Chapter 30 Introduction to Genetics
In the broadest sense, genes are simply units of hereditary information; the genome is the totality of all the hereditary information in a cell or organism; and genetics may be defined as the study of genes and genomes. With the advent of modern molecular biology and the Human Genome Project, all aspects of genetics have come to play a more prominent role in the day-to-day evaluation and management of children with neurologic diseases, most of which have a genetic basis. This chapter presents a brief synopsis of the most important principles of genetics, to serve as background for information presented elsewhere in this text. More detailed information on genetics is available in many excellent textbooks, such as Genetics in Medicine [Nussbaum et al., 2007], Genes IX [Lewin, 2007], and Human Molecular Genetics [Strachan and Read, 2010]. Other resources are available from the National Center for Biotechnology Information website (Table 30-1).
Table 30-1 Genetic Information Websites
| Site | Internet Address |
|---|---|
| NCBI1 Genetic Disease Websites | |
| GeneTests, GeneReviews2 | http://www.ncbi.nlm.nih.gov/sites/GeneTests/ |
| OMIM3 | http://www.ncbi.nlm.nih.gov/omim/ |
| NCBI1 Genome Data Websites | |
| NCBI1 homepage (Entrez) | http://www.ncbi.nlm.nih.gov/ |
| dbGaP Genotypes and Phenotypes | http://www.ncbi.nlm.nih.gov/gap |
| dbSNP (SNP database) | http://www.ncbi.nlm.nih.gov/snp/ |
| Other Genome Data Websites | |
| Ensembl Human Genome Browser | http://uswest.ensembl.org/index.html |
| HUGO4 | http://www.genenames.org/index.html |
| DOE5 Genomics Websites, includes Human Genome Project | http://genomics.energy.gov/ |
| UCSC Genome Bioinformatics6 | http://genome.ucsc.edu/ |
1 National Center for Biotechnology Information.
2 Disease summaries in GeneReviews are authored by experts and peer-reviewed, and so are typically highly accurate and up to date.
3 Disease summaries in OMIM (Online Mendelian Inheritance in Man) are done by staff with oversight and contain both dated and new data; all information from OMIM should be confirmed from a second source.
4 The HUGO Gene Nomenclature Committee website established accepted names for human genes.
5 U.S. Department of Energy Office of Science websites, which include the Human Genome Project website.
6 University of California–Santa Clara Genome Bioinformatics site, which contains the most widely used human genome browser, sometimes called “Golden Path.”
Molecular Basis of Heredity
Structure and Function of DNA
DNA is a large polymer or macromolecule composed of linear sequences of simple repeating units. The specific sequence of these units contains all of the genetic information of an individual cell or organism. The structure of DNA in its native state was deduced by Watson and Crick in 1953 [Watson and Crick, 1953]. The basic repeating unit of DNA is the nucleotide, which consists of a five-carbon sugar known as deoxyribose; a phosphate group; and a nitrogen-containing base, which may be either a purine or a pyrimidine (Figure 30-1A). In DNA, the purine base may be either adenine (A) or guanine (G), and the pyrimidine base may be either thymine (T) or cytosine (C). Nucleotides polymerize into long chains by formation of phosphodiester bonds between the 5′ carbon position of one deoxyribose molecule and the 3′ carbon of the preceding deoxyribose molecule (Figure 30-1B).
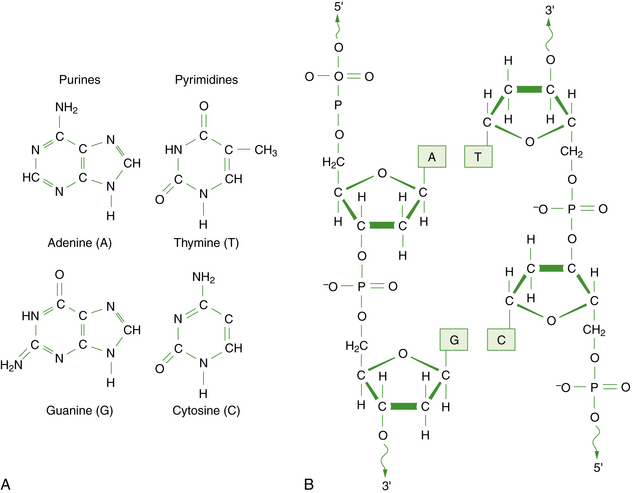
Fig. 30-1 The chemical structure of DNA.
A, The four bases of DNA. B, The sugar-phosphate backbone and 3′–5′ phosphodiester bonds.
Each DNA molecule consists of two strands of nucleotides that are held together by weak hydrogen bonds between pairs of bases: A pairs only with T, and G pairs only with C. These paired units are known as basepairs (bp). In the native state, the two strands wind around each other to form a double helix that resembles a right-hand spiral staircase, with two unequal grooves known as the major and minor grooves (Figure 30-2). A single turn of the helix measures 3.4 nm and contains ten nucleotides. Each strand has a directionality imparted by the deoxyribose sugar backbone. Adjacent nucleotides are linked by phosphodiester bonds between the 5′ and 3′ carbon atoms of the sugar residues, so that one end of the DNA strand has an unlinked 5′ carbon (the 5′ end) and the other end of the strand has an unlinked 3′ carbon atom (the 3′ end). The two strands are antiparallel – that is, they run in opposite directions so that the 5′ end of one strand is paired with the 3′ end of the other. Within living cells, DNA is associated with proteins and supercoiled into more complex structures known as chromosomes, which are described later in the chapter.
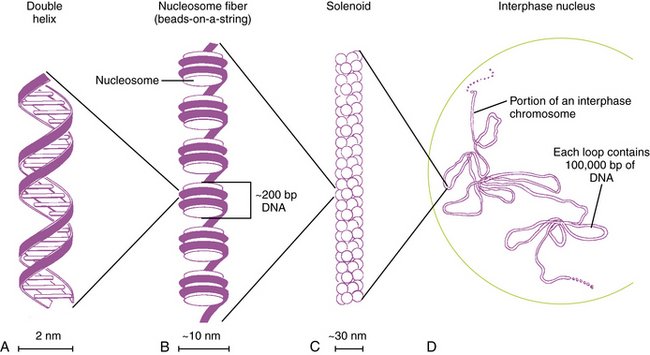
Fig. 30-2 Packaging of DNA by structural proteins.
(Modified from Thompson MR et al. Genetics in medicine, 5th edn. Philadelphia: WB Saunders, 1991.)
Five distinct DNA polymerases have been isolated in mammalian systems, including human cell cultures (Table 30-2). They are able to copy DNA only by adding nucleotides to the 3′ end of the growing chain, so DNA can elongate only in the 5′ to 3′ direction. Thus, the template DNA can be read only in the reverse, or 3′ to 5′, direction. As DNA is unwound, the replication fork necessarily unwinds one strand in the 3′ to 5′ direction and the other in the 5′ to 3′ direction. The 3′ to 5′ or leading strand is replicated in a continuous fashion at the replication fork by DNA polymerases α(I), which primes the reaction, and δ(III), which synthesizes the DNA chain. The new strand is complementary and so elongates in the opposite, or 5′ to 3′, direction.

Structure and Function of RNA
RNA differs chemically from DNA in the substitution of ribose for deoxyribose in the sugar backbone of the molecule, and of uridine (U) for thymine as one of the pyrimidine bases. Also, RNA normally exists as a single-stranded rather than double-stranded molecule. Recent advances have demonstrated far more diverse functions for RNA than were previously appreciated, particularly involving genes that produce functional RNA products that do not code for proteins. These probably represent at least 5 percent of all human genes, as suggested by current knowledge [Strachan and Read, 2010]. Several distinct classes of RNA molecules have been recognized, most of which are involved with regulating or assisting gene expression.
MicroRNA
MicroRNAs (miRNAs) are another class of small noncoding genes that regulate the expression of protein-encoding genes at the post-transcriptional RNA level [Denli et al., 2004]. The process begins with transcription (synthesis) of primary RNA transcripts that range in size from several hundred to several thousand kb. These transcripts are recognized and cut into precursor miRNAs in the nucleus by a protein known as Dicer, moved to the cytoplasm, and processed into mature miRNAs. The mature miRNAs join the RNA-induced silencing complex (RISC), which recognizes and cleaves (or otherwise silences) a target gene. This process has been demonstrated in many organisms, including mammals, and appears likely to play a key role in regulation of many genes.
Structure and Function of Polypeptides and Proteins
Proteins are composed of one or more polypeptide chains. Polypeptides are large polymers or macromolecules composed of linear sequences of repeating units known as amino acids, which are more complex than the repeating units of DNA or RNA. Amino acids consist of a three-carbon backbone, with an amino group attached to carbon 1 and a carboxyl group to carbon 3. They differ in the composition of a side chain attached to carbon 2. With rare exceptions, all polypeptides and proteins in nature are built from different sequences of 20 amino acids (Table 30-3). The side chains may be neutral and hydrophobic, neutral and polar, basic, or acidic. The simplest amino acid is valine, which has a hydrogen ion as the side chain.
| Amino Acid | 3-letter Code | 1-letter Code |
|---|---|---|
| Neutral and Hydrophobic | ||
| Alanine | Ala | A |
| Isoleucine | Ile | I |
| Leucine | Leu | L |
| Methionine | Met | M |
| Phenylalanine | Phe | F |
| Proline | Pro | P |
| Tryptophan | Trp | W |
| Valine | Val | V |
| Neutral and Polar | ||
| Asparagine | Asn | N |
| Cysteine | Cys | C |
| Glutamine | Glu | Q |
| Glycine | Gly | G |
| Serine | Ser | S |
| Threonine | Thr | T |
| Tyrosine | Tyr | Y |
| Acidic | ||
| Aspartic acid | Asp | D |
| Glutamic acid | Glu | E |
| Basic | ||
| Arginine | Arg | R |
| Histidine | His | H |
| Lysine | Lys | K |
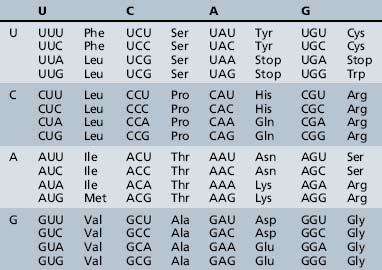
The process of information transfer from RNA polypeptides to proteins is known as translation. It relies on the genetic code, the system by which the nucleotide sequence of mRNA specifies the amino acid sequence of a polypeptide chain. In this nearly universal code, each set of three adjacent bases in the mRNA transcript constitutes a codon, and different combinations of bases within the codon specify the individual amino acids (Table 30-4). The small tRNA molecules serve as the molecular link between mRNA codons and amino acids. One segment of each tRNA transcript contains a three-base anticodon that is complementary to a specific codon on the mRNA, whereas another segment contains a binding site for one of the 20 amino acids.
Gene Structure and Organization
As noted earlier, a gene traditionally has been defined as a unit of genetic information. This concept has gradually progressed to a more useful definition, which states that a gene is a sequence of DNA on a chromosome that is required for production of a functional product, which can be either a protein or a functional RNA molecule [Nussbaum et al., 2007]. By convention, genetic information is always read in the 5′ to 3′ direction, whether encoded in DNA or RNA – in an upstream to downstream direction. The nomenclature regarding the 5′ and 3′ positions of the sugar backbone can be confusing. The 5′ carbon of the first nucleotide of a sequence is joined by a phosphodiester bond to a nucleotide not involved in the sequence, whereas its 3′ carbon is joined to the 5′ carbon of the second nucleotide, and so on. The last nucleotide of the sequence has a 3′ carbon, which joins another uninvolved nucleotide.
Genes
A model of a typical human gene is shown in Figure 30-3. Promoter sequences required for regulation and initiation of RNA transcription (red diamonds in Figure 30-3) are present at the 5′ end of the gene, such as the CAT and TATA boxes whose sequences are tightly conserved among many different genes and species. Downstream from the promoter sequences is a specific sequence that signals the start of transcription. A short way further downstream is an initiator codon, AUG, which codes for methionine. This triplet is the translation start site, which signals the start of the coding sequence for the polypeptide product. The region between the transcription and translation start sites is the 5′ UTR.
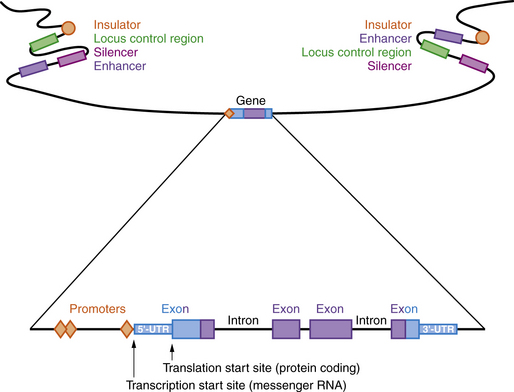
Regulatory Regions
Many genes have highly conserved sequences, a longer distance upstream and downstream of the transcribed gene, that are involved in regulating expression, including enhancers, silencers, locus control regions, and insulators (see Figure 30-3). Enhancer elements function to increase gene expression, while silencers reduce gene expression. Locus control regions may regulate expression of several genes within a chromosome region, while insulators prevent co-regulation of more distant genes and gene regions. All of these are sequences that bind proteins called transcription factors, which can be ubiquitous, tissue-specific, and/or temporally expressed. Promoters are located immediately 5′ of the gene and bind to RNA polymerase II, a necessary step for transcription. Other transcription factors bind upstream of the promoter and activate transcription. Enhancers and silencers are often located at a distance from the promoter, and increase or decrease transcription in a tissue-specific or temporal manner. Overall, the transcription of each gene is tightly regulated, with multiple transcription factors involved.
RNA Processing
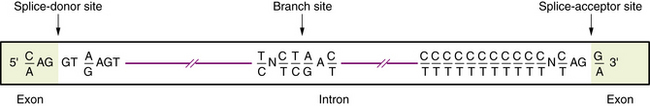
Transcription of DNA gives rise to a precursor RNA that corresponds exactly to the genome sequence but must be modified in several ways to become functional, especially for mRNA. The first modification to mRNA is the addition of a CAP structure to the 5′ end and this is followed by the removal or splicing of introns. The mechanism of mRNA splicing depends on the specific nucleotide sequences at the exon/intron boundaries called splice junctions (Figure 30-4). The most important of these is the GT-AG rule: introns almost always start with GT (actually GU, because this occurs in RNA), which is therefore called the splice-donor site, and end with AG, which is called the splice-acceptor site. Several additional specific sequences are also needed, including sequences within the intron just after the GT splice-donor site, at a highly conserved branch site located about 40 bp before the end of the intron and just before the AG splice-acceptor site. The splicing mechanism produces the following:
Imprinting and X-Inactivation
Imprinting
The process by which certain genes in specific chromosomal regions are expressed from only one chromosome, depending on the parental origin of the chromosome, is known as “imprinting.” Although the mechanism is only partly understood, a key component involves allele-specific DNA methylation, found predominantly at the carbon 5 position of about 80 percent of all cytosines that are part of symmetrical cytosine-guanine (CpG) dinucleotides [Jiang et al., 2004; Strachan and Read, 2010; Weksberg et al., 2003].
This process is controlled by regulatory imprinting “centers,” located nearby on the same chromosome as that of the silenced or “imprinted” gene. In effect, then, two alleles of the same gene that are identical in nucleotide sequence but derived from opposite parents are regulated differently in the same nucleus. This process is reversible, so that the silent, imprinted allele can be reactivated and the active allele silenced when passed through the germline of the opposite-sex parent. Most imprinted genes are found in large clusters of greater than 1 Mb (megabase pairs) in length. Imprinted clusters have been identified in chromosomes 6q24, 7p11.2, 11p15.5, 14q32, 15q11–q13, and 20q13.2, and others may exist as well [Cavaille et al., 2002; Gardner et al., 2000; Hall, 1990; Jiang et al., 2004; Weksberg et al., 2003; Wylie et al., 2000]. Imprinted regions share several common characteristics, including differential DNA methylation, allele-specific RNA transcription, antisense transcripts, histone modifications, and differences in timing of replication.
X-Inactivation
In mammalian cells with two (or more) X chromosomes, all but one undergo widespread gene silencing by methylation. This phenomenon, known as X-chromosome inactivation (Xi), causes one of the two X chromosomes in cells of female mammals to become transcriptionally inactive early in embryonic development, a phenomenon known as the Lyon hypothesis [Lyon, 1961, 2002]. In mutant cells with more than two X chromosomes, all but one become inactivated. This has the effect of balancing gene dosage of X-linked genes between male and female cells. The process of Xi is random, so that on average the maternally and paternally derived X chromosomes are each inactivated in approximately 50 percent of cells. Changes in this pattern are seen in female carriers of some X-linked diseases, resulting in skewing of Xi. This alteration can be favorable, with decreased severity of the phenotype, or unfavorable, with increased severity of the phenotype [Dobyns et al., 2004].
Cell Cycle and Chromosomal Basis of Heredity
Cell Cycle
Mitosis
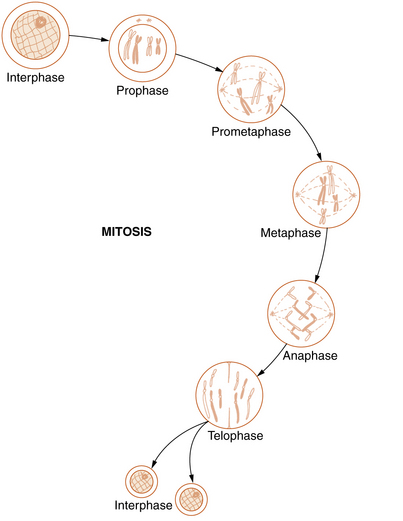
Somatic cell division, or mitosis, is an elaborate mechanism that distributes one chromatid of each duplicated chromosome to each of the two daughter cells. The process is continuous but has been divided into the following five stages: prophase, prometaphase, metaphase, anaphase, and telophase (Figure 30-5).
Meiosis
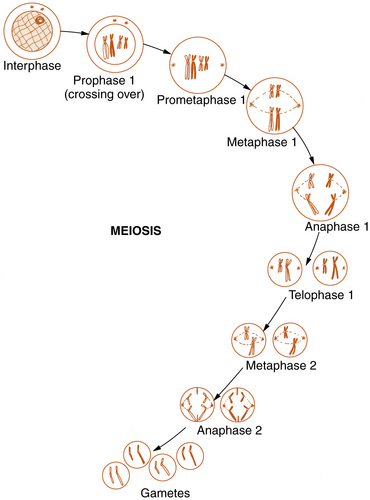
Reproductive cell division, or meiosis, is an even more complex mechanism in which two successive cell divisions, known as meiosis 1 and meiosis 2, give rise to the haploid germ cells (Figure 30-6). Meiosis is of critical importance in understanding many of the methods of modern molecular genetics and the pathogenesis of many genetic diseases.
Chromosomal Basis of Heredity
Chromosome Structure
In humans, the nuclear DNA is dispersed among 46 separate linear structures or chromosomes, each of which consists of a single, uninterrupted double helix that contains 50–250Mb of DNA, and a group of associated proteins that form the support structure or scaffolding. The scaffolding consists of five basic proteins called histones and several more acidic nonhistone proteins. Two copies of each of four histones – H2A, H2B, H3, and H4 – join to form an octamer. The DNA double helix wraps almost twice around the octamer, which involves about 140 bp. Adjacent octamers are separated by a short spacer segment of 20–60bp that is associated with histone H1. The complex of DNA and core histones is known as a nucleosome (see Figure 30-2).
Strings of nucleosomes are further compacted into a secondary helical structure known as a solenoid. These structures have a diameter of about 30 nm (see Figure 30-2) and contain six nucleosomes per turn. The solenoids are packed into large loops of 10–100 kb of DNA, which are attached to a nonhistone protein scaffolding. These loops pack together loosely to form interphase chromosomes. During early prophase, they pack together more closely to form knoblike thickenings known as chromomeres, which then coalesce further to form the bands observed in prometaphase and metaphase chromosomes when stained with appropriate dyes.
Specialized Regions
Origins of replication are specialized sequences where DNA replication begins, and thus are important in maintaining chromosome number and integrity. They consist of autonomously replicating sequence elements that contain a core consensus sequence and some imperfect copies with a length of about 50 nucleotides. A consensus human autonomously replicating sequence has been identified [Strachan and Read, 2010].
Chromosome Number
Each human somatic cell contains 46 chromosomes that consist of 22 matched pairs known as autosomes and two sex chromosomes: XX in females and XY in males (Figure 30-7). In contrast, human germ cells contain only 23 chromosomes, consisting of 22 unpaired autosomes and a single sex chromosome. The former is known as the diploid or 2n number, and the latter is known as the haploid or 1n number. The autosomes were numbered according to length, with chromosome 1 the longest and chromosome 22 thought to be the shortest. Although chromosome 21 later proved to be shorter than chromosome 22, the numbers were retained for historical reasons. The two members of each pair of autosomes and the two X chromosomes in females carry the same genes and are known as homologous chromosomes, or homologs. Although they appear similar under the microscope, homologs are not strictly identical. They contain the same genes, but the nucleotide sequence differs at thousands of positions.

Organization of the Human Genome
The human genome comprises the total of all genetic information in the cell. It is divided into two separate compartments – a large and complex nuclear genome and a much smaller and simpler mitochondrial genome. The mitochondrial genome consists of a single circular DNA molecule that is present in many copies in each mitochondrion, while the nuclear genome is distributed among the 46 nuclear chromosomes. The available data regarding the genome have become much more extensive and accurate with completion of the Human Genome Project. A few of the most useful Human Genome Project-related websites are listed in Table 30-1.
The Nuclear Genome
The human nuclear genome consists of approximately 3 × 109 bp, or 3000 Mb of DNA. About 75 percent of this represents unique or single-copy DNA, which includes genes and some important regulatory elements. The remaining 25 percent consists of several classes of repetitive DNA [Lander et al., 2001; Nussbaum et al., 2007; Venter et al., 2001].
Genes and Conserved Noncoding DNA
Somewhat surprisingly, recent estimates predict that the human genome contains less than 30,000 protein-coding genes (possibly closer to 20,000) and an uncertain number of other genes producing functional RNA products. This is far fewer than earlier estimates, and accounts for only about 1.2 percent of nuclear DNA [Lander et al., 2001; Venter et al., 2001]. Another 5 percent of the human genome is more conserved than would be expected from estimates of neutral evolution, which suggests that many of these regions have specific, regulatory functions [Chiaromonte et al., 2003; Waterston et al., 2002]. Studies of these highly conserved regions of DNA have used different thresholds, such as stretches of more than 100 bp with 70–80 percent conservation between mouse and human. Some of these regions have been found to contain important noncoding elements [Dermitzakis et al., 2002, 2003; Frazer et al., 2004; Hardison, 2000]. More stringent analysis demonstrates that the human genome contains 481 sequences of 200 or more bp that are 100 percent conserved among human, mouse, and rat [Bejerano et al., 2004]. These segments were designated “ultra-conserved elements,” and are preferentially located near genes involved in RNA processing or regulation of transcription and development. Similarly, about 5000 sequences of 100 bp or more are conserved among these three species, which emphasizes that noncoding sequences are common and important.
Low Copy Repeats
Segmental duplications, also known as low copy repeats (LCRs), are DNA sequences of 10–250 kb, present in multiple copies with greater than 95 percent sequence identity, that make up approximately 5 percent of the human genome [Babcock et al., 2003; Bailey et al., 2002; Cheung et al., 2001; Stankiewicz and Lupski, 2002]. LCRs are dynamic regions of the genome because specific repeats tend to cluster within the same genomic regions, where they mediate unequal nonhomologous recombination events, producing segmental deletions and duplications that are collectively designated “copy number variants” (CNVs). Several of these have been associated with well-known developmental disorders in humans, such as Williams’ syndrome in 7q11.23, Angelman’s syndrome and Prader–Willi syndrome in 15q12, hereditary neuropathy with predisposition to pressure palsies and Charcot–Marie–Tooth neuropathy type 1A in 17p12, Smith–Magenis syndrome in 17p11.2, and DiGeorge’s syndrome in 22q11.2 [Babcock et al., 2003]. Many new CNV-associated devlopmental brain disorders have been described over the past few years.
Polymorphisms
A mutation is a permanent change in the DNA of an individual organism, specifically a change in the nucleotide sequence anywhere in the genome [Nussbaum et al., 2007]. Genetic diseases and many cancers are caused by mutations that adversely affect function of one or more genes, although most mutations have little or no effect on gene function and therefore do not change the survival or reproductive fitness of an individual. Some of these persist in the population as morphologic variants known as polymorphisms. Sequence changes that have frequencies of less than 1 percent are known as rare variants, whereas those with frequencies of 1 percent or more are known as polymorphisms. By convention, a genetic polymorphism is defined as the occurrence of two or more variants or alleles in a region of DNA where at least two alleles appear with frequencies greater than 1 percent. Several different classes of polymorphisms occur in the genome, and several methods in molecular biology take advantage of the normal variation between individuals.
Microsatellites
The most common microsatellite family consists of 50,000–100,000 cytosine-adenine (CA) repeats, which consist of short tandem repeats of the dinucleotide CA on one strand and guanine-thymine on the complementary strand. They thus take the form (CA)n/(GT)n, with n in the range of 6–30 [Weber and May, 1989]. The number of repeats within a (CA)n block varies greatly among different members of a species, producing a set of alleles that always differ in size by multiples of two bases. About 70 percent of the human population is heterozygous at any given (CA)n repeat locus, making these highly polymorphic. The human genome contains about 50,000–100,000 interspersed (CA)n blocks, which is enough to place 1 block every 30–60 kb, if evenly spaced.
Single-Nucleotide Polymorphisms
Single-nucleotide polymorphisms (SNPs, pronounced “snips”) are DNA sequence variations that occur when a single nucleotide (A, T, C, or G) in the genome sequence is changed. For example, a SNP might change the DNA sequence TCACG to TTACG. The most common sequence change involves replacement of cytosine (C) with thymidine (T), which accounts for about two-thirds of all SNPs. As with other types of sequence variation, a SNP must occur in at least 1 percent of the population to be classified as a polymorphism. SNPs occur in both unique-sequence (coding and noncoding) and repetitive DNA, and are responsible for about 90 percent of human genetic variation. On average, SNPs are found approximately every 100–300 bases along the entire human genome. Although most SNPs likely have no function, some are known to influence disease predisposition or responses to drugs, and thus are proving to be very valuable in studying the causes of common human diseases. The current inventory of known SNPs can be found in the Human SNP database (dbSNP) on the NCBI Entrez website (see Table 30-1).
Mitochondrial Genome
Mitochondria are cellular organelles that are primarily responsible for cellular respiration and production of adenosine triphosphate. Each cell contains numerous mitochondria, and each mitochondrion contains many copies of a small 16.5-kb circular chromosome, adding up to thousands per cell. The mitochondrial chromosome contains 37 genes that code for two types of rRNA, 22 types of tRNA, and 13 polypeptides. The two DNA strands differ significantly in base composition, with a heavy strand rich in guanines that codes for 28 genes, and a light strand rich in cytosines that codes for 9 genes. It is very densely packed, with 93 percent comprising coding sequence [Strachan and Read, 2010].
Human Genome Project
The importance of DNA, including both genes and noncoding regions, became increasingly apparent during the 1970s and 1980s, leading to one of the most ambitious scientific research projects ever undertaken – a plan to sequence the entire human genome. This project, which was begun in 1990, came to be known as the Human Genome Project. The goals of the project, as taken from the Human Genome Project website (see Table 30-1), were as follows:






Technology of Cytogenetics
Chromosome Analysis
When methods for examining chromosomes under the microscope were first developed, individual chromosomes could not be identified because of solid staining. Instead, they were separated into seven groups (A to G), based on their length and centromere position. It is now possible to identify all 24 human chromosomes individually, using several different staining techniques that take advantage of differences in chromatin structure and composition to produce a recognizable pattern of bands, as shown in the diagram in Figure 30-7. These methods are now used to examine the entire chromosome complement of an individual, which is known as the karyotype. The same term is used to describe the normal chromosome complement of a species.
A uniform system of human chromosome classification and nomenclature was developed at a series of international conferences, and most recently revised in 2009 [ISCN, 2009]. In this system, the chromosomes are separated into regions and subregions, based on the banding pattern. For example, band 17p13.3 (read as “17-p-one-three-point-three”) is found near the telomere of the short arm of chromosome 17. During the past decade, computer image analysis systems have been developed that can locate chromosome spreads on the slide, recognize and automatically sort chromosomes, and help with analysis. However, review by trained cytogenetic technicians is still required.
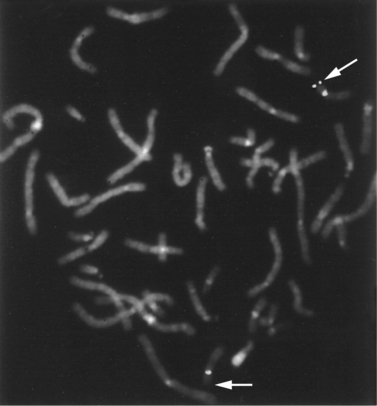
Fluorescence In Situ Hybridization
This study usually is performed on metaphase chromosomes (Figure 30-8) but also can be used on cells in interphase. Interphase FISH often is used for rapid detection of specific types of aneuploidy in fetal cells and for detection of certain deletions, duplications, and other abnormalities in tumor cells. In contrast with metaphase FISH, interphase FISH does not permit visualization of the actual chromosomes, so that most types of structural rearrangements cannot be detected. FISH can be used to examine a small set of chromosomal regions at once, usually 1 or 2, although study of 8–10 is possible with special fluorescent markers. Telomere-specific FISH analysis is an example of hybridization with multiple probes simultaneously. Telomere-specific probes that correspond to the telomeres of all of the chromosomes are hybridized to metaphase chromosomes in groups and used to detect abnormalities at the ends of chromosomes that are not visible by routine chromosome analysis.
Chromosome Microarrays
Array formats for CGH have been developed that have increased the resolution of this technique for detecting smaller deletions and duplications. Instead of hybridizing on to metaphase chromosome spreads, a set of DNA probes across the entire genome is used. The probes are placed on microarrays that can detect DNA fragments with the same sequence as for the probe. Several different methods have been developed using different probes, such as bacterial artificial chromosome (BAC) DNA, complementary DNA (cDNA), or DNA fragments produced by cleavage of genomic DNA by the restriction enzyme BglII [Ishkanian et al., 2004; Lucito et al., 2003; Sebat et al., 2004]. These offer different resolution, with probes every 15–100 kb approximately. The total number of probes has increased from approximately 40,000 to 1 million on commericially available arrays. CGH technology has numerous advantages over FISH, including coverage of the entire genome, finer resolution, and lower cost per probe tested. Thus, clinical tests based on CGH technology have begun to replace FISH technology.
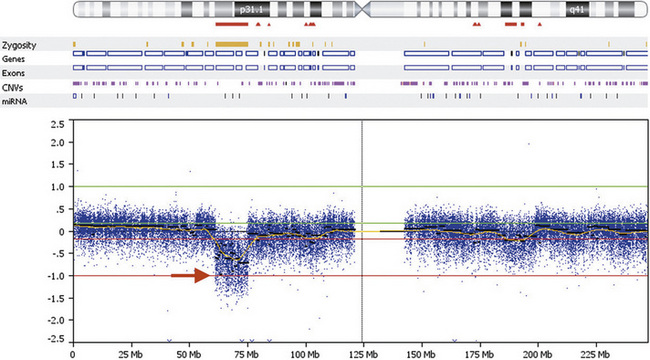
DNA gains and losses may also be detected using the same SNP-based microarrays in common use for genotyping. They are used to measure intensity differences and ratios of alleles at up to 1 million single nucleotides across the genome, detecting many CNVs, as well as detecting intercellular mosaicism and copy-neutral loss of heterozygosity, as occurs with uniparental disomy and other rare mechanisms. In general, the cost per probe is lower with SNP-based microarrays than for CGH-based microarrays, but the results are easier to interpret for CGH-based arrays due to more favorable signal-to-noise ratio. An example of a 14-Mb deletion of human chromosome 1 is shown in Figure 30-9.
Technology of Molecular Genetics
Molecular genetics is that branch of genetics concerned with the structure and function of genes at the molecular DNA level. The rapid gains in this field during the past decade have resulted from discovery of several new techniques that have made detailed analysis of both normal and abnormal genes possible. These discoveries have in turn led to better understanding of many important biologic processes, as well as the molecular basis for many genetic diseases. Several of these methods have proved to be of particular importance and are commonly used in research studies. Some familiarity with these procedures is helpful in understanding the nature and significance of new discoveries in this area. This section presents a brief introduction to some of the more important procedures. More detailed information can be found in several laboratory manuals, especially Current Protocols in Human Genetics [Haines et al., 2010].
DNA Clones
Several common types of vectors have been used, including phage (bacterial virus) (up to 20-kb insert DNA), plasmids (accessory circular bacterial chromosomes, used to clone several kb of DNA), cosmids (approximately 35–45 kb), BACs (approximately 100–150 kb), P1 plasmid artificial chromosomes (PACs, approximately 100–150 kb), and yeast artificial chromosomes (YACs, up to 1000 kb). The most commonly used at present are BACs and PACs [Stein, 1997], which have proved to be useful for FISH, CGH, and many other technologies. Creation of chromosome-specific DNA libraries by cloning followed by mapping and sequencing is the basis of the information obtained through the efforts of the Human Genome Project.
Restriction Enzymes
Restriction enzymes or endonucleases are bacterial enzymes that recognize short, double-stranded DNA sequences and cut the DNA molecule at or near the recognition site [Lewin, 2007]. When a mutation occurs that changes as few as one of the basepairs in the sequence, it is no longer recognized and cut by the enzyme. Several hundred restriction endonucleases have been isolated. Most of the recognition sites are palindromes, which means that they read the same in the 5′ to 3′ direction on both strands, and most of the enzymes leave short overhangs of single-stranded DNA that are known as sticky ends. For example, the enzyme BamHI recognizes the sequence GGATCC and cuts it between the two G bases, leaving the following 4-base overhang:

Polymerase Chain Reaction
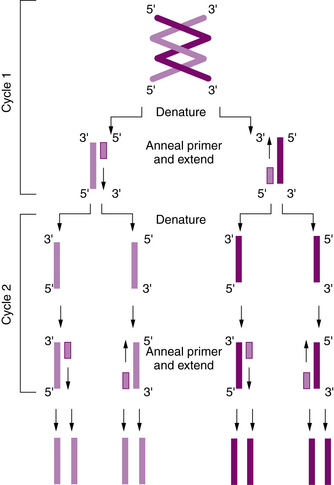
To construct the primers, a short sequence of about 20–25 bp just upstream or 5′ of the target sequence on the DNA “sense” strand is chosen as a starting site, and an oligonucleotide (or primer) that is complementary to this short upstream sequence is synthesized. Another short sequence upstream or 5′ of the target sequence on the complementary (or “antisense”) strand also is chosen, and a second complementary oligonucleotide is synthesized. The two primers thus flank the region of interest on opposite strands. The DNA is denatured to separate the strands, after which the oligonucleotides are hybridized to the complementary sequences. The short oligonucleotides then serve as primers for synthesis of a complete complementary DNA strand with appropriate deoxynucleotide triphosphate molecules (adenosine, cytosine, guanosine, and thymidine triphosphate [dATP, dCTP, dGTP, and dTTP]) being added; this is mediated by the enzyme DNA polymerase. Because both strands are copied, one round of amplification results in a complete second copy of the original target sequence. Repeated cycles of heat denaturation, hybridization of the primers, and DNA synthesis result in the exponential amplification of the target sequence. Within a few hours, more than a million copies of the sequence may be made (Figure 30-10).
Methods of General Mutation Detection
DNA Sequence Analysis
Sanger sequencing
DNA sequence analysis is the most sensitive and direct method to detect mutations at the level of individual nucleotides [Haines et al., 2010]. The most widely used method of DNA sequencing is the Sanger method, also known as dideoxy sequencing or chain termination. It is based on the use of synthetic nucleotide analogs – 2,3-dideoxynucleoside triphosphates (ddNTPs). Dideoxy NTPs differ from nucleotides found in natural DNA in that they lack the 3′-hydroxyl group. When integrated into a sequence, they prevent the addition of further nucleotides as phosphodiester bonds cannot form between a dideoxynucleotide and the next incoming nucleotide. Thus, the DNA chain is terminated.
High-throughput sequencing
High-throughput sequencing, also known as next-generation or second-generation sequencing, is a much more high-throughput form of DNA sequencing that is revolutionizing the field of genetics and resulting in the ability to sequence entire genomes at a fraction of the cost and time compared to Sanger-based sequencing [Haines et al., 2010]. The basis of second-generation sequencing is cyclic-array sequencing, which is the sequencing of a dense array of DNA features by repetitive cycles of enzymatic reactions and imaging-based data collection. At the time of writing, there are three main commercially available platforms for second-generation sequencing, which include: Solexa technology (used by Illumina), 454 sequencing (used by Roche Applied Science), and the SOLiD platform (used by Life Technologies). While each of these platforms differs with regard to the biochemistry of the sequencing reaction and the generation of the array, the overall concept is similar. This is as follows:
Mutation Scanning
Various methods of mutation scanning exist that differ in their sensitivities of mutation detection [Cotton, 1997; Eng and Vijg, 1997; Grompe, 1993]. The general basis of most mutation scanning methods is the abnormal migration of a DNA fragment that contains a sequence change from a normal “wild-type” sequence. All are PCR-based methods – the DNA region to be studied is amplified by PCR before the different mutation scanning methods are performed.
Methods for Detecting Specific Sequence Changes (Genotyping)
Clinical Cytogenetics
Abnormalities of Chromosome Structure
Structural chromosome rearrangements consist of loss, gain, or altered position of segments of chromosomes, and many different types have been recognized. The estimated frequency is about 1 per 1700 cell divisions, making them much less frequent than aneuploidy. Rearrangements are termed balanced if the chromosome complement has a normal amount of genetic information, regardless of its location. They are termed unbalanced if there has been either loss or gain of DNA sequence. The phenotypic effects often are severe. The chromosomes involved in the reconfiguration are known as derivative chromosomes. Many different types of rearrangements have been reported, as described in the following sections. Only those derivative chromosomes that contain a functioning centromere and telomeres, however, are stable and capable of being transmitted unaltered to daughter cells during mitosis or meiosis. Derivatives lacking a centromere or telomere are unstable and are lost during cell division. Some regions of the genome, such as 8p, contain a noncentromeric sequence that is sufficiently similar to function as a “neocentromere” during cell division [Giglio et al., 2001].
Mechanisms
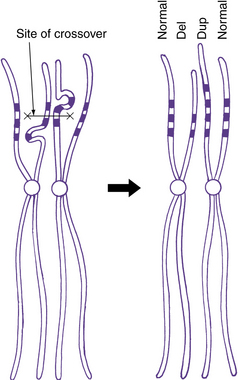
Some progress has been made recently in current understanding of the mechanisms causing or at least predisposing to structural chromosome rearrangements. Many appear to occur randomly, with no evidence of recurrent breakpoints. For example, no consistent breakpoints have been found for the interstitial deletions and reciprocal translocations involving chromosome band 17p13.3 [Cardoso et al., 2003]. In many other locations, the small duplicated regions known as LCRs can mediate several different types of rearrangements. These were first identified as the cause of common deletion or microdeletion syndromes, such as deletion (del) 2q13 with juvenile nephronophthisis and Joubert’s syndrome-related disorder [Parisi et al., 2004; Saunier et al., 2000], 7q11.23 in Williams’ syndrome [Osborne et al., 2001; Urban et al., 1996], del 15q11.2–q13 in Angelman’s syndrome and Prader–Willi syndrome [Amos-Landgraf et al., 1999], del17p12 in hereditary neuropathy with predisposition to pressure palsies [Chance et al., 1994], del17p11.2 in Smith–Magenis syndrome [Chen et al., 1997], and del22q11.2 in DiGeorge’s syndrome/velocardiofacial syndrome [McDermid and Morrow, 2002]. The same LCRs are associated with duplications of the same regions, including duplication 15q11.2–q13 [Mohandas et al., 1999], 17p12 [Pentao et al., 1992], and 17p11.2 [Potocki et al., 2000]. A simple diagram of this mechanism is shown in Figure 30-11.
Many of the LCRs are inverted with respect to each other, which can lead to very complex combinations of deletions and duplications. This can occur for LCRs on the same chromosome, as seen with Williams’ syndrome and some X chromosome rearrangements [Giglio et al., 2000; Osborne et al., 2001], or between homologous chromosomes. The latter appears to be more common when two matching LCRs on homologous chromosomes are inverted with respect to each other, a novel type of polymorphism [Giglio et al., 2001]. Similar mechanisms also can predispose to structural rearrangements between completely different chromosomes involving either an LCR or a gene cluster, such as olfactory receptor clusters [Giglio et al., 2002; Spiteri et al., 2003].
Balanced and Unbalanced Chromosomal Rearrangements
As noted previously, structural chromosome rearrangements that result in no net loss or gain of genomic sequence are balanced, whereas those that do result in a net loss or gain of material are unbalanced. Persons with balanced rearrangements usually are normal, unless one of the chromosomal breaks disrupts an important gene. More recent studies, however, have found that chromosome rearrangements that appear balanced often have submicroscopic loss or gain of material and so are actually unbalanced [Astbury et al., 2004b], and some chromosome rearrangements are more complex than standard chromosome analysis suggests [Astbury et al., 2004a].
Specific Types of Chromosome Rearrangements
The most common structural rearrangements include terminal and interstitial deletions and duplications, reciprocal and robertsonian translocations, inversions, and rings. Examples of most of these are shown in Figure 30-12.
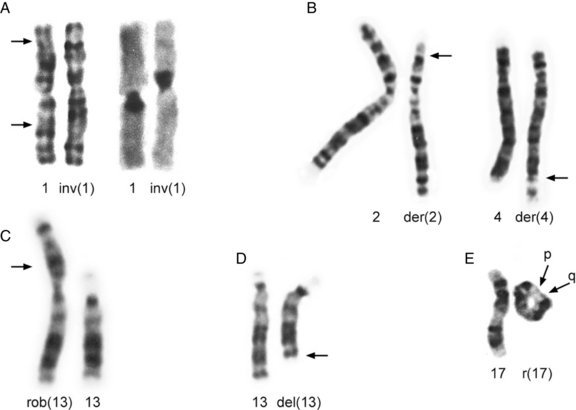
Fig. 30-12 Partial karyotypes of structural chromosome rearrangements.
(Karyotypes in A from Johnson DD et al. Hum Genet 1988;78:315; those in B, C, and D courtesy of BA Hirsch, Department of Laboratory Medicine and Pathology, University of Minnesota Medical School; karyotype in E from Dobyns WB et al. J Pediatr 1983;102:552.)
Deletions and Duplications
A deletion consists of loss or gain of a chromosome segment. Deletions may be either interstitial or terminal, with terminal deletions including the telomere (see Figure 30-8 and Figure 30-12D), whereas most duplications are interstitial. Most interstitial deletions and duplications result from unequal crossing over in LCRs (see Figure 30-11). Terminal deletions are more likely to result from simple chromosome breakage, although some apparently terminal deletions prove to be interstitial deletions in which one breakpoint happens to be close to the telomere. Any carrier of a deletion is hemizygous for the information on the corresponding segment of the normal homolog. Thus, small but cytogenetically visible deletions involving critical genes occasionally produce single-gene phenotypes, such as lissencephaly or retinoblastoma.
Inversions
Inversions are segments within a chromosome that are inverted with respect to the normal orientation; they result from crossovers within existing duplicated segments (LCRs) or from two breaks within a single chromosome, followed by inversion of the intervening segment and repair of the breaks. When the inverted segment includes the centrosome, the rearrangement is described as a pericentric inversion (see Figure 30-12A), whereas rearrangements in which the inverted segment does not include the centrosome are designated as paracentric inversions. Both types of inversions may result in production of unbalanced gametes because of the effects of recombination within the inverted segment. With pericentric inversions, a loop is formed between the inverted chromosome and its homolog during meiosis 1 (Figure 30-13). Recombination is somewhat, but not completely, suppressed within inversion loops, so crossovers are common in larger loops.
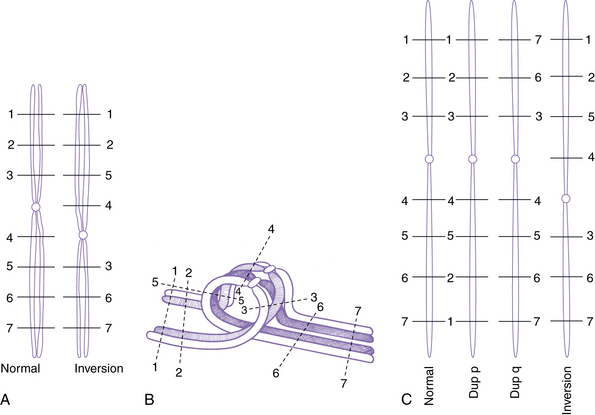
Recombination within a pericentric inversion loop produces derivative chromosomes in which segments distal to the breaks are either duplicated or deleted (see Figure 30-13C). The effects on the phenotype are inversely proportional to the size of the inversion. Thus, the distal segments typically are large with small pericentric inversions, and most unbalanced offspring are spontaneously aborted. Liveborn children with birth defects are more likely with larger inversions that lead to relatively small distal segments.
Reciprocal Translocations
Reciprocal translocations consist of breaks in nonhomologous chromosomes, with a reciprocal exchange of the broken segments (see Figure 30-12B). Usually, only two chromosomes are involved, but complex translocations involving three or more chromosomes have been described and likely are more common than standard chromosome analysis has suggested [Astbury et al., 2004a]. Population studies have detected reciprocal or robertsonian translocations in about 1 in 500 newborns.
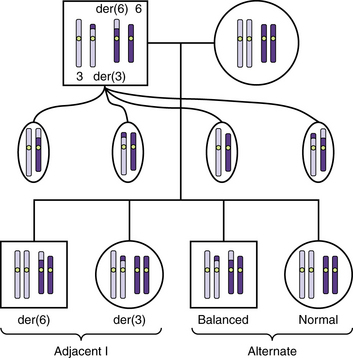
Reciprocal translocations often result in the production of unbalanced gametes. During meiosis 1, the derivative chromosomes and their normal homologs form a quadriradial shape that may separate into pairs in one of three ways: alternate, adjacent 1, and adjacent 2 segregation. Alternate segregation produces balanced gametes that have either normal chromosomes or both derivatives, which are therefore balanced. Adjacent 1 segregation produces unbalanced gametes in which homologous centromeres separate into different daughter cells. It results in duplication of the distal segment of one derivative chromosome and deletion of the distal tip of the other. In most translocation carriers, alternate and adjacent 1 segregation account for a large majority of the gametes (Figure 30-14). Adjacent 2 segregation also produces unbalanced gametes. In this uncommon mechanism, homologous centromeres pass to the same daughter cell. The resulting nondisjunction produces 3:1 and even a 4:0 segregation.
Robertsonian Translocations
Robertsonian translocations involve two acrocentric chromosomes that fuse in or near the centromere region, with loss of the short arms (see Figure 30-12C). Because the short arms contain repetitive DNA elements, especially rRNA, no phenotypic effects result. Carriers of a robertsonian translocation on chromosome 21 have a high risk of producing a child with translocation Down syndrome.
Rings
Rings – ring chromosomes – are formed when a chromosome undergoes two breaks, usually one in each arm, and the broken ends are rejoined (see Figure 30-12E). The two segments distal to the breaks are lost, resulting in deletion of both telomeres and adjacent regions of both the short and the long arms of the chromosome. Rings may not segregate properly during mitosis and meiosis, especially if a crossover occurs. Crossover often results in breakage followed by fusion, which may produce larger or smaller rings.
Cytogenetic Nomenclature
Detailed rules regarding nomenclature of chromosomes and chromosomal abnormalities have been published [ISCN, 2009]. Examples of most of the major types of abnormalities are listed in Table 30-5 using standard nomenclature. Note that breakpoints on the same chromosome are not separated by any punctuation, whereas breakpoints on different chromosomes are separated by a semicolon.
Table 30-5 Examples of Chromosomal Abnormalities Using Standard Nomenclature (Short System)
| Rearrangement | Karyotype |
|---|---|
| Genome Mutations | |
| Triploidy | 69,XXX |
| Monosomy | 45,X (Turner’s syndrome) |
| Trisomy | 47,XX,+21 (Down syndrome) |
| Deletion | |
| Terminal | 46,XY,del(8)(p21.1) |
| Interstitial | 46,XX,del(17)(p11.2p11.2) |
| Ring | 46,XY,r(17)(p13.3q22.3) |
| Duplication | |
| Direct | 46,XY,dir dup (2)(p14p23) |
| Inverted | 46,XX,inv dup (11)(p12p15) |
| Paracentric Inversion | |
| Balanced | 46,XX,inv(1)(p32p36.1) |
| Unbalanced | 46,XX, dup q, inv(1)(p32p36.1) |
| Pericentric Inversion | |
| Balanced | 46,XX,inv(1)(p36.1q32) |
| Unbalanced | 46,XY,dup(q),inv(1)(p36.1q32)mat |
| Reciprocal Translocation | |
| Balanced | 46,XY,t(−17,+der(17),t(7;17)(p22.3;p13.3)pat |
| Robertsonian Translocation | |
| Balanced | 46,XX,rob(13;21) |
| Unbalanced | 46,XY, −13,+der(13),rob(13;21)mat |
del, deletion; der, derivative; dir, direct; dup, duplication; inv, inversion; mat, maternal; pat, paternal; r, ring; rob, robertsonian translocation; t, translocation.
Mutations and Genetic Diseases
Classes of Mutations
DNA Replication Errors
DNA replication normally is an accurate process. The DNA polymerases (see Table 30-2) insert an incorrect base only once in every 10 million bp. A series of DNA repair enzymes exist that are able to recognize and replace noncomplementary bases, correcting more than 99.9 percent of errors. The overall mutation rate is therefore only 10−10 per basepair per cell division. The human genome consists of about 6 × 109 bp, so this mutation rate results in less than 1 bp mutation per cell division. Nevertheless, an estimated 1015 cell divisions occur during the lifetime of an adult human. Thus, thousands of new mutations occur at virtually every position in the genome. Not surprisingly, inherited defects in DNA replication and repair enzymes lead to a striking increase in the frequencies of all types of mutations.
Specific Types of Gene Mutations
The development and widespread use of modern molecular techniques have led to the discovery of specific mutations at many different loci. From among these, many different types of mutations have been recognized, all of which have the potential for causing genetic diseases. They may be divided by size into single- and multiple-base changes. The latter may involve only one or a few bases or may involve millions of basepairs. In any specific gene, mutations are almost always heterogeneous, although some types may be more common than others. Thus, the specific mutations in unrelated persons with the same genetic disease often are different. A few notable exceptions to this rule have been identified, such as achondroplasia, which almost always is caused by a specific single-base change in the fibroblast growth factor receptor 3 gene [Bellus et al., 1995].
Effects of Mutations on Gene Function
Nonsense (Chain Termination) Mutations
Mutations that generate one of the three stop codons result in premature termination of translation, whereas those that alter a stop codon allow translation to continue until the next stop codon is reached. Those mutations that result in a premature stop codon are called nonsense mutations. In general, these mutations have no effect on transcription (DNA to RNA), but the shortened polypeptide may have lost critical functional domains of the protein, or the mRNA may be so unstable that it is rapidly degraded in the cell. The latter process is known as nonsense-mediated mRNA decay, a process by which mRNA species containing premature termination codons are recognized and degraded before translation, although this typically spares truncation mutations in the last coding exon [Frischmeyer and Dietz, 1999]. Both base substitutions and nucleotide loss or gain mutations may result in nonsense mutations.
RNA Splicing Mutations
The sequence surrounding intron splice sites is highly conserved, and mutations of key nucleotides frequently prevent or reduce efficiency of splicing. The key nucleotide sequences at most splice junctions are shown in Figure 30-4.
One example of the first type is a G to C transition in the first position of the intron at the donor splice site in the hexosaminidase A gene found in many Ashkenazi Jewish patients with Tay–Sachs disease [Nussbaum et al., 2007]. In this example, the bases in the exon are capitalized, whereas those in the intron are not, and the mutation is underlined.
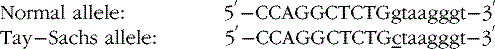
Principles of Medical Genetics
Patterns of Inheritance
The genetic constitution of an individual is the genotype. At any given locus, the normal genotype consists of either a single allele or a pair of alleles. Only a single allele is present for most genes on the X chromosome in males, who have only one X chromosome. A pair of alleles is present for all genes on the autosomes, and for a subset of genes on the X chromosome located in “pseudoautosomal” regions, which have functional homologs on the Y chromosome. The observable expression of the genotype is the phenotype. Penetrance has been defined as the percentage of persons with a particular genotype who have the expected phenotype; this is an all-or-none phenomenon. Expressivity is defined as the extent to which a genetic trait or disease is expressed and may vary greatly between affected persons. The proband is the affected family member through whom a family is identified; the consultand is the person in the family who seeks advice, regardless of whether affected or not. A pedigree is a diagram of the family history that shows the family members, their relationships to the proband, and their status with regard to the hereditary condition. Some of the symbols used for pedigrees in medical genetics are illustrated in Figure 30-15. A more detailed standardized nomenclature has been proposed for publication of pedigrees [Bennet et al., 1995].
The most widely recognized patterns of inheritance are single-gene or “mendelian” patterns, which include autosomal-dominant, autosomal-recessive, and X-linked modes of inheritance; example pedigrees are shown in Figure 30-16. All of these result from mutations in a single gene. The disorder or trait is autosomal if located on human chromosomes 1–22, and X-linked when located on the X chromosome. The only true Y-linked trait is male sex determination (the SRY gene). Autosomal traits are considered dominant when expressed in both heterozygotes and homozygotes, and recessive when expressed only in homozygotes; neither of these terms really fits with X-linked inheritance, as reviewed later on. The pattern of single-gene inheritance is modified in special cases in which mutations involve genes subject to imprinting or X-inactivation, or involve only a proportion of cells in the body or affected tissue. Finally, many diseases have more complicated inheritance.
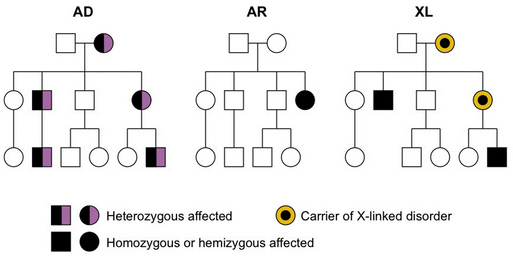
Fig. 30-16 Examples of autosomal-dominant (AD), autosomal-recessive (AR), and X-linked (XL) pedigrees.
Autosomal-Dominant Inheritance
The most important attributes of autosomal-dominant inheritance are expression of the trait in heterozygotes and male-to-male transmission (see Figure 30-16). The autosomal-dominant pattern may be recognized because:
Most persons affected by a disorder of autosomal-dominant inheritance are heterozygous, but rarely a homozygous person is encountered. Generally, the phenotype in homozygous persons is significantly more severe than in heterozygous persons. For example, one child born to parents who each had hereditary motor and sensory neuropathy type I had a much more severe neuropathy consistent with Dejerine–Sottas disease, or hereditary motor and sensory neuropathy type III [Killian and Kloepfer, 1979]. The best-known exception to this rule is Huntington’s disease, in which persons heterozygous for this trait cannot be distinguished from homozygotes.
Autosomal-Recessive Inheritance
The most important attributes of autosomal-recessive inheritance are expression in homozygotes and equal gender distribution (see Figure 30-16). This pattern may be recognized by the following four characteristics:
X-Linked Inheritance
The important characteristics of X-linked inheritance result from differential segregation of the X and Y chromosomes in males and females, and the differences in gene dosage. The most consistent characteristics include more severe phenotype in males than in females, transmission of disease through carrier females who are unaffected or less affected than males, and lack of male-to-male transmission (see Figure 30-16). The last is explained by transmission of the Y chromosome from fathers to sons, whereas the disease genes are located on the X chromosome. X-linked disorders traditionally have been divided into dominant and recessive subtypes, just as for autosomal single-gene disorders. This distinction was first made in fruit flies under experimental conditions but has never worked very well for human disorders. In a recent survey of more than 30 X-linked diseases, a remarkably wide range of penetrance was found, with many disorders intermediate between so-called X-linked dominant and recessive patterns [Dobyns et al., 2004]. On the basis of these and other arguments, use of these subtypes should be discontinued. The rules for X-linked inheritance have been modified to reflect this change (Box 30-1).
Box 30-1 Rules for X-Linked Inheritance in Humans
Rules Related to Segregation of the X and Y Chromosomes
Hemizygous males transmit X chromosomes to daughters and Y chromosomes to sons




Heterozygous females transmit X chromosomes to both sons and daughters






Genomic Imprinting
This differential silencing means that imprinted genes will be expressed from the maternally derived gene or from the paternally derived gene, but not from both. So if the functioning copy of an imprinted gene is lost owing to a deletion or mutation, the affected person is left with no functioning gene, and a disease phenotype will result. The underlying mechanisms are under study but still are not well understood [Hall, 1990; Jiang et al., 2004]. Imprinting disorders result from deletions or other types of mutations of genes within imprinted regions (or in the imprinting control regions). The most common of these result in infantile developmental disorders relevant for pediatric neurologists, such as Beckwith–Wiedemann syndrome due to defects of imprinted genes on 11p15.5 [Weksberg et al., 2003], and Angelman’s and Prader–Willi syndromes due to defects of imprinted genes on 15q11.2–q13 [Amos-Landgraf et al., 2006]. These disorders are reviewed elsewhere in this book.
Uniparental Disomy
When UPD involves a chromosome with an imprinted region, problems occur. For example, a child may inherit two paternally derived chromosomes, in which case no maternally derived chromosome will be present. Any genes that are normally expressed only from the maternally derived gene will not be expressed at all. This is one cause of Angelman’s syndrome. The same type of problem occurs in reverse if the child inherits two maternally inherited chromosomes. No paternally derived genes will be present, and a disease will occur. This is one cause of Prader–Willi syndrome [Amos-Landgraf et al., 2006].
Mitochondrial Inheritance
The mitochondria in any one person are derived almost exclusively from the mother through the ovum. Each ovum contains hundreds of mitochondria, and each mitochondrion contains many copies of the circular mitochondrial chromosome. Sperm contain a few mitochondria, most of which are degraded rapidly by the proteasome-dependent protein degradation pathway of the ubiquitin system within the ovum after fertilization [Sutovsky et al., 2003]. A small paternal contribution of mitochondria, however, has been demonstrated in several species, such as sheep [Zhao et al., 2004]. The same likely is true for humans, as suggested by one example of paternal inheritance of a mitochondrial disorder [Schwartz and Vissing, 2002]. This must be very rare, however, owing to the small proportion of paternal compared with maternal mitochondria, and as indicated by studies in humans with mitochondrial diseases [Filosto et al., 2003].
In patients with mutations of mitochondrial genes, a variable proportion of the mitochondrial chromosomes carry the mutation. Thus, diseases caused by mutations in mitochondrial DNA exhibit strict maternal inheritance (with very rare exceptions) and usually will exhibit phenotypic variation within a family owing to variation in the proportion of mutant and normal mitochondria between individuals [Zeviani et al., 1989].
Genetic Counseling
Standard of Care
All physicians have a professional responsibility to make certain that genetic counseling has been provided in appropriate situations and to ensure that the counseling meets current standards of practice [Directors and Directors, 1995; Parker, 2010]. Failure to provide this information may have tragic results. Perhaps the best example for pediatric neurologists is the birth of a second or even a third male with Duchenne muscular dystrophy in a family. Courts have upheld the principle of physician responsibility to provide accurate counseling on several occasions. For example, the parents of a child with Down syndrome claimed negligence because the mother had not been referred for prenatal diagnosis. In another case, parents who were tested for Tay–Sachs disease carrier status both were told that they were not carriers. They later had an affected child and filed a claim. Cases of this type are known as wrongful life claims.
Genetic Risk
One of the most crucial steps in offering accurate genetic counseling is to estimate the risk of recurrence of a genetic disorder in other family members. Estimation is not difficult for most diseases with a single-gene pattern of inheritance, but even these may be complex because of late age at onset or incomplete penetrance. For many other disorders, empirical recurrence risk estimates are used. These risk figures are derived from previous experience with the same disorder. Although such figures generally are accurate, exceptions occur because of causal heterogeneity and lack of knowledge regarding many rare disorders. The recurrence risks for some of the more common diseases seen in pediatric neurology clinics were reviewed by Baraitser [Baraitser, 1997]. In any given family, the recurrence risk may be different; consultation with a geneticist or genetic counselor may be helpful in providing this information.
 The complete list of references for this chapter is available online at
The complete list of references for this chapter is available online at Amos-Landgraf J.M., Ji Y., Gottlieb W., et al. Chromosome breakage in the Prader-Willi and Angelman syndromes involves recombination between large, transcribed repeats at proximal and distal breakpoints. Am J Hum Genet. 1999;65:370-386.
Amos-Landgraf J.M., Cottle A., Plenge R.M., et al. X chromosome-inactivation patterns of 1,005 phenotypically unaffected females. Am J Hum Genet. 2006;79(3):493-499.
Astbury C., Christ L.A., Aughton D.J., et al. Delineation of complex chromosomal rearrangements: evidence for increased complexity. Hum Genet. 2004;114:448-457.
Astbury C., Christ L.A., Aughton D.J., et al. Detection of deletions in de novo “balanced” chromosome rearrangements: further evidence for their role in phenotypic abnormalities. Genet Med. 2004;6:81-89.
Babcock M., Pavlicek A., Spiteri E., et al. Shuffling of genes within low-copy repeats on 22q11 (LCR22) by Alu-mediated recombination events during evolution. Genome Res. 2003;13(12):2519-2532.
Bailey J.A., Gu Z., Clark R.A., et al. Recent segmental duplications in the human genome. Science. 2002;297(5583):1003-1007.
Baraitser M. The genetics of neurological disorders. Oxford: Oxford University Press; 1997.
Bejerano G., Pheasant M., Makunin I., et al. Ultraconserved elements in the human genome. Science. 2004;304(5675):1321-1325.
Bellus G.A., Hefferon T.W., Ortiz de Luna R.I., et al. Achondroplasia is defined by recurrent G380R mutations of FGFR3. Am J Hum Genet. 1995;56:368-373.
Bennet R.L., Steinhaus K.A., Uhrich S.B., et al. Recommendations for standardized human pedigree nomenclature. Am J Hum Genet. 1995;56:745-752.
Cardoso C., Leventer R.J., Ward H.L., et al. Refinement of a 400-kb critical region allows genotypic differentiation between isolated lissencephaly, Miller-Dieker syndrome, and other phenotypes secondary to deletions of 17p13.3. Am J Hum Genet. 2003;72:918-930.
Cavaille J., Seitz H., Paulsen M., et al. Identification of tandemly-repeated C/D snoRNA genes at the imprinted human 14q32 domain reminiscent of those at the Prader-Willi/Angelman syndrome region. Hum Mol Genet. 2002;11(13):1527-1538.
Chance P.F., Abbas N., Lensch M.W., et al. Two autosomal dominant neuropathies result from reciprocal DNA duplication/deletion of a region on chromosome 17. Hum Mol Genet. 1994;3:223-228.
Chen K.S., Manian P., Koeuth T., et al. Homologous recombination of a flanking repeat gene cluster is a mechanism for a common contiguous gene deletion syndrome. Nat Genet. 1997;17:154-163.
Cheung V.G., Nowak N., Jang W., et al. Integration of cytogenetic landmarks into the draft sequence of the human genome. Nature. 2001;409(6822):953-958.
Chiaromonte F., Weber R.J., Roskin K.M., et al. The share of human genomic DNA under selection estimated from human-mouse genomic alignments. Cold Spring Harb Symp Quant Biol. 2003;68:245-254.
Cotton R.G. Slowly but surely towards better scanning for mutations. Trends Genet. 1997;13:43-46.
Denli A.M., Tops B.B.J., Plasterk R.H.A., et al. Processing of primary microRNAs by the microprocessor complex. Nature. 2004;432(11 Nov):231-235.
Dermitzakis E.T., Reymond A., Lyle R., et al. Numerous potentially functional but non-genic conserved sequences on human chromosome 21. Nature. 2002;420(6915):578-582.
Dermitzakis E.T., Reymond A., Scamuffa N., et al. Evolutionary discrimination of mammalian conserved non-genic sequences (CNGs). Science. 2003;302(5647):1033-1035.
Directors ASoHGBo, Directors ACoMGBo. Points to consider: ethical, legal, and psychosocial implications of genetic testing in children and adolescents. Am J Hum Genet. 1995;57(5):1233-1241.
Dobyns W.B., Stratton R.F., Parke J.T., et al. Miller-Dieker syndrome and monosomy 17p. J Pediatr. 1983;102:552-558.
Dobyns W.B., Filauro A., Tomson B.N., et al. Inheritance of most X-linked traits is not dominant or recessivve, just X-linked. Am J Med Genet. 2004;129A(2):136-143.
Eng C., Vijg J. Genetic testing: the problems and the promise. Nat Biotechnol. 1997;15:422-426.
Filosto M., Mancuso M., Vives-Bauza C., et al. Lack of paternal inheritance of muscle mitochondrial DNA in sporadic mitochondrial myopathies. Ann Neurol. 2003;54(4):524-526.
Frazer K.A., Tao H., Osoegawa K., et al. Noncoding sequences conserved in a limited number of mammals in the SIM2 interval are frequently functional. Genome Res. 2004;14(3):367-372.
Frischmeyer P.A., Dietz H.C. Nonsense-mediated mRNA decay in health and disease. Hum Mol Genet. 1999;8(10):1893-1900.
Gardner R.J., Mackay D.J., Mungall A.J., et al. An imprinted locus associated with transient neonatal diabetes mellitus. Hum Mol Genet. 2000;9(4):589-596.
Giglio S., Broman K.W., Matsumoto N., et al. Olfactory receptor-gene clusters, genomic-inversion polymorphisms, and common chromosome rearrangements. Am J Hum Genet. 2001;68:874-883.
Giglio S., Calvari V., Gregato G., et al. Heterozygous submicroscopic inversions involving olfactory receptor-gene clusters mediate the recurrent t(4;8)(p16;p23) translocation. Am J Hum Genet. 2002;71:276-285.
Giglio S., Pirola B., Arrigo G., et al. Opposite deletions/duplications of the X chromosome: two novel reciprocal rearrangements. Eur J Hum Genet. 2000;8:63-70.
Grompe M. The rapid detection of unknown mutations in nucleic acids. Nat Genet. 1993;5:111-117.
Haines J.L., Korf B.R., Morton C.C., et al. Current Protocols in Human Genetics. United States: John Wiley & Sons; 2010.
Hall J.G. Genomic imprinting: review and relevance to human diseases. Am J Hum Genet. 1990;46(5):857-873.
Hardison R.C. Conserved noncoding sequences are reliable guides to regulatory elements. Trends Genet. 2000;16(9):369-372.
ISCN. An International System for Human Cytogenetic Nomenclature (2009): Recommendations of the International Standing Committee on Human Cytogenetic Nomenclature. L.G. Shaffer, M.L. Slovak, L.J. Campbell. Basel: Karger; 2009:138.
Ishkanian A.S., Malloff C.A., Watson S.K., et al. A tiling resolution DNA microarray with complete coverage of the human genome. Nat Genet. 2004;36(3):299-303.
Jiang Y.H., Bressler J., Beaudet A.L. Epigenetics and human disease. Annu Rev Genomics Hum Genet. 2004;5:479-510.
Johnson D.D., Dobyns W.B., Gordon H., et al. Familial pericentric and paracentric inversions of chromosome 1. Hum Genet. 1988;79:315-320.
Killian J.M., Kloepfer H.W. Homozygous expression of a dominant gene for Charcot-Marie-Tooth neuropathy. Ann Neurol. 1979;5:515-xxx.
Lander E.S., Linton L.M., Birren B., et al. Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):860-921.
Lewin B. Genes IX. Sudbury, MA: Jones and Bartlett Publishers, Inc; 2007.
Lucito R., Healy J., Alexander J., et al. Representational oligonucleotide microarray analysis: a high-resolution method to detect genome copy number variation. Genome Res. 2003;13(10):2291-2305.
Lyon M.F. Gene action in the X-chromosome of the mouse (Mus musculus L.). Naturwissenschaften. 1961;190:372-373.
Lyon M.F. X-chromosome inactivation and human genetic disease. Acta Paediatr Suppl. 2002;91(439):107-112.
McDermid H.E., Morrow B.E. Genomic disorders on 22q11. Am J Hum Genet. 2002;70:1077-1088.
Mohandas T.K., Park J.P., Spellman R.A., et al. Paternally derived de novo interstitial duplication of proximal 15q in a patient with developmental delay. Am J Med Genet. 1999;82:294-300.
Nussbaum R.L., McInnes R.R., Willard H.F. Thompson & Thompson Genetics in Medicine. Philadelphia: Saunders; 2007.
Osborne L.R., Li M., Pober B., et al. A 1.5 million-base pair inversion polymorphism in families with Williams-Beuren syndrome. Nat Genet. 2001;29:321-325.
Parker M. Genetic testing in children and young people. Fam Cancer. 2010;9(1):15-18.
Parisi M.A., Bennett C.L., Eckert M.L., et al. The NPHP1 gene deletion associated with juvenile nephronophthisis is present in a subset of individuals with Joubert syndrome. Am J Hum Genet. 2004;75:82-91.
Pentao L., Wise C.A., Chinault A.C., et al. Charcot-Marie-Tooth type 1A duplication appears to arise from recombination at repeat sequences flanking the 1.5 Mb monomer unit. Nat Genet. 1992;2:292-300.
Potocki L., Chen K.S., Park S.S., et al. Molecular mechanism for duplication 17p11.2-the homologous recombination reciprocal of the Smith-Magenis microdeletion. Nat Genet. 2000;24:84-87.
Saunier S., Calado J., Benessy F., et al. Characterization of the NPHP1 locus: mutational mechanism involved in deletions in familial juvenile nephronophthisis. Am J Hum Genet. 2000;66:778-789.
Schwartz M., Vissing J. Paternal inheritance of mitochondrial DNA. N Engl J Med. 2002;347(8):576-580.
Sebat J., Lakshmi B., Troge J., et al. Large-scale copy number polymorphism in the human genome. Science. 2004;305(5683):525-528.
Spiteri E., Babcock M., Kashork C.D., et al. Frequent translocations occur between low copy repeats on chromosome 22q11.2 (LCR22s) and telomeric bands of partner chromosomes. Hum Mol Genet. 2003;12:1823-1837.
Stankiewicz P., Lupski J.R. Genome architecture, rearrangements and genomic disorders. Trends Genet. 2002;18(2):74-82.
Stein L.D. Strategies for large-insert cloning and analysis. In: Dracopoli N.C., Haines J.L., Korf B.R., Moir D.T., Morton C.C., Seidman C.E., Seidman J.G., Smith D.R., editors. Current Protocols in Human Genetics. United States: John Wiley & Sons; 1997:5.0.1-5.16.24. Quarterly Supplements ed
Strachan T., Read A. Human Molecular Genetics. Oxford, UK: Taylor & Francis, Inc; 2010.
Sutovsky P., McCauley T.C., Sutovsky M., et al. Early degradation of paternal mitochondria in domestic pig (Sus scrofa) is prevented by selective proteasomal inhibitors lactacystin and MG132. Biol Reprod. 2003;68(5):1793-1800.
Urban Z., Helms C., Fekete G., et al. 7q11.23 deletions in Williams syndrome arise as a consequence of unequal meiotic crossover. Am J Hum Genet. 1996;59:958-962.
Venter J.C., Adams M.D., Myers E.W., et al. The sequence of the human genome. Science. 2001;291(5507):1304-1351.
Waterston R.H., Lindblad-Toh K., Birney E., et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420(6915):520-562.
Watson J.D., Crick F.H. Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature. 1953;171(4356):737-738.
Weber J.L., May P.E. Abundant class of human DNA polymorphisms which can be typed using the polymerase chain reaction. Am J Hum Genet. 1989;44:388-xxx.
Weksberg R., Smith A.C., Squire J., et al. Beckwith-Wiedemann syndrome demonstrates a role for epigenetic control of normal development. Hum Mol Genet. 2003;12(Spec No 1):R61-R68.
Wylie A.A., Murphy S.K., Orton T.C., et al. Novel imprinted DLK1/GTL2 domain on human chromosome 14 contains motifs that mimic those implicated in IGF2/H19 regulation. Genome Res. 2000;10(11):1711-1718.
Zeviani M., Bonilla E., DeVivo D.C., et al. Mitochondrial diseases. Neurol Clin. 1989;7(1):123-156.
Zhao X., Li N., Guo W., et al. Further evidence for paternal inheritance of mitochondrial DNA in the sheep (Ovis aries). Heredity. 2004;93(4):399-403.