Chapter 7 Ion Channelopathies
Mechanisms and Genotype-Phenotype Correlations
Recent years have witnessed an explosion of knowledge contributing to the understanding of ion channelopathies associated with inherited cardiac arrhythmia syndromes that are responsible for the sudden death of infants, children, and young adults. These ion channelopathies are the consequences of genetic variations giving rise to primary electrical diseases, including long QT syndrome (LQTS), short QT syndrome (SQTS), and Brugada syndrome (BrS), as well as catecholaminergic ventricular tachycardia (VT) (Table 7-1).1–3 This review focuses on the molecular, genetic, cellular, and ionic mechanisms underlying the arrhythmogenesis associated with these syndromes and the genotype-phenotype correlation.
Brugada Syndrome
Arrhythmogenesis in BrS is believed to be the result of amplification of heterogeneities in the action potential characteristics among the different transmural cell types in the right ventricular (RV) myocardium.4,5 A decrease in sodium (Na+) or calcium (Ca2+) channel current, INa or ICa, or augmentation of any one of a number of outward currents, including rapidly activating delayed rectifier potassium (K+) current (IKr) or transient outward current (Ito), can cause preferential abbreviation of the right ventricular epicardial action potential; this, in turn, leads to the development of spatial dispersion of repolarization and, thus, the substrate and trigger for VT, which is usually polymorphic and less frequently monomorphic.6–12
BrS displays an autosomal dominant mode of inheritance. For many years, the only gene linked to BrS was SCN5A, the gene encoding for the α-subunit of the cardiac Na+ channel gene.6 However, recent evidence has shown that mutations in other genes are linked to the development of BrS.
BrS1, SCN5A
Mutations in SCN5A were the first to be associated with BrS.6 Over 293 mutations in SCN5A have now been linked to the syndrome.13 About three dozen of these have been studied in expression systems and shown to result in loss of function of the Na+ channel because of the following reasons: (1) failure of the sodium channel to express; (2) a shift in the voltage dependence and time dependence of INa activation, inactivation, or reactivation; (3) entry of the Na channel into an intermediate state of inactivation from which it recovers more slowly, or (4) accelerated inactivation of the Na+ channel.14–16 Premature inactivation of the Na+ channel has been observed at physiological temperatures but not at room temperature.17 Because this characteristic of the mutant channel is exaggerated at temperatures above the physiological range, it was suggested that the syndrome may be unmasked and that patients with BrS may be at an increased risk during a febrile state.17
BrS2, GPD1L
Weiss et al described a second locus on chromosome 3, close to but distinct from SCN5A, linked to the syndrome in a large pedigree in which the syndrome is associated with progressive conduction disease, a low sensitivity to procainamide, and a relatively good prognosis. The gene was recently identified as the glycerol-3-phosphate dehydrogenase 1-like (GPD1L) gene, and the mutation was found in this gene.18–20 Interestingly, it was also found that both GPD1L RNA and protein are abundant in the heart. Furthermore, the mutation was present in all affected individuals and absent in more than 500 control subjects. Coexpression studies of mutant GPD1L (A280V) with SCN5A in human embryonic kidney (HEK) cells resulted in a reduction in the magnitude of INa by approximately 50%.19 These studies provided evidence that mutations in GPD1L lead to a reduction in INa and cause BrS.19 Valdivia et al recently demonstrated that mutations in GPD1L related to BrS and sudden infant death syndrome (SIDS) cause a loss of enzymatic function, which results in glycerol-3-phosphate PKC-dependent phosphorylation of SCN5A at serine 1503 (S1503) through a GPD1L-dependent pathway. The direct phosphorylation of S1503 markedly decreases INa. These findings therefore show a function for GPD1L in cellular physiology and a mechanism linking mutations in GPD1L to sudden cardiac arrest. Because the enzymatic step catalyzed by GPD1L depends on nicotinamide adenine dinucleotide (NAD), this GPD1L pathway links the metabolic state of the cell to INa and excitability and may be important more generally in cardiac ischemia and heart failure.21
BrS3 and BrS4, CACNA1c and CACNB2b
The third and fourth genes associated with BrS were recently identified and were shown to encode the α1-subunit (CACNA1c) and the β-subunit (CACNB2b) of the L-type cardiac Ca channel.8 This new clinical entity, which exhibits electrocardiogram (ECG) and arrhythmic manifestations of both BrS and SQTS, was shown to be associated with loss of function mutations in the α1-subunit (CACNA1c) and the β-subunit (CACNB2b) of the L-type cardiac Ca2+ channel.8 Alterations in L-type Ca2+ current have been implicated in the development of BrS both clinically and experimentally.5,8 In both of those studies, the BrS phenotype was the result of a loss in peak ICa. More recently, a study identified a case of BrS in which the disease phenotype was observed as a result of accelerated inactivation of the L-type Ca2+ current without significantly affecting peak current.9 The accelerated inactivation was caused by a mutation in CACNB2b, which encodes the β-subunit of the cardiac L-type Ca2+ current. The carrier of this mutation exhibited ST-segment elevation in only one precordial lead and converted to a more typical BrS phenotype with a procainamide challenge. VT/VF (ventricular fibrillation) was inducible and subsequently detected on interrogation of the implanted implantable cardiac defibrillator (ICD), corroborating the diagnosis of a potentially life-threatening syndrome.
BrS5 and BrS7, SCN1B and SCN3B
Genes that encode cardiac channel β-subunit proteins have long been appealing candidates for the treatment of ion channelopathies such as BrS because of their significant role in modulating channel expression and function.22 The role of β1-subunits has been studied most extensively. Wild-type (WT) β1 coexpression has been reported to have no observable effect on SCN5A function, result in increased Na+ current density with no detectable effects on channel kinetics or voltage-dependence, modulate channel sensitivity to lidocaine blockade with subtle changes in channel kinetics and gating properties, and shift the voltage dependence of steady-state inactivation or alter the rate of recovery from inactivation.23–31 Coexpression of SCN5A with WT β3 results in either (1) increased current density, a depolarizing shift in the voltage-dependence of inactivation, and an increased rate of recovery from inactivation in Xenopus oocytes or (2) a hyperpolarizing shift of inactivation, slowed recovery from inactivation, and reduced late Na+ channel current.30,31
Mutations in SCN1B and SCN3B have recently been identified as the fifth and seventh genes associated with BrS. Mutations in β1-subunits (Navβ1 and Navβ1b) have been shown to be associated with combined BrS and cardiac conduction disease phenotype in humans.32 Another recent study by the authors of this chapter provided evidence that SCN3B is a BrS-susceptible gene.33 An L10P missense mutation in a highly conserved residue was shown to produce a major reduction in INa secondary to both functional and trafficking defects in cardiac Na+ channel expression. These results indicate that a mutation in the extracellular domain can impair trafficking of SCN5A to the membrane. These results suggest that WT β3 plays a role in facilitating SCN5A transport to the plasma membrane, since a mutation in the extracellular domain of β3 is capable of disrupting trafficking of SCN5A to the plasma membrane.33
BrS6, KCNE3
The role of the transient outward K+ current (Ito) is thought to be central to the development of BrS. This hypothesis comes from several lines of evidence. First, since BrS is characterized by ST-segment elevations in the right precordial leads, a more prominent Ito in RV epicardium has been suggested to underlie the much greater prevalence of the Brugada phenotype in males.34 The more prominent Ito causes the end of phase 1 of the RV epicardial action potential to repolarize to more negative potentials in tissue and arterially perfused wedge preparations obtained from male patients; this facilitates the loss of the action potential dome and the development of phase 2 re-entry and polymorphic VT. A link between mutations in genes responsible for the Ito current and the development of BrS was recently reported by Delpón and coworkers. KCNE3 was identified as the seventh gene associated with BrS.11 KCNE3 normally interacts with Kv4.3 to suppress Ito; and a mutation in KCNE3 was shown to result in a gain of function in Ito.11,35 Experimentally, the Brugada phenotype can be produced in arterially perfused wedge preparations by the Ito activator NS5806. This compound has been shown to increase peak Ito amplitude and slow inactivation in isolated cardiomyocytes, which results in a more prominent phase 1 repolarization and loss of the action potential (AP) dome in mid- and epicardial cells.36 The results of the study using the Ito activator are consistent with the clinical observations that an enhancement of Ito can lead to the development of BrS.
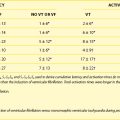
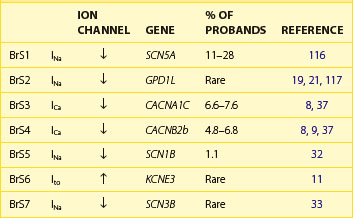
Table 7-2 lists the seven genotypes thus far associated for BrS and their yields. Four of the genes identified produced a loss of function of Na+ channel current; two led to a reduction in Ca2+ channel current; and one gene was associated with a gain of function of transient outward current. SCN5A mutations were identified in 11% to 28% of probands (average of 21%).13 Ca2+ channel mutations are found in approximately 12% to 15% of probands.8,37 Variations in the other genes were relatively rare.
Genotype-Phenotype Correlation
Patients with SCN5A-positive BrS exhibit conduction slowing characterized by prolonged PR intervals and QRS duration.38,39 PR interval and QRS duration are prolonged more prominently, and the QRS axis deviates more to the left with aging in those patients with BrS with SCN5A mutations. Smits et al observed significantly longer conduction intervals at baseline in patients with SCN5A mutations (PR and HV interval) and greater prolongation after the administration of Na+ channel blockers.38 These results concur with the observed loss of function of mutated BrS-related Na+ channels.
Patients with BrS who have calcium channel mutations are phenotypically distinct from those with mutations in other genes.8 A large fraction of CACNA1c– and CACNB2b-positive patients display a shorter-than-normal QTc interval (<360 ms) in addition to an ST-segment elevation in the right precordial leads, thus manifesting a combination of BrS and SQTS. Patients with mutations in Ca2+ channel genes also exhibit a diminished rate adaptation of QT interval.8,40
Mechanism of Arrhythmia in Brugada Syndrome
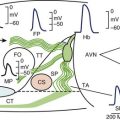
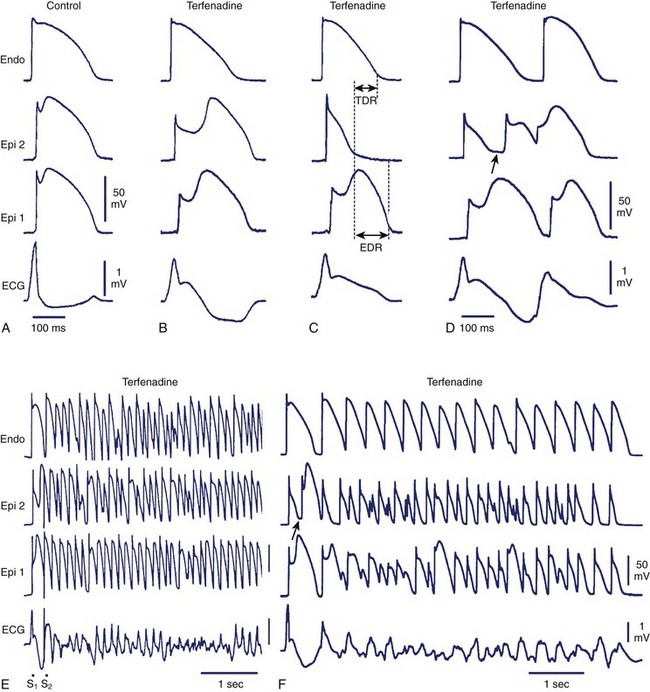
The arrhythmogenic substrate responsible for the development of extrasystoles and polymorphic VT in BrS is believed to be secondary to the amplification of heterogeneities intrinsic to the early phases (phase 1–mediated notch) of the action potential of cells residing in different layers of the right ventricular wall of the heart. Rebalancing of the currents active at the end of phase 1 is thought to underlie the accentuation of the action potential notch in the right ventricular epicardium, which is responsible for the augmented J wave and ST segment elevation associated with BrS (see41–43 for references). The presence of an Ito-mediated spike and dome morphology, or notch, in the ventricular epicardium but not in the endocardium creates a transmural voltage gradient that is responsible for the inscription of the electrocardiographic (ECG) J wave (Figure 7-1, A).5,44 The ST segment is normally isoelectric because of the absence of transmural voltage gradients at the level of the action potential plateau. Accentuation of the right ventricular action potential notch under pathophysiological conditions leads to exaggeration of transmural voltage gradients and thus to accentuation of the J wave or to J point elevation (Figure 7-1, B). If the epicardial action potential continues to repolarize before that of the endocardium, the T wave remains positive, giving rise to a saddleback configuration of the ST-segment elevation. Further accentuation of the notch is accompanied by a prolongation of the epicardial action potential causing it to repolarize after the endocardium, thus leading to inversion of the T wave. The down-sloping ST segment elevation, or accentuated J wave, observed in experimental wedge models often appears as an R, mimicking a right bundle branch block (RBBB) morphology of the ECG, largely because of early repolarization of the right ventricular (RV) epicardium, rather than major delays in impulse conduction in the right bundle.45 Despite the appearance of a typical Brugada sign, the electrophysiological changes shown in Figure 7-1, B, do not give rise to an arrhythmogenic substrate. The arrhythmogenic substrate may develop with a further shift in the balance of current leading to loss of the action potential dome at some epicardial sites but not others (Figure 7-1, C). A marked transmural dispersion of repolarization develops as a consequence, creating a vulnerable window, which can trigger a re-entrant arrhythmia when captured by a premature extrasystole. Because loss of the action potential dome in epicardium is generally heterogeneous, epicardial dispersion of repolarization develops as well. Conduction of the action potential dome from sites at which it is maintained to sites at which it is lost causes local re-excitation via phase 2 re-entry (Figure 7-1, D); this leads to the development of a closely coupled extrasystole that is capable of capturing the vulnerable window across the ventricular wall, thus triggering a circus movement re-entry in the form of VT/VF (Figures. 7-1, E and F).4,46 Support for these hypotheses comes from experiments involving the arterially perfused RV wedge preparations and from recent studies in which monophasic action potential (MAP) electrodes were positioned on the epicardial and endocardial surfaces of the right ventricular outflow tract (RVOT) in patients with BrS.4,5,41,47–51
Long QT Syndrome
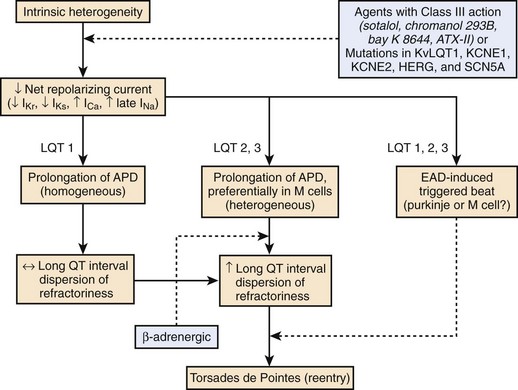
LQTS is characterized by the appearance of long QT intervals on the ECG, an atypical polymorphic VT known as torsades de pointes (TdP), and a high risk for sudden cardiac death.52–54 A reduction of net repolarizing current secondary to loss of function of outward ion channel currents or a gain of function of inward currents underlies the prolongation of the myocardial action potential and QT interval that attend both congenital and acquired LQTS.55,56
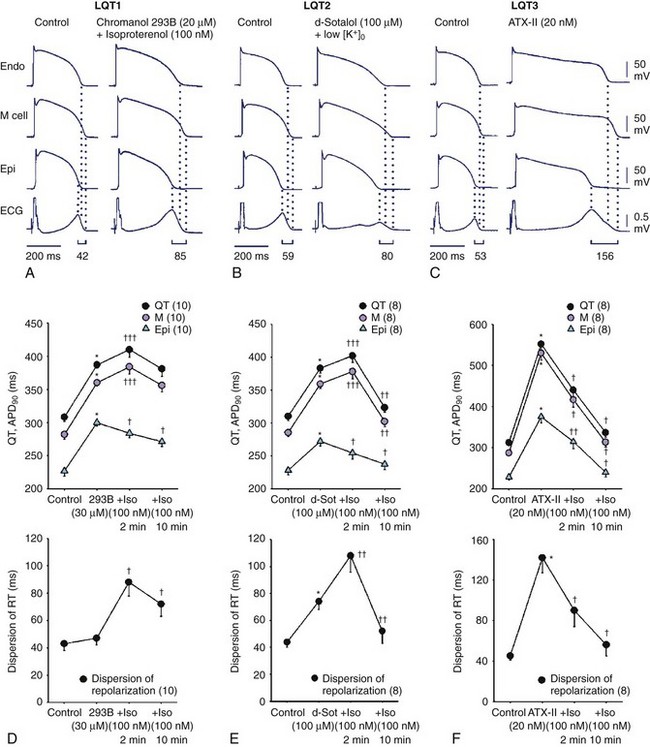
Here again, amplification of spatial dispersion of repolarization is thought to generate the principal arrhythmogenic substrate. The accentuation of spatial dispersion is secondary to an increase of transmural and trans-septal dispersion of repolarization. Early after-depolarization (EAD)–induced triggered activity also contributes to the development of the substrate and provides the triggering extrasystole that precipitates TdP arrhythmias observed under LQTS conditions (Figures 7-2 and 7-3).57,58 In vivo and in vitro models of LQTS have contributed to the understanding of the mechanisms involved in arrhythmogenesis.59,60 Models of the LQT1, LQT2, and LQT3 forms of LQTS have been developed by using arterially perfused left ventricular (LV) wedge preparations (see Figure 7-2) in canine models.61 These models have shown that in these three forms of LQTS, preferential prolongation of the M cell action potential duration (APD) leads to an increase in the QT interval as well as an increase in the transmural dispersion of repolarization (TDR), the latter providing the substrate for the development of spontaneous as well as stimulation-induced TdP.
LQT1, KCNQ1
LQT1 is the most prevalent of congenital LQTS.62 Loss of function of the slowly activating delayed rectifier (IKs) underlies congenital LQT1. Inhibition of IKs using chromanol 293B leads to uniform prolongation of APD in all three cell types (epicardial, endocardial, and M cell) in the wedge, causing little change in TDR. Although the QT interval is prolonged, TdP never occurs under these conditions, nor can it be induced. Addition of isoproterenol results in abbreviation of epicardial and endocardial APD, and the M cell APD either prolongs or remains the same. The dramatic increase in TDR provides the substrate for the development of spontaneous as well as stimulation-induced TdP.63 These results support the thesis that the problem with LQTS is not the long QT interval but, rather, the increase in TDR that often accompanies the prolongation of the QT interval. The combination of IKs block and β-adrenergic stimulation creates a broad-based T wave in the perfused wedge, similar to that observed in patients with LQT1. These findings provide an understanding of the great sensitivity of patients with LQT1 to sympathetic influences (see Figures 7-2, A and D).52,64
LQT2, KCNH2
The second most prevalent form of congenital LQTS is LQT2, which is caused by loss of function of the rapidly activating delayed rectifier (IKr). IKr inhibition is also responsible for most cases of acquired LQTS. In the wedge, inhibition of IKr with d-sotalol produces a preferential prolongation of M cells, resulting in accentuation of TDR and spontaneous as well as stimulation-induced TdP. If IKr block is accompanied by hypokalemia, a deeply notched or bifurcated T wave is observed in the wedge preparation, similar to that seen in patients with LQT2. Isoproterenol further exaggerates TDR and increases the incidence of TdP in this model, but only transiently (see Figures 7-2, B and E).
LQT3, SCN5A
LQT3, which is encountered far less often, is caused by a gain in the function of the late Na+ current (late INa). Augmentation of late INa using the sea anemone toxin ATX-II also produces a preferential prolongation of the M cell action potential in the wedge, which results in a marked increase in TDR and development of TdP. Because epicardial APD is also significantly prolonged, delay in the onset of the T wave in the wedge occurs, as observed in the clinical syndrome.65 β-Adrenergic stimulation abbreviates APD of all cell types under these conditions, reducing TDR and suppressing TdP (see Figures 7-2, C and F).66
Sympathetic activation displays a very different time course in the case of LQT1 and LQT2, both in experimental models (see Figure 7-2) and in the clinic.58,67 In LQT1, isoproterenol produces an increase in TDR that is most prominent during the first 2 minutes but which persists, although to a lesser extent, during the steady state. TdP incidence is enhanced during the initial period as well as during the steady state. In LQT2, isoproterenol produces only a transient increase in TDR that persists for less than 2 minutes. TdP incidence is therefore enhanced only for a brief period. These differences in time course may explain the important differences in the autonomic activity and other gene-specific triggers that contribute to events in patients with different LQTS genotypes.62,64
While β-blockers are considered the first line of therapy in patients with LQT1, they have not been shown to be beneficial in LQT3. Preliminary data suggest that patients with LQT3 might benefit from Na+ channel blockers, such as mexiletine and flecainide, but long-term data are not yet available.68,69 Experimental data have shown that mexiletine reduces transmural dispersion and prevents TdP in LQT3 as well as in LQT1 and LQT2, which suggests that agents that block the late Na+ current may be effective in all forms of LQTS.63,65 Clinical trial data are not currently available.
LQT7, KCNJ2
Andersen-Tawil syndrome (ATS1), also known as LQT7, is a clinical disorder consisting of K-sensitive periodic paralysis, prolonged QT intervals, ventricular arrhythmias, and dysmorphic features caused by mutations in the KCNJ2 gene.70,71 An experimental model of this syndrome has been developed.72
LQT8, CACNA1c
Timothy syndrome, also known as LQT8, is a multisystem disease secondary to mutations in the Ca2+ channel Cav1.2 encoded by the CACNA1c. Because the Ca channel Cav1.2 is present in many tissues, patients with Timothy syndrome have many clinical manifestations, including congenital heart disease, autism, syndactyly, and immune deficiency.73,74 An experimental model of this syndrome has been developed.75
Mutations in seven other genes have been associated with LQTS in recent years (see Table 7-1). These genetic variations, which include structural proteins as well as other ion channel proteins, are thought to be fairly rare.
Genotype-Phenotype Correlation
Genotype–phenotype studies have demonstrated that there are significant differences among patients with LQT1, LQT2, and LQT3 forms of LQTS, which account for 95% of all genotyped patients. Gene-specific ECG patterns have been identified (see Figure 7-2), and the trigger for cardiac events has been shown to be locus specific.62 Patients with LQT1 experience 97% of cardiac events during physical activity as opposed to those with LQT3 who experience the majority of cardiac events at rest or during sleep. Auditory stimuli and arousal have been identified as relatively specific triggers for patients with LQT2 while swimming has been identified as a predisposing setting for cardiac events in those with LQT1.76–79
The first risk stratification scheme based on genotype was proposed by Priori et al in 2003.80 QT interval, genotype, and gender were significantly associated with outcome. A QTc interval longer than 500 ms in LQT2 or LQT3 indicated a worse prognosis. In 2004, the same authors reported that the response to β-blockers is also affected by the genotype, and patients with LQT1 showed greater protection by response to β-blockers than did those with LQT2 and LQT3.81
Patients with LQT2 harboring pore mutations were shown to exhibit a more severe clinical course and to experience a higher frequency of arrhythmia-related cardiac events occurring at an earlier age than do subjects with nonpore mutations.82 KCNH2 missense mutations located in the transmembrane S5-loop-S6 region were again shown to be associated with the greatest risk in a recent study.83
Mechanism of Arrhythmia in Long QT Syndrome
Accentuation of spatial dispersion of refractoriness within the ventricular myocardium, secondary to exaggerated transmural or trans-septal dispersion of repolarization, has been identified as the principal arrhythmogenic substrate in both acquired and congenital LQTS.84,85 This exaggerated intrinsic heterogeneity and triggered activity induced by EADs and delayed after-depolarizations (DADs), both caused by a reduction in net repolarizing current, underlie the substrate and trigger for the development of TdP arrhythmias observed under LQTS conditions.58,86 Experimental models of LQTS suggest that preferential prolongation of the M cell APD leads to an increase in the QT interval as well as an increase in transmural dispersion of repolarization (TDR), which contributes to the development of spontaneous as well as stimulation-induced TdP (Figure 7-3).63,65,66,85,87,88 The spatial dispersion of repolarization is further exaggerated by sympathetic influences in LQT1 and LQT2, which accounts for the great sensitivity of patients with these genotypes to adrenergic stimuli (see Figure 7-2).
Short QT Syndrome
SQTS, a clinical entity recently described in 2000, is characterized by a short QT interval on the ECG, episodes of paroxysmal atrial fibrillation, and sudden cardiac death (SCD) in patients with structurally normal hearts.89 A distinctive ECG feature of SQTS is the appearance of tall peaked symmetrical T waves. The augmented Tpeak to Tend interval associated with this ECG feature of the syndrome suggests that here, as in LQTS, a transmural dispersion of repolarization underlies the arrhythmogenic substrate in the ventricles. To date, three different K+ channel genes and two different Ca2+ channel genes have been linked to SQTS.8,90–92
SQT1, KCNH2
The KCNH2 gene (HERG) encodes for the rapidly activating delayed rectifier K+ channel (IKr). The authors of this chapter and their group identified two different missense mutations in the same residue in KCNH2 in two unrelated families.90 Both mutations resulted in the same substitution of asparagine for lysine at codon 588 (N588K), an area at the outer mouth of the channel pore. Patch clamp studies of N588K channels expressed in TSA201 mammalian cells revealed that the mutation abolished the inactivation, thereby increasing the IKr current. Analysis of the current–voltage relation showed that N588K channels failed to rectify over a physiological range of voltages.93,94 During action potential clamp experiments, N588K currents were larger during all phases of the action potential compared with WT KCNH2 channels.93 The biophysical analysis therefore showed that the mutation induced a “gain of function” in the IKr current, thus shortening the action potential. The presence of paroxysmal atrial fibrillation in some affected patients suggests that the increased heterogeneity would also be present at the atrial level and may be responsible for the arrhythmia. Experimental studies support this observation as well.95,96 In one family, the N588K mutation is associated only with atrial fibrillation with no occurrence of ventricular arrhythmias in any of the family members displaying short QT intervals.97
SQT2, KCNQ1
A second inherited form of SQTS (SQT2) has been linked to a gain of function in the slow delayed rectifier K+ current (IKs) secondary to mutations in KCNQ1.92 This form of SQTS appears to be quite rare. The KCNQ1 gene encodes the α-subunit responsible for IKs. The mutation was first identified in a 70-year-old man with ventricular fibrillation and a QT interval of 290 ms after resuscitation.92 Biophysical analysis showed that mutation in the KCNQ1 gene produced an outward K+ current of comparable magnitude compared with WT channels. However, since the half-activation voltage was markedly shifted to negative potentials, the mutated channel activated at more negative potentials and displayed accelerated activation kinetics.92 These observations demonstrate a gain of function of IKs, which explains the SQTS phenotype.
A second mutation in KCNQ1 was found in a female infant born at 38 weeks. Delivery was induced because the infant was experiencing bradycardia and an irregular rhythm.98 The ECG revealed atrial fibrillation with slow ventricular response and a short QT interval. Genetic analysis identified a de novo missense mutation in the KCNQ1 gene. Voltage clamp experiments to characterize the physiological consequences of this mutation revealed an instantaneous and voltage-independent K+-selective current. Mathematical modeling experiments confirmed a shortening of the action potential duration in ventricular myocytes.98 A recent preliminary report has identified another novel KCNQ1 mutation (R259H) associated with SQT2.99
SQT3
Finally, mutations in the KCNJ2 gene have also been associated with SQTS. The KCNJ2 gene encodes a protein (Kir2.1) responsible for the inward rectifier K+ current (IK1). The proband and her father, in whom the mutation was discovered, displayed short QT correction intervals of 315 and 320 ms, respectively, and ECG recordings showed asymmetrical T waves with an abnormally rapid terminal phase. Expression of the mutant channel in a mammalian cell line revealed that the mutated Kir2.1 channels generated ionic currents in which rectification was reduced, compared with WT channels. The hallmark of IK1 is a region of outward current and negative slope conductance at membrane potentials between –80 mV and –30 mV. Because rectification was reduced in the mutant Kir2.1 channel, a larger outward current was observed over this range of potentials. Functionally, IK1 is responsible for terminal repolarization of the ventricular action potential.100,101 Mathematical modeling of the effects of the mutated channel on the ventricular action potential showed an increase of terminal repolarization and shortening of the APD.
SQT4 and SQT5
The fourth and fifth genes associated with BrS were recently identified and shown to encode the α1-subunit (CACNA1c) and the β-subunit (CACNB2b) of the L-type cardiac Ca2+ channel.8 This new clinical entity, which exhibits ECG and arrhythmic manifestations of both BrS and SQTS, was associated with loss of function mutations in the α1-subunit (CACNA1c) and the β-subunit (CACNB2b) of the L-type cardiac Ca2+ channel.8
Mechanism of Arrhythmia in Short QT Syndrome
An increase in net outward current caused by either a reduction in inward depolarizing current such as INa, ICa, an augmentation of outward repolarizing current like Ito, IK1, IK-ATP, IACh, IKr, IKs, or a combination of both favors early repolarization leading to abbreviation of the action potential and the QT interval (Figure 7-4). Experimental studies suggest that abbreviation of the action potential in SQTS is heterogeneous with preferential abbreviation of either the epicardium or the endocardium, giving rise to an increase in TDR. Dispersion of repolarization and refractoriness serves as the substrate for re-entry in that it promotes unidirectional block. Marked abbreviation of wavelength (product of refractory period and conduction velocity) is an additional factor promoting the maintenance of re-entry. Mutations giving rise to a gain of function of outward K+ currents has been identified in SQT1–SQT3 and a loss of function in inward ICaL have been identified in SQT4-SQT5 (see Table 7-1).8,90–92 Moreover, the Tpeak to Tend interval and the Tpeak to Tend/QT ratio, an electrocardiographic index of spatial dispersion of repolarization, and perhaps TDR, are significantly augmented in cases of SQTS.102,103 This ratio is larger in patients who are symptomatic.104
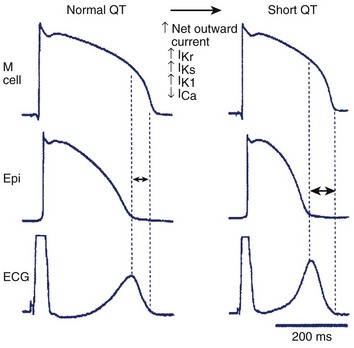
Evidence supporting the role of augmented TDR in arrhythmogenesis in SQTS comes from the experimental models involving use of the KATP activator pinacidil or the selective IKr agonist PD-118057 to abbreviate repolarization time and thus mimic the cellular conditions created by the of gene mutation responsible for SQT1.105–107
Abbreviation of APD, and effective refractory period (ERP), and amplification of spatial dispersion of repolarization have also been shown to predispose to the development of atrial fibrillation by creating the substrate for re-entry.96
Transmural Dispersion of Repolarization as a Common Link in the Development of Arrhythmias
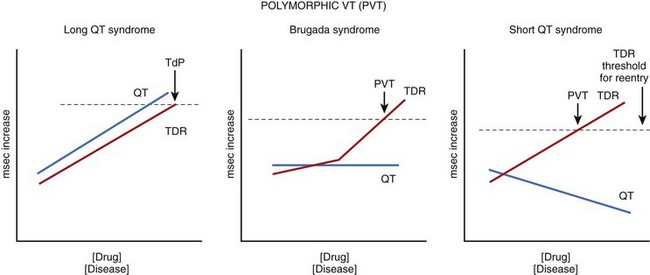
The three inherited sudden cardiac death syndromes thus far discussed differ with respect to the characteristics of the QT interval (Figure 7-5). In LQTS, QT increases as a function of disease or drug concentration. In SQTS, the QT interval decreases as a function of disease or drug, whereas in BrS, the QT interval remains largely unchanged. What these three syndromes have in common is an amplification of TDR, which results in the development of polymorphic VT and fibrillation when dispersion of repolarization and refractoriness reaches the threshold for re-entry. When polymorphic VT occurs in the setting of long QT, it is referred to as TdP. The threshold for re-entry decreases as APD and refractoriness are reduced and the pathlength required for establishing a re-entrant wave is progressively reduced.
Catecholaminergic Polymorphic Ventricular Tachycardia
Catecholaminergic, or familial, polymorphic ventricular tachycardia (CPVT) is a rare, autosomal dominant inherited disorder, predominantly affecting children or adolescents with structurally normal hearts. It is characterized by bidirectional VT (BiVT), polymorphic VT (PVT), and a high risk of sudden cardiac death (30% to 50% by the age of 20 to 30 years).108,109 Molecular genetic studies have identified mutations in genes encoding for the cardiac ryanodine receptor 2 (RyR2) or calsequestrin 2 (CASQ2) in patients with this phenotype.110–113
Mechanisms of Arrhythmias in Catecholaminergic Polymorphic Ventricular Tachycardia
Several lines of evidence point to DAD-induced triggered activity (TA) as the mechanism underlying monomorphic or bidirectional VT in patients with CPVT. These include the identification of genetic mutations involving Ca2+ regulatory proteins, a similarity of the ECG features to those associated with digitalis toxicity, and the precipitation by adrenergic stimulation. The cellular mechanisms underlying the various ECG phenotypes and the transition of monomorphic VT to polymorphic VT or VF were recently elucidated with the help of the wedge preparation.114 The wedge was exposed to low-dose caffeine to mimic the defective Ca2+ homeostasis encountered under conditions that predispose to CPVT. The combination of isoproterenol and caffeine led to the development of DAD-induced TA arising from the epicardium, the endocardium, or the M region. Migration of the source of ectopic activity was responsible for the transition from monomorphic to slow polymorphic VT. Alternation of epicardial and endocardial sources of ectopic activity gave rise to a bidirectional VT. Epicardial VT was associated with an increased Tpeak to Tend interval and transmural dispersion of repolarization caused by reversal of the normal transmural activation sequence; thus, this created the substrate for re-entry, which permitted the induction of a more rapid polymorphic VT with programmed electrical stimulation. Propranolol or verapamil suppressed arrhythmic activity.114
Recently, Cerrone and coworkers used a transgenic murine model to demonstrate that the His–Purkinje system is an important source of DAD-induced triggered activity that gives rise to focal arrhythmias in CPVT.115
1 Kaufman ES. Mechanisms and clinical management of inherited channelopathies: Long QT syndrome, Brugada syndrome, catecholaminergic polymorphic ventricular tachycardia, and short QT syndrome. Heart Rhythm. 2009;6:S51-S55.
2 Patel U, Pavri BB. Short QT syndrome: A review. Cardiol Rev. 2009;17:300-303.
3 Antzelevitch C. The role of spatial dispersion of repolarization in inherited and acquired sudden cardiac death syndromes. Am J Physiol Heart Circ Physiol. 2007;293:H2024-H2038.
4 Yan GX, Antzelevitch C. Cellular basis for the Brugada syndrome and other mechanisms of arrhythmogenesis associated with ST segment elevation. Circulation. 1999;100:1660-1666.
5 Fish JM, Antzelevitch C. Role of sodium and calcium channel block in unmasking the Brugada syndrome. Heart Rhythm. 2004;1:210-217.
6 Chen Q, Kirsch GE, Zhang D, et al. Genetic basis and molecular mechanisms for idiopathic ventricular fibrillation. Nature. 1998;392:293-296.
7 Vatta M, Dumaine R, Varghese G, et al. Genetic and biophysical basis of sudden unexplained nocturnal death syndrome (SUNDS), a disease allelic to Brugada syndrome. Hum Mol Genet. 2002;11:337-345.
8 Antzelevitch C, Pollevick GD, Cordeiro JM, et al. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation. 2007;115:442-449.
9 Cordeiro JM, Marieb M, Pfeiffer R, et al. Accelerated inactivation of the L-type calcium due to a mutation in CACNB2b due to a mutation in CACNB2b underlies Brugada syndrome. J Mol Cell Cardiol. 2009;46:695-703.
10 Verkerk AO, Wilders R, Schulze-Bahr E, et al. Role of sequence variations in the human ether-a-go-go-related gene (HERG, KCNH2) in the Brugada syndrome. Cardiovasc Res. 2005;68:441-453.
11 Delpón E, Cordeiro JM, Núñez L, et al. Functional effects of KCNE3 mutation and its role in the development of Brugada syndrome. Circ Arrhythm Electrophysiol. 2008;1:209-218.
12 Antzelevitch C, Brugada P, Borggrefe M, et al. Brugada syndrome: Report of the second consensus conference: endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation. 2005;111:659-670.
13 Kapplinger JD, Tester DJ, Alders M, et al. An international compendium of mutations in the SCN5A encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm. 2010;7(1):33-46. Epub October 8, 2009
14 Tan HL. Sodium channel variants in heart disease: Expanding horizons. J Cardiovasc Electrophysiol. 2006;17(Suppl 1):S151-S157.
15 Smits JP, Blom MT, Wilde AA, Tan HL. Cardiac sodium channels and inherited electrophysiologic disorders: A pharmacogenetic overview. Expert Opin Pharmacother. 2008;9:537-549.
16 Ruan Y, Liu N, Priori SG. Sodium channel mutations and arrhythmias. Nat Rev Cardiol. 2009;6:337-348.
17 Dumaine R, Towbin JA, Brugada P, et al. Ionic mechanisms responsible for the electrocardiographic phenotype of the Brugada syndrome are temperature dependent. Circ Res. 1999;85:803-809.
18 Weiss R, Barmada MM, Nguyen T, et al. Clinical and molecular heterogeneity in the Brugada syndrome: A novel gene locus on chromosome 3. Circulation. 2002;105:707-713.
19 London B, Michalec M, Mehdi H, et al. Mutation in glycerol-3-phosphate dehydrogenase 1 like gene (GPD1-L) decreases cardiac Na+ current and causes inherited arrhythmias. Circulation. 2007;116:2260-2268.
20 Van Norstrand DW, Valdivia CR, Tester DJ, et al. Molecular and functional characterization of novel glycerol-3-phosphate dehydrogenase 1 like gene (GPD1-L) mutations in sudden infant death syndrome. Circulation. 2007;116:2253-2259.
21 Valdivia CR, Ueda K, Ackerman MJ, Makielski JC. GPD1L links redox state to cardiac excitability by PKC-dependent phosphorylation of the sodium channel SCN5A. Am J Physiol Heart Circ Physiol. 2009;297:H1446-H1452.
22 Meadows LS, Isom LL. Sodium channels as macromolecular complexes: Implications for inherited arrhythmia syndromes. Cardiovasc Res. 2005;67:448-458.
23 Makita N, Bennett PBJr, George ALJr. Voltage-gated Na+ channel b1 subunit mRNA expressed in adult human skeletal muscle, heart, and brain is encoded by a single gene. J Biol Chem. 1994;269:7571-7578.
24 Yang JS, Bennett PB, Makita N, George AL, Barchi RL. Expression of the sodium channel b1 subunit in rat skeletal muscle is selectively associated with the tetrodotoxin-sensitive a subunit isoform. Neuron. 1993;11:915-922.
25 Qu Y, Isom LL, Westenbroek RE, et al. Modulation of cardiac Na+ channel expression in Xenopus oocytes by b1 subunits. J Biol Chem. 1995;270:25696-25701.
26 Nuss HB, Chiamvimonvat N, Perez-Garcia MT, Tomaselli GF, Marban E. Functional association of the b1 subunit with human cardiac (hH1) and rat skeletal muscle (m 1) sodium channel a subunits expressed in Xenopus oocytes. J Gen Physiol. 1995;106:1171-1191.
27 Makielski JC, Limberis JT, Chang SY, Fan Z, Kyle JW. Coexpression of b1 with cardiac sodium channel a subunits in oocytes decreases lidocaine block. Mol Pharmacol. 1996;49:30-39.
28 Malhotra JD, Chen C, Rivolta I, et al. Characterization of sodium channel a- and b-subunits in rat and mouse cardiac myocytes. Circulation. 2001;103:1303-1310.
29 An RH, Wang XL, Kerem B, et al. Novel LQT-3 mutation affects Na+ channel activity through interactions between a- and b1-subunits. Circ Res. 1998;83:141-146.
30 Ko SH, Lenkowski PW, Lee HC, Mounsey JP, Patel MK. Modulation of Nav1.5 by b1- and b3-subunit co-expression in mammalian cells. Pflugers Arch. 2005;449:403-412.
31 Fahmi AI, Patel M, Stevens EB, et al. The sodium channel b-subunit SCN3b modulates the kinetics of SCN5a and is expressed heterogeneously in sheep heart. J Physiol. 2001;537:693-700.
32 Watanabe H, Koopmann TT, Le Scouarnec S, et al. Sodium channel b1 subunit mutations associated with Brugada syndrome and cardiac conduction disease in humans. J Clin Invest. 2008;118:2260-2268.
33 Hu D, Barajas-Martinez H, Burashnikov E, et al. A mutation in the b3 subunit of the cardiac sodium channel associated with Brugada ECG phenotype. Circ Cardiovasc Genet. 2009;2:270-278.
34 Di Diego JM, Cordeiro JM, Goodrow RJ, et al. Ionic and cellular basis for the predominance of the Brugada syndrome phenotype in males. Circulation. 2002;106:2004-2011.
35 Lundby A, Olesen SP. KCNE3 is an inhibitory subunit of the Kv4.3 potassium channel. Biochem Biophys Res Commun. 2006;346:958-967.
36 Calloe K, Cordeiro JM, Di Diego JM, et al. A transient outward potassium current activator recapitulates the electrocardiographic manifestations of Brugada syndrome. Cardiovasc Res. 2009;81:686-694.
37 Burashnikov E, Pfeifer R, Borggrefe M, et al. Mutations in the cardiac L-type calcium channel associated with inherited sudden cardiac death syndromes (abstract). Circulation. 2009;120:S573.
38 Smits JP, Eckardt L, Probst V, et al. Genotype-phenotype relationship in Brugada syndrome: electrocardiographic features differentiate SCN5A-related patients from non-SCN5A-related patients. J Am Coll Cardiol. 2002;40:350-356.
39 Yokokawa M, Noda T, Okamura H, et al. Comparison of long-term follow-up of electrocardiographic features in Brugada syndrome between the SCN5A-positive probands and the SCN5A-negative probands. Am J Cardiol. 2007;100:649-655.
40 Wolpert C, Schimpf R, Giustetto C, et al. Further insights into the effect of quinidine in short QT syndrome caused by a mutation in HERG. J Cardiovasc Electrophysiol. 2005;16:54-58.
41 Antzelevitch C. Brugada syndrome. PACE. 2006;29:1130-1159.
42 Antzelevitch C. The Brugada syndrome: Ionic basis and arrhythmia mechanisms. J Cardiovasc Electrophysiol. 2001;12:268-272.
43 Antzelevitch C, Yan GX. J wave syndromes. Heart Rhythm. 2010;7(4):549-558. Epub December 11, 2009
44 Yan GX, Antzelevitch C. Cellular basis for the electrocardiographic J wave. Circulation. 1996;93:372-379.
45 Gussak I, Antzelevitch C, Bjerregaard P, Towbin JA, Chaitman BR. The Brugada syndrome: Clinical, electrophysiologic and genetic aspects. J Am Coll Cardiol. 1999;33:5-15.
46 Lukas A, Antzelevitch C. Phase 2 re-entry as a mechanism of initiation of circus movement re-entry in canine epicardium exposed to simulated ischemia. Cardiovasc Res. 1996;32:593-603.
47 Morita H, Zipes DP, Fukushima-Kusano K, et al. Repolarization heterogeneity in the right ventricular outflow tract: Correlation with ventricular arrhythmias in Brugada patients and in an in vitro canine Brugada model. Heart Rhythm. 2008;5:725-733.
48 Morita H, Zipes DP, Wu J. Brugada syndrome: Insights of ST elevation, arrhythmogenicity, and risk stratification from experimental observations. Heart Rhythm. 2009;6:S34-S43.
49 Aiba T, Shimizu W, Hidaka I, et al. Cellular basis for trigger and maintenance of ventricular fibrillation in the Brugada syndrome model: High-resolution optical mapping study. J Am Coll Cardiol. 2006;47:2074-2085.
50 Antzelevitch C, Brugada P, Brugada J, et al. Brugada syndrome: A decade of progress. Circ Res. 2002;91:1114-1119.
51 Kurita T, Shimizu W, Inagaki M, et al. The electrophysiologic mechanism of ST-segment elevation in Brugada syndrome. J Am Coll Cardiol. 2002;40:330-334.
52 Schwartz PJ. The idiopathic long QT syndrome: Progress and questions. Am Heart J. 1985;109:399-411.
53 Moss AJ, Schwartz PJ, Crampton RS, et al. The long QT syndrome: Prospective longitudinal study of 328 families. Circulation. 1991;84:1136-1144.
54 Zipes DP. The long QT interval syndrome: A Rosetta stone for sympathetic related ventricular tachyarrhythmias. Circulation. 1991;84:1414-1419.
55 Roden DM. Drug-induced prolongation of the QT interval. N Engl J Med. 2004;350:1013-1022.
56 Dumaine R, Antzelevitch C. Molecular mechanisms underlying the long QT syndrome. Curr Opin Cardiol. 2002;17:36-42.
57 Antzelevitch C. Heterogeneity of cellular repolarization in LQTS: The role of M cells. Eur Heart J. 2001;Suppl 3:K-2-K-16.
58 Antzelevitch C, Shimizu W. Cellular mechanisms underlying the long QT syndrome. Curr Opin Cardiol. 2002;17:43-51.
59 Fenichel RR, Malik M, Antzelevitch C, et al. Drug-induced torsade de pointes and implications for drug development. J Cardiovasc Electrophysiol. 2004;15:475-495.
60 Kozhevnikov DO, Yamamoto K, Robotis D, Restivo M, El-Sherif N. Electrophysiological mechanism of enhanced susceptibility of hypertrophied heart to acquired torsade de pointes arrhythmias: Tridimensional mapping of activation and recovery patterns. Circulation. 2002;105:1128-1134.
61 Shimizu W, Antzelevitch C. Effects of a K+ channel opener to reduce transmural dispersion of repolarization and prevent torsade de pointes in LQT1, LQT2, and LQT3 models of the long-QT syndrome. Circulation. 2000;102:706-712.
62 Schwartz PJ, Priori SG, Spazzolini C, et al. Genotype-phenotype correlation in the long-QT syndrome: Gene-specific triggers for life-threatening arrhythmias. Circulation. 2001;103:89-95.
63 Shimizu W, Antzelevitch C. Cellular basis for the ECG features of the LQT1 form of the long QT syndrome: Effects of b-adrenergic agonists and antagonists and sodium channel blockers on transmural dispersion of repolarization and torsade de pointes. Circulation. 1998;98:2314-2322.
64 Ali RH, Zareba W, Moss A, et al. Clinical and genetic variables associated with acute arousal and nonarousal-related cardiac events among subjects with long QT syndrome. Am J Cardiol. 2000;85:457-461.
65 Shimizu W, Antzelevitch C. Sodium channel block with mexiletine is effective in reducing dispersion of repolarization and preventing torsade de pointes in LQT2 and LQT3 models of the long-QT syndrome. Circulation. 1997;96:2038-2047.
66 Shimizu W, Antzelevitch C. Differential effects of beta-adrenergic agonists and antagonists in LQT1, LQT2 and LQT3 models of the long QT syndrome. J Am Coll Cardiol. 2000;35:778-786.
67 Noda T, Takaki H, Kurita T, et al. Gene-specific response of dynamic ventricular repolarization to sympathetic stimulation in LQT1, LQT2 and LQT3 forms of congenital long QT syndrome. Eur Heart J. 2002;23:975-983.
68 Windle JR, Geletka RC, Moss AJ, Zareba W, Atkins DL. Normalization of ventricular repolarization with flecainide in long QT syndrome patients with SCN5A: DeltaKPQ mutation. Ann Noninvasive Electrocardiol. 2001;6:153-158.
69 Roden DM. Pharmacogenetics and drug-induced arrhythmias. Cardiovasc Res. 2001;50:224-231.
70 Tristani-Firouzi M, Jensen JL, Donaldson MR, et al. Functional and clinical characterization of KCNJ2 mutations associated with LQT7 (Andersen syndrome). J Clin Invest. 2002;110:381-388.
71 Andelfinger G, Tapper AR, Welch RC, Vanoye CG, George ALJr, Benson DW. KCNJ2 mutation results in Andersen syndrome with sex-specific cardiac and skeletal muscle phenotypes. Am J Hum Genet. 2002;71:663-668.
72 Tsuboi M, Antzelevitch C. Cellular basis for electrocardiographic and arrhythmic manifestations of Andersen-Tawil syndrome (LQT7). Heart Rhythm. 2006;3:328-335.
73 Splawski I, Timothy KW, Sharpe LM, et al. Cav1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 2004;119:19-31.
74 Splawski I, Timothy KW, Decher N, et al. Severe arrhythmia disorder caused by cardiac L-type calcium channel mutations. Proc Natl Acad Sci U S A. 2005;102:8089-8096.
75 Sicouri S, Timothy KW, Zygmunt AC, et al. Cellular basis for the electrocardiographic and arrhythmic manifestations of Timothy syndrome: Effects of ranolazine. Heart Rhythm. 2007;4:638-647.
76 Moss AJ, Zareba W, Benhorin J, et al. ECG T-wave patterns in genetically distinct forms of the hereditary long QT syndrome. Circulation. 1995;92:2929-2934.
77 Zhang L, Timothy KW, Vincent GM, et al. Spectrum of ST-T-wave patterns and repolarization parameters in congenital long-QT syndrome: ECG findings identify genotypes. Circulation. 2000;102:2849-2855.
78 Moss AJ, Robinson JL, Gessman L, et al. Comparison of clinical and genetic variables of cardiac events associated with loud noise versus swimming among subjects with the long QT syndrome. Am J Cardiol. 1999;84:876-879.
79 Ackerman MJ, Tester DJ, Porter CJ. Swimming, a gene-specific arrhythmogenic trigger for inherited long QT syndrome. Mayo Clin Proc. 1999;74:1088-1094.
80 Priori SG, Schwartz PJ, Napolitano C, et al. Risk stratification in the long-QT syndrome. N Engl J Med. 2003;348:1866-1874.
81 Priori SG, Napolitano C, Schwartz PJ, et al. Association of long QT syndrome loci and cardiac events among patients treated with beta-blockers. JAMA. 2004;292:1341-1344.
82 Moss AJ, Zareba W, Kaufman ES, et al. Increased risk of arrhythmic events in long-QT syndrome with mutations in the pore region of the human ether-a-go-go-related gene potassium channel. Circulation. 2002;105:794-799.
83 Shimizu W, Moss AJ, Wilde AA, et al. Genotype-phenotype aspects of type 2 long QT syndrome. J Am Coll Cardiol. 2009;54:2052-2062.
84 Antzelevitch C. Heterogeneity and cardiac arrhythmias: An overview. Heart Rhythm. 2007;4:964-972.
85 Sicouri S, Glass A, Ferreiro M, Antzelevitch C. Transseptal dispersion of repolarization and its role in the development of torsade de pointes arrhythmias. J Cardiovasc Electrophysiol. 2010;21(4):441-447. Epub November 10, 2009
86 Belardinelli L, Antzelevitch C, Vos MA. Assessing predictors of drug-induced torsade de pointes. Trends Pharmacol Sci. 2003;24:619-625.
87 Ueda N, Zipes DP, Wu J. Prior ischemia enhances arrhythmogenicity in isolated canine ventricular wedge model of long QT 3. Cardiovasc Res. 2004;63:69-76.
88 Ueda N, Zipes DP, Wu J. Functional and transmural modulation of M cell behavior in canine ventricular wall. Am J Physiol Heart Circ Physiol. 2004;287:H2569-H2575.
89 Gussak I, Brugada P, Brugada J, et al. Idiopathic short QT interval: A new clinical syndrome? Cardiology. 2000;94:99-102.
90 Brugada R, Hong K, Dumaine R, et al. Sudden death associated with short-QT syndrome linked to mutations in HERG. Circulation. 2004;109:30-35.
91 Priori SG, Pandit SV, Rivolta I, et al. A novel form of short QT syndrome (SQT3) is caused by a mutation in the KCNJ2 gene. Circ Res. 2005;96:800-807.
92 Bellocq C, Van Ginneken AC, Bezzina CR, et al. Mutation in the KCNQ1 gene leading to the short QT-interval syndrome. Circulation. 2004;109:2394-2397.
93 Cordeiro JM, Brugada R, Wu YS, Hong K, Dumaine R. Modulation of IKr inactivation by mutation N588K in KCNH2: A link to arrhythmogenesis in short QT syndrome. Cardiovasc Res. 2005;67:498-509.
94 McPate MJ, Duncan RS, Milnes JT, Witchel HJ, Hancox JC. The N588K-HERG K+ channel mutation in the “short QT syndrome”: Mechanism of gain-in-function determined at 37°C. Biochem Biophys Res Commun. 2005;334:441-449.
95 McPate MJ, Zhang H, Adeniran I, Cordeiro JM, Witchel HJ, Hancox JC. Comparative effects of the short QT N588K mutation at 37° C on hERG K+ channel current during ventricular, Purkinje fibre and atrial action potentials: An action potential clamp study. J Physiol Pharmacol. 2009;60:23-41.
96 Nof E, Burashnikov A, Antzelevitch C. Basis for atrial fibrillation in an experimental model of short QT1: Implications for a pharmacologic approach to therapy. Heart Rhythm. 2010;7(2):251-257. Epub October 17, 2009
97 Hong K, Bjerregaard P, Gussak I, Brugada R. Short QT syndrome and atrial fibrillation caused by mutation in KCNH2. J Cardiovasc Electrophysiol. 2005;16:394-396.
98 Hong K, Piper DR, Diaz-Valdecantos A, et al. De novo KCNQ1 mutation responsible for atrial fibrillation and short QT syndrome in utero. Cardiovasc Res. 2005;68:433-440.
99 Li Y, Memmi M, Denegri M, et al. Characterization of a novel KCNQ1 mutation (R259H) that abbreviates repolarization and causes short QT syndrome 2 (abstract). Circulation. 2009;120:S627.
100 Shimoni Y, Clark RB, Giles WR. Role of an inwardly rectifying potassium current in rabbit ventricular action potential. J Physiol (Lond). 1992;448:709-727.
101 Cordeiro JM, Spitzer KW, Giles WR. Repolarizing K+ currents in rabbit heart Purkinje cells. J Physiol. 1998;508(Pt 3):811-823.
102 Anttonen O, Vaananen H, Junttila J, Huikuri HV, Viitasalo M. Electrocardiographic transmural dispersion of repolarization in patients with inherited short QT syndrome. Ann Noninvasive Electrocardiol. 2008;13:295-300.
103 Gupta P, Patel C, Patel H, et al. Tp-e/QT ratio as an index of arrhythmogenesis. J Electrocardiol. 2008;41:567-574.
104 Anttonen O, Junttila MJ, Maury P, et al. Differences in twelve-lead electrocardiogram between symptomatic and asymptomatic subjects with short QT interval. Heart Rhythm. 2009;6:267-271.
105 Extramiana F, Antzelevitch C. Amplified transmural dispersion of repolarization as the basis for arrhythmogenesis in a canine ventricular-wedge model of short QT syndrome. Circulation. 2004;110:3661-3666.
106 Milberg P, Tegelkamp R, Osada N, et al. Reduction of dispersion of repolarization and prolongation of postrepolarization refractoriness explain the antiarrhythmic effects of quinidine in a model of short QT syndrome. J Cardiovasc Electrophysiol. 2007;18:658-664.
107 Patel C, Antzelevitch C. Cellular basis for arrhythmogenesis in an experimental model of the SQT1 form of the short QT syndrome. Heart Rhythm. 2008;5:585-590.
108 Leenhardt A, Lucet V, Denjoy I, Grau F, Ngoc DD, Coumel P. Catecholaminergic polymorphic ventricular tachycardia in children: A 7-year follow-up of 2 patients. Circulation. 1995;91:1512-1519.
109 Swan H, Piippo K, Viitasalo M, et al. Arrhythmic disorder mapped to chromosome 1q42-q43 causes malignant polymorphic ventricular tachycardia in structurally normal hearts. J Am Coll Cardiol. 1999;34:2035-2042.
110 Priori SG, Napolitano C, Memmi M, et al. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation. 2002;106:69-74.
111 Priori SG, Napolitano C, Tiso N, et al. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation. 2001;103:196-200.
112 Laitinen PJ, Brown KM, Piippo K, et al. Mutations of the cardiac ryanodine receptor (RyR2) gene in familial polymorphic ventricular tachycardia. Circulation. 2001;103:485-490.
113 Postma AV, Denjoy I, Hoorntje TM, et al. Absence of calsequestrin 2 causes severe forms of catecholaminergic polymorphic ventricular tachycardia. Circ Res. 2002;91:e21-e26.
114 Nam GB, Burashnikov A, Antzelevitch C. Cellular mechanisms underlying the development of catecholaminergic ventricular tachycardia. Circulation. 2005;111:2727-2733.
115 Cerrone M, Noujaim SF, Tolkacheva EG, et al. Arrhythmogenic mechanisms in a mouse model of catecholaminergic polymorphic ventricular tachycardia. Circ Res. 2007;101:1039-1048.
116 Kapplinger JD, Tester DJ, Salisbury BA, et al. Spectrum and prevalence of mutations from the first 2,500 consecutive unrelated patients referred for the FAMILION long QT syndrome genetic test. Heart Rhythm. 2009;6:1297-1303.
117 Makiyama T, Akao M, Haruna Y, et al. Mutation analysis of the glycerol-3 phosphate dehydrogenase-1 like (GPD1L) gene in Japanese patients with Brugada syndrome. Circ J. 2008:1705-1706.