[level-membership-for-endocrinology-diabetes-and-metabolism-category]Chapter 16
Lipoprotein Metabolism and the Treatment of Lipid Disorders
Atherosclerosis is a chronic inflammatory disease of large and medium-sized arteries that results from the deposition of lipids and lipoproteins in the intima of these vessels.1,2 This process is a major cause of morbidity and mortality in the developed world, and the toll is particularly high in the diabetic population, accounting for approximately two-thirds to three-quarters of all diabetic deaths.3 When combined with significant morbidity arising from ischemic injury and loss of limb perfusion, the impact of atherosclerosis on human health is enormous.5 Intensive lipid-lowering therapy has demonstrated remarkable therapeutic value in the treatment and prevention of coronary heart disease (CHD) in patients with and without diabetes. This chapter will review basic principles of lipoprotein metabolism, focusing on the impact diabetes has on these lipid pathways. Data that address the value of lipid-altering treatments in reducing the burden of atherosclerosis in patients with and without diabetes will be presented. Finally, an approach to the management of patients with dyslipidemia will be outlined.
Lipoprotein Metabolism
The clinically reported serum triglyceride and cholesterol values represent the sum of the cholesterol and triglyceride contents of the circulating lipoproteins present in a volume of blood (expressed either as mg/dL or mmol). While knowledge of these total lipid values is very helpful in assessing cardiovascular risk, it is the distribution of cholesterol among the different lipoprotein fractions that is critical to any sophisticated understanding of atherosclerosis risk. Lipoproteins are structured to solve the problem of transporting highly hydrophobic lipids, such as cholesterol ester and triglyceride, in the aqueous environment of the blood. Since the bulk of lipoproteins are synthesized in the liver or gut, they must be moved via the bloodstream from these sites of synthesis to tissues where lipids will be taken up and utilized. By complexing neutral lipids with both proteins and more polar lipids such as unesterified cholesterol and phospholipids, the lipoprotein structure accomplishes this task. The more hydrophobic lipids are sequestered on the inside of the spherical lipoprotein, whereas more polar lipids and proteins decorate the outer surface (Fig. 16-1). In addition to solving the biophysical transport problem, the lipoprotein structure also enables particles to utilize a class of proteins known as apoproteins (Table 16-1) that serve as targeting molecules which interact with lipid-modifying enzymes (Table 16-2) or receptors and transporters (Table 16-3) critical for the normal functioning of the disparate lipoprotein classes. Mutations in the genes encoding apoproteins or the enzymes and receptors with which apoproteins interact (see Tables 16-1 to 16-3) are rare, but they can have a profound influence on the circulating levels of lipoproteins in individuals with and without diabetes. Because lipids play fundamental roles in a host of other cellular processes, mutations in lipid enzymes, receptors, transporters, and apoproteins can have profound effects on human biology beyond the serum lipoproteins, some of which are cited in Tables 16-1 to 16-3.
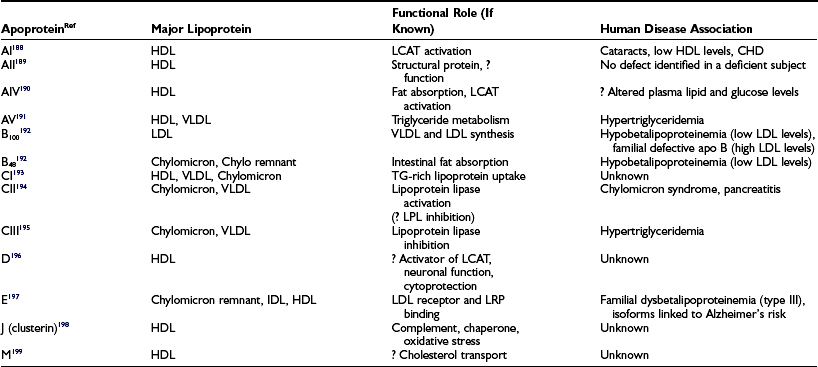
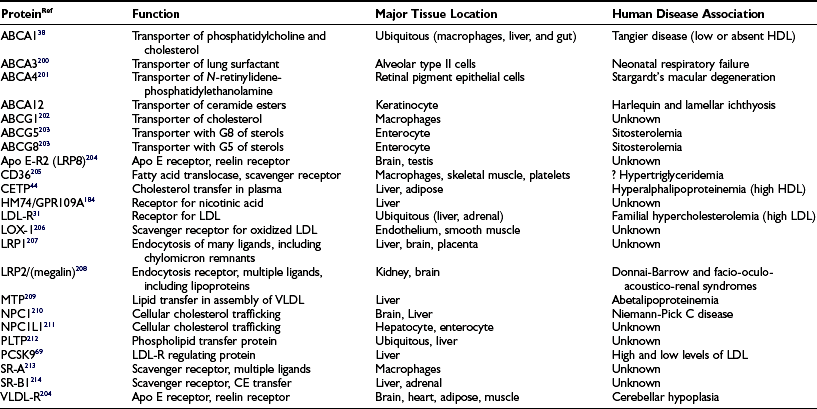
Table 16-3
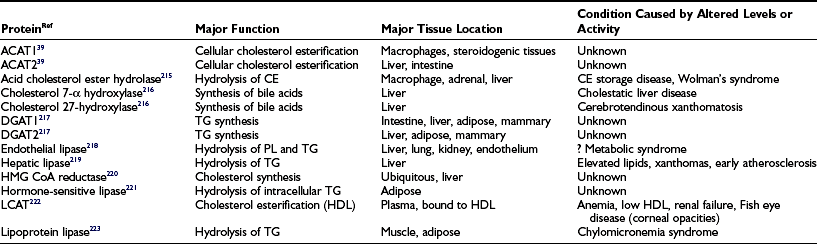
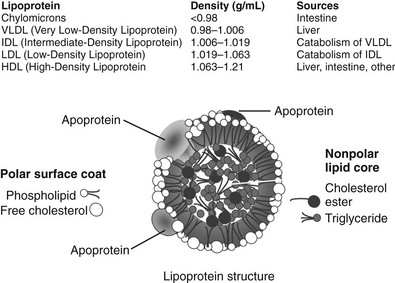
FIGURE 16-1 The upper panel of the figure lists the density range that encompasses each lipoprotein class and the major tissue in which the lipoprotein is synthesized. The bottom panel is a graphic representation of a generic lipoprotein, emphasizing the spherical shape of the particle, the use of amphipathic apoproteins and polar lipids on the particle surface that interact with cells and the blood environment, and the sequestration of nonpolar lipids in the core of the lipoprotein. All lipoproteins share these general features, but they differ in size, lipid composition, and in the specific apoproteins embedded.
Although lipoproteins constitute a continuous spectrum of density within the plasma, they are commonly divided into five major classes (see Fig. 16-1). These are: (1) chylomicrons; (2) very low-density lipoproteins (VLDLs); (3) intermediate-density lipoproteins (IDLs); (4) low-density lipoproteins (LDLs); and (5) high-density lipoproteins (HDLs). Within these classes, further subdivision of lipoproteins can be made. In diabetics, a higher proportion of LDLs is carried in a denser, smaller-diameter subfraction that is called small dense LDL (sdLDL). HDL are commonly divided into HDL2 and HDL3 subclasses, but some investigators have adopted a gel separation method to segregate HDL into a much larger number of subtypes.6 It is important for the clinician to understand that the different laboratory methods of identifying subpopulations of lipoproteins yield different or overlapping species of particles, causing confusion for those not conversant with the separation techniques. In this chapter, the most widely used classifications will be employed for simplicity, but further research may prove that more sophisticated subclassifications have considerable clinical relevance. Overall, it is helpful to remember that the least dense of the lipoproteins are also the largest in size and the most triglyceride-rich. These are the chylomicrons and VLDLs. IDLs contain substantial amounts of both triglyceride and cholesterol, whereas LDLs and HDLs are predominantly cholesterol-carrying particles with very little associated triglyceride. This information permits the clinician to identify the most likely lipoprotein abnormalities when presented with a patient’s total serum triglyceride and cholesterol values. A brief description of each of the five major lipoprotein classes is summarized in the following paragraphs. A reader interested in a more comprehensive introduction to lipoprotein metabolism should consult the lipid sections of the Metabolic Basis of Inherited Disease.7
Chylomicrons
Chylomicrons derive from dietary fat and carry triglycerides throughout the body. The major structural protein of chylomicrons is apolipoprotein (apo) B48, a protein that is produced from apo B100 RNA by a unique editing process employed by gut cells that generates a protein that is 48% of the length of apo B100.8 Chylomicrons also contain other apoproteins, including members of the apo C and apo A family. These proteins can redistribute to other lipoprotein classes within serum as chylomicrons transfer their lipid content to cells and interacting lipoproteins. Chylomicrons have the lowest density of all lipoproteins and will float to the top of a plasma specimen left in a refrigerator overnight, forming a creamlike layer. Owing to its large size, the chylomicron does not readily enter the artery wall and is therefore thought not to be atherogenic, but the atherosclerotic role of the triglyceride-depleted chylomicron remnant remains controversial. Triglyceride makes up most of the chylomicron and is removed by the action of an enzyme that is bound to the surface of endothelial cells, lipoprotein lipase (LPL). Patients deficient in this enzyme or its apoprotein cofactor (apo CII) have very high serum triglyceride levels and are at increased risk for developing acute pancreatitis. Inasmuch as insulin is also a critical cofactor for LPL activity, the vast majority of patients presenting with hypertriglyceridemic pancreatitis have poorly controlled diabetes as the cause of their delayed chylomicron clearance.
Low-Density Lipoproteins
LDLs are the major carriers of cholesterol in humans, responsible for supplying cholesterol to tissues with the highest sterol demands. LDLs are also the lipoproteins most clearly implicated in causing atherogenic plaque formation.9 Circulating LDL levels can be increased in persons who consume large amounts of saturated fat and/or cholesterol.10 LDL levels are also elevated in those who have genetic defects that affect LDL receptor function (familial hypercholesterolemia, PCSK9 mutations, autosomal recessive hypercholesterolemia), the structure of LDL’s apoprotein, apo B, or who have polygenic disorders affecting LDL metabolism.11 Circulating LDLs are taken up by the endothelium lining the artery wall, traverse it, and are deposited and trapped in the arterial intima. There, they may undergo oxidation or other biochemical modification, be taken up by macrophages, and stimulate atherogenesis. The association of total serum cholesterol with CHD is predominantly a reflection of the role of LDL. Studies in patients with diabetes have produced somewhat conflicting data on the prevalence of increased LDL levels, as defined by general population norms, but the preponderance of the evidence indicates that diabetics have LDL levels that are similar to those seen in well-matched, nondiabetic control populations. Given diabetics’ greater risk of developing coronary artery disease (CAD), however, the National Cholesterol Education Program guidelines now define LDL levels above 100 mg/dL as being undesirable in diabetics. When judged by this stringent criterion, elevated LDL levels are extremely common in patients with diabetes. In addition, the LDL in diabetics is frequently smaller, denser, and more readily oxidized—properties that are associated with a higher degree of atherogenicity.12,13
High-Density Lipoproteins
HDLs are the smallest of the lipoproteins; despite this, they carry a variety of apoproteins, including members of the apo A and C family. HDLs, unlike the other lipoproteins, are believed to function in humans primarily to return lipids from peripheral tissues to the liver and gut for excretion, rather than moving lipids from the gut organs to the periphery.14 This perspective on HDL function is an oversimplification, since cell culture experiments indicate that HDL can donate lipids as well as pick them up. It has proven to be quite difficult to quantitate the net movement of cholesterol carried in HDL in and out of peripheral tissues in whole animals. Further complicating the issue is the fact that many of the standard laboratory animals used in metabolic studies carry most of their serum cholesterol in HDL rather than LDL, diminishing the relevance of their lipoprotein metabolism to that of the LDL-rich human. Nevertheless, a considerable body of evidence suggests that HDL functions to protect tissues from unwanted accumulation of cholesterol. The mechanisms by which HDL can protect against atherosclerosis include its involvement in the reverse cholesterol transport pathway described earlier, as well as through contributions arising from its carriage of proteins that appear to have antiinflammatory and antioxidant properties.15,16 The unesterified cholesterol from tissues that is transferred to HDL is esterified by the action of lecithin cholesterol acyltransferase (LCAT) and stored in the central core of HDL. This esterified cholesterol can be transferred back to lower-density lipoproteins by the action of cholesterol ester transfer protein, or it may be removed at the liver by the action of a plasma membrane receptor called scavenger receptor B-1 (SRB-1). A particularly effective reverse transport system is thought to explain, at least in part, the association of elevated HDL cholesterol levels with a reduced risk of developing CHD. Apolipoprotein AI (apo AI) is the major apoprotein of HDL, and its serum concentration also correlates inversely with the risk of CHD. Women have higher levels of HDL cholesterol than men, and this may partly explain the lower incidence of CHD in premenopausal women. Exercise increases HDL levels; obesity, hypertriglyceridemia, and smoking lower them. Patients with diabetes, having higher body mass indices and hypertriglyceridemia, typically have lower HDL cholesterol levels than do nondiabetics, contributing further to the atherogenic lipid profile of the diabetic.
Metabolism of Lipids and Lipoproteins
Following ingestion of a meal containing fat, the triacylglycerols (TAG) are broken down by pancreatic triacylglycerol lipase to yield sn-2-monoacylglycerol and fatty acids. Ingested cholesterol is solubilized by bile salts into micellar structures, a step which is disrupted by bile acid resin binders. The lipids then transverse the enterocyte brush border membrane and are reassembled in the intestinal cell into a triglyceride-rich lipoprotein, the chylomicron. The role of transport proteins in the passage across the enterocyte membrane remains disputed, with evidence for both a passive diffusion and an active transport mechanism.17,18 For cholesterol, a second transport step translocates the sterol into intracellular pools that can be accessed for lipoprotein packaging.19 Niemann-Pick C1-like 1 protein (NPC1L1) appears to be required for this second step to occur and is the target of the cholesterol absorption inhibitor, ezetimibe.20,21 The internalized cholesterol is moved to the endoplasmic reticulum where the monoacylglycerol and fatty acids are also carried by transport proteins. The latter are recombined into triacylglycerol and bound by microsomal triglyceride transport protein (MTP). A second pathway of triglyceride synthesis uses acylation of glycerol-3-phosphate to phosphatidic acid, dephosphorylation to diacylglycerol (DAG), and then acylation of DAG to TAG. A high-density, protein-rich particle synthesized in the enterocyte, containing apo B48 and apo AIV, is then packaged with the absorbed lipids to generate the chylomicron.22 This is then transported by vesicles to the basolateral membrane for exocytosis into the mesenteric lymph. The lipoproteins in the lymph enter the blood circulation via discharge from the thoracic duct.
Two ABCG half transporters, ABCG5 and G8, contribute to net cholesterol absorption in the gut through their ability to resecrete sterol that has been absorbed by the enterocyte back into the gut lumen, thereby reducing net sterol uptake.23 The total amount of diet-derived TAG that is delivered to the circulation varies and is influenced by multiple factors, including the fat content of the diet, the amount of phosphatidylcholine in the intestinal lumen, and the expression level of apo AIV in the enterocyte. Overall, humans have a remarkable ability to absorb large quantities of both cholesterol and fat; 35% to 60% of ingested cholesterol is absorbed, with the amount inversely correlated with hepatic cholesterol synthesis. Up to 600 g of fat is absorbed with 95% efficiency. In individuals with normal lipid metabolism, the ingested fat is cleared from the enterocyte in less than 14 hours, forming the basis for drawing serum lipids after a fast of this duration. Circulating chylomicrons are subsequently cleared from the plasma by the action of endothelial-bound lipoprotein lipase, which cleaves the TAG, enabling the liberated glycerol and free fatty acids to move into the adjacent adipose or muscle tissue. The resulting chylomicron remnant particle can be further metabolized by lipases and appears to be retained in the hepatic space of Disse, where it can bind to heparan sulfate proteoglycans and acquire additional apo E. The acquisition of the apo E enables the remnant to be cleared efficiently by either the LRP1 or LDL receptor, but patients with diabetes have accumulation of remnants and slower hepatic clearance.24,25
The other major triglyceride-rich lipoprotein, VLDL, is synthesized in the liver and contains apo B100 rather than apo B48 as its major structural protein.26 Like chylomicrons, VLDLs rely on MTP to combine lipids with the apo B protein to produce a nascent, lipid-poor lipoprotein.27,28 This VLDL can be secreted from the liver or further lipidated to produce a mature VLDL. Individuals lacking MTP activity develop abetalipoproteinemia, a disorder characterized by erythrocyte acanthocytosis, anemia, steatorrhea, spinocerebellar degeneration, and fat-soluble vitamin deficiency. Acquisition of lipid by the mature VLDL appears to depend on triglycerides that are stored in the cytosol. Increased fatty acid delivery to the liver, from dietary sources or that released by lipase activity in peripheral tissues, stimulates VLDL production. After secretion of the mature VLDL from the liver, its triglycerides are initially removed by the action of LPL, and the resulting intermediate-density lipoprotein (IDL) can be further catabolized by hepatic lipase to produce mature, cholesterol-rich LDL.
Low-Density Lipoprotein Metabolism
There is still some dispute about whether mature LDL can be secreted from the liver rather than always being derived from the progressive removal of triglyceride from VLDL/IDL precursors.29 The preponderance of data indicate that little if any LDL is directly secreted from the liver. Each LDL particle contains a single apo B molecule, which dictates that individuals heterozygous for apo B mutants will have two populations of LDL, one which carries a wild-type protein and one with a mutant protein. LDLs are utilized throughout the body as a source of exogenous cholesterol and are taken up by LDL receptors present at the plasma membrane. While LDL receptors are ubiquitous, the majority are expressed by the liver. The LDL receptor binds to both apo E– and apo B–containing lipoproteins. Because the affinity of the receptor is higher for apo E than for apo B, E-containing lipoproteins, such as IDL, can compete with LDL for uptake by the LDL receptor. However, there are other receptors that can bind apo E–containing lipoproteins, permitting their clearance but not that of LDL when LDL receptor activity is lost or diminished. Thus, mutations in the LDL receptor result in elevated LDL levels in the plasma, causing the autosomal co-dominant disorder, familial hypercholesterolemia, which is among the most common of Mendelian genetic disorders (see Genetics section later).30 The elucidation of the cholesterol homeostatic mechanisms controlled by the LDL receptor constituted one of the major advances in our understanding of human physiology and disease.31 The internalization of LDL by the LDL receptor leads to trafficking of the internalized lipoprotein to the endosome, where after fusion with lysosomes, the sterol in the lipoprotein is released and shuttled to the endoplasmic reticulum (ER). A sterol-sensing system in the ER regulates the movement of a membrane-bound transcription factor, sterol response element binding protein (SREBP), from the ER to the nucleus.32–37 SREBP works in concert with other transcriptional regulators to influence the expression of many of the cholesterol synthesis, metabolizing, and transport molecules, particularly in the liver. This elegant homeostatic system enables cells to reduce their de novo cholesterol synthesis when adequate sterol supplies are present and to increase synthesis and LDL receptor expression when cellular demands require more cholesterol.
High-Density Lipoprotein Metabolism
Although the relationship between elevated levels of HDL and lower rates of cardiovascular disease was recognized more than 50 years ago, the mechanism by which HDL protects against CHD is still only partially understood. HDL are initially produced as lipid-poor phospholipid discs containing apolipoprotein AI. Following interaction with the phospholipid/cholesterol transporter, apo AI, the particle acquires unesterified cholesterol from cellular stores.38 For macrophages in particular, this cholesterol efflux pathway appears to be critical to their ability to unload the excess cholesterol they acquire by phagocytosis of dead cells and by their unregulated uptake of lipoproteins by scavenger receptor pathways.14 Cholesterol is stored in an intracellular lipid pool, having been esterified at its 3′OH position by the activity of acyl coenzyme A (CoA):cholesterol O-acyltransferase (ACAT).39 A neutral cholesterol hydrolase cleaves the fatty acid from cholesterol ester to convert it back to free (unesterified) cholesterol when cellular signals call for the use or export of the sterol.40 This free cholesterol can be shuttled to the plasma membrane, where it can be transported by the action of ABCA1. The interaction of lipid-poor nascent HDL with ABCA1 appears to involve direct binding of apo AI to the transporter, but the actual lipid transfer process is poorly understood.41–43 Once the unesterified cholesterol transfers to the nascent HDL, it can be reesterified by the action of lecithin cholesterol acyltransferase (LCAT) and centralized in the lipid core of the growing HDL particle. This HDL particle can now interact with a second ABC transporter, ABCG1, and additional cholesterol can be acquired.44 The role of ABCA1 is well established, since loss of its activity leads to Tangier disease (see Genetics section) and the absence or near absence of HDL in the plasma. ABCG1’s precise function is still being elucidated. Once the HDL has acquired sufficient cholesterol and esterified it, it can donate this lipid back to lower-density lipoproteins in the plasma, such as VLDL, through the activity of cholesterol ester transfer protein (CETP). Alternatively, HDL can interact with a cellular receptor, SR-BI, to selectively transfer the cholesterol ester to cells, including the liver, where it could excreted in the bile as cholesterol or one of its bile acid derivatives.45 The protein component of HDL is cleared via the kidney, a process that appears to depend on the HDL being lipid depleted. The pathway described, moving cholesterol from cellular storage pools to HDL and ultimately to the liver, is termed the reverse cholesterol transport pathway. The activity of this pathway is postulated to explain at least in part the link between HDL levels and lower CHD risk. However, HDL also carries antioxidants and a host of proteins linked to complement and inflammation pathways that suggest it may be playing a more general role as a scavenger of molecules that stimulate inflammation. The complexity of its function has made targeting increases in HDL levels as a therapeutic objective challenging. Both animal and human data have established that not all methods of increasing HDL result in improvements in atherosclerosis.
Disorders of Lipid Metabolism in Patients With Diabetes
Data dating back at least 30 years have documented that individuals with type 1 diabetes have increased rates of CHD.46–48 This risk has been estimated to be 10-fold or higher over that seen in age-matched controls without diabetes. In the Diabetes U.K. cohort of 23,751 subjects who were diagnosed with insulin-dependent diabetes prior to the age of 30, for example, those individuals 20 to 29 years of age had a standardized mortality ratio for ischemic heart disease of 11.8 for men and 44.8 for women. For those 30 to 39 years of age, the ratios were 8.0 and 41.6 for men and women, respectively.49 Despite the long and consistent reproducibility of this risk association, the pathophysiology underlying that relationship is still poorly understood. Unlike type 2 diabetes, where obesity and insulin resistance lead to changes in the standard lipid profile that are considered atherogenic, the type 1 diabetic who has not developed nephropathy and whose glucose is well controlled will typically have a normal serum lipid profile.50 In the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Study (DCCT/EDIC), the mean LDL-C in the intensively treated group was 111 ± 29 mg/dL in women and 119 ± 31 in men; HDL-C levels averaged 63 ± 16 and 51 ± 14 for women and men, respectively. Serum triglyceride levels were also in the normal range, with women averaging 76 ± 37 mg/dL, and male values running moderately higher at 98 ± 67 mg/dL. These lipid values did not differ significantly from those of the patients enrolled in the conventional treatment group of the study. These findings make clear that the standard lipid profile provides few insights into the increased risk of CHD in type 1 diabetics.
More sophisticated assessments of serum lipoproteins, using nuclear magnetic resonance (NMR), can reveal differences in lipoprotein sizes that distinguish diabetic lipids from those of nondiabetics. A study of 194 diabetics from London, all diagnosed with type 1 diabetes before the age of 25 and not receiving any renal replacement therapies, demonstrated that LDL size and subclass distribution were similar between the men in both groups.51 In contrast, female diabetics had fewer large and more small LDL, producing an overall reduction in LDL size. However, both men and women diabetics had more large and fewer small HDL than did the nondiabetic cohort, yielding an increased overall HDL size. Whereas the LDL size change in women would be considered proatherosclerotic, the HDL differences are thought to be antiatherogenic. These findings led the authors to conclude that the pathogenic significance of the lipoprotein size differences was uncertain.51 The results of the London study appear to differ from those reported in a similar analysis done in the DCCT cohort, although the comparators used in the two studies make direct comparison of the two reports challenging. In the DCCT analysis, a nondiabetic control group was not included, so the authors compared the NMR analysis of lipoproteins in men to those in women. This approach found higher LDL particle concentrations and more small-sized particles in the men. HDL sized decreased, and fewer large HDL were also found in the men. Although the authors stated that both genders had abnormal levels of several of the NMR-classed lipoproteins, these judgments were based on National Cholesterol Education Program (NCEP) optimal levels rather than population normative values, so it is not clear that matched, nondiabetic cohorts would have differed greatly.50
Surprisingly, epidemiologic risk-factor assessment of the correlates of CHD in type 1 diabetes shows a weak association to the one metabolic risk that predominates in the type 1 population: hyperglycemia.52,53 It was therefore somewhat unexpected when a 17-year follow-up of the DCCT cohorts showed that the intensive treatment group had a 57% reduction in the risk of nonfatal myocardial infarction, stroke, or death from cardiovascular disease compared to the conventional treatment group (95% CI, 0.21 to 0.88; P = 0.02). Unfortunately, the DCCT/EDIC findings, if generalizable to the care of diabetics in the community at large, appear not to have translated yet into that community, since the cumulative incidence of CHD in type 1 diabetics has changed little in the past few decades.54 Unlike CHD, peripheral arterial disease,55 amputation,56 and stroke57 rates have all been reported to correlate with levels of hyperglycemia. Possible explanations for the differences between CHD and other forms of CVD in type 1 diabetes are discussed in further detail in Chapter 25.
Type 2 Diabetes
There are alterations in multiple lipoprotein metabolism pathways in type 2 diabetics that lead to the typical abnormal lipid profile commonly seen. This profile includes hypertriglyceridemia secondary to high VLDL levels, as well as increased amounts of sdLDL and lower levels of HDL cholesterol. LDL cholesterol levels are typically not higher in diabetic populations when compared to well-matched nondiabetic subjects. In an elderly Finish population, for example, approximately 30% of diabetic subjects had serum triglyceride levels greater than 200 mg/dL compared to 13% of nondiabetic subjects. HDL cholesterol levels were less than 35 mg/dL in about 25% of diabetics compared to 12% of nondiabetics. In this study, elevated LDL levels as defined by an LDL greater than 130 mg/dL were actually more prevalent in the nondiabetic population than in the diabetics.58,59
The causes of classic diabetic dyslipidemia are only partly understood. Individuals with insulin resistance and type 2 diabetes overproduce mature VLDL in the liver. This is accompanied by a slower clearance of triglyceride-rich lipoproteins via reduced activity of lipoprotein lipase. Coupled with decreased receptor uptake of the remnant and IDL particles that form after LPL partially depletes VLDL of its triglyceride, diabetes produces substantial elevations in serum triglyceride levels. Production of sdLDL appears to be tightly linked to the resulting hypertriglyceridemia insofar as cholesterol ester transfer protein (CETP) exchanges triglyceride from VLDL for cholesterol ester from LDL, producing a triglyceride-enriched LDL.60 This lipoprotein appears to be a preferred substrate for hepatic lipase, which hydrolyzes the triglyceride to produce an LDL that is smaller, lower in cholesterol ester content, and higher in density than LDL produced in the absence of excess VLDL. The diabetic with increased small dense LDL levels has a higher LDL particle number for an equivalent serum LDL cholesterol concentration than does an individual with normal-density LDL. This increased particle number, whether measured by nuclear magnetic resonance assays or serum apo B levels, is associated with an increased risk of CAD.12
Thus, the central metabolic change that accounts for much of the diabetic dyslipidemia is the overproduction of VLDL by the liver. Higher levels of circulating free fatty acids, arising from increased adipose deposits and insulin deficiency or resistance, contribute to higher storage levels of triglyceride in the liver. Increased fat content of the liver is in turn correlated with increased rates of lipidation and secretion of nascent VLDL. This VLDL appears to be normal in size, indicating that higher VLDL triglyceride levels derive from higher particle numbers rather than larger triglyceride-enriched lipoproteins. Administration of insulin reduces the concentration of free fatty acids in the circulation and also suppresses VLDL production by inhibiting the hepatic generation of the early apo B, lipid-poor particle. The molecular mechanisms accounting for VLDL production changes are not fully elucidated, but there is evidence that insulin activates a phosphatidylinositol-3 (PI-3) kinase pathway that inhibits apo B secretion while activating a mitogen-activated protein kinase (MAPK) that down-regulates MTP expression.61,62 Insulin effects on transcriptional regulators of VLDL secretion may also be involved. In vivo metabolic studies suggest that the prevalence of hypertriglyceridemia and low HDL cholesterol levels is higher in diabetics who are insulin resistant as opposed to insulin deficient, accounting for the higher prevalence of these disorders in type 2 versus type 1 diabetics.
Genetic Basis of Lipid Disorders
Although metabolic disorders associated with diabetes, hypothyroidism, and nephropathy can exacerbate or even cause hyperlipidemia (Table 16-4), many individuals have none of these problems. Individuals with the greatest deviation from normal levels of lipoproteins often have a single gene defect that is responsible for their lipid disorder. The vast majority of hyperlipidemic patients, however, do not have a monogenic defect. Rather, they are most likely to have a polygenic disorder and an additional contribution from environmental factors (e.g., excessive saturated fat intake for high LDL levels; obesity or smoking for lower HDL levels). The application of the NCEP treatment guidelines does not require that a genetic diagnosis be made, because treatment is based on phenotype (lipid levels) and not genotype. Nevertheless, the identification of the causes of monogenic lipid disorders has been critical to our understanding of both normal and abnormal lipoprotein physiology. The most important lipid disorders affecting coronary disease risk are those that increase the serum LDL level or reduce the HDL level, and these are briefly presented next.
Table 16-4
Secondary Causes of Hyperlipidemia and Dyslipidemias
| Metabolic/hormonal | Diabetes |
| Hypothyroidism | |
| Lipodystrophy | |
| Polycystic ovarian disease | |
| Hepatic | Primary biliary cirrhosis |
| Other forms of cirrhosis | |
| Renal | Chronic renal failure |
| Nephrotic syndrome | |
| Dietary | Alcohol |
| Foods highly enriched in cholesterol and saturated fat | |
| Medications | Estrogens |
| Glucocorticoids | |
| Anti-HIV treatments, especially protease inhibitors | |
| Oral androgens and anabolic steroids | |
| Thiazide diuretics | |
| Beta-blockers | |
| Retinoic acid | |
| Antipsychotics |
Monogenic Low-Density Lipoprotein Disorders
Familial Hypercholesterolemia
Familial hypercholesterolemia (FH) is one of the most thoroughly studied and common genetic disorders of mankind. Approximately 1 in 500 individuals carry a mutation in one allele encoding the LDL receptor. These mutations results in defective clearance of LDL from the blood and a rise in serum total and LDL cholesterol levels. The elucidation of this defect and its associated cell biology led to insights into the homeostatic control of cholesterol metabolism that transformed the lipid field.31 Several hundred individual mutations in the LDL receptor have now been identified. The gene, located on chromosome 19 and spanning 45 kb, has 18 exons that encode a mature protein of 839 amino acids. The inheritance of the disorder is autosomal co-dominant. Heterozygous patients have LDL cholesterol levels in the 200 to 500 mg/dL range, and the rare homozygous FH patient typically has an LDL cholesterol above 500 mg/dL. Heterozygous patients commonly have tendon xanthomas and premature CAD, whereas these are universal in the untreated homozygous individual. Mutations in the LDL receptor have been described that affect multiple aspects of receptor biology, ranging from defective LDL binding to abnormal trafficking of the receptor to the plasma membrane. The effect of all of these mutations is to reduce LDL and IDL clearance from the blood. The latter effect results in an enhanced conversion of IDL to LDL. The net result is markedly elevated LDL cholesterol levels in the plasma and a strong predisposition to early CHD if the disorder is not treated. FH heterozygotes typically respond well to 3-hydroxy-3-methylglutaryl coenzyme A (HMG CoA) reductase inhibitors (statins), though additional therapies may be needed to reach desirable LDL levels. FH homozygotes respond poorly to reductase inhibitors and inevitably develop early atherosclerotic vascular disease if LDL apheresis or liver transplantation is not performed.
Familial Defective Apolipoprotein B
Familial defective apo B (FDB) is an autosomal co-dominant disorder caused by mutations near the carboxy terminus of apo B.63,64 Apo B is a 4536-amino-acid protein encoded by a gene on chromosome 2 that is composed of 29 exons. The most commonly identified mutations in FDB affect an arginine at position 3500 that is either mutated to a glutamine or tryptophan. Most individuals with FDB are heterozygous for their mutation, and there is only one molecule of apo B per LDL particle, so the LDL in the serum of these individuals is a mixture of normal LDL and FDB LDL. The LDL containing the defective apo B does not bind normally to the LDL receptor and is therefore cleared more slowly. The explanation for the lower receptor affinity of LDL carrying the FDB mutations is not known with certainty. There is evidence, however, of a molecular interaction between the arginine at position 3500 and tryptophan at 4369 that appears to be necessary for the adoption of an apo B conformation recognized by the LDL receptor. Patients with FDB have elevations in their LDL levels that fall within the range of those seen in FH heterozygotes, but the mean is generally lower than the mean in FH cohorts. There is considerable variability in both genetic populations; however, making generalizations about the diagnostic value of this measurement not very useful. Tendon xanthomas are common but not universal, and premature CAD is prevalent. Treatment with reductase inhibitors or other standard LDL-lowering drugs is typically quite effective in FDB patients.
Autosomal Recessive Hypercholesterolemia
Studies of families in which LDL levels were inherited in an autosomal recessive pattern led to a successful genetic linkage analysis that mapped the genetic defect in these families to the short arm of chromosome 1. Candidate gene sequencing led to the identification of mutations in a gene now designated autosomal recessive hypercholesterolemia (ARH) and comprising nine exons and eight introns. Patients diagnosed with ARH to date have been clinically similar to patients who are homozygous for LDL receptor mutations in that they have markedly elevated LDL cholesterol levels (typically > 500 mg/dL), tendon xanthomas, and premature coronary atherosclerosis.65 The autosomal recessive mode of inheritance is an important differentiating factor between ARH and familial hypercholesterolemia, as is greater LDL receptor activity, measured in cultured fibroblasts taken from the patients. The defect in ARH patients appears to affect liver cholesterol metabolism disproportionately, and current data indicate that the ARH gene product likely serves as an adaptor protein required for LDL receptor internalization via clathrin-coated pits.66,67 ARH patients do respond to HMG CoA reductase inhibitors, but this treatment is usually inadequate to control their markedly elevated LDL cholesterol levels, making them candidates for LDL apheresis.
PCSK9 Mutations
Proprotein convertase subtilisin kexin 9 (PCSK9) is a 72-kD protease that is highly expressed in the liver and regulates the levels of LDL receptors acting at the plasma membrane.68,69 Gain-of-function mutations in PCSK9 result in lower LDL receptor levels, whereas loss-of function mutations increase those levels. Serum LDL levels are inversely related to liver LDL receptor expression; the activity of the latter clears the former from the circulation. Thus, gain-of-function mutations are associated with higher serum LDL levels and increased risk of CHD, and loss-of-function mutations result in lower LDL levels and decreased CHD risk. The role of PCSK9 in lipid metabolism was originally recognized when it was discovered that specific heterozygous mutations in the protein were associated with autosomal dominant hypercholesterolemia. Approximately 1 in 50 African Americans has one of two nonsense mutations in PCSK9, causing LDL cholesterol levels to fall approximately 30%. Caucasians can carry a missense mutation that reduces LDL cholesterol by approximately 15%. Individuals with complete loss of PCSK9 activity have extraordinarily low levels of LDL-C, but appear to be otherwise normal. The mechanism by which PCSK9 regulates LDL receptor activity is still being elucidated, but it appears that the protease can bind to the receptor, targeting it for degradation in the lysosome. This action does not depend on the protein’s enzymatic activity, and exogenously administered PCSK9 can reduce LDL receptor expression and serum LDL-C levels. The mechanism by which PCSK9 works makes it an extremely attractive target for pharmaceutical intervention; therapeutics that block PCSK9 function are being actively pursued.
Monogenic High-Density Lipoprotein Disorders
Tangier Disease
Tangier disease was first recognized in 1960 in a sibling pair living on Tangier Island in the Chesapeake Bay in Virginia. Enlarged, yellow-orange tonsils and little or no circulating HDL cholesterol are the classic findings in the disorder. Subsequently, cases have been identified in which the presenting symptom was a peripheral neuropathy. Patients may also have hepatosplenomegaly. Serum LDL and total cholesterol levels are usually quite low, while serum triglyceride values are moderately elevated. Tangier disease is an autosomal recessive disorder. The cause of Tangier disease was identified by several groups, following mapping of the gene defect to chromosome 9. Candidate gene analysis in the appropriate genetic interval identified mutations in an ATP-binding cassette (ABC) transporter as the cause. The transporter, now called ABCA1 (ATP-binding cassette, subfamily A, member 1), is a full-length ABC transporter transmembrane protein that is predicted to span the plasma membrane 12 times. ABCA1 is a 2261-amino-acid protein encoded by a gene spanning 50 exons. Approximately 50 mutations have been identified to date in the gene.70 ABCA1 mediates the efflux of cholesterol from cholesterol-enriched cells when stimulated by the major apoprotein of HDL, apo AI.71 This activity is lost in Tangier patients and is reduced by approximately half in carriers of one abnormal ABCA1 allele. The mechanism of the movement of cholesterol from inside the cell to outside the cell has not been established. Individuals with Tangier disease and heterozygous carriers of ABCA1 mutations (a disorder called familial hypoalphalipoproteinemia [FHA]) both appear to have increased risk of premature coronary disease. There is no specific therapy for this disorder.
Lecithin Cholesterol Acyltransferase Deficiency
The esterification of free cholesterol in circulating lipoproteins is catalyzed by a plasma enzyme called lecithin cholesterol acyltransferase. Two clinically separable syndromes result from a deficiency of LCAT. Fish eye disease is due to a partial deficiency of LCAT, with patients presenting with dense corneal opacities and very low HDL cholesterol levels. Familial LCAT deficiency arises from a nearly completely absence of LCAT activity and produces a more severe syndrome characterized by corneal opacities, anemia, and proteinuric renal failure.72 The serum lipid and lipoprotein profile in the more severe disorder is characterized by normal or increased triglyceride levels, reduced LDL cholesterol values, and markedly diminished HDL levels. The gene encoding LCAT is located on chromosome 16 and is composed of 6 exons. Cleavage of a signal peptide of 24 amino acids converts the proenzyme from a 440-amino-acid precursor to the final 416-amino-acid glycoprotein that circulates in the plasma. When unesterified cholesterol from tissues is transferred to HDL, either by passive diffusion or by ABCA1-mediated lipid transport, LCAT’s activity esterifies the transferred cholesterol, trapping it in the HDL core. Since cholesterol ester is more hydrophobic than unesterified cholesterol, it is energetically unfavorable for the cholesterol ester to transfer back to the cell of origin. These steps of cholesterol transfer and esterification are the initial events in the reverse cholesterol transport pathway whereby cholesterol is moved from peripheral tissues back to the liver. Apo AI is the major activator of LCAT activity, accounting for the predominant effect of the enzyme deficiency on HDL levels. LCAT does, however, contribute to esterification of cholesterol in lower-density lipoproteins as well. Despite very low HDL levels, patients with either fish eye disease or familial LCAT deficiency do not seem to have a predilection for very early coronary atherosclerosis. The small number of patients with the disease, some of whom have been found to have CHD, make it difficult to determine if the risk of coronary atherosclerosis is substantially altered by the enzyme deficiency. There is no specific treatment for LCAT deficiency. Corneal and kidney transplantation are performed in these patients in order to ameliorate their major clinical disabilities.
Apolipoprotein AI Mutations
Apo AI is the major structural protein of HDL. The gene encoding apo AI is located on chromosome 11 and comprises 4 exons. Following cleavage of the signal and prohormone sequences, a 243-amino-acid mature protein is produced. The protein has multiple repeats of an amphipathic helical structure that enables it to interact with both lipid and aqueous environments. Mutations that cause profound alterations in apo AI structure or expression have been reported, though they are extremely rare.73–76 The individuals carrying these mutations have virtually no circulating HDL and typically develop early CHD. There are, however, a number of case reports of apo AI mutations that are not associated with early CHD. Corneal opacities and xanthomas have been documented in many but not all individuals with major apo AI mutations. Most patients harboring these mutations have been found to be homozygous for the gene defect, usually as a result of consanguinity. Heterozygotes carrying these mutations commonly have half-normal HDL cholesterol levels, although cases with greater reductions in HDL levels have been reported, suggesting that some mutations may exert a dominant-negative effect. HDL typically contains four apo A molecules per particle (either four apo AI or two apo AI and two apo AII proteins), so a heterozygous individual carrying an expressed apo AI mutant would have at least one mutant apo AI on most HDL particles. The atherosclerosis of individuals with structural mutations in apo AI appears to be much more pronounced than that seen in patients with either LCAT deficiency or Tangier disease. No specific therapy is available for this disorder, but aggressive LDL lipid-lowering therapy is justified.

Management/Treatment
Assessing the Need to Treat Patients With Hyperlipidemia
Because the magnitude of the benefit of treating hyperlipidemia is positively correlated with the degree of CHD risk, it is imperative to assess that risk before initiating treatment. Assessment should be comprehensive, extending beyond lipid levels to include consideration of blood pressure, smoking, diabetes, family history of premature CHD, age, sex, and presence of established CHD or other atherosclerotic disease. Although currently premature, it is likely that the growing list of polygenic risk factors associated with lipid disorders will be incorporated into future risk-stratification schema.77 Treatment recommendations follow directly from the degree of estimated CHD risk. Dietary modification is the sole mode of therapy for patients at the lower end of the CHD risk spectrum, while pharmacologic measures are reserved for patients at higher risk or for those who fail dietary intervention. All three of the major lipid parameters (LDL cholesterol, HDL cholesterol, and triglyceride) have target goals, but the NCEP ATPIII guidelines focus heavily on LDL levels, since reduction in these have been associated with the clearest benefit on cardiovascular morbidity and mortality.9 The trend over the past 20 years has been to push LDL levels ever lower, with no evidence to date that a value has been reached below which no further benefit is conferred. For the highest-risk patients, the current LDL target therapeutic value is less than 70 mg/dL.9
The NCEP guidelines are the most widely recognized and utilized treatment algorithms for hyperlipidemia in the United States. The approach recommended in the guidelines involves the assembly of laboratory and clinical data to establish a patient’s risk for future CHD events. The critical information needed to employ the guidelines come from a fasting lipid and metabolic profile and clinical history and physical exam data that establish whether the patient has CHD or its equivalents. In addition, the clinician must determine if nonlipid CHD risk factors are present or absent. The guidelines recognize five conditions to be the equivalent of established coronary heart disease: (1) diabetes mellitus, (2) symptomatic carotid artery disease, (3) peripheral artery disease, (4) abdominal aortic aneurysm, and (5) a confluence of multiple risk factors that in aggregate confer a 10-year risk of CHD that is higher than 20%. To those five, some experts in the field would add a sixth: the presence of impaired renal function (GFR < 60 mL/min).78–80 The concept that diabetes is a risk equivalent for CHD derives from the high risk of CHD in middle-aged and older diabetic populations and was exemplified in the East-West study of Finnish patients.81 In that study, patients with prior histories of myocardial infarction and no diabetes had a 7-year incidence of new CHD events of 18.8%, whereas diabetic patients without prior myocardial infarctions had an incidence of 20.2%. The hazard ratio for death from CHD comparing these two groups did not differ. A follow-up report by the same investigators, extending to 18 years of observation and with more CHD events recorded, reaffirmed this outcome and suggested that female diabetics were at particularly high risk.82 A finding of similar degrees of carotid intimal medial thickness in U.S. patients with diabetes and no CHD, compared to patients with CHD but not diabetes, further strengthened the concept of diabetes as a CHD equivalent.83 Several other studies in different populations have supported this conclusion.84–88 However, not all investigations of this topic have yielded similar conclusions, with some studies indicating lower rates of future CHD events in the diabetic population without CHD than is found in nondiabetics with CHD. Other studies have indicated that gender substantially influences this comparative outcome.89–96 What all studies demonstrate, however, is that diabetes does confer a substantial increase in CHD risk, and that the combination of diabetes and CHD puts individuals at a very high risk of a future coronary event. Treating diabetes as a risk equivalent to prior CHD in middle-aged and older patients does, therefore, appear to be clinically quite appropriate.
The other risks that are factored into the NCEP algorithm are age (men ≥45 years, women ≥55 years), hypertension, cigarette smoking, low levels of HDL cholesterol, and a family history of premature CHD (male first-degree relatives with events before age 55, females before age 65). High HDL cholesterol levels (≥60 mg/dL) are counted as a negative risk factor, permitting one to subtract one of the positive risk factors. HDL is not completely protective, however, and there are small percentages of patients with very high HDL levels who still develop CHD.97 Although not considered one of the NCEP risk factors, most epidemiologic evidence also supports the conclusion that a sedentary lifestyle is a significant risk for CHD.98–101 Other commonly assessed risk factors not included in the NCEP guideline calculations of risk include obesity, impaired fasting glucose, markers of inflammation (e.g., C-reactive protein), homocysteine, endothelial dysfunction, and a thrombosis predilection. The role of these other risk factors in improving prognosis remains controversial, but clinicians may want to modulate the standard NCEP risk-factor treatment guideline based on the presence or absence of these nontraditional risks. Finally, in individuals with serum triglycerides greater than 200 mg/dL, the NCEP guidelines identify non-HDL cholesterol (total cholesterol minus HDL cholesterol = non-HDL cholesterol) as a secondary parameter that can be used to guide therapy. The non-HDL cholesterol goals for each risk-factor category are equal to the LDL goal plus 30 (i.e., if the LDL goal is <100 mg/dL, the non-HDL cholesterol goal is <130 mg/dL).102 Table 16-5 provides the NCEP treatment recommendations based on the risk factor and CHD equivalent concepts outlined in the previous paragraphs.

Dietary and Drug Treatment of Lipid Disorders
With the advent of very potent lipid-lowering therapies, the use of dietary approaches to controlling lipid levels has become deemphasized in much of the developed world. While drug treatments (especially for elevated LDL cholesterol levels) are very effective, the role of diet in promoting hyperlipidemia should not be forgotten. The dietary approach to the treatment of elevated LDL cholesterol levels focuses on reductions in total fat, saturated fat, partially hydrogenated unsaturated fatty acids, and dietary cholesterol. Substituting foods that provide polyunsaturated and monounsaturated fats in place of saturated and trans unsaturated fat is particularly important, whereas the value of reducing the total fat intake is less clear.103 The 2008 clinical practice guidelines of the American Diabetes Association (ADA) recommend that saturated fat intake be restricted to less than 7% of calories, and that cholesterol intake not exceed 200 mg per day in patients with diabetes. These dietary recommendations mimic those proposed by the NCEP ATP III guidelines for individuals without diabetes who have established coronary disease. For nondiabetics without coronary disease, the NCEP liberalizes saturated fat intake to less than 10% of calories and cholesterol to under 300 mg/d. In both diets, it is recommended that trans fat intake be kept to a minimum. The adoption of a Mediterranean-style diet, with substitution of monounsaturated fats for saturated fat, has demonstrated benefits on LDL cholesterol reduction, insulin sensitivity, and endothelial reactivity.104–106 The magnitude of the LDL reduction achieved with dietary interventions is quite variable and depends on the preintervention diet, as well as metabolic and genetic factors that influence diet-dependent lipid responses. LDL reductions of 5% to 20% are typical for most patients adopting the reduced fat and cholesterol diets recommended by the ADA and NCEP, but reductions of over 50% can occur in selected individuals.107
For patients with VLDL elevations, which includes a large portion of the diabetic population, carbohydrate and alcohol consumption may be more important dietary factors to address than the intake of cholesterol or saturated fat. The current FDA-recommended daily allowance of carbohydrate is 130 g/d, but studies of long-term carbohydrate-restricted diets are limited. Recently, investigations comparing low-carbohydrate and low-fat diets have demonstrated that low-carbohydrate diets can produce as good or even modestly better weight reduction than can low-fat diets in 6- to 12-month timeframes.108–110 The lower-carbohydrate, higher-fat diets typically produce greater reductions in VLDL (serum triglycerides) and greater increases in HDL cholesterol than do the fat-restricted, higher-carbohydrate diets. On the low-carbohydrate, higher-fat diets, the LDL cholesterol level can vary significantly, and if a substantial amount of saturated fat is used in these diets, the rise could be undesirable.111 Therefore, the use of low-carbohydrate diets should be accompanied by the use of monounsaturated and polyunsaturated fats as the major sources of fat in the diet.
Weight loss (if obese), aerobic exercise, and smoking cessation can increase HDL levels and contribute to the dietary lowering of lower-density lipoproteins and CHD risk. They also reduce CHD risk by decreasing blood pressure. In the Diabetes Prevention Program (DPP), subjects in the lifestyle intervention group reduced their fat intake to 28% of calories after 1 year (down from 34%), and most were able to maintain the goal of 150 minutes per week of moderate physical activity.112,113 On this program, in which a body weight loss of 7% was targeted, subjects experienced less progression to diabetes and reductions in multiple cardiovascular risk factors, including dyslipidemia and hypertension.114,115 Although the DPP was conducted in an academic environment with substantial investigative resources devoted to ensuring compliance with the program, more recent efforts modeled on DPP suggest that the results can be translated to community-based outreach programs.116,117
The clinical trials data that demonstrate improvements in CAD outcomes as a result of dietary interventions alone have generally employed diets that restrict fat and/or cholesterol intake much more dramatically than the ADA and NCEP diets mentioned. In the St. Thomas’ Regression Study (STARS), cholesterol intake in men was reduced to 100 to 120 mg/d, and excess weight was addressed by prescribing a physical activity program. After 3 years, the group treated with a low-cholesterol diet had a slower rate of progression of coronary disease and a higher rate of regression as determined by angiography.118 The Lifestyle Heart Trial employed an even more stringent diet, reducing cholesterol intake to under 10 mg/d, combined with an exercise and behavioral modification program. This intervention led to regression of coronary lesions as measured by angiography and a reduction in symptoms of ischemia.119 This study was conducted in a very small number of highly motivated volunteers; it is unlikely that the interventions could be replicated in more typical clinical care settings. A longer-term follow-up of a small number of participants in this study demonstrated that further regression of coronary atherosclerosis could be obtained.120 A study with potentially greater applicability than either STARS or the Lifestyle Heart Trial is the Lyon Diet Heart Study.121–123 Conducted using a Mediterranean diet rich in alpha-linolenic acid, with participants who had experienced a prior myocardial infarction, this trial demonstrated improvements in its primary endpoint (myocardial infarction and death) as well as several CV and secondary endpoints. The Lyon Diet Heart Study results, combined with other investigations of the impact of similar diets on lipids and lipoproteins, provide substantial support for the utility of Mediterranean-style diets in the prevention and treatment of CHD.
Outcome Studies of Pharmacologic Therapy of Hyperlipidemia
CHD Outcomes With HMG CoA Reductase Inhibitors (Statins)
The current era of lipid-lowering therapy began in 1984 with the publication of the Lipid Research Clinics–Coronary Prevention Trial. A randomized, double-blind study employing cholestyramine compared to placebo, this trial demonstrated that an 8% reduction in total cholesterol yielded a 19% reduction in CHD death or myocardial infarction.124,125 With the advent of statin therapy in 1987, much more effective LDL cholesterol-lowering agents became available. During the 1990s, a series of landmark cardiovascular prevention trials were completed using these agents. In 1994, the Scandinavian Simvastatin Survival Study (4S) established for the first time that a lipid-lowering agent could prolong overall survival in individuals with preexisting CHD.126 Randomizing 4444 patients with a prior myocardial infarction or angina to simvastatin or placebo, the drug treatment cohort of 4S had baseline LDL cholesterol levels fall from approximately 190 mg/dL to 120 mg/dL. With a trial endpoint of death for 10% of the original study enrollees (444 study subjects), it took a little over 5 years to complete the study. The result was that 8% of the simvastatin patients died versus 12% of the placebo-treated individuals. This 30% reduction in mortality established statin therapy as an essential intervention in CHD patients with significant LDL elevations. In addition to the mortality benefit, 4S also produced substantial reductions in major coronary events, overall CHD death (19% versus 28%, a relative risk reduction of 42%) and cerebrovascular events (2.7% versus 4.3%). Unlike several of the pre-statin era cholesterol intervention studies, there were no increases in other causes of mortality to counterbalance the benefits on the cardiovascular system. When 4S was examined in greater detail and after longer follow-up to determine if the therapy had been confined to any subset of patients, these analyses concluded that the benefit was widespread.127–129 Individuals at the highest risk appeared to benefit the most, particularly those with diabetes.130 Though only 202 of the 4444 patients in 4S were diagnosed with diabetes, these individuals experienced relative risk reductions in mortality (43%) and major CHD events (55%) that exceeded those in the nondiabetic population. An equally impressive outcome was noted in individuals who in addition to their elevated LDL cholesterol levels had lipid phenotypes characteristic of the metabolic syndrome (low HDL cholesterol levels and high triglycerides). Study subjects with this lipid triad had the highest event rates among those receiving placebo (35.9%) and the greatest reduction in relative risk with treatment (0.48; 95% CI, 0.33 to 0.69).131
Subsequent studies have further refined our understanding of the benefits of statins in patients with and without preexisting CHD. The West of Scotland Coronary Prevention Group study (WESCOPS) enrolled hyperlipidemic men who had not experienced a prior myocardial infarct but who had high levels of other CHD risk factors. Pravastatin reduced LDL cholesterols from approximately 190 mg/dL to 140 mg/dL and, compared to the placebo-treated group, lowered CHD death and myocardial infarction by 30%.132
A similar-magnitude improvement in a younger population of lower-risk patients was observed in the Air Force/Texas Coronary Atherosclerosis Prevention Study (AFCAPS/TexCAPS) when lovastatin was given to men and women with starting LDL-C levels of 150 mg/dL.133 When compared to the placebo-treated patients, those receiving lovastatin reduced their LDL on average to 115 mg/dL and decreased their incidence of major coronary events by 30% to 40%. In the Cholesterol and Recurrent Events Trial (CARE), pravastatin was given to patients with myocardial infarctions whose LDL cholesterol levels were substantially below (mean of 139 mg/dL) those of the 4S and WESCOPS patients. With a primary endpoint of a fatal coronary event or a nonfatal myocardial infarction, the pravastatin-treated group had a 24% relative risk reduction compared to placebo-treated patients.134 Stroke was reduced 31%. A post hoc analysis of CARE by the investigators who performed it led to the conclusion that reduction of LDL cholesterol in the range from 174 mg/dL to 125 mg/dL lowered the coronary event rate, but that further reductions below 125 mg/dL did not appear to confer additional benefit.135 The value of LDL reductions to levels substantially below 125 mg/dL has been a topic of considerable controversy ever since CARE was published, but several subsequent studies have indicated that further reductions of LDL do confer benefit. For example, the Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) study group enrolled over 9000 patients with recent evidence of CHD and demonstrated benefits in overall mortality and CHD death using pravastatin.136 When stratified by starting LDL cholesterol, the magnitude of the benefit appeared to be maintained even in those with starting levels below 116 mg/dL.137–139 The Treating to New Targets (TNT) trial enrolled 10,000 patients with stable CHD and randomized treatment to high-dose (80 mg/d) or low-dose (10 mg/d) atorvastatin.140 Pretreatment LDL-C levels were between 130 and 250 mg/dL and posttreatment LDL-C values were 77 and 101, respectively, in the high- and low-dose groups. The primary endpoint of the study, which was major cardiovascular events, declined 22% more in the high-dose group than the low-dose cohort, as did rates of myocardial infarction and stroke. However, overall mortality was not lower in the high-dose group, owing to an increase in noncardiovascular deaths. Cancer deaths accounted for most of these events, a finding not explicable on the basis of other studies using the same drug.
The Heart Protection Study (HPS), like several of its predecessor trials, attempted to examine the question of LDL-C lowering in patients with moderate hyperlipidemia. HPS was a much bigger trial, randomizing 20,536 patients to placebo or simvastatin (up to 40 mg/d). The inclusion criteria required evidence of prior cardiovascular disease and permitted a broad range of LDL-C levels, with a third of the patients having values less than 116 mg/dL. After average follow-up of 5.5 years, all-cause mortality was reduced in the statin-treated cohort, as was CHD death and major cardiovascular events.141 Notably, over 5000 diabetics were included in the study, and they also achieved a substantial relative risk reduction in myocardial infarction and stroke.142,143 Particularly striking was the finding that the benefits of LDL-C lowering were produced in all three of the tertiles into which patients’ baseline LDL-C were stratified. Even those with starting LDL-C levels approximating the current NCEP target goal of 100 mg/dL (for those with CHD) had a benefit when their value was reduced substantially below that number. Both men and women and older and younger patients derived similar benefits from the statin treatment. As with HPS, the Collaborative Atorvastatin Diabetes Study (CARDS) demonstrated a significant benefit in diabetic patients with lower LDL-C levels (<160 mg/dL) randomized to the statin arm of a placebo-controlled trial.144 This benefit appeared in those whose starting LDL-C levels were both above and below 120 mg/dL. After 4 years of follow-up, the rate of cardiovascular events dropped 37% (95% CI, −52% to −17%).
In addition to studies of their effects on major clinical outcomes, many of the statins have been used in trials assessing their effects on atherosclerosis progression or regression in the coronary arteries or the carotids. The Familial Atherosclerosis Treatment Study (FATS) compared three regimens that all contained a bile acid resin binder, colestipol, plus either placebo, niacin, or lovastatin. The lovastatin-treated cohort lowered its LDL by 46% and had more regression and less progression of their CAD, as assessed by angiography, than did the placebo-treated group. The niacin group’s response was equivalent to that seen with the lovastatin treatment.145 The Regression Growth Evaluation Statin Study (REGRESS) utilized pravastatin in over 800 men with total cholesterol levels between 155 and 310 mg/dL to demonstrate delayed progression in CHD as well as a lower rate of progression of femoral and carotid atherosclerosis.146,147 The REVERSAL trial compared atorvastatin (80 mg/d) to pravastatin (40 mg/d) in patients with CHD by assessing plaque volume using intravascular ultrasound measurements (IVUS). Atheroma volume declined slightly in the atorvastatin-treated group while increasing in the pravastatin cohort, the difference between the two being statistically significant.148 ASTEROID employed an open-label treatment with rosuvastatin (40 mg/d) in 507 patients with CHD to lower LDL-C by 53% (from a mean of 130 to 61 mg/dL). With this intervention, atheroma volume, as measured by IVUS, was reduced. No control group was included in ASTEROID, making it difficult to assess what is otherwise an impressive reduction in coronary atherosclerosis.149 Taken together, these statin studies provide impressive evidence that aggressive lipid lowering can reduce the rate of CAD progression in patients with established disease and may, with the most intensive therapy, even cause plaque regression.
In addition to their use in studies of patients with longstanding stable CHD, statins have been employed in the setting of acute or recent coronary syndromes to determine if more aggressive lipid-lowering might provide additional benefit in these settings. The Prove-It study, for example, compared individuals with starting LDL-C levels of approximately 110 mg/dL who were randomized to received pravastatin (40 mg/d) or atorvastatin (80 mg/d) after an acute coronary event.150 The high-dose atorvastatin group lowered LDL-C levels to 62 mg/dL, whereas the mid-dose pravastatin cohort only reduced their LDL-C values to 95 mg/dL. A 16% reduction in coronary events was achieved on the more aggressive therapy. It seems probable that this study established that an LDL-C of 62 mg/dL is better than one of 95 mg/dL in patients experiencing an acute coronary event, but the more rigorous conclusion is that 80 mg of atorvastatin works better for CHD outcomes in this population than does 40 mg of pravastatin. Similarly, the Incremental Decrease in End Points through Aggressive Lipid-lowering study used an open-label/blinded-endpoint study design to randomize nearly 9000 patients with a past history of myocardial infarction to receive either atorvastatin (80 mg/d) or simvastatin (20 mg/d). After 24 weeks, the dose of simvastatin could be raised to 40 mg/d if the LDL-C remained greater than 190 mg/dL. The atorvastatin-treated cohort after nearly 5 years had a mean LDL-C that was lower than the simvastatin-treated cohort (81 versus 104 mg/dL), but no significant reduction in the primary endpoint of major coronary events was demonstrated. While there were reductions in some secondary endpoints on atorvastatin, no benefit on overall mortality was seen. Because these studies employed different drugs and achieved different LDL endpoints using those agents, it is not always easy to interpret the outcomes. Overall, the data do support aggressive lipid-lowering therapy in the acute coronary setting, but it is much less clear whether one drug is superior to another when nearly comparable LDL-C lowering is achieved.
In summary, the outcomes studies presented in this section demonstrate that LDL cholesterol reduction with multiple members of the statin class of drugs results in improvements in CHD events and in overall total mortality in patients with established coronary disease. These benefits are present or even magnified in individuals with diabetes or the metabolic syndrome. Lower rates of CHD events are also produced in patients without established CHD but who have substantial CHD risk factors. In lower-risk populations, statins clearly reduce coronary events, but data are less convincing for benefits on CHD death or overall mortality. These conclusions have been confirmed by several meta-analyses of clinical trials that employed a variety of statins.151–157 Although the precise goal of LDL-C reduction remains controversial, the NCEP guideline targets of progressively lower target LDLs for progressively higher levels of risk (i.e., <130, <100, and <70 mg/dL) are both reasonable and consistent with the findings of outcomes studies published to date.
Whether statin therapy should be initiated in young individuals is a topic of debate with little data available to inform the discussion. Young men (age less than 35) and premenopausal women with no CHD risk factors other than hypercholesterolemia are typically at very low 10-year risks for developing CHD. An argument can be made to wait until the risk level escalates before initiating cholesterol-lowering therapies, inasmuch as the benefits of statin therapy are typically evident in less than 2 to 3 years. An alternative view cites the evidence that individuals with genetically driven low LDL cholesterols have extremely low rates of CHD and that one should try to mimic this lifelong low-cholesterol state with earlier and more aggressive drug therapy.68 Both perspectives have merit. For now, this decision will likely balance the magnitude of the individual risk with preferences for more or less aggressive therapy on the part of both physician and patient. Physicians prescribing statins for young women, however, must not forget the potential teratogenicity of this class of drugs. Nonpharmacologic therapy, primarily focusing on improved diets and physical activity programs, should be adopted regardless of age. As widespread clinic use of statins moves into its third decade and the initially approved agents have become available in generic forms, the safety and cost issues associated with their use may recede in importance, and a greater consensus on earlier use of these remarkable drugs becomes possible. For now, the exercise of good clinical judgment is essential in these circumstances.
Lipid-Lowering Outcomes Trials With Agents Other Than Statins
As cited in the previous section, the current era of lipid-lowering was successfully initiated with a bile acid resin binder, cholestyramine.124,125 Though achieving an LDL-C reduction of only 8%, CHD events were reduced 19%. Thus, it is the belief of most experts in the lipid field that LDL cholesterol lowering achieved by any means will result in reduced CHD events, provided the therapeutic approach does not have off-target consequences that would countervail the benefit of the LDL lowering. This view is supported by a wealth of data on the pathogenesis of atherosclerosis, as well as clinical trials of multiple modalities that lower LDL cholesterol, including nonpharmacologic approaches.1,2,120,158,159 At the current time, statin use supersedes that of all other LDL-C lowering agents as first-line monotherapy, because of the superior efficacy and the wealth of outcomes data establishing its benefit. Many patients, however, either cannot meet the LDL targets set by the NCEP guidelines or cannot achieve the levels obtained in the major outcomes trials using statins alone. Some patients have inadequate therapeutic responses to the drugs, and others cannot tolerate their adverse side effects. When that occurs, the use of other lipid-lowering agents becomes relevant. Moreover, some patients, particularly those with diabetes, may have very low HDL-C levels or dramatic elevations in lipoproteins other than LDL, for which statin therapy is much less effective. The following section addresses the current data on outcomes when these other, nonstatin therapies are employed.
Fibrates and CHD Outcomes
Unlike statin therapy, none of the other lipid-lowering medications have produced an overall mortality benefit in a randomized, prospective, double-blind trial. For the most part, this is because studies employing the agents were not powered for a primary outcome focused on mortality but instead targeted reductions in CHD events. With this endpoint, bile acid resins, gemfibrozil, and niacin have all been demonstrated in long-term prospective studies to reduce CHD and/or major CV events.124,125,160–164 Although not available in the United States, bezafibrate also showed promising results in a secondary prevention study.165 Since fibrates (gemfibrozil, fenofibrate, bezafibrate, clofibrate, and ciprofibrate) are more effective than statins at lowering serum triglyceride/VLDL levels, they would appear to be particularly attractive agents for the treatment of the dyslipidemia associated with diabetes and the metabolic syndrome. Subgroup analyses of many studies employing fibrates support this hypothesis, with greater benefit shown in those with high triglycerides or low HDL.166 Insofar as neither bezafibrate nor ciprofibrate are available in the United States, and clofibrate is no longer used because of a significantly higher mortality rate produced by the drug, these agents will not be discussed in this section. The clofibrate findings, however, have cast a long shadow over other fibrates because of the fear that similar biliary and gastrointestinal diseases might accompany their use.167 To date, this fear seems not to have been realized, though the lack of overall mortality benefit with fibrates continues to raise suspicion.
Of the fibrates, then, only fenofibrate and gemfibrozil are prescribed in the United States. Gemfibrozil was used in two major trials, the Helsinki Heart Study (HHS) and the Veterans Affairs High-Density Lipoprotein Intervention Trial (VA-HIT), with both reporting substantial benefit from the drug. The HHS was a double-blind, placebo-controlled 5-year trial that targeted HDL-C raising and non-HDL cholesterol lowering.160 A group of 4081 asymptomatic men with non-HDL-C levels ≥200 mg/dL were studied over a 5-year period, with approximately half receiving 600 mg twice daily of the fibrate and half on placebo. HDL-C, non-HDL-C, LDL-C, and triglycerides all improved in the drug-treated cohort, which experienced a 34% reduction in CHD. On drug treatment, HDL-C rose 11%, LDL-C fell 10%, and triglycerides dropped 35%. Subsequent subgroup analysis of this study showed that the greatest benefits were achieved in those with BMIs above 26 kg/m2 or in individuals that had higher triglyceride levels or lower HDL-C values.168,169 In the VA-HIT study, 2531 men with established CHD, LDL-C levels ≤140 mg/dL and HDL-C levels ≤40 mg/dL were treated with gemfibrozil (1200 mg/d) or placebo.161 Those who received the fibrate experienced a 22% reduction in the primary outcome of nonfatal myocardial infarction or CHD death. These benefits were associated with a 6% higher HDL-C level, a 31% lower serum triglyceride value, and no significant change in LDL-C. Subgroup analysis in this study again revealed that those benefiting the most from the drug were individuals whose HDL-C rose. Somewhat surprisingly, the improvement in triglyceride was not associated with an improved CHD outcome.170 The serious adverse events seen with clofibrate were not reproduced in the gemfibrozil studies, though overall mortality benefits were also not demonstrated in either study.
The results of the HHS and VA-HIT studies convinced many lipid experts that fibrate treatment was beneficial, and these agents have been widely prescribed in the past decade to treat hypertriglyceridemia and low HDL-C levels. Because gemfibrozil was found to affect the metabolism of multiple statins,171 many lipid experts switched to using fenofibrate whenever a statin/fibrate combination therapy was deemed appropriate in order to reduce the likelihood of a serious myopathy. Thus, the publication of the Fenofibrate Intervention and Event Lowering in Diabetes Study (FIELD) in 2005 was an important event in defining lipid therapeutics.172 This trial randomized 9795 middle-aged and older patients with type 2 diabetes to placebo and micronized fenofibrate (200 mg/d). Patients qualified by having total cholesterol levels between 116 and 252 mg/dL and total/HDL-C ratios ≥4.0 or plasma triglycerides between 88 and 435 mg/dL. An unusual feature of the FIELD trial was that patients were permitted to be treated with other lipid-lowering agents at their own physician’s discretion, so many received simultaneous statin therapy. The primary outcome of the study was CHD events. Triglycerides were reduced by the fibrate by 29%, but HDL-C rose only 2%. While CHD events were reduced in the drug-treated group by 11%, this overall outcome was not statistically significant. More worrisome was the fact that the overall event rate represented the average of a positive and negative outcome, nonfatal infarction falling 24%, but CHD mortality rising by 19%. When total mortality was examined, it was non-significantly higher in the fenofibrate cohort. Overall, the FIELD study was widely perceived to have failed to deliver the expected benefits suggested by the earlier gemfibrozil studies. This outcome has led to vigorous and protracted debates about the utility of fibrate therapy in general and fenofibrate in particular. In a recent consensus statement from the ADA and the American College of Cardiology Foundation, the authors favor niacin over fibrates as an agent to combine with statins in treating combined lipid disorders in which VLDL or HDL-C levels are targeted for treatment.173 Despite the epidemiologic evidence linking high serum triglycerides to CHD events, the data from fibrate intervention studies have not shown convincing correlated reductions in triglycerides with CHD benefit. As pointed out earlier in VA-HIT, the effect on HDL-C levels, though modest, correlated with the desirable CHD outcomes, while the marked triglyceride reduction of 31% did not. Thus, the triglyceride effect of fibrates may be most clinically valuable when treating severely hypertriglyceridemic patients (serum triglycerides > 500 mg/dL) in whom the goal is to prevent triglyceride-induced pancreatitis. Ongoing studies with fibrates may clarify their place in the prevention of CHD, but at present, the currently available agents do not appear to have a great enough therapeutic benefit in most CHD patients to warrant widespread use.
Three other classes of drugs are commonly used to treat patients with hyperlipidemia: (1) nicotinic acid or niacin, (2) cholesterol absorption inhibitors (bile acid resins and ezetimibe), and (3) omega-3 fatty acids or fish oils. Outcomes studies with these agents are quite limited. The decrease in CHD produced with niacin in the Coronary Drug Project, combined with long-term follow-up of those originally assigned the drug, provide substantial confidence in the therapeutic value of using this B vitamin.163,174 There is also evidence for benefit when niacin is combined with a statin.175,176 Niacin can lower triglycerides, raise HDL cholesterol, and reduce LDL cholesterol, so it can provide a very useful adjunct to statin therapy in treating patients with combined lipid disorders. Its side-effect profile and its potential for increasing insulin resistance are the major drawbacks to employing niacin more widely. Bile acid resins were the mainstays of lipid therapy prior to the advent of statins; multiple CHD outcomes and atherosclerosis progression studies were performed with various members of the class. Overall, despite only modest LDL-C lowering performance, these drugs yielded benefits on CHD and coronary atherosclerosis when given as monotherapy or in combination with niacin or statins.118,124,125,145,177 Because bile acid resins can interfere with the absorption of other drugs, cause substantial gastrointestinal bloating and constipation, and often substantially increase serum triglycerides, the development of much more selective cholesterol absorption inhibitors was highly desirable. Ezetimibe, which inhibits cholesterol absorption from the gut via blockage of enterocyte cholesterol trafficking pathways, rapidly replaced bile acid resins in the lipid therapeutic armamentarium because of its ease of use. Unfortunately, ezetimibe has yet to demonstrate any meaningful cardiovascular outcome beyond its nearly 20% LDL-C lowering. In its first major outcome study, the addition of ezetimibe to simvastatin provided no additional benefit beyond simvastatin alone on carotid intimal medical thickness assessed by carotid sonography.178 This controversial study demonstrated substantial additional LDL-C reduction with ezetimibe (16.5% greater than the simvastatin alone), indicating that the drug was effective in its primary action in the cohort that received it. The short duration of treatment, the mild degree of carotid disease at the outset of the study, and the use of patients previously treated with statins for many years may have all combined to make it extremely challenging for this study to have shown a benefit. These caveats notwithstanding, the fact remains that ezetimibe has no major outcomes data to support its use at present. Given the effectiveness of bile acid resins at reducing CHD events, it would be extremely surprising for the more potent ezetimibe not to confer at least the same benefit, unless there is some unexpected consequence of inhibiting the gut cholesterol trafficking pathway, or the drug has an as-yet-unidentified off-target effect. Ongoing studies should be completed in the next few years that provide insights into these issues. At present, ezetimibe should be used as an adjunct to statin therapy only when the latter has failed to achieve the target LDL goal at the maximum tolerated statin dose. In patients who cannot tolerate a statin, ezetimibe can also be considered, but niacin would be the first preferred alternative therapy if appropriate.
The final category of hypolipidemic agents, omega-3 fatty acids, are moderately effective agents in lowering elevated serum triglycerides. At doses of 2 to 4 g/d, omega-3 fatty acids inhibit VLDL production while having little or no effect on HDL-C and LDL-C. There are many epidemiologic studies of fish consumption that suggest a cardiovascular benefit, but there are no major CHD outcomes studies in which these compounds have been used to treat hypertriglyceridemic patients. The best available evidence suggests that omega-3 fatty acids may reduce cardiac arrhythmic events but have a modest effect if any on coronary atherosclerosis.179–181
Prescription Drugs Used for Lipid Lowering
HMG-CoA Reductase Inhibitors (Statins)
These agents have become the first-line drug therapy for treatment of elevated LDL levels because of their effectiveness, patient acceptability, clinical outcomes data, and favorable safety record. They block the rate-limiting enzyme in cholesterol synthesis, HMG-CoA reductase. Serum LDL levels fall by 20% to 60%, depending on dose and specific agent. HDL levels generally stay the same or increase slightly on statins. Statins also influence thrombotic and inflammatory mechanisms, effects whose importance remains to be elucidated. Myalgias, with or without creatine kinase elevations, are the most common side effect of the class. Asymptomatic hepatocellular dysfunction, manifested by an increase in serum levels of transaminases, occurs in approximately 1% of patients taking the medications. Statins that are currently available in the United States and Canada are lovastatin, simvastatin, pravastatin, fluvastatin, atorvastatin, and rosuvastatin. The oldest approved statins—lovastatin, simvastatin, and pravastatin—are all available in generic forms and are typically the approved agents on cost-sensitive formularies. Atorvastatin and rosuvastatin are the two most potent statins, with LDL-C reductions of more than 50% routinely achieved in patients given the higher dosages of either agent. Table 16-5 summarizes some key information about the available statins.
Niacin
Niacin is a B vitamin that must be used in megadoses to lower lipids. The precise mechanism of action remains although uncertain, although recent data indicate that it could block VLDL synthesis by inhibiting the activity of hepatic DGAT-2.182 The discovery of receptors for niacin and the elucidation of the prostaglandin-mediated mechanism by which it evokes flushing have contributed to renewed interest in a drug that has been used to treat lipids for over 50 years.183,184 A new medication that combines niacin with an inhibitor of this flushing pathway has been developed but not yet approved by the FDA. Niacin, at doses of 1000 mg and higher, typically lowers LDL and VLDL levels and raises HDL levels. It is the most effective HDL cholesterol–raising drug available. The clinical trial results presented in the outcomes section of this chapter provide sufficient safety and efficacy data to conclude that niacin is an effective anti-atherosclerotic agent, though there are no long-term, randomized prospective clinical trials establishing an overall mortality benefit with its use. Niacin’s principal disadvantages include a litany of side effects reflecting the large doses required to achieve effective lipid lowering. Niacin can exacerbate gout and diabetes, elevate liver enzymes, and produce rashes, nausea, and vomiting. The impact on diabetes with the long-acting, single daily dosing formulation of niacin (Niaspan) appears to be modest (less than 0.3 unit rise in glycated hemoglobin), so many lipid experts recommend its use in diabetics to treat their hypertriglyceridemia, low HDL-C levels, or as adjunctive therapy to statins for LDL-C lowering.185,186 The acute vasodilation associated with flushing can occasionally lead to postural light-headedness, a reaction that aspirin can often mitigate. Niacin is available in both prescription and nonprescription formulations, but physicians should be cautious in recommending no-flush versions of the drug; these often lack any therapeutic efficacy.187 Many patients can tolerate niacin if pretreated with aspirin and given slowly escalating doses, building weekly in 500-mg increments. Typical effective dosing requires from 1500 mg to 3000 mg/d, with side effects increasing as the dose escalates.
References
1. Lusis, AJ. Atherosclerosis. Nature. 2000;407:233–241.
2. Tabas, I, Williams, KJ, Boren, J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications. Circulation. 2007;116:1832–1844.
3. Almdal, T, Scharling, H, Jensen, JS, et al. The independent effect of type 2 diabetes mellitus on ischemic heart disease, stroke, and death: a population-based study of 13,000 men and women with 20 years of follow-up. Arch Intern Med. 2004;164:1422–1426.
4. Genest, J, Libby, P, Gotto, AMJ. Lipoprotein disorders and cardiovascular disease. In: Zipes DP, Libby P, Bonow RO, Braunwald E, eds. Braunwald’s heart disease. Philadelphia: Elsevier Saunders, 2005.
5. Beckman, JA, Creager, MA, Libby, P. Diabetes and atherosclerosis: epidemiology, pathophysiology, and management. JAMA. 2002;287:2570–2581.
6. Asztalos, BF, Collins, D, Cupples, LA, et al. Value of high-density lipoprotein (HDL) subpopulations in predicting recurrent cardiovascular events in the Veterans Affairs HDL Intervention Trial. Arterioscler Thromb Vasc Biol. 2005;25:2185–2191.
7. Havel, RJ, Kane, JP. Introduction: structure and metabolism of plasma lipoproteins. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The metabolic basis of inherited disease. New York: McGraw-Hill, 2001.
8. Powell, LM, Wallis, SC, Pease, RJ, et al. A novel form of tissue-specific RNA processing produces apolipoprotein-B48 in intestine. Cell. 1987;50:831–840.
9. Grundy, SM, Cleeman, JI, Merz, CN, et al. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation. 2004;110:227–239.
10. Grundy, SM. Comparison of monounsaturated fatty acids and carbohydrates for lowering plasma cholesterol. N Engl J Med. 1986;314:745–748.
11. Tosi, I, Toledo-Leiva, P, Neuwirth, C, et al. Genetic defects causing familial hypercholesterolaemia: identification of deletions and duplications in the LDL-receptor gene and summary of all mutations found in patients attending the Hammersmith Hospital Lipid Clinic. Atherosclerosis. 2007;194:102–111.
12. Veniant, MM, Beigneux, AP, Bensadoun, A, et al. Lipoprotein size and susceptibility to atherosclerosis–insights from genetically modified mouse models. Curr Drug Targets. 2008;9:174–189.
13. Packard, CJ. Small dense low-density lipoprotein and its role as an independent predictor of cardiovascular disease. Curr Opin Lipidol. 2006;17:412–417.
14. Cuchel, M, Rader, DJ. Macrophage reverse cholesterol transport: key to the regression of atherosclerosis? Circulation. 2006;113:2548–2555.
15. Vaisar, T, Pennathur, S, Green, PS, et al. Shotgun proteomics implicates protease inhibition and complement activation in the antiinflammatory properties of HDL. J Clin Invest. 2007;117:746–756.
16. Scanu, AM, Edelstein, C. HDL: bridging past and present with a look at the future. FASEB J. 2008.
17. Drover, VA, Ajmal, M, Nassir, F, et al. CD36 deficiency impairs intestinal lipid secretion and clearance of chylomicrons from the blood. J Clin Invest. 2005;115:1290–1297.
18. Kamp, F, Guo, W, Souto, R, et al. Rapid flip-flop of oleic acid across the plasma membrane of adipocytes. J Biol Chem. 2003;278:7988–7995.
19. Hui, DY, Labonte, ED, Howles, PN. Development and physiological regulation of intestinal lipid absorption. III. Intestinal transporters and cholesterol absorption. Am J Physiol Gastrointest Liver Physiol. 2008;294:G839–843.
20. Altmann, SW, Davis, HR, Jr., Zhu, LJ, et al. Niemann-Pick C1 Like 1 protein is critical for intestinal cholesterol absorption. Science. 2004;303:1201–1204.
21. Garcia-Calvo, M, Lisnock, J, Bull, HG, et al. The target of ezetimibe is Niemann-Pick C1-Like 1 (NPC1L1). Proc Natl Acad Sci U S A. 2005;102:8132–8137.
22. Black, DD. Development and physiological regulation of intestinal lipid absorption. I. Development of intestinal lipid absorption: cellular events in chylomicron assembly and secretion. Am J Physiol Gastrointest Liver Physiol. 2007;293:G519–524.
23. Yu, L, Li-Hawkins, J, Hammer, RE, et al. Overexpression of ABCG5 and ABCG8 promotes biliary cholesterol secretion and reduces fractional absorption of dietary cholesterol. J Clin Invest. 2002;110:671–680.
24. Hussain, MM, Innerarity, TL, Brecht, WJ, et al. Chylomicron metabolism in normal, cholesterol-fed, and Watanabe heritable hyperlipidemic rabbits. Saturation of the sequestration step of the remnant clearance pathway. J Biol Chem. 1995;270:8578–8587.
25. Dane-Stewart, CA, Watts, GF, Barrett, PH, et al. Chylomicron remnant metabolism studied with a new breath test in postmenopausal women with and without type 2 diabetes mellitus. Clin Endocrinol (Oxf). 2003;58:415–420.
26. Kane, JP, Havel, RJ. Disorders of the biogenesis and secretion of lipoproteins containing the B apolipoproteins. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The metabolic basis of inherited disease. New York: McGraw-Hill, 2001.
27. Kang, S, Davis, RA. Cholesterol and hepatic lipoprotein assembly and secretion. Biochim Biophys Acta. 2000;1529:223–230.
28. Olofsson, SO, Asp, L, Boren, J. The assembly and secretion of apolipoprotein B-containing lipoproteins. Curr Opin Lipidol. 1999;10:341–346.
29. Parhofer, KG, Barrett, PH. Thematic review series: patient-oriented research. What we have learned about VLDL and LDL metabolism from human kinetics studies. J Lipid Res. 2006;47:1620–1630.
30. Goldstein, JL, Hobbs, HH, Brown, MS. Familial hypercholesterolemia. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The metabolic basis of inherited disease. New York: McGraw-Hill, 2001.
31. Brown, MS, Goldstein, JL. A receptor-mediated pathway for cholesterol homeostasis. Science. 1986;232:34–47.
32. Radhakrishnan, A, Ikeda, Y, Kwon, HJ, et al. Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: oxysterols block transport by binding to Insig. Proc Natl Acad Sci U S A. 2007;104:6511–6518.
33. Sun, LP, Seemann, J, Goldstein, JL, et al. Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: Insig renders sorting signal in Scap inaccessible to COPII proteins. Proc Natl Acad Sci U S A. 2007;104:6519–6526.
34. Gong, Y, Lee, JN, Lee, PC, et al. Sterol-regulated ubiquitination and degradation of Insig-1 creates a convergent mechanism for feedback control of cholesterol synthesis and uptake. Cell Metab. 2006;3:15–24.
35. Flury, I, Garza, R, Shearer, A, et al. INSIG: a broadly conserved transmembrane chaperone for sterol-sensing domain proteins. EMBO J. 2005;24:3917–3926.
36. Sun, LP, Li, L, Goldstein, JL, et al. Insig required for sterol-mediated inhibition of Scap/SREBP binding to COPII proteins in vitro. J Biol Chem. 2005;280:26483–26490.
37. Adams, CM, Reitz, J, De Brabander, JK, et al. Cholesterol and 25-hydroxycholesterol inhibit activation of SREBPs by different mechanisms, both involving SCAP and Insigs. J Biol Chem. 2004;279:52772–52780.
38. Attie, AD. ABCA1: at the nexus of cholesterol, HDL and atherosclerosis. Trends Biochem Sci. 2007;32:172–179.
39. Chang, C, Dong, R, Miyazaki, A, et al. Human acyl-CoA: cholesterol acyltransferase (ACAT) and its potential as a target for pharmaceutical intervention against atherosclerosis. Acta Biochim Biophys Sin (Shanghai). 2006;38:151–156.
40. Zhao, B, Song, J, Chow, WN, et al. Macrophage-specific transgenic expression of cholesteryl ester hydrolase significantly reduces atherosclerosis and lesion necrosis in Ldlr mice. J Clin Invest. 2007;117:2983–2992.
41. Chroni, A, Liu, T, Fitzgerald, ML, et al. Cross-linking and lipid efflux properties of apoA-I mutants suggest direct association between apoA-I helices and ABCA1. Biochemistry. 2004;43:2126–2139.
42. Fitzgerald, ML, Mendez, AJ, Moore, KJ, et al. ATP-binding cassette transporter A1 contains an NH2-terminal signal anchor sequence that translocates the protein’s first hydrophilic domain to the exoplasmic space. J Biol Chem. 2001;276:15137–15145.
43. Fitzgerald, ML, Morris, AL, Rhee, JS, et al. Naturally occurring mutations in the largest extracellular loops of ABCA1 can disrupt its direct interaction with apolipoprotein A-I. J Biol Chem. 2002;277:33178–33187.
44. Tall, AR, Yvan-Charvet, L, Terasaka, N, et al. HDL, ABC transporters, and cholesterol efflux: implications for the treatment of atherosclerosis. Cell Metab. 2008;7:365–375.
45. Trigatti, BL, Krieger, M, Rigotti, A. Influence of the HDL receptor SR-BI on lipoprotein metabolism and atherosclerosis. Arterioscler Thromb Vasc Biol. 2003;23:1732–1738.
46. Deckert, T, Poulsen, JE, Larsen, M. Prognosis of diabetics with diabetes onset before the age of thirty-one. I. Survival, causes of death, and complications. Diabetologia. 1978;14:363–370.
47. Dorman, JS, Laporte, RE, Kuller, LH, et al. The Pittsburgh insulin-dependent diabetes mellitus (IDDM) morbidity and mortality study. Mortality results. Diabetes. 1984;33:271–276.
48. Orchard, TJ, Costacou, T, Kretowski, A, et al. Type 1 diabetes and coronary artery disease. Diabetes Care. 2006;29:2528–2538.
49. Laing, SP, Swerdlow, AJ, Slater, SD, et al. Mortality from heart disease in a cohort of 23,000 patients with insulin-treated diabetes. Diabetologia. 2003;46:760–765.
50. Jenkins, AJ, Lyons, TJ, Zheng, D, et al. Serum lipoproteins in the diabetes control and complications trial/epidemiology of diabetes intervention and complications cohort: associations with gender and glycemia. Diabetes Care. 2003;26:810–818.
51. Colhoun, HM, Otvos, JD, Rubens, MB, et al. Lipoprotein subclasses and particle sizes and their relationship with coronary artery calcification in men and women with and without type 1 diabetes. Diabetes. 2002;51:1949–1956.
52. Orchard, TJ, Olson, JC, Erbey, JR, et al. Insulin resistance-related factors, but not glycemia, predict coronary artery disease in type 1 diabetes: 10-year follow-up data from the Pittsburgh Epidemiology of Diabetes Complications Study. Diabetes Care. 2003;26:1374–1379.
53. Giunti, S, Bruno, G, Veglio, M, et al. Electrocardiographic left ventricular hypertrophy in type 1 diabetes: prevalence and relation to coronary heart disease and cardiovascular risk factors: the Eurodiab IDDM Complications Study. Diabetes Care. 2005;28:2255–2257.
54. Pambianco, G, Costacou, T, Ellis, D, et al. The 30-year natural history of type 1 diabetes complications: the Pittsburgh Epidemiology of Diabetes Complications Study experience. Diabetes. 2006;55:1463–1469.
55. Olson, JC, Erbey, JR, Forrest, KY, et al. Glycemia (or, in women, estimated glucose disposal rate) predict lower extremity arterial disease events in type 1 diabetes. Metabolism. 2002;51:248–254.
56. Moss, SE, Klein, R, Klein, BE. The 14-year incidence of lower-extremity amputations in a diabetic population. The Wisconsin Epidemiologic Study of Diabetic Retinopathy. Diabetes Care. 1999;22:951–959.
57. Klein, BE, Klein, R, McBride, PE, et al. Cardiovascular disease, mortality, and retinal microvascular characteristics in type 1 diabetes: Wisconsin epidemiologic study of diabetic retinopathy. Arch Intern Med. 2004;164:1917–1924.
58. Mykkanen, L, Laakso, M, Penttila, I, et al. Asymptomatic hyperglycemia and cardiovascular risk factors in the elderly. Atherosclerosis. 1991;88:153–161.
59. Haffner, SM. Lipoprotein disorders associated with type 2 diabetes mellitus and insulin resistance. Am J Cardiol. 2002;90:55i–61i.
60. Packard, CJ. Triacylglycerol-rich lipoproteins and the generation of small, dense low-density lipoprotein. Biochem Soc Trans. 2003;31:1066–1069.
61. Sparks, JD, Phung, TL, Bolognino, M, et al. Insulin-mediated inhibition of apolipoprotein B secretion requires an intracellular trafficking event and phosphatidylinositol 3-kinase activation: studies with brefeldin A and wortmannin in primary cultures of rat hepatocytes. Biochem J. 1996;313(Pt 2):567–574.
62. Au, WS, Kung, HF, Lin, MC. Regulation of microsomal triglyceride transfer protein gene by insulin in HepG2 cells: roles of MAPKerk and MAPKp38. Diabetes. 2003;52:1073–1080.
63. Myant, NB. Familial defective apolipoprotein B-100: a review, including some comparisons with familial hypercholesterolaemia. Atherosclerosis. 1993;104:1–18.
64. Whitfield, AJ, Barrett, PH, van Bockxmeer, FM, et al. Lipid disorders and mutations in the APOB gene. Clin Chem. 2004;50:1725–1732.
65. Soutar, AK, Naoumova, RP. Autosomal recessive hypercholesterolemia. Semin Vasc Med. 2004;4:241–248.
66. He, G, Gupta, S, Yi, M, et al. ARH is a modular adaptor protein that interacts with the LDL receptor, clathrin, and AP-2. J Biol Chem. 2002;277:44044–44049.
67. Garcia, CK, Wilund, K, Arca, M, et al. Autosomal recessive hypercholesterolemia caused by mutations in a putative LDL receptor adaptor protein. Science. 2001;292:1394–1398.
68. Cohen, JC, Boerwinkle, E, Mosley, TH, Jr., et al. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354:1264–1272.
69. Peterson, AS, Fong, LG, Young, SG. PCSK9 function and physiology. J Lipid Res. 2008;49:1595–1599.
70. Singaraja, RR, Brunham, LR, Visscher, H, et al. Efflux and atherosclerosis: the clinical and biochemical impact of variations in the ABCA1 gene. Arterioscler Thromb Vasc Biol. 2003;23:1322–1332.
71. Oram, JF. ATP-binding cassette transporter A1 and cholesterol trafficking. Curr Opin Lipidol. 2002;13:373–381.
72. Calabresi, L, Pisciotta, L, Costantin, A, et al. The molecular basis of lecithin : cholesterol acyltransferase deficiency syndromes: a comprehensive study of molecular and biochemical findings in 13 unrelated Italian families. Arterioscler Thromb Vasc Biol. 2005;25:1972–1978.
73. Sorci-Thomas, MG, Thomas, MJ. The effects of altered apolipoprotein A-I structure on plasma HDL concentration. Trends Cardiovasc Med. 2002;12:121–128.
74. Sasaki, J, Matsunaga, A, Huang, W, et al. Structural and functional properties of apolipoprotein A-I mutants. J Atheroscler Thromb. 2000;7:67–70.
75. Schaefer, EJ. Clinical, biochemical, and genetic features in familial disorders of high density lipoprotein deficiency. Arteriosclerosis. 1984;4:303–322.
76. Hovingh, GK, de Groot, E, van der Steeg, W, et al. Inherited disorders of HDL metabolism and atherosclerosis. Curr Opin Lipidol. 2005;16:139–145.
77. Kathiresan, S, Melander, O, Guiducci, C, et al. Six new loci associated with blood low-density lipoprotein cholesterol, high-density lipoprotein cholesterol or triglycerides in humans. Nat Genet. 2008;40:189–197.
78. Sarnak, MJ, Levey, AS, Schoolwerth, AC, et al. Kidney disease as a risk factor for development of cardiovascular disease: a statement from the American Heart Association Councils on Kidney in Cardiovascular Disease, High Blood Pressure Research, Clinical Cardiology, and Epidemiology and Prevention. Circulation. 2003;108:2154–2169.
79. Collins, AJ, Foley, R, Herzog, C, et al. Excerpts from the United States Renal Data System 2007 annual data report. Am J Kidney Dis. 2008;51:S1–320.
80. Di Angelantonio, E, Danesh, J, Eiriksdottir, G, et al. Renal function and risk of coronary heart disease in general populations: new prospective study and systematic review. PLoS Med. 2007;4:e270.
81. Haffner, SM, Lehto, S, Ronnemaa, T, et al. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med. 1998;339:229–234.
82. Juutilainen, A, Lehto, S, Ronnemaa, T, et al. Type 2 diabetes as a “coronary heart disease equivalent”: an 18-year prospective population-based study in Finnish subjects. Diabetes Care. 2005;28:2901–2907.
83. Haffner, SM, Agostino, RD, Jr., Saad, MF, et al. Carotid artery atherosclerosis in type-2 diabetic and nondiabetic subjects with and without symptomatic coronary artery disease (The Insulin Resistance Atherosclerosis Study). Am J Cardiol. 2000;85:1395–1400.
84. Malmberg, K, Yusuf, S, Gerstein, HC, et al. Impact of diabetes on long-term prognosis in patients with unstable angina and non-Q-wave myocardial infarction: results of the OASIS (Organization to Assess Strategies for Ischemic Syndromes) Registry. Circulation. 2000;102:1014–1019.
85. Yusuf, S, Sleight, P, Pogue, J, et al. Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. The Heart Outcomes Prevention Evaluation Study Investigators. N Engl J Med. 2000;342:145–153.
86. Mukamal, KJ, Nesto, RW, Cohen, MC, et al. Impact of diabetes on long-term survival after acute myocardial infarction: comparability of risk with prior myocardial infarction. Diabetes Care. 2001;24:1422–1427.
87. Natarajan, S, Liao, Y, Sinha, D, et al. Sex differences in the effect of diabetes duration on coronary heart disease mortality. Arch Intern Med. 2005;165:430–435.
88. Hu, FB, Stampfer, MJ, Solomon, CG, et al. The impact of diabetes mellitus on mortality from all causes and coronary heart disease in women: 20 years of follow-up. Arch Intern Med. 2001;161:1717–1723.
89. Lotufo, PA, Gaziano, JM, Chae, CU, et al. Diabetes and all-cause and coronary heart disease mortality among US male physicians. Arch Intern Med. 2001;161:242–247.
90. Evans, JM, Wang, J, Morris, AD. Comparison of cardiovascular risk between patients with type 2 diabetes and those who had had a myocardial infarction: cross sectional and cohort studies. BMJ. 2002;324:939–942.
91. Cho, E, Rimm, EB, Stampfer, MJ, et al. The impact of diabetes mellitus and prior myocardial infarction on mortality from all causes and from coronary heart disease in men. J Am Coll Cardiol. 2002;40:954–960.
92. Becker, A, Bos, G, de Vegt, F, et al. Cardiovascular events in type 2 diabetes: comparison with nondiabetic individuals without and with prior cardiovascular disease. 10-year follow-up of the Hoorn Study. Eur Heart J. 2003;24:1406–1413.
93. Natarajan, S, Liao, Y, Cao, G, et al. Sex differences in risk for coronary heart disease mortality associated with diabetes and established coronary heart disease. Arch Intern Med. 2003;163:1735–1740.
94. Lee, CD, Folsom, AR, Pankow, JS, et al. Cardiovascular events in diabetic and nondiabetic adults with or without history of myocardial infarction. Circulation. 2004;109:855–860.
95. Vaccaro, O, Eberly, LE, Neaton, JD, et al. Impact of diabetes and previous myocardial infarction on long-term survival: 25-year mortality follow-up of primary screenees of the Multiple Risk Factor Intervention Trial. Arch Intern Med. 2004;164:1438–1443.
96. Hu, G, Jousilahti, P, Qiao, Q, et al. Sex differences in cardiovascular and total mortality among diabetic and non-diabetic individuals with or without history of myocardial infarction. Diabetologia. 2005;48:856–861.
97. DeFaria Yeh, D, Freeman, MW, Meigs, JB, et al. Risk factors for coronary artery disease in patients with elevated high-density lipoprotein cholesterol. Am J Cardiol. 2007;99:1–4.
98. Leon, AS, Connett, J, Jacobs, DR, Jr., et al. Leisure-time physical activity levels and risk of coronary heart disease and death. The Multiple Risk Factor Intervention Trial. JAMA. 1987;258:2388–2395.
99. Paffenbarger, RS, Jr., Hyde, RT, Wing, AL, et al. The association of changes in physical-activity level and other lifestyle characteristics with mortality among men. N Engl J Med. 1993;328:538–545.
100. Powell, KE, Thompson, PD, Caspersen, CJ, et al. Physical activity and the incidence of coronary heart disease. Annu Rev Public Health. 1987;8:253–287.
101. Yusuf, S, Hawken, S, Ounpuu, S, et al. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): case-control study. Lancet. 2004;364:937–952.
102. Liu, J, Sempos, C, Donahue, RP, et al. Joint distribution of non-HDL and LDL cholesterol and coronary heart disease risk prediction among individuals with and without diabetes. Diabetes Care. 2005;28:1916–1921.
103. Howard, BV, Van Horn, L, Hsia, J, et al. Low-fat dietary pattern and risk of cardiovascular disease: the Women’s Health Initiative Randomized Controlled Dietary Modification Trial. JAMA. 2006;295:655–666.
104. Mensink, RP, Katan, MB. Effect of a diet enriched with monounsaturated or polyunsaturated fatty acids on levels of low-density and high-density lipoprotein cholesterol in healthy women and men. N Engl J Med. 1989;321:436–441.
105. Mensink, RP, Katan, MB. Effect of dietary fatty acids on serum lipids and lipoproteins. A meta-analysis of 27 trials. Arterioscler Thromb. 1992;12:911–919.
106. Ryan, M, McInerney, D, Owens, D, et al. Diabetes and the Mediterranean diet: a beneficial effect of oleic acid on insulin sensitivity, adipocyte glucose transport and endothelium-dependent vasoreactivity. QJM. 2000;93:85–91.
107. Schaefer, EJ, Lamon-Fava, S, Ausman, LM, et al. Individual variability in lipoprotein cholesterol response to National Cholesterol Education Program Step 2 diets. Am J Clin Nutr. 1997;65:823–830.
108. Foster, GD, Wyatt, HR, Hill, JO, et al. A randomized trial of a low-carbohydrate diet for obesity. N Engl J Med. 2003;348:2082–2090.
109. Stern, L, Iqbal, N, Seshadri, P, et al. The effects of low-carbohydrate versus conventional weight loss diets in severely obese adults: one-year follow-up of a randomized trial. Ann Intern Med. 2004;140:778–785.
110. Gardner, CD, Kiazand, A, Alhassan, S, et al. Comparison of the Atkins, Zone, Ornish, and LEARN diets for change in weight and related risk factors among overweight premenopausal women: the A TO Z Weight Loss Study: a randomized trial. JAMA. 2007;297:969–977.
111. McAuley, KA, Hopkins, CM, Smith, KJ, et al. Comparison of high-fat and high-protein diets with a high-carbohydrate diet in insulin-resistant obese women. Diabetologia. 2005;48:8–16.
112. Mayer-Davis, EJ, Sparks, KC, Hirst, K, et al. Dietary intake in the diabetes prevention program cohort: baseline and 1-year post randomization. Ann Epidemiol. 2004;14:763–772.
113. Wing, RR, Hamman, RF, Bray, GA, et al. Achieving weight and activity goals among diabetes prevention program lifestyle participants. Obes Res. 2004;12:1426–1434.
114. Knowler, WC, Barrett-Connor, E, Fowler, SE, et al. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med. 2002;346:393–403.
115. Ratner, R, Goldberg, R, Haffner, S, et al. Impact of intensive lifestyle and metformin therapy on cardiovascular disease risk factors in the diabetes prevention program. Diabetes Care. 2005;28:888–894.
116. Balagopal, P, Kamalamma, N, Patel, TG, et al. A community-based diabetes prevention and management education program in a rural village in India. Diabetes Care. 2008;31:1097–1104.
117. Seidel, MC, Powell, RO, Zgibor, JC, et al. Translating the Diabetes Prevention Program into an urban medically underserved community: a nonrandomized prospective intervention study. Diabetes Care. 2008;31:684–689.
118. Watts, GF, Lewis, B, Brunt, JN, et al. Effects on coronary artery disease of lipid-lowering diet, or diet plus cholestyramine, in the St Thomas’ Atherosclerosis Regression Study (STARS). Lancet. 1992;339:563–569.
119. Ornish, D, Brown, SE, Scherwitz, LW, et al. Can lifestyle changes reverse coronary heart disease? The Lifestyle Heart Trial. Lancet. 1990;336:129–133.
120. Ornish, D, Scherwitz, LW, Billings, JH, et al. Intensive lifestyle changes for reversal of coronary heart disease. JAMA. 1998;280:2001–2007.
121. de Lorgeril, M, Renaud, S, Mamelle, N, et al. Mediterranean alpha-linolenic acid-rich diet in secondary prevention of coronary heart disease. Lancet. 1994;343:1454–1459.
122. De Lorgeril, M, Salen, P, Martin, JL, et al. Effect of a Mediterranean type of diet on the rate of cardiovascular complications in patients with coronary artery disease. Insights into the cardioprotective effect of certain nutriments. J Am Coll Cardiol. 1996;28:1103–1108.
123. de Lorgeril, M, Salen, P, Martin, JL, et al. Mediterranean diet, traditional risk factors, and the rate of cardiovascular complications after myocardial infarction: final report of the Lyon Diet Heart Study. Circulation. 1999;99:779–785.
124. The Lipid Research Clinics Coronary Primary Prevention Trial results. II. The relationship of reduction in incidence of coronary heart disease to cholesterol lowering. JAMA. 1984;251:365–374.
125. The Lipid Research Clinics Coronary Primary Prevention Trial results. I. Reduction in incidence of coronary heart disease. JAMA. 1984;251:351–364.
126. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet. 1994;344:1383–1389.
127. Miettinen, TA, Pyorala, K, Olsson, AG, et al. Cholesterol-lowering therapy in women and elderly patients with myocardial infarction or angina pectoris: findings from the Scandinavian Simvastatin Survival Study (4S). Circulation. 1997;96:4211–4218.
128. Pedersen, TR, Olsson, AG, Faergeman, O, et al. Lipoprotein changes and reduction in the incidence of major coronary heart disease events in the Scandinavian Simvastatin Survival Study (4S). Circulation. 1998;97:1453–1460.
129. Pedersen, TR, Wilhelmsen, L, Faergeman, O, et al. Follow-up study of patients randomized in the Scandinavian simvastatin survival study (4S) of cholesterol lowering. Am J Cardiol. 2000;86:257–262.
130. Pyorala, K, Pedersen, TR, Kjekshus, J, et al. Cholesterol lowering with simvastatin improves prognosis of diabetic patients with coronary heart disease. A subgroup analysis of the Scandinavian Simvastatin Survival Study (4S). Diabetes Care. 1997;20:614–620.
131. Ballantyne, CM, Olsson, AG, Cook, TJ, et al. Influence of low high-density lipoprotein cholesterol and elevated triglyceride on coronary heart disease events and response to simvastatin therapy in 4S. Circulation. 2001;104:3046–3051.
132. Shepherd, J, Cobbe, SM, Ford, I, et al. Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. West of Scotland Coronary Prevention Study Group. N Engl J Med. 1995;333:1301–1307.
133. Downs, JR, Clearfield, M, Weis, S, et al. Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels: results of AFCAPS/TexCAPS. Air Force/Texas Coronary Atherosclerosis Prevention Study. JAMA. 1998;279:1615–1622.
134. Sacks, FM, Pfeffer, MA, Moye, LA, et al. The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. Cholesterol and Recurrent Events Trial investigators. N Engl J Med. 1996;335:1001–1009.
135. Sacks, FM, Moye, LA, Davis, BR, et al. Relationship between plasma LDL concentrations during treatment with pravastatin and recurrent coronary events in the Cholesterol and Recurrent Events trial. Circulation. 1998;97:1446–1452.
136. Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. The Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group. N Engl J Med. 1998;339:1349–1357.
137. Colquhoun, D, Keech, A, Hunt, D, et al. Effects of pravastatin on coronary events in 2073 patients with low levels of both low-density lipoprotein cholesterol and high-density lipoprotein cholesterol: results from the LIPID study. Eur Heart J. 2004;25:771–777.
138. Simes, RJ, Marschner, IC, Hunt, D, et al. Relationship between lipid levels and clinical outcomes in the Long-term Intervention with Pravastatin in Ischemic Disease (LIPID) Trial: to what extent is the reduction in coronary events with pravastatin explained by on-study lipid levels? Circulation. 2002;105:1162–1169.
139. Tonkin, AM, Colquhoun, D, Emberson, J, et al. Effects of pravastatin in 3260 patients with unstable angina: results from the LIPID study. Lancet. 2000;356:1871–1875.
140. LaRosa, JC, Grundy, SM, Waters, DD, et al. Intensive lipid lowering with atorvastatin in patients with stable coronary disease. N Engl J Med. 2005;352:1425–1435.
141. MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet. 2002;360:7–22.
142. Collins, R, Armitage, J, Parish, S, et al. MRC/BHF Heart Protection Study of cholesterol-lowering with simvastatin in 5963 people with diabetes: a randomised placebo-controlled trial. Lancet. 2003;361:2005–2016.
143. Collins, R, Armitage, J, Parish, S, et al. Effects of cholesterol-lowering with simvastatin on stroke and other major vascular events in 20536 people with cerebrovascular disease or other high-risk conditions. Lancet. 2004;363:757–767.
144. Knopp, RH, d’Emden, M, Smilde, JG, et al. Efficacy and safety of atorvastatin in the prevention of cardiovascular end points in subjects with type 2 diabetes: the Atorvastatin Study for Prevention of Coronary Heart Disease Endpoints in non-insulin-dependent diabetes mellitus (ASPEN). Diabetes Care. 2006;29:1478–1485.
145. Brown, G, Albers, JJ, Fisher, LD, et al. Regression of coronary artery disease as a result of intensive lipid-lowering therapy in men with high levels of apolipoprotein B. N Engl J Med. 1990;323:1289–1298.
146. de Groot, E, Jukema, JW, Montauban van Swijndregt, AD, et al. B-mode ultrasound assessment of pravastatin treatment effect on carotid and femoral artery walls and its correlations with coronary arteriographic findings: a report of the Regression Growth Evaluation Statin Study (REGRESS). J Am Coll Cardiol. 1998;31:1561–1567.
147. Jukema, JW, Bruschke, AV, van Boven, AJ, et al. Effects of lipid lowering by pravastatin on progression and regression of coronary artery disease in symptomatic men with normal to moderately elevated serum cholesterol levels. The Regression Growth Evaluation Statin Study (REGRESS). Circulation. 1995;91:2528–2540.
148. Nissen, SE, Tuzcu, EM, Schoenhagen, P, et al. Effect of intensive compared with moderate lipid-lowering therapy on progression of coronary atherosclerosis: a randomized controlled trial. JAMA. 2004;291:1071–1080.
149. Nissen, SE, Nicholls, SJ, Sipahi, I, et al. Effect of very high-intensity statin therapy on regression of coronary atherosclerosis: the ASTEROID trial. JAMA. 2006;295:1556–1565.
150. Cannon, CP, Braunwald, E, McCabe, CH, et al. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med. 2004;350:1495–1504.
151. Afilalo, J, Duque, G, Steele, R, et al. Statins for secondary prevention in elderly patients: a hierarchical bayesian meta-analysis. J Am Coll Cardiol. 2008;51:37–45.
152. Byington, RP, Jukema, JW, Salonen, JT, et al. Reduction in cardiovascular events during pravastatin therapy. Pooled analysis of clinical events of the Pravastatin Atherosclerosis Intervention Program. Circulation. 1995;92:2419–2425.
153. Hebert, PR, Gaziano, JM, Chan, KS, et al. Cholesterol lowering with statin drugs, risk of stroke, and total mortality. An overview of randomized trials. JAMA. 1997;278:313–321.
154. LaRosa, JC, He, J, Vupputuri, S. Effect of statins on risk of coronary disease: a meta-analysis of randomized controlled trials. JAMA. 1999;282:2340–2346.
155. Marchioli, R, Marfisi, RM, Carinci, F, et al. Meta-analysis, clinical trials, and transferability of research results into practice. The case of cholesterol-lowering interventions in the secondary prevention of coronary heart disease. Arch Intern Med. 1996;156:1158–1172.
156. Thavendiranathan, P, Bagai, A, Brookhart, MA, et al. Primary prevention of cardiovascular diseases with statin therapy: a meta-analysis of randomized controlled trials. Arch Intern Med. 2006;166:2307–2313.
157. Baigent, C, Keech, A, Kearney, PM, et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet. 2005;366:1267–1278.
158. Buchwald, H, Varco, RL, Matts, JP, et al. Effect of partial ileal bypass surgery on mortality and morbidity from coronary heart disease in patients with hypercholesterolemia. Report of the Program on the Surgical Control of the Hyperlipidemias (POSCH). N Engl J Med. 1990;323:946–955.
159. Sachais, BS, Katz, J, Ross, J, et al. Long-term effects of LDL apheresis in patients with severe hypercholesterolemia. J Clin Apher. 2005;20:252–255.
160. Frick, MH, Elo, O, Haapa, K, et al. Helsinki Heart Study: primary-prevention trial with gemfibrozil in middle-aged men with dyslipidemia. Safety of treatment, changes in risk factors, and incidence of coronary heart disease. N Engl J Med. 1987;317:1237–1245.
161. Rubins, HB, Robins, SJ, Collins, D, et al. Gemfibrozil for the secondary prevention of coronary heart disease in men with low levels of high-density lipoprotein cholesterol. Veterans Affairs High-Density Lipoprotein Cholesterol Intervention Trial Study Group. N Engl J Med. 1999;341:410–418.
162. Berge, KG, Canner, PL. Coronary drug project: experience with niacin. Coronary Drug Project Research Group. Eur J Clin Pharmacol. 1991;40(Suppl 1):S49–51.
163. Canner, PL, Berge, KG, Wenger, NK, et al. Fifteen year mortality in coronary drug project patients: long-term benefit with niacin. J Am Coll Cardiol. 1986;8:1245–1255.
164. Stamler, J. The coronary drug project–findings with regard to estrogen, dextrothyroxine, clofibrate and niacin. Adv Exp Med Biol. 1977;82:52–75.
165. Secondary prevention by raising HDL cholesterol and reducing triglycerides in patients with coronary artery disease: the Bezafibrate Infarction Prevention (BIP) study. Circulation. 2000;102:21–27.
166. Barter, PJ, Rye, KA. Is there a role for fibrates in the management of dyslipidemia in the metabolic syndrome? Arterioscler Thromb Vasc Biol. 2008;28:39–46.
167. WHO cooperative trial on primary prevention of ischaemic heart disease with clofibrate to lower serum cholesterol: final mortality follow-up. Report of the Committee of Principal Investigators. Lancet. 1984;2:600–604.
168. Tenkanen, L, Manttari, M, Manninen, V. Some coronary risk factors related to the insulin resistance syndrome and treatment with gemfibrozil. Experience from the Helsinki Heart Study. Circulation. 1995;92:1779–1785.
169. Manninen, V, Tenkanen, L, Koskinen, P, et al. Joint effects of serum triglyceride and LDL cholesterol and HDL cholesterol concentrations on coronary heart disease risk in the Helsinki Heart Study. Implications for treatment. Circulation. 1992;85:37–45.
170. Robins, SJ, Collins, D, Wittes, JT, et al. Relation of gemfibrozil treatment and lipid levels with major coronary events: VA-HIT: a randomized controlled trial. JAMA. 2001;285:1585–1591.
171. Davidson, MH. Statin/fibrate combination in patients with metabolic syndrome or diabetes: evaluating the risks of pharmacokinetic drug interactions. Expert Opin Drug Saf. 2006;5:145–156.
172. Keech, A, Simes, RJ, Barter, P, et al. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): randomised controlled trial. Lancet. 2005;366:1849–1861.
173. Brunzell, JD, Davidson, M, Furberg, CD, et al. Lipoprotein management in patients with cardiometabolic risk: consensus statement from the American Diabetes Association and the American College of Cardiology Foundation. Diabetes Care. 2008;31:811–822.
174. Clofibrate and niacin in coronary heart disease. JAMA. 1975;231:360–381.
175. Brown, BG, Zhao, XQ, Chait, A, et al. Simvastatin and niacin, antioxidant vitamins, or the combination for the prevention of coronary disease. N Engl J Med. 2001;345:1583–1592.
176. Taylor, AJ, Sullenberger, LE, Lee, HJ, et al. Arterial Biology for the Investigation of the Treatment Effects of Reducing Cholesterol (ARBITER) 2: a double-blind, placebo-controlled study of extended-release niacin on atherosclerosis progression in secondary prevention patients treated with statins. Circulation. 2004;110:3512–3517.
177. Cashin-Hemphill, L, Mack, WJ, Pogoda, JM, et al. Beneficial effects of colestipol-niacin on coronary atherosclerosis. A 4-year follow-up. JAMA. 1990;264:3013–3017.
178. Kastelein, JJ, Akdim, F, Stroes, ES, et al. Simvastatin with or without ezetimibe in familial hypercholesterolemia. N Engl J Med. 2008;358:1431–1443.
179. Biscione, F, Pignalberi, C, Totteri, A, et al. Cardiovascular effects of omega-3 free fatty acids. Curr Vasc Pharmacol. 2007;5:163–172.
180. Ebbesson, SO, Roman, MJ, Devereux, RB, et al. Consumption of omega-3 fatty acids is not associated with a reduction in carotid atherosclerosis: the Genetics of Coronary Artery Disease in Alaska Natives study. Atherosclerosis. 2008;199:346–353.
181. von Schacky, C, Angerer, P, Kothny, W, et al. The effect of dietary omega-3 fatty acids on coronary atherosclerosis. A randomized, double-blind, placebo-controlled trial. Ann Intern Med. 1999;130:554–562.
182. Kamanna, VS, Kashyap, ML. Mechanism of action of niacin. Am J Cardiol. 2008;101:20B–26B.
183. Tunaru, S, Kero, J, Schaub, A, et al. PUMA-G and HM74 are receptors for nicotinic acid and mediate its anti-lipolytic effect. Nat Med. 2003;9:352–355.
184. Benyo, Z, Gille, A, Kero, J, et al. GPR109A (PUMA-G/HM74A) mediates nicotinic acid-induced flushing. J Clin Invest. 2005;115:3634–3640.
185. Grundy, SM, Vega, GL, McGovern, ME, et al. Efficacy, safety, and tolerability of once-daily niacin for the treatment of dyslipidemia associated with type 2 diabetes: results of the assessment of diabetes control and evaluation of the efficacy of Niaspan trial. Arch Intern Med. 2002;162:1568–1576.
186. Elam, MB, Hunninghake, DB, Davis, KB, et al. Effect of niacin on lipid and lipoprotein levels and glycemic control in patients with diabetes and peripheral arterial disease: the ADMIT study: A randomized trial. Arterial Disease Multiple Intervention Trial. JAMA. 2000;284:1263–1270.
187. Meyers, CD, Carr, MC, Park, S, et al. Varying cost and free nicotinic acid content in over-the-counter niacin preparations for dyslipidemia. Ann Intern Med. 2003;139:996–1002.
188. Davidson, WS, Thompson, TB. The structure of apolipoprotein A-I in high density lipoproteins. J Biol Chem. 2007;282:22249–22253.
189. Martin-Campos, JM, Escola-Gil, JC, Ribas, V, et al. Apolipoprotein A-II, genetic variation on chromosome 1q21-q24, and disease susceptibility. Curr Opin Lipidol. 2004;15:247–253.
190. Stan, S, Delvin, E, Lambert, M, et al. Apo A-IV: an update on regulation and physiologic functions. Biochim Biophys Acta. 2003;1631:177–187.
191. Calandra, S, Priore Oliva, C, Tarugi, P, et al. APOA5 and triglyceride metabolism, lesson from human APOA5 deficiency. Curr Opin Lipidol. 2006;17:122–127.
192. Olofsson, SO, Boren, J. Apolipoprotein B: a clinically important apolipoprotein which assembles atherogenic lipoproteins and promotes the development of atherosclerosis. J Intern Med. 2005;258:395–410.
193. Gautier, T, Tietge, UJ, Boverhof, R, et al. Hepatic lipid accumulation in apolipoprotein C-I-deficient mice is potentiated by cholesteryl ester transfer protein. J Lipid Res. 2007;48:30–40.
194. Pulawa, LK, Jensen, DR, Coates, A, et al. Reduction of plasma triglycerides in apolipoprotein C-II transgenic mice overexpressing lipoprotein lipase in muscle. J Lipid Res. 2007;48:145–151.
195. Ooi, EM, Barrett, PH, Chan, DC, et al. Apolipoprotein C-III: understanding an emerging cardiovascular risk factor. Clin Sci (Lond). 2008;114:611–624.
196. Rassart, E, Bedirian, A, Do Carmo, S, et al. Apolipoprotein D. Biochim Biophys Acta. 2000;1482:185–198.
197. Hatters, DM, Peters-Libeu, CA, Weisgraber, KH. Apolipoprotein E structure: insights into function. Trends Biochem Sci. 2006;31:445–454.
198. Trougakos, IP, Gonos, ES. Regulation of clusterin/apolipoprotein J, a functional homologue to the small heat shock proteins, by oxidative stress in ageing and age-related diseases. Free Radic Res. 2006;40:1324–1334.
199. Ahnstrom, J, Axler, O, Jauhiainen, M, et al. Levels of apolipoprotein M are not associated with the risk of coronary heart disease in two independent case-control studies. J Lipid Res. 2008;49:1912–1917.
200. Fitzgerald, ML, Xavier, R, Haley, KJ, et al. ABCA3 inactivation in mice causes respiratory failure, loss of pulmonary surfactant, and depletion of lung phosphatidylglycerol. J Lipid Res. 2007;48:621–632.
201. Molday, RS. ATP-binding cassette transporter ABCA4: molecular properties and role in vision and macular degeneration. J Bioenerg Biomembr. 2007;39:507–517.
202. Baldan, A, Gomes, AV, Ping, P, et al. Loss of ABCG1 results in chronic pulmonary inflammation. J Immunol. 2008;180:3560–3568.
203. Kidambi, S, Patel, SB. Sitosterolaemia: pathophysiology, clinical presentation and laboratory diagnosis. J Clin Pathol. 2008;61:588–594.
204. May, P, Herz, J, Bock, HH. Molecular mechanisms of lipoprotein receptor signalling. Cell Mol Life Sci. 2005;62:2325–2338.
205. Rac, ME, Safranow, K, Poncyljusz, W. Molecular basis of human CD36 gene mutations. Mol Med. 2007;13:288–296.
206. Mehta, JL, Sanada, N, Hu, CP, et al. Deletion of LOX-1 reduces atherogenesis in LDLR knockout mice fed high cholesterol diet. Circ Res. 2007;100:1634–1642.
207. Lillis, AP, Van Duyn, LB, Murphy-Ullrich, JE, et al. LDL receptor-related protein 1: unique tissue-specific functions revealed by selective gene knockout studies. Physiol Rev. 2008;88:887–918.
208. Fisher, CE, Howie, SE. The role of megalin (LRP-2/Gp330) during development. Dev Biol. 2006;296:279–297.
209. Smolenaars, MM, Madsen, O, Rodenburg, KW, et al. Molecular diversity and evolution of the large lipid transfer protein superfamily. J Lipid Res. 2007;48:489–502.
210. Chang, TY, Reid, PC, Sugii, S, et al. Niemann-Pick type C disease and intracellular cholesterol trafficking. J Biol Chem. 2005;280:20917–20920.
211. Yu, L. The structure and function of Niemann-Pick C1-like 1 protein. Curr Opin Lipidol. 2008;19:263–269.
212. Samyn, H, Moerland, M, van Gent, T, et al. Plasma phospholipid transfer activity is essential for increased atherogenesis in PLTP transgenic mice: a mutation-inactivation study. J Lipid Res. 2008.
213. Moore, KJ, Freeman, MW. Scavenger receptors in atherosclerosis: beyond lipid uptake. Arterioscler Thromb Vasc Biol. 2006;26:1702–1711.
214. Trigatti, B, Covey, S, Rizvi, A. Scavenger receptor class B type I in high-density lipoprotein metabolism, atherosclerosis and heart disease: lessons from gene-targeted mice. Biochem Soc Trans. 2004;32:116–120.
215. Lohse, P, Maas, S, Sewell, AC, et al. Molecular defects underlying Wolman disease appear to be more heterogeneous than those resulting in cholesteryl ester storage disease. J Lipid Res. 1999;40:221–228.
216. Russell, DW. The enzymes, regulation, and genetics of bile acid synthesis. Annu Rev Biochem. 2003;72:137–174.
217. Yen, CL, Stone, SJ, Koliwad, S, et al. DGAT enzymes and triacylglycerol biosynthesis. J Lipid Res. 2008.
218. Lamarche, B, Paradis, ME. Endothelial lipase and the metabolic syndrome. Curr Opin Lipidol. 2007;18:298–303.
219. Santamarina-Fojo, S, Gonzalez-Navarro, H, Freeman, L, et al. Hepatic lipase, lipoprotein metabolism, and atherogenesis. Arterioscler Thromb Vasc Biol. 2004;24:1750–1754.
220. DeBose-Boyd, RA. Feedback regulation of cholesterol synthesis: sterol-accelerated ubiquitination and degradation of HMG CoA reductase. Cell Res. 2008;18:609–621.
221. Yeaman, SJ. Hormone-sensitive lipase–new roles for an old enzyme. Biochem J. 2004;379:11–22.
222. Peelman, F, Vandekerckhove, J, Rosseneu, M. Structure and function of lecithin cholesterol acyl transferase: new insights from structural predictions and animal models. Curr Opin Lipidol. 2000;11:155–160.
223. Otarod, JK, Goldberg, IJ. Lipoprotein lipase and its role in regulation of plasma lipoproteins and cardiac risk. Curr Atheroscler Rep. 2004;6:335–342.
[/level-membership-for-endocrinology-diabetes-and-metabolism-category][not-level-membership-for-endocrinology-diabetes-and-metabolism-category]Chapter 16
Lipoprotein Metabolism and the Treatment of Lipid Disorders
Atherosclerosis is a chronic inflammatory disease of large and medium-sized arteries that results from the deposition of lipids and lipoproteins in the intima of these vessels.1,2 This process is a major cause of morbidity and mortality in the developed world, and the toll is particularly high in the diabetic population, accounting for approximately two-thirds to three-quarters of all diabetic deaths.3 When combined with significant morbidity arising from ischemic injury and loss of limb perfusion, the impact of atherosclerosis on human health is enormous.5 Intensive lipid-lowering therapy has demonstrated remarkable therapeutic value in the treatment and prevention of coronary heart disease (CHD) in patients with and without diabetes. This chapter will review basic principles of lipoprotein metabolism, focusing on the impact diabetes has on these lipid pathways. Data that address the value of lipid-altering treatments in reducing the burden of atherosclerosis in patients with and without diabetes will be presented. Finally, an approach to the management of patients with dyslipidemia will be outlined.
Lipoprotein Metabolism
The clinically reported serum triglyceride and cholesterol values represent the sum of the cholesterol and triglyceride contents of the circulating lipoproteins present in a volume of blood (expressed either as mg/dL or mmol). While knowledge of these total lipid values is very helpful in assessing cardiovascular risk, it is the distribution of cholesterol among the different lipoprotein fractions that is critical to any sophisticated understanding of atherosclerosis risk. Lipoproteins are structured to solve the problem of transporting highly hydrophobic lipids, such as cholesterol ester and triglyceride, in the aqueous environment of the blood. Since the bulk of lipoproteins are synthesized in the liver or gut, they must be moved via the bloodstream from these sites of synthesis to tissues where lipids will be taken up and utilized. By complexing neutral lipids with both proteins and more polar lipids such as unesterified cholesterol and phospholipids, the lipoprotein structure accomplishes this task. The more hydrophobic lipids are sequestered on the inside of the spherical lipoprotein, whereas more polar lipids and proteins decorate the outer surface (Fig. 16-1). In addition to solving the biophysical transport problem, the lipoprotein structure also enables particles to utilize a class of proteins known as apoproteins (Table 16-1) that serve as targeting molecules which interact with lipid-modifying enzymes (Table 16-2) or receptors and transporters (Table 16-3) critical for the normal functioning of the disparate lipoprotein classes. Mutations in the genes encoding apoproteins or the enzymes and receptors with which apoproteins interact (see Tables 16-1 to 16-3) are rare, but they can have a profound influence on the circulating levels of lipoproteins in individuals with and without diabetes. Because lipids play fundamental roles in a host of other cellular processes, mutations in lipid enzymes, receptors, transporters, and apoproteins can have profound effects on human biology beyond the serum lipoproteins, some of which are cited in Tables 16-1 to 16-3.
Table 16-3
FIGURE 16-1 The upper panel of the figure lists the density range that encompasses each lipoprotein class and the major tissue in which the lipoprotein is synthesized. The bottom panel is a graphic representation of a generic lipoprotein, emphasizing the spherical shape of the particle, the use of amphipathic apoproteins and polar lipids on the particle surface that interact with cells and the blood environment, and the sequestration of nonpolar lipids in the core of the lipoprotein. All lipoproteins share these general features, but they differ in size, lipid composition, and in the specific apoproteins embedded.
Although lipoproteins constitute a continuous spectrum of density within the plasma, they are commonly divided into five major classes (see Fig. 16-1). These are: (1) chylomicrons; (2) very low-density lipoproteins (VLDLs); (3) intermediate-density lipoproteins (IDLs); (4) low-density lipoproteins (LDLs); and (5) high-density lipoproteins (HDLs). Within these classes, further subdivision of lipoproteins can be made. In diabetics, a higher proportion of LDLs is carried in a denser, smaller-diameter subfraction that is called small dense LDL (sdLDL). HDL are commonly divided into HDL2 and HDL3 subclasses, but some investigators have adopted a gel separation method to segregate HDL into a much larger number of subtypes.6 It is important for the clinician to understand that the different laboratory methods of identifying subpopulations of lipoproteins yield different or overlapping species of particles, causing confusion for those not conversant with the separation techniques. In this chapter, the most widely used classifications will be employed for simplicity, but further research may prove that more sophisticated subclassifications have considerable clinical relevance. Overall, it is helpful to remember that the least dense of the lipoproteins are also the largest in size and the most triglyceride-rich. These are the chylomicrons and VLDLs. IDLs contain substantial amounts of both triglyceride and cholesterol, whereas LDLs and HDLs are predominantly cholesterol-carrying particles with very little associated triglyceride. This information permits the clinician to identify the most likely lipoprotein abnormalities when presented with a patient’s total serum triglyceride and cholesterol values. A brief description of each of the five major lipoprotein classes is summarized in the following paragraphs. A reader interested in a more comprehensive introduction to lipoprotein metabolism should consult the lipid sections of the Metabolic Basis of Inherited Disease.7
Chylomicrons
Chylomicrons derive from dietary fat and carry triglycerides throughout the body. The major structural protein of chylomicrons is apolipoprotein (apo) B48, a protein that is produced from apo B100 RNA by a unique editing process employed by gut cells that generates a protein that is 48% of the length of apo B100.8 Chylomicrons also contain other apoproteins, including members of the apo C and apo A family. These proteins can redistribute to other lipoprotein classes within serum as chylomicrons transfer their lipid content to cells and interacting lipoproteins. Chylomicrons have the lowest density of all lipoproteins and will float to the top of a plasma specimen left in a refrigerator overnight, forming a creamlike layer. Owing to its large size, the chylomicron does not readily enter the artery wall and is therefore thought not to be atherogenic, but the atherosclerotic role of the triglyceride-depleted chylomicron remnant remains controversial. Triglyceride makes up most of the chylomicron and is removed by the action of an enzyme that is bound to the surface of endothelial cells, lipoprotein lipase (LPL). Patients deficient in this enzyme or its apoprotein cofactor (apo CII) have very high serum triglyceride levels and are at increased risk for developing acute pancreatitis. Inasmuch as insulin is also a critical cofactor for LPL activity, the vast majority of patients presenting with hypertriglyceridemic pancreatitis have poorly controlled diabetes as the cause of their delayed chylomicron clearance.
Low-Density Lipoproteins
LDLs are the major carriers of cholesterol in humans, responsible for supplying cholesterol to tissues with the highest sterol demands. LDLs are also the lipoproteins most clearly implicated in causing atherogenic plaque formation.9 Circulating LDL levels can be increased in persons who consume large amounts of saturated fat and/or cholesterol.10 LDL levels are also elevated in those who have genetic defects that affect LDL receptor function (familial hypercholesterolemia, PCSK9 mutations, autosomal recessive hypercholesterolemia), the structure of LDL’s apoprotein, apo B, or who have polygenic disorders affecting LDL metabolism.11 Circulating LDLs are taken up by the endothelium lining the artery wall, traverse it, and are deposited and trapped in the arterial intima. There, they may undergo oxidation or other biochemical modification, be taken up by macrophages, and stimulate atherogenesis. The association of total serum cholesterol with CHD is predominantly a reflection of the role of LDL. Studies in patients with diabetes have produced somewhat conflicting data on the prevalence of increased LDL levels, as defined by general population norms, but the preponderance of the evidence indicates that diabetics have LDL levels that are similar to those seen in well-matched, nondiabetic control populations. Given diabetics’ greater risk of developing coronary artery disease (CAD), however, the National Cholesterol Education Program guidelines now define LDL levels above 100 mg/dL as being undesirable in diabetics. When judged by this stringent criterion, elevated LDL levels are extremely common in patients with diabetes. In addition, the LDL in diabetics is frequently smaller, denser, and more readily oxidized—properties that are associated with a higher degree of atherogenicity.12,13
High-Density Lipoproteins
HDLs are the smallest of the lipoproteins; despite this, they carry a variety of apoproteins, including members of the apo A and C family. HDLs, unlike the other lipoproteins, are believed to function in humans primarily to return lipids from peripheral tissues to the liver and gut for excretion, rather than moving lipids from the gut organs to the periphery.14 This perspective on HDL function is an oversimplification, since cell culture experiments indicate that HDL can donate lipids as well as pick them up. It has proven to be quite difficult to quantitate the net movement of cholesterol carried in HDL in and out of peripheral tissues in whole animals. Further complicating the issue is the fact that many of the standard laboratory animals used in metabolic studies carry most of their serum cholesterol in HDL rather than LDL, diminishing the relevance of their lipoprotein metabolism to that of the LDL-rich human. Nevertheless, a considerable body of evidence suggests that HDL functions to protect tissues from unwanted accumulation of cholesterol. The mechanisms by which HDL can protect against atherosclerosis include its involvement in the reverse cholesterol transport pathway described earlier, as well as through contributions arising from its carriage of proteins that appear to have antiinflammatory and antioxidant properties.15,16 The unesterified cholesterol from tissues that is transferred to HDL is esterified by the action of lecithin cholesterol acyltransferase (LCAT) and stored in the central core of HDL. This esterified cholesterol can be transferred back to lower-density lipoproteins by the action of cholesterol ester transfer protein, or it may be removed at the liver by the action of a plasma membrane receptor called scavenger receptor B-1 (SRB-1). A particularly effective reverse transport system is thought to explain, at least in part, the association of elevated HDL cholesterol levels with a reduced risk of developing CHD. Apolipoprotein AI (apo AI) is the major apoprotein of HDL, and its serum concentration also correlates inversely with the risk of CHD. Women have higher levels of HDL cholesterol than men, and this may partly explain the lower incidence of CHD in premenopausal women. Exercise increases HDL levels; obesity, hypertriglyceridemia, and smoking lower them. Patients with diabetes, having higher body mass indices and hypertriglyceridemia, typically have lower HDL cholesterol levels than do nondiabetics, contributing further to the atherogenic lipid profile of the diabetic.
Metabolism of Lipids and Lipoproteins
Following ingestion of a meal containing fat, the triacylglycerols (TAG) are broken down by pancreatic triacylglycerol lipase to yield sn-2-monoacylglycerol and fatty acids. Ingested cholesterol is solubilized by bile salts into micellar structures, a step which is disrupted by bile acid resin binders. The lipids then transverse the enterocyte brush border membrane and are reassembled in the intestinal cell into a triglyceride-rich lipoprotein, the chylomicron. The role of transport proteins in the passage across the enterocyte membrane remains disputed, with evidence for both a passive diffusion and an active transport mechanism.17,18 For cholesterol, a second transport step translocates the sterol into intracellular pools that can be accessed for lipoprotein packaging.19 Niemann-Pick C1-like 1 protein (NPC1L1) appears to be required for this second step to occur and is the target of the cholesterol absorption inhibitor, ezetimibe.20,21 The internalized cholesterol is moved to the endoplasmic reticulum where the monoacylglycerol and fatty acids are also carried by transport proteins. The latter are recombined into triacylglycerol and bound by microsomal triglyceride transport protein (MTP). A second pathway of triglyceride synthesis uses acylation of glycerol-3-phosphate to phosphatidic acid, dephosphorylation to diacylglycerol (DAG), and then acylation of DAG to TAG. A high-density, protein-rich particle synthesized in the enterocyte, containing apo B48 and apo AIV, is then packaged with the absorbed lipids to generate the chylomicron.22 This is then transported by vesicles to the basolateral membrane for exocytosis into the mesenteric lymph. The lipoproteins in the lymph enter the blood circulation via discharge from the thoracic duct.
Two ABCG half transporters, ABCG5 and G8, contribute to net cholesterol absorption in the gut through their ability to resecrete sterol that has been absorbed by the enterocyte back into the gut lumen, thereby reducing net sterol uptake.23 The total amount of diet-derived TAG that is delivered to the circulation varies and is influenced by multiple factors, including the fat content of the diet, the amount of phosphatidylcholine in the intestinal lumen, and the expression level of apo AIV in the enterocyte. Overall, humans have a remarkable ability to absorb large quantities of both cholesterol and fat; 35% to 60% of ingested cholesterol is absorbed, with the amount inversely correlated with hepatic cholesterol synthesis. Up to 600 g of fat is absorbed with 95% efficiency. In individuals with normal lipid metabolism, the ingested fat is cleared from the enterocyte in less than 14 hours, forming the basis for drawing serum lipids after a fast of this duration. Circulating chylomicrons are subsequently cleared from the plasma by the action of endothelial-bound lipoprotein lipase, which cleaves the TAG, enabling the liberated glycerol and free fatty acids to move into the adjacent adipose or muscle tissue. The resulting chylomicron remnant particle can be further metabolized by lipases and appears to be retained in the hepatic space of Disse, where it can bind to heparan sulfate proteoglycans and acquire additional apo E. The acquisition of the apo E enables the remnant to be cleared efficiently by either the LRP1 or LDL receptor, but patients with diabetes have accumulation of remnants and slower hepatic clearance.24,25
The other major triglyceride-rich lipoprotein, VLDL, is synthesized in the liver and contains apo B100 rather than apo B48 as its major structural protein.26 Like chylomicrons, VLDLs rely on MTP to combine lipids with the apo B protein to produce a nascent, lipid-poor lipoprotein.27,28 This VLDL can be secreted from the liver or further lipidated to produce a mature VLDL. Individuals lacking MTP activity develop abetalipoproteinemia, a disorder characterized by erythrocyte acanthocytosis, anemia, steatorrhea, spinocerebellar degeneration, and fat-soluble vitamin deficiency. Acquisition of lipid by the mature VLDL appears to depend on triglycerides that are stored in the cytosol. Increased fatty acid delivery to the liver, from dietary sources or that released by lipase activity in peripheral tissues, stimulates VLDL production. After secretion of the mature VLDL from the liver, its triglycerides are initially removed by the action of LPL, and the resulting intermediate-density lipoprotein (IDL) can be further catabolized by hepatic lipase to produce mature, cholesterol-rich LDL.
Low-Density Lipoprotein Metabolism
There is still some dispute about whether mature LDL can be secreted from the liver rather than always being derived from the progressive removal of triglyceride from VLDL/IDL precursors.29 The preponderance of data indicate that little if any LDL is directly secreted from the liver. Each LDL particle contains a single apo B molecule, which dictates that individuals heterozygous for apo B mutants will have two populations of LDL, one which carries a wild-type protein and one with a mutant protein. LDLs are utilized throughout the body as a source of exogenous cholesterol and are taken up by LDL receptors present at the plasma membrane. While LDL receptors are ubiquitous, the majority are expressed by the liver. The LDL receptor binds to both apo E– and apo B–containing lipoproteins. Because the affinity of the receptor is higher for apo E than for apo B, E-containing lipoproteins, such as IDL, can compete with LDL for uptake by the LDL receptor. However, there are other receptors that can bind apo E–containing lipoproteins, permitting their clearance but not that of LDL when LDL receptor activity is lost or diminished. Thus, mutations in the LDL receptor result in elevated LDL levels in the plasma, causing the autosomal co-dominant disorder, familial hypercholesterolemia, which is among the most common of Mendelian genetic disorders (see Genetics section later).30 The elucidation of the cholesterol homeostatic mechanisms controlled by the LDL receptor constituted one of the major advances in our understanding of human physiology and disease.31 The internalization of LDL by the LDL receptor leads to trafficking of the internalized lipoprotein to the endosome, where after fusion with lysosomes, the sterol in the lipoprotein is released and shuttled to the endoplasmic reticulum (ER). A sterol-sensing system in the ER regulates the movement of a membrane-bound transcription factor, sterol response element binding protein (SREBP), from the ER to the nucleus.32–37 SREBP works in concert with other transcriptional regulators to influence the expression of many of the cholesterol synthesis, metabolizing, and transport molecules, particularly in the liver. This elegant homeostatic system enables cells to reduce their de novo cholesterol synthesis when adequate sterol supplies are present and to increase synthesis and LDL receptor expression when cellular demands require more cholesterol.
High-Density Lipoprotein Metabolism
Although the relationship between elevated levels of HDL and lower rates of cardiovascular disease was recognized more than 50 years ago, the mechanism by which HDL protects against CHD is still only partially understood. HDL are initially produced as lipid-poor phospholipid discs containing apolipoprotein AI. Following interaction with the phospholipid/cholesterol transporter, apo AI, the particle acquires unesterified cholesterol from cellular stores.38 For macrophages in particular, this cholesterol efflux pathway appears to be critical to their ability to unload the excess cholesterol they acquire by phagocytosis of dead cells and by their unregulated uptake of lipoproteins by scavenger receptor pathways.14 Cholesterol is stored in an intracellular lipid pool, having been esterified at its 3′OH position by the activity of acyl coenzyme A (CoA):cholesterol O-acyltransferase (ACAT).39 A neutral cholesterol hydrolase cleaves the fatty acid from cholesterol ester to convert it back to free (unesterified) cholesterol when cellular signals call for the use or export of the sterol.40 This free cholesterol can be shuttled to the plasma membrane, where it can be transported by the action of ABCA1. The interaction of lipid-poor nascent HDL with ABCA1 appears to involve direct binding of apo AI to the transporter, but the actual lipid transfer process is poorly understood.41–43 Once the unesterified cholesterol transfers to the nascent HDL, it can be reesterified by the action of lecithin cholesterol acyltransferase (LCAT) and centralized in the lipid core of the growing HDL particle. This HDL particle can now interact with a second ABC transporter, ABCG1, and additional cholesterol can be acquired.44 The role of ABCA1 is well established, since loss of its activity leads to Tangier disease (see Genetics section) and the absence or near absence of HDL in the plasma. ABCG1’s precise function is still being elucidated. Once the HDL has acquired sufficient cholesterol and esterified it, it can donate this lipid back to lower-density lipoproteins in the plasma, such as VLDL, through the activity of cholesterol ester transfer protein (CETP). Alternatively, HDL can interact with a cellular receptor, SR-BI, to selectively transfer the cholesterol ester to cells, including the liver, where it could excreted in the bile as cholesterol or one of its bile acid derivatives.45 The protein component of HDL is cleared via the kidney, a process that appears to depend on the HDL being lipid depleted. The pathway described, moving cholesterol from cellular storage pools to HDL and ultimately to the liver, is termed the reverse cholesterol transport pathway. The activity of this pathway is postulated to explain at least in part the link between HDL levels and lower CHD risk. However, HDL also carries antioxidants and a host of proteins linked to complement and inflammation pathways that suggest it may be playing a more general role as a scavenger of molecules that stimulate inflammation. The complexity of its function has made targeting increases in HDL levels as a therapeutic objective challenging. Both animal and human data have established that not all methods of increasing HDL result in improvements in atherosclerosis.
Disorders of Lipid Metabolism in Patients With Diabetes
Data dating back at least 30 years have documented that individuals with type 1 diabetes have increased rates of CHD.46–48 This risk has been estimated to be 10-fold or higher over that seen in age-matched controls without diabetes. In the Diabetes U.K. cohort of 23,751 subjects who were diagnosed with insulin-dependent diabetes prior to the age of 30, for example, those individuals 20 to 29 years of age had a standardized mortality ratio for ischemic heart disease of 11.8 for men and 44.8 for women. For those 30 to 39 years of age, the ratios were 8.0 and 41.6 for men and women, respectively.49 Despite the long and consistent reproducibility of this risk association, the pathophysiology underlying that relationship is still poorly understood. Unlike type 2 diabetes, where obesity and insulin resistance lead to changes in the standard lipid profile that are considered atherogenic, the type 1 diabetic who has not developed nephropathy and whose glucose is well controlled will typically have a normal serum lipid profile.50 In the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Study (DCCT/EDIC), the mean LDL-C in the intensively treated group was 111 ± 29 mg/dL in women and 119 ± 31 in men; HDL-C levels averaged 63 ± 16 and 51 ± 14 for women and men, respectively. Serum triglyceride levels were also in the normal range, with women averaging 76 ± 37 mg/dL, and male values running moderately higher at 98 ± 67 mg/dL. These lipid values did not differ significantly from those of the patients enrolled in the conventional treatment group of the study. These findings make clear that the standard lipid profile provides few insights into the increased risk of CHD in type 1 diabetics.
More sophisticated assessments of serum lipoproteins, using nuclear magnetic resonance (NMR), can reveal differences in lipoprotein sizes that distinguish diabetic lipids from those of nondiabetics. A study of 194 diabetics from London, all diagnosed with type 1 diabetes before the age of 25 and not receiving any renal replacement therapies, demonstrated that LDL size and subclass distribution were similar between the men in both groups.51 In contrast, female diabetics had fewer large and more small LDL, producing an overall reduction in LDL size. However, both men and women diabetics had more large and fewer small HDL than did the nondiabetic cohort, yielding an increased overall HDL size. Whereas the LDL size change in women would be considered proatherosclerotic, the HDL differences are thought to be antiatherogenic. These findings led the authors to conclude that the pathogenic significance of the lipoprotein size differences was uncertain.51 The results of the London study appear to differ from those reported in a similar analysis done in the DCCT cohort, although the comparators used in the two studies make direct comparison of the two reports challenging. In the DCCT analysis, a nondiabetic control group was not included, so the authors compared the NMR analysis of lipoproteins in men to those in women. This approach found higher LDL particle concentrations and more small-sized particles in the men. HDL sized decreased, and fewer large HDL were also found in the men. Although the authors stated that both genders had abnormal levels of several of the NMR-classed lipoproteins, these judgments were based on National Cholesterol Education Program (NCEP) optimal levels rather than population normative values, so it is not clear that matched, nondiabetic cohorts would have differed greatly.50
Surprisingly, epidemiologic risk-factor assessment of the correlates of CHD in type 1 diabetes shows a weak association to the one metabolic risk that predominates in the type 1 population: hyperglycemia.52,53 It was therefore somewhat unexpected when a 17-year follow-up of the DCCT cohorts showed that the intensive treatment group had a 57% reduction in the risk of nonfatal myocardial infarction, stroke, or death from cardiovascular disease compared to the conventional treatment group (95% CI, 0.21 to 0.88; P = 0.02). Unfortunately, the DCCT/EDIC findings, if generalizable to the care of diabetics in the community at large, appear not to have translated yet into that community, since the cumulative incidence of CHD in type 1 diabetics has changed little in the past few decades.54 Unlike CHD, peripheral arterial disease,55 amputation,56 and stroke57 rates have all been reported to correlate with levels of hyperglycemia. Possible explanations for the differences between CHD and other forms of CVD in type 1 diabetes are discussed in further detail in Chapter 25.
Type 2 Diabetes
There are alterations in multiple lipoprotein metabolism pathways in type 2 diabetics that lead to the typical abnormal lipid profile commonly seen. This profile includes hypertriglyceridemia secondary to high VLDL levels, as well as increased amounts of sdLDL and lower levels of HDL cholesterol. LDL cholesterol levels are typically not higher in diabetic populations when compared to well-matched nondiabetic subjects. In an elderly Finish population, for example, approximately 30% of diabetic subjects had serum triglyceride levels greater than 200 mg/dL compared to 13% of nondiabetic subjects. HDL cholesterol levels were less than 35 mg/dL in about 25% of diabetics compared to 12% of nondiabetics. In this study, elevated LDL levels as defined by an LDL greater than 130 mg/dL were actually more prevalent in the nondiabetic population than in the diabetics.58,59
The causes of classic diabetic dyslipidemia are only partly understood. Individuals with insulin resistance and type 2 diabetes overproduce mature VLDL in the liver. This is accompanied by a slower clearance of triglyceride-rich lipoproteins via reduced activity of lipoprotein lipase. Coupled with decreased receptor uptake of the remnant and IDL particles that form after LPL partially depletes VLDL of its triglyceride, diabetes produces substantial elevations in serum triglyceride levels. Production of sdLDL appears to be tightly linked to the resulting hypertriglyceridemia insofar as cholesterol ester transfer protein (CETP) exchanges triglyceride from VLDL for cholesterol ester from LDL, producing a triglyceride-enriched LDL.60 This lipoprotein appears to be a preferred substrate for hepatic lipase, which hydrolyzes the triglyceride to produce an LDL that is smaller, lower in cholesterol ester content, and higher in density than LDL produced in the absence of excess VLDL. The diabetic with increased small dense LDL levels has a higher LDL particle number for an equivalent serum LDL cholesterol concentration than does an individual with normal-density LDL. This increased particle number, whether measured by nuclear magnetic resonance assays or serum apo B levels, is associated with an increased risk of CAD.12
Thus, the central metabolic change that accounts for much of the diabetic dyslipidemia is the overproduction of VLDL by the liver. Higher levels of circulating free fatty acids, arising from increased adipose deposits and insulin deficiency or resistance, contribute to higher storage levels of triglyceride in the liver. Increased fat content of the liver is in turn correlated with increased rates of lipidation and secretion of nascent VLDL. This VLDL appears to be normal in size, indicating that higher VLDL triglyceride levels derive from higher particle numbers rather than larger triglyceride-enriched lipoproteins. Administration of insulin reduces the concentration of free fatty acids in the circulation and also suppresses VLDL production by inhibiting the hepatic generation of the early apo B, lipid-poor particle. The molecular mechanisms accounting for VLDL production changes are not fully elucidated, but there is evidence that insulin activates a phosphatidylinositol-3 (PI-3) kinase pathway that inhibits apo B secretion while activating a mitogen-activated protein kinase (MAPK) that down-regulates MTP expression.61,62 Insulin effects on transcriptional regulators of VLDL secretion may also be involved. In vivo metabolic studies suggest that the prevalence of hypertriglyceridemia and low HDL cholesterol levels is higher in diabetics who are insulin resistant as opposed to insulin deficient, accounting for the higher prevalence of these disorders in type 2 versus type 1 diabetics.
Genetic Basis of Lipid Disorders
Although metabolic disorders associated with diabetes, hypothyroidism, and nephropathy can exacerbate or even cause hyperlipidemia (Table 16-4), many individuals have none of these problems. Individuals with the greatest deviation from normal levels of lipoproteins often have a single gene defect that is responsible for their lipid disorder. The vast majority of hyperlipidemic patients, however, do not have a monogenic defect. Rather, they are most likely to have a polygenic disorder and an additional contribution from environmental factors (e.g., excessive saturated fat intake for high LDL levels; obesity or smoking for lower HDL levels). The application of the NCEP treatment guidelines does not require that a genetic diagnosis be made, because treatment is based on phenotype (lipid levels) and not genotype. Nevertheless, the identification of the causes of monogenic lipid disorders has been critical to our understanding of both normal and abnormal lipoprotein physiology. The most important lipid disorders affecting coronary disease risk are those that increase the serum LDL level or reduce the HDL level, and these are briefly presented next.
Table 16-4
Secondary Causes of Hyperlipidemia and Dyslipidemias
| Metabolic/hormonal | Diabetes |
| Hypothyroidism | |
| Lipodystrophy | |
| Polycystic ovarian disease | |
| Hepatic | Primary biliary cirrhosis |
| Other forms of cirrhosis | |
| Renal | Chronic renal failure |
| Nephrotic syndrome | |
| Dietary | Alcohol |
| Foods highly enriched in cholesterol and saturated fat | |
| Medications | Estrogens |
| Glucocorticoids | |
| Anti-HIV treatments, especially protease inhibitors | |
| Oral androgens and anabolic steroids | |
| Thiazide diuretics | |
| Beta-blockers | |
| Retinoic acid | |
| Antipsychotics |
Monogenic Low-Density Lipoprotein Disorders
Familial Hypercholesterolemia
Familial hypercholesterolemia (FH) is one of the most thoroughly studied and common genetic disorders of mankind. Approximately 1 in 500 individuals carry a mutation in one allele encoding the LDL receptor. These mutations results in defective clearance of LDL from the blood and a rise in serum total and LDL cholesterol levels. The elucidation of this defect and its associated cell biology led to insights into the homeostatic control of cholesterol metabolism that transformed the lipid field.31 Several hundred individual mutations in the LDL receptor have now been identified. The gene, located on chromosome 19 and spanning 45 kb, has 18 exons that encode a mature protein of 839 amino acids. The inheritance of the disorder is autosomal co-dominant. Heterozygous patients have LDL cholesterol levels in the 200 to 500 mg/dL range, and the rare homozygous FH patient typically has an LDL cholesterol above 500 mg/dL. Heterozygous patients commonly have tendon xanthomas and premature CAD, whereas these are universal in the untreated homozygous individual. Mutations in the LDL receptor have been described that affect multiple aspects of receptor biology, ranging from defective LDL binding to abnormal trafficking of the receptor to the plasma membrane. The effect of all of these mutations is to reduce LDL and IDL clearance from the blood. The latter effect results in an enhanced conversion of IDL to LDL. The net result is markedly elevated LDL cholesterol levels in the plasma and a strong predisposition to early CHD if the disorder is not treated. FH heterozygotes typically respond well to 3-hydroxy-3-methylglutaryl coenzyme A (HMG CoA) reductase inhibitors (statins), though additional therapies may be needed to reach desirable LDL levels. FH homozygotes respond poorly to reductase inhibitors and inevitably develop early atherosclerotic vascular disease if LDL apheresis or liver transplantation is not performed.
Familial Defective Apolipoprotein B
Familial defective apo B (FDB) is an autosomal co-dominant disorder caused by mutations near the carboxy terminus of apo B.63,64 Apo B is a 4536-amino-acid protein encoded by a gene on chromosome 2 that is composed of 29 exons. The most commonly identified mutations in FDB affect an arginine at position 3500 that is either mutated to a glutamine or tryptophan. Most individuals with FDB are heterozygous for their mutation, and there is only one molecule of apo B per LDL particle, so the LDL in the serum of these individuals is a mixture of normal LDL and FDB LDL. The LDL containing the defective apo B does not bind normally to the LDL receptor and is therefore cleared more slowly. The explanation for the lower receptor affinity of LDL carrying the FDB mutations is not known with certainty. There is evidence, however, of a molecular interaction between the arginine at position 3500 and tryptophan at 4369 that appears to be necessary for the adoption of an apo B conformation recognized by the LDL receptor. Patients with FDB have elevations in their LDL levels that fall within the range of those seen in FH heterozygotes, but the mean is generally lower than the mean in FH cohorts. There is considerable variability in both genetic populations; however, making generalizations about the diagnostic value of this measurement not very useful. Tendon xanthomas are common but not universal, and premature CAD is prevalent. Treatment with reductase inhibitors or other standard LDL-lowering drugs is typically quite effective in FDB patients.
Autosomal Recessive Hypercholesterolemia
Studies of families in which LDL levels were inherited in an autosomal recessive pattern led to a successful genetic linkage analysis that mapped the genetic defect in these families to the short arm of chromosome 1. Candidate gene sequencing led to the identification of mutations in a gene now designated autosomal recessive hypercholesterolemia (ARH) and comprising nine exons and eight introns. Patients diagnosed with ARH to date have been clinically similar to patients who are homozygous for LDL receptor mutations in that they have markedly elevated LDL cholesterol levels (typically > 500 mg/dL), tendon xanthomas, and premature coronary atherosclerosis.65 The autosomal recessive mode of inheritance is an important differentiating factor between ARH and familial hypercholesterolemia, as is greater LDL receptor activity, measured in cultured fibroblasts taken from the patients. The defect in ARH patients appears to affect liver cholesterol metabolism disproportionately, and current data indicate that the ARH gene product likely serves as an adaptor protein required for LDL receptor internalization via clathrin-coated pits.66,67 ARH patients do respond to HMG CoA reductase inhibitors, but this treatment is usually inadequate to control their markedly elevated LDL cholesterol levels, making them candidates for LDL apheresis.
PCSK9 Mutations
Proprotein convertase subtilisin kexin 9 (PCSK9) is a 72-kD protease that is highly expressed in the liver and regulates the levels of LDL receptors acting at the plasma membrane.68,69 Gain-of-function mutations in PCSK9 result in lower LDL receptor levels, whereas loss-of function mutations increase those levels. Serum LDL levels are inversely related to liver LDL receptor expression; the activity of the latter clears the former from the circulation. Thus, gain-of-function mutations are associated with higher serum LDL levels and increased risk of CHD, and loss-of-function mutations result in lower LDL levels and decreased CHD risk. The role of PCSK9 in lipid metabolism was originally recognized when it was discovered that specific heterozygous mutations in the protein were associated with autosomal dominant hypercholesterolemia. Approximately 1 in 50 African Americans has one of two nonsense mutations in PCSK9, causing LDL cholesterol levels to fall approximately 30%. Caucasians can carry a missense mutation that reduces LDL cholesterol by approximately 15%. Individuals with complete loss of PCSK9 activity have extraordinarily low levels of LDL-C, but appear to be otherwise normal. The mechanism by which PCSK9 regulates LDL receptor activity is still being elucidated, but it appears that the protease can bind to the receptor, targeting it for degradation in the lysosome. This action does not depend on the protein’s enzymatic activity, and exogenously administered PCSK9 can reduce LDL receptor expression and serum LDL-C levels. The mechanism by which PCSK9 works makes it an extremely attractive target for pharmaceutical intervention; therapeutics that block PCSK9 function are being actively pursued.
Monogenic High-Density Lipoprotein Disorders
Tangier Disease
Tangier disease was first recognized in 1960 in a sibling pair living on Tangier Island in the Chesapeake Bay in Virginia. Enlarged, yellow-orange tonsils and little or no circulating HDL cholesterol are the classic findings in the disorder. Subsequently, cases have been identified in which the presenting symptom was a peripheral neuropathy. Patients may also have hepatosplenomegaly. Serum LDL and total cholesterol levels are usually quite low, while serum triglyceride values are moderately elevated. Tangier disease is an autosomal recessive disorder. The cause of Tangier disease was identified by several groups, following mapping of the gene defect to chromosome 9. Candidate gene analysis in the appropriate genetic interval identified mutations in an ATP-binding cassette (ABC) transporter as the cause. The transporter, now called ABCA1 (ATP-binding cassette, subfamily A, member 1), is a full-length ABC transporter transmembrane protein that is predicted to span the plasma membrane 12 times. ABCA1 is a 2261-amino-acid protein encoded by a gene spanning 50 exons. Approximately 50 mutations have been identified to date in the gene.70 ABCA1 mediates the efflux of cholesterol from cholesterol-enriched cells when stimulated by the major apoprotein of HDL, apo AI.71 This activity is lost in Tangier patients and is reduced by approximately half in carriers of one abnormal ABCA1 allele. The mechanism of the movement of cholesterol from inside the cell to outside the cell has not been established. Individuals with Tangier disease and heterozygous carriers of ABCA1 mutations (a disorder called familial hypoalphalipoproteinemia [FHA]) both appear to have increased risk of premature coronary disease. There is no specific therapy for this disorder.
Lecithin Cholesterol Acyltransferase Deficiency
The esterification of free cholesterol in circulating lipoproteins is catalyzed by a plasma enzyme called lecithin cholesterol acyltransferase. Two clinically separable syndromes result from a deficiency of LCAT. Fish eye disease is due to a partial deficiency of LCAT, with patients presenting with dense corneal opacities and very low HDL cholesterol levels. Familial LCAT deficiency arises from a nearly completely absence of LCAT activity and produces a more severe syndrome characterized by corneal opacities, anemia, and proteinuric renal failure.72
[/not-level-membership-for-endocrinology-diabetes-and-metabolism-category]
