Chapter 96 Liver and pancreas transplantation immunobiology
Maintenance Minimization, Immunosuppressant Withdrawal, and Tolerance
Over the past 5 decades, the ability to manipulate the immune response has become increasingly selective and less morbid. Through a general understanding of alloimmunity and the unique properties of the liver and pancreas, transplantation results have steadily improved (U.S. Organ Procurement and Transplantation Network, 2009). This chapter provides an overview of the principles governing immune management of liver and pancreatic transplant recipients, highlighting agents and strategies generally available for clinical use.
Physiologic Immunity
The immune system is typically divided into two complementary arms, innate and acquired. The innate immune system recognizes general motifs that universally represent pathologic states, such as ischemia, necrosis, trauma, and nonhuman cell surfaces (Dempsey et al, 1996; Fearon & Locksley, 1996; Matzinger, 1994, 2001). The acquired immune system distinguishes specific pathogens through antigen presentation and recognition. Both systems interact to maintain overall homeostasis. Typically, innate responses localize acquired responses to sites of pathologic processes and are less overtly regulated. In contrast, acquired immune responses lead to carefully regulated destruction of antigen-expressing tissue. The regulatory checks on acquired immunity prevent autoimmunity and uncontrolled lymphocyte proliferation. It is important to recognize that the acquired immune system is tailored for the individual based on each person’s unique MHC makeup. Evolutionarily, this diversity reduces the chance that any single pathogen can evade all individuals within a population; however, this also means that one person’s acquired immune response may not respond appropriately when placed in the context of another person’s MHC.
Innate Immunity
Receptors of innate immunity are cell bound on macrophages, neutrophils, and natural killer cells, and they circulate freely in the form of complement (Dempsey et al, 1996; Fearon & Locksley, 1996; Wright et al, 1990). Innate immunity is limited in specificity, and it retains broad reactivity to lipopolysaccharide (LPS) and components of pathogenic organisms. Importantly, the receptors of innate immunity are conserved between individuals and, in general, function similarly in physiologic and transplant situations. Once activated, the innate immune system initiates cytolytic pathways and recruits the acquired immune response.
The complement cascade is the primary mediator of cytolysis, and the byproducts of complement, along with phagocytic cells, signal initiation of acquired immunity (Baldwin et al, 2010; Wasowska et al, 2007). Platelets also have been increasingly recognized as serving an innate immune role through the release of chemotactic proteins and other immunostimulatory molecules (Kirk et al, 2009). Professional APCs not only engulf cells coated with complement but also those bearing foreign carbohydrate moieties (Hart, 1997).
Toll-like receptors (TLRs) have also been recognized as important for innate APC activation. This highly conserved family of receptors binds to pathogen-associated molecular pattern motifs commonly expressed on invading pathogenic organisms (Akira & Takeda, 2004). Engulfed cells are processed by the APC into protein fragments and are expressed on the cell surface bound to MHC molecules. Subsequently, T cells that are specific for these peptides can recognize their cognate antigens and become activated. Interestingly, the TLRs expressed in the liver differ from those expressed in the periphery and tend to be less responsive to ambient LPS (Hart, 1997). This is likely an adaptation to portal bacteremia and is thought to make the liver more tolerant of minor perturbations that would evoke an innate response in other organs.
Acquired Immunity
Specific recognition is the hallmark of the acquired immune system. The lymphocyte receptors (T-cell receptor and antibody) have evolved to distinguish an extremely diverse group of antigens. Furthermore, antigen recognition induces physiologic changes in the recognizing cell that lowers its threshold for subsequent encounters and leads to the phenomenon of immunologic memory, the more rapid response to subsequent antigen encounters (Ahmed & Gray, 1996). T-cell receptors (TCRs) bind peptide antigens that have been processed and presented in combination with MHC, and B-cell immunoglobulins bind antigens in their native conformation at a site remote from the B cell.
Cellular Immunity
Formation of the TCR is fundamental to understanding its function (Cooper, 1987; Davis & Bjorkman, 1988). T cells are formed in the bone marrow and fetal liver and migrate to the thymus during development. After entering the thymus, T cells undergo rearrangement of the DNA that encodes the TCR (Gill & Gulley, 1994). Each gene rearrangement results in generation of a TCR with specificity restricted to one epitope or structurally similar epitopes. The sum of all random TCR gene rearrangements generates TCRs with approximately 109 specificities, essentially all possible combinations of MHC and peptide antigen; if these T cells were released into the periphery, they would mediate fatal autoimmunity. Accordingly, thymic selection eliminates those cells likely to evoke autoimmunity (Bevan, 1997; Kappler et al, 1987).
Thus all surviving T cells bind to self MHC. The developing T cells then move into the thymic medulla, where either CD4 or CD8 expression is lost. If binding to the self MHC in the medulla results in a high-affinity interaction, these T cells are also eliminated, a process known as negative selection. Therefore, the majority of cells released from the thymus bind to self MHC without becoming activated; however, autoreactive cells occasionally escape thymic selection and serve as the etiology of diseases such as sclerosing cholangitis (see Chapter 41), autoimmune hepatitis (see Chapter 97A), and type 1 diabetes (see Chapter 101); thus additional regulation is required to prevent autoimmunity. In fact, a single interaction of TCR and antigen-bearing MHC is inadequate to trigger T-cell activation; rather, approximately 8000 TCR-MHC interactions over a period of several hours are needed to initiate activation (Kumagai et al, 1987; Rothenberg, 1996; Viola & Lanzavecchia, 1996), which further limits the likelihood of autoimmunity. Costimulatory molecules markedly alter this need for redundancy, and we discuss this below.
Immune responses are also regulated by accessory cell surface molecules that limit the types of cells with which a T cell can interact (Leahy et al, 1992; Saizawa et al, 1987). Parenchymal cells of the body express class I MHC and display internal cellular peptides within the binding groove of this molecule. T cells charged with destruction of diseased or infected parenchymal cells express CD8 that stabilizes TCR ligation with class I MHC. These cells are termed cytotoxic T cells. CD8+ T-cell killing can occur through either Ca2+-dependent secretory mechanisms or Ca2+-independent direct cell-contact mechanisms (Berke, 1995).
Hematopoietic cells express class I and class II MHC. Class II MHC displays peptide fragments that have been phagocytized from the extracellular space (Germain, 1994; Monaco, 1993). CD4 stabilizes the TCR–class II MHC interaction. CD4+ cells interact with dendritic cells, macrophages, and in some cases activated endothelial cells that display antigen. In addition, the resting sinusoidal endothelial cells of the liver have the ability to present antigen to T cells, making the liver an organ with considerable ability to evoke or suppress an immune response (Knolle & Gerken, 2000). The interaction between CD4+ T cells and APCs produces APCs that have the ability to martial CD8+ T cells (Lanzavecchia, 1998; Ridge et al, 1998). This process is mediated through upregulation of APC cell surface molecules known as costimulation receptors, thus APCs initiate an immune response but require CD4+ T cells to activate the primary effector arm of the acquired immune system—the CD8+ T cells.
An additional subset of T cells, regulatory T cells (Treg), further control promiscuous immune responses. Regulatory T cells have the ability to suppress cytokine secretion, adhesion molecule expression, and costimulatory signaling. The most extensively studied population of Treg cells express CD4 and CD25, the high-affinity α-chain of the interleukin (IL)-2 receptor (Wood & Sakaguchi, 2003). Animal models have suggested that these cells play a critical role in controlling immune activation (Baecher-Allan et al, 2001; Wood & Sakaguchi, 2003). The prevailing evidence suggests that Treg is responsive to established inflammation, rather than serving a prophylactic role in preventing inflammation; however, harnessing the power of Treg to quell counteradaptive immune responses such as rejection is an ongoing area of research in autoimmunity and alloimmunity.
Humoral Immunity
B cells recognize antigen in its native, unprocessed form (Cambier et al, 1994). When antigen binds to two cell surface antibodies, the antibodies are brought together in a process known as cross-linking, which stimulates B-cell proliferation and differentiation into an antibody-secreting plasma cell. The activation threshold for a resting B cell is relatively high, as it is for T cells. As with TCR recognition, costimulation can lower this threshold substantially (Tedder et al, 1994). B cells also have the ability to internalize antigen bound to surface immunoglobulins and process them for presentation to T cells along with costimulation molecules (Lederman et al, 1992).
Antibody structure is determined in the bone marrow through mechanisms similar to those that govern the generation of TCR diversity in the thymus (Gill & Gulley, 1994; Hozumi & Tanegawa, 1976). Five different heavy-chain loci (µ, γ, α, ε, and δ) on chromosome 14 and two different light-chain loci (κ and λ) on chromosome 2—each with V, D and/or J, and C regions—are brought together randomly by the RAG-1 and RAG-2 apparatus to form a functional antigen receptor (Kim et al, 2000). The basic antibody structure consists of two identical heavy chains and two identical light chains. The type of heavy chain used dictates the immunoglobulin (Ig) type: IgM, IgG, IgA, IgE, or IgD. The overall structure of the antibody results in two identical antigen-binding sites and a common region, the Fc portion. Bound antibody triggers activation of the complement cascade (Baldwin et al, 1995). In addition, most phagocytic cells have receptors for the Fc portion of IgG, allowing them to actively engulf antibody-coated cells.
Unlike the TCR, B-cell immunoglobulin loci undergo alteration after B-cell stimulation to improve the functionality of the secreted antibody. Isotype switching is the process of shifting from the initial heavy-chain IgM to one of four types to improve function and specialization. IgG is the most significant soluble mediator of opsonization and is the dominant antibody produced in response to alloantigen. IgA is important in mucosal immunity, IgE is involved in mast cell–mediated immunity, and IgD is primarily cell bound. After a B cell is activated, the specific D and J regions of the used heavy- and light-chain genes undergo random alterations of the antigen binding site. The resultant B cell clones have altered antigen affinity, hence the term affinity maturation (Griffiths et al, 1984); those clones that have higher affinity for the target antigen have a selective survival advantage and form the basis for a more vigorous response on reexposure to the antigen.
Mediators of Context: Costimulation and Cytokines
Isolated TCR binding with an MHC-peptide complex or antibody ligation with an antigen is not usually sufficient for lymphocyte activation. Receptor-ligand pairs on T and B cells and APCs, known as costimulation receptors, determine the character of the T-cell response (Fig. 96.1; Allison & Krummel, 1995; Chambers & Allison, 1997; Crawford et al, 2006). The type of costimulatory signal received by the lymphocyte determines whether the cell will become activated, remain quiescent, die, or become resistant to subsequent immune stimulation.
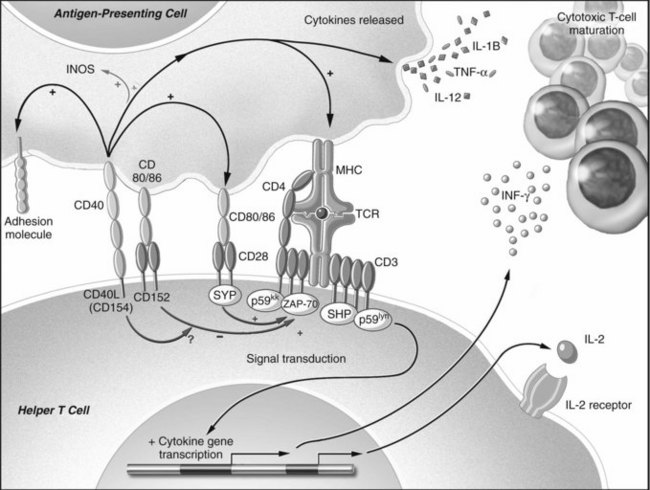
Although the mechanisms of costimulation have not been completely elucidated, it is known that binding of CD28 allows more efficient T-cell signal transduction. Through CD28-B7 interactions, the number of binding events required to trigger activation of a T-cell decrease from 8000 to 1500 (Rothenberg, 1996; Viola & Lanzavecchia, 1996). In contrast, when CTLA-4 binds B7, the T cell becomes incapable of producing IL-2 during the encounter and even in subsequent interactions (Blair et al, 1998). The CD19-CD21 complex provides comparable control of antigen receptor binding for B cells (Tedder et al, 1994).
Additionally, costimulation is mediated through another pair of receptors: CD40 found on dendritic cells, endothelium, B cells and other APCs, and CD154 on T cells and platelets (Armitage et al, 1992a, 1992b; Clark & Ledbetter, 1986; Noelle et al, 1992; Henn et al, 1998). The ability of APCs to stimulate a cytotoxic T-cell response is markedly augmented by the effects of CD40 binding. Following CD40 ligation, activating cytokines are released, and B7 molecules are upregulated (Bennett et al, 1998; Schoenberger et al, 1998). CD154 is upregulated after TCR ligation and provides positive feedback to the APC. In addition, CD154 is found in and released by activated platelets at sites of endothelial injury (Henn et al, 1998); therefore sites of trauma that recruit platelets create an environment of activating costimulatory molecules, thereby bridging the innate and acquired immune systems (Czapiga et al, 2004).
Direct cell-cell contact is not the only means by which immune cells communicate. Soluble mediators of communication known as cytokines, or interleukins, are polypeptides that are released from many cells; they can either activate or suppress adjacent cells (Arai et al, 1990), and the pattern of cytokine expression is thought to influence the resultant type of T-cell response (Mosmann et al, 1986; Mosmann, 1991). Once activated, T cells have been described by one of two cytokine-secretion phenotypes (see Chapter 10): cytotoxic T-cell responses are characterized by expression of interleukin (IL)-2, IL-12, IL-15, and interferon (IFN)-γ and are known as Th1 cells; T cells that promote humoral or eosinophilic responses are characterized by secretion of IL-4, IL-5, IL-10, and IL-13 and are known as Th2 cells.
Transplant Immunity
Most of the significant sequence polymorphism of MHC is located in the areas of the molecule that interact with the TCR, and individual variation in the sequence at the MHC-TCR interface defines alloreactivity. The lack of recipient T-cell thymic education with donor MHC leads to a nonphysiologically high frequency of alloreactive peripheral T cells. Many of these cells are crossreactive with antigen encountered during prior viral exposures, or even with autoantigens, as in the case of autoimmune disease. This is known as heterologous immunity, and it results in a situation whereby recipients have allospecific memory without having been exposed to the alloantigen (Adams et al, 2003). Thus a person’s immune response to a donor is the product of the individual’s MHC makeup and past immune exposures. This can lead to vigorous early rejection in apparently nonsensitized recipients.
T cells recognize alloantigen via their TCR in two distinct ways: either directly, by binding to donor MHC on transplanted tissues in the presence of donor costimulation, or indirectly, through self APC that has phagocytized and processed alloantigens to be presented, bound to self MHC and costimulation (Rogers & Lechler, 2001). In the case of transplanted organs, surgical trauma and ischemia exacerbate the potential for T-cell activation by causing class I and class II MHC upregulation (Gerritsen & Bloor, 1993). In addition, adhesion molecules and costimulation molecules are upregulated perioperatively (Takada et al, 1997; Hoffmann et al, 2002).
Initial T-cell binding to donor cells is nonspecific, mediated by adhesion molecules upregulated during donor cell activation (Fuggle & Koo, 1998). CD40 on donor APCs and endothelial cells is important in mediating cell activation in this setting via CD154 on T cells and activated platelets (Henn et al, 1998). Following nonspecific adhesion, MHC recognition occurs in the relatively high costimulation environment induced by surgical trauma and ischemia (Takada et al, 1997). Once alloreactive T cells are activated, they secrete cytokines, including IL-2 and IFN-γ, and they stimulate APCs to secrete IL-12 (Arai et al, 1990; Kirk et al, 1995; Krams et al, 1992). The resultant cytokine milieu recruits more T cells to the site of injury and potentiates clonal expansion. Secretory and cell contact–dependent mechanisms, perforin/granzyme and Fas mechanisms, respectively, are involved in T-cell cytotoxicity within the graft, resulting in graft destruction (Strehlau et al, 1997). Although acute rejection is the result of T-cell activation, antibody responses accompany many episodes. Cellular and soluble components of immunity mediate multiple distinct clinical rejection syndromes through cytokine mediated toxicity, cellular cytotoxicity, and direct effects of antibody and complement.
Clinical Rejection Syndromes
Hyperacute Rejection
Hyperacute rejection (HAR) is caused by donor-specific antibody that exists at the time of transplantation as a result of prior exposure to donor antigens or to antigens with crossreactivity. It develops precipitously within minutes to hours after graft reperfusion. Typically, HAR is avoided by confirming ABO compatibility and performing a crossmatch using techniques that detect donor-specific antibodies. When clinically relevant donor-specific antibodies are detected, graft survival is significantly decreased for most organ types (Noreen et al, 2003); however, the liver has long been regarded as relatively resistant to HAR, and crossmatching is often done only retrospectively (Neumann et al, 2001). Actually, the rates of acute rejection and long-term liver survival are similar among groups with a positive or negative crossmatch; this does not hold true for ABO-incompatible liver allografts (Egawa et al, 2004).
The role of antibody in liver transplantation remains an area of investigation at present. Unfortunately, the pancreas does not have similar resistance to HAR, and a positive crossmatch represents an absolute contraindication to pancreas transplantation. Although high titer antibodies mediate rapid graft injury, chronic exposure to lower titer antibody results in indolent graft damage, particularly in the case of the pancreas. The role of chronic alloantibody in liver transplantation remains controversial but likely mediates some degree of graft injury over time. Autoantibody directed against nonpolymorphic determinants of a graft is increasingly being recognized as detrimental in pancreas transplantation with recurrent β cell–specific autoimmunity responsible for at least some late graft loss after pancreas transplantation (Vendrame et al, 2010). Recurrent autoimmunity is also relevant in autoimmune hepatitis (Hytiroglou et al, 2009).
Acute Rejection
Acute rejection is most common between 4 days and 6 months after transplantation, and rejection that occurs during this time is considered early acute rejection; acute rejection after 6 months is considered late acute rejection. Liver allografts undergo acute rejection at a rate of approximately 24% to 47% (Fisher et al, 2004; Neuhaus et al, 2002; Wiesner et al, 2001), whereas pancreas allografts in the modern era (post 1995) undergo acute rejection at a rate of approximately 15% to 30%. These differences reflect a tendency toward less immunosuppression in liver patients, because the liver tolerates rejection better, perhaps because damaged parenchyma can regenerate. Also, liver rejection is more easily diagnosed than pancreas rejection through biopsy and serum enzyme measurements.
In both organs, acute rejection evolves over a period of days to weeks. After activation by either direct or indirect allorecognition, T cells infiltrate the allograft and initiate organ destruction through cytolysis and endothelial and ductular damage. Much of the acute graft dysfunction is likely mediated through the effects of macrophage-derived inflammatory cytokines that hinder parenchymal function without necessarily causing cell death (Girlanda et al, 2008), thus prompt treatment has the potential to quell the rejection prior to direct T-cell–mediated cytotoxicity. Usually acute liver allograft rejection is also accompanied by graft and peripheral eosinophilia (Barnes et al, 2003).
Early detection of acute rejection is critical to facilitate intervention prior to permanent T-cell–mediated parenchymal damage; therefore unexplained hepatic dysfunction should prompt a graft biopsy. In the case of pancreas allografts, usually no direct biochemical evidence of rejection is present. If the transplantation was performed with a kidney, rejection of the kidney precedes or signals concomitant pancreas rejection in approximately 80% of patients. In transplantations of pancreas alone, fever, abdominal pain, allograft tenderness, and elevations in serum amylase or lipase may be signs of rejection; however, many rejections occur without early symptoms (Stratta et al, 1996). Hyperglycemia is a late complication in pancreas rejection, because acinar cells reject prior to β cells.
Prompt recognition of acute rejection is imperative, because prolonged rejection allows for recruitment of multiple arms of the immune system and consequently results in decreased efficacy of antirejection therapies directed against T-cell responses. T-cell–specific therapies can resolve acute rejection episodes in most cases, and steroids are the first-line agent in most centers. Typically, early acute rejection episodes have a negative impact on the long-term survival of an allograft, although the liver is relatively resistant to this effect (Dousset et al, 1998). In fact, even late acute rejection in liver transplantation has not been shown to influence long-term graft function (Junge et al, 2005). This greatly contrasts both early and late acute rejection episodes in pancreas transplantation, which have repeatedly been shown to be deleterious to long-term graft survival (Reddy et al, 2001; Tesi et al, 1994).
Chronic Graft Loss
The causes of chronic graft loss remain poorly characterized (Libby & Pober, 2001). Although called chronic rejection, it likely has nonimmune components and immune origins. Evolving over the course of months to years, chronic graft dysfunction remains resistant to conventional immunosuppressive therapies; regardless of the transplanted organ, it develops as fibrosis with modest lymphocytic infiltration. Monocytic and dendritic cell infiltrates predominate, and destruction of epithelial and endothelial structures is progressive.
Many aspects of chronic graft loss relate to the events surrounding transplantation, such as ischemic injury. In the liver, chronic graft loss manifests as ductopenia, or vanishing bile duct syndrome (Inomata & Tanaka, 2001), typically defined as a condition wherein less than 50% of portal triads contain bile ducts (Demetris et al, 1998). The rate of chronic graft loss is far slower for liver grafts than for other organs, likely because of the ability of the liver to regenerate following subtle injury. As the technical challenges of pancreas transplantation have been corralled, more grafts are being lost to chronic rejection (Humar et al, 2003), a significant portion of which may be related to recurrent autoimmune diabetes.
Immunosuppression
To date, no single agent has been discovered that effectively prevents allograft rejection, although liver transplant recipients can often be weaned over time to a single-drug regimen. Similarly, all manipulations that limit rejection also increase susceptibility to infection and malignancy. No immunosuppressant is allograft specific; therefore the rational selection of immunosuppressants involves using multiple synergistic agents to prevent rejection without simultaneously crippling the recipient’s defenses. Characteristically, liver allografts typically require less immunosuppression than other organs (Ramos et al, 1995). This has been proposed to be a function of the APCs in the liver, the sheer size and antigenic load of the liver, and its regenerative capacity. Indeed, recent evidence indicates that 10% to 20% of liver transplant recipients can eventually be withdrawn from all immunosuppressive drugs over a period of years (Martinez-Llordella et al, 2008). Importantly, no immunosuppressive regimen has clearly established itself as superior, and the therapies chosen remain variable from one center to another.
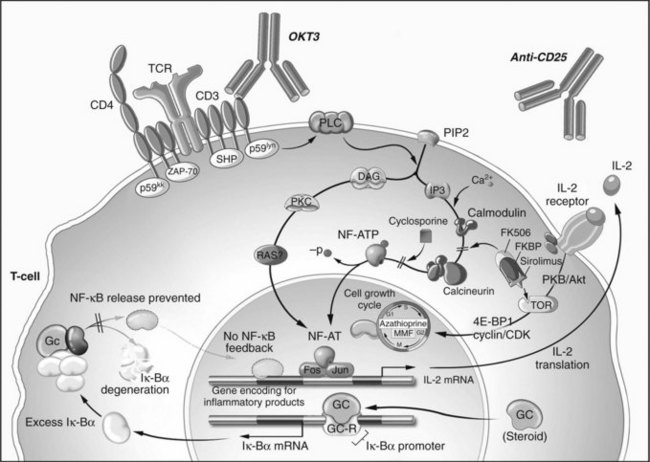
The immune system is most prone to reject an allograft perioperatively because of the surgical and ischemic injury associated with transplantation; therefore the most intense immunosuppression is given during the weeks that follow transplantation. A period of rigorous immunosuppression at the time of transplant is known as induction therapy (Kirk, 2006), which typically consists of T-cell–depleting strategies that, while effective, are too toxic to be administered long term. Maintenance immunosuppression is less potent but can be given chronically to prevent acute rejection for the life of the transplant recipient. Finally, agents used to halt ongoing rejection are known as rescue agents. The sites of action of various immunosuppressive agents are illustrated in Figure 96.2.
Corticosteroids
Corticosteroids have been a mainstay of transplant immunosuppression for over 40 years (Starzl et al, 1963). At low doses, glucocorticosteroids, typically prednisone or methylprednisolone, are used as maintenance immunosuppression; at higher doses, they can be used as rescue therapy. Although steroids are ineffective as monotherapy to prevent rejection, they have been effectively combined with other agents to improve graft survival. Unfortunately, the desirable immunosuppressive effects of steroids are counterbalanced by their contribution to transplant morbidity; therefore many ongoing efforts have sought to minimize or eliminate glucocorticosteroid use for maintenance immunosuppression. Many centers now rapidly wean liver allograft recipients off steroids, whereas most pancreas programs rely on low-dose steroids indefinitely, although steroid-free regimens are increasingly being used (Singh & Stratta, 2008; Tanchanco et al, 2008; Vessal et al, 2007).
The immunosuppressive mechanism of glucocorticosteroids was elucidated long after its clinical introduction (Auphan et al, 1995; Scheinman et al, 1995). After nonspecific cytoplasmic uptake, steroids bind to an intracellular receptor, enter the nucleus as a receptor-ligand pair, and increase the transcription of several genes, notably IκBα. This protein binds to and inactivates NFκB, an important transcription factor of T-cell and APC activation and cytokine production.
Antiproliferative Agents
Azathioprine
The antimetabolite azathioprine (AZA) was the first immunosuppressant used in organ transplantation (Calne & Murray, 1961; Hitchings et al, 1950). AZA undergoes hepatic conversion to 6-mercaptopurine (6-MP) and then to 6-thio-inosine-monophosphate (6-tIMP). These derivatives alkylate DNA precursors and inhibit DNA synthesis. In addition, they can introduce chromosomal breaks and interfere with DNA repair mechanisms, and the ultimate effect is to deplete the cell of adenosine. AZA works on all rapidly dividing cells, not only on lymphocytes; consequently, bone marrow, hepatic, and gastrointestinal toxicity are limiting factors. As monotherapy, AZA is ineffective and is rarely found in modern immunosuppressive regimens.
Mycophenolate Mofetil
Since 1995, mycophenolate mofetil (MMF) has been an approved immunosuppressant for use in adults (Platz et al, 1991). MMF is a morpholinoethyl ester of mycophenolic acid (MPA), a noncompetitive, reversible inhibitor of inosine monophosphate (IMP) dehydrogenase with improved bioavailability. MMF prevents a critical step in RNA and DNA synthesis by blocking guanosine monophosphate (GMP) formation from IMP by IMP dehydrogenase. In all cells except lymphocytes, there exists a “salvage pathway” for GMP formation. This crucial difference is exploited by MMF to produce a relatively lymphocyte-specific immunosuppressant. MMF blocks the proliferation of T and B cells and inhibits the formation of donor-specific antibody. Its introduction dramatically improved the success of pancreas transplantation; therefore it is used as an adjuvant immunosuppressive agent in most pancreas centers.
In liver transplantation, the use of MMF has continued to gain favor, because it allows a reduction in calcineurin inhibitors, the cornerstone of most immunosuppressive regimens. Presumably, this reduction mitigates the deleterious effects on renal function of calcineurin inhibitors in hepatic and pancreatic transplant recipients (Biselli et al, 2009). MMF combined with low-dose calcineurin inhibitors and corticosteroids have also been proven useful in decreasing episodes of acute rejection (Farkas et al, 2009). Its chronic use is typical in pancreas transplantation, and it plays at least a temporary role in most liver transplant regimens.
Calcineurin Inhibitors
Cyclosporine
Cyclosporine A (CyA) is a cyclic endecapeptide isolated from the fungus Tolypocladium inflatum gams (Borel et al, 1976; Kahan, 1994). Its introduction revolutionized transplantation and made extrarenal transplantation a viable reality. The mechanism of action of this T-cell–specific immunosuppressant is mediated through cyclophilin (Cn) binding. The CyA-Cn complex binds to the calcineurin-calmodulin complex and prevents phosphorylation and activation of NF-AT, a transcription regulating factor. Blockade of NF-AT prevents IL-2 gene transcription. In addition, transforming growth factor (TGF)-α transcription is unregulated, and other genes critical for T-cell activation are altered (Khanna et al, 1996; Kirk et al, 1997). These and other effects may be responsible for the toxicity of CyA. Primarily, CyA blocks TCR signal transduction but does not inhibit costimulatory signaling (June et al, 1987). The effects of CyA can be overcome with high levels of IL-2, therefore once IL-2 is present in the graft, as in the case of ongoing rejection, CyA is rendered ineffective. Consequently, CyA is a maintenance immunosuppressant that has no role in rescue therapy.
CyA nephrotoxicity is a constant consideration in its use. Through a TGF-α mediated mechanism, CyA decreases renal blood flow by up to 30% (Khanna et al, 1996; Kirk et al, 1997). Increased transcription of endothelin also activates the renin-angiotensin pathway, leading to hypertension; the vascular effects of CyA may also delay resolution of hepatorenal syndrome; and CyA may lead to neurologic side effects, hypertrichosis, and malignancy (Hojo et al, 1999). In addition, metabolism of CyA through cytochrome P450 enzymes leads to multiple drug interactions.
Tacrolimus
Kino and colleagues (1987) first demonstrated the immunosuppressant effects of tacrolimus in 1987, and it has become the mainstay of most liver and pancreas transplant regimens. Tacrolimus is a macrolide antibiotic produced by Streptomyces tsukubaensis. Like CyA, tacrolimus blocks NF-AT activation and its downstream effects (Fruman et al, 1992). The intracellular target of tacrolimus is known as FK-binding protein (FK-BP). Similar to CyA, tacrolimus increases TGF-α transcription and therefore carries with it the benefits and toxicities of this cytokine (Khanna et al, 1996; Kirk et al, 1997). Although it is 100 times more potent than CyA in preventing IL-2 and IFN-γ transcription, its toxicities limit the dose to approximately 1% of CyA. In addition to its use as a maintenance agent, tacrolimus has been shown to have efficacy at high doses in reversing liver rejection episodes (Starzl et al, 1989).
As compared to CyA, tacrolimus has more pronounced neurologic side effects, such as mental status changes and tremors. Tacrolimus use also results in a higher rate of posttransplant type 2 diabetes, although it has fewer cosmetic side effects. Tacrolimus has been shown to be superior to CyA in preventing acute episodes of rejection, reducing the number of steroid-resistant acute rejections, and improving long-term graft and patient survival. For these reasons it has replaced cyclosporine as the first-line calcineurin inhibitor used in both liver and pancreas transplantation in most centers (Haddad et al, 2006). Because tacrolimus is metabolized by cytochrome P450 enzymes, drug interaction concerns are similar to those with CyA. In recent years, many liver transplant recipients have been successfully weaned to monotherapy tacrolimus, but this trend has not been seen in pancreas transplantation.
mTOR Inhibitors
Sirolimus and Everolimus
Sirolimus and everolimus are macrolide antibiotics developed from Streptomyces hygroscopicus (Baker et al, 1978; Martel et al, 1977; Sehgal et al, 1975). Similar to tacrolimus, these agents bind to immunophilin FKBP12, also known as the mammalian target of rapamycin (mTOR). Unlike tacrolimus, these agents do not affect calcineurin activity (Dumont et al, 1990a, 1990b; Molnar-Kimber 1996). The primary effect of mTOR inhibitors is to inhibit IL-2 receptor signal transduction and not to block NF-AT nuclear translocation; therefore T cells are rendered incompetent against the proliferative effects of exogenous IL-2 (Kuo et al, 1992) but are still capable of IL-2 gene transcription.
The primary toxicities of mTOR inhibitors are hypercholesterolemia, hypertriglyceridemia, poor wound healing, thrombocytopenia, and oral ulcers. Currently, sirolimus carries an FDA black-box warning against its use in liver transplantation in the first 28 days postoperatively because of increased risk of hepatic artery thrombosis, thus its use in liver transplantation is limited to conversion regimens. In addition, sirolimus may have a direct hepatotoxic effect (Neff et al, 2004).
The principal manner in which mTOR inhibitors are being used is as an adjunct or alternative to calcineurin inhibitors, and mTOR inhibitors have been developed in recent years as antiproliferative agents for use in oncologic indications. As such, its use in patients transplanted for the treatment of hepatic malignancies is being investigated (Schnitzbauer et al, 2010; Toso et al, 2010; Vivarelli et al, 2010).
Antilymphocyte Agents
Antilymphocyte agents are commonly used in pancreas transplantation during the induction phase of immunosuppression (Niemeyer et al, 2002; Stratta et al, 2003). Their use in liver transplantation is sparse owing to the reduced immunogenic phenotype typical of liver grafts.
Antilymphocyte/Antithymocyte Globulin
A polyclonal antilymphocyte globulin (ALG) is produced by inoculating an animal, such as a horse or rabbit, with human lymphocytes and then collecting the serum and purifying the resultant IgG. This preparation contains antibodies of multiple epitope specificities directed against lymphocyte and other cell antigens (Gaber et al, 1998; Merion et al, 1998). In the United States, the common immunogen is thymocytes, and the resultant polyclonal antibody is rabbit antithymocyte globulin (RATG). These agents promote T-cell depletion through opsonization and complement-mediated lysis (Merion et al, 1998). They also have multiple nondepletional effects that inhibit T-cell activation and function, such as crosslinking costimulation and adhesion molecules and blocking TCR signal transduction through internalization of other cell surface receptors.
OKT3
Unlike polyclonal antibody preparations, monoclonal antibodies have single-target epitope specificity. Muromonab-CD3, better known as OKT3 (Orthoclone OKT3; Ortho Pharmaceuticals, Raritan, NJ), is a murine antibody directed against the CD3 signal transduction subunit of the TCR; it was the first monoclonal antibody approved for any use (Ortho Multicenter Transplant Study Group, 1985; Wilde & Goa, 1996). OKT3 mediates its effects by binding to CD3 and causing internalization of the TCR complex, thereby preventing antigen recognition (Marano et al, 1989; Wilde & Goa, 1996). Additionally, OKT3 opsonizes T cells, causing their activation and degranulation. This exhausts the cell, making it ineffective as an effector, but it also leads to OKT3’s primary side effect: cytokine release syndrome (Chatenoud et al, 1990). Cytokine release can result in profound hypotension, pulmonary edema, and cardiac depression. Because of the relatively toxic side effect profile and the availability of other agents that accomplish the same purpose, OKT3 is no longer manufactured.
Anti–IL-2 Receptor α-Chain Monoclonal Antibodies
The high affinity α-subunit of the IL-2 receptor (CD25) has been the target of two monoclonal antibodies, daclizumab and basiliximab. CD25 is required for clonal naïve T-cell expansion and is therefore an attractive target for specific elimination of activated T cells (Goebel et al, 2000). Unlike OKT3, binding of the target epitope does not result in cytokine release, thus administration is generally well tolerated, although the antirejection effect is similarly modest. Daclizumab has recently been discontinued for use in the United States and Europe, and simulect is used in approximately a third of all pancreas and liver transplant centers.
Costimulation Blockade
Recently the development of belatacept, a high-affinity fusion protein specific for the B7 molecules CD80 and CD86, has been shown to be a viable costimulatory antagonist for use in renal transplant (Durrbach et al, 2010; Vincenti et al, 2010). It has also been shown to be an effective replacement for calcineurin inhibitors in selected patients and thus may find a role in liver and pancreas transplantation. Currently, trials are underway to investigate belatacept in liver and pancreas transplantation in combination with, and compared with, calcineurin inhibitor–based immunosuppression.
Maintenance Minimization, Immunosuppressant Withdrawal, and Tolerance
The strategy of using depletional agents such as RATG to reduce the requirement for maintenance immunosuppression is gaining traction. Recently, another monoclonal depleting antibody has been investigated in liver transplantation. Alemtuzumab, a CD52-specific antibody, rapidly depletes T cells and, to a lesser degree, B cells and monocytes from the circulation and from secondary lymphoid organs. It is approved for the treatment of lymphoid malignancies and has been investigated off label in transplantation. Recently, it has been used with low-dose tacrolimus to prevent rejection in liver transplant recipients (Marcos et al, 2004; Tzakis et al, 2004). Similar efforts have been made using antithymocyte globulin induction therapy (Tchervenkov et al, 2004). Alemtuzumab depletion has also been used in pancreas (Kaufman et al, 2006), and this strategy has been used effectively in kidney transplantation (Calne et al, 1998; Kirk et al, 2003; Knechtle et al, 2003).
Steroid withdrawal is the goal for many transplant clinicians wishing to spare their patients from the numerous side effects associated with glucocorticosteroid dependence. Steroid withdrawal can be achieved in approximately 85% of liver transplant recipients at 3 months without a significant increase in acute rejection rates (Reding, 2000). Although attempts to withdraw steroids in patients receiving pancreas transplants have been less successful, with appropriate patient selection, up to 70% of pancreas transplant patients may be amenable to steroid withdrawal (Humar et al, 2000).
A modest percentage of liver transplant recipients can be completely withdrawn from all immunosuppressive agents (Benitez et al, 2009; Lee et al, 2009; Martinez-Llordella et al, 2007; Mazariegos et al, 1997). Although the periods of follow-up among withdrawal reports are highly variable, and the long-term incidence of chronic rejection and graft loss has not been defined, some recipients clearly are capable of spontaneously accepting liver allografts. The challenge remains to establish criteria for appropriate withdrawal from immunosuppression and to identify those individuals in whom drug withdrawal is a safe strategy. At present, the best predictor of successful drug withdrawal is long-term rejection-free survival on low-dose immunosuppression. Liver transplant recipients on single-drug therapy for more than 10 years are 10 times more likely to be withdrawn successfully compared with patients in the first 3 years after transplantation.
It is possible that signatures of recipient gene expression or other biologic parameters can identify a protolerant signature in liver transplantation patients (Martinez-Llordella et al, 2007). The majority of these targets are associated with NK and γδTCR+ T cells, which could be used to predict which liver transplant patients might be successfully withdrawn from immune-modulating medications; however, no evidence suggests that pancreas transplant recipients can be withdrawn from immunosuppression.
The “holy grail” of transplantation is tolerance. From its conception (Billingham et al, 1953), acquired allospecific tolerance has been defined as the ability to maintain a functional allograft and an intact immune response without the need for any therapeutic drugs. Numerous strategies have been attempted in reaching this goal, but to date, all prospective attempts to create broadly applicable, reliable, and durable tolerance in humans have failed. Although the reasons are numerous and still in the process of being elucidated, the pursuit of tolerance will bring all the benefits of organ replacement therapy and eliminate its toxicity, namely chronic immunosuppression. Ongoing strategies include the manipulation of costimulation signals, depletional approaches, and techniques designed to induce mixed chimerism, a state in which elements of both the donor and recipient immune systems persist in one individual (Cosimi & Sachs, 2004; Harlan & Kirk 1999; Kirk, 2003).
Many have speculated that liver recipients are more likely to be rendered tolerant than other allograft recipients, and indeed the emerging results from immunosuppressive withdrawal trials suggest this to be the case. Reasons cited include the unique APCs in the liver (see Chapter 9), the liver’s regenerative capacity, its shear antigenic bulk, and it tendency to seed the recipient’s body with donor hematopoietic cells, perhaps overwhelming the recipient immune system into a state of clonal exhaustion (Starzl, 1998). Tolerance attempts in liver transplantation are likely to proceed in the coming years, and their results will be anticipated. In contrast, the pancreas has been considered an immunogenic organ with little capacity to regenerate or deal with immune insults. As such, pancreas allograft tolerance will be much less likely to be achieved in the near future with the current strategies available.
Adams AB, Pearson TC, Larsen CP. Heterologous immunity: an overlooked barrier to tolerance. Immunol Rev. 2003;196:147-160.
Ahmed R, Gray D. Immunological memory and protective immunity: understanding their relation. Science. 1996;272:54-60.
Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499-511.
Allison JP, Krummel MF. The yin and yang of T cell costimulation. Science. 1995;270:932-933.
Arai KI, et al. Cytokines: coordinators of immune and inflammatory responses. Annu Rev Biochem. 1990;59:783-836.
Armitage RJ, et al. Molecular and biological characterization of a murine ligand for CD40. Nature. 1992;357:80-82.
Armitage RJ, et al. Identification of a source of biologically active CD40 ligand. Eur J Immunol. 1992;22:2071-2076.
Auphan N, et al. Immunosuppression by glucocorticoids: inhibition of NF-kappa B activity through induction of I kappa B synthesis. Science. 1995;270:286-290.
Baecher-Allan C, et al. CD4+CD25 high regulatory cells in human peripheral blood. J Immunol. 2001;167:1245-1253.
Baker H, et al. Rapamycin (AY-22,989), a new antifungal antibiotic. III. In vitro and in vivo evaluation. J Antibiot (Tokyo). 1978;31:539-545.
Baldwin WM3rd, et al. Complement in organ transplantation: contributions to inflammation, injury, and rejection. Transplantation. 1995;59:797-808.
Baldwin WMr, Valujskikh A, Fairchild RL. Antibody-mediated rejection: emergence of animal models to answer clinical questions. Am J Transplant. 2010;10:1135-1142.
Barnes EJ, et al. Applications and limitations of blood eosinophilia for the diagnosis of acute cellular rejection in liver transplantation. Am J Transplant. 2003;3:432-438.
Bennett SR, et al. Help for cytotoxic T-cell responses is mediated by CD40 signalling. Nature. 1998;393:478-480.
Benitez C, Lozano JJ, Fueyo AS. Gene expression profiling and transplantation tolerance in the clinic. Transplantation. 2009;88:S50-S53.
Berke G. The CTL’s kiss of death. Cell. 1995;81:9-12.
Bevan MJ. In thymic selection, peptide diversity gives and takes away. Immunity. 1997;7:175-178.
Billingham RE, Brent L, Medawar PB. Activity acquired tolerance of foreign cells. Nature. 1953;172:603-606.
Biselli M, et al. Two yr mycophenolate mofetil plus low-dose calcineurin inhibitor for renal dysfunction after liver transplant. Clin Transplant. 2009;23:191-198.
Blair PJ, et al. CTLA-4 ligation delivers a unique signal to resting human CD4 T cells that inhibits interleukin-2 secretion but allows Bcl-X(L) induction. J Immunol. 1998;160:12-15.
Borel JF, et al. Biological effects of cyclosporin A: a new antilymphocytic agent. Agents Actions. 1976;6:468-475.
Calne R, et al. Prope tolerance, perioperative campath 1H, and low-dose cyclosporin monotherapy in renal allograft recipients. Lancet. 1998;351:1701-1702.
Calne RY, Murray JE. Inhibition of the rejection of renal homografts in dogs by Burroughs Wellcome 57-322. Surg Forum. 1961;12:118-120.
Cambier JC, Pleiman CM, Clark MR. Signal transduction by the B cell antigen receptor and its coreceptors. Annu Rev Immunol. 1994;12:457-486.
Chambers CA, Allison JP. Co-stimulation in T cell responses. Curr Opin Immunol. 1997;9:396-404.
Chatenoud L, et al. In vivo cell activation following OKT3 administration: systemic cytokine release and modulation by corticosteroids. Transplantation. 1990;49:697-702.
Clark EA, Ledbetter JA. Activation of human B cells mediated through two distinct cell surface differentiation antigens, Bp35 and Bp50. Proc Natl Acad Sci U S A. 1986;83:4494-4498.
Cooper MD. Current concepts: B lymphocytes—normal development and function. N Engl J Med. 1987;317:1452-1456.
Cosimi AB, Sachs DH. Mixed chimerism and transplantation tolerance. Transplantation. 2004;77:943-946.
Crawford A, et al. Primary T cell expansion and differentiation in vivo requires antigen presentation by B cells. J Immunol. 2006;176:3498-3506.
Czapiga M, Kirk AD, Lekstrom-Himes J. Platelets deliver costimulatory signals to antigen-presenting cells: a potential bridge between injury and immune activation. Exp Hematol. 2004;32:135-139.
Davis MM, Bjorkman PJ. T-cell antigen receptor genes and T-cell recognition. Nature. 1988;334:395-402.
Demetris AJ, et al. Chronic liver allograft rejection: a National Institute of Diabetes and Digestive and Kidney Diseases interinstitutional study analyzing the reliability of current criteria and proposal of an expanded definition. National Institute of Diabetes and Digestive and Kidney Diseases Liver Transplantation Database. Am J Surg Pathol. 1998;22:28-39.
Dempsey PW, et al. C3d of complement as a molecular adjuvant: bridging innate and acquired immunity. Science. 1996;271:348-350.
Dousset B, et al. Is acute rejection deleterious to long-term liver allograft function? J Hepatol. 1998;29:660-668.
Dumont FJ, et al. Distinct mechanisms of suppression of murine T cell activation by the related macrolides FK-506 and rapamycin. J Immunol. 1990;144:251-258.
Dumont FJ, et al. The immunosuppressive macrolides FK-506 and rapamycin act as reciprocal antagonists in murine T cells. J Immunol. 1990;144:1418-1424.
Durrbach A, et al. A phase III study of belatacept versus cyclosporine in kidney transplants from extended criteria donors (BENEFIT-EXT study). Am J Transplant. 2010;10:547-557.
Egawa H, et al. Impact of recipient age on outcome of ABO-incompatible living-donor liver transplantation. Transplantation. 2004;77:403-411.
Farkas SA, et al. Calcineurin inhibitor minimization protocols in liver transplantation. Transpl Int. 2009;22:49-60.
Fearon DT, Locksley RM. The instructive role of innate immunity in the acquired immune response. Science. 1996;272:50-53.
Fisher RA, et al. Four-year follow-up of a prospective randomized trial of mycophenolate mofetil with cyclosporine microemulsion or tacrolimus following liver transplantation. Clin Transplant. 2004;18:463-472.
Fruman DA, et al. Calcineurin phosphatase activity in T lymphocytes is inhibited by FK 506 and cyclosporin A. Proc Natl Acad Sci U S A. 1992;89:3686-3690.
Fuggle SV, Koo DD. Cell adhesion molecules in clinical renal transplantation. Transplantation. 1998;65:763-769.
Gaber AO, et al. Results of the double-blind, randomized, multicenter, phase III clinical trial of Thymoglobulin versus Atgam in the treatment of acute graft rejection episodes after renal transplantation. Transplantation. 1998;66:29-37.
Germain RN. MHC-dependent antigen processing and peptide presentation: providing ligands for T lymphocyte activation. Cell. 1994;76:287-299.
Gerritsen ME, Bloor CM. Endothelial cell gene expression in response to injury. FASEB J. 1993;7:523-532.
Gill JI, Gulley ML. Immunoglobulin and T-cell receptor gene rearrangement. Hematol Oncol Clin North Am. 1994;8:751-770.
Girlanda R, et al. Monocyte infiltration and kidney allograft dysfunction during acute rejection. Am J Transplant. 2008;8:600-607.
Goebel J, et al. Daclizumab (Zenapax) inhibits early interleukin-2 receptor signal transduction events. Transpl Immunol. 2000;8:153-159.
Griffiths GM, et al. Somatic mutation and the maturation of immune response to 2-phenyl oxazolone. Nature. 1984;312:271-275.
Haddad EM, et al. Cyclosporin versus tacrolimus for liver transplanted patients. Cochrane Database Syst Rev. 4, 2006.
Harlan DM, Kirk AD. The future of organ and tissue transplantation: is a new paradigm on the horizon? JAMA. 1999;282:1076-1082.
Hart DN. Dendritic cells: unique leukocyte populations which control the primary immune response. Blood. 1997;90:3245-3287.
Henn V, et al. CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature. 1998;391:591-594.
Hitchings GH, Elion GB, Falco EA. Antagonists of nucleic acid derivatives. II. Reversal studies with substances structurally related to thymine. J Biol Chem. 1950;185:643-649.
Hoffmann SC, et al. Molecular and immunohistochemical characterization of the onset and resolution of human renal allograft ischemia-reperfusion injury. Transplantation. 2002;74:916-923.
Hojo M, et al. Cyclosporine induces cancer progression by a cell-autonomous mechanism. Nature. 1999;397:530-534.
Hozumi N, Tonegawa S. Evidence for somatic rearrangement of immunoglobulin genes coding for variable and constant regions. Proc Natl Acad Sci U S A. 1976;73:3628-3632.
Humar A, et al. Steroid withdrawal in pancreas transplant recipients. Clin Transplant. 2000;14:75-78.
Humar A, et al. Chronic rejection: the next major challenge for pancreas transplant recipients. Transplantation. 2003;76:918-923.
Hytiroglou P, et al. Recurrence of primary biliary cirrhosis and development of autoimmune hepatitis after liver transplant: a blind histologic study. Hepatol Res. 2009;39:577-584.
Inomata Y, Tanaka K. Pathogenesis and treatment of bile duct loss after liver transplantation. J Hepatobiliary Pancreat Surg. 2001;8:316-322.
June CH, et al. T-cell proliferation involving the CD28 pathway is associated with cyclosporine-resistant interleukin 2 gene expression. Mol Cell Biol. 1987;7:4472-4481.
Junge G, et al. The influence of late acute rejection episodes on long-term graft outcome after liver transplantation. Transplant Proc. 2005;37:1716-1717.
Kahan BD. Role of cyclosporine: present and future. Transplant Proc. 1994;26:3082-3087.
Kappler JW, Roehm N, Marrack P. T cell tolerance by clonal elimination in the thymus. Cell. 1987;49:273-280.
Kaufman DB, et al. Alemtuzumab induction and prednisone-free maintenance immunotherapy in simultaneous pancreas–kidney transplantation comparison with rabbit antithymocyte globulin induction: long-term results. Am J Transplant. 2006;6:331-339.
Khanna A, et al. Immunoregulatory and fibrogenic activities of cyclosporine: a unifying hypothesis based on transforming growth factor-beta expression. Transplant Proc. 1996;28:2015-2018.
Kim DR, Park SJ, Oettinger MA. V(D)J recombination: site-specific cleavage and repair. Mol Cells. 2000;10:367-374.
Kino T, et al. FK-506, a novel immunosuppressant isolated from a Streptomyces. II. Immunosuppressive effect of FK-506 in vitro. J Antibiot (Tokyo). 1987;40:1256-1265.
Kirk AD. Less is more: maintenance minimization as a step toward tolerance. Am J Transplant. 2003;3:643-645.
Kirk AD. Induction immunosuppression. Transplantation. 2006;82:593-602.
Kirk AD, Bollinger RR, Finn OJ. Rapid, comprehensive analysis of human cytokine mRNA and its application to the study of acute renal allograft rejection. Hum Immunol. 1995;43:113-128.
Kirk AD, Morrell CN, Baldwin WM3rd. Platelets influence vascularized organ transplants from start to finish. Am J Transplant. 2009;9:14-22.
Kirk AD, et al. Posttransplant diastolic hypertension: associations with intragraft transforming growth factor-beta, endothelin, and renin transcription. Transplantation. 1997;64:1716-1720.
Kirk AD, et al. Results from a human renal allograft tolerance trial evaluating the humanized CD52-specific monoclonal antibody alemtuzumab (CAMPATH-1H). Transplantation. 2003;76:120-129.
Knechtle SJ, et al. Campath-1H induction plus rapamycin monotherapy for renal transplantation: results of a pilot study. Am J Transplant. 2003;3:722-730.
Knolle PA, Gerken G. Local control of the immune response in the liver. Immunol Rev. 2000;174:21-34.
Krams SM, et al. Cytokine and T cell receptor gene expression at the site of allograft rejection. Transplantation. 1992;53:151-156.
Kumagai N, et al. Requirements for the simultaneous presence of phorbol esters and calcium ionophores in the expression of human T lymphocyte proliferation-related genes. J Immunol. 1987;139:1393-1399.
Kuo CJ, et al. Rapamycin selectively inhibits interleukin-2 activation of p70 S6 kinase. Nature. 1992;358:70-73.
Lanzavecchia A. Immunology: licence to kill. Nature. 1998;393:413-414.
Leahy DJ, Axel R, Hendrickson WA. Crystal structure of a soluble form of the human T cell coreceptor CD8 at 2.6 A resolution. Cell. 1992;68:1145-1162.
Lederman S, et al. Molecular interactions mediating T–B lymphocyte collaboration in human lymphoid follicles: roles of T cell–B-cell-activating molecule (5c8 antigen) and CD40 in contact-dependent help. J Immunol. 1992;149:3817-3826.
Lee JH, et al. Withdrawal of immunosuppression in pediatric liver transplant recipients in Korea. Yonsei Med J. 2009;50:784-788.
Libby P, Pober JS. Chronic rejection. Immunity. 2001;14:387-397.
Marano N, Holowka D, Baird B. Bivalent binding of an anti-CD3 antibody to Jurkat cells induces association of the T cell receptor complex with the cytoskeleton. J Immunol. 1989;143:931-938.
Marcos A, et al. Use of alemtuzumab and tacrolimus monotherapy for cadaveric liver transplantation: with particular reference to hepatitis C virus. Transplantation. 2004;78:966-971.
Martel RR, Klicius J, Galet S. Inhibition of the immune response by rapamycin, a new antifungal antibiotic. Can J Physiol Pharmacol. 1977;55:48-51.
Martinez-Llordella M, et al. Multiparameter immune profiling of operational tolerance in liver transplantation. Am J Transplant. 2007;7:309-319.
Martinez-Llordella M, et al. Using transcriptional profiling to develop a diagnostic test of operational tolerance in liver transplant recipients. J Clin Invest. 2008;118:2845-2857.
Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. 1994;12:991-1045.
Matzinger P. Introduction to the series: danger model of immunity. Scand J Immunol. 2001;54:2-3.
Mazariegos GV, et al. Weaning of immunosuppression in liver transplant recipients. Transplantation. 1997;63:243-249.
Merion RM, Howell T, Bromberg JS. Partial T-cell activation and anergy induction by polyclonal antithymocyte globulin. Transplantation. 1998;65:1481-1489.
Molnar-Kimber KL. Mechanism of action of rapamycin (sirolimus, Rapamune). Transplant Proc. 1996;28:964-969.
Monaco JJ. Structure and function of genes in the MHC class II region. Curr Opin Immunol. 1993;5:17-20.
Mosmann TR. Cytokines: is there biological meaning? Curr Opin Immunol. 1991;3:311-314.
Mosmann TR, et al. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol. 1986;136:2348-2357.
Neff GW, et al. Sirolimus-associated hepatotoxicity in liver transplantation. Ann Pharmacother. 2004;38:1593-1596.
Neuhaus P, et al. Improved treatment response with basiliximab immunoprophylaxis after liver transplantation: results from a double-blind randomized placebo-controlled trial. Liver Transpl. 2002;8:132-142.
Neumann UP, et al. Significance of a T-lymphocytotoxic crossmatch in liver and combined liver-kidney transplantation. Transplantation. 2001;71:1163-1168.
Niemeyer G, et al. Long-term safety, tolerability and efficacy of daclizumab (Zenapax) in a two-dose regimen in liver transplant recipients. Am J Transplant. 2002;2:454-460.
Noelle RJ, et al. A 39-kDa protein on activated helper T cells binds CD40 and transduces the signal for cognate activation of B cells. Proc Natl Acad Sci U S A. 1992;89:6550-6554.
Noreen HJ, et al. Positive remote crossmatch: impact on short-term and long-term outcome in cadaver renal transplantation. Transplantation. 2003;75:501-505.
Ortho Multicenter Transplant Study Group. A randomized clinical trial of OKT3 monoclonal antibody for acute rejection of cadaveric renal transplants. Ortho Multicenter Transplant Study Group. N Engl J Med. 1985;313:337-342.
Platz KP, et al. RS-61443—a new, potent immunosuppressive agent. Transplantation. 1991;51:27-31.
Ramos HC, et al. Weaning of immunosuppression in long-term liver transplant recipients. Transplantation. 1995;59:212-217.
Reddy KS, et al. Long-term survival following simultaneous kidney-pancreas transplantation versus kidney transplantation alone in patients with type 1 diabetes mellitus and renal failure. Transplant Proc. 2001;33:1659-1660.
Reding R. Steroid withdrawal in liver transplantation: benefits, risks, and unanswered questions. Transplantation. 2000;70:405-410.
Ridge JP, Di Rosa F, Matzinger P. A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell. Nature. 1998;393:474-478.
Rogers NJ, Lechler RI. Allorecognition. Am J Transplant. 2001;1:97-102.
Rothenberg EV. How T cells count. Science. 1996;273:78-79.
Saizawa K, Rojo J, Janeway CAJr. Evidence for a physical association of CD4 and the CD3:alpha:beta T-cell receptor. Nature. 1987;328:260-263.
Scheinman RI, et al. Role of transcriptional activation of I kappa B alpha in mediation of immunosuppression by glucocorticoids. Science. 1995;270:283-286.
Schnitzbauer AA, et al. A prospective randomised, open-labeled, trial comparing sirolimus-containing versus mTOR-inhibitor–free immunosuppression in patients undergoing liver transplantation for hepatocellular carcinoma. BMC Cancer. 2010;10:190.
Schoenberger SP, et al. T-cell help for cytotoxic T lymphocytes is mediated by CD40–CD40L interactions. Nature. 1998;393:480-483.
Sehgal SN, Baker H, Vezina C. Rapamycin (AY-22,989), a new antifungal antibiotic. II. Fermentation, isolation and characterization. J Antibiot (Tokyo). 1975;28:727-732.
Singh RP, Stratta RJ. Advances in immunosuppression for pancreas transplantation. Curr Opin Organ Transplant. 2008;13:79-84.
Starzl T. Chimerism and clonal exhaustion. Transplantation. 1998;66:272-273.
Starzl TE, Marchioro TL, Waddell WR. The reversal of rejection in human renal homografts with subsequent development of homograft tolerance. Surg Gynecol Obstet. 1963;117:385-395.
Starzl TE, et al. FK 506 for liver, kidney, and pancreas transplantation. Lancet. 1989;2:1000-1004.
Stratta RJ, Taylor RJ, Gill IS. Pancreas transplantation: a managed cure approach to diabetes. Curr Probl Surg. 1996;33:709-808.
Stratta RJ, et al. Two-dose daclizumab regimen in simultaneous kidney-pancreas transplant recipients: primary endpoint analysis of a multicenter, randomized study. Transplantation. 2003;75:1260-1266.
Strehlau J, et al. Quantitative detection of immune activation transcripts as a diagnostic tool in kidney transplantation. Proc Natl Acad Sci U S A. 1997;94:695-700.
Takada M, et al. The role of the B7 costimulatory pathway in experimental cold ischemia/reperfusion injury. J Clin Invest. 1997;100:1199-1203.
Tanchanco R, et al. Beneficial outcomes of a steroid-free regimen with thymoglobulin induction in pancreas-kidney transplantation. Transplant Proc. 2008;40:1551-1554.
Tchervenkov JI, et al. The impact of thymoglobulin on renal function and calcineurin inhibitor initiation in recipients of orthotopic liver transplant: a retrospective analysis of 298 consecutive patients. Transplant Proc. 2004;36:1747-1752.
Tedder TF, Zhou LJ, Engel P. The CD19/CD21 signal transduction complex of B lymphocytes. Immunol Today. 1994;15:437-442.
Tesi RJ, et al. The frequency of rejection episodes after combined kidney-pancreas transplant: the impact on graft survival. Transplantation. 1994;58:424-430.
Toso C, et al. Sirolimus-based immunosuppression is associated with increased survival after liver transplantation for hepatocellular carcinoma. Hepatology. 2010;51:1237-1243.
Tzakis AG, et al. Preliminary experience with alemtuzumab (Campath-1H) and low-dose tacrolimus immunosuppression in adult liver transplantation. Transplantation. 2004;77:1209-1214.
U.S. Organ Procurement and Transplantation Network and the Scientific Registry of Transplant Recipients. Transplant Data 1999-2008. Rockville, MD: U.S. Department of Health and Human Services, Health Resources and Services Administration, Healthcare Systems Bureau, Division of Transplantation; 2009.
Vendrame F, et al. Recurrence of type 1 diabetes after simultaneous pancreas-kidney transplantation, despite immunosuppression, is associated with autoantibodies and pathogenic autoreactive CD4 T-cells. Diabetes. 2010;59:947-957.
Vessal G, et al. Early steroid withdrawal in solitary pancreas transplantation results in equivalent graft and patient survival compared with maintenance steroid therapy. Clin Transplant. 2007;21:491-497.
Vincenti F, et al. A phase III study of belatacept-based immunosuppression regimens versus cyclosporine in renal transplant recipients (BENEFIT study). Am J Transplant. 2010;10:535-546.
Viola A, Lanzavecchia A. T cell activation determined by T cell receptor number and tunable thresholds. Science. 1996;273:104-106.
Vivarelli M, et al. Effect of different immunosuppressive schedules on recurrence-free survival after liver transplantation for hepatocellular carcinoma. Transplantation. 2010;89:227-231.
Wasowska BA, et al. New concepts of complement in allorecognition and graft rejection. Cell Immunol. 2007;248:18-30.
Wiesner R, et al. A randomized double-blind comparative study of mycophenolate mofetil and azathioprine in combination with cyclosporine and corticosteroids in primary liver transplant recipients. Liver Transpl. 2001;7:442-450.
Wilde MI, Goa KL. Muromonab CD3: a reappraisal of its pharmacology and use as prophylaxis of solid organ transplant rejection. Drugs. 1996;51:865-894.
Wood KJ, Sakaguchi S. Regulatory T cells in transplantation tolerance. Nat Rev Immunol. 2003;3:199-210.
Wright SD, et al. CD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS binding protein. Science. 1990;249:1431-1433.