[level-membership-for-opthalmology-category]
Chapter 155 Leukemias and Lymphomas
Introduction
Leukemias and lymphomas are myeloproliferative disorders that may affect the eye. The ophthalmic symptoms and findings can be the initial manifestation of the systemic illness. Indeed, before the use of bone marrow biopsy, the ophthalmologist was often called upon to assist with the diagnosis of the leukemias.1
Estimates of the frequency of intraocular involvement with leukemia range as high as 90% of cases.1 Although melanoma is the most common primary intraocular tumor in adults and retinoblastoma is the most common primary intraocular tumor in children, when secondary or metastatic neoplastic intraocular disease is considered, intraocular manifestations of hematological malignancies are found to be overwhelmingly more common since the prevalence of myeloproliferative disorders is higher in the general population.
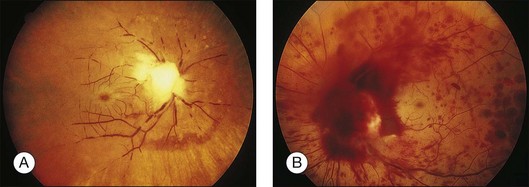
Liebreich first described leukemic retinopathy in the 1860s. Since that time, reports have documented that virtually all intraocular structures may be involved. Patients have been reported with leukemic infiltrates of the optic nerve, choroid, retina, iris, ciliary body, and anterior chamber. A child with a leukemic hypopyon is illustrated in Figure 155.1. Central serous chorioretinopathy overlying areas of choroidal infiltration has been reported, as has retinal vascular sheathing, subconjunctival hemorrhage, anterior chamber hemorrhage, intraretinal hemorrhage, and intravitreal hemorrhage.2 Retinal changes may be related to direct invasion of tissue by neoplastic cells, to manifestations of associated hematological abnormalities such as anemia, thrombocytopenia, or hyperviscosity states, to opportunistic infections, or to unrelated chance findings.
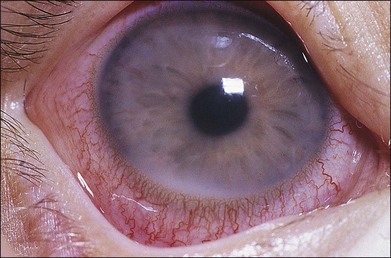
Fig. 155.1 Leukemic iris infiltration in a child with acute lymphocytic leukemia. Note hypopyon inferiorly.
Intraocular infiltrates appear to correlate with associated central nervous system (CNS) involvement and decreased survival. The intraocular manifestations of leukemia are protean, and clinically, it usually is not possible to differentiate the various leukemias and lymphomas on the sole basis of their ophthalmoscopic findings.3
Systemic classification of leukemia and lymphoma
At presentation, the acute leukemias most often have systemic manifestations of anemia, hemorrhage, infection or signs and symptoms related to infiltration of organs. Acute lymphocytic leukemia is the predominant leukemia type in children, and more than 50% of these patients can be cured. In adults, acute myelogenous leukemia is the predominant myeloproliferative disorder and survival rates are lower in adults than children.1
The chronic leukemias often first appear in an indolent manner. Symptoms may be vague. These diseases generally are found in older individuals. Although associated infections or other complications are often treated aggressively, many patients receive no treatment for their underlying disease because treatment has not definitely been shown to prolong survival. Some chronic leukemias degenerate into a “blast” (acute) phase, in which case the signs and symptoms resemble those of the acute leukemias.1
Leukemia
Prevalence and incidence
Scant data are available from prospective series of patients concerning ocular findings at the time of diagnosis.4,5 Even less information can be found about the frequency with which various intraocular manifestations are seen during the course of the illness. Most clinical series contain cross-sections of patients seen both early and late during the course of the illness. Autopsy series, which presumably show the highest prevalence rates, often overstate the frequency with which clinical disease is detected.
Four autopsy series have shown variable prevalence data. Allen and Straatsma3 found that 38 of 76 patients (50%) had ocular involvement by neoplastic disease or pathologic changes that could be directly ascribed to the neoplastic disease. Nelson et al.6 found that 33 of 117 patients (28%) with various types of leukemia had ocular metastases at the time of death. Leonardy et al.7 found leukemic infiltrates in the eyes of 42 of 135 patients (31.1%), with the choroid being the most frequently involved site. Data from Kincaid and Green1 are summarized in Table 155.1, which shows that 75% of patients with chronic leukemias, 82% of patients with acute leukemias, and 80% of affected patients in this study had intraocular involvement at the time of death.
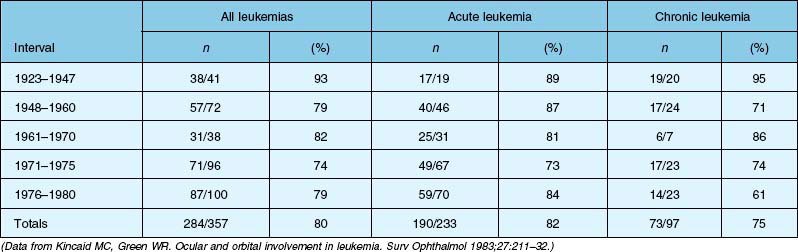
Clinical series show highly variable prevalence rates. A small prospective series was reported by Karesh et al.,4 who examined the fundus findings in 56 newly diagnosed, untreated patients with acute myelogenous leukemia. They found retinopathy at the time of initial examination in 28 of 56 patients (50%) and no cases of leukemic infiltration. Schachat et al.5 reported on the largest series of patients with newly diagnosed leukemia examined within a few days of diagnosis. Among the 120 patients examined (65 with acute myelogenous leukemia, 51 with acute lymphocytic leukemia, and four with other leukemias), 62% had ocular abnormalities due to their underlying myeloproliferative disorder.
Clinical manifestations
Reddy et al. reported on 82 children examined after diagnosis of acute leukemia and before initiation of chemotherapy. Although only 4% of patients complained of visual symptoms, abnormal retinal findings were noted in 16% of all patients examined.8 Reddy and colleagues also reported on the ocular findings in 288 newly diagnosed cases of leukemia in adults and children. Clinical symptoms were present in 10%, but ocular findings were noted in 35.4%. Ocular findings were more common in adults (49%) than in children (16%), and were more frequent in myeloid leukemia (41%) compared with lymphoid leukemia (29.2%).9
Russo et al. evaluated 180 pediatric patients with acute leukemia and found ocular manifestations in 66% of children with acute myeloid leukemia and 11.5% of children with acute lymphocytic leukemia.10 Data from multiple reports suggest the need for routine ocular examination of all patients with newly diagnosed acute leukemia regardless of whether they are symptomatic or not. Other investigators have also noted that chronic lymphocytic leukemia appears to have a very low prevalence of ocular involvement and routine screening of these patients at the time of diagnosis does not appear to be necessary.11
In our series of 120 cases examined at the time of diagnosis, four patients (3%) had leukemic infiltrates. In addition, intraretinal hemorrhages were present in 29 cases (24%), white-centered retinal hemorrhages in 13 cases (11%), vitreous hemorrhage in three cases (2%), and cotton-wool spots in 19 cases (16%). Miscellaneous findings, presumably unrelated to leukemia, were present in 20% of cases.5 The low prevalence of leukemic infiltrates in our series differs from those reported in autopsy series. One potential reason for the lower prevalence is the ability to detect leukemic choroidal infiltrates, which are a frequent finding in autopsy series but are difficult to diagnose clinically.
Leukemic infiltrates
Retinal or preretinal infiltrates
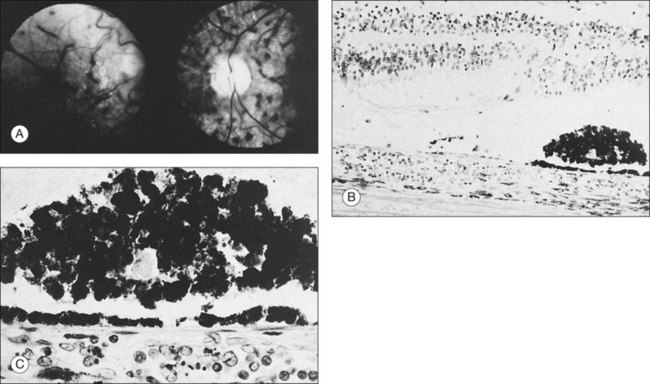
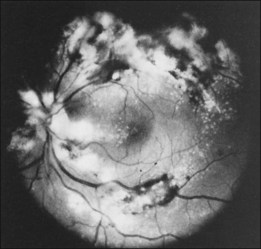
Leukemic infiltrates have been described by several investigators. Kuwabara and Aiello12 reported on a patient with chronic myelogenous leukemia with large gray-white nodules of varying sizes in the retina.12 They suggested that this finding was an ominous prognostic sign and, in general, associated with high blood counts, fulminant disease, and early demise.13 Merle and colleagues have described a subretinal infiltrate with venous vasculitis in a patient with adult T-cell leukemia.14 As the ocular lesions progressed, the general health of the patient declined despite treatment with chemotherapy. Another patient with leukemic infiltrates is illustrated in Figure 155.2. This type of pathologic change in the fundus was seen in 2–3% of patients in our prospective series.5 Optic nerve infiltration may be the initial ocular manifestation of acute leukemia and can result in severe vision loss if not treated.15
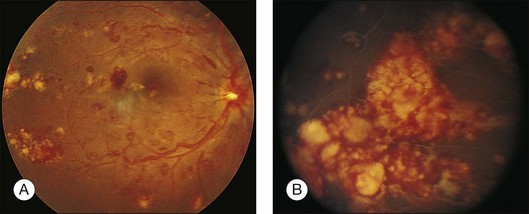
Gray-white streaks along vessels may be caused by local perivascular leukemic infiltrates. A case of diffuse unilateral retinal angiopathy that resembled frosted-branch angiitis that responded to local radiation and intrathecal chemotherapy was reported by Kim et al.16 This case represented the sole manifestation of relapsing acute lymphoblastic leukemia in an 18-year-old man and also involved optic nerve infiltration in the same eye.
Choroidal infiltrates
Leukemia may also infiltrate the choroid; however, clinical signs of choroidal involvement are often subtle unless overlying retinal or retinal pigment epithelial (RPE) changes bring them to attention. Serous retinal detachment overlying choroidal infiltrates17 or overlying frank choroidal masses18 is an important clue. Histopathologic confirmation of choroidal infiltration has been reported in a patient with adult T-cell leukemia.19
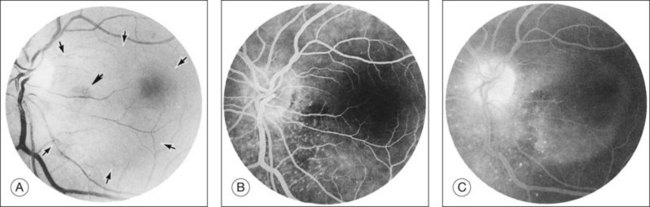
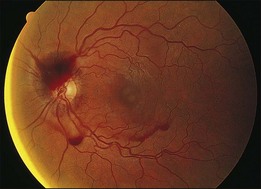
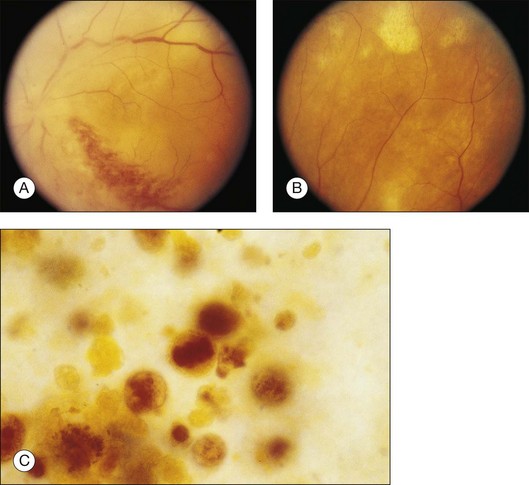
Kincaid et al.20 reported on a 71-year-old woman with an exudative retinal detachment, shifting subretinal fluid, and areas of diffuse pinpoint fluorescein hyperfluorescence with dye leakage into the subretinal space. The patient had a poorly characterized and unusual leukemia. Histopathologic study showed that the choroid was moderately distended by a diffuse cellular infiltrate with an overlying serous retinal detachment. The RPE showed areas of depigmentation and proliferation. There were a few small areas of associated pigment epithelial detachment.20 Gass21 described a patient with myelomonocytic leukemia who had a discrete choroidal mass with an overlying serous retinal detachment. Pinpoint hyperfluorescence was seen on the angiogram in areas overlying the mass (Fig. 155.3).
Burns et al.22 reported on a 37-year-old patient with acute lymphocytic leukemia who appeared to have “bilateral macular edema.” The patient was treated with ocular radiation on the assumption that underlying leukemic infiltration of the choroid was present, and this was confirmed at autopsy. Serous retinal detachment overlying areas of choroidal infiltration has been reported in patients with chronic lymphocytic leukemia, acute lymphocytic leukemia, chronic myelogenous leukemia, and acute myelogenous leukemia.1,23 Serous RPE detachment has also been reported in a patient with acute lymphoblastic leukemia.24 The serous retinal and RPE detachments can be the presenting manifestation of the leukemia.25 Eventually, as the fluid and detachment resolve, coarse clumping of the RPE is seen.17,24
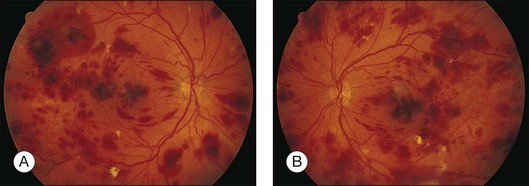
Prominent pigment epithelial changes also may be seen after resolution of what may be retinal infiltrates. Jakobiec and Behrens26 reported on a 3-year-old patient with acute leukemia who was found to have preretinal hemorrhages, some with white centers. A multitude of jet-black spots were seen, most prominently in the posterior pole. Soft white patches were described in the retinal periphery. The authors believed that these patches were the precursor lesions of the black spots. They postulated that the spots represented proliferation of pigment epithelial cells or small pigment epithelial detachments.26 Clayman et al.17 reported on a child with acute lymphocytic leukemia who had massive pigmentary changes simulating leopard spots, most marked in the posterior pole (Fig. 155.4). At autopsy, the retina and the choroid were infiltrated with atypical immature lymphocytes. Areas of RPE hyperplasia were present, including heaped-up masses of pigment epithelium surrounding leukemic cells.
Vitreous infiltrates
Vitreous opacities may be manifestations of an intraocular malignancy. Moribund patients may show massive collections of tumor cells in the vitreous, but most patients with intravitreal hemorrhage have neoplastic cells in the vitreous only because their peripheral blood contains tumor cells (Fig. 155.5). The cells do not appear to be preferentially replicating in the vitreous cavity. Swartz and Schumann2 described a patient with acute lymphocytic leukemia who had a dense vitreous cellular infiltration. Diagnostic vitreous aspiration confirmed the neoplastic nature of the process. Examination of the cerebrospinal fluid was also positive for blast cells. At the time, the peripheral blood was negative for tumor. The patient was treated with systemic and intrathecal chemotherapy and 1200 cGy of cranial radiation therapy. The vitreous cleared after the treatment.2
Belmont et al.27 reported on a 72-year-old patient with chronic unilateral uveitis. Leukemic cells were found in the iris and throughout the vitreous at the time of a pars plana vitrectomy performed to rule out reticulum cell sarcoma. Isolated ocular blast crisis with leukemic hypopyon has been reported to occur in chronic myelogenous leukemia.28 Vitreous cells and leukemic retinopathy have been reported in a single case of hairy cell leukemia (which constitutes less than 2% of adult leukemias).29 A masquerade syndrome with primarily ocular findings of panuveitis in a patient with T-cell prolymphocytic leukemia was reported by Dhar-Munshi et al.30 Additional cases of vitreous involvement in leukemia have been cited by Kincaid and Green1 as well as by Rothova et al.31 Leukemic cells have been found in the vitreous of patients with neovascularization of the disc. Delaney and Kinsella32 reported on a patient with chronic myelogenous leukemia and disc neovascularization who had good peripheral perfusion, although the macula did show areas of nonperfusion. de Juan et al.33 described a 3-year-old patient with acute lymphocytic leukemia who had disc neovascularization, vitreous hemorrhage, and vitreous infiltration. This case was unusual because neither the cell count nor the platelet count were remarkably elevated.
Hattenhauer and Pach34 reported a single case of ocular B-cell lymphoma occurring in a patient with chronic lymphocytic leukemia. The occurrence of diffuse large cell lymphoma in patients with chronic lymphocytic leukemia is known as Richter syndrome. Richter syndrome is estimated to occur in 3–10% of patients with chronic lymphocytic leukemia. It portends an abrupt deterioration in health and carries a poor prognosis for survival.
Possible leukemic infiltrates
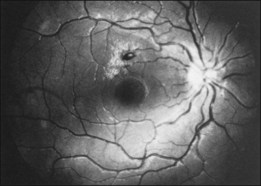
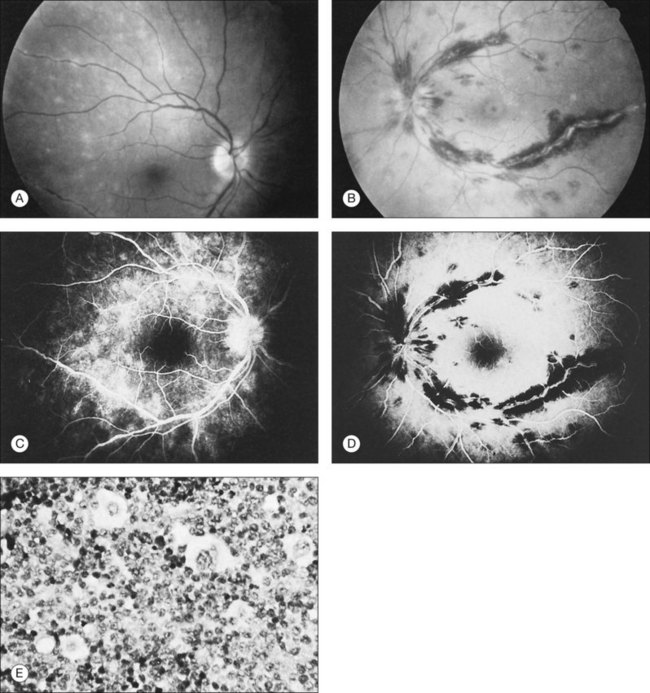
Duane et al.35 reviewed the causes of white-centered retinal hemorrhages (Fig. 155.6) and also described possible pathophysiologic mechanisms. The authors suggested that these lesions should be classified as suspicious for direct intraocular manifestations of leukemia, since aggregates of leukocytes have been reported in the center of white-centered hemorrhages.36,37 However, it should be noted that white-centered retinal hemorrhages can be composed of fibrin-platelet aggregates and are not necessarily a definite sign of neoplasia.
Manifestations of anemia and thrombocytopenia
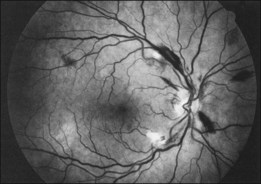
Leukemic retinopathy is the term most often used to denote the fundus manifestations of anemia, thrombocytopenia, and increased blood viscosity seen in patients with leukemia. In general, the term does not necessarily refer to frank leukemic proliferation. The changes of “leukemic retinopathy” may be more commonly seen with the acute leukemias, but the frequency with which they occur has not been adequately studied to be certain. Although perivascular sheathing may be due to actual perivascular infiltrates, tortuous dilation of the retinal veins probably is not. The veins and arteries may assume a yellowish tinge because of both anemia and leukocytosis.1 Retinal hemorrhages may occur, often at the posterior pole, and may be subretinal, deep retinal, superficial retinal, or preretinal, with potential breakthrough bleeding into the vitreous cavity. Hemorrhages may have a blot or blotch shape, flame shape, or they may have white centers (Figs 155.7, 155.8).35
Cotton-wool spots may be the presenting abnormality that precipitates the systemic evaluation leading to the diagnosis of leukemia.38 Cotton-wool spots may be caused by local factors, such as an abnormally large cell or a cluster of cells occluding retinal arterioles, and may not be related to the overall peripheral blood composition. In general, hematologic parameters are not associated with the presence of cotton-wool spots.39 Cotton-wool spots and hemorrhages can spontaneously resolve in patients with chronic leukemic disease.2
Manifestations of hyperviscosity
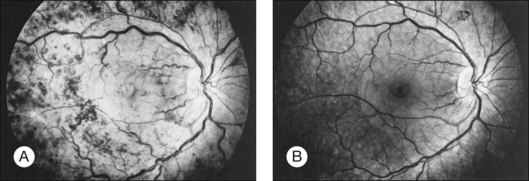
Whole-blood hyperviscosity may lead to veno-occlusive disease, microaneurysm formation, retinal hemorrhages, and retinal neovascularization. The most common manifestation is probably a mild, or “hyperpermeable,” central retinal vein occlusion (Fig. 155.9). A systemic hyperviscosity state should be suspected in patients with simultaneous, bilateral retinal vein occlusion. Also, the very high white cell count may lead to a hyperviscosity state that results in poor absorption of cerebrospinal fluid, creating a clinical picture similar to that of benign intracranial hypertension with bilateral disc swelling.40
Peripheral retinal microaneurysms in leukemic patients were originally described by Duke et al.41 and subsequently by Jampol et al.42 Duke et al.41 found that 50% of patients dying with chronic leukemia had peripheral microaneurysms. None were seen in patients with the acute leukemias. Seven of nine patients with chronic myelogenous leukemia and three of 10 patients with chronic lymphocytic leukemia had this finding, but it was not present in any of the 21 patients with acute leukemia.41 Kincaid and Green1 saw only one case in their large series. They noted that trypsin digest of the retina was essential or else the change would be overlooked on histopathologic examination.
Peripheral retinal neovascularization has been reported in patients with chronic myelogenous leukemia in association with peripheral capillary nonperfusion. Most cases have associated extreme leukocytosis or thrombocytosis.43–46 Presumably, the hyperviscosity state leads to peripheral nonperfusion and subsequent development of retinal neovascularization, as in patients with proliferative sickle retinopathy.
Morse and McCready46 reported on a 32-year-old patient with chronic myelogenous leukemia and retinal neovascularization. The peripheral white blood cell count was 340 500 and subsequently rose to 524 000/mm3. The fasting blood sugar value was normal, as was the hemoglobin electrophoresis study. No paraproteins were present. A fluorescein study highlighted multiple sea fans, and obliteration of the terminal arterioles was apparent. Frank and Ryan43 described a 30-year-old patient with a subhyaloid and vitreous hemorrhage who had a white blood cell count of 250 000/mm3 related to chronic myelogenous leukemia. Numerous sea fans were apparent and a glucose tolerance test and hemoglobin and serum protein electrophoresis studies were negative.46 Like Morse and McCready,46 Frank and Ryan43 believed that the pathogenic mechanism was related to increased blood viscosity, as in patients with complications of Waldenström macroglobulinemia or polycythemia. Kincaid and Green,1 however, did not see any cases of peripheral retinal neovascularization in their series.
Levielle and Morse44 described a patient with chronic myelogenous leukemia who had a relatively low (33 700/mm3) white blood cell count. In general, the blood viscosity begins to increase remarkably only with white blood cell counts of >50 000.47 In Levielle’s case report, the patient had an elevated platelet count of 988 000/mm3, and the peripheral neovascularization was attributed to this elevation. However, the authors did not emphasize that their patient also had an 11-year history of diabetes mellitus; therefore diabetic retinopathy also may have contributed to retinal capillary non-perfusion and formation of peripheral neovascularization.47 Melberg et al.48 described the effect of acute lymphocytic leukemia on the progression of mild diabetic retinopathy in a 16-year-old girl. The patient developed bilateral rubeosis, and after aggressive laser and vitrectomy, her vision declined to 20/200 bilaterally as a result of macular ischemia. The accelerated course of diabetic retinopathy correlated most closely with the anemia accompanying her leukemia and its treatment.
Wiznia et al.49 reported on concurrent optic disc and retinal neovascularization in an 18-year-old woman with acute lymphocytic leukemia who underwent therapy. They described progression of the neovascularization caused by the additive effects of radiation retinopathy and chemotherapy, resulting in macular traction detachment. The authors postulated that toxic effects of chemotherapy when combined with radiation therapy could lead to a more severe form of ischemic retinal vasculopathy than would be encountered with acute lymphocytic leukemia alone.49
The mechanism of the retinal hemorrhage seen in patients with leukemia is not yet known. The hemorrhages may be caused by an associated anemia or thrombocytopenia. Although commonly associated with severe leukocytosis, white-centered hemorrhages may be present regardless of the degree of leukocytosis.50
Some authors believe that the platelet count is more predictive than the hematocrit for the presence or absence of retinal hemorrhages.51 Kincaid and Green1 summarized the issue in 1983 and wrote that there is no close correlation between the degree of retinal involvement and red blood cell, white blood cell, or platelet levels. We have prospectively correlated the ocular findings with hematologic values on presentation in our series of 120 cases examined within a few days of diagnosis.52 We found a strong association between low platelet counts and intraretinal hemorrhages. Acute lymphoblastic leukemia patients with hemorrhages had a mean platelet count of 26 857/mm3, whereas patients without hemorrhage had mean counts of 116 159 (P≤0.0001); acute myelogenous leukemia patients with platelet counts of <15 000 were more likely than those without such low counts to have intraretinal hemorrhages (55% versus 29%). In addition, there was also a statistical difference between hematocrits (a mean of 20.3 mL/dL for patients with hemorrhage and a mean of 26.2 for patients without hemorrhage). However, a two or three point difference in the hematocrit is not of clinical importance. We believe that the platelet count plays a much stronger role in determining the presence or absence of intraretinal hemorrhage.52 Furthermore, on presentation, hematologic values were not found to be correlated with the presence of cotton-wool spots.
The presence of specific retinal manifestations of leukemic retinopathy and the subsequent risk of developing an intracranial hemorrhage was reported by Jackson et al.53 They reported a fivefold relative risk of an intracranial hemorrhage developing in those patients with macular hemorrhages compared with those patients without such hemorrhage. No increased risk of intracranial hemorrhage existed with the presence of non-macular intraretinal hemorrhages, white-centered hemorrhages, or cotton-wool spots. Therefore, patients with macular hemorrhages may require close monitoring for the possible development of intracranial hemorrhages, and these patients may need platelet transfusions if such an intracranial hemorrhage occurs.
Opportunistic infections
Opportunistic infections are common in immunosuppressed patients. Cytomegalovirus (CMV) is one of the most common causes of infectious retinitis in patients with altered immune status (Fig. 155.10). HTLV-1-associated adult T-cell leukemia can also present with a necrotizing retinal vasculitis.54 Various herpesviruses also can cause infectious retinitis.55 An unusual case of mumps uveitis in a patient with acute lymphocytic leukemia has been reported.56 A case of progressive outer retinal necrosis (PORN) after bone marrow transplantation in a 15-year-old boy with acute myelogenous leukemia has been reported by Lewis et al.57 The course of the infection was so rapid that antiviral therapy was unable to save the patient’s vision.
Among parasitic infections, ocular toxoplasmosis is the most common. Fungal intraocular involvement is a frequent and severe problem.58 One should consider opportunistic infection as a cause of retinal infiltration even in the absence of vitritis in immunocompromised patients.59 If a diagnostic dilemma exists, vitreous biopsy with 23- or 25-gauge vitrectomy may be helpful in identifying infectious etiologies.60,61 Hematologic malignancy has been reported as a common predisposing systemic factor for intraocular fungal infections and ophthalmologists should have a high index of suspicion when examining patients with myeloproliferative disorders.62
Prognosis
Leukemic retinopathy usually is seen when the patient is in relapse and is related to coexisting anemia.18 Many authors had previously observed that fundal changes did not appear to carry prognostic significance.63 However, the relationship of leukemic retinopathy to patient survival was evaluated prospectively in 54 patients by Abu el-Asrar et al.64 Among the 35% of patients with leukemic retinopathy, the mean survival rate was significantly shorter for those patients with cotton-wool spots than those without them (169 versus 609 days). Patients with cotton-wool spots were eight times more likely to die in the follow-up period than patients without this finding, possibly because of severe bone marrow dysfunction. The prognostic significance of leukemic retinopathy in childhood leukemia was evaluated in 63 patients by Ohkoshi and Tsiaras.65 The 5-year survival rate was significantly lower in those with leukemic retinopathy on presentation than in those without ophthalmic involvement (21.4% versus 45.7%). These two studies suggest that patients with clinical leukemic retinopathy may have more aggressive systemic disease that might lead to a worse prognosis.
Although peripheral blood counts or retinal hemorrhages and exudates do not seem to be predictors of systemic relapse or mortality, retinal infiltrates defined as whitish irregular patches near or around retinal vessels have been associated with leukemia that has a worse prognosis. Gross leukemic infiltrates, as illustrated in Figure 155.2, have been identified in moribund patients.
Treatment
Intraocular manifestations of leukemia usually are not treated directly. Rather, systemic chemotherapy is administered in an attempt to control the underlying systemic problem.66,67 In addition, recent studies have demonstrated that patients with central nervous system leukemic involvement may benefit from both intravenous and intrathecal chemotherapy without the need for cranial irrdiation.68 General supportive measures (e.g., blood transfusions) may also be recommended for patients with severe anemia or thrombocytopenia.
It is not known whether or not most systemic chemotherapeutic agents penetrate into the eye. The eye does appear to be beyond the reach of intrathecal chemotherapy.69 When definite leukemic infiltrates fail to respond promptly to systemic or intrathecal chemotherapy, ocular radiation may be recommended. Varying doses have been used,1 and consultation with an experienced radiation oncologist is essential. Hoover et al. reported on their follow-up of 82 survivors of leukemia treated at their institution and found only minimal ocular morbidity (posterior subcapsular cataracts in 52%).70 Lopez et al. described the occurrence of radiation retinopathy after low doses of teletherapy in five of eight patients with leukemia who were treated with high-dose chemotherapy in the setting of bone marrow transplantation.71 They suggest that high-dose chemotherapy may increase the susceptibility for the development of radiation retinopathy at otherwise safe radiation doses. Webster et al. reported on a similar case of ischemic retinopathy complicating bone marrow transplantation that was combined with the use of campath-1G (for suppression of graft-versus-host disease).72 Therefore, treatment with cranial or orbital radiation may result in radiation retinopathy and follow-up evaluation is necessary to detect this potential complication. Hyperleukocytic retinopathy may be managed by leukapheresis. Mehta et al. reported on three patients with hemorrhages and exudates in a pattern consistent with a state of severe hyperviscosity.73 They noted retinal venous distention, scattered hemorrhages, and optic disc edema in a pattern of a mild central retinal vein occlusion. The patients had peripheral white blood cell counts of 129 000, 379 000, and 1 043 000/mm3, respectively. Two patients improved rapidly after leukapheresis. In addition, the authors cite a number of other cases in which the procedure has been used successfully.73
Lymphomas
Lymphomas are classified as either Hodgkin lymphoma or non-Hodgkin lymphoma. In addition, malignant lymphomas are divided into primary intraocular lymphoma and secondary intraocular lymphoma. Primary intraocular lymphoma involves primary central nervous system lymphoma whereas secondary intraocular lymphoma involves a metastasis from a primary visceral lymphoma. The incidence of neoplastic intraocular involvement in patients with lymphomas is probably much less than that in patients with various leukemias. Lymphoid infiltration of the uvea, formally termed reactive lymphoid hyperplasia, is rare and usually is not associated with systemic disease.74
Non-Hodgkin lymphoma
Non-Hodgkin lymphomas constitute approximately 5% of all newly diagnosed cancers in the USA. They are predominantly of B-cell lymphocytic origin, although some may be derived from T cells.75 Ocular manifestations in this disease occur in two distinct settings, systemic non-Hodgkin lymphoma and primary central nervous system (CNS) non-Hodgkin lymphoma.76 Systemic non-Hodgkin lymphoma most commonly involves the internal structures of the eye, gaining access by the choroidal tissues.77–80 Ocular involvement is often asymmetric and most commonly affects individuals in the sixth decade of life and beyond (although a case has been reported in a 15-year-old boy79).
Until recently, primary ocular lymphoma has been an uncommon cause of chronic vitritis and has represented one of several sites of multifocal primary central nervous system lymphoma (PCNSL), also referred to as non-Hodgkin lymphoma of the central nervous system (NHL-CNS).81 In up to half of the cases of PCNSL, the eyes were the initial site of disease and it is frequently bilateral.76,79 In a large retrospective study of 221 patients with PCNSL, the median age at diagnosis was 60 years and ocular disturbances and behavioral/cognitive changes were the most common presenting symptoms.82 The correct diagnosis is often not established until late in the disease course, since this condition often masquerades as chronic uveitis, despite the absence of external signs of inflammation.31 Approximately 60–80% of patients who have ocular lymphoma at presentation develop cerebral lymphoma with a long enough follow-up period.77 Metastasis of PCNSL outside the CNS is seen in only up to 8% of autopsy series. In the past decade there has been a steady increase in the frequency of reports of primary intraocular lymphomas.83 This increased incidence of PCNSL has been reported in both immunocompromised patients (from AIDS and from organ transplantation) and immunocompetent patients.79,84 It is postulated that immunocompromised patients manifest with an abnormal response to the Epstein–Barr virus (EBV) through development of a monoclonal B cell proliferation.85 Ocular lymphoma and PCNSL are restricted to the central nervous system and are not a consequence of metastasis of systemic lymphoma.
Retinal hemorrhages and cotton-wool spots related to anemia or thrombocytopenia are common in patients with non-Hodgkin lymphoma, but direct retinal involvement in patients with systemic lymphoma is extremely rare. Lewis and Clark86 described a patient with well-differentiated lymphocytic lymphoma who had massive perivenous infiltrates (Fig. 155.11). The patient was treated with 3000 cGy of external beam radiation, and a partial response occurred. The authors believed that the most likely cause was lymphomatous infiltration, although no pathologic examination was performed.86 The appearance of the retina was similar to that seen in frosted-branch angiitis.87 Topilow et al. described a case of lymphomatous infiltration of the retina simulating progressive outer retinal necrosis.88 Ocular lymphoma should be considered in the differential diagnosis of retinal vasculitis or necrotizing retinitis.75,80,89 Gass et al. described two patients with the presenting symptom of a flecked retina simulating fundus flavimaculatus in one eye months before developing signs and symptoms of the systemic form of non-Hodgkin lymphoma.90 Shah et al. reported the presence of multiple white spots at the level of the outer retina resembling the multiple evanescent white dot syndrome.91 Marmor et al. reported on a patient with poorly differentiated lymphocytic lymphoma who subsequently developed histiocytic lymphoma.92 This patient was thought to have lymphomatous involvement of the optic nerve head, although pathologic study demonstrated cytomegalovirus (CMV) in the optic nerve head.92 Fredrick et al. described a large peripapillary choroidal lymphomatous mass as the presenting manifestation of systemic lymphoma.93 In addition, Jensen et al. reported an intraocular T-cell lymphoma mimicking a ring melanoma as the first manifestation of a systemic lymphoma.94 We examined a patient with non-Hodgkin lymphoma who had massive infiltration of the optic disc with subsequent development of a central retinal artery occlusion (Fig. 155.12).95
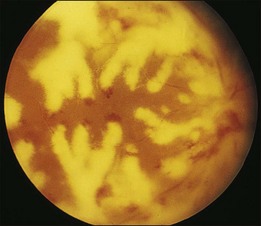
Fig. 155.11 Massive perivascular infiltrates in a patient with well-differentiated lymphocytic lymphoma.
(Reproduced with permission from Lewis RA, Clark RB. Infiltrative retinopathy in systemic lymphoma. Am J Ophthalmol 1975;79:48–52.)
Characteristic yellowish placoid RPE detachments may occur in some cases of ocular lymphoma, and the malignant cells are found in the vitreous, the retina, and between the RPE and Bruch’s membrane.96 These tumorous RPE detachments can resolve spontaneously, resulting in the formation of disciform scars or areolar atrophy of the RPE. Indirect effects of systemic lymphoma on the eye have also been reported. Cohen et al. described an 11-year-old girl with a central retinal artery occlusion in association with a systemic T-cell lymphoma.97 They postulated that a paraneoplastic hypercoagulopathy was responsible, although they could not rule out the possibility of a lesion behind the nerve head. Das et al. reported on a 19-year-old man with retinopathy caused by hypertension, which was caused by renal involvement by non-Hodgkin lymphoma.98
Cytologic examination of vitreous biopsies is the benchmark for diagnosis of intraocular lymphoma.80,81,88,99–105 A diagnostic vitrectomy with prompt and careful cytopreparatory techniques is essential for the detection of lymphoma cells. Lymphoma cells are typically 2–4 times the size of normal lymphocytes and display high nuclear/cytoplasmic ratios, prominent nucleoli, nuclear pleomorphism, and coarse chromatin patterns.99,105,106 The skills and experience of the pathologist interpreting the specimen are critical in making the diagnosis in conjunction with the use of flow cytometry to define the cellular origins. Monoclonal proliferations of B cells through polymerase chain reaction (PCR) have been used to increase the sensitivity of vitreous biopsies performed in patients suspected of having B-cell lymphoma.107 In addition, interleukin-10 and interleukin-6 concentrations in the vitreous may be correlated with the clinical activity and number of malignant cells.108–110 Elevated levels of interleukin-10 therefore should alert the pathologist to the presence of malignant cells in the vitreous specimen.105,110
Other routes for tissue biopsy include subretinal aspiration biopsy,111,112 fine-needle aspiration biopsy,113,114 and transscleral chorioretinal biopsy.115 In some cases, concurrent vitreous biopsies may be cytologically negative, and the diagnosis may be made only with the subretinal biopsy (Fig. 155.13).
Neuroimaging has been shown to have a low sensitivity for differentiating intraocular lymphoma from uveitis or melanoma.116 Ultrasonography has been useful in documenting the extent of posterior segment involvement before therapy and may be useful in evaluating the response to treatment.117 All patients with positive vitreous biopsies should undergo cerebrospinal fluid cytologic examination.
The paraneoplastic syndrome of bilateral diffuse uveal melanocytic proliferation has been reported in the single case of a patient with non-Hodgkin systemic lymphoma.118
Hodgkin lymphoma
Hodgkin disease is a malignant lymphoma with protean manifestations. The disease is characterized by painless swelling of the lymph nodes, and Reed–Sternberg cells are seen on histopathologic examination. Barr and Joondeph78 described a patient whose initial manifestation of Hodgkin disease was periphlebitis, focal chorioretinitis, vitritis, and optic disc edema (Fig. 155.14). The retinitis and vitritis resolved after 4000 cGy of radiation therapy.78 The authors summarized a number of previous reports of ophthalmic manifestations of Hodgkin disease, including bilateral exudative retinal detachment, cotton-wool spots with retinal hemorrhages, and necrotizing retinitis. Patients with “numerous white deposits in the retinal periphery,” chorioretinitis, Roth spots, and perivascular retinitis have been reported.39,78 Histopathologic evidence of cotton-wool spots has been reported in patients with Hodgkin disease.119
Direct demonstration of tumor cells in ocular tissues is rare in patients with Hodgkin disease. Primbs et al. described a patient with primarily anterior uveitis at presentation who had no malignant cells in the vitreous. Reed-Sternberg cells were seen in the anterior chamber and trabecular meshwork.120 Mosteller et al. reported on a patient with anterior and posterior uveitis, pars plana exudates, perivascular sheathing, small, round, discrete white retinal lesions, and cystoid macular edema. A vitrectomy specimen demonstrated only acute and chronic inflammatory cells, and no tumor cells were seen.121
In one published report, a patient with Hodgkin disease had widespread severe destruction of the retina that was believed to represent a manifestation of drug toxicity.122 However, the patient also had concomitant disseminated herpes zoster infection, and viral retinitis of this type is known to occur in immunocompromised hosts.123 This association suggests that many cases previously diagnosed as intraocular Hodgkin disease probably represent secondary viral retinitis. Diddie et al. reported the histopathologic findings of a patient with Hodgkin lymphoma who had herpesvirus infection.124 Toy and Knowlden summarized the findings of a patient with CMV retinitis who had been misdiagnosed as having Hodgkin lymphoma deposits.125
Opportunistic infections are common in patients with Hodgkin lymphoma. Toxoplasmic uveitis and chorioretinitis,126 Nocardia infection,127 and virtually all viral infections of the herpes family have been previously reported.39
Treatment of lymphoma
Management of ocular involvement by non-Hodgkin and Hodgkin lymphoma consists of systemic and/or intrathecal chemotherapy with possible radiation therapy for CNS disease that is recalcitrant to chemotherapy.105 The most common systemic agents used include methotrexate, cytarabine, vincristine, rituximab, cyclophosphamide, and steroids. The optimal treatment for PCNSL is still controversial.
In the past few years, ophthalmologists have employed the use of intravitreal injection of chemotherapeutic agents as an alternative to external beam radiation of the eye. Reports have demonstrated successful use of intravitreal methotrexate and rituximab.128,129
The prognosis for survival in untreated disease is poor, but it may be improved with early and aggressive therapy.77 Even with aggressive modern therapy, the median survival is expected to be about 3 years.130,131
Mycosis fungoides
Mycosis fungoides is a malignant T-cell lymphoma arising in the skin. There are three stages of the disease: (1) a prolonged phase of premycotic/eczematous skin lesions; (2) a phase characterized by infiltrative plaque lesions; and (3) a final phase of frank cutaneous tumor. Histopathologically, mycosis fungoides is characterized by cellular infiltrates of atypical lymphoid cells in the dermis (Pautrier’s microabscesses), clusters of atypical lymphoid cells, and mycosis cells/lymphoid cells with large, irregular, and deeply indented “cerebriform” nuclei.132 Systemic involvement occurs late in the course of the disease and may include virtually all organ systems.133 Commonly these extracutaneous sites of involvement include the lymph nodes, liver, spleen, and CNS. Most affected individuals develop the disease in the fifth decade of life, and many die of unrelated causes before widespread involvement.
Mycosis fungoides involves the eye in up to one third of individuals and tends to involve the external eye and adnexa much more commonly than the intraocular structures.132 Only a few cases with intraocular involvement have been reported. Keltner et al. saw a patient with disc swelling and macular edema. The disc swelling was probably related to papilledema because lethargy, confusion, and focal neurologic signs were observed.132 The patient initially responded to radiation therapy, steroids, and intrathecal methotrexate but subsequently developed swelling and pallor of both discs and infiltration of the retina and vitreous. On histopathologic examination, atypical cells and lymphocytes, as well as polymorphonuclear cells, were seen in the vitreous. Similar atypical cells infiltrated the retina, and a perivascular lymphocytic infiltrate was noted. Rossi reported on a patient with bilateral papilledema, venous stasis, retinal edema, and retinal hemorrhages.134 Gartner described a patient with choroidal involvement chiefly characterized by perivascular granulomas.135 Foerster presented the histopathologic findings from a patient with a blind, painful eye who had diffuse neoplastic subretinal pigment epithelial infiltrates.136 Necrotic tumor cells were also seen in the vitreous. A 16-year-old girl who died of visceral mycosis fungoides had no light perception in either eye or bilateral disc swelling. Malignant cells infiltrated the surrounding peripapillary retina.
The largest series of patients with mycosis fungoides reported in the ophthalmic literature is that of Stenson and Ramsay.137 A total of 30 consecutive patients with mycosis fungoides were examined; 11 had positive findings related to mycosis fungoides, although only four had posterior segment changes (three patients had optic atrophy, and one patient had recurrent panuveitis). Erny et al. reported on the clinical and histopathologic findings in a 48-year-old man with vitritis and subretinal lesions that proved to be malignant T-cell infiltrates in the retina and the vitreous and between the RPE and Bruch’s membrane.138 Leitch et al. established the diagnosis in a 61-year-old woman with the aid of a vitreous biopsy.139 Several of these reports demonstrated intraocular infiltration as an early manifestation of extracutaneous disease.132,138,139
Burkitt lymphoma
Burkitt lymphoma was originally described in 1958 in a review of a series of patients from Kampala, Uganda.140 It is a poorly differentiated lymphocytic lymphoma with characteristic clinical and histological patterns. Burkitt lymphoma is the most common childhood tumor in Africa, but it occurs only rarely in the USA.141
Karp et al.141 described a case in which the choroid and much of the interior of the globe was diffusely infiltrated by tumor. Burkitt lymphoma commonly involves the orbital structures, and the authors did not rule out the possibility that an invasive orbital neoplasm secondarily involved the intraocular structures. Feman et al.142 described a patient who had a white patch over the disc associated with many small retinal hemorrhages. Histopathologic study demonstrated diffuse neoplastic infiltration of the optic disc and peripapillary retina. The infiltrate contained scattered histiocytes (Fig. 155.15). The choroid was uninvolved, but scattered cells were present in the vitreous. Definite primary intraocular Burkitt lymphoma is exceedingly rare.
Multiple myeloma and Waldenström macroglobulinemia
Multiple myeloma is a plasma cell neoplasm that can exhibit a wide range of systemic and ocular signs and symptoms.143 Pars plana cysts are common and can be a striking ophthalmic finding.143 Uveal involvement has been reported by Bronstein.144 The most common retinal findings include manifestations of anemia and thrombocytopenia, such as flame-shaped or white-centered hemorrhages and nerve fiber layer infarcts.145 In one report these findings were seen in 8 of 22 patients with multiple myeloma.36
Microaneurysm formation may be apparent, most frequently in the retinal periphery and midperiphery.146,147 The cause of this change is not certain. It may be related to hyperviscosity,148 although a frank hyperviscosity syndrome is not as common in patients with multiple myeloma as it is in patients with, e.g., Waldenström macroglobulinemia. If hyperviscosity is severe, retinal changes similar to those described in Waldenström macroglobulinemia may be seen.149 Serous and exudative retinal detachments associated with multiple myeloma have also been reported.55,150
A clinicopathologic review of multiple myeloma and its ocular manifestations has been presented by Knapp et al.143 In this report, the patients with retinopathy had lower hematocrit levels and platelet counts than those without retinopathy. In many patients the retinopathy cleared with treatment. The presence of retinal changes did not appear to alter the prognosis.
Patients with Waldenström macroglobulinemia have an abnormal elevation of their IgM fraction of plasma proteins. The retinal findings are chiefly those of systemic hyperviscosity, although manifestations of anemia or thrombocytopenia may be seen. Clinically, patients may present with bilateral venous dilation which is difficult to differentiate from the findings of central retinal vein obstruction.151 In addition to the picture of venous obstruction, microaneurysms associated with peripheral retinal neovascularization may be present. Peripheral capillary non-perfusion of the retina has also been reported.152 Case reports have also documented the formation of serous retinal detachment.153 The findings may be reversible when the hyperviscosity state is normalized (see Figure 155.9). Visually symptomatic patients may benefit from plasmapheresis.146
1 Kincaid MC, Green WR. Ocular and orbital involvement in leukemia. Surv Ophthalmol. 1983;27:211–232.
2 Swartz M, Schumann B. Acute leukemic infiltration of the vitreous diagnosed by pars plana aspiration. Am J Ophthalmol. 1980;90:326–330.
3 Allen RA, Straatsma BR. Ocular involvement in leukemia and allied disorders. Arch Ophthalmol. 1961;66:490–508.
4 Karesh JW, Goldman EJ, Reck K, et al. A prospective ophthalmic evaluation of patients with acute myeloid leukemia: correlation of ocular and hematologic findings. J Clin Oncol. 1989;7:1528.
5 Schachat AP, Markowitz JA, Guyer DR, et al. Ophthalmic manifestations of leukemia. Arch Ophthalmol. 1989;107:697–700.
6 Nelson CC, Hertzberg BS, Klintworth GK. A histopathologic study of 716 selected eyes in patients with cancer at the time of death. Am J Ophthalmol. 1983;95:788–793.
7 Leonardy NJ, Rupani M, Dent G, et al. Analysis of 135 autopsy eyes for ocular involvement in leukemia. Am J Ophthalmol. 1990;109:436–444.
8 Reddy SC, Menon BS. A prospective study of ocular manifestations in childhood acute leukemia. Acta Ophthalmol Scand. 1998;76:700–703.
9 Reddy SC, Jackson N, Menon BS. Ocular involvement in leukemia – a study of 288 cases. Ophthalmologica. 2003;217:441–445.
10 Russo V, Scott IU, Querques G, et al. Orbital and ocular manifestations of acute childhood leukemia: clinical and statistical analysis of 180 patients. Eur J Ophthalmol. 2008;18:619–623.
11 Buchan J, McKibbin M, Burton T. The prevalence of ocular disease in chronic lymphocytic leukemia. Eye. 2003;17:3–4.
12 Kuwabara T, Aiello L. Leukemic miliary nodules in the retina. Arch Ophthalmol. 1964;72:494–497.
13 Robb RM, Ervin LD, Sallan SE. A pathological study of eye involvement in acute leukemia of childhood. Trans Am Ophthalmol Soc. 1978;76:90–101.
14 Merle H, Donnio A, Gonin C, et al. Retinal vasculitis caused by adult T-cell leukemia/lymphoma. Jpn J Ophthalmol. 2005;49:41–45.
15 Bandyopadhyay S, Das D, Das G, et al. Oman J. Unilateral optic nerve infiltration as an initial site of relapse of acute lymphoblastic leukemia in remission. Ophthalmol. 2010;3:153–154.
16 Kim TS, Duker JS, Hedges TR. Retinal angiopathy resembling unilateral frosted branch angiitis in a patient with relapsing acute lymphoblastic leukemia. Am J Ophthalmol. 1994;117:806–808.
17 Clayman HM, Flynn JT, Koch K, et al. Retinal pigment epithelial abnormalities in leukemic disease. Am J Ophthalmol. 1972;74:416–419.
18 Rosenthal AR. Ocular manifestations of leukemia: a review. Ophthalmology. 1983;90:899–905.
19 Liu MM, Furusato E, Cao X, et al. Ocular manifestations and pathology of adult T-cell leukemia/lymphoma associated with human T-lymphotropic virus type 1. Rare tumors. 2010;2:e63.
20 Kincaid MC, Green WR, Kelley JS. Acute ocular leukemia. Am J Ophthalmol. 1979;87:698–702.
21 Gass JDM. Stereoscopic atlas of macular diseases: diagnosis and treatment, 4th ed. St Louis: Mosby; 1997.
22 Burns CA, Blodi FC, Williamson BK. Acute lymphocytic leukemia and central serous retinopathy. Trans Am Acad Ophthalmol Otolaryngol. 1965;69:307–309.
23 Paydas S, Soylu MB, Disel U, et al. Serous retinal detachment in a case with chronic lymphocytic leukemia: no response to systemic and local treatment. Leuk Res. 2003;27:557–559.
24 Tang RA, Vila-Coro AA, Wall S, et al. Acute leukemia presenting as a retinal pigment epithelium detachment. Arch Ophthalmol. 1988;106:21–22.
25 Stewart MW, Gitter KA, Cohen G. Acute leukemia presenting as a unilateral exudative retinal detachment. Retina. 1989;9:110–114.
26 Jakobiec F, Behrens M. Leukemic retinal pigment epitheliopathy with report of a unilateral case. J Pediatr Ophthalmol. 1975;12:10–15.
27 Belmont JB, Michelson JB, Bordin GM. Ocular inflammation associated with chronic lymphocytic leukemia. J Ocul Ther Surg. 1985;4:125–129.
28 Lipton JH, McGowan HD, Payne DG. Ocular masquerade syndrome in lymphoid blast crisis of chronic myeloid leukemia. Leuk Lymphoma. 1995;20:161–163.
29 Robinson A, Eting E, Zeidman A, et al. Manifestations of hairy cell leukemia with dramatic response to 2-chloro-deoxy-adenosine. Am J Ophthalmol. 1996;121:97–98.
30 Dhar-Munshi S, Alton P, Ayliffe WH. Masquerade syndrome: T-cell prolymphocytic leukemia presenting as panuveitis. Am J Ophthalmol. 2001;132:275–277.
31 Rothova A, Ooijman F, Kerkhoff F, et al. Uveitis masquerade syndromes. Ophthalmology. 2001;108:386–399.
32 Delaney WV, Jr., Kinsella G. Optic disc neovascularization in leukemia. Am J Ophthalmol. 1985;99:212–213.
33 de Juan E, Green WR, Rice TA, et al. Optic disc neovascularization associated with ocular involvement in acute lymphocytic leukemia. Retina. 1982;2:61–64.
34 Hattenhauer MG, Pach JM. Ocular lymphoma in a patient with chronic lymphocytic leukemia. Am J Ophthalmol. 1996;122:266–268.
35 Duane TD, Osher RH, Green WR. White centered hemorrhages: their significance. Ophthalmology. 1980;87:66–69.
36 Holt JM, Gordon-Smith EL. Retinal abnormalities in diseases of the blood. Br J Ophthalmol. 1969;53:145–160.
37 Phelps CD. The association of pale-centered retinal hemorrhages with intracranial bleeding in infancy. Am J Ophthalmol. 1971;73:348–350.
38 Brown GC, Brown MM, Hiller T, et al. Cottonwool spots. Retina. 1985;5:206–214.
39 Bishop JE, Salmonsen PC. Presumed intraocular Hodgkin’s disease. Ann Ophthalmol. 1985;17:589–592.
40 Guymer RH, Cairns JD, O’Day J. Benign intracranial hypertension in chronic myeloid leukemia. Aust NZ J Ophthalmol. 1993;21:181–185.
41 Duke JR, Wilkinson CP, Sigelman S. Retinal microaneurysms in leukemia. Br J Ophthalmol. 1968;52:368–374.
42 Jampol LM, Goldberg MF, Busse B. Peripheral microaneurysms in chronic leukemia. Am J Ophthalmol. 1975;80:242–248.
43 Frank RN, Ryan SJ, Jr. Peripheral retinal neovascularization with chronic myelogenous leukemia. Arch Ophthalmol. 1972;87:585–589.
44 Levielle AS, Morse PH. Platelet-induced retinal neovascularization in leukemia. Am J Ophthalmol. 1981;91:640–643.
45 Little HL. The role of abnormal hemorrheodynamics in the pathogenesis of diabetic retinopathy. Trans Am Ophthalmol Soc. 1976;74:573–636.
46 Morse PH, McCready JL. Peripheral retinal neovascularization in chronic myelocytic leukemia. Am J Ophthalmol. 1971;72:975–978.
47 Stephens DJ. Relation of viscosity of blood to leukocyte count with particular reference to chronic myelogenous leukemia. Proc Soc Exp Biol Med. 1936;35:251–256.
48 Melberg NS, Grand MG, Rup D. The impact of acute lymphocytic leukemia on diabetic retinopathy. J Pediatr Hematol Oncol. 1995;17:81–84.
49 Wiznia RA, Rose A, Levy AL. Occlusive microvascular retinopathy with optic disc and retinal neovascularization in acute lymphocytic leukemia. Retina. 1994;14:253–255.
50 Gibson GG. Clinical significance of the retinal changes in leukemia. Arch Ophthalmol. 1938;20:364–370.
51 Culler AM. Fundus changes in leukemia. Trans Am Ophthalmol Soc. 1951;49:445–473.
52 Guyer DR, Schachat AP, Vitale S, et al. Leukemic retinopathy: relationship between fundus lesions and hematologic parameters at diagnosis. Ophthalmology. 1989;96:860–864.
53 Jackson N, Reddy SC, Harun MH, et al. Macular hemorrhage in adult acute leukemia patients at presentation and the risk of subsequent intracranial hemorrhage. Br J Haematol. 1997;98:204–209.
54 Levy-Clarke GA, Buggage RR, Shen D, et al. Human T-cell lymphocytic virus type-1 associated T-cell leukemia/lymphoma masquerading as necrotizing retinal vasculitis. Ophthalmology. 2002;109:1717–1722.
55 Brody JM, Butrus SI, Ashraf MF, et al. Multiple myeloma presenting with bilateral exudative macular detachments. Acta Ophthalmol Scand. 1995;73:81–82.
56 Al-Rashid RA, Cress C. Mumps uveitis complicating the course of acute leukemia. J Pediatr Ophthalmol. 1977;14:100–102.
57 Lewis JM, Nagae Y, Tano Y. Progressive outer retinal necrosis after bone marrow transplantation. Am J Ophthalmol. 1996;122:892–895.
58 Phillips WB, Shields CL, Shields JA, et al. Nocardia choroidal abscess. Br J Ophthalmol. 1992;76:694–696.
59 Gordon KB, Rugo HS, Duncan JL, et al. Ocular manifestations of leukemia: leukemic infiltration versus infectious process. Ophthalmology. 2001;108:2293–2300.
60 Palkovacs EM, Correa Z, Ausburger JJ, et al. Acquired toxoplasmic retinitis in an immunosuppressed patient: diagnosis by transvitreal fine-needle aspiration biopsy. Graefes Arch Clin Exp Ophthalmol. 2008;246:1495–1497.
61 Harper TW, Miller D, Schiffman JC, et al. Polymerase chain reaction analysis of aqueous and vitreous specimens in the diagnosis of posterior segment infectious uveitis. Am J Ophthalmol. 2009;147:140–147.
62 McDonnell PJ, McDonnell JM, Brown RH, et al. Ocular involvement in patients with fungal infections. Ophthalmology. 1985;92:706–709.
63 Marshall RA. A review of lesions in the optic fundus in various diseases of the blood. Blood. 1959;14:882–891.
64 Abu el-Asrar AM, Al-Momen AK, et al. Prognostic importance of retinopathy in acute leukemia. Doc Ophthalmol. 1996;91:273–281.
65 Ohkoshi K, Tsiaras WG. Prognostic importance of ophthalmic manifestations in childhood leukemia. Br J Ophthalmol. 1992;76:651–655.
66 Jabbour E, Branford S, Saglio G, et al. Practical advice for determining the role of BCR-ABL mutations in guiding tyrosine kinase inhibitor therapy in patients with chronic myeloid leukemia. Cancer. 2011;117:1800–1811.
67 Pollyea DA, Kohrt HE, Medeiros BC. Acute myeloid leukaemia in the elderly: a review. Br J Haematol. 2011;152:524–542.
68 Pui CH. Recent research advances in childhood acute lymphoblastic leukemia. J Formos Med Assoc. 2010;109:777–787.
69 Ellis W, Little HL. Leukemic infiltration of the optic nerve head. Am J Ophthalmol. 1983;75:867–871.
70 Hoover DL, Smith LEH, Turner SJ, et al. Ophthalmic evaluation of survivors of acute lymphoblastic leukemia. Ophthalmology. 1988;95:151–155.
71 Lopez PF, Sternberg P, Dabbs CK, et al. Bone marrow transplant retinopathy. Am J Ophthalmol. 1991;112:635–646.
72 Webster AR, Anderson JR, Richards EM, et al. Ischemic retinopathy occurring in patients receiving bone marrow allografts and Campath-1G: a clinicopathological study. Br J Ophthalmol. 1995;79:687–691.
73 Mehta AB, Goldman JM, Kohner E. Hyperleucocytic retinopathy in chronic granulocytic leukaemia: the role of intensive leukapheresis. Br J Haematol. 1984;56:661–667.
74 Jakobiec FA, Sacks E, Kronish JW, et al. Multifocal static creamy choroidal infiltrates. Ophthalmology. 1987;94:397–406.
75 Brown SM, Jampol LM, Cantrill HL. Intraocular lymphoma presenting as retinal vasculitis. Surv Ophthalmol. 1994;39:133–140.
76 Hoffman PM, McKelvie P, Hall AJ, et al. Intraocular lymphoma – a series of 14 patients with clinicopathological features and treatment outcomes. Eye. 2003;17:513–521.
77 Freeman LN, Schachat AP, Knox DL, et al. Clinical features, laboratory investigations and survival in ocular reticulum cell sarcoma. Ophthalmology. 1987;94:1631–1638.
78 Barr CC, Joondeph HC. Retinal periphlebitis as the initial clinical finding in a patient with Hodgkin’s disease. Retina. 1983;3:253–257.
79 Peterson K, Gordon KB, Heinemann MH, et al. The clinical spectrum of ocular lymphoma. Cancer. 1993;72:843–849.
80 Ridley ME, McDonald HR, Sternberg P, et al. Retinal manifestations of ocular lymphoma (reticulum cell sarcoma). Ophthalmology. 1992;99:1153–1161.
81 Whitcup SM, de Smet MD, Rubin BI, et al. Intraocular lymphoma: clinical and histopathologic diagnosis. Ophthalmology. 1993;100:1399–1406.
82 Grimm SA, McCannel CA, Omuro AM, et al. Primary CNV lymphoma with intraocular involvement: International PCNSL Collaborative Group Report. Neurology. 2008;71:1355–1360.
83 Levy-Clarke GA, Chan CC, Nussenblatt RB. Diagnosis and management of primary intraocular lymphoma. Hematol Oncol Clin North Am. 2005;19:739–749.
84 Zimmer-Galler I, Lie JT. Choroidal infiltrates as the initial manifestation of lymphoma in rheumatoid arthritis after treatment with low-dose methotrexate. Mayo Clin Proc. 1994;69:258–261.
85 Mittra RA, Pulido JS, Hanson GA, et al. Ocular Epstein–Barr virus associated non-Hodgkin’s lymphoma in a patient with AIDS: a clinicopathologic report. Retina. 1999;19:45–50.
86 Lewis RA, Clark RB. Infiltrative retinopathy in systemic lymphoma. Am J Ophthalmol. 1975;79:48–52.
87 Geier SA, Nasemann J, Klauss V, et al. Frosted branch angiitis in a patient with the acquired immunodeficiency syndrome. Am J Ophthalmol. 1992;113:203–205.
88 Topilow HW, Ackerman AL, Friedman A. Progressive outer retinal necrosis. Ophthalmology. 1995;102:1737–1738.
89 Kohno T, Uchida H, Inomata H, et al. Ocular manifestations of adult T-cell leukemia/lymphoma: a clinicopathologic study. Ophthalmology. 1993;100:1794–1799.
90 Gass JDM, Weleber RG, Johnson DR. Non-Hodgkin’s lymphoma causing fundus picture simulating fundus flavimaculatus. Retina. 1987;7:209–214.
91 Shah GK, Kleiner R, Augsburger J, et al. Primary intraocular lymphoma presenting as transient white fundus lesions simulating multiple evanescent white dot syndrome. Arch Ophthalmol. 2001;119:617–620.
92 Marmor MF, Egbert PR, Egbert BM, et al. Optic nerve head involvement with cytomegalovirus in an adult with lymphoma. Arch Ophthalmol. 1978;96:1252–1254.
93 Fredrick DR, Char DH, Ljung B, et al. Solitary intraocular lymphoma as an initial presentation of widespread disease. Arch Ophthalmol. 1989;107:395–397.
94 Jensen OA, Johansen S, Kiss K. Intraocular T-cell lymphoma mimicking a ring melanoma: first manifestation of systemic disease. Graefes Arch Clin Exp Ophthalmol. 1994;232:148–152.
95 Guyer DR, Green WR, Schachat AP, et al. Bilateral ischemic optic neuropathy and retinal vascular occlusions associated with lymphoma and sepsis. Ophthalmology. 1990;97:882–888.
96 Dean JM, Novak MA, Chan CC, et al. Tumor detachments of the retinal pigment epithelium in ocular/central nervous system lymphoma. Retina. 1996;16:47–56.
97 Cohen RG, Hedges TR, Duker JS. Central retinal artery occlusion in a child with T-cell lymphoma. Am J Ophthalmol. 1995;120:118–120.
98 Das A, Puklin JE, Spoor TC, et al. Retinopathy due to renovascular hypertension in a patient with non-Hodgkin’s lymphoma. Arch Ophthalmol. 1992;110:1052–1053.
99 Blumenkranz MS, Ward T, Murphy S, et al. Applications and limitations of vitreoretinal biopsy techniques in intraocular large cell lymphoma. Retina. 1992;12:64–70.
100 Buettner H, Bolling JP. Intravitreal large-cell lymphoma. Mayo Clin Proc. 1993;68:1011–1015.
101 Rutzen AR, Ortega-Larrocea G, Dugel PU, et al. Clinicopathologic study of retinal and choroidal biopsies in intraocular inflammation. Am J Ophthalmol. 1995;119:597–611.
102 Siegel MJ, Dalton J, Griedman AH, et al. Ten-year experience with primary ocular “reticulum cell sarcoma” (large cell non-Hodgkin’s lymphoma). Br J Ophthalmol. 1989;73:342–346.
103 Verbraeken HE, Hanssens M, Priem H, et al. Ocular non-Hodgkin’s lymphoma: a clinical study of nine cases. Br J Ophthalmol. 1997;81:31–36.
104 Wilson DJ, Braziel R, Rosenbaum JT. Intraocular lymphoma: immunopathologic analysis of vitreous biopsy specimens. Arch Ophthalmol. 1992;110:1455–1458.
105 Rajagopal R, Jarbour JW. Diagnostic testing and treatment choices in primary vitreoretinal lymphoma. Retina. 2011;31:435–440.
106 Conlon MR, Craig I, Harris JF, et al. Effect of vitrectomy and cytopreparatory techniques on cell survival and preservation. Can J Ophthalmol. 1992;27:168–171.
107 Katai N, Kuroiwa S, Fujimori K, et al. Diagnosis of intraocular lymphoma by polymerase chain reaction. Graefes Arch Clin Exp Ophthalmol. 1997;235:431–436.
108 Chan CC. Molecular pathology of primary intraocular lymphoma. Trans Am Ophthalmol Soc. 2003;101:275–292.
109 Chan CC, Whitcup SM, Solomon D, et al. Interleukin-10 in the vitreous of patients with primary intraocular lymphoma. Am J Ophthalmol. 1995;120:671–673.
110 Whitcup SM, Stark-Vancs V, Wittes RE, et al. Association of interleukin-10 in the vitreous and cerebrospinal fluid and primary central nervous system lymphoma. Arch Ophthalmol. 1997;115:1157–1160.
111 Ciulla TA, Pesavento RD, Yoo S. Subretinal aspiration biopsy of ocular lymphoma. Am J Ophthalmol. 1997;123:420–422.
112 Pavan PR, Oteiza EE, Margo CE. Ocular lymphoma diagnosed by internal subretinal pigment epithelium biopsy. Arch Ophthalmol. 1995;113:1233–1234.
113 Lobo A, Lightman S. Vitreous aspiration needle tap in the diagnosis of intraocular inflammation. Ophthalmology. 2003;110:595–599.
114 Shields JA, Shields CL, Ehya H, et al. Fine-needle aspiration biopsy of suspected intraocular tumors. Ophthalmology. 1993;100:1677–1684.
115 Matsuo K, Nakatuka K, Matsuura T, et al. Primary intraocular lymphoma mimicking late postoperative endophthalmitis. Ophthalmologica. 1995;209:331–335.
116 Kuker W, Herrlinger U, Gronewaller E, et al. J Neurology. 2002;249:1713–1716.
117 Ursea R, Heinemann MH, Silverman RH, et al. Ophthalmic ultrasonographic findings in primary central nervous system lymphoma with ocular involvement. Retina. 1997;17:118–123.
118 Murphy MA, Hart WM, Olk RJ. Bilateral diffuse uveal melanocytic proliferation simulating an arteriovenous fistula. J Neuroophthalmol. 1997;17:166–169.
119 Brihayeian Gerrtruyden M. Retinal lesions in Hodgkin’s disease. Arch Ophthalmol. 1956;56:94–99.
120 Primbs GB, Monsees WE, Irvine AR. Intraocular Hodgkin’s disease. Arch Ophthalmol. 1961;66:477–482.
121 Mosteller MW, Margo CE, Hesse RJ. Hodgkin’s disease and granulomatous uveitis. Ann Ophthalmol. 1985;17:787–790.
122 Kurz GH. Retinopathy of obscure (toxic?) origin in Hodgkin’s disease. Am J Ophthalmol. 1964;57:205–213.
123 Jabs DA, Schachat AP, Liss R, et al. Presumed varicella zoster retinitis in immunocompromised patients. Retina. 1987;7:9–13.
124 Diddie KR, Schanzlin DJ, Mausolf FA, et al. Retinitis caused by opportunistic virus infection in a patient with Hodgkin’s disease. Am J Ophthalmol. 1979;88:668–673.
125 Toy JL, Knowlden RP. Cytomegalovirus retinitis misdiagnosed as Hodgkin’s lymphoma deposits. BMJ. 1978;2:1398–1399.
126 Toussaint D, Vanderhaeghan JJ. Ocular toxoplasmosis, trigeminal herpes zoster and pulmonary tuberculosis in a patient with Hodgkin’s disease. Ophthalmologica. 1975;171:237–243.
127 Lissner GS, O’Grady R, Choromokos E. Endogenous intraocular Nocardia asteroides in Hodgkin’s disease. Am J Ophthalmol. 1978;86:388–394.
128 Kitzmann AS, Pulido JS, Moheney BG, et al. Intraocular use of rituximab. Eye. 2007;21:1524–1527.
129 Itty S, Olson JH, O’Connell DJ, et al. Treatment of primary intraocular lymphoma has involved systemic, intravitreal, or intrathecal chemotherapy and/or radiotherapy. Retina. 2009;29:415–416.
130 Hormigo A, DeAngelis LM. Primary ocular lymphoma – clinical features, diagnosis, and treatment. Clin Lymphoma. 2003;4:30–31.
131 Merchant A, Foster CS. Primary intraocular lymphoma. Int Ophthalmol Clin. 1997;37:101–115.
132 Keltner JL, Fritsch E, Lykiert RC, et al. Mycosis fungoides: intraocular and central nervous system involvement. Arch Ophthalmol. 1977;95:645–650.
133 Long JC. Mycosis fungoides with extracutaneous dissemination: a distinct clinico-pathologic entity. Cancer. 1974;34:1745–1755.
134 Rossi V. Le reticulo-endotheliosi in oftalmologia. Arch Oftalmol. 1946;50:3–18.
135 Gartner J. Mycosis fungoides mit Beteiligung der Aderhaut. Klin Monatsbl Augenheilkd. 1957;131:61–69.
136 Foerster HC. Mycosis fungoides with intraocular involvement. Trans Am Acad Ophthalmol. 1960;64:308–313.
137 Stenson S, Ramsay DL. Ocular findings in mycosis fungoides. Arch Ophthalmol. 1981;99:272–277.
138 Erny BC, Egbert PR, Peat IM, et al. Intraocular involvement with subretinal pigment epithelium infiltrates by mycosis fungoides. Br J Ophthalmol. 1991;75:698–701.
139 Leitch RJ, Rennie IG, Parsons MA. Ocular involvement in mycosis fungoides. Br J Ophthalmol. 1993;77:126–127.
140 Burkitt D. A sarcoma involving the jaws in African children. Br J Surg. 1958;46:218–223.
141 Karp LA, Zimmerman LE, Payne T. Intraocular involvement in Burkitt’s lymphoma. Arch Ophthalmol. 1971;85:295–298.
142 Feman SS, Niwayama G, Hepler R, et al. “Burkitt tumor” with intraocular involvement. Surv Ophthalmol. 1969;14:106–111.
143 Knapp AJ, Gartner S, Henkind P. Multiple myeloma and its ocular manifestations. Surv Ophthalmol. 1987;31:343–351.
144 Bronstein M. Ocular involvement in multiple myeloma. Arch Ophthalmol. 1955;55:188–192.
145 Shami MJ, Uy RN. Isolated cottonwool spots in a 67-year-old woman. Surv Ophthalmol. 1996;40:413–415.
146 Carr R, Henkind P. Retinal findings associated with serum hyperviscosity. Am J Ophthalmol. 1963;56:23–31.
147 Sanders JE, Podos SM, Rosenbaum LJ. Intraocular manifestations of multiple myeloma. Arch Ophthalmol. 1967;77:789–794.
148 Hayasaka S, Ugomori S, Kodama T, et al. Central retinal vein occlusion in two patients with immunoglobulin G multiple myeloma associated with blood hyperviscosity. Ann Ophthalmol. 1993;25:191–194.
149 Knabben H, Wolf S, Remky A, et al. Retinal hemodynamics in patients with hyperviscosity syndrome. Klin Monatsbl Augenheilkd. 1995;206:152–156.
150 Franklin RM, Kenyon KR, Green WR, et al. Epibulbar IgA plasmacytoma occurring in multiple myeloma. Arch Ophthalmol. 1982;100:451–456.
151 Lekhra OP, Sawhney IM, Gupta A, et al. Venous stasis retinopathy in Waldenstrom’s macroglobulinemia. J Assoc Physicians India. 1996;44:61–62.
152 Koustandrea C, Kostsolis A, Gerogalas I, et al. Peripheral capillary non-perfusion in asymptomatic Waldenstrom’s macroglobulinemia. BMC Ophthalmol. 2010;10:30.
153 Quhill F, Khan J, Rashid A. Bilateral serous macular detachments in Waldenström’s macroglobulinaemia. Postgrad Med J. 2009;85:382.
[/level-membership-for-opthalmology-category][not-level-membership-for-opthalmology-category]
Chapter 155 Leukemias and Lymphomas
Introduction
Leukemias and lymphomas are myeloproliferative disorders that may affect the eye. The ophthalmic symptoms and findings can be the initial manifestation of the systemic illness. Indeed, before the use of bone marrow biopsy, the ophthalmologist was often called upon to assist with the diagnosis of the leukemias.1
Estimates of the frequency of intraocular involvement with leukemia range as high as 90% of cases.1 Although melanoma is the most common primary intraocular tumor in adults and retinoblastoma is the most common primary intraocular tumor in children, when secondary or metastatic neoplastic intraocular disease is considered, intraocular manifestations of hematological malignancies are found to be overwhelmingly more common since the prevalence of myeloproliferative disorders is higher in the general population.
Liebreich first described leukemic retinopathy in the 1860s. Since that time, reports have documented that virtually all intraocular structures may be involved. Patients have been reported with leukemic infiltrates of the optic nerve, choroid, retina, iris, ciliary body, and anterior chamber. A child with a leukemic hypopyon is illustrated in Figure 155.1. Central serous chorioretinopathy overlying areas of choroidal infiltration has been reported, as has retinal vascular sheathing, subconjunctival hemorrhage, anterior chamber hemorrhage, intraretinal hemorrhage, and intravitreal hemorrhage.2 Retinal changes may be related to direct invasion of tissue by neoplastic cells, to manifestations of associated hematological abnormalities such as anemia, thrombocytopenia, or hyperviscosity states, to opportunistic infections, or to unrelated chance findings.
Fig. 155.1 Leukemic iris infiltration in a child with acute lymphocytic leukemia. Note hypopyon inferiorly.
Intraocular infiltrates appear to correlate with associated central nervous system (CNS) involvement and decreased survival. The intraocular manifestations of leukemia are protean, and clinically, it usually is not possible to differentiate the various leukemias and lymphomas on the sole basis of their ophthalmoscopic findings.3
Systemic classification of leukemia and lymphoma
At presentation, the acute leukemias most often have systemic manifestations of anemia, hemorrhage, infection or signs and symptoms related to infiltration of organs. Acute lymphocytic leukemia is the predominant leukemia type in children, and more than 50% of these patients can be cured. In adults, acute myelogenous leukemia is the predominant myeloproliferative disorder and survival rates are lower in adults than children.1
The chronic leukemias often first appear in an indolent manner. Symptoms may be vague. These diseases generally are found in older individuals. Although associated infections or other complications are often treated aggressively, many patients receive no treatment for their underlying disease because treatment has not definitely been shown to prolong survival. Some chronic leukemias degenerate into a “blast” (acute) phase, in which case the signs and symptoms resemble those of the acute leukemias.1
Leukemia
Prevalence and incidence
Scant data are available from prospective series of patients concerning ocular findings at the time of diagnosis.4,5 Even less information can be found about the frequency with which various intraocular manifestations are seen during the course of the illness. Most clinical series contain cross-sections of patients seen both early and late during the course of the illness. Autopsy series, which presumably show the highest prevalence rates, often overstate the frequency with which clinical disease is detected.
Four autopsy series have shown variable prevalence data. Allen and Straatsma3 found that 38 of 76 patients (50%) had ocular involvement by neoplastic disease or pathologic changes that could be directly ascribed to the neoplastic disease. Nelson et al.6 found that 33 of 117 patients (28%) with various types of leukemia had ocular metastases at the time of death. Leonardy et al.7 found leukemic infiltrates in the eyes of 42 of 135 patients (31.1%), with the choroid being the most frequently involved site. Data from Kincaid and Green1 are summarized in Table 155.1, which shows that 75% of patients with chronic leukemias, 82% of patients with acute leukemias, and 80% of affected patients in this study had intraocular involvement at the time of death.
Clinical series show highly variable prevalence rates. A small prospective series was reported by Karesh et al.,4 who examined the fundus findings in 56 newly diagnosed, untreated patients with acute myelogenous leukemia. They found retinopathy at the time of initial examination in 28 of 56 patients (50%) and no cases of leukemic infiltration. Schachat et al.5 reported on the largest series of patients with newly diagnosed leukemia examined within a few days of diagnosis. Among the 120 patients examined (65 with acute myelogenous leukemia, 51 with acute lymphocytic leukemia, and four with other leukemias), 62% had ocular abnormalities due to their underlying myeloproliferative disorder.
Clinical manifestations
Reddy et al. reported on 82 children examined after diagnosis of acute leukemia and before initiation of chemotherapy. Although only 4% of patients complained of visual symptoms, abnormal retinal findings were noted in 16% of all patients examined.8 Reddy and colleagues also reported on the ocular findings in 288 newly diagnosed cases of leukemia in adults and children. Clinical symptoms were present in 10%, but ocular findings were noted in 35.4%. Ocular findings were more common in adults (49%) than in children (16%), and were more frequent in myeloid leukemia (41%) compared with lymphoid leukemia (29.2%).9
Russo et al. evaluated 180 pediatric patients with acute leukemia and found ocular manifestations in 66% of children with acute myeloid leukemia and 11.5% of children with acute lymphocytic leukemia.10 Data from multiple reports suggest the need for routine ocular examination of all patients with newly diagnosed acute leukemia regardless of whether they are symptomatic or not. Other investigators have also noted that chronic lymphocytic leukemia appears to have a very low prevalence of ocular involvement and routine screening of these patients at the time of diagnosis does not appear to be necessary.11
In our series of 120 cases examined at the time of diagnosis, four patients (3%) had leukemic infiltrates. In addition, intraretinal hemorrhages were present in 29 cases (24%), white-centered retinal hemorrhages in 13 cases (11%), vitreous hemorrhage in three cases (2%), and cotton-wool spots in 19 cases (16%). Miscellaneous findings, presumably unrelated to leukemia, were present in 20% of cases.5 The low prevalence of leukemic infiltrates in our series differs from those reported in autopsy series. One potential reason for the lower prevalence is the ability to detect leukemic choroidal infiltrates, which are a frequent finding in autopsy series but are difficult to diagnose clinically.
Leukemic infiltrates
Retinal or preretinal infiltrates
Leukemic infiltrates have been described by several investigators. Kuwabara and Aiello12 reported on a patient with chronic myelogenous leukemia with large gray-white nodules of varying sizes in the retina.12 They suggested that this finding was an ominous prognostic sign and, in general, associated with high blood counts, fulminant disease, and early demise.13 Merle and colleagues have described a subretinal infiltrate with venous vasculitis in a patient with adult T-cell leukemia.14 As the ocular lesions progressed, the general health of the patient declined despite treatment with chemotherapy. Another patient with leukemic infiltrates is illustrated in Figure 155.2. This type of pathologic change in the fundus was seen in 2–3% of patients in our prospective series.5 Optic nerve infiltration may be the initial ocular manifestation of acute leukemia and can result in severe vision loss if not treated.15
Gray-white streaks along vessels may be caused by local perivascular leukemic infiltrates. A case of diffuse unilateral retinal angiopathy that resembled frosted-branch angiitis that responded to local radiation and intrathecal chemotherapy was reported by Kim et al.16 This case represented the sole manifestation of relapsing acute lymphoblastic leukemia in an 18-year-old man and also involved optic nerve infiltration in the same eye.
Choroidal infiltrates
Leukemia may also infiltrate the choroid; however, clinical signs of choroidal involvement are often subtle unless overlying retinal or retinal pigment epithelial (RPE) changes bring them to attention. Serous retinal detachment overlying choroidal infiltrates17 or overlying frank choroidal masses18 is an important clue. Histopathologic confirmation of choroidal infiltration has been reported in a patient with adult T-cell leukemia.19
Kincaid et al.20 reported on a 71-year-old woman with an exudative retinal detachment, shifting subretinal fluid, and areas of diffuse pinpoint fluorescein hyperfluorescence with dye leakage into the subretinal space. The patient had a poorly characterized and unusual leukemia. Histopathologic study showed that the choroid was moderately distended by a diffuse cellular infiltrate with an overlying serous retinal detachment. The RPE showed areas of depigmentation and proliferation. There were a few small areas of associated pigment epithelial detachment.20 Gass21 described a patient with myelomonocytic leukemia who had a discrete choroidal mass with an overlying serous retinal detachment. Pinpoint hyperfluorescence was seen on the angiogram in areas overlying the mass (Fig. 155.3).
Burns et al.22 reported on a 37-year-old patient with acute lymphocytic leukemia who appeared to have “bilateral macular edema.” The patient was treated with ocular radiation on the assumption that underlying leukemic infiltration of the choroid was present, and this was confirmed at autopsy. Serous retinal detachment overlying areas of choroidal infiltration has been reported in patients with chronic lymphocytic leukemia, acute lymphocytic leukemia, chronic myelogenous leukemia, and acute myelogenous leukemia.1,23 Serous RPE detachment has also been reported in a patient with acute lymphoblastic leukemia.24 The serous retinal and RPE detachments can be the presenting manifestation of the leukemia.25 Eventually, as the fluid and detachment resolve, coarse clumping of the RPE is seen.17,24
Prominent pigment epithelial changes also may be seen after resolution of what may be retinal infiltrates. Jakobiec and Behrens26 reported on a 3-year-old patient with acute leukemia who was found to have preretinal hemorrhages, some with white centers. A multitude of jet-black spots were seen, most prominently in the posterior pole. Soft white patches were described in the retinal periphery. The authors believed that these patches were the precursor lesions of the black spots. They postulated that the spots represented proliferation of pigment epithelial cells or small pigment epithelial detachments.26 Clayman et al.17 reported on a child with acute lymphocytic leukemia who had massive pigmentary changes simulating leopard spots, most marked in the posterior pole (Fig. 155.4). At autopsy, the retina and the choroid were infiltrated with atypical immature lymphocytes. Areas of RPE hyperplasia were present, including heaped-up masses of pigment epithelium surrounding leukemic cells.
Vitreous infiltrates
Vitreous opacities may be manifestations of an intraocular malignancy. Moribund patients may show massive collections of tumor cells in the vitreous, but most patients with intravitreal hemorrhage have neoplastic cells in the vitreous only because their peripheral blood contains tumor cells (Fig. 155.5). The cells do not appear to be preferentially replicating in the vitreous cavity. Swartz and Schumann2 described a patient with acute lymphocytic leukemia who had a dense vitreous cellular infiltration. Diagnostic vitreous aspiration confirmed the neoplastic nature of the process. Examination of the cerebrospinal fluid was also positive for blast cells. At the time, the peripheral blood was negative for tumor. The patient was treated with systemic and intrathecal chemotherapy and 1200 cGy of cranial radiation therapy. The vitreous cleared after the treatment.2
Belmont et al.27 reported on a 72-year-old patient with chronic unilateral uveitis. Leukemic cells were found in the iris and throughout the vitreous at the time of a pars plana vitrectomy performed to rule out reticulum cell sarcoma. Isolated ocular blast crisis with leukemic hypopyon has been reported to occur in chronic myelogenous leukemia.28 Vitreous cells and leukemic retinopathy have been reported in a single case of hairy cell leukemia (which constitutes less than 2% of adult leukemias).29 A masquerade syndrome with primarily ocular findings of panuveitis in a patient with T-cell prolymphocytic leukemia was reported by Dhar-Munshi et al.30 Additional cases of vitreous involvement in leukemia have been cited by Kincaid and Green1 as well as by Rothova et al.31 Leukemic cells have been found in the vitreous of patients with neovascularization of the disc. Delaney and Kinsella32 reported on a patient with chronic myelogenous leukemia and disc neovascularization who had good peripheral perfusion, although the macula did show areas of nonperfusion. de Juan et al.33 described a 3-year-old patient with acute lymphocytic leukemia who had disc neovascularization, vitreous hemorrhage, and vitreous infiltration. This case was unusual because neither the cell count nor the platelet count were remarkably elevated.
Hattenhauer and Pach34 reported a single case of ocular B-cell lymphoma occurring in a patient with chronic lymphocytic leukemia. The occurrence of diffuse large cell lymphoma in patients with chronic lymphocytic leukemia is known as Richter syndrome. Richter syndrome is estimated to occur in 3–10% of patients with chronic lymphocytic leukemia. It portends an abrupt deterioration in health and carries a poor prognosis for survival.
Possible leukemic infiltrates
Duane et al.35 reviewed the causes of white-centered retinal hemorrhages (Fig. 155.6) and also described possible pathophysiologic mechanisms. The authors suggested that these lesions should be classified as suspicious for direct intraocular manifestations of leukemia, since aggregates of leukocytes have been reported in the center of white-centered hemorrhages.36,37 However, it should be noted that white-centered retinal hemorrhages can be composed of fibrin-platelet aggregates and are not necessarily a definite sign of neoplasia.
Manifestations of anemia and thrombocytopenia
Leukemic retinopathy is the term most often used to denote the fundus manifestations of anemia, thrombocytopenia, and increased blood viscosity seen in patients with leukemia. In general, the term does not necessarily refer to frank leukemic proliferation. The changes of “leukemic retinopathy” may be more commonly seen with the acute leukemias, but the frequency with which they occur has not been adequately studied to be certain. Although perivascular sheathing may be due to actual perivascular infiltrates, tortuous dilation of the retinal veins probably is not. The veins and arteries may assume a yellowish tinge because of both anemia and leukocytosis.1 Retinal hemorrhages may occur, often at the posterior pole, and may be subretinal, deep retinal, superficial retinal, or preretinal, with potential breakthrough bleeding into the vitreous cavity. Hemorrhages may have a blot or blotch shape, flame shape, or they may have white centers (Figs 155.7, 155.8).35
Cotton-wool spots may be the presenting abnormality that precipitates the systemic evaluation leading to the diagnosis of leukemia.38 Cotton-wool spots may be caused by local factors, such as an abnormally large cell or a cluster of cells occluding retinal arterioles, and may not be related to the overall peripheral blood composition. In general, hematologic parameters are not associated with the presence of cotton-wool spots.39 Cotton-wool spots and hemorrhages can spontaneously resolve in patients with chronic leukemic disease.2
Manifestations of hyperviscosity
Whole-blood hyperviscosity may lead to veno-occlusive disease, microaneurysm formation, retinal hemorrhages, and retinal neovascularization. The most common manifestation is probably a mild, or “hyperpermeable,” central retinal vein occlusion (Fig. 155.9). A systemic hyperviscosity state should be suspected in patients with simultaneous, bilateral retinal vein occlusion. Also, the very high white cell count may lead to a hyperviscosity state that results in poor absorption of cerebrospinal fluid, creating a clinical picture similar to that of benign intracranial hypertension with bilateral disc swelling.40
Peripheral retinal microaneurysms in leukemic patients were originally described by Duke et al.41 and subsequently by Jampol et al.42 Duke et al.41 found that 50% of patients dying with chronic leukemia had peripheral microaneurysms. None were seen in patients with the acute leukemias. Seven of nine patients with chronic myelogenous leukemia and three of 10 patients with chronic lymphocytic leukemia had this finding, but it was not present in any of the 21 patients with acute leukemia.41 Kincaid and Green1 saw only one case in their large series. They noted that trypsin digest of the retina was essential or else the change would be overlooked on histopathologic examination.
Peripheral retinal neovascularization has been reported in patients with chronic myelogenous leukemia in association with peripheral capillary nonperfusion. Most cases have associated extreme leukocytosis or thrombocytosis.43–46 Presumably, the hyperviscosity state leads to peripheral nonperfusion and subsequent development of retinal neovascularization, as in patients with proliferative sickle retinopathy.
Morse and McCready46 reported on a 32-year-old patient with chronic myelogenous leukemia and retinal neovascularization. The peripheral white blood cell count was 340 500 and subsequently rose to 524 000/mm3. The fasting blood sugar value was normal, as was the hemoglobin electrophoresis study. No paraproteins were present. A fluorescein study highlighted multiple sea fans, and obliteration of the terminal arterioles was apparent. Frank and Ryan43 described a 30-year-old patient with a subhyaloid and vitreous hemorrhage who had a white blood cell count of 250 000/mm3 related to chronic myelogenous leukemia. Numerous sea fans were apparent and a glucose tolerance test and hemoglobin and serum protein electrophoresis studies were negative.46 Like Morse and McCready,46 Frank and Ryan43 believed that the pathogenic mechanism was related to increased blood viscosity, as in patients with complications of Waldenström macroglobulinemia or polycythemia. Kincaid and Green,1 however, did not see any cases of peripheral retinal neovascularization in their series.
Levielle and Morse44 described a patient with chronic myelogenous leukemia who had a relatively low (33 700/mm3) white blood cell count. In general, the blood viscosity begins to increase remarkably only with white blood cell counts of >50 000.47 In Levielle’s case report, the patient had an elevated platelet count of 988 000/mm3, and the peripheral neovascularization was attributed to this elevation. However, the authors did not emphasize that their patient also had an 11-year history of diabetes mellitus; therefore diabetic retinopathy also may have contributed to retinal capillary non-perfusion and formation of peripheral neovascularization.47 Melberg et al.48 described the effect of acute lymphocytic leukemia on the progression of mild diabetic retinopathy in a 16-year-old girl. The patient developed bilateral rubeosis, and after aggressive laser and vitrectomy, her vision declined to 20/200 bilaterally as a result of macular ischemia. The accelerated course of diabetic retinopathy correlated most closely with the anemia accompanying her leukemia and its treatment.
Wiznia et al.49 reported on concurrent optic disc and retinal neovascularization in an 18-year-old woman with acute lymphocytic leukemia who underwent therapy. They described progression of the neovascularization caused by the additive effects of radiation retinopathy and chemotherapy, resulting in macular traction detachment. The authors postulated that toxic effects of chemotherapy when combined with radiation therapy could lead to a more severe form of ischemic retinal vasculopathy than would be encountered with acute lymphocytic leukemia alone.49
The mechanism of the retinal hemorrhage seen in patients with leukemia is not yet known. The hemorrhages may be caused by an associated anemia or thrombocytopenia. Although commonly associated with severe leukocytosis, white-centered hemorrhages may be present regardless of the degree of leukocytosis.50
Some authors believe that the platelet count is more predictive than the hematocrit for the presence or absence of retinal hemorrhages.51 Kincaid and Green1
[/not-level-membership-for-opthalmology-category]
