[level-membership-for-endocrinology-diabetes-and-metabolism-category]Chapter 12
Lipodystrophy Syndromes
Lipodystrophies: Definition and Diagnosis
Pathophysiology of Lipodystrophy
Classifications of Lipodystrophies and Their Clinical Manifestations
Mechanisms Responsible for Severe Insulin Resistance
Lipodystrophies: Definition and Diagnosis
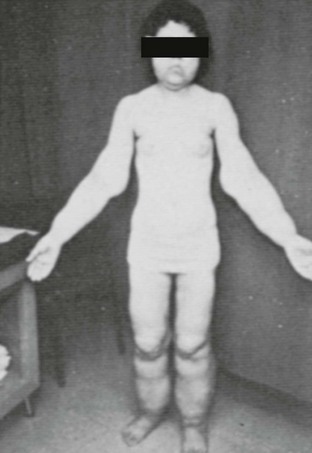
The lipodystrophic states are a diverse group of clinical disorders, the central feature of which is either a congenital or acquired, complete or partial lack of adipose tissue (lipoatrophy), and/or a combination of lack of adipose tissue in certain body areas, with excess of adipose tissue (lipohypertrophy) elsewhere. Insulin resistance and associated clinical features are present in nearly all varieties of lipodystrophies. In addition, patients with lipodystrophies also manifest a group of unique features such as severe hyperlipidemia, progressive liver disease, and increased metabolic rate (Fig. 12-1).
Dual-energy x-ray absorptiometry (DEXA), magnetic resonance imaging (MRI), and computed tomography (CT) scan are additional modalities that have been developed to allow direct quantification of fat within specific tissues and/or body mass, as well as fat distribution. Although all these techniques are accurate and noninvasive, they are also expensive, have limited availability, and are apparently not cost-effective for use in everyday clinical practice. Their use, therefore, has been limited to the research field.1
In the past few years, ultrasound (US) has emerged as a promising alternative to assess body fat changes. Although it demonstrates good accuracy and accessibility, more studies are needed to elucidate its value and to comparatively evaluate ultrasound (US) with other imaging modalities. Standardization of the US techniques and cost-effectiveness analyses will also be essential for its potential wider clinical applications.2
Pathophysiology of Lipodystrophy
Much of the knowledge on the mechanisms underlying the pathogenesis and manifestations of lipodystrophies has been obtained through the performance of mouse studies and from the human genome sequencing. It is now understood that patients with lipodystrophy have primarily a loss of mature, functional adipocytes, as opposed to an absence of lipids in otherwise normal adipocytes.3–5 The underlying defects could be associated with failure of adipogenesis, adipocyte apoptosis, or a failure to store triglycerides in existing adipocytes because of ineffective lipogenesis or excessive lipolysis.
Classifications of Lipodystrophies and Their Clinical Manifestations
Generalized Lipodystrophy
Generalized lipodystrophy encompasses rare but clinically striking disorders that may be congenital (Berardinelli-Seip syndrome)6,7 or acquired (Lawrence syndrome).
Congenital Generalized Lipodystrophy
Congenital generalized lipodystrophy (CGL), or Berardinelli-Seip congenital lipodystrophy (BSCL), is a rare syndrome characterized by near complete absence of body fat. It is inherited in an autosomal recessive fashion and is observed in the highest frequency with parental consanguinity. To date, it has been reported in approximately 250 patients with various ethnic backgrounds.8,9
Babies with CGL are noted to have an abnormal appearance due to absence of body fat within the first 2 years of life and frequently soon after birth. Adipose tissue is absent from not only subcutaneous but also from intraabdominal sites. Magnetic resonance (MR) imaging of the abdomen shows complete absence of intraabdominal, retroperitoneal, and subcutaneous fat but a prominent fatty liver and presence of fat in certain anatomic sites such as orbits, palms, and soles. Thus this genetic defect results in poor development of metabolically active but not mechanically important adipose tissue.10–12
Other somatic abnormalities that contribute to the abnormal appearance are acanthosis nigricans, a protuberant abdomen associated with hepatomegaly and/or splenomegaly, and prominent musculature. Congenital muscular weakness and cervical spine instability has also been reported in occasional cases.13 Females may present with enlarged clitoris, increased body hair, absence of or irregular menstrual cycles, and polycystic ovaries. Only a few affected women have had successful pregnancies, whereas affected men have normal fertility. The patients with CGL tend to have voracious appetite. The basal metabolic rate of their body may also be increased. Although patients may have accelerated linear growth and advanced bone age during their childhood, they normally have normal or reduced heights as adults.
Although both boys and girls are affected at similar rates, the metabolic features tend to be more severe and develop earlier in girls. Hypertriglyceridemia is characterized by increased concentrations of very low–density lipoproteins (VLDLs) and chylomicrons, whereas serum high-density lipoprotein (HDL) is usually low. Severely elevated triglycerides may provoke acute pancreatitis and is frequently related to fatty liver. This commonly progresses to cirrhosis, which in many cases may be fatal. Insulin resistance has been noted at an early age and may be present even at birth. Clinical diabetes usually develops in the early teens, is rarely ketotic, and is usually refractory to insulin therapy. Serum adipocytokines, the hormones produced by adipose tissue (e.g., leptin and adiponectin), circulate in extremely low levels in CGL.14 CGL may also be associated with focal lytic lesions in the long bones,15–17 mild mental retardation,18 cardiomyopathy,19–21 generalized muscle weakness, and cervical spine instability.13
Type 1 CGL (CGL1) is due to AGPAT2 gene mutations. This gene has been mapped to chromosome 9q34.22,23 It encodes the enzyme acyltransferase 1-acylglycerol-3-phosphate O-acyltransferase 2 (AGPAT2) which catalyzes the acylation of lysophosphatidic acid to form phosphatidic acid, a key intermediate in the biosynthesis of triacylglyceride and glycerophospholipids. AGPAT2 mutations are found predominantly in patients of African ancestry.
Type 2 CGL (CGL2) is due to BSCL2 gene mutations. This gene is located in chromosome 11q13 and encodes a 398-amino-acid protein called seipin.24 The BSCL2 gene mutation has been found in patients of European and Middle Eastern origins but has also been reported as a causative gene in Japanese patients with CGL. Seipin is expressed diffusely in many tissues but predominantly in testis and brain; its function in humans is largely unknown. A recent study in yeast suggests that seipin is important for lipid droplet morphology and perhaps assembly.9 Another study in cultured murine and human adipocytes also indicates that BSCL2 expression is critical for normal adipogenesis in vitro, as cells lacking BSCL2 failed to induce expression of key lipogenic transcription factors (peroxisome proliferator–activated receptor gamma [PPARG] and CCAAT/enhancer binding protein alpha [C/EBP-α]), as well as enzymes (AGPAT2, DGAT2, and lipin 1). BSCL2 mutations are usually related to more severe adipose tissue loss than in CGL1.9
Recently a third gene mutation has been identified in one individual with CGL25–27 who had a homozygous nonsense mutation of CAV1, probably as a result of a consanguineous union. CAV1 is located on chromosome 7q31. Its end product, caveolin 1, is a highly conserved 22-KD protein and a crucial component of plasma membrane microdomains known as caveolae. These plasma membrane domains have important roles in regulating signaling pathways and processes such as cell migration, polarization, and proliferation. Caveolin 1 has also been identified as a major fatty acid binder on the plasma membranes. Mutated function of CAV1 may induce lipodystrophy by interfering with lipid handling, lipid droplet formation, and adipocyte differentiation.9,28
Acquired Generalized Lipodystrophy (Lawrence Syndrome)
The acquired syndrome of total lipoatrophy is similar to that of the congenital disorder, except that it develops in a previously healthy individual over days to weeks, often after a nonspecific febrile illness. The syndrome is very rare. It commonly develops during childhood and aldolence in patients who are predominantly white, with a male-to-female ratio of 1 : 3.26,29
In addition to the generalized loss of fat that has an active metabolic function, as seen in CGL, fat loss in acquired generalized lipodystrophy (AGL) also occurs in palms, soles, and genital areas. However, retroorbital and bone marrow fat may be preserved.27
The median time to develop diabetes after loss of fat tissue is approximately 4 years.27 Diabetic ketoacidosis has been reported, and hypertriglyceridemia, hepatic steatosis, acanthosis nigricans, menstrual irregularities, and polycystic ovary syndrome (PCOS) are also common findings. Patients with AGL also have markedly reduced adiponectin levels and moderately reduced leptin levels.14
Several autoimmune diseases and inflammatory conditions have shown a temporal relationship to AGL. These include juvenile-onset dermatomyositis (JDM), rheumatoid arthritis, systemic sclerosis, systemic lupus erythematosus, Sjögren syndrome, and panniculitis.27,30 JDM shows a particularly strong correlation with lipodystrophy; 8% to 40% of patients with JDM develop acquired lipodystrophy.31–35 The chronicity and severity of JDM, as well as the high frequency of calcinosis, have been shown to predict the onset of lipodystrophy.31 AGL following autoimmune diseases is also termed AGL type 2 or the autoimmune disease variety. Panniculitis is another inflammatory condition that frequently heralds the onset of acquired generalized lipodystrophy. It is estimated to be present in approximately 25% of affected patients.27,36 Panniculitis manifests as subcutaneous inflammatory nodules which show a mixed infiltrate of lymphocytes and mononucleated macrophages in adipose tissue. The course of AGL is frequently protracted in patients with panniculitis and linked to less fat loss and less severe metabolic disorders.32,33 The panniculitis variety is also known as AGL type 1. Up to 50% of AGL patients have no clear history of autoimmune disease or panniculitis, however. These lipodystrophies are known as AGL type 3 or the idiopathic type.
The pathogenesis of AGL is unknown. The autoimmune-mediated destruction of adipocytes or preadipocytes has been hypothesized to be the underlying mechanism. Autoantibodies against adipocyte membranes may also impair fat uptake and adipocyte differentiation.30,31,37 Several antibodies have been found to be present in AGL, but no causative relationship has been established.31 Cytokines, including tumor necrosis factor alpha (TNF-α) and interleukin 1 (IL-1), are also likely to play important roles in the immunopathogenesis of lipodystrophy. They can potentially lead to lipodystrophy by inhibiting adipogenesis38 or increasing receptor-mediated apoptosis of adipocytes and preadipocytes.39
Partial Lipodystrophy
Dunnigan-Variety (Face-Sparing) Lipodystrophy
It is characterized by gradual loss of almost all subcutaneous fat from the extremities, commencing at puberty. This gives rise to the characteristic phenotype of “increased muscularity” in the arms and legs. Variable and progressive loss of fat from the anterior abdomen and chest occurs later. Excess fat may subsequently accumulate in the face and neck and in the intraabdominal region, resulting in a Cushingoid appearance.26
Affected females tend to have more recognizable phenotypes. Although questions for gender differences have been raised, anthropometric measures and MRI data demonstrated that both affected men and women have similar patterns of fat loss. In comparison to the affected men, women may have more severe hypoleptinemia and metabolic sequelae of insulin resistance, and they may also have higher prevalence of diabetes and atherosclerotic vascular disease as well as higher serum triglycerides and lower high-density lipoproteins. The prevalence of hypertension and fasting serum insulin concentrations are similar in men and women.27,36 The prevalence of diabetes is not related to age, menopausal status, or family history of type 2 diabetes.37
Patients with the Dunnigan variety are more prone to develop PCOS, infertility, and gestational diabetes. The prevalence of gestational diabetes and miscarriage is significantly higher than in women with similar body mass index (BMI) and PCOS.40
The gene for Dunnigan variety, LMNA, is located on chromosome 1q21-22. It encodes for lamins A and C, which are essential components of nuclear lamina and provide structural integrity of the nuclear envelope. Most FPLD2 mutations in LMNA are missense mutations within the 3′ end of the gene.41 The mutant gene products may disrupt interaction with chromatin or other nuclear lamina proteins, resulting in apoptosis and premature death of adipocytes.42 The accumulation of prelamin A may also impair adipogenesis by interfering with the key adipocyte transcription factors/regulators, including sterol response element–binding protein 1 (SREBP-1) and PPARG.42–45 It is interesting to note that there is a lack of difference in the levels of lamin A and lamin C expression in different adipose depots even though the fat loss of FPLD2 is regionally selective, suggesting that the downstream effects of LMNA mutations are differentially regulated in different areas of the body.40
Köbberling-Type Lipodystrophy
Also known as FPLD type 1 (FPLD1), Köbberling-type lipodystrophy was first reported by Köbberling et al. in 1971. In comparison to Dunnigan variety, the loss of adipose tissue is restricted to the extremities. The distribution of fat on the face and neck is normal or increased in association to frequently observed significant central obesity. The hallmark anthropomorphic feature of this syndrome includes a palpable “ledge” formed between the normal and lipodystrophic areas and high triceps-to-forearm and abdomen-to-thigh skinfold ratios.46 Only women have been diagnosed with Köbberling-type lipodystrophy to date. It is hypothesized that males have a very gentle clinical presentation that does not allow early detection, but this remains to be proven. FPLD1 tends to have a childhood onset.
Metabolic syndrome, especially hypertriglyceridemia, is common in Köbberling-type lipodystrophy. This correlates with high incidence of pancreatitis and premature coronary artery disease. Leptin concentrations are low and correspond to the BMI and the level of fat loss of individual patients.46
Familial Partial Lipodystrophy Due to PPARG Mutations
Also referred as FPLD3, familial partial lipodystrophy due to PPARG mutations is associated with heterozygous PPARG gene mutations. The phenotype is similar to the Dunnigan variety, with the exception that fat accumulation in the head and neck may be spared.47–50 Patients with FPLD3 appear to have more severe metabolic abnormalities than those with FDLP2.45
Familial Partial Lipodystrophy Due to AKT2 Mutation
Familial partial lipodystrophy due to AKT2 mutation has been reported in a single family by George et al.51 It is inherited in an autosomal dominant fashion and manifests as severe insulin resistance and partial lipodystrophy confined to extremities.51
AKT, also known as protein kinase B, is a serine/threonine protein kinase and plays multiple roles in cell signaling, cell growth, and glycogen synthesis, as well as insulin-stimulated glucose transport.52 Lipodystrophy in patients with AKT2 mutations is thought to be due to reduced adipocyte differentiation and dysfunctional postreceptor insulin signaling.
Partial Lipodystrophy Due to CAV1 Mutation
Partial lipodystrophy due to CAV1 mutation was recently identified as a rare cause for partial lipodystrophy.53 Two cases with different frameshift CAV1 mutations have been reported. Both patients were described to have partial lipodystrophy with subcutaneous fat loss in the face and upper body, micrognathia, and congenital cataracts. One case was also associated with abnormal neurologic findings. Diabetes, hypertriglyceridemia, and recurrent pancreatitis were reported in both cases.53
Other Syndromes With a Component of Lipodystrophy
Mandibuloacral dysplasia (MAD) is an extremely rare autosomal recessive progeroid syndrome which has been reported in approximately 40 case reports. MAD is characterized by postnatal growth retardation, craniofacial and skeletal abnormalities (mandibular and clavicular hypoplasia, delayed closure of the cranial sutures, acroosteolysis, joint contractures, birdlike face, dental abnormalities), cutaneous changes (restrictive dermatopathy, skin atrophy, alopecia, and mottled cutaneous pigmentation), and lipodystrophy. Although MAD is present at birth, dysmorphic manifestations and progeroid features become more prominent with time, and the full clinical phenotype is recognizable during the early school years. The patients have normal intelligence,54 and their serum leptin concentration can be low or normal. Hyperinsulinemia, insulin resistance, impaired glucose tolerance, diabetes mellitus, and hyperlipidemia have been reported in some patients.
There are two distinctive phenotypes of MAD: type A involves the loss of subcutaneous fat from the arms and legs but normal or excessive deposition of fat in the face and neck, and type B is characterized by more generalized loss of subcutaneous fat. Mandibular dysplasia type A (MADA) is also considered to be due to mutations of the LMNA gene which result in accumulation of prelamin A and lead to alterations of nuclear architecture and chromatin defects. It remains unclear how different mutations in the same gene lead to a variety of phenotypes. Patients with mandibular dysplasia type B have been reported to carry compound heterozygous mutations in the gene encoding an endoprotease, zinc metalloprotease (ZMPSTE24), on chromosome 1q34. The enzyme is important in posttranslational processing of prelamin A to mature lamin A. As in MADA, the accumulation of farnesylated prelamin A is proposed to be responsible for the phenotype.54 Focal segmental glomerulosclerosis has been reported in patients with ZMPSTE24 deficiency.55
Multiple other syndromes are also linked to lipodystrophy. Several of them have also been identified as laminopathies, including Hutchinson-Gilford progeria syndrome (HGPS, a very rare and uniformly fatal segmental progeroid syndrome with progressive and generalized fat loss), restrictive dermopathy (RD), progeria-associated arthropathy, and atypical progeroid syndrome (also referred to as atypical Werner’s syndrome).54
Acquired Partial Lipodystrophy (Barraquer-Simons Syndrome)
First reported in 1885 by Mitchell, acquired partial lipodystrophy (APL) was further characterized by Barraquer-Roviralta in 1907. There have been approximately 250 cases reported in the English-language literature.26
Patients with APL are primarily of European descent; however, cases have also been reported in Asian Indian, Vietnamese, and Samoan populations. The disease shows a female dominance, and most patients have clinical manifestations in early puberty or early adulthood. The characteristic fat loss progresses in a “cephalocaudal” fashion, with fat loss appearing first in the face and spreading to the upper part of the body. Fat under the umbilicus is rarely affected. Excess fat accumulation is seen over the lower abdomen, gluteal region, thighs, and calves. Breasts may lose fat and consist of firm glandular tissue only.27 Hepatomegaly is common among patients with APL.
In contrast to other types of lipodystrophies, acanthosis nigricans, hirsutism, and hypertrichosis are rare. Female patients normally have regular menses and intact fertility. The prevalence of the metabolic syndrome is also significantly lower in patients with APL. Insulin resistance is uncommon, and the prevalence of diabetes is much reduced compared to other types of lipodystrophies. In the series of case reports by Misra and Garg,27 35% of APL patients had hypertriglyceridemia, and a third had low concentrations of HDL. Serum leptin levels were normal in the majority of patients.14
A strong association has been proposed to exist between acquired partial lipodystrophy and membranoproliferative glomerulonephritis (MPGN) type 2. The spectrum of presentations range from acute glomerulonephritis, hematuria, nocturia, urinary casts, albuminuria, and nephritic syndrome to chronic glomerulonephritis and uremia.53,56 The serum C3 complement levels are usually low with the presence of C3 nephritic factor.53 Patients with low C3 levels tend to have an earlier onset of lipodystrophy than those with normal serum C3 levels. The median time interval between the onset of lipodystrophy and the development of MPGN is approximately 5 to 10 years but could be as long as 20 years.53,57
Similar to acquired generalized lipodystrophy, APL is also frequently seen in the context of autoimmunity or infections.30 The most frequently cited infection preceding APL is measles. The low C3 levels may also render APL patients susceptible to recurrent pyogenic infections, particularly due to Neisseria.58 Systemic lupus erythematosus and dermatomyositis/polymyositis are autoimmune diseases most frequently associated with acquired partial lipodystrophy.27
The precise mechanisms leading to adipose-tissue atrophy in APL remain unclear. The C3 nephritic factor has been shown to induce lysis of adipocytes expressing factor D (adipsin).27 Lamin B gene mutations have also been reported in some cases of APL.59 In a recent study by Guallar et al., PPARG gene down-regulation and mitochondrial toxicity were observed in a patient with APL, suggesting that impaired adipogenesis and adipocyte metabolism may also underlie the pathogenesis of APL.60
HIV-Associated Lipodystrophy Syndrome
Most HIV-infected patients with lipodystrophy are otherwise relatively healthy, but dyslipidemia, especially hypertriglyceridemia, is common among HIV-infected patients receiving HAART. HIV viremia has been linked to decreased plasma concentrations of total, LDL, and HDL cholesterol, and at later stages elevated triglyceride levels. HAART has been shown to cause a worsening lipid profile, with increased plasma triglyceride, increased total and LDL cholesterol, and decreased HDL, which can be further accompanied by increases in small, dense LDL particles, lipoprotein (a), and apolipoproteins B, C-III, E, and H.61 HAART-associated dyslipidemia is associated with accelerated atherosclerosis and signs of endothelial dysfunction.62–65 Frank diabetes and insulin resistance are more prevalent in HIV subjects with lipodystrophy, but acanthosis nigricans seems to be extremely rare. Hepatic steatosis may also develop. Both leptin and adiponectin levels are decreased in patients with HALS. The reduction of leptin levels correlates with decreased subcutaneous fat mass,66 whereas decreased adiponectin levels are more closely associated with intraabdominal fat accumulation.67
Fat atrophy and fat deposition appear to be associated with different risk factors in HALS. Low baseline fat mass and increased disease severity are associated with a higher incidence of fat atrophy.68 Epidemiology studies have also shown that co-infection with hepatitis C can increase the chance of fat atrophy in HIV-infected individuals.69 On the other hand, older age, female sex, high baseline body fat, and longer duration of HAART are associated with a higher risk of fat accumulation in HIV patients.68
The frequency and manifestations of lipodystrophy also differ with respect to the drugs used. Nucleoside reverse transcriptase inhibitors (NRTIs), particularly zidovudine and stavudine, are commonly associated with morphologic changes, particularly fat loss from the extremities, whereas protease inhibitors (PIs) are more frequently linked to hypertriglyceridemia, insulin resistance, and localized fat accumulation.70
NRTIs have been shown to suppress adipogenesis either through mitochondrial toxicity (by inhibiting DNA polymerase gamma) or by induction of genes that inhibit adipogenesis. In-vitro studies suggest that zalcitabine, didanosine, and stavudine have the worst effects in a reducing order of magnitude, whereas tenofovir and lamivudine show minimal or no mitochondrial toxicity.60 Combinations of drugs can act synergistically and lead to mitochondrial depletion. In addition, zidovudine, emtricitabine, and abacavir can also impair cell proliferation and increase lactate and lipid production. NRTIs may also contribute to insulin resistance by altering the levels of IL-6, TNF-α, and adiponectin levels.70
PIs can lead to adipose-tissue changes through several potential mechanisms: (1) Impairment of adipocyte differentiation by down-regulation of the expression of master adipogenic transcription factors, such as C/EBP-α and C/EBP-β, PPARG, and SREBP-171; (2) increase of adipocyte apoptosis, leading to a reduction in cell numbers; and (3) decrease of lipid accumulation in adipocytes through reactive oxygen species (ROS) production. Therapy with PIs has also been implicated in the causation of metabolic abnormalities by inhibiting glucose transport-4 (GLUT-4)-mediated glucose transport, by suppressing insulin signaling, and through activation of lipolysis, induction of IL-6 and TNF-α, reduction in gene expression, and secretion of adiponectin, as well as proteosome dysfunction. Lopinavir, ritonavir, saquinavir, and nelfinavir are the worst offenders. The newer PI, atazanavir, has a much milder effect. Indinavir does not have much effect on cell viability or lipogenesis but inhibits glucose uptake to a greater extent than the other PIs.70
Non-nucleoside reverse transcriptase inhibitors (NNRTIs), including efavirenz and nevirapine, appear to have more favorable safety profiles in terms of lipodystrophy complications. Although an in-vitro study showed that efavirenz may interfere with adipogenesis by reducing the expression of SREBP-1, a key adipogenic transcription factor,72 and a prospective randomized trial suggested that efavirenz could have greater potential for causing lipoatrophy than the combination of lopinavir plus ritonavir,73 the results from several clinical studies imply that the potential role of efavirenz in the development of lipodystrophy is minimal, and that it may depend on the NRTIs that form the backbone of the regimen.74
There is also increasing evidence that HIV-1 infection itself, regardless of HAART, may induce inflammatory and pro-apoptotic pathways in adipose tissue and thus contribute to lipoatrophy. This could be either occurring through the direct HIV-1 infection of cells in adipose tissue or may be mediated by HIV-1-encoded proteins.75 Inflammatory cytokines, including interferon alpha (IFN-α), TNF-α, IFN-γ, IL-1, IL-6, and IL-12, may also contribute to or mediate the clinical manifestations of this syndrome,66,75 and this is an active area of research.
Mechanisms Responsible for Severe Insulin Resistance
Fat Redistribution and Fat Metabolism
Changes in fat distribution (reduced subcutaneous fat with or without increased visceral fat) may cause increased insulin resistance. The lack of adipose tissue can result in inadequate storage of and therefore increased levels of free fatty acids (FFAs). Intracellular fatty acid accumulation can directly inhibit insulin-mediated glucose transport in skeletal muscle76,77; excess FFAs can also lead to lipotoxicity by inducing ectopic fat accumulation in the liver and muscle, where the adipose tissue is considered to have more “pathogenic” potential. The deposition of fat in the pancreas can also impair beta-cell response and further contribute to insulin resistance.78 Moreover, because the fat tissue at different depots shows different degrees of metabolic activity, such as lipolysis and inflammation, lipodystrophic states that are associated with higher volume of visceral fat and abdominal fat are likely to display a higher degree of insulin resistance.78
Adipocytokines
Alterations of adipocytokine levels can affect metabolic homeostasis and insulin resistance. Leptin and adiponectin are two of the most abundant adipocytokines produced by adipocytes, and their levels decrease in the lipodystrophic states.14 Serum adiponectin levels correlate positively with insulin sensitivity79,80; adiponectin levels are up-regulated by PPARG agonists.81 It acts by reducing hepatic gluconeogenesis (mainly via adiponectin receptor 2 and activation of AMPK phosphorylation) and increasing fatty acid oxidation in muscle (mainly via adiponectin receptor 1).82 A recent study in animals also shows that adiponectin may also act in the hypothalamus (via adiponectin receptor 1) to activate insulin and leptin signaling pathways, thus promoting reduction of food intake.83 Antiinflammatory effects of adiponectin have also been proven in various animal models of liver inflammation81 and have been suggested by several observational studies in humans.84
Serum leptin levels reflect the overall amount of adipose tissue in the body and are positively correlated with adiposity.78 In addition to regulating food intake and increasing energy expenditure, leptin also plays an important role in the regulation of glucose homeostasis, possibly independently of its weight-reducing effects.85 Aside from its actions in the central nervous system (CNS), leptin may exert its insulin-sensitizing effects peripherally by decreasing gluconeogenesis in the liver and adipose tissue and/or by increasing glucose utilization in skeletal muscle.85 Leptin may also prevent the “lipotoxic” effects of intramyocellular lipid accumulation by activating fatty acid oxidation in skeletal muscle.85,86 We have recently shown that leptin levels are decreased in patients with HALS67 and that leptin administration improves metabolic manifestations of HALS in humans.87
Inflammation
Inflammation in adipose tissue is likely to contribute to increased insulin resistance in the lipodystrophic state. Altered innate immunity and chronic inflammation appear to be strongly associated with insulin resistance in obesity and type 2 diabetes.88 Inflammatory adipocytokines, such as TNF-α, IL-6, IL-8, macrophage inflammatory protein (MIP)-1α and 1β, monocyte chemotactic protein-1 (MCP-1; also known as CCL-2), plasminogen activator inhibitor-1 (PAI-1), angiotensinogen, retinol-binding protein-4 (RBP-4), and others have been implicated as the key regulators of insulin sensitivity.81,89 The expression of several of them, including TNF-α, IL-6, and IL-8, as well as macrophage markers (CD 68, ITGAM, EMR1, ADAM8) and chemokines (MCP-1 and CCL-3), is increased in subcutaneous tissue of patients with HALS.90 In a small study of HALS, plasma PAI-1 was also found to be elevated, although the level of its adipose tissue expression was not.91
Accumulating evidence supports an association between inflammation and insulin resistance. TNF-α mediates insulin resistance via reduction of insulin receptor kinase activity, induction of lipolysis and down-regulation of GLUT-4.92,93 It may also induce apoptosis of adipocytes.93 The impact of IL-6 on insulin resistance is less clear, but the majority of evidence indicates that chronic elevation of IL-6 promotes hepatic insulin resistance and impedes differentiation of adipose tissue,94 whereas an acute elevation of IL-6 after exercise may promote improved glucose and lipid metabolism.94 MCP-1 has been shown to induce insulin resistance by down-regulation of GLUT-4, beta-adrenergic receptors, and PPARG in mice. It is also associated with increased levels of atherosclerosis. Two pathways, the NF-κB pathway or the c-Jun NH2-terminal (JNK) pathway, are essential to mediate insulin resistance. Pharmacologic inhibition of the pathways has resulted in improved insulin resistance.81,89
Endoplasmic Reticulum and Mitochondrial Stress
Endoplasmic reticulum (ER) plays an important role in regulating lipid, glucose, cholesterol, and protein metabolism. Stress on the ER luminal environment may generate an increased load of unfolded or misfolded proteins and may lead to adipocyte apoptosis, inflammation, and insulin resistance.76 Seipinopathy has recently been identified as an ER stress–associated disease95 and may contribute to insulin resistance in patients with CGL2.
Mitochondrial defects have been considered as a central factor in NRTI-induced lipodystrophy. Mitochondrial dysfunction will lead to oxidative phosphorylation defects and reactive oxygen species (ROS) accumulation. Observational studies have shown that clinical conditions associated with increased ROS levels are also associated with increased insulin resistance.76 Oxidative stress may also trigger beta-cell apoptosis and may contribute further to insulin resistance.96,97 Angiotensin receptor blockers can attenuate oxidative stress and prevent further progression of insulin resistance.98
Treatment of Syndromes of Lipodystrophies
Lifestyle Modification
There are limited data on the effectiveness of diet and nutrition support on body composition and metabolic abnormalities in patients with lipodystrophy. The general clinical recommendations have been to follow the standard dietary advice regarding management of dyslipidemia, obesity, insulin resistance, and impaired glucose tolerance, with the goal of attaining ideal body weight. Supplementation with dietary fiber and fish oil containing high doses of omega-3 fatty acids should be encouraged.101,102 For patients with severe hypertriglyceridemia, an extremely low-fat diet (preferably <15% of daily caloric intake coming from fat) should be advised. Patients should also avoid alcohol consumption to prevent chylomicronemia and acute pancreatitis. For patients with fat atrophy, increased caloric intake may be necessary.102
Studies evaluating the effect of exercise regimens involving resistance training, aerobic exercise, or stretching and relaxation techniques have all shown only modest benefit in improving body composition and metabolic abnormalities in HIV-infected patients.103,104 Although both strength and endurance training improve peripheral insulin sensitivity, only strength training reduces total body fat in HIV-infected patients with lipodystrophy.105
Management of Insulin Resistance
The treatment strategy for the management of insulin resistance in patients with lipodystrophy is in general not different from that in other states of insulin resistance. Traditional insulin sensitizers such as metformin and/or thiazolidinediones can be considered along with lifestyle modifications. There are no known pharmacologic interactions between antihyperglycemic agents and antiretroviral agents.106
Metformin
Metformin, acting by inhibiting gluconeogenesis in the liver and increasing peripheral glucose utilization, has shown efficacy in improving insulin sensitivity in patients with lipodystrophies.107 It may also potentially improve fat redistribution in HALS, as indicated by a randomized controlled trial.108 Other studies have cast doubt on the potential usefulness of metformin, however, by suggesting that metformin may lead to no change in waist-to-hip ratio and may possibly cause further loss in limb fat.109–111 Nevertheless, metformin, particularly in combination with exercise training, may be useful in HIV-infected patients with significant lipohypertrophy and minimal lipoatrophy.
Thiazolidinediones
The thiazolidinediones (TZDs), acting by stimulating nuclear transcription factor PPARG, have shown promise in treating patients with lipodystrophies. TZDs result in improved insulin sensitivity, but data on fat redistribution and lipid profiles have been variable and conflicting. While some studies show that TZDs may increase subcutaneous fat in patients with lipodystrophy,109,112–115 other studies on HIV-associated lipodystrophy showed no statistically significant difference in limb-fat gain between rosiglitazone and placebo-treated groups.116–118 Rosiglitazone may also be associated with increased LDL and triglyceride levels, a fact that makes it a less desirable drug to treat lipodystrophies. Pioglitazone, with its favorable lipid profile, may have a more central place in the control of metabolic abnormalities associated with HALS. A recent randomized study has demonstrated that treatment with pioglitazone can improve limb fat in HIV-infected patients, although clinical benefits may not be perceived by the patients.114 TZDs can also lower blood pressure, improve endothelial function, and increase serum adiponectin levels and therefore may reduce overall cardiovascular risk in the HALS population.119 More, larger, and longer randomized studies are needed in this area.
Management of Dyslipidemia
The treatment of dyslipidemia in lipodystrophy should follow the same guidelines as in the general population,120 with the goal of total cholesterol level being less than 200 mg/dL, HDL 60 mg/dL or higher, LDL cholesterol less than 70 mg/dL, and triglyceride levels less than 150 mg/dL.121 Lifestyle modification should be emphasized and attempted first. If ineffective, a change of antiretroviral treatment may be considered initially, followed by starting lipid-lowering medications in high-risk HALS patients. Lipodystrophy-related dyslipidemia can be difficult to treat, and multiple agents may be necessary to lower lipids to target ranges.106 Extra caution may be necessary, given the potential drug-drug interactions among lipid-lowering agents and HIV-specific treatments.
Statins
Statins, as 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors, are normally used as first-line agents for hypercholesterolemia, especially for those patients who have fasting triglycerides less than 500 mg/dL. They have antiinflammatory, antithrombotic, and endothelial effects that contribute to their overall beneficial effects on reducing mortality from cardiovascular diseases. Extensive studies have been conducted on the use of statins to treat the hyperlipidemia associated with HAART, and these have demonstrated efficacy in lowering total and LDL cholesterol and triglycerides. Among statins, pravastatin may also increase subcutaneous and limb fat.122 However, caution has to be exercised in cases of co-administrations of statins and HAART. Coadministration of protease inhibitors normally results in increased levels of statins, except for pravastatin. Therefore, simvastatin and lovastatin should be avoided in HIV-infected patients receiving protease inhibitors, whereas atorvastatin should be used with caution. A higher dose of pravastatin may be necessary to achieve optimal lipid-lowering activity in HALS,123,124 and co-administration of statins with efavirenz reduces atorvastatin, simvastatin, and pravastatin serum concentrations, necessitating statins to be administered at higher doses.125
Fibrates
Fibrates are normally reserved for patients with elevated trigly-cerides, and particularly when triglycerides are higher than 500 mg/dL. They are normally well tolerated and efficacious in HALS.61 Combinations of fibrates and statins appear to provide additional benefit in lipid lowering; however, caution is advised given the increased risk of skeletal toxicity, especially in patients with renal insufficiency.
Nicotinic Acid
Nicotinic acid, or niacin, is effective for hypertriglyceridemia, but its use may be limited by adverse effects including flushing, rashes, pruritus, and exacerbation of insulin resistance and hyperuricemia. Extended-release niacin (Niaspan) is generally better tolerated in HIV lipodystrophy. The side effects can be controlled with daily aspirin intake.61 Acipimox, a long-acting niacin analog, may also improve triglycerides and improve insulin sensitivity.126
Other Lipid-Lowering Agents
Ezetimibe, a cholesterol absorption inhibitor, might be useful in the treatment of statin-intolerant patients or in severe dyslipidemia associated with lipodystrophies.61 Studies have shown that ezetimibe can provide incremental reduction in LDL cholesterol levels when combined with a statin, but a recent study suggested that the combination treatment may not provide additional reduction in cardiovascular risk.127 Omega-3 fatty acids have also proven to be effective in reducing triglycerides in HIV-associated lipodystrophy. They are generally well tolerated, but their use may be associated with increased LDL levels.61,128 Tetradecylthioacetic acid, Cholestin, and L-carnitine have also shown efficacy in controlling dyslipidemia via unknown mechanisms,61 but their use in HALS remains limited.
Leptin
The administration of recombinant leptin (r-metHuLeptin) has been tested in the treatment of congenital and acquired non-HIV-related lipodystrophies and has shown amelioration of the metabolic abnormalities. Several small, open-labeled studies show that subcutaneous injection of leptin (0.04 to 0.08 mg/kg/day) in patients with severe generalized lipodystrophy results in significant and sustained weight loss with decreased fat and lean body mass. The weight loss is associated with a decrease in appetite, calorie intake, and resting energy expenditure.129–132 Leptin therapy improves insulin sensitivity and leads to decreased fasting glucose, improved glucose tolerance, reduced hemoglobin A1c, and decreased requirement for insulin or oral hypoglycemics133–135 in these uncontrolled, open-labeled studies. Hypertriglyceridemia, usually refractory to traditional lipid-lowering agents, is commonly responsive to leptin treatment.129 The liver volume decreases with leptin treatment, most likely as a result of decreased intrahepatic lipid content.136,137 Transaminases and hepatocellular injuries that are associated with nonalcoholic steatohepatitis (NASH) are also reduced.136 In comparison to patients with generalized lipodystrophies, patients with familial partial lipodystrophy may have a less dramatic response to leptin treatment.138,139 Since all these studies are uncontrolled and open labeled, it remains to be proven beyond any doubt whether the beneficial effects observed are r-metHuLeptin specific.
The mechanisms through which leptin exerts its role remains under intensive investigation. Animal studies indicate that leptin acts mainly at the hypothalamus, particularly at the neurons containing pro-opiomelanocortin (POMC) and neuropeptide Y (NPY), to regulate food intake and fuel partitioning.140 Functional magnetic resonance imaging (fMRI) studies in humans confirm that leptin mediates its “adipo-static” effect through hypothalamic and other brain areas that are important in emotional and cognitive control.141,142 Although animal and human studies indicate that leptin may also work peripherally (i.e., in the liver, muscle, and white adipose tissue) to affect lipid metabolism,143,144 a recent study by Prieur et al. suggests that these effects are largely dependent on the anorectic effects of leptin on the CNS.145 Long-term leptin treatment may also attenuate beta-cell function and decrease glucose-induced insulin secretion,140,146 but whether these effects are independent from reduction of insulin resistance remains to be seen. All the above areas are the subject of intensive research efforts that are expected to lead to significant breakthroughs in the not-so-distant future.
In addition to generalized lipodystrophies, we have reported a modest effect of leptin therapy on metabolic abnormalities in a randomized, placebo-controlled study in patients with HALS. Compared with placebo, r-metHuLeptin therapy administered at 0.02 mg/kg twice a day decreases body weight mass and truncal fat mass but not peripheral fat or lean body mass, and it improves fasting insulin levels, insulin resistance, and levels of high-density lipoprotein.87 These results have been confirmed by Mulligan et al. (unpublished observations) in an independent open-labeled study in a different population with HALS.
Replacement of r-metHuLeptin may provide additional benefits beyond ameliorating metabolic abnormalities, which may include alleviation of the glomerular injury in humans with lipodystrophies135,147 and improvement of pituitary-gonadal function in lipodystrophic patients with severe leptin deficiency.148,149 It remains inconclusive, however, whether r-metHuLeptin therapy will affect bone density in the long term. Small human studies suggest that long-term r-metHuLeptin treatment may not affect bone density in patients with generalized lipodystrophies with regular menses.131,135,150
Leptin has been demonstrated to be a safe medication when administered in normal replacement doses. No side effects have been observed with long-term treatment up to 36 months.135 However, the effects of leptin appear not to be sustainable after the therapy is discontinued.129,132
Management of HIV-Infected Patients with Haart-Induced Metabolic Syndrome
Growth Hormone and Growth Hormone–Releasing Hormone Analogs
Growth hormone (GH) replacement has been proposed to have a promising role in HALS treatment in that this group of patients is more prone to growth hormone deficiency (GHD). It has been observed that basal GH concentrations, overnight GH secretion, and pulse amplitude are reduced in patients with HIV lipodystrophy,151 although normal pulse frequency is maintained.152 Relative GHD also appears to be common in HALS, as evidenced by decreased response to a standardized GH-releasing hormone arginine stimulation test.153 Although the causative relationship of GHD and metabolic abnormalities is not well understood, interventions that normalize GH concentrations have demonstrated efficacy in improving metabolic abnormalities associated with visceral fat accumulation in the HIV population.152
Treatment with high doses of GH (2 to 6 mg/day) has shown to effectively reduce visceral fat and improve lipid parameters.154–156 However, its application is hampered by significant side effects, including fluid retention, arthralgia, myalgia, carpal tunnel syndrome, and worsening glucose control. There is also the theoretical concern that long-term GH therapy may increase the risk for cancer. In addition, high-dose GH replacement may lead to a further decrease in peripheral subcutaneous fat,157 which is undesirable in this patient population from the cosmetic point of view. The effect of GH generally diminishes after discontinuation of treatment.
Increasing evidence demonstrates that physiologic GH replacement, with doses as low as 2 to 6 mcg/kg/day, can also effectively raise circulating IGF-1 levels and may provide similar benefits in visceral fat reduction but fewer adverse effects.158–161 Although low-dose GH treatment did not result in worsened insulin sensitivity,152 this potential side effect and the lack of durability with GH treatment limit its clinical application. GH therapy is not currently FDA approved for treatment of HALS.
Growth hormone–releasing hormone (GHRH) has been recently proposed as an alternative treatment option for HALS. It can augment endogenous GH pulsatility and may preserve the negative feedback of IGF-1 on the pituitary.152 Subcutaneous injection of the GHRH analog tesamorelin (1 to 2 mg/day in single or divided doses) has been shown to decrease visceral fat while preserving insulin sensitivity.162,163 Treatment with tesamorelin is also associated with improvement of triglycerides and total cholesterol levels,164 but its effect on HDL seems to be variable162,164 and needs to be confirmed by long-term controlled studies. The treatment with GHRH and analogs appears to be well tolerated. No adverse effects associated with GH were observed, although long-term data are lacking.
Modification of HAART
Interruption of antiretroviral therapy is associated with increased mortality and opportunistic infections. Therefore, it should be avoided. Instead, a careful evaluation for cardiovascular risk factors should be conducted before antiretroviral therapy is initiated, and the drug with the least metabolic implications and a comparable efficacy should be selected. Once lipoatrophy or metabolic complications develop, switching from the offending drug to another agent may be an important strategy.165 However, the reversal is normally slow and gradual.
In the case of lipoatrophy, the treatment regimen with two thymidine analogs should generally be avoided. Switching to abacavir or tenofovir may partially restore subcutaneous fat.166 Switching from a PI to an NNRTI or abacavir has not shown any beneficial effects in terms of improvement of lipoatrophy.
As regards to metabolic abnormalities, switching from a PI to nevirapine or abacavir has generally resulted in improved total cholesterol and triglycerides, whereas switching to efavirenz has generated less consistent results.165 Newer-generation protease inhibitors (e.g., atazanavir, darunavir, and saquinavir) are associated with favorable lipid profiles. However, because antiretroviral regimens containing PIs normally require low-dose ritonavir for its boosting effect, the impact of switching from one ritonavir-boosted PI to another may be modest.106
Uridine
Uridine is a pyridine precursor which reverses the mitochondrial toxicity. It may have clinical value in treating HIV-associated lipodystrophy induced by pyrimidines such as zalcitabine and stavudine. Uridine can also reverse the cell depletion and lactic acidosis seen with zidovudine and lamivudine combination. It has no effect on lipoatrophy caused by didanosine, a purine analog. Uridine is generally well tolerated.70
Management of Cosmetic Appearance
Autologous fat transplant and implantation of synthetic bulking agents have been used for the cosmetic correction of facial lipoatrophy and appears to be associated with improvements in quality of life. However, long-term, well-designed studies are needed to assess their efficacy and safety.167 Occasionally, the fat harvested from HIV-infected patients with buffalo hump can cause hypertrophies in the transplanted sites (cheeks), and cause a disfiguring “hamster” appearance. The intradermal injections of synthetic agents (polylactic acid or New-Fill) have been shown to result in a more durable increase in total cutaneous thickness (TCT) persisting up to 48 weeks.
Future Perspective
Adiponectin and its receptors AdiopoR1 and AdipoR2 are attractive future targets for drug development. Because of its complex molecular structure, synthetic adiponectin is not yet available for therapy in humans. However, it is likely to become a valuable addition to our armamentarium as a future treatment option for lipodystrophy. Based on the fact that adiponectin is decreased in lipodystrophic states168 and the evidence from mice studies showing that adiponectin administration can improve insulin sensitivity, dyslipidemia, sustained weight loss without reducing food intake, and production of proinflammatory cytokines,169,170 adiponectin analog administration in replacement doses may prove to be effective treatment options in the not-so-distant future.
References
1. Goodpaster, BH. Measuring body fat distribution and content in humans. Curr Opin Clin Nutr Metab Care. 2002;5(5):481–487.
2. Gulizia, R, Vercelli, A, Gervasoni, C, et al. Comparability of echographic and tomographic assessments of body fat changes related to the HIV associated adipose redistribution syndrome (HARS) in antiretroviral treated patients. Ultrasound Med Biol. 2008;34(7):1043–1048.
3. Bastard, JP, Caron, M, Vidal, H, et al. Association between altered expression of adipogenic factor SREBP1 in lipoatrophic adipose tissue from HIV-1-infected patients and abnormal adipocyte differentiation and insulin resistance. Lancet. 2002;359(9311):1026–1031.
4. Domingo, P, Matias-Guiu, X, Pujol, RM, et al. Subcutaneous adipocyte apoptosis in HIV-1 protease inhibitor-associated lipodystrophy. AIDS. 1999;13(16):2261–2267.
5. Reitman, ML, Arioglu, E, Gavrilova, O, et al. Lipoatrophy revisited. Trends Endocrinol Metab. 2000;11(10):410–416.
6. Berardinelli, W. An undiagnosed endocrinometabolic syndrome: report of 2 cases. J Clin Endocrinol Metab. 1954;14(2):193–204.
7. Seip, M. Lipodystrophy and gigantism with associated endocrine manifestations. A new diencephalic syndrome? Acta Paediatr. 1959;48:555–574.
8. Garg, A, Misra, A. Lipodystrophies: rare disorders causing metabolic syndrome. Endocrinol Metab Clin North Am. 2004;33(2):305–331.
9. Szymanski, KM, Binns, D, Bartz, R, et al. The lipodystrophy protein seipin is found at endoplasmic reticulum lipid droplet junctions and is important for droplet morphology. Proc Natl Acad Sci U S A. 2007;104(52):20890–20895.
10. Agarwal, AK, Simha, V, Oral, EA, et al. Phenotypic and genetic heterogeneity in congenital generalized lipodystrophy. J Clin Endocrinol Metab. 2003;88(10):4840–4847.
11. Garg, A, Fleckenstein, JL, Peshock, RM, et al. Peculiar distribution of adipose tissue in patients with congenital generalized lipodystrophy. J Clin Endocrinol Metab. 1992;75(2):358–361.
12. Simha, V, Garg, A. Phenotypic heterogeneity in body fat distribution in patients with congenital generalized lipodystrophy caused by mutations in the AGPAT2 or seipin genes. J Clin Endocrinol Metab. 2003;88(11):5433–5437.
13. Simha, V, Agarwal, AK, Aronin, PA, et al. Novel subtype of congenital generalized lipodystrophy associated with muscular weakness and cervical spine instability. Am J Med Genet A. 2008;146A(18):2318–2326.
14. Haque, WA, Shimomura, I, Matsuzawa, Y, et al. Serum adiponectin and leptin levels in patients with lipodystrophies. J Clin Endocrinol Metab. 2002;87(5):2395.
15. Brunzell, JD, Shankle, SW, Bethune, JE. Congenital generalized lipodystrophy accompanied by cystic angiomatosis. Ann Intern Med. 1968;69(3):501–516.
16. Fleckenstein, JL, Garg, A, Bonte, FJ, et al. The skeleton in congenital, generalized lipodystrophy: evaluation using whole-body radiographic surveys, magnetic resonance imaging and technetium-99m bone scintigraphy. Skeletal Radiol. 1992;21(6):381–386.
17. Guell-Gonzalez, JR, Mateo, dA, Alavez-Martin, E, et al. Bone lesions in congenital generalised lipodystrophy. Lancet. 1971;2(7715):104–105.
18. Van Maldergem, L, Magre, J, Khallouf, TE, et al. Genotype-phenotype relationships in Berardinelli-Seip congenital lipodystrophy. J Med Genet. 2002;39(10):722–733.
19. Bhayana, S, Siu, VM, Joubert, GI, et al. Cardiomyopathy in congenital complete lipodystrophy. Clin Genet. 2002;61(4):283–287.
20. Bjornstad, PG, Foerster, A, Ihlen, H. Cardiac findings in generalized lipodystrophy. Acta Paediatr Suppl. 1996;413:39–43.
21. Rheuban, KS, Blizzard, RM, Parker, MA, et al. Hypertrophic cardiomyopathy in total lipodystrophy. J Pediatr. 1986;109(2):301–302.
22. Agarwal, AK, Arioglu, E, De Almeida, S, et al. AGPAT2 is mutated in congenital generalized lipodystrophy linked to chromosome 9q34. Nat Genet. 2002;31(1):21–23.
23. Garg, A, Wilson, R, Barnes, R, et al. A gene for congenital generalized lipodystrophy maps to human chromosome 9q34. J Clin Endocrinol Metab. 1999;84(9):3390–3394.
24. Magre, J, Delepine, M, Khallouf, E, et al. Identification of the gene altered in Berardinelli-Seip congenital lipodystrophy on chromosome 11q13. Nat Genet. 2001;28(4):365–370.
25. Kim, CA, Delepine, M, Boutet, E, et al. Association of a homozygous nonsense caveolin-1 mutation with Berardinelli-Seip congenital lipodystrophy. J Clin Endocrinol Metab. 2008;93(4):1129–1134.
26. Garg, A. Acquired and inherited lipodystrophies. N Engl J Med. 2004;350(12):1220–1234.
27. Misra, A, Garg, A. Clinical features and metabolic derangements in acquired generalized lipodystrophy: case reports and review of the literature. Medicine (Baltimore). 2003;82(2):129–146.
28. Garg, A, Agarwal, AK. Caveolin-1: a new locus for human lipodystrophy. J Clin Endocrinol Metab. 2008;93(4):1183–1185.
29. Misra, A, Garg, A. Clinical features and metabolic derangements in acquired generalized lipodystrophy: case reports and review of the literature. Medicine (Baltimore). 2003;82(2):129–146.
30. Pope, E, Janson, A, Khambalia, A, et al. Childhood acquired lipodystrophy: a retrospective study. J Am Acad Dermatol. 2006;55(6):947–950.
31. Bingham, A, Mamyrova, G, Rother, KI, et al. Predictors of acquired lipodystrophy in juvenile-onset dermatomyositis and a gradient of severity. Medicine (Baltimore). 2008;87(2):70–86.
32. Huemer, C, Kitson, H, Malleson, PN, et al. Lipodystrophy in patients with juvenile dermatomyositis–evaluation of clinical and metabolic abnormalities. J Rheumatol. 2001;28(3):610–615.
33. McCann, LJ, Juggins, AD, Maillard, SM, et al. The Juvenile Dermatomyositis National Registry and Repository (UK and Ireland)–clinical characteristics of children recruited within the first 5 yr. Rheumatology (Oxford). 2006;45(10):1255–1260.
34. Singh, S, Bansal, A. Twelve years experience of juvenile dermatomyositis in North India. Rheumatol Int. 2006;26(6):510–515.
35. Verma, S, Singh, S, Bhalla, AK, et al. Study of subcutaneous fat in children with juvenile dermatomyositis. Arthritis Rheum. 2006;55(4):564–568.
36. Billings, JK, Milgraum, SS, Gupta, AK, et al. Lipoatrophic panniculitis: a possible autoimmune inflammatory disease of fat. Report of three cases. Arch Dermatol. 1987;123(12):1662–1666.
37. Arioglu, E, Andewelt, A, Diabo, C, et al. Clinical course of the syndrome of autoantibodies to the insulin receptor (type B insulin resistance): a 28-year perspective. Medicine (Baltimore). 2002;81(2):87–100.
38. Feve, B. Adipogenesis: cellular and molecular aspects. Best Pract Res Clin Endocrinol Metab. 2005;19(4):483–499.
39. Fischer-Posovszky, P, Hebestreit, H, Hofmann, AK, et al. Role of CD95-mediated adipocyte loss in autoimmune lipodystrophy. J Clin Endocrinol Metab. 2006;91(3):1129–1135.
40. Vantyghem, MC, Vincent-Desplanques, D, Defrance-Faivre, F, et al. Fertility and obstetrical complications in women with LMNA-related familial partial lipodystrophy. J Clin Endocrinol Metab. 2008;93(6):2223–2229.
41. Hegele, RA, Joy, TR, Al Attar, SA, et al. Thematic review series: Adipocyte Biology. Lipodystrophies: windows on adipose biology and metabolism. J Lipid Res. 2007;48(7):1433–1444.
42. Agarwal, AK, Barnes, RI, Garg, A. Genetic basis of congenital generalized lipodystrophy. Int J Obes Relat Metab Disord. 2004;28(2):336–339.
43. Araujo-Vilar, D, Lattanzi, G, Gonzalez-Mendez, B, et al. Site-dependent differences in both prelamin A and adipogenic genes in subcutaneous adipose tissue of patients with type 2 familial partial lipodystrophy. J Med Genet. 2008.
44. Capanni, C, Mattioli, E, Columbaro, M, et al. Altered pre-lamin A processing is a common mechanism leading to lipodystrophy. Hum Mol Genet. 2005;14(11):1489–1502.
45. Lloyd, DJ, Trembath, RC, Shackleton, S. A novel interaction between lamin A and SREBP1: implications for partial lipodystrophy and other laminopathies. Hum Mol Genet. 2002;11(7):769–777.
46. Herbst, KL, Tannock, LR, Deeb, SS, et al. Köbberling type of familial partial lipodystrophy: an underrecognized syndrome. Diabetes Care. 2003;26(6):1819–1824.
47. Agarwal, AK, Garg, A. A novel heterozygous mutation in peroxisome proliferator-activated receptor-gamma gene in a patient with familial partial lipodystrophy. J Clin Endocrinol Metab. 2002;87(1):408–411.
48. Al Shali, K, Cao, H, Knoers, N, et al. A single-base mutation in the peroxisome proliferator-activated receptor gamma4 promoter associated with altered in vitro expression and partial lipodystrophy. J Clin Endocrinol Metab. 2004;89(11):5655–5660.
49. Ludtke, A, Buettner, J, Wu, W, et al. Peroxisome proliferator-activated receptor-gamma C190S mutation causes partial lipodystrophy. J Clin Endocrinol Metab. 2007;92(6):2248–2255.
50. Monajemi, H, Zhang, L, Li, G, et al. Familial partial lipodystrophy phenotype resulting from a single-base mutation in deoxyribonucleic acid-binding domain of peroxisome proliferator-activated receptor-gamma. J Clin Endocrinol Metab. 2007;92(5):1606–1612.
51. George, S, Rochford, JJ, Wolfrum, C, et al. A family with severe insulin resistance and diabetes due to a mutation in AKT2. Science. 2004;304(5675):1325–1328.
52. Chehab, FF. Obesity and lipodystrophy–where do the circles intersect? Endocrinology. 2008;149(3):925–934.
53. Cao, H, Alston, L, Ruschman, J, et al. Heterozygous CAV1 frameshift mutations (MIM 601047) in patients with atypical partial lipodystrophy and hypertriglyceridemia. Lipids Health Dis. 2008;7:3.
54. Miyoshi, Y, Akagi, M, Agarwal, AK, et al. Severe mandibuloacral dysplasia caused by novel compound heterozygous ZMPSTE24 mutations in two Japanese siblings. Clin Genet. 2008;73(6):535–544.
55. Agarwal, AK, Zhou, XJ, Hall, RK, et al. Focal segmental glomerulosclerosis in patients with mandibuloacral dysplasia owing to ZMPSTE24 deficiency. J Investig Med. 2006;54(4):208–213.
56. Eisinger, AJ, Shortland, JR, Moorhead, PJ. Renal disease in partial lipodystrophy. Q J Med. 1972;41(163):343–354.
57. Sissons, JG, West, RJ, Fallows, J, et al. The complement abnormalities of lipodystrophy. N Engl J Med. 1976;294(9):461–465.
58. Allen, BR. Partial lipodystrophy. Br J Dermatol. 1978;99(Suppl 16):48–49.
59. Hegele, RA, Cao, H, Liu, DM, et al. Sequencing of the reannotated LMNB2 gene reveals novel mutations in patients with acquired partial lipodystrophy. Am J Hum Genet. 2006;79(2):383–389.
60. Guallar, JP, Rojas-Garcia, R, Garcia-Arumi, E, et al. Impaired expression of mitochondrial and adipogenic genes in adipose tissue from a patient with acquired partial lipodystrophy (Barraquer-Simons syndrome): a case report. J Med Case Reports. 2008;2:284.
61. Oh, J, Hegele, RA. HIV-associated dyslipidaemia: pathogenesis and treatment. Lancet Infect Dis. 2007;7(12):787–796.
62. Depairon, M, Chessex, S, Sudre, P, et al. Premature atherosclerosis in HIV-infected individuals–focus on protease inhibitor therapy. AIDS. 2001;15(3):329–334.
63. Maggi, P, Serio, G, Epifani, G, et al. Premature lesions of the carotid vessels in HIV-1-infected patients treated with protease inhibitors. AIDS. 2000;14(16):F123–F128.
64. Seminari, E, Pan, A, Voltini, G, et al. Assessment of atherosclerosis using carotid ultrasonography in a cohort of HIV-positive patients treated with protease inhibitors. Atherosclerosis. 2002;162(2):433–438.
65. Stein, JH, Klein, MA, Bellehumeur, JL, et al. Use of human immunodeficiency virus-1 protease inhibitors is associated with atherogenic lipoprotein changes and endothelial dysfunction. Circulation. 2001;104(3):257–262.
66. Sweeney, LL, Brennan, AM, Mantzoros, CS. The role of adipokines in relation to HIV lipodystrophy. AIDS. 2007;21(8):895–904.
67. Nagy, GS, Tsiodras, S, Martin, LD, et al. Human immunodeficiency virus type 1-related lipoatrophy and lipohypertrophy are associated with serum concentrations of leptin. Clin Infect Dis. 2003;36(6):795–802.
68. Jacobson, DL, Knox, T, Spiegelman, D, et al. Prevalence of, evolution of, and risk factors for fat atrophy and fat deposition in a cohort of HIV-infected men and women. Clin Infect Dis. 2005;40(12):1837–1845.
69. Duong, M, Petit, JM, Piroth, L, et al. Association between insulin resistance and hepatitis C virus chronic infection in HIV-hepatitis C virus-coinfected patients undergoing antiretroviral therapy. J Acquir Immune Defic Syndr. 2001;27(3):245–250.
70. Mallewa, JE, Wilkins, E, Vilar, J, et al. HIV-associated lipodystrophy: a review of underlying mechanisms and therapeutic options. J Antimicrob Chemother. 2008;62(4):648–660.
71. Bastard, JP, Caron, M, Vidal, H, et al. Association between altered expression of adipogenic factor SREBP1 in lipoatrophic adipose tissue from HIV-1-infected patients and abnormal adipocyte differentiation and insulin resistance. Lancet. 2002;359(9311):1026–1031.
72. El Hadri, K, Glorian, M, Monsempes, C, et al. In vitro suppression of the lipogenic pathway by the nonnucleoside reverse transcriptase inhibitor efavirenz in 3T3 and human preadipocytes or adipocytes. J Biol Chem. 2004;279(15):15130–15141.
73. Haubrich, RH, Kemper, CA, Hellmann, NS, et al. A randomized, prospective study of phenotype susceptibility testing versus standard of care to manage antiretroviral therapy: CCTG 575. AIDS. 2005;19(3):295–302.
74. Perez-Molina, JA, Domingo, P, Martinez, E, et al. The role of efavirenz compared with protease inhibitors in the body fat changes associated with highly active antiretroviral therapy. J Antimicrob Chemother. 2008;62(2):234–245.
75. Villarroya, F, Domingo, P, Giralt, M. Lipodystrophy in HIV 1-infected patients: lessons for obesity research. Int J Obes (Lond). 2007;31(12):1763–1776.
76. Mittra, S, Bansal, VS, Bhatnagar, PK. From a glucocentric to a lipocentric approach towards metabolic syndrome. Drug Discov Today. 2008;13(5–6):211–218.
77. Petersen, KF, Shulman, GI. Etiology of insulin resistance. Am J Med. 2006;119(5 Suppl 1):S10–S16.
78. Bays, HE, Gonzalez-Campoy, JM, Bray, GA, et al. Pathogenic potential of adipose tissue and metabolic consequences of adipocyte hypertrophy and increased visceral adiposity. Expert Rev Cardiovasc Ther. 2008;6(3):343–368.
79. Matsubara, M, Maruoka, S, Katayose, S. Inverse relationship between plasma adiponectin and leptin concentrations in normal-weight and obese women. Eur J Endocrinol. 2002;147(2):173–180.
80. Weyer, C, Funahashi, T, Tanaka, S, et al. Hypoadiponectinemia in obesity and type 2 diabetes: close association with insulin resistance and hyperinsulinemia. J Clin Endocrinol Metab. 2001;86(5):1930–1935.
81. Tilg, H, Moschen, AR. Adipocytokines: mediators linking adipose tissue, inflammation and immunity. Nat Rev Immunol. 2006;6(10):772–783.
82. Sheng, T, Yang, K. Adiponectin and its association with insulin resistance and type 2 diabetes. J Genet Genomics. 2008;35(6):321–326.
83. Coope, A, Milanski, M, Araujo, EP, et al. AdipoR1 mediates the anorexigenic and insulin/leptin-like actions of adiponectin in the hypothalamus. FEBS Lett. 2008;582(10):1471–1476.
84. Mantzoros, CS, Li, T, Manson, JE, et al. Circulating adiponectin levels are associated with better glycemic control, more favorable lipid profile, and reduced inflammation in women with type 2 diabetes. J Clin Endocrinol Metab. 2005;90(8):4542–4548.
85. Ceddia, RB. Direct metabolic regulation in skeletal muscle and fat tissue by leptin: implications for glucose and fatty acids homeostasis. Int J Obes (Lond). 2005;29(10):1175–1183.
86. Minokoshi, Y, Kahn, BB. Role of AMP-activated protein kinase in leptin-induced fatty acid oxidation in muscle. Biochem Soc Trans. 2003;31(Pt 1):196–201.
87. Lee, JH, Chan, JL, Sourlas, E, et al. Recombinant methionyl human leptin therapy in replacement doses improves insulin resistance and metabolic profile in patients with lipoatrophy and metabolic syndrome induced by the highly active antiretroviral therapy. J Clin Endocrinol Metab. 2006;91(7):2605–2611.
88. Guest, CB, Park, MJ, Johnson, DR, et al. The implication of proinflammatory cytokines in type 2 diabetes. Front Biosci. 2008;13:5187–5194.
89. Shoelson, SE, Herrero, L, Naaz, A. Obesity, inflammation, and insulin resistance. Gastroenterology. 2007;132(6):2169–2180.
90. Sevastianova, K, Sutinen, J, Kannisto, K, et al. Adipose tissue inflammation and liver fat in patients with highly active antiretroviral therapy-associated lipodystrophy. Am J Physiol Endocrinol Metab. 2008;295(1):E85–E91.
91. He, G, Andersen, O, Haugaard, SB, et al. Plasminogen activator inhibitor type 1 (PAI-1) in plasma and adipose tissue in HIV-associated lipodystrophy syndrome. Implications of adipokines. Eur J Clin Invest. 2005;35(9):583–590.
92. Cawthorn, WP, Heyd, F, Hegyi, K, et al. Tumour necrosis factor-alpha inhibits adipogenesis via a beta-catenin/TCF4(TCF7L2)-dependent pathway. Cell Death Differ. 2007;14(7):1361–1373.
93. Cawthorn, WP, Sethi, JK. TNF-alpha and adipocyte biology. FEBS Lett. 2008;582(1):117–131.
94. Glund, S, Krook, A. Role of interleukin-6 signalling in glucose and lipid metabolism. Acta Physiol (Oxf). 2008;192(1):37–48.
95. Ito, D, Suzuki, N. Seipinopathy: a novel endoplasmic reticulum stress-associated disease. Brain. 2008.
96. Rhodes, CJ. Type 2 diabetes: a matter of beta-cell life and death? Science. 2005;307(5708):380–384.
97. Donath, MY, Ehses, JA, Maedler, K, et al. Mechanisms of beta-cell death in type 2 diabetes. Diabetes. 2005;54(Suppl 2)):S108–S113.
98. White, M, Lepage, S, Lavoie, J, et al. Effects of combined candesartan and ACE inhibitors on BNP, markers of inflammation and oxidative stress, and glucose regulation in patients with symptomatic heart failure. J Card Fail. 2007;13(2):86–94.
99. Samaras, K. Metabolic consequences and therapeutic options in highly active antiretroviral therapy in human immunodeficiency virus-1 infection. J Antimicrob Chemother. 2008;61(2):238–245.
100. Noor, MA, Seneviratne, T, Aweeka, FT, et al. Indinavir acutely inhibits insulin-stimulated glucose disposal in humans: a randomized, placebo-controlled study. AIDS. 2002;16(5):F1–F8.
101. Shah, M, Tierney, K, Adams-Huet, B, et al. The role of diet, exercise and smoking in dyslipidaemia in HIV-infected patients with lipodystrophy. HIV Med. 2005;6(4):291–298.
102. Dong, KR, Hendricks, KM. The role of nutrition in fat deposition and fat atrophy in patients with HIV. Nutr Clin Care. 2005;8(1):31–36.
103. Terry, L, Sprinz, E, Stein, R, et al. Exercise training in HIV-1-infected individuals with dyslipidemia and lipodystrophy. Med Sci Sports Exerc. 2006;38(3):411–417.
104. Dolan, SE, Frontera, W, Librizzi, J, et al. Effects of a supervised home-based aerobic and progressive resistance training regimen in women infected with human immunodeficiency virus: a randomized trial. Arch Intern Med. 2006;166(11):1225–1231.
105. Lindegaard, B, Hansen, T, Hvid, T, et al. The effect of strength and endurance training on insulin sensitivity and fat distribution in human immunodeficiency virus-infected patients with lipodystrophy. J Clin Endocrinol Metab. 2008;93(10):3860–3869.
106. Brown, TT. Approach to the human immunodeficiency virus-infected patient with lipodystrophy. J Clin Endocrinol Metab. 2008;93(8):2937–2945.
107. Aboud, M, Elgalib, A, Kulasegaram, R, et al. Insulin resistance and HIV infection: a review. Int J Clin Pract. 2007;61(3):463–472.
108. Kohli, R, Shevitz, A, Gorbach, S, et al. A randomized placebo-controlled trial of metformin for the treatment of HIV lipodystrophy. HIV Med. 2007;8(7):420–426.
109. Mulligan, K, Yang, Y, Wininger, DA, et al. Effects of metformin and rosiglitazone in HIV-infected patients with hyperinsulinemia and elevated waist/hip ratio. AIDS. 2007;21(1):47–57.
110. Kohli, R, Shevitz, A, Gorbach, S, et al. A randomized placebo-controlled trial of metformin for the treatment of HIV lipodystrophy. HIV Med. 2007;8(7):420–426.
111. Driscoll, SD, Meininger, GE, Ljungquist, K, et al. Differential effects of metformin and exercise on muscle adiposity and metabolic indices in human immunodeficiency virus-infected patients. J Clin Endocrinol Metab. 2004;89(5):2171–2178.
112. Hadigan, C, Yawetz, S, Thomas, A, et al. Metabolic effects of rosiglitazone in HIV lipodystrophy: a randomized, controlled trial. Ann Intern Med. 2004;140(10):786–794.
113. Macallan, DC, Baldwin, C, Mandalia, S, et al. Treatment of altered body composition in HIV-associated lipodystrophy: comparison of rosiglitazone, pravastatin, and recombinant human growth hormone. HIV Clin Trials. 2008;9(4):254–268.
114. Slama, L, Lanoy, E, Valantin, MA, et al. Effect of pioglitazone on HIV-1-related lipodystrophy: a randomized double-blind placebo-controlled trial (ANRS 113). Antivir Ther. 2008;13(1):67–76.
115. van Wijk, JP, de Koning, EJ, Cabezas, MC, et al. Comparison of rosiglitazone and metformin for treating HIV lipodystrophy: a randomized trial. Ann Intern Med. 2005;143(5):337–346.
116. Carr, A, Workman, C, Carey, D, et al. No effect of rosiglitazone for treatment of HIV-1 lipoatrophy: randomised, double-blind, placebo-controlled trial. Lancet. 2004;363(9407):429–438.
117. Cavalcanti, RB, Raboud, J, Shen, S, et al. A randomized, placebo-controlled trial of rosiglitazone for HIV-related lipoatrophy. J Infect Dis. 2007;195(12):1754–1761.
118. Sutinen, J, Hakkinen, AM, Westerbacka, J, et al. Rosiglitazone in the treatment of HAART-associated lipodystrophy–a randomized double-blind placebo-controlled study. Antivir Ther. 2003;8(3):199–207.
119. Kovacic, JC, Martin, A, Carey, D, et al. Influence of rosiglitazone on flow-mediated dilation and other markers of cardiovascular risk in HIV-infected patients with lipoatrophy. Antivir Ther. 2005;10(1):135–143.
120. Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation. 2002;106(25):3143–3421.
121. Aberg, JA. Management of dyslipidemia and other cardiovascular risk factors in HIV-infected patients: case-based review. Top HIV Med. 2006;14(4):134–139.
122. Mallon, PW, Miller, J, Kovacic, JC, et al. Effect of pravastatin on body composition and markers of cardiovascular disease in HIV-infected men—a randomized, placebo-controlled study. AIDS. 2006;20(7):1003–1010.
123. Aberg, JA, Rosenkranz, SL, Fichtenbaum, CJ, et al. Pharmacokinetic interaction between nelfinavir and pravastatin in HIV-seronegative volunteers: ACTG Study A5108. AIDS. 2006;20(5):725–729.
124. Fichtenbaum, CJ, Gerber, JG. Interactions between antiretroviral drugs and drugs used for the therapy of the metabolic complications encountered during HIV infection. Clin Pharmacokinet. 2002;41(14):1195–1211.
125. Gerber, JG, Rosenkranz, SL, Fichtenbaum, CJ, et al. Effect of efavirenz on the pharmacokinetics of simvastatin, atorvastatin, and pravastatin: results of AIDS Clinical Trials Group 5108 Study. J Acquir Immune Defic Syndr. 2005;39(3):307–312.
126. Hadigan, C, Liebau, J, Torriani, M, et al. Improved triglycerides and insulin sensitivity with 3 months of acipimox in human immunodeficiency virus-infected patients with hypertriglyceridemia. J Clin Endocrinol Metab. 2006;91(11):4438–4444.
127. Kastelein, JJ, Akdim, F, Stroes, ES, et al. Simvastatin with or without ezetimibe in familial hypercholesterolemia. N Engl J Med. 2008;358(14):1431–1443.
128. Wohl, DA, Tien, HC, Busby, M, et al. Randomized study of the safety and efficacy of fish oil (omega-3 fatty acid) supplementation with dietary and exercise counseling for the treatment of antiretroviral therapy-associated hypertriglyceridemia. Clin Infect Dis. 2005;41(10):1498–1504.
129. Javor, ED, Cochran, EK, Musso, C, et al. Long-term efficacy of leptin replacement in patients with generalized lipodystrophy. Diabetes. 2005;54(7):1994–2002.
130. McDuffie, JR, Riggs, PA, Calis, KA, et al. Effects of exogenous leptin on satiety and satiation in patients with lipodystrophy and leptin insufficiency. J Clin Endocrinol Metab. 2004;89(9):4258–4263.
131. Moran, SA, Patten, N, Young, JR, et al. Changes in body composition in patients with severe lipodystrophy after leptin replacement therapy. Metabolism. 2004;53(4):513–519.
132. Oral, EA, Simha, V, Ruiz, E, et al. Leptin-replacement therapy for lipodystrophy. N Engl J Med. 2002;346(8):570–578.
133. Andreev, VP, Paz-Filho, G, Wong, ML, et al. Deconvolution of insulin secretion, insulin hepatic extraction post-hepatic delivery rates and sensitivity during 24-hour standardized meals: time course of glucose homeostasis in leptin replacement treatment. Horm Metab Res. 2008.
134. Beltrand, J, Beregszaszi, M, Chevenne, D, et al. Metabolic correction induced by leptin replacement treatment in young children with Berardinelli-Seip congenital lipoatrophy. Pediatrics. 2007;120(2):e291–e296.
135. Ebihara, K, Kusakabe, T, Hirata, M, et al. Efficacy and safety of leptin-replacement therapy and possible mechanisms of leptin actions in patients with generalized lipodystrophy. J Clin Endocrinol Metab. 2007;92(2):532–541.
136. Javor, ED, Ghany, MG, Cochran, EK, et al. Leptin reverses nonalcoholic steatohepatitis in patients with severe lipodystrophy. Hepatology. 2005;41(4):753–760.
137. Simha, V, Szczepaniak, LS, Wagner, AJ, et al. Effect of leptin replacement on intrahepatic and intramyocellular lipid content in patients with generalized lipodystrophy. Diabetes Care. 2003;26(1):30–35.
138. Guettier, JM, Park, JY, Cochran, EK, et al. Leptin therapy for partial lipodystrophy linked to a PPAR-gamma mutation. Clin Endocrinol (Oxf). 2008;68(4):547–554.
139. Park, JY, Javor, ED, Cochran, EK, et al. Long-term efficacy of leptin replacement in patients with Dunnigan-type familial partial lipodystrophy. Metabolism. 2007;56(4):508–516.
140. Park, S, Hong, SM, Sung, SR, et al. Long-term effects of central leptin and resistin on body weight, insulin resistance, and beta-cell function and mass by the modulation of hypothalamic leptin and insulin signaling. Endocrinology. 2008;149(2):445–454.
141. Farooqi, IS, Bullmore, E, Keogh, J, et al. Leptin regulates striatal regions and human eating behavior. Science. 2007;317(5843):1355.
142. Rosenbaum, M, Sy, M, Pavlovich, K, et al. Leptin reverses weight-loss-induced changes in regional neural activity responses to visual food stimuli. J Clin Invest. 2008;118(7):2583–2591.
143. Farooqi, IS, Matarese, G, Lord, GM, et al. Beneficial effects of leptin on obesity, T cell hyporesponsiveness, and neuroendocrine/metabolic dysfunction of human congenital leptin deficiency. J Clin Invest. 2002;110(8):1093–1103.
144. Gallardo, N, Bonzon-Kulichenko, E, Fernandez-Agullo, T, et al. Tissue-specific effects of central leptin on the expression of genes involved in lipid metabolism in liver and white adipose tissue. Endocrinology. 2007;148(12):5604–5610.
145. Prieur, X, Tung, YC, Griffin, JL, et al. Leptin regulates peripheral lipid metabolism primarily through central effects on food intake. Endocrinology. 2008;149(11):5432–5439.
146. Maedler, K, Schulthess, FT, Bielman, C, et al. Glucose and leptin induce apoptosis in human beta-cells and impair glucose-stimulated insulin secretion through activation of c-Jun N-terminal kinases. FASEB J. 2008;22(6):1905–1913.
147. Javor, ED, Moran, SA, Young, JR, et al. Proteinuric nephropathy in acquired and congenital generalized lipodystrophy: baseline characteristics and course during recombinant leptin therapy. J Clin Endocrinol Metab. 2004;89(7):3199–3207.
148. Musso, C, Cochran, E, Javor, E, et al. The long-term effect of recombinant methionyl human leptin therapy on hyperandrogenism and menstrual function in female and pituitary function in male and female hypoleptinemic lipodystrophic patients. Metabolism. 2005;54(2):255–263.
149. Oral, EA, Ruiz, E, Andewelt, A, et al. Effect of leptin replacement on pituitary hormone regulation in patients with severe lipodystrophy. J Clin Endocrinol Metab. 2002;87(7):3110–3117.
150. Simha, V, Zerwekh, JE, Sakhaee, K, et al. Effect of subcutaneous leptin replacement therapy on bone metabolism in patients with generalized lipodystrophy. J Clin Endocrinol Metab. 2002;87(11):4942–4945.
151. Rietschel, P, Hadigan, C, Corcoran, C, et al. Assessment of growth hormone dynamics in human immunodeficiency virus-related lipodystrophy. J Clin Endocrinol Metab. 2001;86(2):504–510.
152. Stanley, TL, Grinspoon, SK. GH/GHRH axis in HIV lipodystrophy. Pituitary. 2009;12(2):143–152.
153. Koutkia, P, Canavan, B, Breu, J, et al. Growth hormone (GH) responses to GH-releasing hormone-arginine testing in human immunodeficiency virus lipodystrophy. J Clin Endocrinol Metab. 2005;90(1):32–38.
154. Bickel, M, Zangos, S, Jacobi, V, et al. A randomized, open-label study to compare the effects of two different doses of recombinant human growth hormone on fat reduction and fasting metabolic parameters in HIV-1-infected patients with lipodystrophy. HIV Med. 2006;7(6):397–403.
155. Grunfeld, C, Thompson, M, Brown, SJ, et al. Recombinant human growth hormone to treat HIV-associated adipose redistribution syndrome: 12 week induction and 24-week maintenance therapy. J Acquir Immune Defic Syndr. 2007;45(3):286–297.
156. He, Q, Engelson, ES, Albu, JB, et al. Preferential loss of omental-mesenteric fat during growth hormone therapy of HIV-associated lipodystrophy. J Appl Physiol. 2003;94(5):2051–2057.
157. Engelson, ES, Glesby, MJ, Mendez, D, et al. Effect of recombinant human growth hormone in the treatment of visceral fat accumulation in HIV infection. J Acquir Immune Defic Syndr. 2002;30(4):379–391.
158. Andersen, O, Haugaard, SB, Flyvbjerg, A, et al. Low-dose growth hormone and human immunodeficiency virus-associated lipodystrophy syndrome: a pilot study. Eur J Clin Invest. 2004;34(8):561–568.
159. Lo, J, You, SM, Canavan, B, et al. Low-dose physiological growth hormone in patients with HIV and abdominal fat accumulation: a randomized controlled trial. JAMA. 2008;300(5):509–519.
160. Lo, JC, Mulligan, K, Noor, MA, et al. The effects of low-dose growth hormone in HIV-infected men with fat accumulation: a pilot study. Clin Infect Dis. 2004;39(5):732–735.
161. Luzi, L, Meneghini, E, Oggionni, S, et al. GH treatment reduces trunkal adiposity in HIV-infected patients with lipodystrophy: a randomized placebo-controlled study. Eur J Endocrinol. 2005;153(6):781–789.
162. Falutz, J, Allas, S, Blot, K, et al. Metabolic effects of a growth hormone-releasing factor in patients with HIV. N Engl J Med. 2007;357(23):2359–2370.
163. Koutkia, P, Canavan, B, Breu, J, et al. Growth hormone-releasing hormone in HIV-infected men with lipodystrophy: a randomized controlled trial. JAMA. 2004;292(2):210–218.
164. Falutz, J, Allas, S, Mamputu, JC, et al. Long-term safety and effects of tesamorelin, a growth hormone-releasing factor analogue, in HIV patients with abdominal fat accumulation. AIDS. 2008;22(14):1719–1728.
165. Dube, MP, Stein, JH, Aberg, JA, et al. Guidelines for the evaluation and management of dyslipidemia in human immunodeficiency virus (HIV)-infected adults receiving antiretroviral therapy: recommendations of the HIV Medical Association of the Infectious Disease Society of America and the Adult AIDS Clinical Trials Group. Clin Infect Dis. 2003;37(5):613–627.
166. Lundgren, JD, Battegay, M, Behrens, G, et al. European AIDS Clinical Society (EACS) guidelines on the prevention and management of metabolic diseases in HIV. HIV Med. 2008;9(2):72–81.
167. Carey, D, Liew, S, Emery, S. Restorative interventions for HIV facial lipoatrophy. AIDS Rev. 2008;10(2):116–124.
168. Haque, WA, Shimomura, I, Matsuzawa, Y, et al. Serum adiponectin and leptin levels in patients with lipodystrophies. J Clin Endocrinol Metab. 2002;87(5):2395.
169. Duntas, LH, Popovic, V, Panotopoulos, G. Adiponectin: novelties in metabolism and hormonal regulation. Nutr Neurosci. 2004;7(4):195–200.
170. Xu, A, Yin, S, Wong, L, et al. Adiponectin ameliorates dyslipidemia induced by the human immunodeficiency virus protease inhibitor ritonavir in mice. Endocrinology. 2004;145(2):487–494.
[/level-membership-for-endocrinology-diabetes-and-metabolism-category][not-level-membership-for-endocrinology-diabetes-and-metabolism-category]Chapter 12
Lipodystrophy Syndromes
Lipodystrophies: Definition and Diagnosis
Pathophysiology of Lipodystrophy
Classifications of Lipodystrophies and Their Clinical Manifestations
Mechanisms Responsible for Severe Insulin Resistance
Lipodystrophies: Definition and Diagnosis
The lipodystrophic states are a diverse group of clinical disorders, the central feature of which is either a congenital or acquired, complete or partial lack of adipose tissue (lipoatrophy), and/or a combination of lack of adipose tissue in certain body areas, with excess of adipose tissue (lipohypertrophy) elsewhere. Insulin resistance and associated clinical features are present in nearly all varieties of lipodystrophies. In addition, patients with lipodystrophies also manifest a group of unique features such as severe hyperlipidemia, progressive liver disease, and increased metabolic rate (Fig. 12-1).
Dual-energy x-ray absorptiometry (DEXA), magnetic resonance imaging (MRI), and computed tomography (CT) scan are additional modalities that have been developed to allow direct quantification of fat within specific tissues and/or body mass, as well as fat distribution. Although all these techniques are accurate and noninvasive, they are also expensive, have limited availability, and are apparently not cost-effective for use in everyday clinical practice. Their use, therefore, has been limited to the research field.1
In the past few years, ultrasound (US) has emerged as a promising alternative to assess body fat changes. Although it demonstrates good accuracy and accessibility, more studies are needed to elucidate its value and to comparatively evaluate ultrasound (US) with other imaging modalities. Standardization of the US techniques and cost-effectiveness analyses will also be essential for its potential wider clinical applications.2
Pathophysiology of Lipodystrophy
Much of the knowledge on the mechanisms underlying the pathogenesis and manifestations of lipodystrophies has been obtained through the performance of mouse studies and from the human genome sequencing. It is now understood that patients with lipodystrophy have primarily a loss of mature, functional adipocytes, as opposed to an absence of lipids in otherwise normal adipocytes.3–5 The underlying defects could be associated with failure of adipogenesis, adipocyte apoptosis, or a failure to store triglycerides in existing adipocytes because of ineffective lipogenesis or excessive lipolysis.
Classifications of Lipodystrophies and Their Clinical Manifestations
Generalized Lipodystrophy
Generalized lipodystrophy encompasses rare but clinically striking disorders that may be congenital (Berardinelli-Seip syndrome)6,7 or acquired (Lawrence syndrome).
Congenital Generalized Lipodystrophy
Congenital generalized lipodystrophy (CGL), or Berardinelli-Seip congenital lipodystrophy (BSCL), is a rare syndrome characterized by near complete absence of body fat. It is inherited in an autosomal recessive fashion and is observed in the highest frequency with parental consanguinity. To date, it has been reported in approximately 250 patients with various ethnic backgrounds.8,9
Babies with CGL are noted to have an abnormal appearance due to absence of body fat within the first 2 years of life and frequently soon after birth. Adipose tissue is absent from not only subcutaneous but also from intraabdominal sites. Magnetic resonance (MR) imaging of the abdomen shows complete absence of intraabdominal, retroperitoneal, and subcutaneous fat but a prominent fatty liver and presence of fat in certain anatomic sites such as orbits, palms, and soles. Thus this genetic defect results in poor development of metabolically active but not mechanically important adipose tissue.10–12
Other somatic abnormalities that contribute to the abnormal appearance are acanthosis nigricans, a protuberant abdomen associated with hepatomegaly and/or splenomegaly, and prominent musculature. Congenital muscular weakness and cervical spine instability has also been reported in occasional cases.13 Females may present with enlarged clitoris, increased body hair, absence of or irregular menstrual cycles, and polycystic ovaries. Only a few affected women have had successful pregnancies, whereas affected men have normal fertility. The patients with CGL tend to have voracious appetite. The basal metabolic rate of their body may also be increased. Although patients may have accelerated linear growth and advanced bone age during their childhood, they normally have normal or reduced heights as adults.
Although both boys and girls are affected at similar rates, the metabolic features tend to be more severe and develop earlier in girls. Hypertriglyceridemia is characterized by increased concentrations of very low–density lipoproteins (VLDLs) and chylomicrons, whereas serum high-density lipoprotein (HDL) is usually low. Severely elevated triglycerides may provoke acute pancreatitis and is frequently related to fatty liver. This commonly progresses to cirrhosis, which in many cases may be fatal. Insulin resistance has been noted at an early age and may be present even at birth. Clinical diabetes usually develops in the early teens, is rarely ketotic, and is usually refractory to insulin therapy. Serum adipocytokines, the hormones produced by adipose tissue (e.g., leptin and adiponectin), circulate in extremely low levels in CGL.14 CGL may also be associated with focal lytic lesions in the long bones,15–17 mild mental retardation,18 cardiomyopathy,19–21 generalized muscle weakness, and cervical spine instability.13
Type 1 CGL (CGL1) is due to AGPAT2 gene mutations. This gene has been mapped to chromosome 9q34.22,23 It encodes the enzyme acyltransferase 1-acylglycerol-3-phosphate O-acyltransferase 2 (AGPAT2) which catalyzes the acylation of lysophosphatidic acid to form phosphatidic acid, a key intermediate in the biosynthesis of triacylglyceride and glycerophospholipids. AGPAT2 mutations are found predominantly in patients of African ancestry.
Type 2 CGL (CGL2) is due to BSCL2 gene mutations. This gene is located in chromosome 11q13 and encodes a 398-amino-acid protein called seipin.24 The BSCL2 gene mutation has been found in patients of European and Middle Eastern origins but has also been reported as a causative gene in Japanese patients with CGL. Seipin is expressed diffusely in many tissues but predominantly in testis and brain; its function in humans is largely unknown. A recent study in yeast suggests that seipin is important for lipid droplet morphology and perhaps assembly.9 Another study in cultured murine and human adipocytes also indicates that BSCL2 expression is critical for normal adipogenesis in vitro, as cells lacking BSCL2 failed to induce expression of key lipogenic transcription factors (peroxisome proliferator–activated receptor gamma [PPARG] and CCAAT/enhancer binding protein alpha [C/EBP-α]), as well as enzymes (AGPAT2, DGAT2, and lipin 1). BSCL2 mutations are usually related to more severe adipose tissue loss than in CGL1.9
Recently a third gene mutation has been identified in one individual with CGL25–27 who had a homozygous nonsense mutation of CAV1, probably as a result of a consanguineous union. CAV1 is located on chromosome 7q31. Its end product, caveolin 1, is a highly conserved 22-KD protein and a crucial component of plasma membrane microdomains known as caveolae. These plasma membrane domains have important roles in regulating signaling pathways and processes such as cell migration, polarization, and proliferation. Caveolin 1 has also been identified as a major fatty acid binder on the plasma membranes. Mutated function of CAV1 may induce lipodystrophy by interfering with lipid handling, lipid droplet formation, and adipocyte differentiation.9,28
Acquired Generalized Lipodystrophy (Lawrence Syndrome)
The acquired syndrome of total lipoatrophy is similar to that of the congenital disorder, except that it develops in a previously healthy individual over days to weeks, often after a nonspecific febrile illness. The syndrome is very rare. It commonly develops during childhood and aldolence in patients who are predominantly white, with a male-to-female ratio of 1 : 3.26,29
In addition to the generalized loss of fat that has an active metabolic function, as seen in CGL, fat loss in acquired generalized lipodystrophy (AGL) also occurs in palms, soles, and genital areas. However, retroorbital and bone marrow fat may be preserved.27
The median time to develop diabetes after loss of fat tissue is approximately 4 years.27 Diabetic ketoacidosis has been reported, and hypertriglyceridemia, hepatic steatosis, acanthosis nigricans, menstrual irregularities, and polycystic ovary syndrome (PCOS) are also common findings. Patients with AGL also have markedly reduced adiponectin levels and moderately reduced leptin levels.14
Several autoimmune diseases and inflammatory conditions have shown a temporal relationship to AGL. These include juvenile-onset dermatomyositis (JDM), rheumatoid arthritis, systemic sclerosis, systemic lupus erythematosus, Sjögren syndrome, and panniculitis.27,30 JDM shows a particularly strong correlation with lipodystrophy; 8% to 40% of patients with JDM develop acquired lipodystrophy.31–35 The chronicity and severity of JDM, as well as the high frequency of calcinosis, have been shown to predict the onset of lipodystrophy.31 AGL following autoimmune diseases is also termed AGL type 2 or the autoimmune disease variety. Panniculitis is another inflammatory condition that frequently heralds the onset of acquired generalized lipodystrophy. It is estimated to be present in approximately 25% of affected patients.27,36 Panniculitis manifests as subcutaneous inflammatory nodules which show a mixed infiltrate of lymphocytes and mononucleated macrophages in adipose tissue. The course of AGL is frequently protracted in patients with panniculitis and linked to less fat loss and less severe metabolic disorders.32,33 The panniculitis variety is also known as AGL type 1. Up to 50% of AGL patients have no clear history of autoimmune disease or panniculitis, however. These lipodystrophies are known as AGL type 3 or the idiopathic type.
The pathogenesis of AGL is unknown. The autoimmune-mediated destruction of adipocytes or preadipocytes has been hypothesized to be the underlying mechanism. Autoantibodies against adipocyte membranes may also impair fat uptake and adipocyte differentiation.30,31,37 Several antibodies have been found to be present in AGL, but no causative relationship has been established.31 Cytokines, including tumor necrosis factor alpha (TNF-α) and interleukin 1 (IL-1), are also likely to play important roles in the immunopathogenesis of lipodystrophy. They can potentially lead to lipodystrophy by inhibiting adipogenesis38 or increasing receptor-mediated apoptosis of adipocytes and preadipocytes.39
Partial Lipodystrophy
Dunnigan-Variety (Face-Sparing) Lipodystrophy
It is characterized by gradual loss of almost all subcutaneous fat from the extremities, commencing at puberty. This gives rise to the characteristic phenotype of “increased muscularity” in the arms and legs. Variable and progressive loss of fat from the anterior abdomen and chest occurs later. Excess fat may subsequently accumulate in the face and neck and in the intraabdominal region, resulting in a Cushingoid appearance.26
Affected females tend to have more recognizable phenotypes. Although questions for gender differences have been raised, anthropometric measures and MRI data demonstrated that both affected men and women have similar patterns of fat loss. In comparison to the affected men, women may have more severe hypoleptinemia and metabolic sequelae of insulin resistance, and they may also have higher prevalence of diabetes and atherosclerotic vascular disease as well as higher serum triglycerides and lower high-density lipoproteins. The prevalence of hypertension and fasting serum insulin concentrations are similar in men and women.27,36 The prevalence of diabetes is not related to age, menopausal status, or family history of type 2 diabetes.37
Patients with the Dunnigan variety are more prone to develop PCOS, infertility, and gestational diabetes. The prevalence of gestational diabetes and miscarriage is significantly higher than in women with similar body mass index (BMI) and PCOS.40
The gene for Dunnigan variety, LMNA, is located on chromosome 1q21-22. It encodes for lamins A and C, which are essential components of nuclear lamina and provide structural integrity of the nuclear envelope. Most FPLD2 mutations in LMNA are missense mutations within the 3′ end of the gene.41 The mutant gene products may disrupt interaction with chromatin or other nuclear lamina proteins, resulting in apoptosis and premature death of adipocytes.42 The accumulation of prelamin A may also impair adipogenesis by interfering with the key adipocyte transcription factors/regulators, including sterol response element–binding protein 1 (SREBP-1) and PPARG.42–45 It is interesting to note that there is a lack of difference in the levels of lamin A and lamin C expression in different adipose depots even though the fat loss of FPLD2 is regionally selective, suggesting that the downstream effects of LMNA mutations are differentially regulated in different areas of the body.40
Köbberling-Type Lipodystrophy
Also known as FPLD type 1 (FPLD1), Köbberling-type lipodystrophy was first reported by Köbberling et al. in 1971. In comparison to Dunnigan variety, the loss of adipose tissue is restricted to the extremities. The distribution of fat on the face and neck is normal or increased in association to frequently observed significant central obesity. The hallmark anthropomorphic feature of this syndrome includes a palpable “ledge” formed between the normal and lipodystrophic areas and high triceps-to-forearm and abdomen-to-thigh skinfold ratios.46 Only women have been diagnosed with Köbberling-type lipodystrophy to date. It is hypothesized that males have a very gentle clinical presentation that does not allow early detection, but this remains to be proven. FPLD1 tends to have a childhood onset.
Metabolic syndrome, especially hypertriglyceridemia, is common in Köbberling-type lipodystrophy. This correlates with high incidence of pancreatitis and premature coronary artery disease. Leptin concentrations are low and correspond to the BMI and the level of fat loss of individual patients.46
Familial Partial Lipodystrophy Due to PPARG Mutations
Also referred as FPLD3, familial partial lipodystrophy due to PPARG mutations is associated with heterozygous PPARG gene mutations. The phenotype is similar to the Dunnigan variety, with the exception that fat accumulation in the head and neck may be spared.47–50 Patients with FPLD3 appear to have more severe metabolic abnormalities than those with FDLP2.45
Familial Partial Lipodystrophy Due to AKT2 Mutation
Familial partial lipodystrophy due to AKT2 mutation has been reported in a single family by George et al.51 It is inherited in an autosomal dominant fashion and manifests as severe insulin resistance and partial lipodystrophy confined to extremities.51
AKT, also known as protein kinase B, is a serine/threonine protein kinase and plays multiple roles in cell signaling, cell growth, and glycogen synthesis, as well as insulin-stimulated glucose transport.52 Lipodystrophy in patients with AKT2 mutations is thought to be due to reduced adipocyte differentiation and dysfunctional postreceptor insulin signaling.
Partial Lipodystrophy Due to CAV1 Mutation
Partial lipodystrophy due to CAV1 mutation was recently identified as a rare cause for partial lipodystrophy.53 Two cases with different frameshift CAV1 mutations have been reported. Both patients were described to have partial lipodystrophy with subcutaneous fat loss in the face and upper body, micrognathia, and congenital cataracts. One case was also associated with abnormal neurologic findings. Diabetes, hypertriglyceridemia, and recurrent pancreatitis were reported in both cases.53
Other Syndromes With a Component of Lipodystrophy
Mandibuloacral dysplasia (MAD) is an extremely rare autosomal recessive progeroid syndrome which has been reported in approximately 40 case reports. MAD is characterized by postnatal growth retardation, craniofacial and skeletal abnormalities (mandibular and clavicular hypoplasia, delayed closure of the cranial sutures, acroosteolysis, joint contractures, birdlike face, dental abnormalities), cutaneous changes (restrictive dermatopathy, skin atrophy, alopecia, and mottled cutaneous pigmentation), and lipodystrophy. Although MAD is present at birth, dysmorphic manifestations and progeroid features become more prominent with time, and the full clinical phenotype is recognizable during the early school years. The patients have normal intelligence,54 and their serum leptin concentration can be low or normal. Hyperinsulinemia, insulin resistance, impaired glucose tolerance, diabetes mellitus, and hyperlipidemia have been reported in some patients.
There are two distinctive phenotypes of MAD: type A involves the loss of subcutaneous fat from the arms and legs but normal or excessive deposition of fat in the face and neck, and type B is characterized by more generalized loss of subcutaneous fat. Mandibular dysplasia type A (MADA) is also considered to be due to mutations of the LMNA gene which result in accumulation of prelamin A and lead to alterations of nuclear architecture and chromatin defects. It remains unclear how different mutations in the same gene lead to a variety of phenotypes. Patients with mandibular dysplasia type B have been reported to carry compound heterozygous mutations in the gene encoding an endoprotease, zinc metalloprotease (ZMPSTE24), on chromosome 1q34. The enzyme is important in posttranslational processing of prelamin A to mature lamin A. As in MADA, the accumulation of farnesylated prelamin A is proposed to be responsible for the phenotype.54 Focal segmental glomerulosclerosis has been reported in patients with ZMPSTE24 deficiency.55
Multiple other syndromes are also linked to lipodystrophy. Several of them have also been identified as laminopathies, including Hutchinson-Gilford progeria syndrome (HGPS, a very rare and uniformly fatal segmental progeroid syndrome with progressive and generalized fat loss), restrictive dermopathy (RD), progeria-associated arthropathy, and atypical progeroid syndrome (also referred to as atypical Werner’s syndrome).54
Acquired Partial Lipodystrophy (Barraquer-Simons Syndrome)
First reported in 1885 by Mitchell, acquired partial lipodystrophy (APL) was further characterized by Barraquer-Roviralta in 1907. There have been approximately 250 cases reported in the English-language literature.26
Patients with APL are primarily of European descent; however, cases have also been reported in Asian Indian, Vietnamese, and Samoan populations. The disease shows a female dominance, and most patients have clinical manifestations in early puberty or early adulthood. The characteristic fat loss progresses in a “cephalocaudal” fashion, with fat loss appearing first in the face and spreading to the upper part of the body. Fat under the umbilicus is rarely affected. Excess fat accumulation is seen over the lower abdomen, gluteal region, thighs, and calves. Breasts may lose fat and consist of firm glandular tissue only.27 Hepatomegaly is common among patients with APL.
In contrast to other types of lipodystrophies, acanthosis nigricans, hirsutism, and hypertrichosis are rare. Female patients normally have regular menses and intact fertility. The prevalence of the metabolic syndrome is also significantly lower in patients with APL. Insulin resistance is uncommon, and the prevalence of diabetes is much reduced compared to other types of lipodystrophies. In the series of case reports by Misra and Garg,27 35% of APL patients had hypertriglyceridemia, and a third had low concentrations of HDL. Serum leptin levels were normal in the majority of patients.14
A strong association has been proposed to exist between acquired partial lipodystrophy and membranoproliferative glomerulonephritis (MPGN) type 2. The spectrum of presentations range from acute glomerulonephritis, hematuria, nocturia, urinary casts, albuminuria, and nephritic syndrome to chronic glomerulonephritis and uremia.53,56 The serum C3 complement levels are usually low with the presence of C3 nephritic factor.53 Patients with low C3 levels tend to have an earlier onset of lipodystrophy than those with normal serum C3 levels. The median time interval between the onset of lipodystrophy and the development of MPGN is approximately 5 to 10 years but could be as long as 20 years.53,57
Similar to acquired generalized lipodystrophy, APL is also frequently seen in the context of autoimmunity or infections.30 The most frequently cited infection preceding APL is measles. The low C3 levels may also render APL patients susceptible to recurrent pyogenic infections, particularly due to Neisseria.58 Systemic lupus erythematosus and dermatomyositis/polymyositis are autoimmune diseases most frequently associated with acquired partial lipodystrophy.27
The precise mechanisms leading to adipose-tissue atrophy in APL remain unclear. The C3 nephritic factor has been shown to induce lysis of adipocytes expressing factor D (adipsin).27 Lamin B gene mutations have also been reported in some cases of APL.59 In a recent study by Guallar et al., PPARG gene down-regulation and mitochondrial toxicity were observed in a patient with APL, suggesting that impaired adipogenesis and adipocyte metabolism may also underlie the pathogenesis of APL.60
HIV-Associated Lipodystrophy Syndrome
Most HIV-infected patients with lipodystrophy are otherwise relatively healthy, but dyslipidemia, especially hypertriglyceridemia, is common among HIV-infected patients receiving HAART. HIV viremia has been linked to decreased plasma concentrations of total, LDL, and HDL cholesterol, and at later stages elevated triglyceride levels. HAART has been shown to cause a worsening lipid profile, with increased plasma triglyceride, increased total and LDL cholesterol, and decreased HDL, which can be further accompanied by increases in small, dense LDL particles, lipoprotein (a), and apolipoproteins B, C-III, E, and H.61 HAART-associated dyslipidemia is associated with accelerated atherosclerosis and signs of endothelial dysfunction.62–65 Frank diabetes and insulin resistance are more prevalent in HIV subjects with lipodystrophy, but acanthosis nigricans seems to be extremely rare. Hepatic steatosis may also develop. Both leptin and adiponectin levels are decreased in patients with HALS. The reduction of leptin levels correlates with decreased subcutaneous fat mass,66 whereas decreased adiponectin levels are more closely associated with intraabdominal fat accumulation.67
Fat atrophy and fat deposition appear to be associated with different risk factors in HALS. Low baseline fat mass and increased disease severity are associated with a higher incidence of fat atrophy.68 Epidemiology studies have also shown that co-infection with hepatitis C can increase the chance of fat atrophy in HIV-infected individuals.69 On the other hand, older age, female sex, high baseline body fat, and longer duration of HAART are associated with a higher risk of fat accumulation in HIV patients.68
The frequency and manifestations of lipodystrophy also differ with respect to the drugs used. Nucleoside reverse transcriptase inhibitors (NRTIs), particularly zidovudine and stavudine, are commonly associated with morphologic changes, particularly fat loss from the extremities, whereas protease inhibitors (PIs) are more frequently linked to hypertriglyceridemia, insulin resistance, and localized fat accumulation.70
[/not-level-membership-for-endocrinology-diabetes-and-metabolism-category]
