[level-membership-for-basic-science-category]
Chapter 38 Introduction to Endocrine Pharmacology and Hormones of the Hypothalamus and Pituitary Gland
| Abbreviations | |
|---|---|
| ACTH | Adrenocorticotropic hormone |
| AVP | Arginine vasopressin, antidiuretic hormone |
| cAMP | Cyclic adenosine monophosphate |
| CNS | Central nervous system |
| CRH | Corticotropin-releasing hormone |
| DHEA | Dehydroepiandrosterone |
| DHT | Dihydrotestosterone |
| DI | Diabetes insipidus |
| DNA | Deoxyribonucleic acid |
| Epi | Epinephrine |
| FDA | United States Food and Drug Administration |
| FSH | Follicle-stimulating hormone |
| GH | Growth hormone |
| GHRH | Growth hormone-releasing hormone |
| GI | Gastrointestinal |
| GnRH | Hypothalamic gonadotropin-releasing hormone |
| hCG | Human chorionic gonadotropin |
| hGH | Human growth hormone |
| hMG | Human menopausal gonadotropin |
| IGF-1 | Insulin-like growth factor-1 |
| IM | Intramuscular |
| IV | Intravenous |
| LH | Luteinizing hormone |
| RNA | Ribonucleic acid |
| SC | Subcutaneous |
| SRIF | Somatostatin, somototropin-release inhibiting hormone |
| TRH | Thyrotropin-releasing hormone |
| TSH | Thyroid-stimulating hormone |
The endocrine system is a complex communication system responsible for maintaining homeostasis throughout the body, and it is vital to individual and species survival and propagation as well as adaptation to the environment. The system consists of a diverse group of ductless glands that secrete chemical messengers called hormones into the circulation. The secreted hormones are transported in the bloodstream to target organs, where they act to regulate cellular activities. For a hormone to elicit a response, it must interact with specific receptors on the cells of the target organ, much like the interaction between neurotransmitters and receptors involved in the process of neurotransmission in the central and peripheral nervous systems (see Chapters 9 and 27). Receptors play a key role in the mechanisms of action of endocrine hormone systems; key receptor mechanisms pertinent to endocrine systems are summarized in Chapter 1.
Hormones are chemically and structurally diverse compounds and can be divided into three main classes based on chemical composition, viz., the amino acid analogs, the peptides, and the steroids. The amino acid analogs, often termed amine hormones, are all derived from tyrosine and include epinephrine (Epi) and the iodothyronines or thyroid hormones. The peptide hormones are subclassified on the basis of size and glycosylation state and may be single- or double-chain peptides. The steroid hormones are all derived from cholesterol and may be subclassified as adrenal steroids or sex steroids, the former synthesized primarily in the adrenal cortex and the latter synthesized in the ovaries or testes. The major endocrine glands and their associated hormones are listed in Box 38-1.
Hormones are generally distinguished from other types of modulatory factors (i.e., neurotransmitters) by a longer duration of effect and more extensive circulation in the body. While in the circulation, a hormone is frequently associated with one or more types of transport proteins from which it must dissociate to interact with responsive receptors. In addition, availability to tissues is dependent upon membrane exclusion mechanisms, susceptibility to tissue modification, and ultimately the rate of renal or hepatic metabolism, inactivation, and excretion. As mentioned, hormones exert their effects by binding to and activating receptors on target cells. These receptors can be located on the cell surface, as for peptide hormones, or within the cell, as in the case of steroids and thyroid hormones. After receptor activation, intracellular signaling pathways (e.g., second messenger systems or ligand-activated transcription factors) are modulated, which acutely or chronically alter cellular physiology and potentially whole organism physiology.
Excessive levels of endogenous hormones may result from excessive organ secretion or unregulated ectopic formation. Hormone overproduction by a secreting organ is commonly associated with excessive stimulation or a malignancy (or hyperplasia). Successful management of this situation includes blockade of the stimulatory agent, if identifiable, or interference with hormone formation, secretion, or action. Ectopic production of a biologically active form of the hormone by tissues is complicated by the lack of feedback mechanisms to regulate hormone production and is typically associated with tissue malignancy or infection. The primary determinant of successful intervention frequently requires a combination of ablation of the secreting tissue and pharmacological agents to antagonize the effects of elevated hormone levels. The success of this technique hinges on the ability of the responsible tissue to respond to pharmacological intervention. If it is not possible or detrimental to directly reduce hormone levels, a situation often encountered before or immediately after surgery, or when the cause of the elevated hormone levels is unknown or uncorrectable, alternative, patient-specific strategies to reduce the effects of elevated hormone levels must be used. A summary of strategies to manage the levels and action of hormones is presented in Box 38-2. A list of drugs that affect hormonal balance and their mechanisms of action are in Box 38-3.
BOX 38–2 Strategies to Manage the Levels and Action of Hormones
BOX 38–3 Drugs Known to Affect Hormonal Balance
Effectors of Hormone Release/Reuptake
Steroid Biochemistry and Physiology
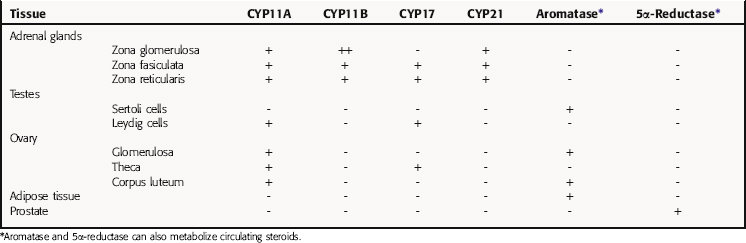
All secreted steroids are synthesized from cholesterol, which can be synthesized de novo or derived from circulating lipoproteins. Similar metabolic pathways mediate steroid synthesis in all organs (Fig. 38-1). The organ-specific formation of secreted steroids depends on the presence of specific catalytic enzymes (Table 38-1).
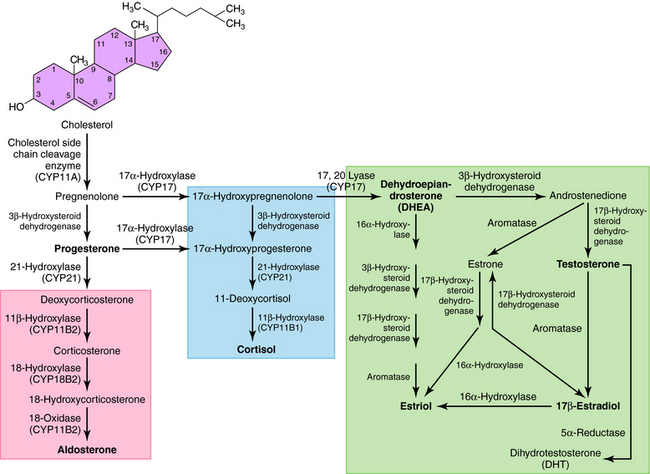
TABLE 38–1 Enzymes Present in Different Tissues Mediating the Organ- or Tissue-Specific Formation of Steroid Hormones
The action of steroids is mediated largely by altering gene transcription through interaction with promoter deoxyribonucleic acid (DNA) of genes. Steroid receptors are dimeric and coupled with accessory proteins until activated by ligands outside the nucleus. The steroid-receptor complex is phosphorylated and translocated to the nucleus through a nuclear pore, facilitated by the importin protein. The interaction with the gene promoter region occurs through steroid-specific palindromic nucleotide sequences within the receptor. The interaction of DNA and the steroid-receptor complex is dependent on steroid structural differences, amino acid sequence of the DNA binding domain, the nucleotide sequence of the DNA binding site, and the architecture of the gene promoter. The structures of the primary circulating steroids are shown in Figure 38-2.
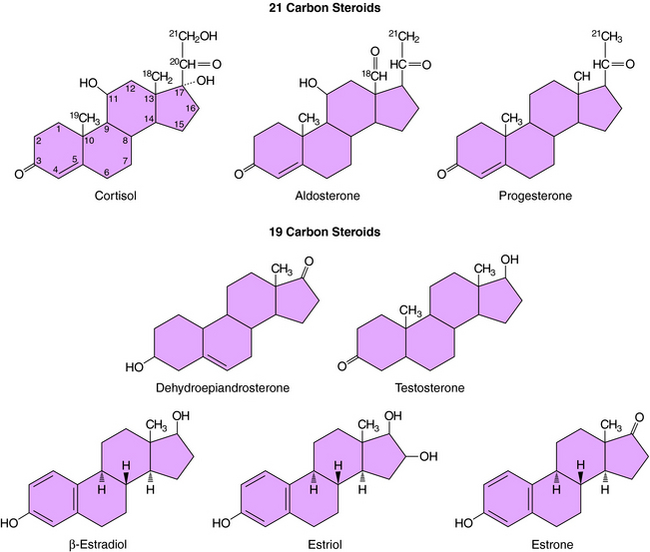
In the adrenal gland, the primary secreted steroids are aldosterone, cortisol, and dehydroepiandrosterone (DHEA) (see Fig. 38-2). Aldosterone is the primary mineralocorticoid and acts at the luminal epithelia to promote renal reuptake of Na+, which conserves Na+ and can elevate blood pressure. In the zona glomerulosa, the lack of CYP17 is associated with nearly exclusive formation of aldosterone. Further, the release of aldosterone from the zona glomerulosa is regulated by the renin-angiotensin pathway as a result of activation angiotensin II-receptors, which are linked to the formation of 1,4,5-inositol triphosphate. The amount of aldosterone released is relatively low (50 to 150 µg/day); aldosterone is transported in the blood through an interaction with albumin with a bound/free ratio of 70/30.
The complement of enzymes in the zona fasciculata and zona reticularis permits the formation of cortisol, the primary circulating glucocorticoid (see Chapter 39). The release of cortisol is dependent on a tightly regulated hypothalamic-anterior pituitary-adrenal cortex axis. The biological role of glucocorticoids is complex and temporal. The liver has the greatest level of nuclear receptors or steroid-activated transcription factors, although they are present in many tissues. The primary systems affected by cortisol include self-regulation of formation via suppression of corticotropin-releasing hormone (CRH) and adrenocorticotropic hormone (ACTH) secretion, storage of hepatic glycogen, response to stress, and suppression of the immune system. The daily production of cortisol ranges from 10 to 20 mg, and plasma levels follow a diurnal pattern with the highest levels in the morning. In the blood, cortisol is bound to a specific hepatic protein, corticosteroid-binding protein (aka transcortin), which promotes its transport and increases its duration of action.
The secretion of estrogen (β-estradiol) and progesterone from the ovary is regulated by hypothalamic gonadotropin-releasing hormone (GnRH), anterior pituitary follicle-stimulating hormone (FSH), and leutenizing hormone (LH). Release of the gonadotropic hormones and the ovarian steroids during the menstrual cycle is episodic, and the highest levels of β-estradiol or progesterone occur during the late follicular phase or midleutal phase, respectively (see Chapter 40). Most of these circulating steroids (98%) are bound to specific steroid hormone-binding globins.
The primary testicular androgen, testosterone, is converted to dihydrotestosterone (DHT) in tissues expressing 5α-reductase. The actions of androgens include development of male reproductive tract and accessory tissues, stimulation of secondary sexual traits, growth, and development of the central nervous system (CNS) (see Chapter 41). As shown in Figure 38-1, the expression of steroid metabolizing enzymes promotes the formation of DHEA and androstenedione leading to the formation of testosterone; the expression of aromatase in ovarian cells permits conversion of testosterone to β-estradiol.
Therapeutic Overview
Pharmacology of Hypothalamic and Pituitary Hormones
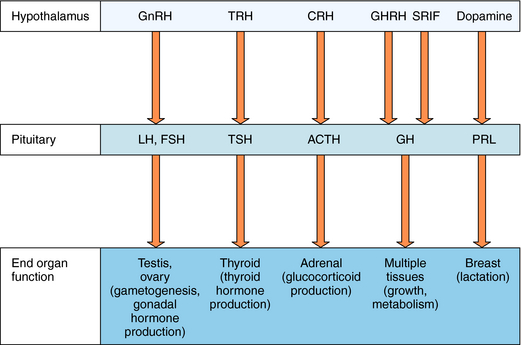
The hypothalamus and pituitary gland work in concert to regulate endocrine systems throughout the body. Peptides and biogenic amines synthesized and secreted by specialized neurons within the hypothalamus are transported to the anterior pituitary by the hypothalamic-hypophyseal portal circulation, where they act through specific receptors to stimulate or inhibit hormone secretion (Fig. 38-3). Anterior pituitary hormones trigger peripheral endocrine organs to produce hormones, which have individual functions and provide feedback to the hypothalamus and pituitary to regulate the synthesis and release of their tropic hormones. As mentioned, GnRH (also called luteinizing hormone releasing hormone) stimulates the secretion of LH and FSH by the pituitary. LH and FSH promote gametogenesis and gonadal hormone production by the ovaries and testes (see Chapters 40 and Chapter 41). Thyrotropin-releasing hormone (TRH) stimulates secretion of thyroid-stimulating hormone (TSH), which in turn controls thyroid function (see Chapter 42) CRH stimulates the secretion of ACTH, which promotes the secretion of cortisol by the adrenal cortex (see Chapter 39). Growth hormone-releasing hormone (GHRH) stimulates and somatostatin (also called somatotropin-release inhibiting factor, SRIF) inhibits the production of growth hormone (GH), which has numerous effects on growth and metabolism. Hypothalamic dopamine functions to tonically inhibit secretion of prolactin, the hormone primarily responsible for lactation and suppression of fertility while nursing.
| Therapeutic Overview |
|---|
| Hypothalamic Hormones |
| GnRH |
| Replacement therapy for idiopathic hypogonadotropic hypogonadism |
| GnRH analogs |
| Prostate and breast cancer |
| Idiopathic precocious puberty |
| Endometriosis |
| Fertility/contraception |
| Dopamine agonists |
| Pathological hyperprolactinemia |
| Acromegaly |
| Parkinson’s disease |
| Somatostatin and analogs |
| Acromegaly |
| Carcinoid and vasoactive intestinal peptide-secreting tumors |
| Pituitary Hormones |
| LH and FSH |
| Infertility in women |
| Infertility in men with hypogonadotropic hypogonadism |
| GH agonists |
| Adult GH deficiency |
| Growth failure |
| AVP agonists and antagonists |
| Diabetes insipidus |
| Syndrome of inappropriate antidiuretic hormone |
Mechanisms of Action
GnRH-receptor interaction initiates secretion of LH and FSH. The GnRH receptor gene consists of a 327-amino acid protein with seven transmembrane domains but lacks the typical intracellular C-terminus of a G protein-coupled receptor. Microaggregation stimulates up regulation of GnRH receptors and is followed by internalization of the hormone-receptor complex (see Chapter 1). Receptor activation results in increased intracellular Ca++.
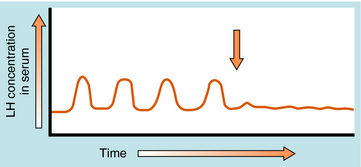
GnRH is released in a pulsatile manner by the so-called “hypothalamic GnRH pulse generator.” This pattern of intermittent bursts is essential for normal function. Continuous administration of GnRH will initially produce an increase in serum gonadotropin concentrations. However, this is followed by a decrease in gonadotropin secretion caused by pituitary GnRH receptor down regulation, a decrease in expression of GnRH receptors, and desensitization of pituitary gonadotrophs. GnRH analog agonists and antagonists have been synthesized through selective substitution of amino acids in the GnRH peptide (Fig. 38-4). These GnRH analogs have greater receptor binding and reduced susceptibility to enzymatic degradation, resulting in prolonged biological activity.

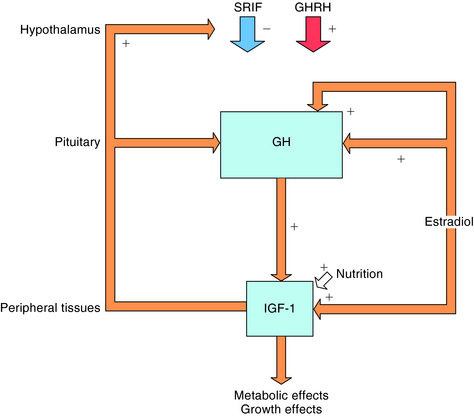
Secretion of GH is regulated by two opposing hypothalamic hormones: GHRH and somatostatin (Fig. 38-5). Somatostatin is a cyclic peptide that is processed from a preprohormone into two molecular forms: SRIF-14 and SRIF-28. The 14-amino acid sequence at the carboxyl terminal of SRIF-28 is identical to SRIF-14. In addition to its presence in the hypothalamus, somatostatin is widely distributed throughout the CNS, the gastrointestinal (GI) tract, pancreas, thyroid, thymus, heart, skin, and eye. Somatostatin has multiple actions including inhibition of GI hormone secretion (e.g., gastrin, vasoactive intestinal peptide, motilin, and secretin), pancreatic exocrine secretion (e.g., gastric acid, pepsin, pancreatic bicarbonate), pancreatic endocrine secretion (e.g., insulin, glucagon), GI motility, gastric emptying, and gallbladder contraction. Somatostatin also decreases GI absorption and mesenteric blood flow. In the CNS, somatostatin acts as both a neurotransmitter and a neuromodulator.
In addition to regulating estrogen production, gonadotropins have multiple effects on ovarian follicles. FSH directly stimulates follicular growth and maturation and enhances granulosa cell responsiveness to LH. LH is essential for the breakdown of the follicular wall, resulting in ovulation, and for the subsequent resumption of oocyte meiosis.
GH is a 191-amino acid polypeptide belonging to a family of structurally similar hormones, including prolactin and chorionic somatomammotropin (also known as human placental lactogen). GH is synthesized by somatotropes of the anterior pituitary. The major product is a peptide with two disulfide bonds. The precise signaling mechanism by which GH exerts its intracellular effects likely involves its interaction with specific plasma membrane receptors and activation of the JAK family of intracellular tyrosine kinases and the STAT family of nuclear transcription factors (see Chapter 1). In addition, GH binds to proteins in both the cytosol and plasma. The specificity of the circulating binding protein is similar to that of the GH receptor.
AVP, also known as antidiuretic hormone, is a polypeptide that functions as the primary antidiuretic hormone in humans (Fig. 38-6). Synthesized primarily in the magnocellular neuronal systems of the supraoptic and paraventricular nuclei of the hypothalamus, the AVP precursor molecule contains a signal peptide, a neurophysin, and a glycosylated moiety, in addition to the AVP sequence. After translation of the messenger RNA to form a preprohormone (166 amino acids), the signal peptide is cleaved, forming a prohormone. The prohormone is stored in neurosecretory granules that travel down the supraoptico-hypophyseal tract to the posterior pituitary. The primary stimuli for AVP release are hyperosmolarity, as measured by osmoreceptors in the supraoptic and paraventricular nuclei, and volume depletion, detected by baroreceptors in the vascular bed and heart. Nausea, emesis, and hypoglycemia may also stimulate AVP release.
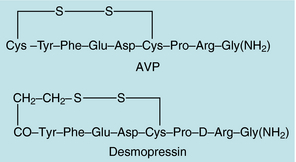
FIGURE 38–6 Amino acid sequences of arginine vasopressin (AVP) and 1-desamino-8-D-arginine-vasopressin (desmopressin).
AVP acts via V1 and V2 receptors in smooth muscle and renal collecting tubules, respectively. V1 receptors mediate vasoconstriction, while V2 receptors mediate antidiuretic effects. Specifically, AVP binding to V2 receptors activates adenylyl cyclase and a subsequent cascade resulting in fusion of the water channel, aquaporin-2, with the luminal membrane, thereby allowing water reabsorption.
Pharmacokinetics
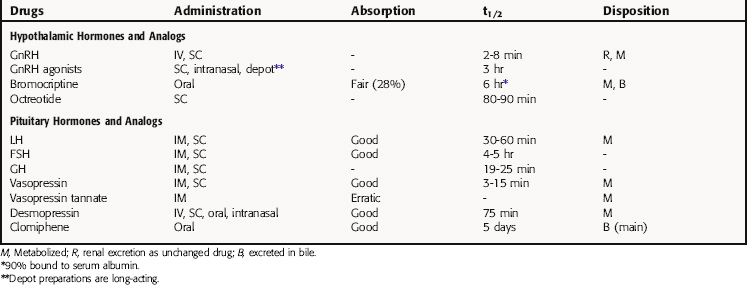
The pharmacokinetic parameters for the hypothalamic and pituitary hormones and analogs are summarized in Table 38-2.
Dopamine, in addition to being a neurotransmitter, is a sympathomimetic and is commonly used to treat cardiogenic shock, septic shock, acute myocardial infarction, and renal failure (see Chapter 23). Because of its vasoconstrictor properties, it is not administered SC or IM. It has a short half-life (2 minutes) and must be administered by continuous IV infusion. Therefore it is not used in treatment of hyperprolactinemia, although it does effectively decrease serum prolactin levels.
Bromocriptine is a long-acting dopamine agonist (see Chapter 28). After oral administration, approximately 28% of bromocriptine is absorbed, with peak plasma levels reached in 1 hour. It is transported primarily by serum albumin (90% to 96%) and has a half-life of approximately 6 hours. An intravaginal route can be used effectively to avoid drug sensitivity. Bromocriptine is metabolized by the liver and excreted in bile (84.6% within 120 hours).
Relationship of Mechanisms of Action to Clinical Response
Faulty GnRH secretion in men is referred to as idiopathic hypogonadotropic hypogonadism. A small clinical study using long-term pulsatile administration of GnRH for at least 3 months demonstrated significant increases of serum testosterone concentrations and testicular size. Mature spermatogenesis was achieved in 50% of patients, and men with unfused epiphyses experienced linear bone growth. Idiopathic or surgically induced hypogonadotropic hypogonadism is treated with testosterone (see Chapter 41) to promote masculinization and to preserve bone mineral density. Human chorionic gonadotropin and human menopausal gonadotropin are used to promote spermatogenesis and restore fertility in male hypogonadotropic hypogonadism.
The association of orchiectomy and regression of prostate cancer led to the development of approaches to decrease serum androgen concentrations in men with metastatic prostate cancer. Methods to induce androgen deprivation include orchiectomy, estrogen therapy, GnRH analogs, and antiandrogens (see Chapter 41). Combined androgen blockade, in which orchiectomy or GnRH analogs are combined with an antiandrogen, is also used in treating metastatic hormone-dependent prostate cancer.
Orchiectomy is an effective and relatively safe surgical procedure that significantly lowers testosterone levels (90%). The emotional impact of orchiectomy decreases its desirability for men with metastatic prostate cancer. Another approach is to use estrogens to suppress LH secretion, which promotes decreased serum androgen levels in men. However, estrogen therapy in men has been linked with an increased incidence of deep venous thrombosis and gynecomastia.
Long-acting GnRH agonists can be used to down regulate pituitary gonadotropin receptors and suppress release of LH (Fig. 38-7), resulting in reduction of serum testosterone concentrations comparable to that seen with orchiectomy. However, continuous GnRH agonist therapy will initially increase LH secretion from the pituitary, causing a transient increase in serum testosterone. This “flare” response occurs approximately 72 hours after initiating therapy and can exacerbate symptoms of metastatic prostate cancer, such as bone pain and ureteral obstruction. Coadministration of the antiandrogen flutamide with a GnRH agonist can prevent these negative effects. Pituitary gonadotroph desensitization occurs 1 to 2 weeks after starting the GnRH agonist, with castrate levels of testosterone seen in 2 to 4 weeks.
Dopamine agonists are used to treat hyperprolactinemia caused by both prolactinomas and lactotroph hyperplasia. Dopamine agonists bind to D2 receptors on the lactotrophs, resulting in decreased prolactin synthesis and secretion. Decreases in prolactin concentration can be seen within 2 to 3 weeks of initiating therapy. Dopamine agonists also decrease the size of the lactotroph, leading to shrinkage of the prolactinoma. Within a few days, significant abatement of the clinical signs and symptoms of the intracranial tumor are noted. For many patients a significant reduction of tumor size can be seen upon imaging within 6 weeks of initiating the dopamine agonist. Prolactinomas are the only type of pituitary adenoma in which medical therapy, as opposed to transsphenoidal resection, is first-line treatment. With reduction of the serum prolactin concentration to normal, galactorrhea is abolished and gonadal function restored. Patients who do not respond to one dopamine agonist may respond to another, and cabergoline may be more effective than bromocriptine.
Clomiphene is a compound with both estrogenic and antiestrogenic activity that is indicated for women with normogonadotropic anovulation (see Chapter 40). The use of clomiphene results in lower rates of multiple gestation births (~ 5%), compared with the incidence using gonadotropins.
Central (or neurogenic) DI is characterized by polyuria and polydipsia and results from inadequate secretion of AVP from the posterior pituitary. Nephrogenic DI results from failure of the kidney to respond to secreted AVP. The diagnosis of DI is confirmed by using AVP during a water deprivation test. The water deprivation test is also used to distinguish between central and nephrogenic DI. During water deprivation and subsequent elevation of plasma osmolality, patients with DI exhibit an inability to retain water or concentrate their urine. Patients with central DI exhibit an increase in urine osmolality after administration of AVP, whereas patients with nephrogenic DI exhibit little to no response.
AVP is also used for treatment of certain bleeding disorders such as mild hemophilia A and mild to moderate von Willebrand’s disease. AVP increases circulating concentrations of factor VIII (antihemophilic factor; see Chapter 26), perhaps by stimulating its release from cells in the vascular endothelium. Desmopressin is used for treatment of acute bleeding in patients with platelet dysfunction caused by uremia and is preferred to AVP because of its lack of vasopressor activity.
Pharmacovigilance: Side Effects, Clinical Problems, and Toxicity
| GnRH | Breast tenderness, decreased sex drive; hot flashes/sweating; impotence |
| Occasional nausea or vomiting, headache, abdominal discomfort; difficulty sleeping | |
| Anaphylaxis (rare) with IV use | |
| Localized problems at injection site | |
| Somatostatin analogs | Hyperglycemia, loose stools, gallstones |
| Dopamine agonists | Nausea, orthostatic hypotension initially |
| Confusion, headache, dizziness, drowsiness, faintness |
| LH and FSH | Multiple gestation pregnancy |
| Gynecomastia in men | |
| Occasional febrile reactions | |
| GH | Antibodies |
| Blurred vision, unusual tingling feelings, dizziness, nervousness, severe headache, altered heartbeat | |
| Abuse in athletics | |
| AVP | Nausea, vertigo, headache |
| Anaphylaxis | |
| Angina, myocardial infarction | |
| DDAVP | Rare side effects include chills, confusion, drowsiness, convulsions, fever, breathing problems, skin rash |
and age must be used to prevent gigantism. Because hGH is potentially diabetogenic, care must be given when administering to a patient with a personal or family history of abnormal glucose tolerance.
Anonymous. Cool.Click: A needle-free device for growth hormone delivery. Med Lett. 2001;43:2-3.
Anonymous. Pegvisomant (Somavert) for acromegaly. Med Lett. 2003;45:55-56.
Anonymous. Growth hormone for normal short children. Med Lett. 2003;45:89-90.
Additional information on this topic: http://www.aspet.org/AMSPC/Knowledge_Objectives/files/11-Endocrine.htm.
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
Chapter 38 Introduction to Endocrine Pharmacology and Hormones of the Hypothalamus and Pituitary Gland
| Abbreviations | |
|---|---|
| ACTH | Adrenocorticotropic hormone |
| AVP | Arginine vasopressin, antidiuretic hormone |
| cAMP | Cyclic adenosine monophosphate |
| CNS | Central nervous system |
| CRH | Corticotropin-releasing hormone |
| DHEA | Dehydroepiandrosterone |
| DHT | Dihydrotestosterone |
| DI | Diabetes insipidus |
| DNA | Deoxyribonucleic acid |
| Epi | Epinephrine |
| FDA | United States Food and Drug Administration |
| FSH | Follicle-stimulating hormone |
| GH | Growth hormone |
| GHRH | Growth hormone-releasing hormone |
| GI | Gastrointestinal |
| GnRH | Hypothalamic gonadotropin-releasing hormone |
| hCG | Human chorionic gonadotropin |
| hGH | Human growth hormone |
| hMG | Human menopausal gonadotropin |
| IGF-1 | Insulin-like growth factor-1 |
| IM | Intramuscular |
| IV | Intravenous |
| LH | Luteinizing hormone |
| RNA | Ribonucleic acid |
| SC | Subcutaneous |
| SRIF | Somatostatin, somototropin-release inhibiting hormone |
| TRH | Thyrotropin-releasing hormone |
| TSH | Thyroid-stimulating hormone |
The endocrine system is a complex communication system responsible for maintaining homeostasis throughout the body, and it is vital to individual and species survival and propagation as well as adaptation to the environment. The system consists of a diverse group of ductless glands that secrete chemical messengers called hormones into the circulation. The secreted hormones are transported in the bloodstream to target organs, where they act to regulate cellular activities. For a hormone to elicit a response, it must interact with specific receptors on the cells of the target organ, much like the interaction between neurotransmitters and receptors involved in the process of neurotransmission in the central and peripheral nervous systems (see Chapters 9 and 27). Receptors play a key role in the mechanisms of action of endocrine hormone systems; key receptor mechanisms pertinent to endocrine systems are summarized in Chapter 1.
Hormones are chemically and structurally diverse compounds and can be divided into three main classes based on chemical composition, viz., the amino acid analogs, the peptides, and the steroids. The amino acid analogs, often termed amine hormones, are all derived from tyrosine and include epinephrine (Epi) and the iodothyronines or thyroid hormones. The peptide hormones are subclassified on the basis of size and glycosylation state and may be single- or double-chain peptides. The steroid hormones are all derived from cholesterol and may be subclassified as adrenal steroids or sex steroids, the former synthesized primarily in the adrenal cortex and the latter synthesized in the ovaries or testes. The major endocrine glands and their associated hormones are listed in Box 38-1.
Hormones are generally distinguished from other types of modulatory factors (i.e., neurotransmitters) by a longer duration of effect and more extensive circulation in the body. While in the circulation, a hormone is frequently associated with one or more types of transport proteins from which it must dissociate to interact with responsive receptors. In addition, availability to tissues is dependent upon membrane exclusion mechanisms, susceptibility to tissue modification, and ultimately the rate of renal or hepatic metabolism, inactivation, and excretion. As mentioned, hormones exert their effects by binding to and activating receptors on target cells. These receptors can be located on the cell surface, as for peptide hormones, or within the cell, as in the case of steroids and thyroid hormones. After receptor activation, intracellular signaling pathways (e.g., second messenger systems or ligand-activated transcription factors) are modulated, which acutely or chronically alter cellular physiology and potentially whole organism physiology.
Excessive levels of endogenous hormones may result from excessive organ secretion or unregulated ectopic formation. Hormone overproduction by a secreting organ is commonly associated with excessive stimulation or a malignancy (or hyperplasia). Successful management of this situation includes blockade of the stimulatory agent, if identifiable, or interference with hormone formation, secretion, or action. Ectopic production of a biologically active form of the hormone by tissues is complicated by the lack of feedback mechanisms to regulate hormone production and is typically associated with tissue malignancy or infection. The primary determinant of successful intervention frequently requires a combination of ablation of the secreting tissue and pharmacological agents to antagonize the effects of elevated hormone levels. The success of this technique hinges on the ability of the responsible tissue to respond to pharmacological intervention. If it is not possible or detrimental to directly reduce hormone levels, a situation often encountered before or immediately after surgery, or when the cause of the elevated hormone levels is unknown or uncorrectable, alternative, patient-specific strategies to reduce the effects of elevated hormone levels must be used. A summary of strategies to manage the levels and action of hormones is presented in Box 38-2. A list of drugs that affect hormonal balance and their mechanisms of action are in Box 38-3.
BOX 38–2 Strategies to Manage the Levels and Action of Hormones
BOX 38–3 Drugs Known to Affect Hormonal Balance
Effectors of Hormone Release/Reuptake
Steroid Biochemistry and Physiology
All secreted steroids are synthesized from cholesterol, which can be synthesized de novo or derived from circulating lipoproteins. Similar metabolic pathways mediate steroid synthesis in all organs (Fig. 38-1). The organ-specific formation of secreted steroids depends on the presence of specific catalytic enzymes (Table 38-1).
TABLE 38–1 Enzymes Present in Different Tissues Mediating the Organ- or Tissue-Specific Formation of Steroid Hormones
The action of steroids is mediated largely by altering gene transcription through interaction with promoter deoxyribonucleic acid (DNA) of genes. Steroid receptors are dimeric and coupled with accessory proteins until activated by ligands outside the nucleus. The steroid-receptor complex is phosphorylated and translocated to the nucleus through a nuclear pore, facilitated by the importin protein. The interaction with the gene promoter region occurs through steroid-specific palindromic nucleotide sequences within the receptor. The interaction of DNA and the steroid-receptor complex is dependent on steroid structural differences, amino acid sequence of the DNA binding domain, the nucleotide sequence of the DNA binding site, and the architecture of the gene promoter. The structures of the primary circulating steroids are shown in Figure 38-2.
In the adrenal gland, the primary secreted steroids are aldosterone, cortisol, and dehydroepiandrosterone (DHEA) (see Fig. 38-2). Aldosterone is the primary mineralocorticoid and acts at the luminal epithelia to promote renal reuptake of Na+, which conserves Na+ and can elevate blood pressure. In the zona glomerulosa, the lack of CYP17 is associated with nearly exclusive formation of aldosterone. Further, the release of aldosterone from the zona glomerulosa is regulated by the renin-angiotensin pathway as a result of activation angiotensin II-receptors, which are linked to the formation of 1,4,5-inositol triphosphate. The amount of aldosterone released is relatively low (50 to 150 µg/day); aldosterone is transported in the blood through an interaction with albumin with a bound/free ratio of 70/30.
The complement of enzymes in the zona fasciculata and zona reticularis permits the formation of cortisol, the primary circulating glucocorticoid (see Chapter 39). The release of cortisol is dependent on a tightly regulated hypothalamic-anterior pituitary-adrenal cortex axis. The biological role of glucocorticoids is complex and temporal. The liver has the greatest level of nuclear receptors or steroid-activated transcription factors, although they are present in many tissues. The primary systems affected by cortisol include self-regulation of formation via suppression of corticotropin-releasing hormone (CRH) and adrenocorticotropic hormone (ACTH) secretion, storage of hepatic glycogen, response to stress, and suppression of the immune system. The daily production of cortisol ranges from 10 to 20 mg, and plasma levels follow a diurnal pattern with the highest levels in the morning. In the blood, cortisol is bound to a specific hepatic protein, corticosteroid-binding protein (aka transcortin), which promotes its transport and increases its duration of action.
The secretion of estrogen (β-estradiol) and progesterone from the ovary is regulated by hypothalamic gonadotropin-releasing hormone (GnRH), anterior pituitary follicle-stimulating hormone (FSH), and leutenizing hormone (LH). Release of the gonadotropic hormones and the ovarian steroids during the menstrual cycle is episodic, and the highest levels of β-estradiol or progesterone occur during the late follicular phase or midleutal phase, respectively (see Chapter 40). Most of these circulating steroids (98%) are bound to specific steroid hormone-binding globins.
The primary testicular androgen, testosterone, is converted to dihydrotestosterone (DHT) in tissues expressing 5α-reductase. The actions of androgens include development of male reproductive tract and accessory tissues, stimulation of secondary sexual traits, growth, and development of the central nervous system (CNS) (see Chapter 41). As shown in Figure 38-1, the expression of steroid metabolizing enzymes promotes the formation of DHEA and androstenedione leading to the formation of testosterone; the expression of aromatase in ovarian cells permits conversion of testosterone to β-estradiol.
Therapeutic Overview
Pharmacology of Hypothalamic and Pituitary Hormones
The hypothalamus and pituitary gland work in concert to regulate endocrine systems throughout the body. Peptides and biogenic amines synthesized and secreted by specialized neurons within the hypothalamus are transported to the anterior pituitary by the hypothalamic-hypophyseal portal circulation, where they act through specific receptors to stimulate or inhibit hormone secretion (Fig. 38-3). Anterior pituitary hormones trigger peripheral endocrine organs to produce hormones, which have individual functions and provide feedback to the hypothalamus and pituitary to regulate the synthesis and release of their tropic hormones. As mentioned, GnRH (also called luteinizing hormone releasing hormone) stimulates the secretion of LH and FSH by the pituitary. LH and FSH promote gametogenesis and gonadal hormone production by the ovaries and testes (see Chapters 40 and Chapter 41). Thyrotropin-releasing hormone (TRH) stimulates secretion of thyroid-stimulating hormone (TSH), which in turn controls thyroid function (see Chapter 42) CRH stimulates the secretion of ACTH, which promotes the secretion of cortisol by the adrenal cortex (see Chapter 39). Growth hormone-releasing hormone (GHRH) stimulates and somatostatin (also called somatotropin-release inhibiting factor, SRIF) inhibits the production of growth hormone (GH), which has numerous effects on growth and metabolism. Hypothalamic dopamine functions to tonically inhibit secretion of prolactin, the hormone primarily responsible for lactation and suppression of fertility while nursing.
