Chapter 25 Lipid-Lowering Drugs and Atherosclerosis
| Abbreviations | |
|---|---|
| Acetyl-CoA | Acetyl coenzyme A |
| Apo | Apolipoproteins |
| CVD | Cardiovascular disease |
| HDL | High density lipoprotein |
| HDL-C | High density lipoprotein cholesterol |
| HMG-CoA | Hydroxy-3-methyl-glutaryl coenzyme A |
| IDL | Intermediate density lipoprotein |
| LDL | Low density lipoprotein |
| LDL-C | Low density lipoprotein cholesterol |
| Lp(a) | Lipoprotein (a) |
| PPAR-α | Peroxisome proliferator-activated receptor alpha |
| VLDL | Very low density lipoprotein |
Therapeutic Overview
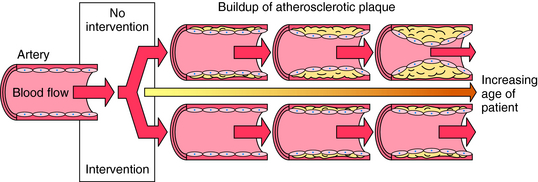
The major risk factors for atherosclerosis and its complications are known and are targets for treatment (Box 25-1). Lowering low-density lipoprotein (LDL) cholesterol (LDL-C) with hydroxy-3-methyl-glutaryl coenzyme A (HMG-CoA) reductase inhibitors, collectively known as the statins, is associated with decreased rate of death, acute coronary syndromes, strokes, and need for coronary artery revascularization by bypass surgery or angioplasty in patients at risk and with established congestive heart disease. Smoking cessation, diet and exercise, controlling blood pressure, daily low-dose aspirin, and increasing levels of high-density lipoprotein (HDL) cholesterol (HDL-C) also reduce the risk for atherosclerosis-related events (Fig. 25-1). Lipid-altering strategies shown to be effective include statins, fibric acid derivatives, bile acid sequestrants (resins), cholesterol absorption inhibitors, niacin, intestinal bypass, and removal of LDL by plasma apheresis. In general, for every 1% lowering of cholesterol, there is a 2% reduced risk of coronary artery disease.
Pathobiology of Atherosclerosis and Therapeutic Targets
Histological evidence suggests that plaque growth may be gradual over years, with bursts of growth from periodic intraplaque hemorrhage and repair. Gradual buildup of plaque over decades can lead to the gradual narrowing of coronary and other conduit arteries (carotid, femoral, popliteal), and, alternatively, can rupture, leading to sudden occlusion, resulting in an acute ischemic syndrome (unstable angina, myocardial infarction, stroke, death, critical limb ischemia). Fibrous plaques are prevalent in the 4th and 5th decades of life, and symptoms (angina, claudication) from occlusive plaques peak in the 7th decade. Ruptures and fissures of plaques, which can lead to sudden occlusion with a superimposed thrombus resulting in acute ischemic events, occur predominantly in nonocclusive lesions. The prevalence of acute coronary syndromes in healthy men and women increases with age, from very rare in the 30s to more than 1% in men over 60 years and women over 70 years of age.
Lipid-lowering therapy can improve endothelial function, reduce coronary events and strokes, relieve symptoms, prevent new plaque formation, reduce rate of progression, and even induce regression of focal narrowing. Raising HDL-C enhances endothelial function and results in removal of cholesterol from cells and lipid pools, known as reverse cholesterol transport. In established coronary heart disease and for primary prevention, serum lipids are one of many interactive risk factors requiring lifestyle changes and drug therapy (see Fig. 25-1). Aspirin and other platelet antagonists (see Chapter 26) reduce risks of acute coronary syndrome and strokes by reducing thrombosis. Antihypertensive strategies (see Chapter 20) reduce wall stress and plaque rupture by various mechanisms.
| Therapeutic Overview |
|---|
| Reduce formation and rate of progression in coronary and peripheral atherosclerosis from childhood to old age |
| Prevention of coronary events and strokes in apparently healthy persons at risk, particularly middle-aged and elderly |
| Prevention of heart attacks, strokes, need for revascularization in persons with established atherosclerosis |
| Prevention and treatment of pancreatitis in hypertriglyceridemia |
Therapeutic uses of lipid-lowering drugs are summarized in the Therapeutic Overview Box.
Mechanisms of Action
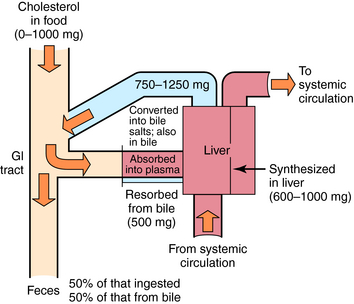
The dynamics of cholesterol ingestion, synthesis, and elimination are depicted in Figure 25-2. The sole sources of exogenous cholesterol are ingested animal-based food substances, including meats and dairy products. Dietary intake can vary from 0 to 1000 mg/day, with 30% to 75% typically absorbed.
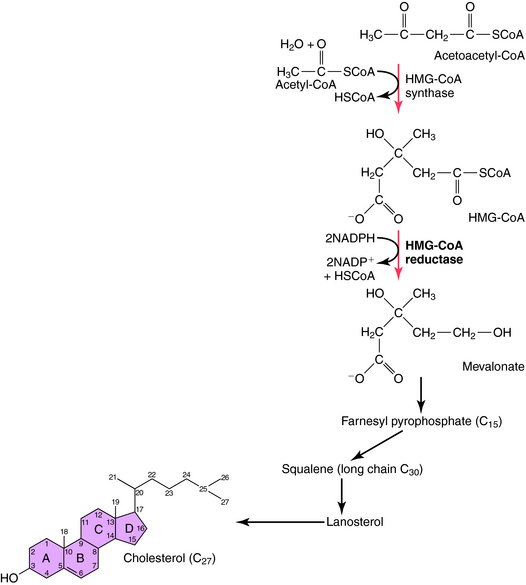
Synthesis of cholesterol originates with a reaction between acetyl coenyzmeA (acetyl-CoA), a key intermediate for glycolysis, the citric acid cycle, and fatty acid degradation, and acetoacetyl-CoA to produce HMG-CoA. The next step is rate-limiting for cholesterol synthesis and involves the irreversible conversion of HMG-CoA to mevalonic acid (Fig. 25-3). The rate of this reaction is influenced by several factors, including time of day (predominantly at night), diet composition, excessive food intake, or obesity. Diets rich in saturated fats increase serum cholesterol primarily by down regulating hepatic clearance, whereas a diet of predominantly unsaturated fats or carbohydrates is generally associated with lower serum cholesterol. In addition, many other factors affect the rate of cholesterol synthesis, including a dynamic equilibrium with certain lipoproteins.
Cholesterol, triglycerides, and phospholipids are transported in plasma and other fluids as lipoproteins, which have a lipid core encased in a protein coat. Triglycerides are assembled in the liver from fatty acids and glycerol. The largest plasma lipoprotein is the chylomicron, composed of triglyceride:cholesterol in 10:1 ratio. The shell is composed of phospholipid, cholesterol, and several apolipoproteins (Apo). Chylomicrons are usually present only after eating, especially a meal with a high fat content, but may be present in fasting persons with inadequate chylomicron metabolism. They are synthesized in the intestine, and their principal role is to transport dietary fats to adipose tissue, muscle, and liver. The apolipoproteins associated with chylomicrons in the intestine are ApoB-48 and ApoA-I (Fig. 25-4). After chylomicrons have been secreted and enter the plasma, they acquire ApoE, ApoC-I, ApoC-II, and ApoC-III. ApoC-II is critical and acts with insulin to activate lipoprotein lipase in the capillary wall, liberating free fatty acids and glycerol from released triglycerides. The chylomicron remnants, containing ApoA, ApoB, and ApoE, but having lost ApoC-II and ApoC-III, continue to circulate and are eventually removed by specific hepatic remnant receptors. This is the principal route by which dietary fat is transported and is referred to as the exogenous pathway. Fasting chylomicronemia resulting from inherited or acquired deficiency of lipoprotein lipase is usually associated with triglyceride levels greater than 2000 mg/dL, which may result in life-threatening pancreatitis.
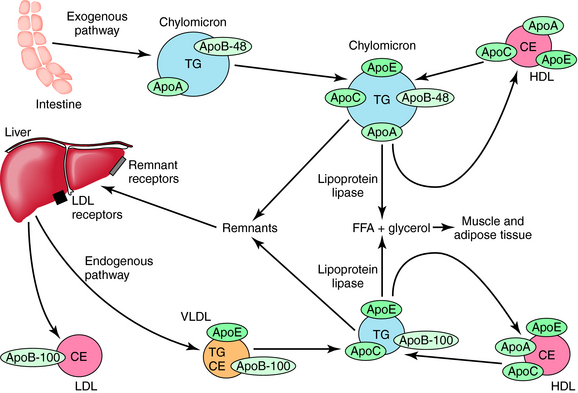
The endogenous formation and transport of triglycerides is accomplished by very-low-density lipoprotein (VLDL) particles (see Fig. 25-4). Synthesized principally by the liver and to a lesser extent by the intestine, these particles are much smaller than chylomicrons. In contrast to chylomicrons, triglycerides in VLDL are obtained from fatty acids synthesized by the liver or released by adipose tissue and circulate to the liver. In addition to cholesterol and phospholipid, the wall of VLDL particles contains ApoB-100, ApoE, and ApoC-I, ApoC-II, and ApoC-III, the latter obtained from HDL. The internal composition is 5:1, triglyceride:cholesterol. ApoC-II on the VLDL surface results in lipoprotein lipase activation, releasing free fatty acids to muscle and adipose tissues and resulting in smaller, increasingly dense intermediate-density lipoprotein (IDL) particles. The surface ApoE on these VLDL remnants results in clearance of some particles via the same hepatic remnant receptors that bind chylomicron remnants.
Other IDL particles continue to lose triglycerides via lipoprotein lipase and hepatic lipase, resulting in contracted particles known as LDLs, which are approximately 2% the size of VLDL particles and 0.02% the volume of a chylomicron. LDL particles contain 50% to 60% cholesterol and less than 10% triglyceride and have one molecule of ApoB-100 on their surface. LDL particles vary in density and size. The small, dense particles are usually found in association with higher levels of serum triglycerides, are highly atherogenic because they readily cross the endothelial barrier, are more easily oxidized, and are more readily taken up by scavenger receptors. Atherogenicity is related to both LDL particle number and size. Every 1% increase in LDL-C increases the rate of coronary events by approximately 2%. In addition, both VLDL remnants and IDL particles have been found in atheroma. Lipoprotein(a) [Lp(a)] is a small particle formed in the liver, the size of LDL particles, containing Apo(a) linked to ApoB-100. Apo(a) has a homology with plasminogen, resulting in competition for plasminogen receptors and decreasing thrombolysis.
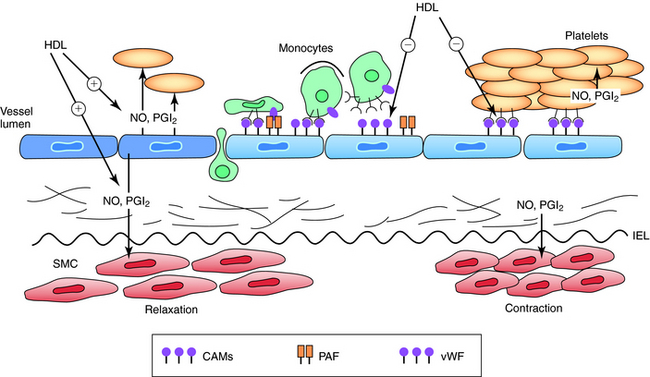
VLDL and IDL particles also exchange triglycerides for cholesterol with HDL particles, which is facilitated by cholesterol ester transfer protein. The clearance of these particles via remnant and LDL receptors facilitates hepatic clearance of cholesterol. Each of these mechanisms contributes to the antiatherosclerotic process known as reverse cholesterol transport. Treatment strategies designed to raise HDL levels alone or in association with decreasing triglycerides have been associated with less atherosclerosis progression, disease regression, and decreased coronary event rates in persons with atherosclerosis. Low levels of HDL-C (<45 mg/dL in men and 50 mg/dL in women) are associated with increasing risk for coronary disease and strokes at normal and low levels of LDL-C. Figure 25-5 demonstrates how HDL particles impact vascular endothelial function and tone. Total and HDL-C are measured in the nonfasting state to assess the risk of coronary heart disease in adults.
A low total cholesterol (<200 mg/dL) and a normal or high HDL-C (>45 mg/dL in men and 55 mg/dL in women) in the absence of other risk factors or a family history of atherosclerosis generally infers a low risk. In persons with other risk factors and an increase in total cholesterol and less than average HDL-C, lipids and lipoproteins are measured in the fasting state (approximately 12 hours) to eliminate the postprandial increase in triglycerides. Total cholesterol, triglycerides, and HDL-C are measured, and the LDL-C is calculated by the Fredrickson equation:
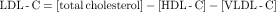
Although ideal total cholesterol is less than 150 mg/dL and LDL-C is less than 100 mg/dL, the average person in the United States, with or without coronary heart disease, has cholesterol of 205 mg/dL and an LDL-C of 135 mg/dL. The major cause of elevated cholesterol in the United States and industrialized world is an increased intake of saturated fats. Approximately 5% of persons have primary hypercholesterolemia, which can be divided into polygenic hypercholesterolemia (3.8%), familial combined hyperlipidemia (1.5%), and familial hypercholesterolemia (0.2%), the latter caused by a decreased number or activity of LDL receptors. The heterozygous form of familial hypercholesterolemia appears in approximately 0.2% of people, whereas the homozygous form is rare (0.000001%). Heterozygotes have serum cholesterol concentrations approximately twice normal and LDL-C levels greater than 240 mg/dL. Homozygotes have cholesterol concentrations six times normal and may show evidence of coronary heart disease in childhood and adolescence. Many homozygotes die before 10 to 12 years of age, and almost all experience a myocardial infarction by 20 years of age. Polygenic hypercholesterolemia and familial combined or mixed hyperlipidemia (increased cholesterol and triglycerides) are much more common, involving increased absorption of dietary fats, overproduction of lipids, or decreased clearance of lipoprotein particles. A deficiency in lipoprotein lipase activity, either inherited or acquired with obesity, excess dietary carbohydrates, and diabetes, results in high levels of fasting triglycerides (200 to 10,000 mg/dL) and chylomicronemia. The lipoproteins that promote atherosclerosis and the vasculoprotective effect of HDL particles are depicted in Figure 25-6.
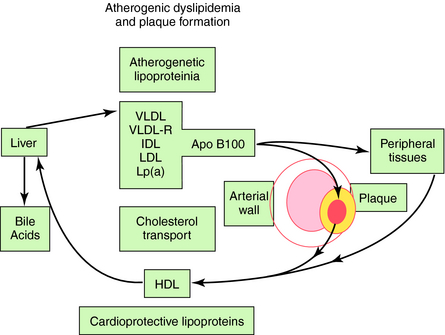
A classic inheritable atherogenic lipoprotein phenotype is present in 5% to 10% of the population, nearly all diabetics, the metabolic syndrome associated with insulin resistance (hypertension, truncal obesity, elevated insulin), and nearly 50% of persons with premature coronary artery disease. The phenotype is characterized by increased small dense LDL particles, decreased HDL particles with less of the larger buoyant type, and moderately increased VLDL remnant particles rich in triglycerides and cholesterol. Elevated triglyceride levels increase the risk in men and, particularly, women with elevated cholesterol. This is likely due to the association with small LDL particles and low HDL-C, and the atherogenicity of VLDL remnant particles. Elevated Lp(a) is the most common abnormal lipid in patients with a family history of premature coronary heart disease. In addition to increasing thrombosis and decreasing thrombolysis, Lp(a) is a small LDL-like particle that is easily oxidized and atherogenic. Low HDL-C and increased triglycerides and isolated low HDL-C are common causes of premature coronary artery disease. Ironically, hypercholesterolemia accounts for less than 10% of premature coronary artery disease.
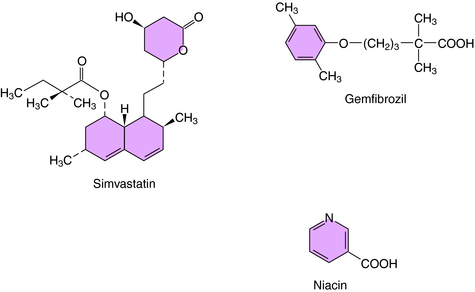
The statins inhibit the enzyme HMG-CoA reductase, the initial rate-limiting step in cholesterol synthesis (see Fig. 25-3). Statins act as competitive inhibitors for the active site on the reductase enzyme, with a higher affinity than HMG-CoA. Inhibition of cholesterol synthesis, particularly in hepatocytes, decreases intracellular pools, which triggers an increase in LDL receptor number and activity. This leads to an increased clearance of LDL particles. Plasma concentrations of LDL-C and the number of LDL particles decrease, and less LDL is available to react with cells in blood and vessel walls. Statins also lower plasma lipids, including LDL-C and triglycerides, by inhibition of hepatic VLDL synthesis, resulting in decreased numbers of VLDL, IDL, and LDL particles. The structure of simvastatin, a typical HMG-CoA reductase inhibitor, is shown in Figure 25-7.
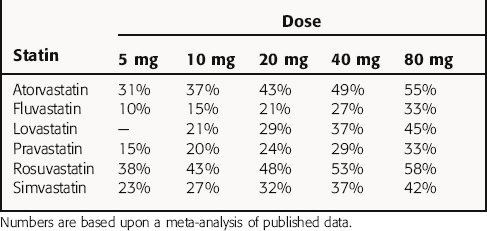
The statins vary in their abilities to increase LDL receptors and hence increase the clearance of LDL particles. The LDL-C-lowering effect of comparable doses of each of the statins is summarized in Table 25-1, although all doses are not necessarily available commercially. Note that a doubling of dose results in only a 5% to 6% further reduction in LDL-C. The effects of the statins on ApoB, VLDL-C, IDL-C, and triglycerides are proportionate to the decrease in LDL-C. With cessation of therapy, lipids return to pretreatment levels within 4 weeks.
Phenoxyisobutyric acid, or fibric acid, is the parent compound for several drugs that lower plasma cholesterol and triglyceride concentrations, known collectively as the fibrates or fibric acid derivatives, and include gemfibrozil, fenofibrate, and clofibrate. The structure of gemfibrozil is shown in Figure 25-7.
Fibrates have a broad spectrum of lipid-modulating and pleiotropic effects related to their capacity to mimic the structure and biological functions of free fatty acids. Their mechanisms of action are only partially understood but appear to activate transcription factors belonging to the nuclear hormone receptor superfamily, the peroxisome proliferator-activated receptors (PPARs). PPAR-α mediates the action of fibrates on HDL-C levels via transcriptional induction of synthesis of major HDL apolipoproteins (ApoA-I and ApoA-II) and increased synthesis of lipoprotein lipase. Fibrates decrease hepatic ApoC-III transcription, reducing inhibition of lipoprotein lipase and enhancing clearance of triglyceride-rich lipoproteins. Other functions altered by the actions of fibrates on PPAR-α result in increased fatty acid uptake, decreased fibrinogen and high sensitivity C-reactive protein, and increased cholesterol efflux. The effect of fibrates on raising HDL and reducing triglyceride-rich chylomicron and VLDL particles and lipid content is mediated by decreasing production of hepatic VLDL containing less ApoC-III, and induction of hepatic and systemic expression of lipoprotein lipase. Increasing the activity of endothelial lipoprotein lipase enhances release of VLDL surface fragments to form nascent HDL and increases production of ApoA-1. Decreasing triglyceride content and number of VLDL remnants reduces the cholesterol ester transfer protein transfer of triglycerides to HDL particles in exchange for cholesterol.
Niacin (see Fig. 25-7) was observed in the mid-1950s to lower serum triglycerides and cholesterol. It was the first drug with lipid-modulating effects shown to reduce recurrent coronary events and mortality in men who had recovered from a myocardial infarction. After absorption, niacin is enzymatically converted to nicotinamide adenine dinucleotide. However, nicotinamide does not have hypolipidemic activity.
Pharmacokinetics
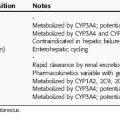
Selected pharmacokinetic parameters of individual drugs are listed in Table 25-2. All the lipid-lowering drugs are taken by oral administration; thus their absorption may be affected by the presence of food.
| Drug* | t1/2 (hrs) | Disposition |
|---|---|---|
| Statins | ||
| Atorvastatin | 14 | F, R |
| Rosuvastatin | 20 | F, R |
| Lovastatin | 3-4 | F, R |
| Pravastatin | 1.8 | F, R |
| Simvastatin | 3 | F, R |
| Fibric Acid Derivatives | ||
| Gemfibrozil | 1.5 | M |
| Fenofibrate | 20 | M |
| Bile Acid Sequestrants | ||
| Cholestyramine | NA | F |
| Colestipol | NA | F |
| Others | ||
| Niacin† | 1 | M |
| Ezetimibe | 22 | M |
F, Fecal excretion; R, renal excretion; M, metabolized; NA, not absorbed.
* All agents are administered orally.
† Extended-release preparations are available with substantially different pharmacokinetics.
Atorvastatin and rosuvastatin, the two most potent statins, have a t1/2 of 14 to 20 hours, compared with less than 4 hours for the other statins. Because of their long t1/2, atorvastatin and rosuvastatin can be taken once in the morning. The statins differ in their bioavailability and in the effect of food on their absorption. Lovastatin absorption is enhanced by food, whereas fluvastatin and pravastatin absorption is reduced by food; thus they are taken at bedtime. The absorption of simvastatin, rosuvastatin, and atorvastatin is unaffected by food. All statins are subject to high (approximately 60%) first-pass metabolism by the liver. Their varying hydrophilic or lipophilic natures do not appear to correlate with lipid-lowering effects, side effects, or toxicity.
Relationship of Mechanisms of Action to Clinical Response
The fibrates also reduce the magnitude of both fasting and postprandial hyperlipidemia. Although epidemiological evidence implicating lipids in atherosclerosis is predominantly in fasting states, there is considerable evidence that postprandial increases in triglycerides and VLDL receptors and IDL particles help explain the increase in coronary artery disease risk in diabetes and the metabolic syndrome. Impaired postprandial triglyceride metabolism is associated with endothelial dysfunction, possibly related to cytotoxicity of triglycerides.
The major antiatherosclerotic effect of niacin appears to be its ability to raise HDL-C, which is considerable and greater than that of the fibrates. In contrast to the fibrates, niacin is very effective in individuals with isolated low HDL-C. Like the statins, nonlipid pleiotropic effects of niacin are also important for preventing coronary events and progression of atherosclerosis. Table 25-3 summarizes the effects of niacin. Clinical trials have shown that niacin reduces nonfatal and recurrent myocardial infarctions and is associated with relatively less new lesion formation and more coronary plaque regression.

The effect of niacin is also highly dependent on fasting lipids. In patients with elevated triglycerides and low HDL-C, niacin will usually reduce triglycerides by 15% to 25%, increase HDL-C by 20% to 30%, and reduce LDL-C minimally. Average reduction in LDL-C by 2 g of niacin is approximately 15%. A slow-release product that reduces the side effect of flushing without a loss of efficacy is available. The cholesterol-lowering effect of niacin can be enhanced by coadministration with statins and resins.
Pharmacovigilance: Side Effects, Clinical Problems, and Toxicity
Adverse effects of the various drug classes are listed in the Clinical Problems Box.
Clofibrate use has diminished because of the increased incidence of cholelithiasis and a possible increased incidence of carcinoma. Both clofibrate and gemfibrozil have been associated with increasing suicidal and accidental deaths.
| Drug class | Side effects |
| HMG-CoA reductase inhibitors (statins) | Muscle pain and weakness, myositis, increased liver enzymes, rosuvastatin-increased microalbuminuria |
| Bile acid sequestrants |
Because they are not absorbed, the resins can be safely administered to children and during pregnancy and breastfeeding.
Lifestyle Interventions and Drugs in Atherosclerosis
Prevention and treatment of atherosclerosis need to be comprehensive and targeted to each of the major and contributing risk factors (see Box 25-1). Both food choices and calories consumed should be tailored for weight control, lipid management, and hypertension. Because cholesterol levels increase with dietary fat and age, a basic recommendation is to decrease caloric intake and lower the proportion of dietary fat to less than 30% and saturated fat to less than 7% to 10%. This requires a shift to foods rich in monounsaturated fats, such as olive oil, lean meat, and certain vegetables.
The effect of dietary intervention varies widely. Diets high in fiber, antioxidant-containing fruits and vegetables, and cold water fish rich in omega-3 polyunsaturated fats have been shown to reduce first and recurrent coronary events independent of drugs. Patients with established atherosclerosis should be placed on drug therapy with appropriate dietary advice. The argument for dietary change as a principal component in prevention of atherosclerosis is based on the following:
Anonymous. Drugs for lipids. Treat Guidel Med Lett. 2008;6:9-16.
Chapman MJ. Fibrates in 2003: Therapeutic action in atherogenic dyslipidemia and future perspectives. Atherosclerosis. 2003;171:1-13.
Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: A randomized placebo-controlled trial. Lancet. 2002;360:7-22.
Rosenson RS. Antiatherothrombotic effects of nicotinic acid. Atherosclerosis. 2003;171:87-96.