CHAPTER 11 Iron deficiency anemia, anemia of chronic disorders and iron overload
Introduction
Disturbances of iron metabolism are among the commonest disorders affecting human populations. This high frequency reflects the combination of an essential requirement for iron by all living organisms, with a relatively precarious human iron balance. Iron in heme is maintained in the reduced ferrous state (Fe2+) for oxygen transport by hemoglobin and for storage and use of oxygen in muscles by myoglobin. It is also essential for a wide range of cellular heme and iron-sulfur proteins that are responsible for electron transport and energy generation in mitochondrial respiration and the citric acid cycle, and for ribonucleotide reductase, responsible for DNA synthesis.1 To be available for absorption, dietary iron needs to be in a soluble form, but in an oxygen-rich environment it is readily converted to insoluble ferric (Fe3+) hydroxide (‘rust’),2 or bound as insoluble ferric iron complexes, particularly in a predominantly vegetarian diet. This places an upper limit on the capacity of dietary iron to meet increased iron needs, whether physiologic or due to blood loss. As a result, iron deficiency anemia affects, at a conservative estimate, at least 500 million of the world’s population,3 and this takes no account of the additional, greater numbers of people who will have borderline iron status with depleted iron stores.4,5 In this context it is perhaps not surprising that humans conserve iron rigorously, with no mechanism for active iron excretion.6
Spectrum of pathology related to disorders of iron metabolism
Iron overload
Iron accumulating within macrophages is relatively non-toxic, these cells being specialized to deal with the high throughput of iron derived from hemoglobin in senescent red cells. By contrast, iron loading of parenchymal cells is associated with saturation of the plasma transferrin, the appearance of more labile and toxic non-transferrin-bound iron in the plasma,7 accumulation of iron in hepatocytes, cardiac myocytes and endocrine cells, and increased degradation of ferritin to insoluble hemosiderin within cellular lysosomes:8 the end result is the pattern of organ damage that is characteristic of hemochromatosis. There are no clear direct effects on bone marrow function, but iron-induced liver damage and cirrhosis may have indirect effects on blood cells (e.g. red cell macrocytosis, or cytopenias related to hypersplenism). By contrast, disturbed bone marrow function with massive ineffective erythropoiesis (e.g. in the β-thalassemia intermedia syndromes and some patients with sideroblastic or congenital dyserythropoietic anemias – see Chapters 9, 14, and 15), is definitely associated with inappropriately increased iron absorption and eventual iron overload. The hematologist is likely to be involved in the diagnosis and treatment of iron overload, including phlebotomy in hereditary hemochromatosis and iron chelation in iron-loading anemias.
Major pathways of iron exchange
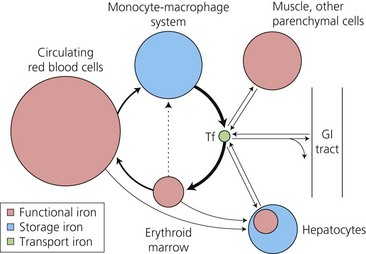
These were delineated many years ago by use of ferrokinetic studies, following the tissue uptake of 59Fe after its intravenous injection bound to plasma transferrin.9 The pathways (Fig. 11.1) are dominated (80–90% of plasma iron turnover) by the supply of plasma iron, bound to the circulating transport protein, transferrin, to bone marrow erythroid precursors for hemoglobin synthesis. At the end of their life span, red cells are phagocytosed by tissue macrophages. Heme oxygenase releases the iron from heme for recycling back to plasma transferrin, or alternatively diversion to intracellular ferritin, a protein cage made up of 24 H- (Heavy) and L- (Light) subunits which can store up to 4500 atoms of iron. A smaller uptake by the liver hepatocytes is the main alternative site of transferrin iron uptake (approximately 10%), and reflects expression of transferrin receptors on hepatocytes as well as erythroblasts. Whereas macrophages gain nearly all their iron in the unidirectional flow from senescent red cells, hepatocytes are able to take up iron in a variety of forms (see below) as well as to release the iron in times of increased need. This makes the liver the major ‘buffer’ within the system, and the prime target for iron loading and damage in iron overload conditions. There is very limited exchange of iron with the exterior. Obligatory losses (skin and gastrointestinal mucosal cell loss) of approximately 1 mg/day in males (rather more in women of child-bearing age with the additional losses of menstruation, pregnancy and lactation)10 are normally balanced by absorption of a similar amount from the diet.
Over recent years, understanding of the processes of cellular iron uptake through transferrin receptors, and regulation of intracellular iron homeostasis, has greatly increased. The discovery of the genetic basis of HFE-related hemochromatosis in the mid 1990s, combined with the use of molecular genetic studies of animal models with altered iron metabolism or erythropoiesis, led to rapid advances, with identification of additional proteins involved in regulating iron absorption and internal exchange.11 An understanding of these molecular processes underpins discussion of the pathophysiology of the iron disorders and their diagnosis.
Molecular mechanisms in iron metabolism
Cellular uptake of iron from transferrin
Cell surface expression of the classical transferrin receptor (TfR1) is greatest on rapidly dividing cells such as erythroid precursors, though it is also required for normal development of lymphoid cells and neuroepithelial differentiation in the developing nervous system. The receptor has a greater affinity for fully saturated, diferric, transferrin than for monoferric transferrin,12 and does not bind apotransferrin at the neutral pH of plasma. These affinities in part account for the importance of measuring the transferrin saturation as a vital part of a screen for the risk of an iron loading condition,13 and the fact that transferrin saturation is a better guide to iron supply to the tissues in iron deficiency than the serum iron value.14 At a high transferrin saturation most plasma iron is present as diferric transferrin, and iron uptake via transferrin receptors is enhanced. By contrast, the increased concentrations of serum transferrin which are found in iron deficiency mean that the small amount of iron present is in the form of monoferric transferrin, with its reduced rate of uptake by transferrin receptors.
After receptor-mediated endocytosis of the transferrin/receptor complex (Fig. 11.2), acidification of the endosome releases the iron from the transferrin, which at low pH even after releasing its iron still has a high affinity for the receptor.15 The apotransferrin recycles with the receptor back to the cell membrane, where it dissociates and is released into the plasma to continue its role in iron delivery to the tissues. The iron is reduced to Fe2+ by a ferrireductase, STEAP3,16 before being transported into the cytosol from the endosome by divalent metal transporter, DMT1 (previously known as NRAMP2, or DCT1).17
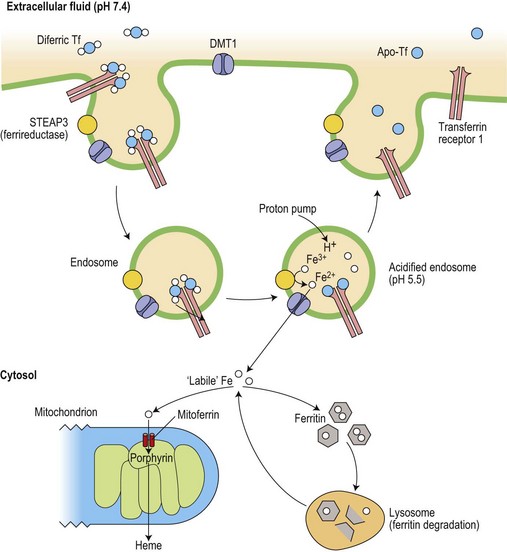
Fig. 11.2 Cellular uptake of transferrin iron and its intracellular utilization. The iron released into the cytoplasm plays a role in the translational regulation of transferrin receptor, ferritin and erythroid ALA-synthase (see Fig. 11.3). Heme synthesis dominates in erythroid cells, whereas ferritin metabolism is crucial in iron storage cells (macrophages and hepatocytes) Ferritin is constantly catabolized and the iron is either released back to the cytosol or remains in lysosomes as insoluble hemosiderin.
Regulation of cellular iron homeostasis
The intracellular iron content is finely regulated at the level of translation of the mRNA of several key iron-related proteins.18 Two iron-regulatory proteins (IRP1, coded on chromosome 9, and IRP2, coded on chromosome 15), are able to bind to sequences which form stem loop structures called iron responsive elements (IREs) in the untranslated regions of mRNAs for transferrin receptor and both H- and L-subunits of ferritin (Fig. 11.3). When IRP1 contains an iron-sulfur (4Fe-4S) cluster it has a low affinity for the IRE (and functions as cytoplasmic aconitase), while IRP2 is unstable in the presence of iron. However, when intracellular ‘labile’ (metabolically active) iron is at a low level, IRP binding to IREs that are present in the 3′ untranslated region of the mRNA for transferrin receptor protects the message from cytoplasmic degradation and allows its translation: at the same time, binding of IRP to the single stem loop in the 5′ untranslated region of the ferritin mRNA inhibits ferritin protein synthesis. The relative rates of synthesis of the two proteins are reversed in conditions of iron excess, and this reciprocal relationship serves to stabilize the intracellular labile iron content. Other iron related proteins also have IRE structures in their mRNAs.19 Those with 5′ IREs include the cellular iron exporter protein, ferroportin, (see below) and erythroid delta-aminolevulinic acid synthase (ALAS). IRP binding to the latter enables heme synthesis to be matched to the iron supply: the initial part of the heme synthetic pathway is switched off when the cell is deficient in iron. At least one isoform of DMT1 has an IRE in its 3′ untranslated mRNA, and IRP binding may stabilize its mRNA in an iron depleted cell, leading to increased expression and enhanced iron uptake.
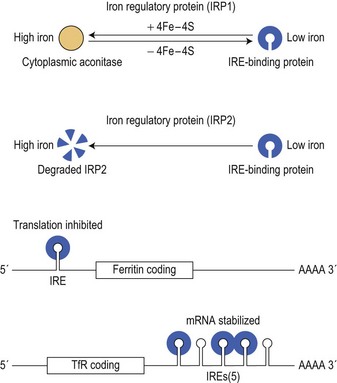
Fig. 11.3 Coordinate regulation of synthesis of ferritin and transferrin receptor-1 (TfR1) by the interaction of iron binding proteins (IRPs) with mRNA iron responsive elements (IRE). When cytoplasmic iron (see Fig. 11.2) is low, IRPs bind to IRE stem-loop structures to inhibit ferritin translation but to increase translation of TfR1 by preventing degradation of its mRNA. When iron levels are high, IRP1 functions as a cytoplasmic aconitase and no longer binds to IREs and IRP2 is degraded: this allows increased ferritin synthesis but reduces TfR1 synthesis.
Iron entering the cell cytosol becomes available for a variety of functional iron compounds or can be taken up by the iron storage protein, ferritin (Fig. 11.2). However, control of these intracellular movements is only just beginning to be explored. The H-subunits of ferritin have an intrinsic ferroxidase activity which is important for their storage function. Mitochondrial iron importers (mitoferrin-1 and -2) have been shown to be essential for heme and iron-sulfur cluster synthesis in the mitochondria of a variety of cells. Mitoferrin-1, itself stabilized by a mitochondrial ATP binding cassette transporter protein,20 is essential for iron utilization by the mitochondria of erythroid cells.21 The amount of mitoferrin may thus regulate the delivery of iron for incorporation into protoporphyrin by the enzyme ferrochelatase in the final step of heme synthesis.
Disturbances of intracellular iron distribution resulting from abnormal mitochondrial iron homeostasis are associated with a number of inherited diseases. For example, mutations in erythroid ALAS are responsible for X-linked sideroblastic anemia, with iron accumulation within mitochondria related to impaired protoporphyrin synthesis.22 Mutations in two other mitochondrial proteins give rise to mitochondrial iron accumulation, and decreased cytosolic iron with defects in iron-sulfur cluster formation and heme synthesis. Mutations in the gene for ATP-binding cassette 7 (ABCB7) underlie the sideroblastic anemia with spinocerebellar ataxia that is linked to Xq13 (see Chapter 14).23 Autosomal recessive inheritance of increased numbers of trinucleotide repeats in the gene coding for the mitochondrial iron chaperone, frataxin, is accompanied by the neurological and cardiac problems of Friedreich’s ataxia and by up-regulation of the mitochondrial iron importer, mitoferrin, with mitochondrial iron loading.24
Regulation of iron uptake in specific tissues
Developmental (transcriptional) regulation of iron uptake in the erythron
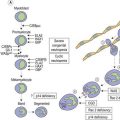
Committed erythroid progenitors express erythropoietin receptors maximally at the late BFU-E and CFU-E stages, declining from the proerythroblast stage. Erythropoietin prevents apoptosis of these progenitors and allows their proliferation. It also activates IRP and thus up-regulates synthesis of transferrin receptors25 which reach a peak in the basophilic erythroblasts. Uptake of iron thus precedes the onset of maximum heme synthesis in later polychromatic erythroblasts.26 Any iron not subsequently used appears in cytoplasmic ferritin and siderotic granules (giving rise to normal sideroblasts on Perls’ staining of marrow smears, or to Pappenheimer bodies in mature red cells on Romanowsky staining of peripheral blood films). These are more prominent where transferrin saturation, and thus the proportion of diferric transferrin, which has a high affinity for the receptor (see above), is increased, and are absent when a low saturation gives rise to iron-deficient erythropoiesis.
Hepatocyte iron uptake
The hepatocyte is able to take up iron in many forms, including transferrin- and non-transferrin-bound iron, hemoglobin–haptoglobin and heme–hemopexin complexes (which provide a potential direct shunt from the erythron to the hepatocyte in conditions associated with hemolysis or ineffective erythropoiesis), and any tissue ferritin which has been released into the circulation as the result of cell damage. The hepatocyte has a low level of expression of classical transferrin receptors (TfR1), but expresses a homolog, transferrin receptor 2 (TfR2).27,28 This differs from TfR1 in having no 3′ IRE in its mRNA, and it is thus not down-regulated in the presence of iron overload.29 Homozygous inheritance of a mutation in the TfR2 gene underlies some cases of hemochromatosis, implicating this receptor in the pathway that regulates iron absorption in relation to iron stores (see below).30 Iron uptake by hepatocytes from transferrin may occur by the receptor-mediated endocytosis described above or, following release of the iron at the hepatocyte surface, by the route taken by non-transferrin-bound iron.15 Although the detailed mechanisms of iron uptake (and release) by hepatocytes remain somewhat uncertain, it is clear that the liver may continue to take in iron from various sources, even when there are already increased iron stores, and it is thus highly vulnerable to damage in iron-loading disorders.
Macrophage iron uptake
The phagocytic cells of the reticuloendothelial system normally recycle approximately 20 mg iron a day from hemoglobin in senescent red cells and the normal small proportion of ineffective erythropoiesis.9,26 Although they express transferrin receptors, these are at a low level, and do not account for a significant proportion of the plasma iron turnover derived from transferrin. Increased uptake of iron from lactoferrin produced by neutrophils is no longer thought to be a significant source of the increased macrophage storage iron that is seen with inflammation.31
Uptake of iron by duodenal mucosal cells
Duodenal mucosal cells are specialized to take up soluble iron at their apical brush border and to transfer this across their basolateral membrane to circulating plasma transferrin (Fig. 11.4). Stomach acid has an important role in the solubilization of non-heme iron, and the detailed make-up of the diet influences its availability for absorption. For example, phytates and phosphates in vegetarian diets inhibit non-heme iron absorption by forming insoluble complexes, whereas ascorbate and meat protein digestion products form soluble complexes and enhance absorption. Heme iron from animal products is also relatively well absorbed.
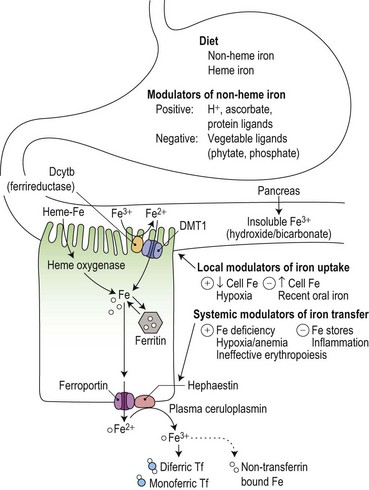
Reduction from Fe3+ to Fe2+ is an essential initial step in the uptake of dietary non-heme iron.32 It is mediated by a heme-containing ferrireductase, duodenal cytochrome b (Dcytb) which is expressed in the apical brush border membrane of duodenal enterocytes and induced by iron deficiency.33 Ascorbate could have a role as an electron donor in this process, contributing to its ability to promote the availability of iron for absorption. An apical mucosal iron transporter, DMT1, then transfers the Fe2+ into the enterocyte.34 Alternative splicing generates two isoforms of intestinal DMT1, only one of which has a 3′ IRE that could potentially be regulated by the IRE/IBP mechanism: the presence of further isoforms containing an additional 5′ exon may be required for up-regulation in iron deficiency.35 Dietary heme is taken up at the apical surface by a separate ill-understood mechanism, and the iron released by intracellular heme oxygenase is thought to join the same metabolic pool as that derived from non-heme iron.36 Once inside the cell, the iron may either be incorporated into ferritin stores (to be shed with the enterocyte into the gut lumen at the end of the cell’s life span) or transferred across the basolateral membrane by another iron transporter protein, ferroportin (also known as Ireg1).37
Duodenal enterocytes also express TfRs at their basolateral membrane, and iron uptake by this route during their maturation from duodenal crypt cells into absorptive enterocytes may contribute to modulation of the expression of transporter proteins through IRE/IBP mechanisms or through transcriptional regulation.38 The observation that a large oral dose of iron may result in a ‘mucosal block’ to absorption of a subsequent oral dose has been attributed to a rapid and selective down-regulation of Dcytb and DMT1 with no effect on the mRNA of basolateral iron transport molecules.39
Iron release from ‘donor’ cells
As well as being found in the basolateral membrane of the duodenal enterocyte, ferroportin is also found in macrophages and hepatocytes.40 Ferroportin transports reduced (Fe2+) iron and is coupled with the copper oxidases, plasma ceruloplasmin and its membrane bound homolog, hephaestin:41 the iron released as Fe3+ is taken up by plasma transferrin. A ceruloplasmin defect impairs macrophage and hepatocyte iron release,42 giving rise to liver iron overload that is resistant to phlebotomy.43 Transferrin is not essential for release of iron at the plasma membrane, since its absence in rare cases of congenital atransferrinemia does not prevent increased iron uptake from the duodenum and subsequent development of liver parenchymal iron overload.44
Regulation of iron absorption and internal iron exchange
Iron absorption in health and disease
Iron absorption is normally extremely sensitive to changes in body iron status, and both heme and non-heme iron absorption show an inverse relationship to iron stores.45 However, pathological disturbances also influence iron absorption (Fig. 11.4). Inflammatory disease reduces absorption contributing to the impaired iron supply which is characteristic of the anemia of chronic disorders.46 It has also long been recognized that grossly expanded erythropoiesis, particularly when the latter is ineffective as, for example, in the β-thalassemia disorders, can be associated with a marked increase in iron absorption, even in the face of pre-existing iron overload and an increased transferrin saturation.47,48 Hypoxia is known to potentiate mucosal iron uptake in animal studies,49 and in normal humans, stimulation of erythropoiesis by injection of recombinant erythropoietin markedly enhanced non-heme iron absorption.50 Iron absorption studies carried out sequentially during treatment with erythropoietin and phlebotomy of iron-loaded patients with chronic renal failure suggested that anemic hypoxia enhanced mucosal iron uptake, while reduction in iron stores and increased erythropoiesis independently increased the transfer of iron to the plasma.51 The concept of ‘iron store’ and ‘erythroid’ regulators of iron absorption gained currency, but their mechanisms remained obscure.52 It is now known that hepcidin, a 25 amino-acid peptide hormone derived from a propeptide produced by the liver,53 plays a key role in mediating these effects.
Role of hepcidin
In 2001 it was reported that expression of hepcidin was increased in iron loaded mice,54 and that mice in which HAMP, coding for hepcidin, had been knocked out developed hepatic but not macrophage iron overload.55 This pattern of disturbed iron metabolism in hepcidin deficient mice mirrors that seen in human hemochromatosis56 and it became clear that the various types of hemochromatosis (see Table 11.3, below) are associated with inappropriately low production of hepcidin except in rare cases with a ferroportin mutation that prevents a response to hepcidin. Conversely, hepcidin over-expression in mice led to iron deficiency,57 including in mice with inactivation of TMPRSS6 (coding for matriptase-2, a liver transmembrane serine protease).58,59 A human inherited iron-refractory iron deficiency anemia (IRIDA) was then shown to be associated with over-expression of hepcidin resulting from recessive inheritance of inactivating mutations of TMPRSS6.60,61
It is now clear that hepcidin is the major physiological regulator of iron absorption and internal iron exchange (Fig. 11.5), and that inappropriate hepcidin production underlies much iron pathophysiology.11 Hepcidin binds to ferroportin at the ‘donor’ cell surface and promotes the transporter’s internalization followed by ubiquitin-mediated lysosomal degradation.62,63,64 Hepcidin-induced degradation of ferroportin prevents iron release to circulating plasma transferrin and leads to reduced iron absorption, a block on the release of iron derived from senescent red cells within macrophages, and reduced serum iron concentration. It is therefore a negative regulator of iron release from cells. Hepcidin is excreted rapidly into the urine consistent with its regulation at the level of production. Decreased hepcidin production is seen with hypoxia65 and in association with ineffective erythropoiesis,66 as well as in iron deficiency. Conversely increased hepcidin production, occurs with inflammation65 as well as with increased iron stores.
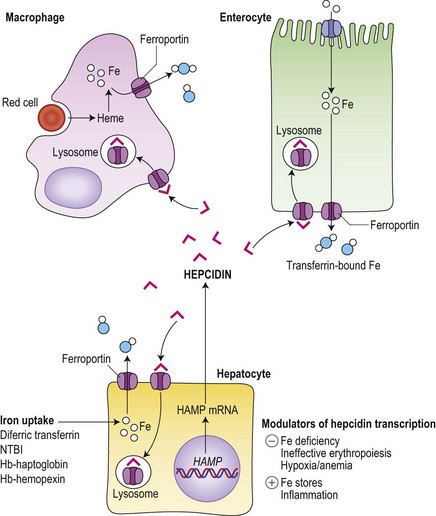
Fig. 11.5 Central role of hepcidin in iron metabolism. Hepcidin produced by hepatocytes down-regulates iron export to circulating transferrin from iron ‘donor’ cells (hepatocytes, macrophages and duodenal enterocytes) by promoting the internalization and lysosomal degradation of ferroportin. Hepatocytes take up iron in a number of forms, whereas enterocytes obtain their iron predominantly from the gut lumen (see Fig. 11.4) and macrophages are specialized to deal with the high throughput of iron from senescent red cells.
Regulation of hepcidin production
Uncovering the genetic basis of the various types of hemochromatosis (see Table 11.3, below) and of IRIDA stimulated exploration of signal pathways that act on the liver to regulate hepcidin production.67 Iron status signals via the bone morphogenetic protein (BMP) pathway (Fig. 11.6).68,69 Hemojuvelin (HJV), mutated in most cases of juvenile hemochromatosis, is a glycosylphosphoinositol (GPI)-linked cell surface protein which acts as a co-receptor for BMP6 (a key endogenous regulator of hepcidin synthesis).69 It is thought that cell surface HFE (commonly mutated in adult hemochromatosis) and TfR2 (mutated in other cases of hemochromatosis) associates with HJV on the cell surface to allow signaling via BMP receptors and a SMAD pathway to the hepcidin promoter. It is possible that the link to iron status may be provided by competitive interactions between HFE and diferric transferrin for binding to TfR1: only the HFE remaining would then be available to associate with TfR2, the complex being stabilized by diferric transferrin.70 A physiological increase in diferric transferrin would signal increased hepcidin transcription, reducing iron absorption and release from macrophages, while pathological disruption of HFE, HJV or TfR2 (in hemochromatosis) would be accompanied by reduced hepcidin synthesis and increased iron absorption. Negative regulation of hepcidin production by matriptase-2 (TMPRSS6)71 is mediated by proteolysis of membrane HJV,72 and its absence accounts for the over-expression of hepcidin in familial iron-refractory iron deficiency anemia.
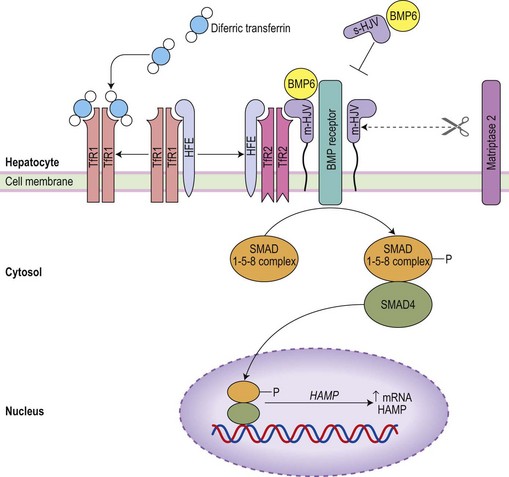
The increase in hepcidin in response to inflammation is predominantly mediated by interleukin-6 (IL-6)73 and activation of signal transduction via STAT3.74 Suppression of hepcidin production in association with ineffective erythropoiesis in thalassemia major occurs independently of iron stores.75 A number of potential mechanisms for the ‘erythroid regulator’ are emerging. Erythropoietin can directly down-regulate hepcidin production through the erythropoietin receptor and C/EBPα transcription factor,76,77 but this cannot account for a dependence of the erythroid regulator on the presence of an expanded erythroid marrow. Growth differentiation factor 15 (a member of the transforming growth factor β family) is up-regulated in serum from patients with thalassemia and congenital dyserythropoietic anemia type 1, and suppresses hepcidin production in vitro.78,79 Its release from apoptotic erythroblasts would explain the greater iron loading in anemias with expanded erythroid marrow due to ineffective erythropoiesis rather than hemolysis. A further candidate molecule derived from erythroblasts is ‘twisted gastrulation’ which has been shown to interfere with BMP signaling via its receptor80 and would thus suppress hepcidin synthesis. Finally, hypoxia may inhibit hepcidin production through involvement of the hypoxia inducible factor (HIF)/von Hippel–Lindau pathway. HIF-1α, induced by hypoxia and stabilized by iron deficiency, up-regulates furin-mediated cleavage of HJV to produce a soluble form of HJV: it is likely that this competes with membrane HJV for the co-receptors for BMP, thus down-regulating hepcidin production.81
Assessment of iron status
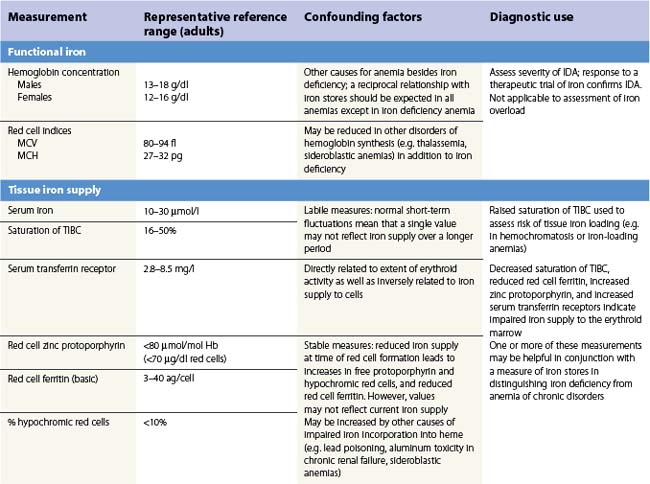
There is no single measure of iron status that is applicable in every situation. Combinations of measures of iron stores (macrophage and hepatocyte), iron supply to the tissues, and functional hemoglobin iron are often needed to arrive at a clear assessment of iron status.82 The measures used are summarized in Table 11.1. All are subject to potential confounding factors.
Serum transferrin receptors
Serum transferrin receptors are truncated soluble receptors that are shed into the circulation mainly from the erythroblasts in the marrow.83 The measurement reflects both the iron status of individual eythroblasts and the total mass of the erythron.84 It is likely to be of most value in distinguishing iron deficiency from the anemia of chronic disorders.85
Serum ferritin
Serum ferritin is apoferritin made up from glycosylated ferritin light chains, the release of which from cells reflects current ferritin protein synthesis.86 It is thus related to the intracellular labile iron that determines IRP affinity for the IRE, and only indirectly to iron stores through the release of ferritin iron either within the cytosol or through lysosomal degradation.87 Ferritin protein synthesis also increases in response to inflammatory cytokines, behaving as an acute phase protein independently of iron stores. Damage to ferritin-rich tissues can release iron-containing ferritin into the circulation giving high ferritin values, e.g. in hepatitis, splenic infarction, or bone marrow infarction in sickle cell disease. Its use as a guide to the presence of increased iron stores is thus limited,88 though a low serum ferritin is a clear indication that iron stores are absent. The dependence of ferritin protein synthesis on translational regulation by the IRP/IRE mechanism is illustrated by the rare hereditary hyperferritinemia/cataract syndrome, where autosomal dominant mutations in critical parts of the IRE stem loop are accompanied by uncontrolled synthesis of ferritin light chain: high serum ferritin values are seen with the development of cataracts, but there is no iron overload and transferrin saturations are not increased.89 Other uncommon autosomal dominant causes of a high serum ferritin but normal transferrin saturation include loss of function ‘ferroportin disease’ and a benign hyperferritinemia associated with a point mutation in the coding sequence of L-ferritin.90
The reciprocal relationship between the synthesis of transferrin receptors and ferritin within cells that is mediated by the IRE/IBP mechanism (Fig. 11.3) has its counterpart in values for serum transferrin receptors (increased) and serum ferritin (reduced) during the development of iron deficiency. The sensitivity of these measures for assessing iron status is increased by expressing them as a ratio.91,92
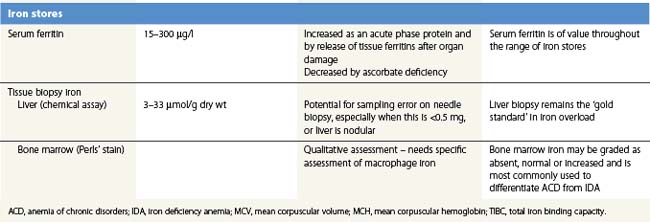
Tissue biopsy
Bone marrow biopsy is used to examine macrophage iron stores. It is primarily used for supporting a diagnosis of the anemia of chronic disorders rather than iron deficiency. Bone marrow biopsy touch preparations may give results comparable to aspirates.93 Positive identification of the absence of iron stores may be inaccurate94 unless careful examination of macrophages is made.
Liver biopsy provides the opportunity for histological examination of increased hepatocyte iron and any fibrotic or cirrhotic changes. The periportal distribution of iron accumulation in hemochromatosis may reflect selective expression of ferroportin in periportal hepatocytes and its degradation when hepcidin production is low.64 In addition, chemical iron determination allows a quantitative assessment of the degree of iron loading. Quantitative phlebotomy in patients with thalassemia major who had undergone curative allogeneic bone marrow transplantation has confirmed that the liver iron concentration is a reliable indicator of total body iron stores in secondary iron overload resulting from red cell transfusions.95 Magnetic resonance quantitative measurements (particularly T2*) of liver (and cardiac) iron96 may reduce the future need for liver biopsy in iron overload disorders.
Potential clinical use of hepcidin assay
Difficulties in measuring urinary and serum hepcidin with lack of standardization97 have so far restricted investigation of potential diagnostic uses. Plasma hepcidin concentrations are highly correlated with serum ferritin values in normal men,98 and identification of inappropriately low hepcidin levels for existing iron stores might be diagnostically helpful in predicting the severity of the clinical course in iron loading disorders. However, like many other measures of iron status, confounding by the effects of inflammation is likely to make interpretation difficult. The possibility that hepcidin measurement might help to identify patients with anemia of chronic disease who also have an element of iron deficiency requires investigation.99
Iron deficiency
Causes of iron deficiency
Iron deficiency anemia is extremely common3 particularly among women of child-bearing age and pre-school children (who have increased physiological requirements for iron and whose intake is more likely to fall below the reference nutrient intake). In the UK in 2000–2001 the mean daily intake was 14.0 mg for men, and 11.6 mg for women,100 and can be compared with the estimated average requirements of 6.7 mg for men and 11.4 mg for the majority of pre-menopausal women.101
The combination of a diet with poorly available iron and physiological increased iron requirements is probably the commonest cause of iron deficiency on a world scale. However, diet alone is very seldom the cause of iron deficiency in men or post-menopausal women in whom pathological blood loss should be suspected. Blood loss of more than about 6 ml (3 mg iron)/day, added to obligatory losses of about 1 mg/day through shedding of skin and intestinal cells, is likely to exceed the maximum iron absorptive capacity, hookworm infestation being a major cause in many parts of the world. In women of reproductive age, menstruation adds an average of 20 mg/month, and menorrhagia is a likely cause of anemia. In men or postmenopausal women, occult gastrointestinal blood loss must be considered, and in these patients, as well as younger women who have symptoms suggestive of gastrointestinal disease, endoscopic and/or radiological investigation of the gut is likely to be required.102 This should be considered whether or not fecal occult blood tests are positive, since such bleeding may be intermittent. Less commonly, malabsorption of iron may be responsible for negative iron balance. Iron deficiency is a predictable complication after gastrectomy, where loss of the stomach acid and more rapid transit past the duodenal absorptive area of the gut, combine to reduce dietary iron availability. Celiac disease may present with isolated iron deficiency and features of hyposplenism on the blood film. Autoimmune gastritis, often without cobalamin deficiency, and Helicobacter pylori infection may account for many patients whose iron deficiency is otherwise unexplained.103 The causes of iron deficiency are summarized in Table 11.2.
| Increased physiological requirements | |
| Growth | Pre-term and low birthweight |
| Pre-school children | |
| Adolescents | |
| Reproduction | Menstruation |
| Pregnancy | |
| Lactation | |
| Dietary insufficiency or poor bioavailability | |
| Early introduction of cow’s milk (low iron content) in infancy | |
| Vegetarian diet (insoluble phytate iron complexes) | |
| Antacids/protein pump inhibitors | |
| Clay eating (pica) | |
| Blood loss | |
| Gastrointestinal | Epistaxes |
| Varices | |
| Erosive gastritis | |
| Peptic ulcer | |
| Aspirin or other NSAIDS | |
| Carcinoma of stomach, colon | |
| Meckel’s diverticulum | |
| Angiodysplasia | |
| Inflammatory bowel disease | |
| Diverticulosis | |
| Hemorrhoids | |
| Pulmonary | Hemoptysis |
| Pulmonary hemosiderosis | |
| Genitourinary | Menorrhagia |
| Post-menopausal bleeding | |
| Parturition | |
| Hematuria (e.g. renal or bladder origin) | |
| Hemoglobinuria (e.g. paroxysmal nocturnal hemoglobinuria) | |
| Other blood loss | Trauma |
| Widespread bleeding disorder | |
| Self-inflicted | |
| Malabsorption | |
| Post gastrectomy | |
| Autoimmune gastritis | |
| Helicobacter pylori infection | |
| Chronic systemic inflammatory disease | |
| Gluten-induced enteropathy (celiac disease) | |
| Genetic | Congenital iron-refractory iron deficiency anemia (IRIDA) |
There remain a minority of patients in whom the cause remains uncertain. In older patients, bleeding from angiodysplastic lesions in the gut may be suspected, but careful follow-up is required since re-investigation may be needed if there are new symptoms or worsening of the negative iron balance. One of the rare genetic defects affecting iron metabolism may be suspected: iron-refractory iron deficiency anemia has already been discussed as a disorder with inappropriately high hepcidin production caused by inactivating mutation of the TMPRSS6 gene; three patients with autosomal recessive inheritance of mutations in DMT1 had an iron deficiency anemia but this was associated with increased transferrin saturation and hepatic iron loading.104
Development and pathological effects of iron deficiency
The development of iron deficiency can be considered in three stages105 corresponding to the sequential involvement of storage iron, iron supply to the tissues, and the functional compartment of hemoglobin iron.
Exhaustion of iron stores
The first response to a negative iron balance is the mobilization of any iron stores from macrophages and hepatocytes, and an up-regulation of iron absorption. Assessment of iron stores will show declining values (Table 11.1), but there is no good evidence that depletion of iron stores has any harmful effects apart from reducing the ability to respond to increased demands for iron whether these are physiologic (e.g. pregnancy) or pathologic (e.g. hemorrhage). Erythropoiesis remains unaffected at this stage.
Iron-deficient erythropoiesis
A continued negative iron balance after iron stores are exhausted, leads to a decline in serum iron concentration and transferrin saturation to below the value of 16% found necessary to support normal erythropoieses.14 The reduced iron supply to the erythron leads to up-regulation of transferrin receptors on the erythroblasts, with a rise in serum transferrin receptor concentration (Table 11.1). Other measures also begin to reflect the impaired iron supply, with increase in red cell protoporphyrin, and detection of poorly hemoglobinized reticulocytes and hypochromic red cells.82,106,107 At this stage the hemoglobin concentration, mean red cell volume (MCV) and mean corpuscular hemoglobin (MCH) may still be in the reference range, though the blood film may show occasional hypochromic red cells.
Iron deficiency anemia
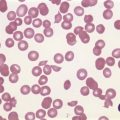
Further depletion of body iron leads to the development of iron deficiency anemia. The hemoglobin concentration drops below the threshold for definition of anemia (13.0 g/dl in men and 12.0 g/dl in women), and the red cell MCV and MCH are reduced. On the blood film, the red cells become more obviously hypochromic and variable in size, and poikilocytosis may be marked often with elongated ‘pencil’ forms (Fig. 11.7). Target cells may be visible. Reticulocytes are not increased appropriately for the degree of anemia, though serum erythropoietin concentrations are markedly raised. Platelet counts are usually increased, but there are case reports of associated thrombocytopenia.108 The serum transferrin saturation is likely to be very low: this effect is exacerbated by a rising serum transferrin concentration – transcriptional regulation of transferrin synthesis by the liver is inhibited by iron excess and stimulated by its absence.109
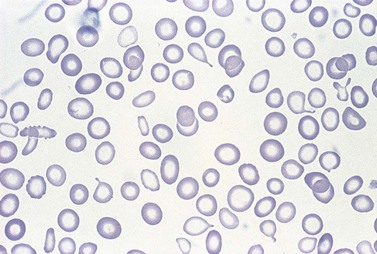
A bone marrow examination is rarely needed to confirm the diagnosis. Marrow sideroblasts disappear early in the development of iron deficient erythropoiesis.14 Erythroblasts show delayed hemoglobinization with a ragged, vacuolated cytoplasm, and relatively pyknotic nucleus for the stage of hemoglobinization. The white cell series is usually normal. Absence of stainable marrow iron stores in a randomly selected population of 38-year-old women could be predicted by a serum ferritin concentration of <16 µg/l (specificity 98%; sensitivity 75%).110
Mechanism of iron deficiency anemia
Impaired heme synthesis within individual erythroid precursors clearly accounts for many of the marrow and peripheral blood findings, but not why the anemia is hypoproliferative, with an inappropriately low reticulocyte count despite raised erythropoietin concentrations. Studies in iron deficient rats111 suggest that iron deficiency produces a maturation defect between CFU-E and early normoblasts.
Non-hematological effects of iron deficiency
The epithelial abnormalities described above are poorly correlated with tissue enzyme levels112 and may respond slowly or not at all to iron therapy. Gastric atrophy and esophageal webs are also associated with circulating parietal cell antibodies, achlorhydria and an increased risk of pernicious anemia,113 suggesting additional predisposing genetic factors.
Iron deficiency may impair immune function, with reduced T-cell and neutrophil function, while there are inconsistent data on whether iron supplements may increase the severity of some infections (e.g. malaria).31,114 It remains uncertain whether iron has any significant effects in protecting against or potentiating the risk of infection. However, the links between immunity and iron metabolism have been re-emphasized by the discovery of the central role of hepcidin which down-regulates iron supply in response to infection and inflammation.
Work performance, aerobic capacity and endurance capacity is impaired in the presence of iron deficiency.115 The effects can be attributed to anemia, though additional effects of tissue iron depletion on muscle oxidative metabolism and function are possible.
Iron deficiency anemia in children has been associated with impaired cognitive, motor and behavioral development, but separation from confounding socioeconomic disadvantage has been difficult.116 Iron is taken up from transferrin by brain capillary endothelial cells, transferred by less certain mechanisms to the different brain cells.117 It accumulates throughout childhood reaching highest concentrations in the basal ganglia. Since iron deficiency is common in pre-school children even in developed nations, there remains a need to determine whether it causes any psychomotor impairment and whether this is reversible.
Diagnosis of iron deficiency
The identification of a microcytic hypochromic anemia in a patient with a good reason for iron deficiency (e.g. menorrhagia) may require no further immediate investigation, diagnosis being confirmed by the correction of the hematological abnormalities with iron therapy. Lack of response to iron therapy is most likely to be due to a failure to take the treatment, or to continued occult blood loss. The diagnosis should be reviewed to make sure that other causes of microcytic anemia (e.g. defects of globin chain synthesis, severe and chronic underlying inflammatory disease or sideroblastic anemia) have been excluded: low serum ferritin combined with anemia will confirm the presence of depleted total body iron. However, because the serum ferritin is an acute phase protein, it may be within the reference range in the presence of an inflammatory disorder, even when there is coexistent depletion of iron stores (see discussion of the diagnosis of anemia of chronic disorders). Mild iron deficiency anemia may still have a red cell MCV within the reference range, and here the main differential diagnosis is with the anemia of chronic disease.
Functional iron deficiency
Pathophysiology
A functional iron deficiency develops when the demands of the erythron for iron outstrip the ability to deliver iron to the marrow. For example, this balance is disturbed in thalassemia intermedia syndromes, when blood supply to the expanded marrow may be insufficient, even with increased plasma transferrin saturation, to satisfy the demands of the erythoblasts.118 When erythropoietin therapy for the anemia of chronic renal disease was introduced, it was noted that even in patients with adequate or increased amounts of storage iron from previous red cell transfusions, the stimulation of erythropoiesis was typically accompanied by a drop in transferrin saturation119 and the appearance of hypochromic red cells.107 The impaired iron supply limited the hemoglobin response to erythropoietin, an effect which could be corrected by iron therapy.
Treatment
During erythropoietin therapy for the anemia of renal disease, the emphasis has moved away from avoiding iron therapy and its associated risk of iatrogenic iron overload.120 The aim now is to maintain the iron supply (assessed by transferrin saturation or the percentage of hypochromic red cells), and thus the response to erythropoietin, through regular small doses of intravenous iron (e.g. 100 mg iron sucrose each month), provided the serum ferritin does not exceed 500–1000 µg/l.121
Anemia of chronic disorders
A normochromic normocytic anemia with an inappropriately low reticulocyte response develops in patients with a variety of inflammatory disorders provided these last more than a few days. Where the inflammatory stimulus is prolonged the anemia may become more microcytic. The severity of the hypoproliferative anemia is proportional to that of the underlying disorder, which may be infection, malignancy or an autoimmune disease such as rheumatoid arthritis. Inflammatory stimulation of the synthesis of acute phase proteins is reflected in an increase in plasma viscosity or erythrocyte sedimentation rate, and may be obvious on a blood film by the presence of increased formation of red cell rouleaux. The pathogenesis of the anemia is multifactorial: it involves the activation of cellular immunity and the production of a range of inflammatory cytokines by monocytes/macrophages, T lymphocytes and hepatocytes,122 and is accompanied by an alteration in the handling of iron by macrophages (and other iron-donating cells) that is mediated by hepcidin and results in characteristic changes in measures of iron status.
Pathogenesis
Impaired production of erythropoietin
Patients with the anemia of chronic disorders generally have lower serum concentrations of erythropoietin compared with patients with other causes for anemia.123,124 However, this is not a universal finding, and patients with juvenile chronic arthritis may retain a normal erythopoietin drive to the marrow:125 this may account for the more profound red cell microcytosis often encountered in these patients, in whom limitations on the iron supply to the erythron appear to play a more dominant role. Pro-inflammatory cytokines interleukin-1 (IL1) and tumor necrosis factor-alpha (TNFα) are the likely mediators of reduced erythropoietin production by the kidney in response to anemic hypoxia.122
Inhibition of erythropoiesis
The inflammatory cytokines IL1 and TNFα also inhibit the growth of erythroid progenitors,122 particularly CFU-E. These cytokine effects, mediated by promotion of apoptosis,126 require the presence of marrow stromal cells (mainly macrophages), which are stimulated by IL1 to release interferon-γ (IFNγ), and by TNFα to release IFNβ.127,128 The suppressive effect of IL1 and IFNγ could be reversed by high doses of exogenous erythropoietin, but the effects of TNFα may be more resistant to erythropoietin. Hepcidin has now also been found to inhibit erythroid colony growth.129 The effects of reduced erythropoietin production and inhibition of eythropoiesis are thus likely to be the main cause of the hypoproliferative nature of the anemia in inflammatory disorders.
Decreased red cell survival
Activation of macrophages and enhanced erythophagocytosis is responsible for a modest reduction in red cell survival in inflammation.130 Although the reduction in red cell survival would not, on its own, be sufficient to produce significant anemia, it compounds the impaired erythroid response (see above) mediated via inflammatory cytokines.
Reduction in iron supply to the erythroid marrow
Inflammation, and the anemia of chronic disorders, is associated with reduced serum iron concentrations and retention of iron within macrophages, as the result of increased hepcidin production induced by interleukin-6 (IL6).65,73 In addition, the inflammatory cytokines IL1 and TNFα stimulate ferritin synthesis within macrophages and hepatocytes,131,132 through an effect on transcription and translation of the mRNA which is independent of the IRP/IRE regulatory mechanism. Ferritin thus behaves as an acute phase protein providing a repository for the iron ‘blocked’ within macrophages. The plasma concentration of transferrin also tends to be reduced, and the reduction of transferrin saturation is thus often less severe than in iron deficiency anemias. The end result of these changes is the redistribution of iron from circulating red cells to macrophage iron stores (Fig. 11.8). Despite increased marrow iron staining and raised serum ferritin values, there is a modestly reduced iron supply to the erythroblasts. The finding that serum concentrations of transferrin receptor are not generally increased in the anemia of chronic disorders (unlike iron deficiency)85 may relate to cytokine-induced inhibition of receptor synthesis. Alternatively, cytokine-induced inhibition of erythropoiesis may lead to a balanced decrease in both erythropoiesis and iron supply, so that the iron requirements of individual erythroblasts continue to be met and there is no activation of transferrin receptor synthesis through the IRP/IRE regulatory mechanism. In patients with inflammatory disorders who develop more severe red cell microcytosis there may be either more severe impairment of iron supply to the erythron, or the suppression of erythropoiesis may be less marked. Both these factors appear to play a part in the microcytic anemia of juvenile chronic arthritis where abnormal cytokine production is dominated by an increase in IL6,125 which stimulates hepcidin production but has little inhibitory effect on erythropoietin production or the response of the erythron. This suggests that differences in the amount and pattern of cytokine expression between different inflammatory disorders or between individual patients may account for variation in the effects on iron metabolism and erythropoiesis, and the corresponding degree of anemia and red cell microcytosis.
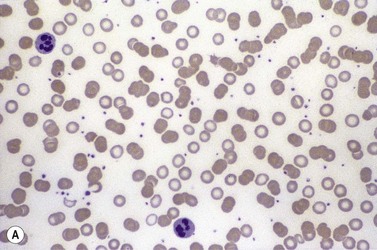
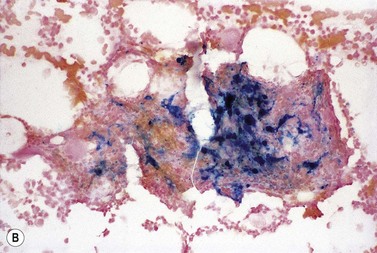
Diagnosis
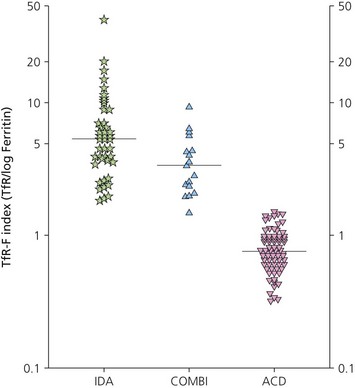
The finding of a mild to moderate normochromic anemia in association with an obvious underlying disorder usually makes the clinical diagnosis of the anemia of chronic disorders, and the pattern of disturbance of the measures of iron status usually helps to confirm the diagnosis. Difficulty may arise when there is a possibility of iron deficiency coexisting with the anemia of chronic disorders. Here the inflammatory increase in ferritin synthesis by macrophages and hepatocytes (see above) can lead to serum ferritin values which are within the normal reference range, even where iron stores are absent. Interpretation must take into account that values up to 70 µg/l may still be associated with absent iron stores where there is an acute phase response.133 Furthermore these values must be considered in relation to the accompanying anemia, since there may still be inadequate iron stores to permit full regeneration of the hemoglobin once the inflammatory stimulus is removed. An increase in serum transferrin receptor concentration considered on its own may not reliably indicate a coincidental absolute iron deficiency where erythropoiesis is maintained despite a hepcidin-induced reduction in iron supply to the erythron, e.g. in juvenile chronic arthritis.125 Under these circumstances the transferrin receptor/log ferritin ratio may yield a better distinction between anemia of chronic disorders alone and combined with iron deficiency (Fig. 11.9).92,134 However, bone marrow examination, and Perls’ stain for hemosiderin iron, may still be the simplest way to be certain whether there are iron stores present.
Treatment
The main approach is to treat the underlying inflammatory disorder: in many cases it is the underlying disease that gives rise to the main symptoms rather than the accompanying, generally modest, anemia. It may be possible to target one or more of the inflammatory cytokines: for example in rheumatoid arthritis an antibody to TNFα (infliximab) reversed excessive apoptosis among marrow erythroid progenitors and partially corrected the anemia.135
Where the underlying disease is intractable and the anemia is impairing quality of life (e.g. in association with cancer, where patients undergoing chemotherapy are especially at risk), red cell transfusion may help. Erythropoietic stimulating agents (ESAs) have been trialled in an attempt to obtain a more sustained rise in hemoglobin and avoid the need for, and potential complications of, transfusion. However, the outcomes of these trials have been disappointing: significant improvement in quality of life has not been shown consistently and there are concerns about increased venous thromboembolic disease and mortality in ESA-treated patients with a range of solid tumors.136 Erythropoietin-induced down-regulation of hepcidin synthesis76,77 might also be expected to improve iron supply to the erythron, but a hemoglobin rise is achieved more quickly if parenteral iron is given with the ESA.137 Intravenous iron supplementation alone is likely to have limited benefit as it bypasses only one of the several restrictions on erythropoiesis in inflammatory disease, i.e. the hepcidin-induced limitation on iron supply. The possibility that iron chelators might be used to treat the anemia by ‘shuttling’ iron out of macrophage iron stores has been raised,138 but has not been taken further.
Iron overload
Iron overload may be primary, resulting from a genetically determined increase in iron absorption, or secondary to a disturbance of erythropoiesis or other diseases (Table 11.3). The distinction is not always clear-cut. For example, coinheritance of the C282Y mutation of the HFE gene (see below) may contribute to iron loading in diseases such as porphyria cutanea tarda,139 and an unidentified genetic component may be required to allow the dietary iron loading in sub-Saharan iron overload.140 However, the severe secondary iron overload that accompanies some genetic or acquired anemias is either the result of regular transfusion of red cells or a marked increase in iron absorption mediated by ineffective erythropoiesis: an additional contribution from a hemochromatosis gene is not required.
| Condition | Features of iron overload | Prevalence of iron overload |
| Primary iron overload | ||
| Disordered hepcidin/ferroportin regulatory axis with parenchymal iron loading | ||
| • Type 1 hemochromatosis (HFE-related) | Autosomal recessive. Adult onset. HFE mutations. Hepcidin inappropriately low. |
Common in those of northern European origin. |
| • Type 2 hemochromatosis (juvenile) | Autosomal recessive. Presentation in 2nd or 3rd decade of life. Type 2a HJV mutations. Hepcidin low. Type 2b HAMP mutations. Hepcidin absent. |
Rare |
| • Type 3 hemochromatosis | Autosomal recessive. Adult onset. TFR2 mutations. Hepcidin low. |
Rare – small proportion of adult hemochromatosis. |
| • Type 4 hemochromatosis (ferroportin ‘gain of function’) | Autosomal dominant SLC40A1 ‘gain of function’ mutations Hepcidin insensitivity – levels high |
Rare |
| Impaired cellular iron export with macrophage and parenchymal iron loading | ||
| Type 4 ‘hemochromatosis’ (ferroportin disease) | Autosomal dominant. SLC40A1 ‘loss of function’ mutations. Mainly macrophage iron accumulation with low risk of damage. Hepcidin high. |
Uncommon |
| Aceruloplasminemia | Autosomal recessive. CP mutations with loss of plasma ferroxidase. Brain and liver iron accumulation and damage. |
Rare |
| Impaired iron utilization by the erythron with parenchymal iron loading | ||
| • Congenital atransferrinemia | Autosomal recessive. TF mutations. Microcytic anemia with hepatocyte iron loading. |
Very rare |
| • DMT1 mutations | Autosomal recessive SLC11A2 mutations. Microcytic anemia with hepatocyte iron loading. |
Very rare |
| Neonatal hemochromatosis | Fatal liver damage with iron loading. Mechanisms unknown. Possible autosomal recessive component. | Rare |
| Secondary iron overload | ||
| Iron-loading anemias | ||
| • Massive ineffective erythropoiesis (severe β-thalassemia syndromes, sideroblastic anemias, congenital dyserythropoietic anemias) | Increased iron absorption and/or blood transfusion. Hepcidin low. | Common, as a result of thalassemia, in those of Mediterranean, Middle-Eastern, and Asian origin. Other causes rare. |
| • Refractory hypoplastic anemias (e.g. chronic renal failure, pure red cell aplasia, aplastic and myelodysplastic syndromes) | Blood transfusion. Hepcidin variable. | Relatively common where adequate transfusion services available. |
| • Severe chronic hemolytic anemias (e.g. pyruvate kinase deficiency, congenital spherocytosis, sickle cell disease) | Mainly blood transfusion. | Rare as causes of severe iron overload. |
| Sub-Saharan dietary iron overload | Increase in both dietary iron and iron absorption. Macrophage and parenchymal iron loading. | Common in sub-Saharan Africa |
| Causes of modest iron overload | ||
| • Chronic liver disease (alcoholic cirrhosis, portocaval anastamosis) | Increased iron absorption. | Common |
| • Metabolic syndrome | Mainly macrophage iron loading. | Relatively common |
| • Porphyria cutanea tarda | Increased iron absorption. Increased frequency of HFE mutations. | Relatively uncommon |
| Local iron overload | ||
| • Lung (idiopathic pulmonary hemosiderosis) | Pulmonary hemorrhage. | Rare |
| • Renal (e.g. paroxysmal nocturnal hemoglobinuria) | Hemoglobinuria with renal tubular hemosiderosis. | Rare |
Iron overload disorders – definitions and pathophysiology
Severe iron overload (Table 11.3), whether primary (due to an inherited disorder of iron metabolism) or secondary (most commonly to disorders with expanded ineffective erythropoiesis), is arbitrarily defined as greater than 5 g excess iron.141 The phenotype of hemochromatosis is the combination of such iron overload with iron-induced tissue damage. Hereditary hemochromatosis now encompasses several genotypes that result in excessive iron absorption with eventual parenchymal iron overload.142 HFE mutations are common in populations of northern European origin,143 while the other mutations are rare (Table 11.3). These genetic changes all involve disruption of the hepcidin/ferroportin axis (Fig. 11.5): mutations of HAMP directly prevent hepcidin production; mutations of HFE, TFR2 and HJV disrupt the signaling pathway that up-regulates hepcidin production in response to iron (Fig. 11.6), and ‘gain of function’ mutations of ferroportin are insensitive to hepcidin.
Primary iron overload
HFE-related (Type 1) hereditary hemochromatosis
Genetics. The disorder is inherited as an autosomal recessive: the HFE gene is located on the short arm of chromosome 6 and codes for an HLA-class-1-like protein.144 In populations of northern European origin nearly all patients with hereditary hemochromatosis are homozygous for a point mutation responsible for a cysteine to tyrosine substitution at amino acid 282 (C282Y). Around 9% of people in the UK carry the C282Y mutation, but this becomes less frequent on moving south through Europe, being only 0.5% in Italy.145 Mild increases in body iron are seen in less than a quarter of heterozygotes, but they are not at risk of clinically important iron overload.146 A second mutation, substituting a histidine with aspartic acid at amino acid 63 (H63D), is more common in the general population and has a less defined role in predisposing towards iron loading: 4% of patients with hemochromatosis were found to be compound heterozygotes (C282Y/H63D), though most individuals with this genotype are not iron loaded.
Clinical features. A sustained positive iron balance leads to progressive accumulation of iron, and presentation, usually in the 4th or 5th decade, with evidence of iron-induced organ damage. A variety of clinical presentations mean that a high degree of clinical suspicion is necessary. Weakness and fatigue are prominent, while arthralgia, and impotence in males are common.147,148 Arthritis particularly affects the second and third metacarpal-phalangeal joints, and a destructive arthropathy of the hip and knee joints may also occur. Late onset diabetes, abnormal liver function tests or skin pigmentation may all trigger suspicion. In younger patients cardiac failure and arrhythmias are more common at presentation, perhaps the result of more rapid accumulation of iron. Abdominal pain may result from hepatic enlargement or hepatocellular carcinoma.
Diagnosis. An increase in transferrin saturation is the first biochemical abnormality in the development of iron overload, and a fasting saturation of greater than 55% (men and post-menopausal women) or 50% (pre-menopausal women) suggests a risk of increased parenchymal iron uptake: a parallel measurement of serum ferritin allows an estimate of the current accumulation of excess iron stores. Genetic testing, initially for the common mutations of HFE, allows consideration of liver biopsy to be restricted to: 1) those patients with a raised ferritin as well as transferrin saturation but who lack pathogenic mutations (where the question is whether there is an alternative explanation, e.g. hepatitis C, alcoholic or fatty liver); 2) patients with homozygous C282Y or C282Y/H63D, who have a serum ferritin >1000 µg/l, or abnormal liver function tests (where the question is whether fibrosis has progressed to cirrhosis, with the need to monitor for potential complications of the latter). Where patients have raised fasting transferrin saturation, but no evidence of increased iron stores (i.e. normal serum ferritin), the measures of iron status should be repeated at annual intervals: regular phlebotomy can then be started if the serum ferritin becomes elevated. Patients with pathogenic mutations and a raised serum ferritin, but to a level of <1000 µg/l and with no evidence of liver damage, do not require a diagnostic liver biopsy.149 Quantitative phlebotomy – estimating the total amount of iron removed (at approximately 200 mg/unit of blood) before iron stores are exhausted, provides both treatment and confirmation of the diagnosis of iron overload.
Patients diagnosed and treated before cirrhosis has developed have a normal life expectancy.147 Phlebotomy at a rate of 450 ml/week is usually well tolerated, and continuation until iron stores are exhausted (serum ferritin <20 µg/l) allows calculation of the total iron load. Weekly phlebotomy may need to be continued for many months or years, since the excess iron is usually greater than 10 g in those with established tissue damage. The rate of phlebotomy may need to be reduced if the hemoglobin drops below 12 g/l. Thereafter current practice is to carry out phlebotomy at less frequent intervals (usually 2–6 times each year) to maintain the serum ferritin below 50 µg/l. However, this target is arbitrary, and some relaxation may allow the frequency of phlebotomy to be further reduced: iron absorption is still regulated by iron stores in hemochromatosis, albeit at a higher level, and maintenance of very low iron stores will increase the rate at which iron re-accumulates.150
Population screening for HFE mutations remains controversial, since although there are clear benefits in early identification of those at risk, many of the approximately 1 in 200 people of northern European origin who have a hemochromatosis genotype will never develop the phenotype of severe iron loading and symptomatic disease151,152 In the clinic it is currently difficult to predict which C282Y homozygote patients have already reached a stable relatively low level of iron accumulation and do not require phlebotomy treatment,153 though elderly patients with serum ferritin values well below 1000 µg/l are most unlikely to progress. Phenotypic expression of the disease is likely to be dependent on an interaction with other genetic modifiers (e.g. cis and trans-acting polymorphisms that may impact on hepcidin expression154) and with physiological and environmental factors (e.g. symptomatic iron overload is less frequent in women before the menopause, while a high alcohol intake, enhancing the bioavailability of dietary iron,155 is common in men with symptomatic disease).
Non HFE-related hereditary hemochromatosis
Type 2 hemochromatosis (juvenile hemochromatosis) is a more severe form of iron overload which presents before the age of 30 years with cardiomyopathy and gonadal failure.156 The majority of cases (type 2a) are due to autosomal recessive inheritance of mutations of the gene coding for hemojuvelin (HJV), but in a minority (type 2b) it is the hepcidin gene itself (HAMP) that is mutated.157
Type 3 hemochromatosis has a similar clinical picture to HFE-related hemochromatosis. It results from autosomal recessive inheritance of mutations of the gene coding for transferrin receptor 2 (TFR2).157
Type 4 hemochromatosis arises from one of two broad types of mutation of the ferroportin gene, both inherited as autosomal dominants.11 Mutations that prevent ferroportin regulation by hepcidin give rise to ‘gain of function’ and the typical clinical picture of hemochromatosis with parenchymal iron loading. By contrast, ‘loss of function’ mutations give rise to ‘ferroportin disease’, characterized by iron accumulation in macrophages, a high serum ferritin, and a low tolerance of phlebotomy. It is thus mainly a disorder of iron maldistribution rather than typical hemochromatosis.
Iron loading in other inherited disorders of iron metabolism
Hereditary atransferrinemia is a very rare disorder characterized by severe microcytic anemia and parenchymal liver iron overload.158 It illustrates the essential role for transferrin in delivering iron to the erythron, and the lack of dependence on transferrin for iron release to plasma and uptake by the liver. Increased iron absorption may reflect the absence of diferric transferrin from the signaling pathway that up-regulates hepcidin production in response to iron (Fig. 11.6).
Divalent metal transporter-1 (DMT1) defects, also exceedingly rare, give rise to a microcytic hypochromic anemia consistent with the role of the transporter in uptake of transferrin-bound iron by the erythron (Fig. 11.2). Accompanying hepatic iron overload implies an excessive iron absorption by a route that bypasses DMT1 for intestinal mucosal uptake.104
Aceruloplasminemia is characterized by iron accumulation in liver, spleen and brain with neurodegeneration.158,159 Hepatic iron loading resistant to phlebotomy43 and reduced serum iron with microcytic hypochromic anemia159 are consistent with the ferroxidase role of ceruloplasmin in promoting cellular iron efflux via ferroportin.
Neonatal hemochromatosis
This is a rare severe form of hemochromatosis, characterized by perinatal liver failure, widespread tissue iron loading and high mortality.160 In some cases it may be related to maternal infection, but others show features consistent with autosomal recessive inheritance of unknown basis.
Secondary iron overload
Parenteral iron loading
In transfusion-dependent anemias, each unit of red cells delivers approximately 200 mg iron. In congenital anemias such as β-thalassemia major, this can add up to 100 g iron by the end of the second decade of life, by which time most patients will have died from the toxic effects of the excess iron.161 These include cardiac arrhythmias and heart failure, diabetes and failure of puberty, and cirrhosis. The rate of iron loading through regular transfusions is considerably greater than the maximum possible through increased iron absorption, and pathologic changes may therefore occur somewhat earlier than where the latter route predominates.162
Increased dietary iron intake
Increased oral intake of iron does not normally result in significant iron loading.163 In sub-Saharan iron overload a combination of unusually high dietary iron bioavailability with an underlying (unknown) genetic defect is required to produce the disorder.140 A ferroportin polymorphism common in African people is associated with higher serum ferritin concentrations, but there is no increased prevalence in affected families.164
Increased iron absorption
In patients with iron loading anemias associated with massive erythroid expansion (e.g. the β-thalassemia intermedia syndromes,47 congenital dyserythropoietic anemias165) the rate of iron loading is directly related to the degree of erythroid expansion (Fig. 11.10).162 For a given degree of erythroid expansion increased absorption is more pronounced with dyserythropoiesis than hemolysis.48 However, even in HbH disease, where the defect is primarily hemolytic, iron overload from excessive iron absorption can be a problem.166 The risk of iron loading via the gastrointestinal route may be concealed in some patients in whom a mild and asymptomatic anemia coexists with a marked expansion of dyserythropoietic marrow. Such patients may be at particular risk of misguided oral iron therapy,167 and liver biopsy may be needed to determine the true extent of iron loading.
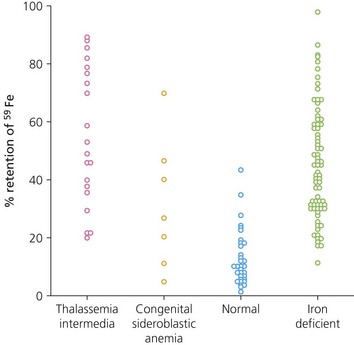
Fig. 11.10 Iron absorption from a test dose of 5 mg 59Fe ferrous sulphate measured by whole body counting. Patients with massive ineffective erythropoiesis had iron retention similar to that of patients with iron deficiency, despite the fact that they had already accumulated substantial amounts of excess iron.
(From Pippard MJ, Weatherall DJ. Iron absorption in non-transfused iron loading anaemias: prediction of risk for iron loading, and response to iron chelation treatment, in thalassaemia intermedia and congenital sideroblastic anaemias. Haematologia 1984;17:407–14).
Mild degrees of iron excess are seen in some cases of chronic liver disease, and the metabolic syndrome (associated with diabetes, hypertension, hypertriglyceridemia, and obesity) can be a cause of high serum ferritin, though usually with only modest increase in iron stores and no increase in transferrin saturation.168,90
Pathogenesis of iron-induced damage
Changes of liver fibrosis are seen very early in the development of iron overload in transfusion-dependent thalassemia.169 In the liver, lipid peroxidation by iron-catalyzed hydroxyl radicals170 results in impairment of membrane-dependent functions of mitochondria, including mitochondrial respiration, and damage to lysosomes resulting in their increased fragility. In iron overload, the lysosomal uptake of ferritin results in partial ferritin degradation to insoluble hemosiderin, and this may also increase the lysosomal fragility.8 Pathologic fibrogenesis is mediated by the activation and proliferation of hepatic stellate cells, probably as the result of iron-induced oxidative stress. The degree of damage in various organs is not always clearly correlated with their iron content. In particular, the heart may show functional impairment despite relatively low and patchy iron deposition.
Treatment
Management of secondary iron overload in the iron-loading anemias is mainly dependent on the use of iron chelation therapy. It is the metabolically active labile iron within cells which is both the main catalyst for free radical formation and the major source of chelatable iron.171 Deferoxamine is the most longstanding iron chelating agent in clinical use and has to be given as a continuous infusion on a regular daily basis to produce effective iron removal. Nevertheless, when used conscientiously in this way, maintenance of serum ferritin below 2500 µg/l prevents cardiac complications of iron overload,172 and allows improved pubertal growth and prolonged survival. Its main disadvantage is the need for parenteral administration, and there are now two alternative oral chelators that have had significant clinical exposure, deferiprone and deferasirox.173,174 The sites of action of the chelators may differ and this has provided a rationale for combined therapies where the iron overload is particularly severe.175
1 Dallman PR, Beutler E, Finch CA. Annotation: effects of iron deficiency exclusive of anaemia. British Journal of Haematology. 1978;40:179-184.
2 Aisen P. Iron metabolism: an evolutionary perspective. In: Brock JH, Halliday JW, Pippard MJ, Powell LW, editors. Iron metabolism in health and disease. London: Saunders; 1994:1-30.
3 DeMaeyer E, Adiels-Tegman M. The prevalence of anaemia in the world. Rapport Trimestriel de Statistiques Sanitaires Mondiales. 1985;38:302-316.
4 Cook JD, Finch CA, Smith NJ. Evaluation of the iron status of a population. Blood. 1976;48:449-455.
5 Expert Scientific Working Group. Summary of a report on assessment of the iron nutritional status of the United States population. American Journal of Clinical Nutrition. 1985;42:1318-1330.
6 McCance RA, Widdowson EM. Absorption and excretion of iron. Lancet. 1937;ii:680-684.
7 Esposito BP, Breur W, Sirankapracha P, et al. Labile plasma iron in iron overload: redox activity and susceptibility to chelation. Blood. 2003;102:2670-2677.
8 Selden C, Owen M, Hopkins JM, Peters TJ. Studies on the concentration and intracellular localization of iron proteins in liver biopsy specimens from patients with iron overload with special reference to their role in lysosomal disruption. British Journal of Haematology. 1980;44:593-603.
9 Finch CA, Duebelbeiss K, Cook JD, et al. Ferrokinetics in man. Medicine. 1970;49:17-53.
10 Green R, Charlton R, Seftel H, et al. Body iron excretion in man: a collaborative study. American Journal of Medicine. 1968;45:336-353.
11 Andrews NC. Forging a field: the golden age of iron biology. Blood. 2008;112:219-230.
12 Huebers H, Csiba E, Huebers E, Finch CA. Competitive advantage of diferric transferrin in delivering iron to reticulocytes. Proceedings of the National Academy of Sciences, USA. 1983;80:300-304.
13 Edwards CQ, Kushner JP. Screening for hemochromatosis. New England Journal of Medicine. 1993;328:1616-1620.
14 Bainton DF, Finch CA. The diagnosis of iron deficiency anemia. American Journal of Medicine. 1964;37:62-70.
15 Baker E, Morgan EH. Iron transport. In: Brock JH, Halliday JW, Pippard MJ, Powell LW, editors. Iron metabolism in health and disease. London: Saunders; 1994:63-95.
16 Ohgami RS, Campagna DR, Greer EL, et al. Identification of a ferrireductase required for efficient transferrin-dependent iron uptake in erythroid cells. Nature Genetics. 2005;37:1264-1269.
17 Fleming MD, Romano MA, Su MA, et al. Nramp2 is mutated in the anemic Belgrade (b) rat: evidence of a role for Nramp2 in endosomal iron transport. Proceedings of the National Academy of Sciences, USA. 1998;95:1148-1153.
18 Kuhn LC. Molecular regulation of iron proteins. Baillière’s Clinical Haematology. 1994;7:763-785.
19 Cairo G, Peitrangelo A. Iron regulatory proteins in pathobiology. Biochemistry Journal. 2000;352:241-250.
20 Chen W, Paradkar PN, Li L, et al. Abcb10 physically interacts with mitoferrin-1 (Slc25a37) to enhance its stability and function in the erythroid mitochondria. Proceedings of the National Academy of Sciences, USA. 106, 2009. 16263–16238
21 Shaw GC, Cope JJ, Li L, et al. Mitoferrin is essential for erythroid iron assimilation. Nature. 2006;440:96-100.
22 May A, Bishop DF. The molecular biology and pyridoxine responsiveness of X-linked sideroblastic anaemia. Haematologica. 1998;83:56-70.
23 Bekri S, Kispal G, Lange H, et al. Human ABC7 transporter: gene structure and mutation causing X-linked sideroblastic anemia with ataxia with disruption of cystosolic iron-sulfur protein maturation. Blood. 2000;96:3256-3264.
24 Huang ML, Becker EM, Whitnall M, et al. Elucidation of the mechanism of mitochondrial iron loading in Friedreich’s ataxia by analysis of a mouse mutant. Proceedings of the National Academy of Sciences, USA. 2009;106:16381-16386.
25 Weiss G, Houston T, Kastner S, et al. Regulation of cellular iron metabolism by erythropoietin: activation of iron-regulatory protein and upregulation of transferrin receptor expression in erythroid cells. Blood. 1997;89:680-687.
26 Brittenham GM. The red cell cycle. In: Brock JH, Halliday JW, Pippard MJ, Powell LW, editors. Iron metabolism in health and disease. London: Saunders; 1994:31-62.
27 Kawabata H, Yang R, Hirama T, et al. Molecular cloning of transferrin receptor 2. A new member of the transferrin receptor-like family. Journal of Biological Chemistry. 1999;274:20826-20832.
28 West APJr, Bennett MJ, Sellers VM, et al. Comparison of the interactions of transferrin receptor and transferrin receptor 2 with transferrin and the hereditary hemochromatosis protein HFE. Journal of Biological Chemistry. 2000;275:38135-38138.
29 Fleming RE, Migas MC, Holden CC, et al. Transferrin receptor 2: continued expression in mouse liver in the face of iron overload and in hereditary hemochromatosis. Proceedings of the National Academy of Sciences, USA. 2000;97:2214-2219.
30 Camaschella C, Roetto A, Cali A, et al. The gene TFR2 is mutated in a new type of haemochromatosis mapping to 7q22. Nature Genetics. 2000;25:14-15.
31 Brock JH. Iron in infection, immunity, inflammation and neoplasia. In: Brock JH, Halliday JW, Pippard MJ, Powell LW, editors. Iron metabolism in health and disease. Saunders; 1994:353-389.
32 Raja KB, Simpson RJ, Peters TJ. Investigation of a role for reduction in ferric iron uptake by mouse duodenum. Biochimica et Biophysica Acta. 1992;1135:141-146.
33 McKie AT, Barrow D, Latunde-Dada GO, et al. An iron-regulated ferric reductase associated with the absorption of dietary iron. Science. 2001;291:1755-1759.
34 Gunshin H, Mackenzie B, Berger UV, et al. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature. 1997;388:482-488.
35 Hubert N, Hentze MW. Previously uncharacterised isoforms of divalent metal transporter (DMT)-1: implications for regulation and cellular function. Proceedings of the National Academy of Sciences, USA. 2002;99:12345-12350.
36 Uzel C, Conrad ME. Absorption of heme iron. Seminars in Hematology. 1998;35:27-34.
37 McKie AT, Marciani P, Rolfs A, et al. A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Molecular Cell. 2000;5:299-309.
38 Zoller H, Theurl I, Koch R, et al. Mechanisms of iron mediated regulation of the duodenal iron transporters divalent metal transporter 1 and ferroportin 1. Blood Cells, Molecules, and Diseases. 2002;29:488-497.
39 Frazer DM, Wilkins SJ, Becker EM, et al. A rapid decrease in the expression of DMT1 and Dcytb, but not Ireg1 or hephaestin explains the mucosal block phenomenon of iron absorption. Gut. 2003;52:340-346.
40 Fleming MD. The regulation of hepcidin and its effects on systemic and cellular iron metabolism Hematology. American Society of Hematology Education Program; 2009. p. 151–8
41 Vulpe CD, Kuo YM, Murphy TL, et al. Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nature Genetics. 1999;21:195-199.
42 Harris ZL, Durley AP, Man TK, Gitlin JD. Targeted gene disruption reveals an essential role for ceruloplasmin in cellular iron efflux. Proceedings of the National Academy of Sciences, USA. 1999;96:10812-10817.
43 Hellman NE, Schaefer M, Gehrke S, et al. Hepatic iron overload in aceruloplasminaemia. Gut. 2000;47:858-860.
44 Beutler E, Gelbart T, Lee P, et al. Molecular characterization of a case of atransferrinemia. Blood. 2000;96:4071-4074.
45 Lynch SR, Skikne BS, Cook JD. Food iron absorption in idiopathic hemochromatosis. Blood. 1989;74:2187-2193.
46 Weber J, Were JM, Julius HW, Marx JJ. Decreased iron absorption in patients with active rheumatoid arthritis, with and without iron deficiency. Annals of the Rheumatic Diseases. 1988;47:404-409.
47 Pippard MJ, Callender ST, Warner GT, Weatherall DJ. Iron absorption and loading in beta-thalassaemia intermedia. Lancet. 1979;2:819-821.
48 Pootrakul P, Kitcharoen K, Yansukon, et al. The effect of erythroid hyperplasia on iron balance. Blood. 1988;71:1124-1129.
49 Raja KB, Simpson RJ, Pippard MJ, Peters TJ. In vivo studies on the relationship between intestinal iron (Fe3+) absorption, hypoxia and erythropoiesis in the mouse. British Journal of Haematology. 1988;68:373-378.
50 Skikne BS, Cook JD. Effect of enhanced erythropoiesis on iron absorption. Journal of Laboratory and Clinical Medicine. 1992;120:746-751.
51 Hughes RT, Smith T, Hesp R, et al. Regulation of iron absorption in iron loaded subjects with end stage renal disease: effects of treatment with recombinant human erythropoietin and reduction of iron stores. British Journal of Haematology. 1992;82:445-454.
52 Finch CA. Regulators of iron balance in humans. Blood. 1994;84:1697-1702.
53 Park CH, Valore EV, Waring AJ, Ganz T. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. Journal of Biological Chemistry. 2001;276:7806-7810.
54 Pigeon C, Ilyin G, Courselaud B, et al. A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. Journal of Biological Chemistry. 2001;276:7811-7819.
55 Nicolas G, Bennoun M, Devaux I, et al. Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proceedings of the National Academy of Sciences, USA. 2001;98:8780-8785.
56 Fillet G, Beguin Y, Baldelli L. Model of reticuloendothelial iron metabolism in humans: abnormal behaviour in idiopathic hemochromatosis and in inflammation. Blood. 1989;74:844-851.
57 Nicolas G, Bennoun M, Porteu A, et al. Severe iron deficiency anemia in transgenic mice expressing liver hepcidin. Proceedings of the National Academy of Sciences, USA. 2002;99:4596-4601.
58 Du X, She E, Gelbart T, et al. The serine protease TMPRSS6 is required to sense iron deficiency. Science. 2008;320:1088-1092.
59 Folgueras AR, de Lara FM, Pendas AM, et al. Membrane-bound serine protease matriptase-2 (Tmprss6) is an essential regulator of iron homeostasis. Blood. 2008;112:2539-2545.
60 Finberg KE, Heeney MM, Campagna DR, et al. Mutations in TMPRSS6 cause iron-refractory iron deficiency anemia (IRIDA). Nature Genetics. 2008;40:569-571.
61 Ramsay AJ, Hooper JD, Folgueras AR, et al. Matriptase-2 (TMPRSS6): a proteolytic regulator of iron homeostasis. Haematologica. 2009;94:840-849.
62 Nemeth E, Tuttle MS, Powelson J, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306:2090-2093.
63 De Domenico I, Ward DM, Langelier C, et al. The molecular mechanism of hepcidin-mediated ferroportin down-regulation. Molecular Biology of the Cell. 2007;18:2569-2578.
64 Ramey G, Deschemin J-C, Durel B, et al. Hepcidin targets ferroportin for degradation in hepatocytes. Haematologica. 2009;95:501-504.
65 Nicolas G, Chauvet C, Viatte L, et al. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. Journal of Clinical Investigation. 2002;110:1037-1044.
66 Origa R, Galanello R, Ganz T, et al. Liver iron concentrations and urinary hepcidin in β-thalassemia. Haematologica. 2007;92:583-588.
67 Camaschella C, Silvestri L. New and old players in the hepcidin pathway. Haematologica. 2008;98:1441-1444.
68 Babitt JL, Huang FW, Xia Y, et al. Modulation of bone morphogenetic protein signaling in vivo regulates systemic iron balance. Journal of Clinical Investigation. 2007;117:1933-1999.
69 Andriopoulos B, Corradini E, Xia Y, et al. BMP6 is a key endogenous regulator of hepcidin expression and iron metabolism. Nature Genetics. 2009;41:482-487.
70 Fleming MD. The regulation of hepcidin and its effects on systemic and cellular iron metabolism. Hematology: American Society of Hematology Education Program; 2008. p. 151–8
71 Finberg KE, Whittlesey RL, Fleming MD, Andrews NC. Down-regulation of Bmp/Smad signaling by Tmprss6 is required for maintenance of systemic iron homeostasis. Blood. 2010;115:3817-3826.
72 Silvestri L, Pagani A, Nai A, et al. The serine protease matriptase-2 (TMPRSS6) inhibits hepcidin activation by cleaving membrane hemojuvelin. Cell Metabolism. 2008;8:502-511.
73 Nemeth E, Rivera S Gabayan V, et al. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. Journal of Clinical Investigation. 2004;113:1271-1276.
74 Wrighting DM, Andrews NC. Interleukin-6 induces hepcidin expression through STAT3. Blood. 2006;108:3204-3209.
75 Kattamis A, Papassotiriou I, Palaiologou D, et al. The effects of erythropoietic activity and iron burden on hepcidin expression in patients with thalassemia major. Haematologica. 2006;91:809-812.
76 Ashby DR, Gale DP, Busbridge M, et al. Erythropoietin administration in humans causes a marked and prolonged reduction in circulating hepcidin. Haematologica. 2010;95:505-508.
77 Pinto JP, Ribeiro S, Pontes H, et al. Erythropoietin mediates hepcidin expression in hepatocytes through EPOR signaling and regulation of C/EBPα. Blood. 2008;111(12):5727-5733.
78 Tanno T, Bhanu NV, Oneal PA, et al. High levels of GDF15 in thalassemia suppress expression of the iron regulatory protein hepcidin. Nature Medicine. 2007;13:1096-1101.
79 Tamary H, Shalev H, Perez-Avraham G, et al. Elevated growth differentiation factor 15 expression in patients with congenital dyserythropoietic anemia type 1. Blood. 2008;112:5241-5244.
80 Tanno T, Porayette P, Sripichai O, et al. Identification of TWSG1 as a second novel erythroid regulator of hepcidin expression in murine and human cells. Blood. 2009;114:181-186.
81 Silvestri L, Pagani A, Camaschella C. Furin-mediated release of soluble hemojuvelin: a new link between hypoxia and iron homeostasis. Blood. 2008;111:924-931.
82 Worwood M. Laboratory determination of iron status. In: Brock JH, Halliday JW, Pippard MJ, Powell LW, editors. Iron metabolism in health and disease. London: Saunders; 1994:449-476.
83 Cook JD. The measurement of serum transferrin receptor. American Journal of Medical Sciences. 1999;318:269-276.
84 Cazzola M, Beguin Y. New tools for clinical evaluation of erythron function in man. British Journal of Haematology. 1992;80:278-284.
85 Ferguson BJ, Skikne BS, Simpson KM, et al. Serum transferrin receptor distinguishes the anemia of chronic disease from iron deficiency anemia. Journal of Laboratory and Clinical Medicine. 1992;19:385-390.
86 Worwood M. Serum ferritin. Clinical Science. 1986;70:215-220.
87 De Domenico I, Ward DM, Kaplan J. Specific iron chelators determine the route of ferritin degradation. Blood. 2009;114:4546-4551.
88 Worwood M, Cragg SJ, Jacobs A, et al. Binding of serum ferritin to concanavalin A: patients with homozygous β thalassaemia and transfusional iron overload. British Journal of Haematology. 1980;46:409-416.
89 Cazzola M, Bergamaschi G, Tonon L, et al. Hereditary hyperferritinemia-cataract syndrome: relationship between phenotypes and specific mutations in the iron-responsive element of ferritin light-chain mRNA. Blood. 1997;90:814-821.
90 Camaschella C, Poggiali E. Towards explaining ‘unexplained hyperferritinemia’. Haematologica. 2009;94:307-309.
91 Skikne BS, Flowers CH, Cook JD. Serum transferrin receptor: a quantitative measure of tissue iron deficiency. Blood. 1990;75:1870-1876.
92 Punnonen K, Irjala K, Rajamaki A. Serum transferrin receptor and its ratio to serum ferritin in the diagnosis of iron deficiency. Blood. 1997;89:1052-1057.
93 Pasquale D, Chikkappa G. Bone marrow biopsy imprints (touch preparations) for assessment of iron stores. American Journal of Hematology. 1995;48:201-202.
94 Barron BA, Hoyer JD, Tefferi A. A bone marrow report of absent stainable iron is not diagnostic of iron deficiency. Annals of Hematology. 2001;80:166-169.
95 Angelucci E, Brittenham GM, McLaren CE, et al. Hepatic iron concentration and total body iron stores in thalassemia major. New England Journal of Medicine. 2000;343:327-331.
96 Anderson LJ, Holden S, Davis B, et al. Cardiovascular T2-star (T2*) magnetic resonance for the early diagnosis of myocardial iron overload. European Heart Journal. 2001;22:2171-2179.
97 Kroot JJ, Kemna EH, Bansal SS, et al. Results of the first international round robin for the quantification of urinary and plasma hepcidin assays: need for standardization. Haematologica. 2009;94:1631-1633.
98 Roe MA, Collings R, Dainty JR. Plasma hepcidin concentrations significantly predict interindividual variation in iron absorption in health men. American Journal of Clinical Nutrition. 2009;89:1088-1091.
99 Theurl I, Aigner E, Theurl M, et al. Regulation of iron homeostasis in anemia of chronic disease and iron deficiency anemia: diagnostic and therapeutic implications. Blood. 2009;113:5277-5286.
100 Henderson L, Irving K, Gregory J, et al. The National Diet and Nutrition Survey: adults aged 19 to 64 years. Volume 3: Vitamin and mineral intake and urinary analytes. London: HMSO; 2003.
101 Department of Health. Dietary reference values for food energy and nutrients for the United Kingdom. London: HMSO; 1989.
102 Rockey DC, Cello JP. Evaluation of the gastrointestinal tract in patients with iron-deficiency anemia. New England Journal of Medicine. 1993;329:1691-1695.
103 Hershko C, Hoffbrand AV, Keret D. Role of autoimmune gastritis, Helicobacter pylori and celiac disease in refractory or unexplained iron deficiency anemia. Haematologica. 2005;90:585-595.
104 Iolascon A, De Falco L, Beaumont C. Molecular basis of inherited microcytic anemia due to defects in iron acquisition or heme synthesis. Haematologica. 2009;94:395-408.
105 Charlton RW, Bothwell TH. Definition, prevalence and prevention of iron deficiency. Clinics in Haematology. 1982;11:309-325.
106 Brugnara C. Reticulocyte cellular indices: a new approach in the diagnosis of anemias and monitoring of erythropoietic function. Critical Reviews in Clinical Laboratory Sciences. 2000;37:93-130.
107 MacDougall IC, Cavill I, Hulme B, et al. Detection of functional iron deficiency during erythropoietin treatment: a new approach. British Medical Journal. 1992;304:225-226.
108 Berger M, Brass LF. Severe thrombocytopenia in iron deficiency anemia. American Journal of Hematology. 1987;24:425-428.
109 McKnight GS, Lee DC, Hemmaplardh D, et al. Transferrin gene expression. Effects of nutritional iron deficiency. Journal of Biological Chemistry. 1983;255:144-147.
110 Hallberg L, Bengtsson C, Lapidus, et al. Screening for iron deficiency: an analysis based on bone marrow examinations and serum ferritin determinations in a population sample of women. British Journal of Haematology. 1993;85:787-798.
111 Kimura H, Finch CA, Adamson JW. Hematopoiesis in the rat: quantitation of hematopoietic progenitors and the response to iron deficiency anemia. Journal of Cell Physiology. 1986;126:298-306.
112 Jacobs A. Iron-containing enzymes in the buccal epithelium. Lancet. 1961;ii:1331-1333.
113 Jacobs A, Kilpatrick GS. The Paterson–Kelly syndrome. British Medical Journal. 1964;2:79-82.
114 Prentice AM. Iron metabolism, malaria, and other infections: what is all the fuss about? Journal of Nutrition. 2008;138:2537-2541.
115 Haas JD, Brownlie T. Iron deficiency and reduced work capacity: a critical review of the research to determine a causal relationship. Journal of Nutrition. 2001;131:676S-688S.
116 Grantham-Mcgregor S, Ani C. A review of studies on the effect of iron deficiency on cognitive development in children. Journal of Nutrition. 2001;131:649S-666S.
117 Moos T, Rosengren Nielson T, Skjorringe T, Morgan EH. Iron trafficking inside the brain. Journal of Neurochemistry. 2007;103:1730-1740.
118 Pootrakul P, Wattanasaree J, Anuwatanakulchai M, Wasi P. Increased red blood cell protoporphyrin in thalassemia: a result of relative iron deficiency. American Journal of Clinical Pathology. 1984;82:289-293.
119 Eschbach JW, Egrie JC, Downing MR, et al. Correction of the anemia of end-stage renal disease with recombinant human erythropoietin: results of a combined Phase I and II clinical trial. New England Journal of Medicine. 1987;316:73-78.
120 Gokal R, Millard PR, Weatherall DJ, et al. Iron metabolism in haemodialysis patients. Quarterly Journal of Medicine. 1979;48:369-391.
121 Drueke TB, Barany P, Cazzola M, et al. Management of iron deficiency in renal anemia: guidelines for the optimal therapeutic approach in erythropoietin-treated patients. Clinical Nephrology. 1997;48:1-8.
122 Weiss G, Goodnough LT. Anemia of chronic disease. New England Journal of Medicine. 2005;352:1011-1023.
123 Baer AN, Dessypris EN, Goldwasser E, Krantz SB. Blunted erythropoietin response to anaemia in rheumatoid arthritis. British Journal of Haematology. 1987;66:559-564.
124 Miller CB, Jones RJ, Piantadosi S, et al. Decreased erythropoietin response in patients with the anemia of cancer. New England Journal of Medicine. 1990;322:1689-1692.
125 Cazzola M, Ponchio L, de Benedetti F, et al. Defective iron supply for erythropoiesis and adequate endogenous erythropoietin production in the anemia associated with systemic-onset juvenile chronic arthritis. Blood. 1996;87:4824-4830.
126 Selleri C, Sato T, Anderson S, et al. Interferon-gamma and tumor necrosis factor-alpha suppress both early and late stages of hematopoiesis and induce programmed cell death. Journal of Cell Physiology. 1995;165:538-546.
127 Means RT, Krantz SB. Inhibition of human erythroid colony formation by IFN-gamma can be corrected by human recombinant erythropoietin. Blood. 1991;78:2564-2570.
128 Means RT, Krantz SB. Inhibition of human erythroid colony-forming units by tumor necrosis factor requires beta interferon. Journal of Clinical Investigation. 1993;91:416-419.
129 Dallalio G, Law E, Means RT. Hepcidin inhibits in vitro erythroid colony formation at reduced erythropoietin concentrations. Blood. 2006;107:2702-2704.
130 Dinant HJ, De Maat CEM. Erythropoiesis and mean red cell lifespan in normal subjects and in patients with the anaemia of active rheumatoid arthritis. British Journal of Haematology. 1992;39:437-444.
131 Konijn AM, Carmel N, Levy R, Hershko C. Ferritin synthesis in inflammation II. Mechanism of increased ferritin synthesis. British Journal of Haematology. 1981;49:361-370.
132 Rogers JT, Bridges KR, Durmowicz GP, et al. Translational control during the acute phase response. Ferritin synthesis in response to interleukin-1. Journal of Biological Chemistry. 1990;265:14572-14578.
133 Coenen JLLM, van Dieijen-Visser MP, van Pelt J. Measurements of serum ferritin used to predict concentrations of iron in bone marrow in anemia of chronic disease. Cinical Chemistry. 1991;37:560-563.
134 Punnonen K, Kaipiainen-Seppanen O, Riittinen L, et al. Evaluation of iron status in anemic patients with rheumatoid arthritis using an automated immunoturbidimetric assay for transferrin receptor. Clinical Chemistry and Laboratory Medicine. 2000;38:1297-1300.
135 Papadaki HA, Kritikos HD, Valatas V, et al. Anemia of chronic disease in rheumatoid arthritis is associated with increased apoptosis of bone marrow erythroid cells: improvement following anti-tumor necrosis factor-α antibody therapy. Blood. 2002;100:474-482.
136 Bennett CL, Silver SM, Djulbegovic B, et al. Venous thromboembolism and mortality associated with recombinant erythropoietin and darbepoetin administration for the treatment of cancer-associated anemia. Journal of the American Medical Association. 2008;299:914-924.
137 Adamson JW. The anemia of inflammation/malignancy: mechanisms and management. Hematology: American Society of Hematology Educational Program; 2008. p. 159–65
138 Vreugdenhill G, Swaak AJG, De Jeu-Jaspars C, van Eijk HG. Correlation of iron exchange between the oral iron chelator 1,2-dimethyl-3-hydroxypyrid-4-one(L1) and transferrin and possible antianaemic effects of L1 in rheumatoid arthritis. Annals of the Rheumatic Diseases. 1990;49:956-957.
139 Roberts AG, Whatley SD, Morgan RR, et al. Increased frequency of the haemochromatosis Cys282Tyr mutation in sporadic porphyria cutanea tarda. Lancet. 1997;349:321-323.
140 Gordeuk V, Mukiibi J, Hasstedt SJ, et al. Iron overload in Africa. Interaction between a gene and dietary iron content. New England Journal of Medicine. 1992;326:95-100.
141 Pippard MJ. Secondary iron overload. In: Brock JH, Halliday JW, Pippard MJ, Powell LW, editors. Iron metabolism in health and disease. London: Saunders; 1994:271-309.
142 Pietrangelo A. Hereditary hemochromatosis – a new look at an old disease. New England Journal of Medicine. 2004;350:2383-2397.
143 Adams PC, Reboussin DM, Barton JC. Hemochromatosis and iron-overload screening in a racially diverse population. New England Journal of Medicine. 352, 2005. 1769–1678
144 Feder JN, Gnirke A, Thomas W, et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nature Genetics. 1996;13:399-408.
145 Merryweather-Clarke AT, Pointon JJ, Shearman JD, Robson KJ. Global prevalence of putative haemochromatosis mutations. Journal of Medical Genetics. 1997;34:275-278.
146 Bulaj ZJ, Griffen LM, Jorde LB, et al. Clinical and biochemical abnormalities in people heterozygous for hemochromatosis. New England Journal of Medicine. 1996;335:1799-1805.
147 Niederau C, Fischer R, Purschel A, et al. Long-term survival and causes of death in patients with hereditary hemochromatosis. Gastroenterology. 1991;110:1107-1119.
148 Adams PC, Deugnier Y, Moirand R, Brissot P. The relationship between iron overload, clinical symptoms, and age in 410 patients with genetic hemochromatosis. Hepatology. 1997;25:162-166.
149 Dooley J, Worwood M. Genetic haemochromatosis. Guidelines on diagnosis and therapy compiled on behalf of the British Committee for Standards in Haematology. Abingdon: Darwin Medical Communications; 2000.
150 Piperno A, Girelli, Nemeth E, et al. Blunted hepcidin response to oral iron challenge in HFE-related hemochromatosis. Blood. 2007;110:4096-4100.
151 Jackson HA, Carter K, Darke C, et al. HFE mutations, iron deficiency and overload in 10,500 blood donors. British Journal of Haematology. 2001;114:474-484.
152 Beutler E, Felitti VJ, Koziol JA, et al. Penetrance of 845G –> A (C282Y) HFE hereditary haemochromatosis mutation in the USA. Lancet. 2002;359:211-218.
153 Adams PC, Barton JC. How I treat hemochromatosis. Blood. 2010;116:317-325.
154 Bayele HK, Srai SKS. Genetic variation in hepcidin expression and its implications for phenotypic differences in iron metabolism. Haematologica. 2009;94:1185-1188.
155 Charlton RW, Jacobs P, Seftel H, Bothwell TH. Effect of alcohol on iron absorption. British Medical Journal. 1964;2:1427-1429.
156 Kelly AL, Rhodes DA, Roland JM, et al. Hereditary juvenile haemochromatosis: a genetically heterogenous life-threatening iron-storage disease. Quarterly Journal of Medicine. 1998;91:607-618.
157 Camaschella C. Understanding iron homeostasis through genetic analysis of hemochromatosis and related disorders. Blood. 2005;106:3710-3717.
158 Jeong SY, David S. Glycophosphotidylinositol-anchored ceruloplasmin is required for iron efflux from cells in the central nervous system. Journal of Biological Chemistry. 2003;278:27144-27148.
159 Bosio S, De Gobbi M, Roetto A, et al. Anemia and iron overload due to compound heterozygosity for novel ceruloplasmin mutations. Blood. 2002;100:2246-2248.
160 Kelly AL, Lunt PW, Rodrigues F, et al. Classification and genetic features of neonatal haemochromatosis: a study of 27 affected pedigrees and molecular analysis of genes implicated in iron metabolism. Journal of Medical Genetics. 2001;38:599-610.
161 Modell B. Advances in the use of iron-chelating agents for the treatment of iron overload. Progress in Hematology. 1979;11:267-312.
162 Pippard MJ, Weatherall DJ. Iron absorption in non-transfused iron loading anaemias: prediction of risk for iron loading, and response to iron chelation treatment, in β thalassaemia intermedia and congenital sideroblastic anaemias. Haematologia. 1984;17:407-414.
163 Sayers MH, English G, Finch C. Capacity of the store-regulator in maintaining iron balance. American Journal of Hematology. 1994;47:194-197.
164 McNamara L, Gordeuk VR, MacPhail AP. Ferroportin (Q248H) mutations in African families with dietary iron overload. Journal of Gastroenterology and Hepatology. 2005;20:1855-1858.
165 Wickramasinghe SN, Thein SL, Srichairatanakool S, Porter JB. Determinants of iron status and bilirubin levels in congenital dyserythropoietic anaemia type I. British Journal of Haematology. 1999;107:522-525.
166 Chen FE, Ooi C, Ha SY, et al. Genetic and clinical features of hemoglobin H disease in Chinese patients. New England Journal of Medicine. 2000;343:544-550.
167 Peto TEA, Pippard MJ, Weatherall DJ. Iron overload in mild sideroblastic anaemias. Lancet. 1983;i:375-378.
168 Moirand R, Moertaji AM, Loreal O, et al. A new syndrome of liver iron overload with normal transferrrin saturation. Lancet. 1997;349:95-97.
169 Iancu TC, Neustein HB, Landing BH. The liver in thalassaemia major: ultrastructural observation. In: Iron Metabolism. Ciba Foundation Symposium 51. North Holland: Elsevier; 1977:293-309.
170 McCord JM. Iron, free radicals, and oxidative injury. Seminars in Hematology. 1998;35:5-12.
171 Pippard MJ, Callender ST, Finch CA. Ferrioxamine excretion in iron-loaded man. Blood. 1982;60:288-294.
172 Olivieri NF, Nathan DG, MacMillan JH, et al. Survival in medically treated patients with homozygous α-thalassemia. New England Journal of Medicine. 1994;331:574-578.
173 Maggio A. Light and shadows in the iron chelation treatment of haematological diseases. British Journal of Haematology. 2007;138:407-421.
174 Cappellini MD, Porter JD, El-Beshlawy A, et al. Tailoring iron chelation by iron intake and serum ferritin: the prospective EPIC study of deferasirox in 1744 patients with transfusion-dependent anemias. Haematologica. 2010;95:557-566.
175 Angelucci E, Barosi G, Camaschella C, et al. Italian Society of Hematology practice guidelines for the management of iron overload in thalassemia major and related disorders. Haematologica. 2008;93:741-752.