Chapter 27 Is the Manifestation of the Restless Legs Syndrome Related to a Pain Mechanism?
Pain is a sensory and emotional experience that is distinctly subjective and personalized according to each individual patient’s beliefs, past pain experience, and genetic background.1,2 Physiologically, pain sensations are triggered by a complex cascade of events referred to as nociception that arises as a consequence of tissue injury or immune reaction. This sensory information can be carried from the spinal cord to the brain via six major ascending pathways: the spinothalamic, spinoreticular, spinomesencephalic, spinoparabrachial, spinohypothalamic and spinocervical tracts.3 Briefly, nociception comprises three steps: (1) activation of sensory nerve endings of Aδ and C first-order neurons; (2) activation of ascending projection neurons located in the dorsal horn of the spinal cord or trigeminal nucleus, that project to higher brain areas (several nuclei in the thalamus, reticular formation of the pons and periaqueductal gray matter of the midbrain); and (3) activation of third-order neurons that project to the cortical mantle (e.g., primary and secondary somatosensory, insular, anterior cingulate cortices).4
A myriad of diverse chemical entities are known to activate (e.g., K+, serotonin, histamine, bradykinin) or sensitize (e.g., prostaglandins, substance P, leukotrienes) nociceptors.5,6 These events are important players in initial pain transmission and maintenance. From a physiological viewpoint, perceived pain results from the balance of the activity in nociceptive and non-nociceptive afferents. This concept is referred to as the gate control theory. This hypothesis is based on the involvement of inhibitory interneurons able to modulate projection neurons, by integrating the respective activity of the myelinated non-nociceptive afferent and nonmyelinated nociceptors.7
Pain perception can also be modulated via descending pathways originating from the dopaminergic periaqueductal gray of the midbrain, noradrenergic locus ceruleus, and serotoninergic neurons of the nucleus raphe magnus.8 These descending pathways can inhibit projection neurons directly or indirectly by activation of enkephaline-containing interneurons in the superficial layers of the dorsal horn. A more diffuse way of modulating pain has also been identified and referred to as the DNIC (diffuse noxious inhibitory controls).9–12
Restless legs syndrome (RLS) is a sleep-related sensorimotor disorder that is often associated with pain complaints and severe insomnia.13,14 Recently, some evidence proposed a dysfunction of the endogenous opioid system in RLS sufferers as a possible primary cause of RLS.15,16 On the other hand, the abnormalities in somatosensory processing observed in RLS patients could be mediated by other consequences of sleep. Both the sensory discomforts of RLS and the associated periodic movements in sleep can lead to frequent arousals or awakenings, with a consequence of major sleep deprivation. Sensory hyperexcitability in patients as measured by hyperalgesia16 could be a consequence of this sleep loss.17 In this chapter, we review some of the emerging knowledge on sleep and pain interaction to better understand the mechanisms by which pain becomes prevalent in RLS, and to evaluate the possible involvement of the opioid system as a causal factor in the pathogenesis of RLS.
Restless Legs Syndrome and Pain
RLS is characterized by an often irresistible urge to move usually accompanied by or caused by unpleasant, sometimes even painful, sensations in the lower limbs. These symptoms are worse at night, or during periods of inactivity, and can be partially or totally relieved by movement.18 RLS patients often exhibit stereotyped and recurring movements of the lower limbs and these periodic limb movements (PLMs) occur during both sleep and wakefulness.19 Recent clinical evidence suggests that pain is a relatively frequent complaint of patients suffering from RLS. In an international study performed in a primary care setting, 21.4% of 23,052 patients reported pain as a concomitant symptom of their RLS.13 In a genetic study encompassing 300 patients, pain was reported among RLS-related symptoms by 61% of cases with a positive familial history of RLS and by 85% of patients with no family history of RLS.20 In another study of over 218 RLS patients, painful disorders such as rheumatoid arthritis, disorders of the back, and arthropathy were concurrently reported by 2.6% to 49.6% of the female subjects and by 1.0% to 35.9% of male subjects.18 To our knowledge, none of these reports estimated the pain intensity quantitatively (e.g., using a numerical scale of 0 to 100 mm, a visual analogue scale [VAS]) nor the time course of worst pain, from onset to its peak. To diagnose RLS patients, sleep clinicians are using the terms dysesthesia (i.e., altered perception of sensory inputs) and paresthesia (i.e., partial lost of sensation, numbness or tingling sensations), which are typically used for neuropathic pain assessments.21
Pain: Definition and Sleep Interaction
Pain manifestations can be described according to two major components. The sensory-discriminative aspect of pain is a function of the intensity of the perceived stimuli, and the emotional dimension is related to the unpleasantness of the painful experience. These qualities can be investigated separately in a clinical as well as an experimental setting with the use of simple objective and subjective psychophysical tools.9 Pain can be acute (days, weeks) or chronic (>3 months) or can be recurrent with variable durations of pain-free periods. The International Association for the Study of Pain (IASP) developed an extensive classification similar to the International Classification of Sleep Disorders.9 However, for practical reasons in clinics, the day-to-day classification of pain is categorized into three groups: musculoskeletal pain, neuropathic pain, and pain with psychological components.22 The prevalence of chronic pain in the general population is between 11% and 29%, depending on the population surveyed and types of questions used.23–25
According to a survey from the National Sleep Foundation in 2000, “20% of American adults reported that pain or physical discomfort disrupted their sleep” a few nights a week.26 Poor sleep quality, frequently defined as an unrefreshing sleep or fatigue on awakening, is reported by two thirds of chronic pain patients, and pain is most often the primary trigger of poor sleep complaints.27–35
Because sleep is a state with altered vigilance and with a partial disconnection from the external milieu, pain perception and processing during sleep are probably better described by the term nociception, because no voluntary reaction is expected if no sleep arousal toward a behavioral wake state is present.10,35,36
Pain Assessment Tools in the Clinic and Laboratory
Questionnaires
In the clinical setting, patients use words that describe both the intensity and emotional aspects of the noxious stimulus (e.g., Melzack scale).37 When a patient reports pain with words such as dull, diffuse, or cramp-like, this typically refers to muscle pain. In opposition, when pain is reported with words such as sharp, pins and needles, electric shock, or burning, clinicians are prone to classify the complaints as neuropathic pain. However, in the clinic, it is the use of a 0 (no pain)-to-10 (most intense pain imaginable) numerical scale that is most frequently used by clinicians to estimate pain intensity. This scale is easily understood by patients and is an effective means of collecting data rapidly by clinicians, but it is not very accurate in estimating pain relief, since pretreatment pain intensity can be overestimated or underestimated by patients.38 Moreover, with patients who are too young or too old, under medication (e.g., psychoactive), or with cognitive or mental dysfunction, scales with graded colors or faces (from smiling to unhappy) are very useful. In the clinical research environment, the gold standard is the use of the VAS with anchor words at both extremities of the scale: from no pain to the most intense pain imaginable. Sophisticated questionnaires also assess the impact of pain in relation to quality of life, mood alterations, and fatigue (see the McGill Pain Questionnaire [MPQ], the Multidimensional Pain Inventory [MPI], and Neuropathic Pain Symptom Inventory [NPSI] for examples).22,39,40 For neuropathic pain, 10 descriptors were used in a recent study to evaluate the intensity of neuropathic pain and the efficacy of treatments. These descriptors included burning, pressure, squeezing, electrical shocks, stabbing, pin and needles, tingling, and pain induced by brushing, pressure, or cold.41 The descriptors are presented with numerical 0-to-100 scales. Such scales need further validation and replication but add a quantitative dimension rarely used in RLS symptom assessments.
Psychophysical Quantitative Testing
Several reviews describe the psychophysical methods to assess sensory changes in humans, for various sensory disorders, and at different body sites.10,42–46 These methods are known under the name of Quantitative Sensory Testing (QST). In summary, the pain threshold is estimated experimentally from stimulations that cause pain in 50% of trials, and the pain tolerance is estimated as the maximal pain a subject can tolerate (Table 27-1). The measured variables are pain intensity, unpleasantness scores, tolerance measures, amplitude of stimulation (e.g., current, temperature), and/or latency of response. Pain can be evoked experimentally using stimuli of various nature, to access the reactivity of different sensory pathways. Thermal stimulations (cold or heat) are used to assess either Aδ (heat) or C (cold) sensory fibers, mechanical pressure tests (pressure, von Frey hair, two points, brush, vibration) are used to assess tactile reactivity of all A and C fibers, and chemical stimulation (hypertonic saline, glutamate, capsaicin) usually challenge the roles of free nerve endings and C fibers. Superficial thermal cold (0° to 4° C) or heat (43° to 50° C) pain test is often administered by a water bath or a Peltier bridge. Laser stimulation is another way of causing superficial heat pain. The duration of thermal stimulation ranges from 60 milliseconds (laser) up to 60 seconds (Peltier bridge) and is usually harmless. Deep tissue pain is estimated with a mechanical pain device or graded finger palpation displaced slowly; data are expressed in Newton or in kg/cm2. Touch discrimination and two-point discrimination using von Frey hairs, grade scale, or pin-prick tests are used to assess the presence of allodynia. Subcutaneous pain perception and muscle pain can be mimicked by injection of algesic substances such as capsaicin, glutamate, or hypertonic saline (5%). Electrical stimulations that trigger motor reflexes or brain-evoked potentials have also been used during sleep; however, these are not considered as being representative of clinical pain in comparison with other means of stimulation.36,47–52 Even though the data obtained with these assessment techniques are quite reproducible, the function of sensory fibers is modified in conditions like sensitization and allydonia, where activation of a non-nociceptive touch fiber may be interpreted as a painful stimulus.
| Algesia | Any pain experience following a stimulus |
| Allodynia | Pain due to an innocuous stimulus that does not normally provoke pain (e.g., skin touch after a sunburn) |
| Causalgia | Pain after a trauma to a nerve (could be associated with vasomotor dysfunction) |
| Habituation | A decrease or loss of response in nerve receptors or nerve cells following repetitive stimulations |
| Hyperalgesia | Increased pain response to a nociceptive stimulus |
| Hypoalgesia | Diminished pain response to a nociceptive stimulus |
| Hypoesthesia | Decreased sensitivity to stimulation that feels similar to the effect of local anesthesia |
| Neuroma | The mass of a peripheral neurons formed by a healing scar at the level of a damaged nerve preventing regeneration. It can cause hyperexcitability of neurons or spontaneous discharge also termed ectopic discharge. |
| Neuropathic pain | The aberrant pain reaction induced by an injury to a sensory nerve or neuron. The aberrant reaction may be evoked by thermal, mechanical, or chemical stimuli or may be secondary to a disease (e.g., diabetes, post-herpetic neuralgia), or may also be central. |
| Nociception | The reception, transmission, and perception of noxious sensory input |
| Pain | An unpleasant sensory, emotional, and motivational experience associated with actual or potential tissue damage |
| Pain threshold | The lowest level of stimulation perceived as painful by a subject (in 50% of trials) |
| Pain tolerance | The highest level of pain that a subject is able to tolerate |
| Paresthesia/dysesthesia | An abnormal sensation that is termed dysesthesia when it becomes unpleasant |
| Sensitization | Nerve receptors or nerve cells that, over time, become activated or hyperexcitable by stimulation of a lower magnitude. Can be peripheral, central, or both. |
| Sprouting | The excessive regeneration of nerve endings over surrounding tissue following nerve damage |
Adapted from Lavigne GJ, McMillan D, Zucconi M. Pain and sleep. In: Kryger MH, Roth T, Dement WC (eds). Principles and Practice of Sleep Medicine. Philadelphia, WB Saunders, 2005, pp 1246-1255.
Assessment of Sensory Dysfunction in Restless Legs Syndrome During Wakefulness
The estimation of the leg discomfort of RLS patients, as assessed on the VAS in the evening, discriminated RLS patients from age-matched asymptomatic subjects.53 Over a period of 60 minutes of immobilization, the leg discomfort rose up to 50 mm over the 100-mm scale, and this was highly correlated with the number of periodic leg movements (PLMs). An early study investigated peripheral neuropathy in RLS patients and found significant changes in electrical, psychophysiological, and/or morphological characteristics of axonal neuropathy. These results were confirmed for secondary RLS in a recent study, where sensory deficits seem at least in part caused by small fiber neuropathy.54 In opposition, idiopathic RLS seems to depend more on central somatosensory processing impairments.55 In addition, pin-prick ratings in 11 patients with RLS were significantly elevated, compared with 11 age- and gender-matched healthy control subjects, whereas pain to light touch (allodynia) showed no difference.16 Furthermore, hyperalgesia in RLS patients can be reduced by long-term dopamineric treatment.16 In this light, dysfunction of supraspinal pain modulatory pathways involving the basal ganglia and/or descending dopaminergic pathways might be involved in the pathophysiology of RLS.
Although the pin-prick test is a validated method to assess tactile sensory dysfunction, it is not frequently used in the clinical assessment of pain.56,57 Use of thermal (heat or cold), touch (pin-prick [so far, the only one tested]), deep tissue chemical, or mechanical (hypertonic saline or pressure, respectively) tests may be more comprehensive approaches to further discriminate sensory symptoms of RLS subjects and, eventually, to predict treatment efficacy (sensitivity changes).
Assessment of Sensory Dysfunction in Restless Legs Syndrome During Sleep
To our knowledge, only a few studies have assessed RLS sensory perception in patients during sleep. One sleep study tested motor excitability with a flexion reflex and revealed an increase in spinal cord excitability in RLS patients.58 A similar reflex-testing paradigm was used to assess whether changes in pain perception occurred during sleep in normal subjects.49 The threshold of the RIII polysynaptic nociceptive flexion reflex is higher in all stages of sleep compared with wakefulness, and its latency is prolonged during stage 4 and REM (rapid eye movement) sleep, suggesting that sleep decreased pain perception.49 However, electrical stimulation is considered an aversive method that may trigger a hypervigilance reaction and does not represent the usual sensory complaints of pain patients or of RLS patients. Hence, the question of “ecological validity” is raised. Moreover, during sleep, the duration of stimulation seems critical to trigger a clinically relevant response. Brief pain stimulations (6 to 12 seconds) trigger microarousal, whereas longer ones (90 to 120 seconds), such as infusion of hypertonic saline, seem more representative of natural painful experiences and often induce awakenings—in 40% of hypertonic infusions in stage 2, 26.7% in slow-wave sleep (SWS) and 45.8% in REM sleep.36,52 In other words, the reactivation of the reticular system needed to interpret inputs as painful in a sleeping brain needs sufficient time to assess a threat to the body’s integrity. It seems apparent that during sleep, the presence of pain can trigger either a brief and unconscious microarousal response accompanied by a sleep stage shift, or an awakening with a rise in consciousness/vigilance, that may interfere with sleep continuity.
Fatigue
The assessment of pain in patients with sleep complaints needs to be controlled for other confounding influences. In clinics, it is important to assess the type of fatigue that is reported. A fatigue complaint due to sleep fragmentation in patients with RLS or sleep apnea is probably very different from the one experienced in a context of low motivation associated with lack of outside stimulations. Several questionnaires are available to assess fatigue,59–62 but again their validity in investigating the sleep and pain interaction in RLS patients needs to be evaluated.
Moreover, because some patients with chronic pain tend to report more impairment in memory and attention,63–65 RLS patients might present cognitive alterations due to fatigue and these changes need to be further estimated. To our knowledge, the influence of RLS on cognitive functions has not yet been systematically studied; however, one study has shown that the sleep deprivation of RLS can cause frontal lobe dysfunction.66
Circadian Pattern for Pain
In normal volunteers, some studies shows circadian variation in objective and subjective measures of sensitivity to experimental acute pain,67,68 whereas others fails to reveal any differences.69,70 A more recent study by Roehrs and colleagues17 showed a time-of-test difference in finger withdrawal latency, being shorter at 2:30 P.M. than at 10:30 A.M. These divergent findings may be due to methodological differences, as pain was produced by different techniques and many parameters were used to quantify pain intensity. In addition, the presence of circadian fluctuation in pain sensitivity could be linked to altered vigilance and/or motivation, and not to changes in pain processing per se. On the other hand, a circadian pattern of pain is often found in chronic pain patients suffering from different diseases.71 Symptoms can be low in the morning or in the evening depending on the pain syndrome. Musculoskeletal pain complaints show two main patterns: arthritic pain that is worst at waking time, with subsequent movement alleviating discomfort, and fibromyalgia and various myofascial pains that gradually increase in intensity from morning to evening.28,70,72–74
Interestingly, RLS symptoms, leg or limb discomfort, and motor restlessness increase in intensity from morning to evening and are paroxysmal around midnight.53,75 The study from Michaud53 suggested that the downregulation of central dopamine secretion by melatonin may play a role in the nocturnal manifestation of RLS symptoms. Furthermore, dysfunction or atrophy of dopaminergic A11 neurons could be involved in the generation of circadian rhythms of symptoms, because these cells are in close proximity to the hypothalamic circadian pacemaker.
Sleep and Pain Interaction
In humans, acute pain perception during sleep depends mainly on the intensity, the duration and the nature of the sensory stimulation, as well as the depth of sleep. Indeed, although short (60 milliseconds) laser stimulation is ineffective in producing an evoked cortical response in stage 2 sleep,47 a mid-duration (6 to 12 seconds) thermal pain stimulation triggers more microarousals during stage 2 sleep than during SWS or REM sleep.36 Furthermore, infusion of hypertonic saline (5%) that mimics a muscle cramp for more than 70 seconds causes clear awakening responses with possible recall the following morning, similarly across all sleep stages.52 These experimental models underlie the involvement of pain in poor sleep quality, with respect to fragmentation of sleep continuity, lower sleep efficacy, and numerous sleep stage shifts (stages 3 and 4 toward stages 1 and 2). In addition, certain chronic pain patients present abnormal sleep architecture, intrusions of arousal in clusters, or cardiac sympathetic overactivation in sleep.35
RLS as an undeniable influence on the sleep quality of patients, sometimes leading to severe insomnia.14,76 Because sleep loss has consequences on many physiological processes and cognitive functions, the morbidity of RLS, including altered pain perception, could be a result of modified sleep architecture, sleep fragmentation, or sleep stage shifts. Indeed, studies of total or specific sleep stage deprivation underlie the effect of sleep on subsequent noxious stimuli processing. REM sleep deprivation in rats has been shown to increase the behavioral responses to painfull stimuli.77 In the human, Roehrs and colleagues17 observed a linear decrease in finger withdrawal latency after radiant heat stimulus, as a function of either total or REM sleep deprivation. Early studies showed that SWS deprivation influences the magnitude of pain on the following day.78 On the other hand, delta wave sleep interruption in healthy subjects has been shown, in another study, to cause no significant lowering of pain thresholds compared with a control group.79 Interestingly, a recent publication showed that healthy female subjects with the shortest REM sleep latency and longest REM duration reported highest suprathreshold pain ratings, suggesting that a REM-related mechanism may be associated to hyperalgesia.80
Patients reporting chronic pain seem to have been free of major sleep complaints before the initial occurrence of pain, suggesting a linear relation in which pain preceded poor sleep. Indeed, with acute pain, the poor sleep complaints do not usually continue after pain clearance.35 This linear relation was noted in approximately two thirds of patients with various musculoskeletal types of pain.29,30,32 However, more evidence now supports the view that when pain becomes persistent, patients may report that a day with intense pain is followed by a night of poor sleep quality, and that a night of poor sleep is followed by a day with exacerbated pain. This circular interaction was first noted in fibromyalgia patients, but similar findings were also observed in post-trauma patients with severe skin burns.81–83 The existence of such a sequence for pain and dysesthesia symptoms in RLS patients is unknown.
Another popular concept that persists in sleep medicine is the importance of the so-called alpha EEG intrusion. In the 1970s, these electrographic alterations were found in several sleep studies of fibromyalgia patients; however, such findings have been questioned by polysomnographic studies performed under controlled conditions with automated analysis systems to prevent any scoring bias.84–89 The observation that sleep patients with pain have arousals in clusters or in a sequence of phasic events, or cyclic alternating pattern (CAP), offers an alternative interpretation of the previous findings—that is, the presence of cyclic are brain and autonomic-cardiac reactivity may be concomitantly responsible of the sleep lightening in pain patients.33,90–92 Moreover, pain and fibromyalgia patients seem to have a deregulated balance of the parasympathetic/sympathetic cardiac activity. In fact, fibromyalgia patients present a high sympathetic activity over the total sleep period, and recent analysis revealed that the usual reactivation of the sympathetic cardiac system in REM sleep is blunted in female patients.93,94
Allodynia and Hyperalgesia
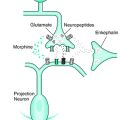
Allodynia is a sensory phenomenon in which a normally non-noxious stimulus is perceived as painful, and hyperalgesia refers to an increase response to a noxious stimuli. A multitude of changes in the peripheral nervous system, spinal cord, brainstem, and brain may contribute to abnormal pain processing. At the periphery, increased excitability of sensory neurons can be triggered by an upregulation of voltage-gated sodium channels, a down regulation of potassium channels, and possibly of a reduction in the threshold of TRP (transient receptor potential) transducer heat sensitive channels.95 Subsequently, augmented inflow may generate central sensitization characterized by activity-dependent changes in excitability of dorsal horn neurons and eventually a more persistent transcription-dependent sensitization.96 Another hypothesis used to explain touch-evoked pain is based on the notion that low-threshold Aβ mechanoreceptors may presynaptically modulate nociceptive neurons in the spinal cord, resulting in an increased activity of wide dynamic range or nociceptor-specific cells.97 In RLS patients, a recent finding suggested that a central sensitization to Aδ fiber high-threshold mechanoreceptor input may occur.16 In this study, a tactile pin-prick test was used to assess sensory thresholds in normal individuals and in RLS patients. RLS patients demonstrated static mechanical hyperalgesia, without tactile allodynia, which was decreased with dopaminergic drug therapy.16
Several authors have reviewed the changes in neuronal plasticity in the presence of persistent pain, which support the view that such abnormal pain sensations may result from an altered transmission system and/or a dysfunctional endogenous analgesic pathways.5,11 The descending inhibitory influence from the upper brainstem areas (i.e., periaquaductal gray [PAG], locus ceruleus, and nuclus raphe magnus) are among the candidate regions involved in the endogenous analgesic pathways that could be disrupted in abnormal pain perception. In addition, dysfunction of the “diffuse noxious inhibitory controls” (DNIC) has been suggested to explain, in part, the hyperexcitability associated with the sensory changes observed in chronic pain patients.98–101 This analgesic system is mediated by endogenous opioids and originates from supraspinal sources.40,102 This system normally reduces bodily sensations in a diff-use nonspecific manner, as demonstrated by experimental manipulations involving “sensory conditioning” to alter pain perception. Dysfunction of the DNIC activation has been proposed to be partly responsible for the establishment of persistent pain in fibromyalgia patients.85,103 The dysfunction of endogenous analgesic systems, including DNIC, does not appear to have been adequately assessed in RLS patients.
The complexity of the potential interactions between various systems, involving both peripheral and central processes, could serve to explain RLS symptomatology. Although some results support the presence of peripheral neuropathy in idiopathic and secondary RLS,54,104,105 central sensitization and abnormal recruitment of the opioidergic system seem to be major players in the disease.15,16,55
Restless Legs Syndrome Periodic Limb Movements and Pain: Opioids and Dopamine
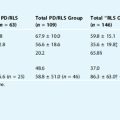
Children and adults with fibromyalgia, rheumatoid arthritis, or chronic back pain have elevated numbers of PLMS in their sleep.90,106–108 In a comparative analysis of matched RLS patients to fibromyalgia and chronic pain or control subjects, the PLM measurement was high in RLS patients (mean of 33 per hour of sleep), lower in fibromyalgia and pain patients (11 and 10.2, respectively), and very low in control subjects (2.0).109 The comorbidity between chronic pain and motor dysfunctions suggests that the opioid and dopamine systems might interact in the pathogenesis of RLS.
In addition, patients with RLS who are refractory to dopaminergic medications are treated with opioids, such as oxycodone, propoxyphene, and codeine; although tolerance and dependence do occur, the risk of addiction seems to be low.110–114 Even though the mechanism of action of opioids in RLS remains unknown, evidence suggests that the efficacy of opioids may not be related to their analgesic properties, as relief produce by methadone is delayed by up to 1 week.115 Furthermore, because naloxone, a specific opiate antagonist, could block the therapeutic effect of opioids in two RLS patients116,117 but not the effect of dopamine agonists in one patient118 and was unable to induce sensory or motor symptoms of the disease,119 the involvement of a dysfunctional opioidergic system in RLS is controversial. Although the evidence is scarce, these findings suggest that the therapeutic effects of opioids in RLS are likely mediated by the dopamine system, and not the opposite.14 In addition, it has been shown that activation of the opioidergic system with exogenous agonists can lead to increased dopamine release in healthy subjects.120 This effect could account for the therapeutic benefit observed after opioid treatment in RLS patients. Also, when pimozide, a dopamine receptor antagonist, was used in a case study, it was showed to block the effects of opioids,121 further incriminating the pivotal role of dysfunctional dopaminergic system in RLS physiopathology. Additionally, a positron emission tomography scan study revealed no difference in opioid receptor binding between RLS patients and control subjects.15 On the other hand, it is important to note that the radioligand used in this study is a nonselective opioid receptor ligand and that normal receptor availability may not account for a functional deficit.
The same study also revealed a positive correlation between severity of symptoms and the release of endogenous opioids within the medial affective pain system, that is, the medial thalamus, amygdala, caudate nucleus, anterior cingulate gyrus, insular cortex, and orbitofrontal cortex.15 Although the etiology of RSL is likely related to a dysfunction of the dopaminergic system, these results suggest a possible role of opioids in RLS. Indeed, considering the high density of opioid receptor and peptides in many dopaminergic neurons,122 and the influence of exogenous and endogenous opioids on the dopaminergic function,123,124 the role of opioids in primary RLS could be very complex. On the other hand, the recruitment of the opioidergic system seen in positron emission tomography with opioid receptor radioligand15 might be secondary to pain due to leg discomfort and motor restlessness. Alternatively, dysfunctional dopamine function might influence the liberation of opioids, as the disruption of the D2 receptor in mice was demonstrated to increase the preproenkephalin levels by about 50%.125
Clinically, the role of pain with the dysfunctional “reward” system and its interaction with dopamine, has been proposed to explain the persistence of chronic pain and of headaches due to overuse of medication.126–128 Pain is also reported in patients with basal ganglia-related disorders such as tardive dyskinesia or Parkinson’s disease,129 but an analysis of patient with tardive dyskinesia secondary to Parkinson’s disease failed to reveal any difference in pain perception in comparison with age- and gender-matched control subjects (R. E. Popovici, P. Blanchet, and G. Lavigne, unpublished observations). Human imaging studies revealed that dopaminergic D2 receptors are increased in the putamen of atypical facial pain patients.130 Other studies in animals have shown that morphine reduces dyskinetic responses in 1-methyl-4-phenyl-1,2,3,6-tetra hydropyridine (MPTP)-treated Parkinsonian-like monkeys131 and that activation of dopamine systems diminishes hindlimb autotomy in rats following sciatic nerve sectioning.132 These converging lines of evidence suggest that opioids and dopamine might share complex interactions in RLS, which further complicates the description of a single mechanism. Moreover, variations in animal and human pain perception in relation to ethnicity, gender, animal strain, and genetic polymorphisms need to be considered in the attempt to elucidate the possible disruption of the dopaminergic and opioidergic systems involved in RLS.133–138
Structures and Pathways That May Contribute to Pain Symptoms in Restless Legs Syndrome Patients
The neural pathways and mechanisms that may underlie the dysesthesias and paraestheisas associated with RLS are not known. Imaging studies conducted by Bucher and colleagues139 suggest that the thalamus is activated contralaterally during the sensory discomfort that is associated with RLS. Although this study cannot differentiate whether the activation of the thalamus precedes RLS-associated motor symptoms, it suggests that the thalamus could be one of the generators of RLS sensory discomfort. Considering the absence of cortical activity before myoclonic jerks in RLS and that the thalamus is the subcortical structure controlling sensory input closest to the somatosensory cortex, the sensory and motor symptoms of RLS might originate from the thalamus.140,141 Additionally, RLS patients exhibit bilateral thalamic gray matter changes.142 An MRI voxel-based morphometric analysis was used to test the hypothesis that subtle changes in brain structure occur in idiopathic RLS patients. They reported a statistically significant increase in gray matter in the pulvinar of the thalamus.142 How this change in thalamic structure relates to the pathogenesis of RLS (i.e., whether it may be consequential to an increase in afferent input from disinhibited spinal cord sensory pathways) is not clearly understood at this time.
The magnocellular red nucleus, the inferior olive, and the cerebellum are three brainstem structures activated during RLS motor symptoms in wakefulness.139 The inferior olive serves as a filter of sensory inputs during movement and therefore may provide a neural substrate for the reduction of the paraesthesia in RLS patients during periods of active movements.141 The red nucleus is an integral part of the network regulating the afferent information from the cerebellum to either the inferior olive or directly to the spinal cord. Other components, namely spinal neurons, can generate PLMS by activation of central locomotor pattern generators and transmission to the red nucleus. Indeed, the presence of PLMS in paraplegic patients raises the possibility that an interruption of supraspinal inhibitory influences could evoke rhythmic coordinated activity in the spinal cord143,144 and this mechanism could account for spontaneous movements in RLS. Noxious stimulation can also induce a rythmic pattern of action potentials in the magnocellular division of the red nucleus, potentially leading to locomotor-like activity generated in the spinal cord, ultimatly correlating with the motor events in RLS.141
The dysesthesias and paraesthesias of RLS are the typical triggers that cause an irresistible urge to move one’s lower limbs in an effort to quell the uncomfortable sensations. RLS symptoms are ameliorated with dopamine (dopamine D2 receptor) agonists and exacerbated with DA 2 antagonists.112,145–147 Hence, dysfunctional corticofugal dopamine inhibition of spinal cord sensory neurons may also contribute to the dysesthesias of RLS.148 Clinically, RLS patients display reduced sensory thresholds that are then normalized by L-DOPA administration.16
The reduction of hyperalgesia in RLS patients following dopaminergic treatment suggests that supraspinal pain modulation involving the basal ganglia and/or descending dopaminergic pathways from ventral periaqueductal gray matter (A11 cell group) might be involved in the pathophysiology of RLS.149 In addition, the lack of correlation between sensory impairments and the extent of peripheral small fiber neuropathy, reinforces the assumption that a central dysfunction of somatosensory processing is involved in idiopathic RLS.55 Moreover, the precise sensory pathways conveying RLS discomfort of lower limb origin have not yet been identified.139,141,150 Behavioral observations of animal models with impaired dopaminergic corticofugal function and electrophysiological protocols could help to identify the precise dopaminergic cell population involved in the pathophysiology of RLS.
Spinal cord spinothalamic tract (STT) and dorsal spinocerebellar tract neurons (DSCT) represent likely targets of abnormal corticofugal dopaminergic influences in human RLS.139 The sole source of DA in the spinal cord originates from centripetally projecting neurons located in the diencephalon, namely the A11 cell group.151,152 Intrathecal dopamine injection produces antinociception in awake behaving animals,153,154 and microiontophoretic administration of dopamine antagonists block the suppression of nociceptor-driven responses of rat STT neurons following electrical stimulation of the A11 cell groups.155 A study of post-mortem brain tissue of RLS patients failed to define any loss of nigrostriatal DA neuron cell groups; however, more rostrally located diencephalospinal DA neurons were not analyzed.156
Whether neurodegeneration or dysfunction (e.g., poor iron utilization) of dopaminergic A11 neurons could lead to downstream disinhibition of lumbar sensory tract neurons remains a critical open question regarding the trigger mechanism(s) underlying RLS dysesthesias. However, specific destruction of dopaminergic A11 neurons may represent a useful animal model. Insofar as the phenomenological analogy to sensory dysesthesia versus PLM of the RLS state is not yet clearly differentiated,157 a very recent investigation demonstrated that selective A11 DA cell group lesions resulted in dramatic reductions in both sensory thermal and mechanical thresholds of both forelimbs and hindlimbs in awake rats that were accompanied by a long-lasting marked reduction in lumbar spinal dopamine levels.158 Systemic or intrathecal administration of dopaminergic drugs reversed the mechanical and thermal sensitivities toward baseline values taken before 6-hydroxydopamine ablation of the A11 cell groups. Stereotypical periodic limb movements were absent.158,159 Such findings suggest that the diencephalospinal dopamine neurons may provide a selective tonic inhibition of spinal cord sensory inflow. These findings also partly satisfy criteria of face validity, construct validity, and predictive validity160–162 as applied to RLS and warrant further studies using chemical ablation of A11 as a key approach for elucidating the mechanism and trigger(s) for the sensory dysesthesias of RLS.158
The dopamine D3 receptor knock-out mouse, which shows hyperactivity and increased locomotor activity that peaks at night and are relieved by dopamine agonists, might be a another useful tool to model the motor aspects of RLS.163 Although the development of animal models emulating sensory dysesthesias and/or PLMS of RLS would be very useful for understanding and treating the disease, the subjective nature of the symptoms makes it particularly difficult, and hence, similar but quantifiable measures need to be employed in additional human and animal studies.
During wakefulness and NREM sleep, DSCT neurons and spinoreticular tract neurons are subjected to tonic GABAergic and glycinergic inhibition that is further enhanced during the state of active REM sleep. Recent microdialysis studies in the cat have demonstrated that reciprocal changes occured between the spinal cord levels of GABA and glycine and those of dopamine as a function of behavioral state. That is, during REM sleep, GABA and glycine levels increased significantly by 68% and 35%, respectively, whereas dopamine levels decreased significantly by about 22% compared with control states of wakefulness and NREM sleep states.164–166 Therefore, it appears that sensory inflow to higher brain centers via the DSCT and SRT may be governed, under normal conditions, by an intricate balance of multiple neurotransmitters such as GABA, glycine, and dopamine. During the pathological condition of RLS, this balance may be disrupted and lead to disinhibition and consequent hypersensitivity of STT, DSCT, SRT, or other sensory tract neurons in the spinal cord. The future of research on motor and sensory dysfunction in RLS, in relation to pain mechanisms, will ensure the development of new therapeutic avenues.
1. Zubieta JK, Heitzeg MM, Smith YR, et al. COMT avl158met Genotype affects μ-opioid neurotransmitter responses to a pain stressor. Science. 2003;299:1240-1243.
2. Kim H, Neubert JK, San Miguel A, et al. Genetic influence on variability in human acute experimental pain sensitivity associated with gender, ethnicity and psychological temperament. Pain. 2004;109:488-496.
3. Almeida T, Roizenblatt S, Tufik S. Afferent pain pathways: A neuroanatomical review. Brain Res. 2004;1000:40-56.
4. Wager TD, Rilling JK, Smith EE, et al. Placebo-induced changes in fMRI in the anticipation and experience of pain. Science. 2004;303:1162-1167.
5. Julius D, Basbaum AI. Molecular mechanisms of nociception. Nature. 2001;413:203-210.
6. Scholz J, Woolf CJ. Can we conquer pain? Nature Neuroscience. 2002;5(suppl):1062-1067.
7. Melzack R. From the gate to the neuromatrix. Pain. (suppl 6. 1999:S121-S126.
8. Willis WD, Westlund KN. Neuroanatomy of the pain system and of the pathways that modulate pain. J Clin Neurophysiol. 1997;14(1):2-31.
9. Merskey H, Bogduk N. Classification of Chronic Pain, 2. IASP Press, 1994.
10. Bromm B, Lorenz J. Neurophysiological evaluation of pain. Electroencephalogr Clin Neurophysiol. 1998;107:227-253.
11. Woolf CJ, Salter MW. Neuronal plasticity: Increasing the gain in pain. Science. 2000;288:1765-1768.
12. Julius D, Basbaum AI. Molecular mechanisms of nociception. Nature. 2001;413:203-210.
13. Hening W, Walters AS, Allen RP, et al. Impact, diagnosis and treatment of restless legs syndrome (RLS) in a primary care population: The REST (RLS Epidemiology, Symptoms and Treatment) primary care study. Sleep Med. 2004;5:237-246.
14. Barrière G, Cazalets JR, Bioulac B, et al. The restless legs syndrome. Prog Neurobiol. 2005;77:139-165.
15. von Spiczak S, Whone AL, Hammers A, et al. The role of opioids in restless legs syndrome: An [11C]diprenorphine PET study. Brain. 2005;128:906-917.
16. Stiasny-Kolster K, Magerl W, Oertel WH, et al. Static mechanical hyperalgesia without dynamic tactile allodynia in patients with restless legs syndrome. Brain. 2004;127:773-782.
17. Roehrs T, Hyde H, Blasdell B, et al. Sleep loss and REM sleep loss are hyperalgesic. Sleep. 2006;29(2):145-151.
18. Banno K, Delaive K, Walld R, et al. Restless legs syndrome in 218 patients: Associated disorders. Sleep Med. 2000;1:221-229.
19. Michaud M, Dumont M, Selmaoui B, et al. Circadian rhythm of restless legs syndrome: Relationship with biological markers. Ann Neurol. 2004;3:372-380.
20. Winkelmann J, Wetter TC, Collado-Seidel V, et al. Clinical characteristics and frequency of the hereditary restless legs syndrome in a population of 300 patients. Sleep. 2000;23(5):597-602.
21. Bentley AJ, Rosman KD, Mitchell D. Can the sensory symptoms of restless legs syndrome be assessed using a qualitative pain questionnaire. Clin J Pain. 2007;23:62-66.
22. Turk DC, Melzack R. Handbook of Pain Assessment, 2. The Guilford Press, 2001.
23. Harstall C, Ospina M. How prevalent is chronic pain? Pain Clin Updates (IASP). 2003;11(2):14.
24. Ohayon MM, Schatzberg AF. Using chronic pain to predict depressive morbidity in the general population. Arch Gen Psychiatry. 2003;60:39-47.
25. Moulin DE, Clarke AJ, Speechley M, et al. Chronic pain in Canada—Prevalence, treatment, impact and the role of opioid analgesia. Pain Res Manage. 2002;7:179-184.
26. National Sleep Foundation. 2000. Available at www.sleep.foundation.org/publications/sleepandpain.cfm.
27. Atkinson JH, Ancoli-Israel S, Slater MA, et al. Subjective sleep disturbance in chronic back pain. Clin J Pain. 1988;4:225-232.
28. Dao TTT, Lavigne GJ, Feine JS, et al. Quality of life and pain in myofascial pain patients and bruxers. J Dent Res. 1994;73:2047.
29. Morin CM, Gibson D, Wade J. Self-reported sleep and mood disturbance in chronic pain patients. Clin J Pain. 1998;14:311-314.
30. Smith MT, Perlis ML, Smith MS, et al. Sleep quality and pre sleep arousal in chronic pain. J Behav Med. 2000;23:1-13.
31. Dauvilliers Y, Touchon J. Le sommeil du fibromyalgique: Revue des données cliniques et polygraphiques. Neurophysiol Clin. 2001;31:18-33.
32. Riley JL, Benson MB, Gremillion HA, et al. Sleep disturbances in orofacial pain patients: Pain-related or emotional distress? J Craniomandib Pract. 2001;19:106-113.
33. Roizenblatt S, Moldofsky H, Benedito-Silva AA, Tufik S. Alpha sleep characteristics in fibromyalgia. Arthritis Rheum. 2001;44:222-230.
34. McCracken LM, Iverson GL. Disrupted sleep patterns and daily functioning in patients with chronic pain. Pain Res Manage. 2002;7:75-79.
35. Lavigne GJ, McMillan D, Zucconi M. Pain and sleep. In: Kryger MH, Roth T, Dement WC, editors. Principles and Practice of Sleep Medicine. Philadelphia: WB Saunders; 2005:1246-1255.
36. Lavigne GJ, Zucconi M, Castronovo C, et al. Sleep arousal response to experimental thermal stimulation during sleep in human subjects free of pain and sleep problems. Pain. 2000;84:283-290.
37. Melzack R. The McGill Pain Questionnaire: Major properties and scoring methods. Pain. 1975;1(3):277-299.
38. Feine JS, Lavigne GJ, Thuan Dao TT, et al. Memories of chronic pain and perception of relief. Pain. 1998;77:137-141.
39. McDowell I, Newell C. Measuring Health: A Guide to Rating Scales and Questionnaires, 2. New York, 1996.
40. Bouhassira D, Le Bars D, Bolgert F, et al. Diffuse noxious inhibitory controls in humans: A neurophysiological investigation of a patient with a form of Brown-Séquard syndrome. Ann Neurol. 1993;34:536-543.
41. Bouhassira D, Attal N, Fermanian J, et al. Development and validation of the neuropathic pain symptom inventory. Pain. 2004;108:248-257.
42. Handwerker HO, Kobal G. Psychophysiology of experimentally induced pain. Physiol Rev. 1993;3:639-671.
43. Meh D, Denislic M. Quantitative assessment of thermal and pain sensitivity. J Neurol Sci. 1994;127:164-169.
44. Gruener G, Dyck PJ. Quantitative sensory testing: Methodology, applications, and future directions. J Clin Neurophysiol. 1994;6:568-583.
45. Yarnitsky D. Quantitative sensory testing. Muscle Nerve. 1997;2:198-204.
46. Cruccu G, Anand P, Attal N, et al. EFNS guidelines on neuropathic pain assessment. Eur J Neurol. 2004;3:153-162.
47. Beydoun A, Morrow TJ, Shen J, et al. Variability of laser-evoked potentials: Attention, arousal and laterized differences. Electroencephalogr Clin Neurophysiol. 1993;88:173-181.
48. Drewes AM, Nielsen KM, Arendt-Nielsen L, et al. The effect of cutaneous and deep pain on the electroencephalogram during sleep—An experimental study. Sleep. 1997;20:623-640.
49. Sandrini G, Milanov I, Rossi B, et al. Effects of sleep on spinal nociceptive reflexes in humans. Sleep. 2001;24:13-17.
50. Bentley A, Newton S, Zio CD. Sensitivity of sleep stages to painful thermal stimuli. J Sleep Res. 2003;12:143-147.
51. Wang X, Inni K, Qiu Y, et al. Effects of sleep on pain-related somatosensory evoked potentials in humans. Neurosci Res. 2003;45:53-57.
52. Lavigne G, Brousseau M, Kato T, et al. Experimental pain perception remains equally active over all sleep stages. Pain. 2004;110:646-655.
53. Michaud M, Lavigne G, Desautels A, et al. Effects of immobility on sensory and motor symptoms of restless legs syndrome. Mov Disord. 2002;17:112-115.
54. Polydefkis M, Allen RP, Hauer P, et al. Subclinical sensory neuropathy in late onset restless legs syndrome. Neurology. 2000;8:1115-1121.
55. Schattschneider J, Bode A, Wasner G, et al. Idiopathic restless legs syndrome: Abnormalities in central somatosensory processing. J Neurol. 2004;251:977-982.
56. Graven-Nielsen T, Arendt-Nielsen L, Svensson P, et al. Stimulus-response functions in areas with experimentally induced referred muscle pain: A psychophysical study. Brain Res. 1997;744:121-128.
57. Baumgärtner U, Magerl W, Klein T, et al. Neurogenic hyperalgia versus painful hypoalgesia: Two distinct mechanisms of neuropathic pain. Pain. 2002;96:141-151.
58. Bara-Jimenez W, Aksu M, Graham B, et al. Periodic limb movements in sleep. State-dependent excitability of the spinal flexor reflex. Neurology. 2004;54:1609-1615.
59. Kurt TL. In response to Dr. May’s letter. Vet Hum Toxicol. 1989;2:181-182.
60. Schwartz JE, Jandorf L, Krupp LB. The measurement of fatigue: A new instrument. J Psychosom Res. 1993;37:753-762.
61. Dawson D, Fletcher A. A quantitative model of work-related fatigue: Background and definition. Ergonomics. 2001;44:144-163.
62. Fisk JD, Doble SE. Construction and validation of a fatigue impact scale for daily administration (D-FIS). Qual Life Res. 2002;11:263-272.
63. Kewman DG, Vaishampayan N, Zald D, et al. Cognitive impairment in musculoskeletal pain patients. Int J Psychiat Med. 1991;21:253-262.
64. Landro NI, Stiles TC, Sletvold H. Memory functioning in patients with primary fibromyalgia and major depression and healthy controls. J Psychosom Res. 1997;42:297-306.
65. Côté KA, Moldofsky H. Sleep, daytime symptoms and cognitive performance in patients with fibromyalgia. J Rheumatol. 1997;24:2014-2023.
66. Pearson VE, Allen RP, Dean T, et al. Cognitive deficits associated with restless legs syndrome (RLS). Sleep Med. 2006;7:25-30.
67. Bourdallé-Badie C, Andre M, Pourquier P, et al. Circadian rhythm of pain in man: Study by measure of nociceptive flexion reflex. Annu Rev Chronopharmacol. 1990;7:249-252.
68. Davis GC, Buchsbaum MS, Bunney WEJr. Naloxone decreases diurnal variation in pain sensitivity and somatosensory evoked potentials. Life Sci. 1978;23:1449-1460.
69. Göbel H, Cordes P. Circadian variation of pain sensitivity in pericranial musculature. Headache. 1990;30:418-422.
70. Strian F, Lautenbacher S, Galfe G, et al. Diurnal variations in pain perception and thermal sensitivity. Pain. 1989;36:125-131.
71. Labrecque G, Vanier MC. Biological rhythms in pain and in the effects of opioid analgesics. Pharmacol Ther. 1995;68:129-147.
72. Rogers EJ, Vilkin B. Diurnal variation in sensory and pain thresholds correlated with mood states. J Clin Psych. 1978;39:431-438.
73. Bellamy N, Sothern RB, Campbell J, et al. Circadian rhythm in pain, stiffness, and manual dexterity in rheumatoid arthritis: Relation between discomfort and disability. Ann Rheum Dis. 1991;50:243-248.
74. Reilly PA, Littlejohn GO. Diurnal variation in the symptoms and signs of the fibromyalgia syndrome (FS). J Musculoskeletal Pain. 1993;1:237-243.
75. Hening WA, Walters AS, Wagner M, et al. Circadian rhythm of motor restlessness and sensory symptoms in the idiopathic restless legs syndrome. Sleep. 1999;22:901-912.
76. Trenkwalder C, Paulus W, Walters AS. The restless legs syndrome. Neurology. 2005;4:465-475.
77. Onen SH, Alloui D, Jourdan D, et al. Effects of rapid eye movement (REM) sleep deprivation on pain sensitivity in the rat. Brain Res. 2001;900:261-267.
78. Moldofsky H, Scarisbrick P. Induction of neurasthenic musculoskeletal pain syndrome by selective sleep stage deprivation. Psychosom Med. 1976;38:35-44.
79. Older SA, Battafarano DF, Danning CL, et al. The effects of delta wave sleep interruption on pain thresholds and fibromyalgia like symptoms in healthy subjects; correlations with insulin like growth factor. J Rheumatol. 1998;25:1180-1186.
80. Smith MT, Edwards RR, Stonerock GL, et al. Individual variation in rapid eye movement sleep is associated with pain perception in healthy women: Preliminary data. Sleep. 2005;28(7):809-812.
81. Affleck G, Urrows S, Tennen H, et al. Sequential daily relations of sleep, pain intensity, and attention to pain among women with fibromyalgia. Pain. 1996;68:363-368.
82. Nicassio PM, Moxham EG, Schuman CE, et al. The contribution of pain, reported sleep quality and depressive symptoms to fatigue in fibromyalgia. Pain. 2002;100:271-279.
83. Raymond I, Nielsen TA, Lavigne GJ, et al. Quality of sleep and its daily relationship to pain intensity in hospitalized adult burn patients. Pain. 2001;92:381-388.
84. Pillemer SR, Bradley LA, Crofford LJ, et al. The neuroscience and endocrinology of fibromyalgia. Arthritis Rheum. 1997;40:1928-1939.
85. Bennett RM. Emerging concepts in the neurobiology of chronic pain: Evidence of abnormal sensory processing in fibromyalgia. Mayo Clin Proc. 1999;74:385-398.
86. Carette S, Dakson G, Guimont C, et al. Sleep electroencephalography and the clinical response to amitriptyline in patients with fibromyalgia. Arthritis Rheum. 1995;38:1211-1217.
87. Mahowald ML, Mahowald MW. Nighttime sleep and daytime functioning (sleepiness and fatigue) in less well-defined chronic rheumatic diseases with particular reference to the “alpha-delta NREM sleep anomaly”. Sleep Med. 2000;1:195-207.
88. Menefee LA, Cohen M, Anderson WR, et al. Sleep disturbance and nonmalignant chronic pain: A comprehensive review of the literature. Pain Med. 2000;1:156-172.
89. Rains JC, Penzien DB. Sleep and chronic pain challenges to the α-EEG sleep pattern as a pain specific sleep anormaly. J Psychosom Res. 2003;54:77-83.
90. Moldofsky H. Sleep and pain: Clinical review. Sleep Med Rev. 2001;5:387-398.
91. Rizzi M, Sarzi-Puttini P, Atzeni F, et al. Cyclic alternating pattern: A new marker of sleep alteration in patients with fibromyalgia? J Rheumatol. 2004;31:1193-1199.
92. Staedt J, Stoppe G, Hajak G, et al. Sleep disorders—What can be done when hypnotics no longer help? Overview and case report. Fortschr Neurol Psychiatr. 1995;9:368-372.
93. Okura K, Lavigne GJ, Montplaisir JY, et al. Slow wave activity and heart rate variation are less dominant in fibromyalgia and chronic pain patients than in controls. Sleep. (28. 2005:A302.
94. Martinez-Lavin M, Hermosillo AG, Rosas M, et al. Circadian studies of autonomic nervous balance in patients with fibromyalgia: A heart rate variability analysis. Arthri Rheum. 1998;41:1966-1971.
95. Waxman SG, Dib-Hajj S, Cummins TR, et al. Sodium channels and pain. Proc Natl Acad Sci USA. 1999;96:7635-7639.
96. Woolf CJ. Dissecting out mechanisms responsible for peripheral neuropathic pain: Implications for diagnosis and therapy. Life Sci. 2004;74:2605-2610.
97. Cervero F, Laird JMA. Mechanisms of touch-evoked pain (allodynia): A new model. Pain. 1996;68:13-23.
98. Farrell MJ. Pain & aging. APS Bull. 2000;10:8-11.
99. Edwards RR, Fillingim RB. Effects of age on temporal summation and habituation of thermal pain: Clinical relevance in healthy older and younger adults. J Pain. 2001;2:307-317.
100. Staud R. Evidence of involvement of central neural mechanisms in generating fibromyalgia pain. Curr Rheumatol Rep. 2002;4(4):299-305.
101. Washington LL, Gibson SJ, Helme RD. Age-related differences in the endogenous analgesic response to repeated cold water immersion in human volunteers. Pain. 2000;89:89-96.
102. Le Bars D, Willer JC, De Broucker T. Morphine blocks descending pain inhibitory controls in humans. Pain. 1992;48:13-20.
103. Lautenbacher S, Rollman GB. Possible deficiencies of pain modulation in fibromyalgia. Clin J Pain. 1997;13:189-196.
104. Iannaccone S, Zucconi M, Marchettini P, et al. Evidence of peripheral axonal neuropathy in primary restless legs syndrome. Mov Disord. 1995;10:2-9.
105. Rutkove SB, De Girolami U, Preston DC, et al. Myotonia in colchicine myoneuropathy. Muscle Nerve. 1996;7:870-875.
106. Mahowald MW, Mahowald SR, Bundlie SR, et al. Sleep fragmentation in rheumatoid arthritis. Arthritis Rheum. 1989;32:974-983.
107. Moldofsky H. Sleep and musculoskeletal pain. Am J Med. 1986;81:85-89.
108. Tayag-Kier CE, Keenan GF, Scalzi LV, et al. Sleep and periodic limb movement in sleep in juvenile fibromyalgia. Pediatrics. 2000;106:1-4.
109. Okura K, Rompré S, Manzini C, et al. Sleep duration and quality in chronic pain patients in comparison to the sleep of fibromyalgia, insomnia, RLS and control subjects [abstract]. Sleep. 2004:A329.
110. Kaplan PW, Allen RP, Buchholz DW, et al. A double-blind, placebo controlled study of the treatment of periodic limb movements in sleep using carbidopa/levodopa and propoxyphene. Sleep. 1993;16:717-723.
111. Hening W, Allen R, Earley C, et al. The treatment of restless legs syndrome and periodic limb movement disorder. An American Academy of Sleep Medicine review. Sleep. 1999;22:970-999.
112. Montplaisir J, Allen RP, Walters AS, et al. Restless legs syndrome and periodic limb movements during sleep. In: Kryger MH, Roth T, Dement WC, editors. Principles and Practice of Sleep Medicine. Philadelphia: WB Saunders; 2005:839-852.
113. Walters AS, Wagner ML, Hening WA, et al. Successful treatment of the idiopathic restless legs syndrome in a randomized double-blind trial of oxycodone versus placebo. Sleep. 1993;16:327-332.
114. Walters AS, Winkelmann J, Trenkwalder C, et al. Long-term follow-up on restless legs syndrome patients treated with opioids. Mov Disord. 2001;16:1105-1109.
115. Ondo WG. Methadone for refractory restless legs syndrome. Mov Disord. 2005;20:345-348.
116. Walters A, Hening W, Cote L, et al. Dominantly inherited restless legs with myoclonus and periodic movements of sleep: A syndrome related to the endogenous opiates? Adv Neurol. 1986;43:309-319.
117. Hening W, Walters A, Kavey N, et al. Dyskinesias while awake and periodic movements in sleep in restless legs syndrome: Treatment with opioids. Neurology. 1986;36:1363-1366.
118. Akpinar S. Restless legs syndrome treatment with dopaminergic drugs. Clin Neuropharmacol. 1987;10:69-79.
119. Winkelmann J, Schadrack J, Wetter TC, et al. Opioid and dopamine antagonist drug challenges in untreated restless legs syndrome patients. Sleep Med. 2001;2:57-61.
120. Hagelberg N, Aalto S, Kajander J, et al. Alfentanil increases cortical dopamine D2/D3 receptor binding in healthy subjects. Pain. 2004;109:86-93.
121. Montplaisir J, Lorrain D, Godbout R. Restless legs syndrome and periodic leg movements in sleep: The primary role of dopaminergic mechanism. Eur Neurol. 1991;31(1):41-43.
122. Cordonnier L, Sanchez M, Roques BP, et al. Facilitation of enkephalins-induced delta-opioid behavioral responses by chronic amisulpride treatment. Neuroscience. 2005;135:1-10.
123. Di Chiara G, Imperato A. Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc Natl Acad Sci USA. 1988;85:5274-5278.
124. Kalivas PW, Richardson-Carlson R. Endogenous enkephalin modulation of dopamine neurons in ventral tegmental area. Am J Physiol. 1986;251:R243-R249.
125. Baik JH, Picetti R, Salardi A, et al. Parkinsonian-like locomotor impairment in mice lacking dopamine D2 receptors. Nature. 1995;377:424-428.
126. Zubieta JK, Smith YR, Bueller JA, et al. Regional mu opioid receptor regulation of sensory and affective dimensions of pain. Science. 2001;293:311-315.
127. Wood PB. Mesolimbic dopaminergic mechanisms and pain control. Pain. 2006;120(3):230-234.
128. Calabresi P, Cupini LM. Medication-overuse headache: Similarities with drug addiction. Trends Pharmacol Sci. 2005;26(2):62-68.
129. Clarke IMC. Tardive dyskinesia and chronic pain. Pain. 1991;45:167-170.
130. Hagelberg N, Forssell H, Aalto S, et al. Altered dopamine D2 receptor binding in atypical facial pain. Pain. 2003;106:43-48.
131. Samadi P, Gregoire L, Bedard PJ. The opioid agonist morphine decreases the dyskinetic response to dopaminergic agents in parkinsonian monkeys. Neurobiol Dis. 2004;16(1):246-253.
132. Lopez-Avila A, Coffeen U, Ortega-Legaspi JM, et al. Dopamine and NMDA systems modulate long term nociception in the rat anterior cingulate cortex. Pain. 2004;111:136-143.
133. Xu XJ, Pleasan A, Yu W, et al. Possible impact of genetic differences on the development of neuropathic pain like behaviors after unilateral sciatic nerve ischemic injury in rats. Pain. 2001;89:135-145.
134. Taheri S, Mignot E. The genetics of sleep disorders. Lancet Neurol. 2002;1:242-250.
135. Raber P, Devor M. Social variables affect phenotype in the neuroma model of neuropathic pain. Pain. 2002;97:139-150.
136. Mogil JS, Wilson SG, Chesler EJ, et al. The melanocortin-1 receptor gene mediates female specific mechanisms of analgesia in mice and humans. Proc Natl Acad Sci USA. 2003;8:4867-4872.
137. Kim HK, Park SK, Zhou J-L, et al. Reactive oxygen species (ROS) play an important role in a rat model of neuropathic pain. Pain. 2004;111:116-124.
138. Ferini-Strambi L, Bonati MT, Oldani A, et al. Genetics in restless legs syndrome. Sleep Med. 2004;5:301-304.
139. Bucher SF, Seelos KC, Oertel WH, et al. Cerebral generators involved in the pathogenesis of the restless legs syndrome. Ann Neurol. 1997;41:639-645.
140. Trenkwalder C, Bucher SF, Oertel WH, et al. Bereitschaftspotential in idiopathic and symptomatic restless legs syndrome. Electroencephalogr Clin Neurophysiol. 1993;89:95-103.
141. Trenkwalder C, Paulus W. Why do restless legs occur at rest? Pathophysiology of neuronal structures in RLS. Neurophysiology of RLS (part 2). Clin Neurophysiol. 2004;115:1975-1988.
142. Etgen T, Draganski B, Ilg C, et al. Bilateral thalamic gray matter changes in patients with restless legs syndrome. Neuroimage. 2005;24(4):1242-1247.
143. Hartmann M, Pfister R, Pfadenhauer K. Restless legs syndrome associated with spinal cord lesions. J Neurol Neurosurg Psychiatry. 1999;66:688-689.
144. de Mello MT, Lauro FA, Silva AC, Tufik S. Incidence of periodic leg movements and of the restless legs syndrome during sleep following acute physical activity in spinal cord injury subjects. Spinal Cord. 1996;34:294-296.
145. Allen RP, Earley CJ. Restless legs syndrome: A review of clinical and pathophysiologic features. J Clin Neurophysiol. 2001;18:128-147.
146. Happe S, Trenkwalder C. Role of dopamine receptor agonists in the treatment of restless legs syndrome. CNS Drugs. 2004;18:27-33.
147. Wetter TC, Winkelmann J, Eisensehr I. Current treatment options for restless legs syndrome. Exp Opin Pharmacother. 2003;4:1727-1738.
148. Rye DB. Parkinson’s disease and RLS: The dopaminergic bridge. Sleep Med. 2004;5:317-328.
149. Mignot E, Taheri S, Nishino S. Sleeping with the hypothalamus: Emerging therapeutic targets for sleep disorders. Nat Neurosci. 2002;5(suppl):1071-1075.
150. Wetter TC, Eisensehr I, Trenkwalder C. Functional neuroimaging studies in restless legs syndrome. Sleep Med. 2004;5:401-406.
151. Hokfelt T, Phillipson O, Goldstein M. Evidence for a dopaminergic pathway in the rat descending from the A11 cell group to the spinal cord. Acta Physiol Scand. 1979;107:393-435.
152. Skagerberg G, Lindvall O. Organization of diencephalic dopamine neurons projecting to the spinal cord in the rat. Brain Res. 1985;342:340-351.
153. Liu QS, Qiao JT, Dafny N. D2 dopamine receptor involvement in spinal dopamine-produced antinociception. Life Sci. 1992;51:1485-1492.
154. Barasi S, Duggal KN. The effect of local and systemic application of dopaminergic agents on tail flick latency in the rat. Eur J Pharmacol. 1985;117:287-294.
155. Fleetwood-Walker SM, Hope PJ, Mitchell R. Antinociceptive actions of descending dopaminergic tracts on cat and rat dorsal horn somatosensory neurones. J Physiol. 1988;399:335-348.
156. Connor JR, Wang XS, Patton SM, et al. Decreased transferrin receptor expression by neuromelanin cells in restless legs syndrome. Neurology. 2004;62:1563-1567.
157. Ondo WG, He Y, Rajasekaran S, et al. Clinical correlates of 6-hydroxydopamine injections into A11 dopaminergic neurons in rats: A possible model for restless legs syndrome. Mov Disord. 2000;15:154-158.
158. Wang D, Sutherland D, McErlane SA, et al. Long-lasting reduction in rat sensory thresholds following ablation of diencephalic A11 dopamine (DA) nuclei [abstract]. Washington, DC: Society for Neuroscience, 2005.
159. Wang D. Dopaminergic basis for the dysesthesias of the restless legs syndrome: Development of an animal model. Thesis: University of British Columbia, 2006.
160. Willner P, Mitchell PJ. The validity of animal models of predisposition to depression. Behav Pharmacol. 2002;13:169-188.
161. Willner P. Animal models of depression: Validity and applications. Psychopharmacol. 1995;49:19-41.
162. Willner P. Validity, reliability and utility of the chronic mild stress model of depression: A 10-year review and evaluation. Psychopharmacology (Berl). 1997;134:319-329.
163. Clemens S, Rye D, Hochman S. Restless legs syndrome: Revisiting the dopamine hypothesis from the spinal cord perspective. Neurology. 2006;67:125-130.
164. Taepavarapruk N, McErlane SA, Soja PJ. State-related inhibition by GABA and glycine of transmission in Clarke’s column. J Neurosci. 2002;22:5777-5788.
165. Taepavarapruk N, McErlane SA, Chan A, et al. State-dependent GABAergic inhibition of sciatic nerve-evoked responses of dorsal spinocerebellar tract neurons. J Neurophysiol. 2004;92:1479-1490.
166. Taepavarapruk N, Taepavarapruk P, John J, et al. State-dependent changes in glutamate, glycine, GABA, and dopamine levels in cat lumbar spinal cord. J Neurophysiol. 2008;100:598-608.