Introduction to Increased Destruction of Erythrocytes
After completion of this chapter, the reader will be able to:
1. Define hemolysis and recognize its hallmark clinical findings.
2. Differentiate a hemolytic disorder from hemolytic anemia by definition and recognition of laboratory findings.
3. Discuss methods of classifying hemolytic anemias and apply the classification to an unfamiliar anemia.
4. Describe the processes of intravascular (fragmentation) and extravascular (macrophage-mediated) hemolysis, emphasizing sites of hemolysis for each and associating the clinical findings of each.
5. Differentiate intravascular and extravascular hemolysis when given clinical and laboratory findings.
6. Describe the process of normal red blood cell (RBC)/hemoglobin catabolism, emphasizing the process for catabolism of protoporphyrin.
7. Identify laboratory tests that indicate accelerated RBC destruction, explain the physiologic mechanisms involved, and interpret the results of each test.
8. Identify, explain, and interpret the results of laboratory tests that indicate increased erythropoiesis.
9. Differentiate between hemolytic anemias and other causes of increased erythropoiesis given laboratory or clinical information.
10. Describe the mechanisms that salvage hemoglobin during intravascular hemolysis.
11. Explain the rationale for tests of the hemoglobin salvage mechanisms and interpret the results of each.
Case Study
1. What process is indicated by the root beer–colored urine?
2. What laboratory tests can be used to differentiate the cause of the hemolysis?
3. Based on the patient’s clinical presentation, predict the results expected for each test listed for question 2.
This chapter presents an overview of the hemolytic process and provides a foundation that is applicable in the following chapters on red blood cell (RBC) disorders. The term hemolysis or hemolytic disorder refers to increased destruction (i.e., lysis) of RBCs, shortening their life span. The reduced number of cells results in reduced tissue oxygenation and increased erythropoietin production by the kidney. When the patient is otherwise healthy, the bone marrow responds by accelerating erythrocyte production, which leads to reticulocytosis. A hemolytic process is present without anemia if the bone marrow is able to compensate by accelerating RBC production sufficiently to replace the RBCs lost through hemolysis. Healthy bone marrow can increase its production of RBCs by six to eight times normal1; therefore, significant RBC destruction must occur before an anemia develops. A hemolytic anemia results when the rate of RBC destruction exceeds the increased rate of RBC production.
Classification
When hemolysis is the primary feature, anemias can be classified as follows:
Every hemolytic condition can be classified according to each of these descriptors. Table 22-1 shows this and provides a noncomprehensive list of hemolytic anemias. This chapter focuses on the distinction between intravascular and extravascular hemolytic conditions. The other classifying schemes are summarized here briefly and are used to organize the chapters that follow.
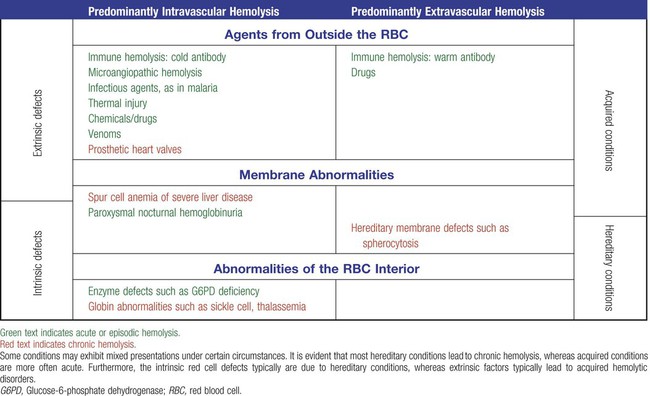
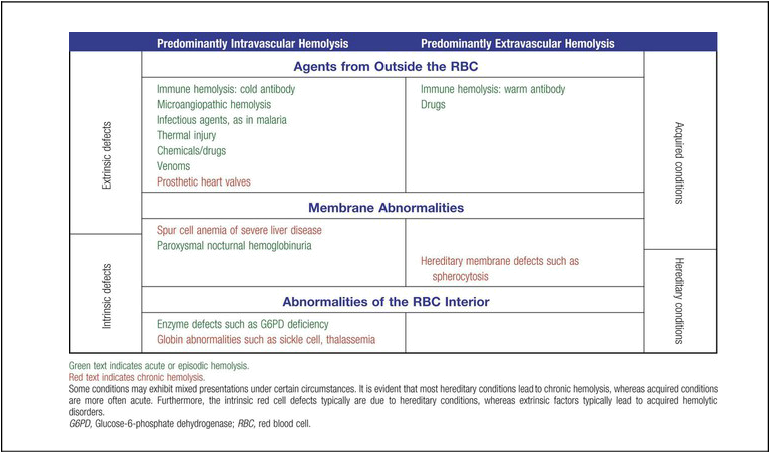
Extrinsic hemolytic conditions are those that arise from outside the RBC, typically substances in the plasma or conditions affecting the anatomy of the circulatory system. Even though malaria protozoa and other infectious agents are within the RBC, they are classified as extrinsic because the RBC was normal until it was invaded by an outside agent. An antibody against RBC antigens and a prosthetic heart valve are examples of noninfectious extrinsic agents. In extrinsic hemolysis, cross-transfusion studies have shown that the patient’s RBCs have a normal life span in the bloodstream of a normal individual, but normal cells are lysed more rapidly in the patient’s circulation. These studies confirm that something outside the RBCs is causing the hemolysis. (Of course, in the case of intracellular parasites, the cross-transfusion study is not applicable.) Most intrinsic defects are inherited; most extrinsic ones are acquired (see Table 22-1). A few exceptions exist, such as paroxysmal nocturnal hemoglobinuria, an acquired disorder involving an intrinsic defect (see Chapter 23).
Intrinsic disorders are subclassified as membrane defects, enzyme defects, and hemoglobinopathies. Extrinsic hemolysis may be immunohemolytic, traumatic, or microangiopathic, or may be caused by infectious agents, chemical agents (drugs and venoms), or physical agents (see Table 22-1).
Hemolysis
Normal Bilirubin Metabolism
RBCs live approximately 120 days. During this time, they undergo various metabolic and chemical changes, which result in a loss of deformability. Under normal circumstances, macrophages of the mononuclear phagocyte system (or reticuloendothelial system) recognize these changes and phagocytize the aged erythrocytes (see Chapter 8). The organs of this system include the spleen, bone marrow, liver, lymph nodes, and circulating monocytes, but it is primarily the spleen and liver that process senescent RBCs. The macrophages in the spleen (littoral cells) are especially sensitive to subtle RBC changes, whereas the macrophages of the liver (Kupffer cells) detect and destroy more severely damaged RBCs. This is an extravascular hemolytic process. Hemolysis occurs within macrophages; thus macrophage-mediated hemolysis is a more precise term.
The majority of RBC degradation occurs extravascularly as enzymes of the macrophage lyse the phagocytized erythrocytes (Figure 22-1). Hemoglobin is hydrolyzed into heme and globin; the latter is further degraded into amino acids that return to the amino acid pool. Iron is released from the heme, returned to the plasma via ferroportin, bound to its protein carrier molecule (transferrin), and recycled to developing RBCs. The remaining protoporphyrin is degraded through a series of biochemical reactions in different tissues and organs.
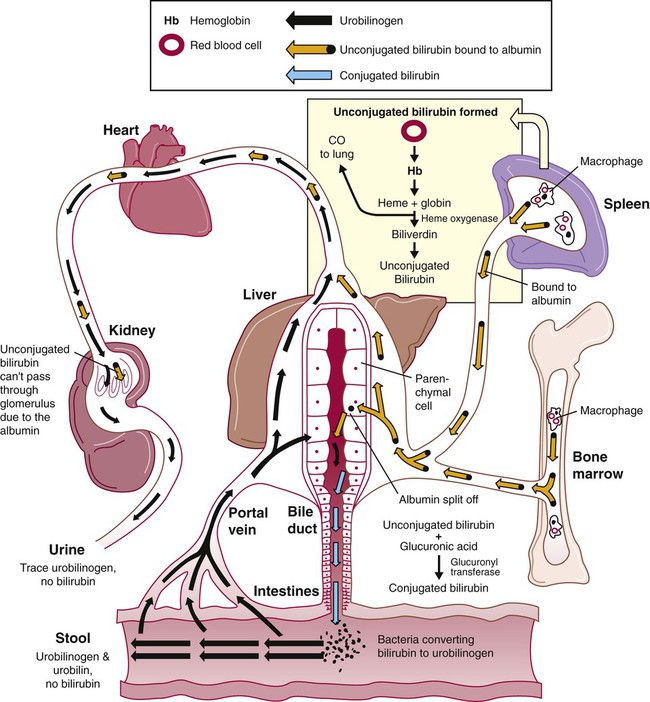
Figure 22-1 illustrates protoporphyrin catabolism. While protoporphyrin is inside the macrophage, heme oxygenase acts on it, breaking the porphyrin ring to yield a linear molecule, biliverdin. The lungs excrete a by-product of that reaction, carbon monoxide. The green biliverdin is reduced to bilirubin, a nonpolar yellow molecule that is secreted into the plasma (Box 22-1). Because it is hydrophobic, this bilirubin must bind to albumin to be transported in plasma to the liver. The bilirubin-albumin complex enters the liver parenchymal cells, where the bilirubin dissociates from the albumin and is joined (i.e., conjugated) with two molecules of glucuronic acid by glucuronyl transferase to form bilirubin diglucuronide.2 The addition of the two sugar acid molecules makes the molecule polar and water soluble. Bilirubin diglucuronide is also called conjugated bilirubin. The bilirubin originally released from macrophages that lacks these sugars is termed unconjugated.
Normal Plasma Hemoglobin Salvage During Intravascular Hemolysis
Intravascular hemolysis is trauma to the RBC membrane that causes a breach sufficient for the cell contents, chiefly hemoglobin, to spill directly into plasma (Box 22-2). Approximately 10% to 20% of normal RBC destruction is intravascular,3 secondary to turbulence and anatomic restrictions in the vasculature.
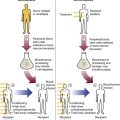
Because hemoglobin is filtered by the kidney, iron could be lost in normal intravascular hemolysis. In addition, free hemoglobin and iron can cause oxidative damage to cells. Several mechanisms exist to salvage hemoglobin iron and prevent oxidation reactions, and are collectively called the haptoglobin-hemopexin-methemalbumin system (Figure 22-2).
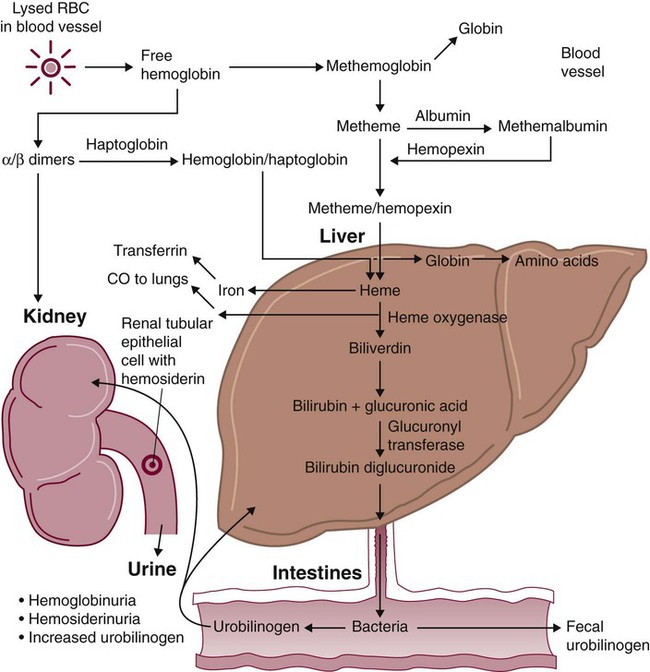
When free in the plasma, hemoglobin exists as α/β dimers bound to a liver-produced plasma protein called haptoglobin. This is the first mechanism of hemoglobin iron salvage. By binding to haptoglobin, hemoglobin avoids filtration at the glomerulus and is saved from urinary loss. The complex is carried to the liver, where the haptoglobin-hemoglobin complex is bound to macrophage receptors and internalized into the macrophage.4 Inside the macrophage, iron is salvaged, and the remaining protoporphyrin is converted to bilirubin, just as though the RBC had been ingested by the macrophage. If lysis is accelerated, haptoglobin is depleted, because the liver’s production of haptoglobin does not increase in response to the increased hemolysis and is typically adequate to salvage only a normal amount of plasma hemoglobin.
A secondary mechanism of iron salvage involves hemopexin. In hemoglobin that is not bound by haptoglobin, the iron becomes oxidized, forming methemoglobin. The heme molecule (actually metheme) dissociates from the globin and binds to another liver-produced plasma protein, hemopexin.5,6 This binding also saves the iron from urinary loss and prevents oxidation of cell membranes. Hemopexin-metheme binds to hepatocyte receptors and is internalized. There the iron is salvaged, and the protoporphyrin is converted to bilirubin, as in a macrophage. In contrast to haptoglobin, hemopexin is recycled to the plasma from the hepatocyte, so a single hemopexin molecule collects and salvages more free metheme molecules. Although its production is constant, because it is recycled to the plasma, levels of hemopexin do not fall dramatically during periods when there is an increase in intravascular hemolysis.
A third mechanism of iron salvage is the metheme-albumin system. Albumin acts a carrier for many molecules. Metheme can bind to albumin when hemopexin is saturated, which further reduces urinary loss. This is probably a temporary holding state for the metheme, which transfers to hemopexin because it has a higher binding affinity for metheme than does albumin.7 It is then transported to the liver for processing.
If the previous systems are overloaded, the excess hemoglobin or heme iron will be filtered into the urine. Iron still can be retained in the kidney, however.8,9 Some hemoglobin or (met)heme that enters the filtrate is reabsorbed, not by specific carriers but as part of the general filtrate in the proximal tubule. When inside the renal tubular cell, the iron is separated from the protoporphyrin. There it is stored as ferritin or hemosiderin. The role of the kidney in salvaging iron and returning it to the circulation is uncertain at this time. Although carrier proteins like divalent metal transporter 1 and ferroportin have been identified in the kidney of experimental mice, their localization, function, and regulation are still not clear. When the amount of hemoglobin presented to the kidney exceeds what can be reabsorbed, the excess is then detectable in the urine.
Excessive Extravascular Hemolysis
Many hemolytic anemias are a result of increased extravascular hemolysis (Figure 22-3) during which more than the usual number of RBCs are removed from the circulation daily. Under normal circumstances, senescent RBCs display surface markers that identify them to macrophages as aged cells requiring removal (see Chapter 8). Pathologic processes also lead to expression of abnormal markers, so that cells are identified and removed. If the number of affected cells increases beyond the quantity normally removed each day due to senescence, and if the bone marrow cannot compensate, then anemia develops. As an example, Heinz bodies, aggregates of denatured hemoglobin formed in various anemias, bind to the inner surface of the RBC membrane, producing changes to the exterior of the membrane that can be detected by macrophages. When something causes increased formation of Heinz bodies or other intracellular inclusions, the cells are removed from the circulation prematurely by macrophages. A similar process occurs when intracellular parasites are present or when complement or immunoglobulins are on the surface of the RBC.
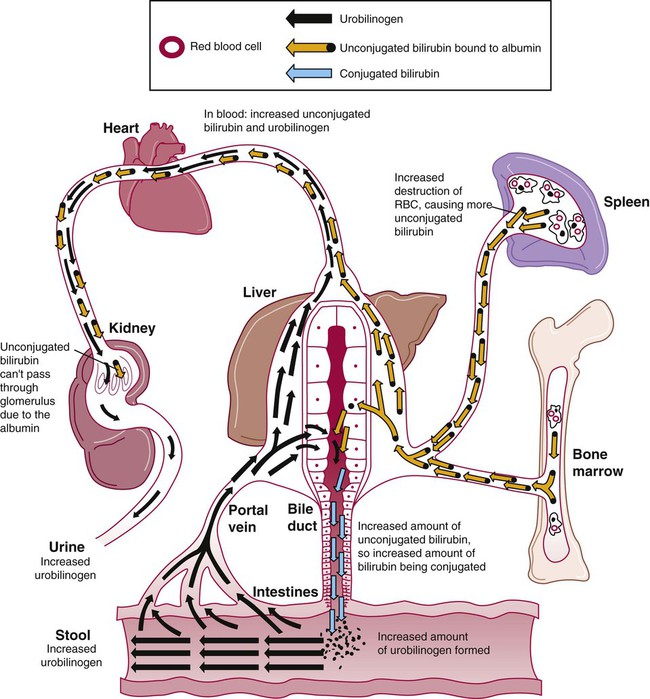
In extravascular hemolytic anemias, the plasma bilirubin level rises as RBCs lyse prematurely. The plasma bilirubin is largely unconjugated. As long as the liver is healthy, it processes the increased load of unconjugated bilirubin, producing conjugated bilirubin that enters the intestine. Increased urobilinogen forms in the intestines and is subsequently absorbed by the portal circulation and excreted by the kidney. As a result, increased urobilinogen is detectable in the urine. Although there is an increase in unconjugated bilirubin in the plasma, none of it appears in the urine because it is bound to albumin and cannot pass through the glomerulus. These findings are summarized in Table 22-2.
TABLE 22-2
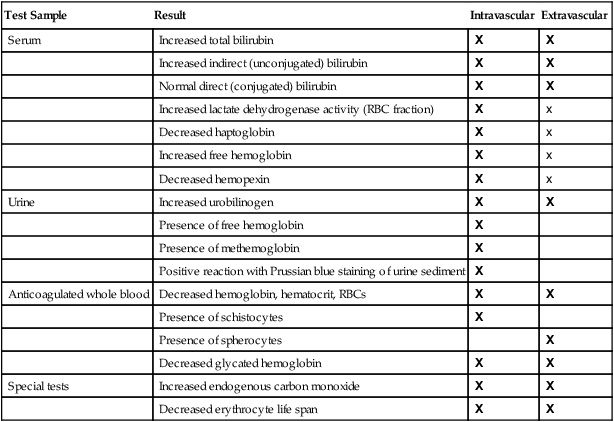
| Test Sample | Result | Intravascular | Extravascular |
| Serum | Increased total bilirubin | X | X |
| Increased indirect (unconjugated) bilirubin | X | X | |
| Normal direct (conjugated) bilirubin | X | X | |
| Increased lactate dehydrogenase activity (RBC fraction) | X | x | |
| Decreased haptoglobin | X | x | |
| Increased free hemoglobin | X | x | |
| Decreased hemopexin | X | x | |
| Urine | Increased urobilinogen | X | X |
| Presence of free hemoglobin | X | ||
| Presence of methemoglobin | X | ||
| Positive reaction with Prussian blue staining of urine sediment | X | ||
| Anticoagulated whole blood | Decreased hemoglobin, hematocrit, RBCs | X | X |
| Presence of schistocytes | X | ||
| Presence of spherocytes | X | ||
| Decreased glycated hemoglobin | X | X | |
| Special tests | Increased endogenous carbon monoxide | X | X |
| Decreased erythrocyte life span | X | X |
Excessive Intravascular Hemolysis
Although intravascular hemolysis is a minor component of normal RBC destruction, it can be a major feature of pathologic processes (Figure 22-4). Dramatic examples of intravascular hemolysis are the traumatic, physical lysis of RBCs caused by prosthetic heart valves and the exit of mature intracellular RBC parasites, such as malaria protozoa, by bursting out of the cell. In these instances, the intravascular destruction of RBCs causes profound anemias.
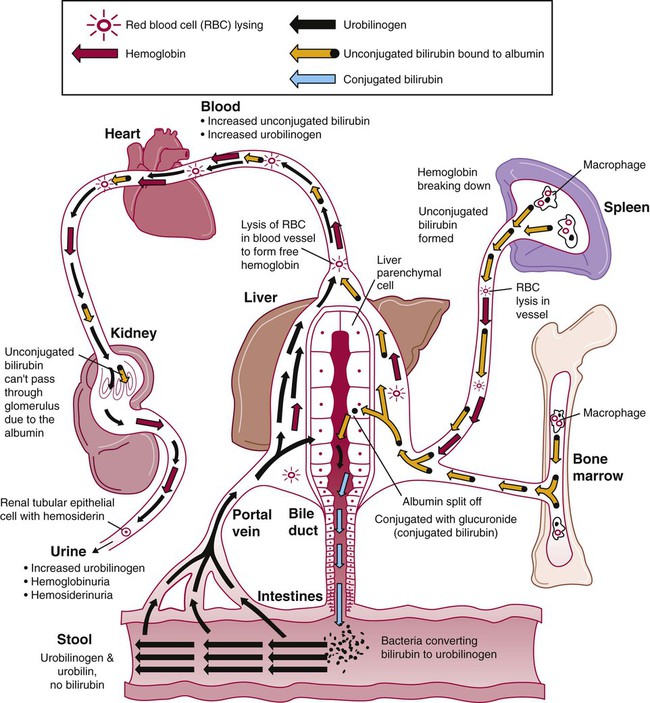
Intravascular hemolysis is fragmentation of the RBCs that releases detectable hemoglobin into the plasma. Characteristic laboratory findings are hemoglobinemia, hemoglobinuria, and hemosiderinuria (see Table 22-2). In addition, methemalbumin and hemopexin-heme are detected in plasma, and the haptoglobin level is decreased. Plasma bilirubin (largely unconjugated) and elevated levels of urinary urobilinogen are also measurable but not immediately, because time is needed to produce these products. The time course of these findings assists with the differential diagnosis (see later).
In a properly collected specimen, normal intravascular hemolysis produces a plasma hemoglobin level of less than 1 mg/dL.10 Plasma does not become visibly red until the plasma hemoglobin level is at 50 mg/dL.10 Typical values during hemolytic processes can range from 15 to 100 mg/dL, so an increase in plasma hemoglobin may not always be visible.11
During excess intravascular hemolysis, plasma haptoglobin is rapidly depleted. Levels of hemopexin also decline, but not as significantly as haptoglobin levels, because of its recycling. The hemopexin level is decreased during severe hemolysis, but it is not often measured. Levels of the hemopexin-heme complex, however, increase. Methemalbumin can also be detected in the plasma, although it too is seldom measured. The presence of methemalbumin and hemopexin-heme, which impart a coffee-brown color to plasma, strongly suggests intravascular hemolysis.12,13 Classically, these pigments have been detected directly in plasma spectrophotometrically at 620 to 630 nm and do not disappear with the addition of hydrogen peroxide, as does methemoglobin. If ammonium sulfide is added (Schumm test), the 620- to 630-nm band disappears, and a band at 558 nm forms.14 More recently, enzyme-linked immunosorbent assays have been developed for hemopexin.
In addition, if the plasma hemoglobin is sufficiently elevated, hemoglobin is detectable in the urine (hemoglobinuria). For example, when an acute intravascular hemolytic transfusion reaction is suspected, a urine sample is tested promptly for hemoglobin using the urinalysis reagent strip. Hemoglobin may give a dark red or brown color (due to methemoglobin) to the urine if the amount is sufficiently high, and thus the urine may be described as looking like root beer or beer. In chronic intravascular hemolysis, iron absorbed into renal tubular epithelial cells may be visualized. These cells slough into the urine and appear in the sediment, and they can be stained with Prussian blue. This is an easy and inexpensive way to detect intravascular hemolysis.11 These findings are summarized in Table 22-2.
Clinical Features
Some signs differentiate chronic from acute hemolysis. Splenomegaly can develop, particularly with chronic extravascular hemolytic processes. Gallstones (cholelithiasis) can occur whenever hemolysis is chronic; the constantly increased amount of bilirubin in the bile leads to the formation of the stones.15 When hemolysis is chronic in children, the persistent compensatory bone marrow hyperplasia can lead to bone deformities, because the bones are still forming (see Figure 27-3). For patients in whom an acquired, acute hemolytic process develops, the associated malaise, aches, vomiting, and possible fever may cause it to be confused with an acute infectious process. Profound prostration and shock may develop, particularly with acute intravascular hemolysis. Flank pain, oliguria, or anuria develops, which leads to acute renal failure.
Laboratory Findings
Tests of Accelerated Red Blood Cell Destruction
For a patient with a hemolytic process, a complete blood count (CBC) may provide clues to the cause. Spherocytes can be expected to be seen with extravascular hemolysis (see Table 22-2), whereas fragmented cells, or schistocytes, are noted with intravascular hemolysis. Other clues to the particular cause of the hemolysis may be present in the morphology, such as ring forms in malaria, sickle cells in sickle cell anemia, target cells and microcytes in thalassemia, and spherocytes in hereditary spherocytosis or immune-related hemolysis.
In either intravascular or extravascular hemolysis, the increased rate of hemoglobin catabolism results in increased amounts of the principal by-products, bilirubin and carbon monoxide. Most of the bilirubin in bilirubinemia is unconjugated (also called indirect bilirubin) (Box 22-3). If liver function is normal, conjugated bilirubin is formed and excreted as urobilinogen in the stool, and the serum level of conjugated bilirubin (also called direct bilirubin) remains within the reference range. No bilirubin is detected in the urine, because unconjugated bilirubin is bound to albumin and the complex is not filtered by the glomerulus. Urinary urobilinogen level may be increased, however, because there is increased urobilinogen in the stool, and more than usual amounts are absorbed by the portal circulation. Serum bilirubin values can be misleadingly low, because the amount of bilirubin in the blood depends on the rate of RBC catabolism and hepatic function. In some patients with hemolytic anemia, if the rate of hemolysis is low and liver function is normal, the total serum bilirubin level is within the reference range. Quantitative measurements of fecal urobilinogen would demonstrate an increase, however.
A substantial decline in serum haptoglobin level indicates intravascular hemolysis. In what is mostly an extravascular hemolysis, there can be a minor component of intravascular lysis involving spherical cells that are fragile, so a modest decline in haptoglobin level can be seen. Suffice it to say that, whenever the level of hemoglobin in the plasma increases, haptoglobin level declines. In one study, a low haptoglobin level indicated an 87% probability of hemolytic disease.16 Haptoglobin tests are prone to false positive and false negative results, however. Low values suggest hemolysis but may be due instead to impaired synthesis caused by liver disease. Alternatively, a patient with hemolysis may have a relatively normal haptoglobin level if there is also a complicating infection or inflammation, because haptoglobin is an acute phase reactant.
Tests to determine the rate of endogenous carbon monoxide production have been developed, because carbon monoxide is produced in the first step of heme breakdown. Values of 2 to 10 times the normal rate have been detected in some patients with hemolytic anemia, but the methods are too complex for most clinical laboratories.17
Other laboratory test results are incidentally abnormal. Serum lactate dehydrogenase activity is often increased in patients with intravascular hemolysis, but other conditions, such as myocardial infarction or liver disease, also can cause increases.18 Although enzyme fractionation points to an RBC origin of the increase, this test is generally not needed. Rather, when other results point to hemolysis, one should expect an increase in the levels of lactate dehydrogenase and other RBC enzymes and should not be misled into assuming that there is liver damage (see Table 22-2).
General evidence of reduced RBC survival can be gleaned by measuring glycated hemoglobin (by the Hb A1c test).19 Glycated hemoglobin increases over the life of a cell as it is exposed to plasma glucose. Glycated hemoglobin level is usually decreased in hemolytic disease, because the cells have less exposure to plasma glucose before lysis. The mean reference value for glycated hemoglobin is 6.7%; in a hemolytic process, the mean value is 3.9%.19 The magnitude of the decrease is related to the magnitude of the hemolytic process over the previous 4- to 8-week period. Glycated hemoglobin level is not a reliable indicator of shortened RBC survival in patients with diabetes mellitus because of the increased rate of glycation with elevated blood glucose levels.
A more exact RBC survival assay uses random labeling of blood with chromium radioisotope. This is the reference method for RBC survival studies published by the International Committee for Standardization in Haematology.20 A sample of blood is collected, mixed with the isotope, and returned to the patient. The labeled cells are of all ages, reflecting normal peripheral blood. This method differs from cohort labeling in which RBCs are labeled with radioactive iron as they are produced in the bone marrow, so that the labeled cells are generally of the same age. In both methods, the disappearance of the label from the blood is measured over time. As measured using the random chromium labeling technique, the normal half-time of chromium is 25 to 32 days.20 A half-time of 20 to 25 days suggests mild hemolysis; 15 to 20 days, moderate hemolysis; and less than 15 days, severe hemolysis. Erythrocyte life span determinations are time consuming and expensive, require the use of radioactive isotopes, and are rarely necessary for diagnosis except in patients with difficult diagnostic problems.
Tests of Increased Erythropoiesis
If the bone marrow is healthy, the hypoxia associated with hemolysis leads to increased erythropoiesis. Recognition of this increase may be a first clue to the presence of a hemolytic process. Laboratory findings indicating increased erythropoiesis are listed in Table 22-3. These findings are persistently present in chronic hemolytic disease and are evident within 3 to 6 days after an acute hemolytic episode. Increased erythropoiesis is not unique to hemolytic anemias and is not diagnostic. Similar results are expected after hemorrhage and with successful specific therapy for anemia caused by iron, folate, or vitamin B12 deficiency. An assessment of erythropoiesis can determine the effectiveness of the bone marrow response, however, and should be factored into the differential diagnosis (see Differential Diagnosis).
TABLE 22-3
Hematologic Findings Indicating Accelerated Red Blood Cell (RBC) Production
| Specimen | Findings |
| Anticoagulated peripheral blood | Increased reticulocyte count Rising mean cell volume (compared with baseline) Polychromasia, nucleated RBCs |
| Bone marrow | Erythroid hyperplasia |
Reticulocyte Count
The reticulocyte count is the most commonly used test to detect accelerated erythropoiesis and is useful for this purpose. An increased reticulocyte count and an increased reticulocyte production index (see Chapter 14) support a diagnosis of anemia secondary to blood loss or destruction of RBCs. When hemolysis is severe enough to produce anemia, the reticulocyte count is expected to be substantially increased. The association is so strong that if a patient has an elevated reticulocyte count, a cause of hemolysis should be sought. The increase usually correlates well with the severity of the hemolysis. Exceptions occur during aplastic crises of hemolytic anemias and in some immunohemolytic anemias with hypoplastic marrow, which suggests that the autoantibodies were directed against the bone marrow RBC precursors and circulating erythrocytes.10 Chapters 14 and 18 describe the interpretation of reticulocyte indices in patients with anemia.
Bone Marrow Examination
Bone marrow examination is usually not necessary to diagnose hemolytic anemia. If conducted, however, bone marrow examination will reveal erythroid hyperplasia. The myeloid-to-erythroid ratio decreases in hemolytic disease from a normal of about 3 : 1 as the erythroid component (the denominator) increases. (The myeloid-to-erythroid ratio is defined in Chapter 16.) As always, the cellularity of the bone marrow should be determined on a core biopsy specimen, rather than an aspirated specimen, for a more accurate judgment.
Laboratory Tests to Determine Specific Hemolytic Processes
A well-made blood film is helpful in determining the cause of hemolytic anemia. Certain abnormalities found on the film, such as spherocytes, elliptocytes, acanthocytes, burr cells, sickle cells, target cells, agglutination, erythrophagocytosis, or parasites, may help reveal the specific disorder causing the hemolysis (Table 22-4).
TABLE 22-4
Morphologic Abnormalities Associated with Hemolytic Anemia
| RBC Morphology | Hemolytic Disorders |
| Spherocytes | Hereditary spherocytosis, warm hemolytic anemia, thermal injury to RBCs |
| Elliptocytes (ovalocytes) | Hereditary elliptocytosis |
| Acanthocytes | Abetalipoproteinemia, severe liver disease |
| Burr cells | Pyruvate kinase deficiency, uremia |
| Schistocytes | Microangiopathic hemolytic anemia |
| Erythrophagocytosis | Damage to RBC surface, especially due to complement-fixing antibodies |
| RBC agglutination | Cold agglutinins, immunohemolytic disease |
Differential Diagnosis
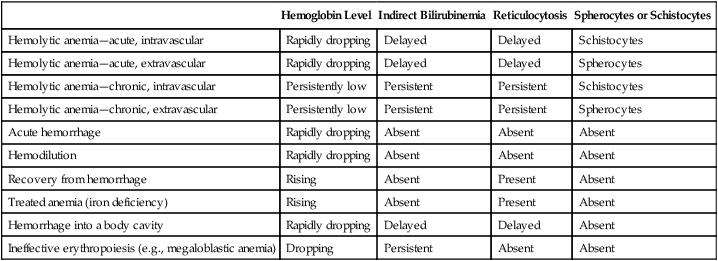
The differential diagnosis of hemolytic anemias incorporates several intersecting lines of deduction. The first is to establish the hemolytic nature of the anemia. A rapid decrease in hemoglobin concentration (e.g., 1 g/dL per week) from previously normal levels can signal hemolysis when hemorrhage and hemodilution have been ruled out. Jaundice and reticulocytosis provide additional confirmation of a hemolytic cause for an anemia of at least several days’ duration. When the indirect fraction of the total serum bilirubin is elevated, hemolytic jaundice is confirmed. An elevated urinary urobilinogen level strengthens the conclusion. The rapid decrease in hemoglobin during an acute hemolytic episode, however, usually is apparent before reticulocytosis and bilirubinemia develop. For acute hemolysis, hemoglobinemia and hemoglobinuria are expected with intravascular causes; therefore their absence suggests an extravascular cause. RBC morphology and haptoglobin levels can assist in differentiating an intravascular from an extravascular cause. Figure 22-5 is a graphic representation of the general timeline of the events in acute intravascular and extravascular hemolysis. In chronic hemolysis, persistence of hemoglobinemia, hemoglobinuria, decreased serum haptoglobin level, indirect bilirubinemia, and elevated reticulocyte count is expected, depending on the cause of the hemolysis.
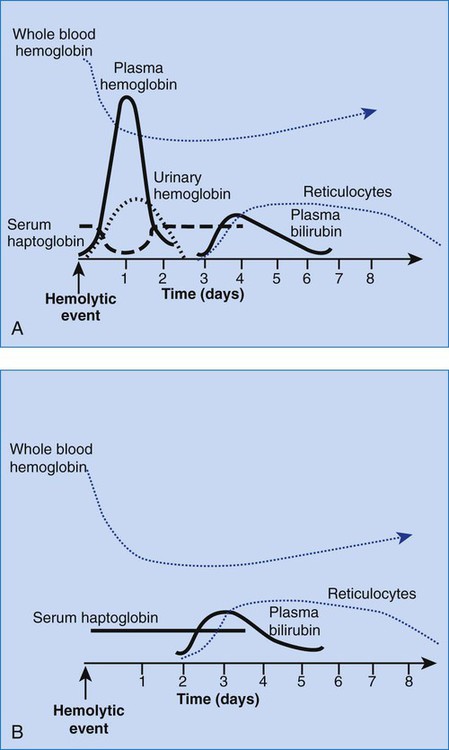
Hemolytic anemias must be differentiated from other anemias associated with bilirubinemia, reticulocytosis, or both. Anemia with reticulocytosis but without bilirubinemia is expected during recovery from hemorrhage not treated with transfusion or with effective treatment of deficiencies such as iron deficiency. Anemia that results from hemorrhage into a body cavity is characterized by reticulocytosis during recovery and bilirubinemia due to catabolism of the hemoglobin in the hemorrhaged cells. The RBC morphology should remain normal throughout the event. Anemias associated with ineffective erythropoiesis, such as megaloblastic anemia, are essentially hemolytic, with the cell death occurring in the bone marrow. Bilirubinemia and elevated serum lactate dehydrogenase levels are to be expected, but the reticulocyte count is low. Because most of the RBCs never reach the periphery, such anemias are typically classified as anemias of diminished production rather than as hemolytic anemias. As summarized in Table 22-5, the differential diagnosis in each of these instances may rely on negative results on tests for increased cell destruction or accelerated production.
TABLE 22-5
| Hemoglobin Level | Indirect Bilirubinemia | Reticulocytosis | Spherocytes or Schistocytes | |
| Hemolytic anemia—acute, intravascular | Rapidly dropping | Delayed | Delayed | Schistocytes |
| Hemolytic anemia—acute, extravascular | Rapidly dropping | Delayed | Delayed | Spherocytes |
| Hemolytic anemia—chronic, intravascular | Persistently low | Persistent | Persistent | Schistocytes |
| Hemolytic anemia—chronic, extravascular | Persistently low | Persistent | Persistent | Spherocytes |
| Acute hemorrhage | Rapidly dropping | Absent | Absent | Absent |
| Hemodilution | Rapidly dropping | Absent | Absent | Absent |
| Recovery from hemorrhage | Rising | Absent | Present | Absent |
| Treated anemia (iron deficiency) | Rising | Absent | Present | Absent |
| Hemorrhage into a body cavity | Rapidly dropping | Delayed | Delayed | Absent |
| Ineffective erythropoiesis (e.g., megaloblastic anemia) | Dropping | Persistent | Absent | Absent |
Summary
• A hemolytic disorder is a condition in which there is increased destruction of erythrocytes and a compensatory acceleration in erythrocyte production by the bone marrow.
• A hemolytic anemia develops when the bone marrow is unable to compensate for the shortened survival of the RBCs.
• Hemolytic anemias can be classified as acute or chronic, intravascular or extravascular, acquired or inherited, and intrinsic or extrinsic.
• Normally, most erythrocyte catabolism occurs extravascularly in the macrophages of the spleen, liver, and bone marrow. A small amount occurs intravascularly secondary to mechanical trauma.
• Hemoglobin from the RBCs is converted to heme and globin within macrophages. Heme is further degraded to iron, carbon monoxide, and unconjugated bilirubin. The bilirubin is secreted into the plasma, where it binds to albumin and is transported to the liver. In the liver, the bilirubin is conjugated with glucuronic acid, excreted as bile into the intestines, and converted to urobilinogen. Some urobilinogen is reabsorbed into the portal circulation and reexcreted through the liver. A small amount of urobilinogen remains in the plasma, and it is excreted by the kidney into the urine.
• In extravascular (macrophage-mediated) hemolytic anemia, there is an increase in unconjugated bilirubin in the plasma and an increase in urobilinogen in the stool and urine. Spherocytes may be seen on the blood film.
• Signs of intravascular hemolysis include hemoglobinemia, (met)hemoglobinuria, and hemosiderinuria. Serum haptoglobin is markedly decreased or absent. Schistocytes may be seen on the blood film.
• Jaundice results from increased serum unconjugated bilirubin and is associated with all hemolytic anemias.
• The major clinical features of chronic inherited hemolytic anemia are varying degrees of anemia, jaundice, the occurrence of crises, splenomegaly, and the development of cholelithiasis. In children, bone abnormalities develop as a result of accelerated erythropoiesis.
• Laboratory studies providing evidence of hemolytic anemia include tests for increased erythrocyte destruction and compensatory increase in the rate of erythropoiesis. The reticulocyte count is the most commonly used laboratory test to identify accelerated erythropoiesis. Elevated serum indirect (unconjugated) bilirubin level, with a normal serum direct (conjugated) bilirubin level and a decreased haptoglobin level, indicate accelerated RBC destruction. Other tests that are specific to a particular diagnosis also may be needed.
• The peripheral blood film evaluation can identify morphologic changes that point to an intravascular or extravascular etiology and to specific anemias. It is a valuable assessment in the differential diagnosis of anemia.
• Hemolytic anemias must be differentiated from other anemias with reticulocytosis, including the posthemorrhage state and recovery from iron deficiency.
Review Questions
1. The term hemolytic disorder in general refers to a disorder in which there is:
a. Increased destruction of RBCs after they enter the bloodstream
b. Excessive loss of RBCs from the body
2. RBC destruction that occurs when macrophages ingest and destroy RBCs is termed:
3. Signs of hemolysis that are typically associated with intravascular hemolysis only (and not extravascular hemolysis) include all of the following except:
4. An elderly white woman is evaluated for worsening anemia, with a decrease of approximately 0.5 mg/dL of hemoglobin each week. The patient is pale, and her skin and eyes are slightly yellow. She complains of extreme fatigue and is unable to complete the tasks of daily living without napping in mid-morning and mid-afternoon. She also tires with exertion, finding it difficult to climb even five stairs. Which of the features of this description points to a hemolytic cause for her anemia?
5. Which of the following tests provides a good indication of accelerated erythropoiesis?
6. A 5-year-old girl was seen by her physician several days ago and was diagnosed with pneumonia. Her mother has brought her to the physician again because the girl’s urine began to darken after the first visit and now is alarmingly dark. The girl has no history of anemia, and there is no family history of any hematologic disorder. The CBC shows a mild anemia, polychromasia, and a few schistocytes. This anemia could be categorized as:
7. A patient has a personal and family history of a mild hemolytic anemia. The patient has consistently elevated levels of total and indirect serum bilirubin and urinary urobilinogen. The serum haptoglobin level is consistently decreased, whereas the reticulocyte count is elevated. The latter can be seen as polychromasia on the patient’s blood film along with spherocytes. Which of the findings reported for this patient is inconsistent with a diagnosis of intravascular hemolysis?
8. Select the statement that is true about bilirubin metabolism.
a. Indirect bilirubin is formed in the liver by the addition of two sugar molecules to direct bilirubin.
b. Macrophages of the spleen liberate bilirubin during hemoglobin catabolism.
c. Urobilinogen is not water soluble and is not excreted in the urine.
d. Normally, the major fraction of bilirubin in the blood is the direct (conjugated) form released from macrophages.
9. A patient has anemia that has been worsening over the last several months. The hemoglobin level has been declining slowly with a drop of 1.5 g/dL of hemoglobin over about 6 weeks. Polychromasia and anisocytosis are seen on the blood film, consistent with the elevated reticulocyte count and RBC distribution width (RDW). Serum levels of total bilirubin and indirect fractions are normal. Urinary urobilinogen level also is normal. When these findings are evaluated, the conclusion is drawn that the anemia does not have a hemolytic component. Based on the data given here, why was hemolysis ruled out as the cause of the anemia?
a. The decline in hemoglobin is too gradual to be associated with hemolysis.
b. The elevation of the reticulocyte count suggests a malignant cause.
c. Evidence of increased protoporphyrin catabolism is lacking.
d. Elevated RDW points to an anemia of decreased production.
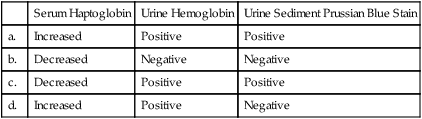
10. Which of the following sets of test results is typically expected with intravascular hemolysis?
| Serum Haptoglobin | Urine Hemoglobin | Urine Sediment Prussian Blue Stain | |
| a. | Increased | Positive | Positive |
| b. | Decreased | Negative | Negative |
| c. | Decreased | Positive | Positive |
| d. | Increased | Positive | Negative |