Sogawa Y, Monokoshi M, Silveira DC, et al. Timing of cognitive deficits following neonatal seizures: relationship to histological changes in the hippocampus. Brain Res Dev Brain Res. 2001;131:73.
53. Sayin U, Sutula TP, Stafstrom CE. Seizures in the developing brain cause adverse long-term effects on spatial learning and anxiety. Epilepsia. 2004;45:1539.
54. Stafstrom CE, Chronopoulos A, Thurber S, et al. Age-dependent cognitive and behavioral deficits after kainic acid seizures. Epilepsia. 1993;34:420.
55. Nitecka L, Tremblay E, Charton G, et al. Maturation of kainic acid seizure-brain damage syndrome in the rat. II. Histopathological sequelae. Neuroscience. 1984;13:1073.
56. Cavalheiro EA, Silva DF, Turski WA, et al. The susceptibility of rats to pilocarpine-induced seizures is age-dependent. Brain Res. 1987;465:43.
57. Sperber EF, Haas KZ, Romero MT, et al. Flurothyl status epilepticus in developing rats: behavioral, electrographic histological and electrophysiological studies. Brain Res Dev Brain Res. 1999;116:59.
58. Holmes GL, Sarkisian M, Ben-Ari Y, et al. Mossy fiber sprouting after recurrent seizures during early development in rats. J Comp Neurol. 1999;404:537.
59. Kubova H, Druga R, Lukasiuk K, et al. Status epilepticus causes necrotic damage in the mediodorsal nucleus of the thalamus in immature rats. J Neurosci. 2001;21:3593.
60. Vreugdenhil M, Wadman WJ. Potassium currents in isolated CA1 neurons of the rat after kindling epileptogenesis. Neuroscience. 1995;66:805.
61. Hendriksen H, Kamphius W, Lopes da Silva FH. Changes in voltage-dependent calcium channel alpha1-subunit mRNA levels in the kindling model of epileptogenesis. Brain Res Mol Brain Res. 1997;50:257.
62. Faas GC, Vreugdenhil M, Wadman WJ. Calcium currents in pyramidal CA1 neurons in vitro after kindling epileptogenesis in the hippocampus of the rat. Neuroscience. 1996;75:57.
63. Blalock EM, Chen K-C, Vanaman TC, et al. Epilepsy-induced decrease of L-type Ca2+ channel activity and coordinate regulation of subunit mRNA in single neurons of rat hippocampal “zipper” slices. Epilepsy Res. 2001;43:211.
64. Bernstein GM, Medonca A, Wadia J, et al. Kindling induces an asymmetric enhancement of N-type Ca2+ channel density in the dendritic fields of the rat hippocampus. Neurosci Lett. 1999;268:155.
65. Bernstein GM, Medonca A, Wadia J, et al. Kindling induces a long-term enhancement in the density of N-type calcium channels in the rat hippocampus. Neuroscience. 1999;94:1083.
66. Evans MS, Cady CJ, Disney KE, et al. Three brief epileptic seizures reduce inhibitory synaptic currents, GABAA currents, and GABAA-receptor subunits. Epilepsia. 2006;47:1655.
67. Tauck DL, Nadler JV. Evidence of functional mossy fiber sprouting in hippocampal formation of kainic acid-treated rats. J Neurosci. 1985;5:1016.
68. Sutula T, Cascino G, Cavazos J, et al. Mossy fiber synaptic reorganization in the epileptic human temporal lobe. Ann Neurol. 1989;26:321.
69. Gowers WR. Epilepsy and Other Chronic Convulsive Disorders: Their Causes, Symptoms and Treatment. London: J&A Churchill, 1881.
70. Pinel JPJ, Rovner LI. Experimental epileptogenesis: kindling-induced epilepsy in rats. Experimental Neurol. 1978;58:190.
71. Sayin U, Osting S, Hagen, et al. Spontaneous seizures and loss of axo-axonic and axo-somatic inhibition induced by repeated brief seizures in kindled rats. J Neurosci. 2003;23:2759.
72. Wada JA, Mizoguchi T, Osawa T. Secondarily generalized convulsive seizures induced by daily amygdaloid stimulation in rhesus monkeys. Neurology. 1978;28:1026.
73. Wada JA, Osawa T. Spontaneous recurrent seizure state induced by daily electrical amygdaloid stimulation in Senegalese baboons (Papio papio). Neurology. 1976;26:273.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Chapter 4 Long-Term Effects of Seizures on Brain Structure and Function
Perhaps one of the most common questions asked by patients with epilepsy or by parents of children with epilepsy is, “What are the seizures doing to the brain?” Implicit in the question is a concern that the seizures are having a negative effect. The number of questions that derive from this one are almost endless: “Are the seizures causing brain damage?” “Will my child’s cognitive development be impaired by having seizures?” “Will the seizures cause more seizures?” The answers to these and other related questions are as varied as the causes of epilepsy, the types of seizure, and the ages of the patient when the seizures occur. In short, there is no single, simple answer. The physiology associated with seizures varies from a short run of rhythmic spike and wave activity to prolonged tonic discharges. Some types of epilepsy are seemingly benign and self-limited, whereas others are inexorably progressive. Seizures can occur in a neonatal brain with years of development to come or in aging brains that have suffered the ravages of time and are hanging by their fingertips over the abyss of oblivion. In approaching these questions, we therefore have to consider, among other things, which kind of seizure, what cause, and what type of brain. Many of the answers are often colored by the physician’s interpretation of imperfect data, an interpretation that can be further clouded by attempts to extrapolate data across seizure types, developmental state, and species. In this chapter, we will provide an overview of some of the relevant data. However, we cannot offer clarity where none exists. Thus, readers will find answers that contain enough caveats to cast doubt on any attempt at certainty. Our goal is to provide a perspective on the complexities involved in answering your patients’ and their families’ legitimate questions.
Before we enter the discussion regarding the effect of seizures on the brain, it is essential that we differentiate among the different types of epilepsy and seizures because it is clear that the pathophysiology of the many syndromes and situations are quite varied in both cause and consequence. These are important distinctions to make, as one of the problems that we encounter in determining the effect of seizures on the brain is that many of the causes involve preexisting abnormalities or destructive injuries. Thus, claiming any change found as a potential consequence of seizure activity cannot be supported. The primary distinction to be made between seizure types is between status epilepticus (SE) and intermittent spontaneous seizures. Even though SE is often caused by events such as stroke, infection, or trauma, there is ample evidence, especially from the laboratory, that after a duration of as short as 30 minutes, the seizure activity itself can induce neuronal loss.1,2 However, it is likely that even in the general area of SE, there are types that cause injury and types that do not. Convulsive SE or SE associated with high-frequency discharges on electroencephalogram (EEG) clearly can cause damage, even when the high-intensity physical convulsions are controlled.3,4 Absence SE or spontaneous episodes of prolonged partial SE associated with lower frequency discharges (3 c/s spike and wave or rhythmic delta/theta activity) may not be, as there appear to be no long-term consequences to these episodes.
Neonatal Seizures
The developing brain is predisposed to seizures,5,6 and seizures are common in the neonate with an estimated incidence of 1 to 6 per 1000 live births.7–11 The term neonatal seizures encompasses seizures of diverse etiologies ranging from acute symptomatic causes (e.g., hypoxia-ischemia, intracranial hemorrhage, infections, and metabolic derangements such as hypocalcemia) to remote symptomatic causes (e.g., cerebral dysgenesis and inborn errors of metabolism) to idiopathic, genetically determined epilepsies (e.g., early myoclonic encephalopathy of infancy and familial neonatal convulsions).
As a group, the outcome for children with neonatal seizures, although improving, is poor.12 The mortality rate for neonates with seizures is high, with values ranging from 15 to 30%. In survivors, approximately 30% will develop neurological sequelae that include epilepsy, mental retardation, and cerebral palsy. Those most at risk for mortality and neurological morbidity are those neonates with persistent, difficult-to-treat seizures.13–17 It has been proposed that these seizures produce neurological injury as the result of energy depletion and cerebral metabolism dysfunction.18 However, because these prolonged, persistent seizures tend to be observed in those neonates with the most severe CNS involvement and injury (e.g., severe hypoxic ischemic encephalopathy), it is difficult to distinguish between the secondary damage, if any, imposed by the seizures and the primary injury imposed on the brain by the underlying etiology.
There are good examples in which neonatal seizures tend to be associated with a more favorable outcome. One example is the genetically determined benign familial neonatal convulsions, an autosomal dominantly inherited epilepsy syndrome resulting from mutations in voltage-gated potassium channel. These convulsions are brief events commencing as tonic posturing with apnea and other autonomic features, which can evolve to clonic movements in otherwise healthy neonates. These convulsions typically commence during the first week of life, can be observed multiple times per day, and may be difficult to treat. In most cases, the seizures will remit by 16 months of age. Although there is a increased risk of febrile and afebrile seizures later in life, psychomotor development tends to be normal.19 For those with psychomotor delay, it is likely that this outcome is the result of genetic factors and not the seizures.20
In summary, caveats abound when attempting to answer the question of whether seizures damage the neonatal brain. Although all recognize that the answer to this question is important, clinical data are limited.21 It is assumed that few would argue against the concept that in some circumstances seizures in the neonatal brain impose additional injury, but not all would agree on the circumstances or the extent of the injury, especially as some neonatal seizures have a benign outcome.
The Childhood Epilepsies: Good versus Bad
For many children with epilepsy, the outcome is good. The seizures remit and may not recur, even after medication is discontinued. Although cognitive and behavioral disorders may be observed transiently during the time period when the epilepsy was most active and required treatment with an antiepileptic medication, these children have no apparent evidence of a significant, long-term disability resulting from their epilepsy. In contrast, a group of children with epilepsy will have an associated psychomotor slowing or regression that persists even in the absence of seizures. In some of these children, data suggest that the seizures or the interictal EEG abnormalities contributed to the regression. Most childhood epilepsies fall somewhere between these two extremes (Table 4-1).
| Benign Epilepsies |
|---|
| Benign familial neonatal convulsions |
| Benign myoclonic epilepsy of infancy |
| Childhood absence epilepsy |
| Juvenile absence epilepsy |
| Benign rolandic epilepsy |
| Panayiotopoulos syndrome |
| Juvenile myoclonic epilepsy |
| Malignant Epilepsies |
|---|
| Ohtahara syndrome |
| Early myoclonic encephalopathy |
| West syndrome |
| Dravet syndrome |
| Lennox-Gastaut syndrome |
| Landau-Kleffner syndrome |
BENIGN ROLANDIC EPILEPSY
Given that the seizure frequency associated with this syndrome is low,22 and that the seizures most commonly occur during sleep, the majority of children with this syndrome do not require antiepileptic therapy.23 In addition, these children outgrow the epilepsy, and the interictal EEG abnormality resolves during their teenage years.24 That the seizures remit and the rate of epilepsy in these children in adulthood is no greater than that in the general population24 is in contrast to the adage that seizures beget seizures.
Although BRE is considered a benign epilepsy, cognitive and behavioral difficulties may be observed.25–28 However, for the most part, these difficulties are transient and appear to be related to the severity of the interictal discharges and not the result of a permanent injury to the brain.29 Indeed, as a group, the developmental outcome for these children, including those with cognitive and behavioral difficulties, whether treated or not, is good. A potential alternative explanation is that an underlying genetic predisposition underlies both the epilepsy and cognitive dysfunction.
WEST SYNDROME
West syndrome is a cryptogenic or symptomatic generalized epilepsy syndrome composed of infantile spasms, a hypsarrhythmic EEG, and mental retardation. The spasms, which tend to occur in clusters, are characterized by a brief, massive flexion, extension, or mixed contraction of the axial musculature coincident with an electrodecrement on the EEG. The incidence for this syndrome ranges from 3 to 4.5 per 10,000 live births, and the majority of children present during the first year of life.30–32 Causes of West syndrome are multiple and include the neurocutaneous syndromes (e.g., tuberous sclerosis complex), brain malformation and tumors, inborn disorders of metabolism, and genetic disorders, as well as acquired injuries from fetal infections, trauma, and hypoxic-ischemic injury.
As a group, the long-term prognosis is poor and includes medically refractory epilepsy, cognitive impairments ranging from learning disorders to mental retardation, and autism. To a large extent, like neonatal seizures, the prognosis is dependent on the etiology.33 However, medical and surgical reports demonstrating improved developmental outcome in those children with the shortest epilepsy duration and prompt response to therapy suggest that the seizure activity itself may be detrimental. But once again, caution in interpretation is required, as those with a short treatment lag or who responded quickly to therapy or potentially those most amenable to surgical resection may be those who would have had a better developmental outcome, even in the absence of effective treatment.34
Febrile Seizures and Mesial Temporal Lobe Epilepsy
On the other hand, a number of epidemiological studies indicate that the risk for developing epilepsy (or any other negative consequence) following febrile seizures is not greater than for children who never experienced them. However, the studies have also revealed that febrile seizures are not a single entity. Some are classified as simple and last less than 5 minutes, and these do not carry any additional risk for a poor long-term outcome. Complex febrile seizures (focal ictal behavior or seizures lasting more than 30 minutes), especially those that are prolonged, have a much higher risk for poor long-term outcome and the development of chronic epilepsy.
[/not-level-membership-for-neurology-category]
