Chapter 4 Intradural Extramedullary Benign Tumors
MENINGIOMA
HISTOLOGY/GRADING
GENERAL
• Meningiomas are dural-based neoplastic proliferations of meningothelial cells (arachnoid cap cells).
• The classic histologic findings include lamellar (psammomatous) calcifications and nuclear pseudoinclusions, which can be found in all subtypes of this tumor.
• The neoplastic cells demonstrate uniform vimentin immunoreactivity and patchy to widespread epithelial membrane antigen (EMA) immunoreactivity.
• Adhering to the World Health Organization 2000 classification of central nervous system tumors,1 grading is based on histologic subtype and other features (e.g., necrosis, mitotic rate).
Grade I
MENINGOTHELIAL
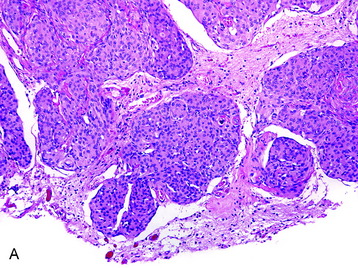
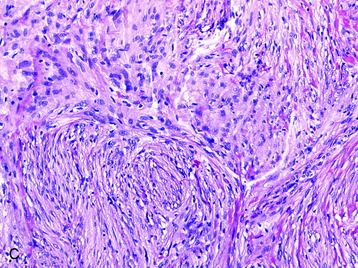
• The meningothelial meningioma is composed of syncytial lobules of uniform cells with oval nuclei (Fig. 4-1, A).
• The histologic differential (in small specimens) includes benign meningothelial proliferations associated with a neighboring process (e.g., a reactive response).
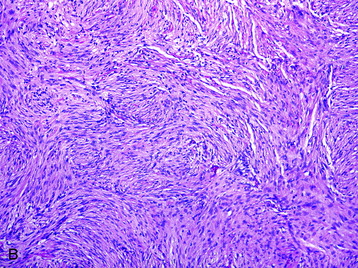
FIBROUS
TRANSITIONAL
PSAMMOMATOUS
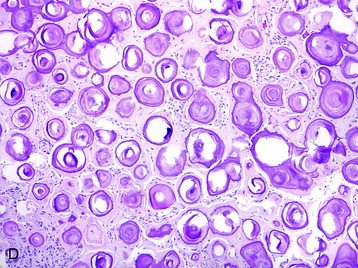
• This subtype is composed mainly of psammomatous calcifications with a paucity of cellular elements (Fig. 4-1, D).
Grade II
CLEAR CELL
CHORDOID
Atypical
• Irrespective of the histologic subtypes previously described, a meningioma is assigned a grade II if three of the following five criteria are fulfilled:
• A grade of II is assigned if four or more mitotic figures per 10 high-power fields are identified.
• Clinicopathological studies3 have indicated that the presence of brain invasion confers a more aggressive clinical course, and this finding alone justifies a diagnosis of grade II meningioma (Fig. 4-2, D).
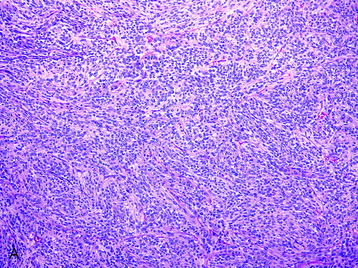
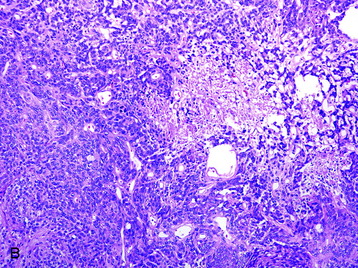
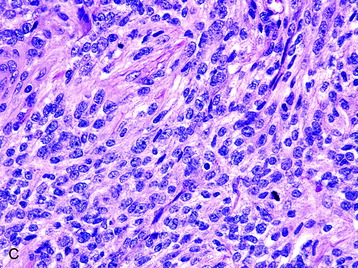
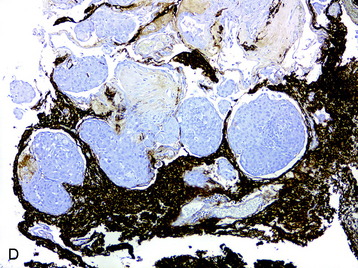
Fig. 4-2 Atypical features in meningiomas. A, Patternless or sheet-like growth pattern. B, Focus of spontaneous necrosis. C, A hypercellular area containing cells with prominent nucleoli and mitotic figures. D, Glial fibrillary acidic protein immunohistochemical stain of tumor in Fig. 4-1, A highlighting brain parenchyma between the infiltrating tumor nests (×100). (A-C, hematoxylin–eosin stain; A and B, ×100; C, ×400.)
Grade III
PAPILLARY
RHABDOID
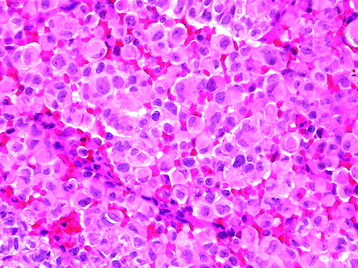
• The rhabdoid subtype is composed of cells with abundant eosinophilic cytoplasm and eccentrically positioned nuclei (Fig. 4-3). This meningioma subtype typically has an association with high mitotic rate and other cytoarchitectural anaplastic features, as well as evidence of brain invasion.4,5
RADIOLOGY
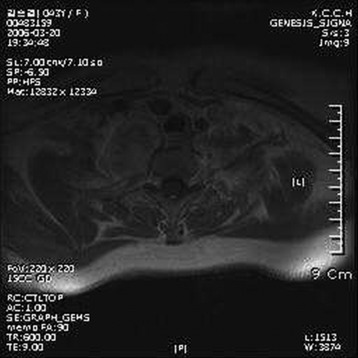
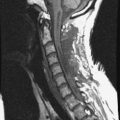
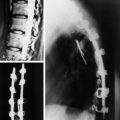
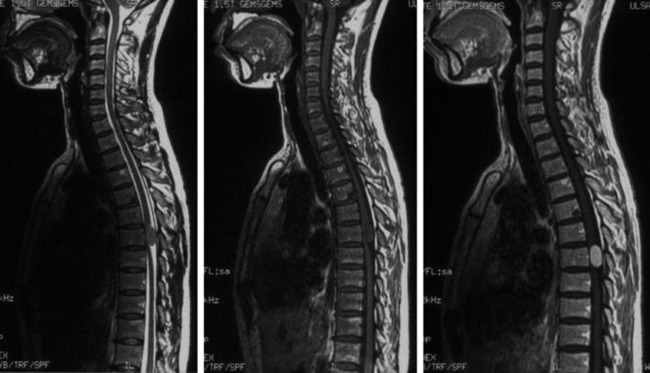
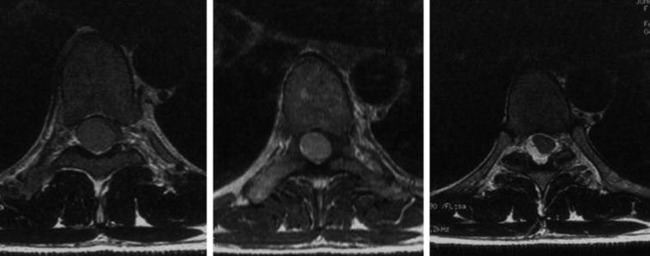
• The location of meningioma is typically intradural, extramedullary. However, it is rarely located in intraosseous, extradural, or even paraspinous areas. On T1-weighted images (T1WI), the signal of the mass is isointense to the spinal cord (Figs. 4-4 and 4-5).
• On T2-weighted images (T2WI), most parts of the mass show iso-signal to the spinal cord (see Figs. 4-4 and 4-5).
• If a calcified area is included, the signal may change to low. When the meningioma is very vascular, prominent flow void can be displayed.
NERVE SHEATH TUMOR—SCHWANNOMA
DISTRIBUTION
• Schwannomas may be found throughout all levels of the spinal cord and involve the sensory nerve roots.
HISTOLOGY/GRADING
• Schwannomas are proliferations of neoplastic Schwann cells, which may have variable appearances (e.g., spindled, epithelioid, melanotic).
• Classic schwannoma (grade I)
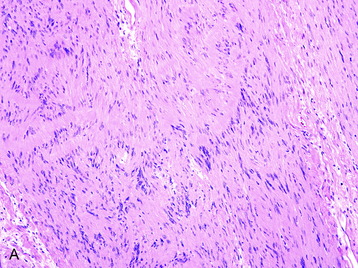
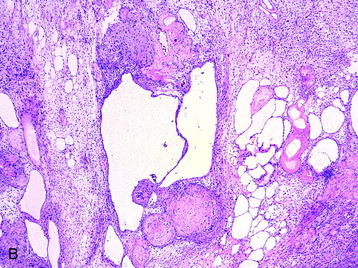
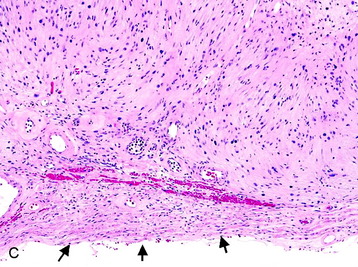
• Histologic sections show Antoni A (densely cellular) and Antoni B (less cellular, sometimes cystic) areas (Fig. 4-7, A).
• Within Antoni A regions, Verocay bodies (small groups of fibrils surrounded by parallel rows of neoplastic cells) are commonly found.
• Cellular atypia alone (usually in Antoni B areas) in otherwise unremarkable schwannomas reflect degenerative (so-called “ancient”) changes and confers no prognostic significance. Other reactive and degenerative features, including cyst formation, macrophage and lymphocyte infiltration, and hemosiderin-laden cells, are commonly found in Antoni B areas (Fig. 4-7, B).
• The nerve from which the schwannoma arises may be identified in the specimen and, if so, is compressed to the edge of the tumor (Fig. 4-7, C).
• A histologic differential diagnosis for schwannomas usually only arises when less-than-classic microscopic features are present. Meningiomas, particularly of the fibrous subtype, may enter the differential and can be distinguished from schwannomas by the presence of diffuse EMA and patchy S100 protein immunoreactivity in the former lesion. Tumors with concurrent schwannoma and meningioma features have been reported.7,8
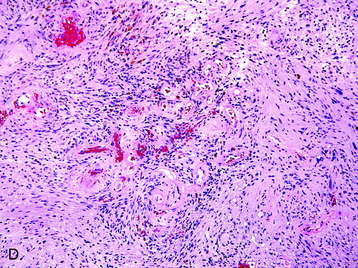
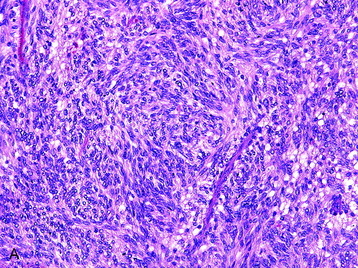
• Cellular schwannomas (grade I [Fig. 4-8, A])
• Cellular schwannomas have an overall greater degree of cellularity than classic schwannomas and are composed almost exclusively of Antoni A regions with an absence of Verocay bodies.
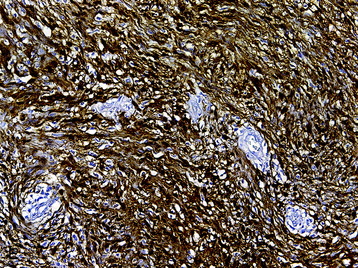
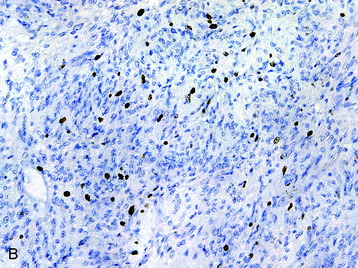
• Modest mitotic activity and increased Ki-67 labeling indices (Fig. 4-8, B), cellular atypia, and a fascicular growth pattern are typical histologic findings in cellular schwannomas.
RADIOLOGY
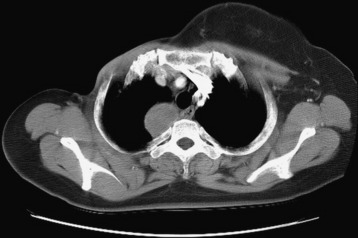
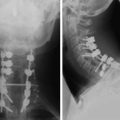
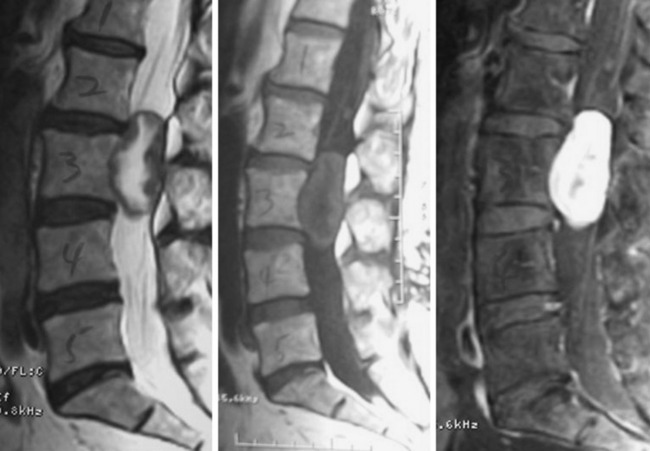
• Typical morphology of the schwannoma is a well-circumscribed, round, or lobulated mass in the spinal canal or extraspinal location.
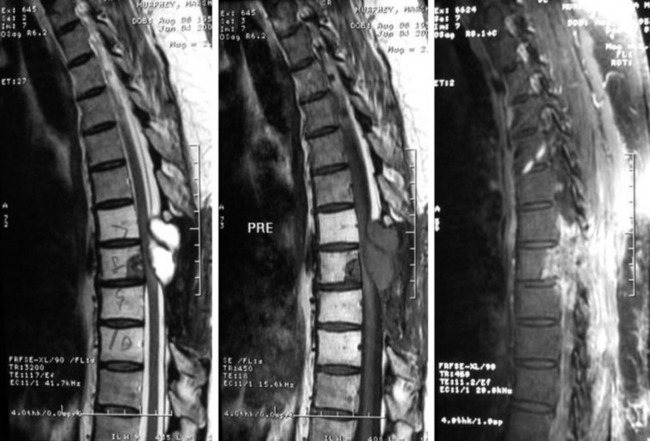
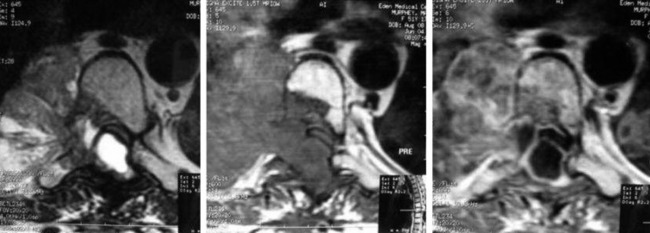
• Seventy percent of the schwannomas are located in the intradural space (Fig. 4-9), and 15% of them occupy the extradural space. The rest of them take on a dumbbell shape (combined intradural and extradural; Figs. 4-10 and 4-11).
• On simple radiography, benign bony change may be seen. Interpedicular distance can be widened, and posterior vertebral margin may be eroded. The width of the pedicle can be thinned, and the intervertebral foramen may be enlarged.
• On computed tomography (CT) scan, the density is isodense to the spinal cord. Cystic change may commonly be seen.
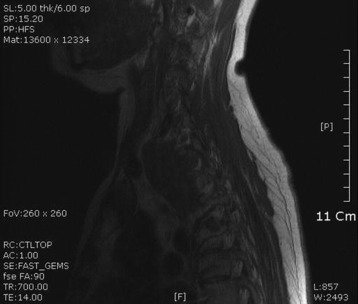
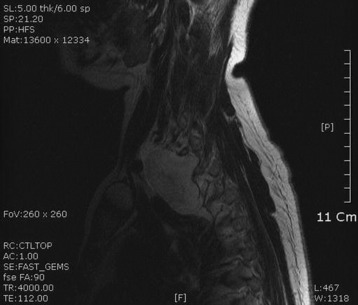
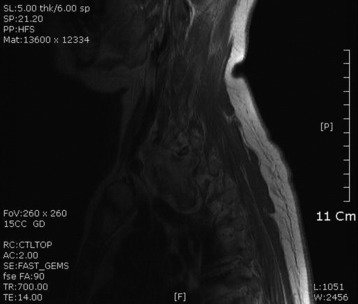
• On magnetic resonance imaging (MRI), the signal of the mass is isointense or hypointense to spinal cord on T1WI. The signal is hyperintense in most cases on T2WI. The enhancement pattern is uniform or heterogeneous (Figs. 4-10 to 4-13).
NEUROFIBROMA
EPIDEMIOLOGY
• Neurofibromas of the spine can occur spontaneously but are found at a high frequency in patients with neurofibromatosis type 1 (NF-1).
• Spinal nerve sheath tumors constitute one-fourth of all spinal tumors and more than one-third of intradural extramedullary tumors of the spinal cord.9
DISTRIBUTION
• Neurofibromas are typically extradural lesions that may extend proximally to an intradural location.
• Neurofibromas can be located entirely inside the dural sac (intradural), entirely outside the dural sac (extradural), or partly inside and partly outside the dural sac, according to the location of the tumor origin along the spinal nerve root.
• In the literature, 70–80% of spinal neurofibromas are intradural, and those extending through the dural aperture as a dumbbell mass with both intradural and extradural components account for another 15%.9
• Tumors with an extraforaminal component decrease from the cervical region to the lumbar region of the spinal axis.
• A spinal nerve sheath tumor originates from the spinal nerve root and grows along the spinal nerve root.
• The ratio of the intradural segment to the extradural segment of the spinal nerve root within the spinal canal gradually increases from the high cervical region to the sacral region. Similarly, the length of the nerve root within the spinal canal gradually increases from the rostral region to the caudal region.11
HISTOLOGY/GRADING
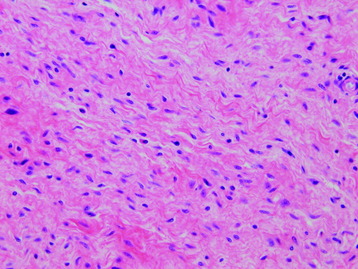
• Neurofibromas are grade I lesions, which represent a proliferation of all components of a nerve, including Schwann cells, collagen, and perineurial cells, resulting in a fusiform expansion (Fig. 4-14).
• Unlike schwannomas, fragments of the axons in the native nerve are “peppered” throughout the neoplasm rather than eccentrically compressed.
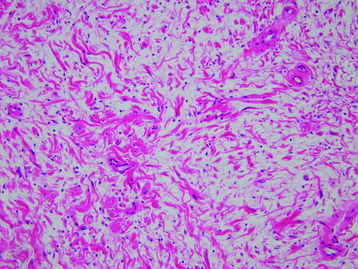
• Neurofibromas often have a myxoid matrix in which mast cells are common. The appearance of small, haphazardly arrayed aggregates of collagen has been likened to “shredded carrots” (Fig. 4-15).
• Plexiform neurofibromas are visually complex appearing lesions with interdigitating fascicles. This form of neurofibroma typically involves larger nerve trunks and is nearly pathognomonic for NF-1.
• In contrast to schwannomas, neurofibromas demonstrate less dense S100 protein immunoreactivity (Fig. 4-16).
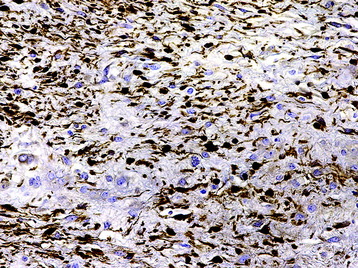
RADIOLOGY
• The best diagnostic clue is bulky, multilevel spinal nerve root tumors in patients with stigmata of NF-1.
• The location of the tumor is variable: intradural extramedullary, extradural intraspinal, or extraspinal (paraspinal). In the extraspinal area, the tumor can develop from the spinal nerve root, neural plexus, peripheral nerve, or end organs.
• On CT, the mass shows isodense image to the spinal cord. Neural foramen enlargement can accompany the mass.
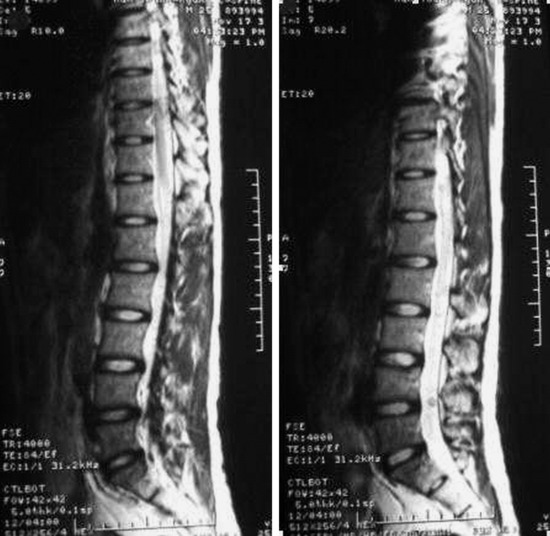
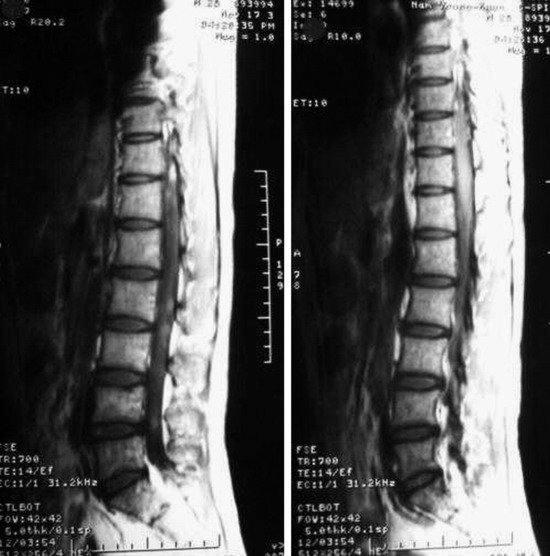
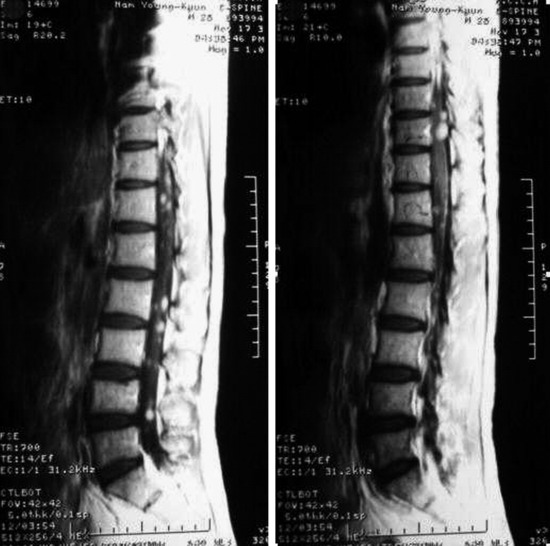
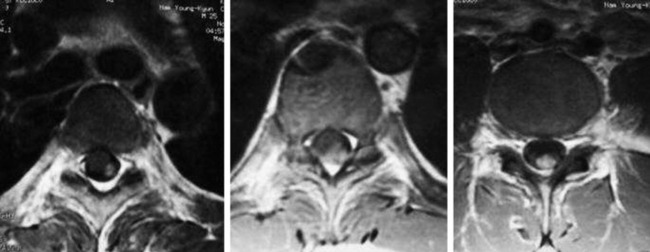
• On T2WI MRI, neurofibroma can be of isointense or hyperintense signal intensity. Sometimes, central high signal and peripheral low signal may be seen (Target sign) (Fig. 4-17).
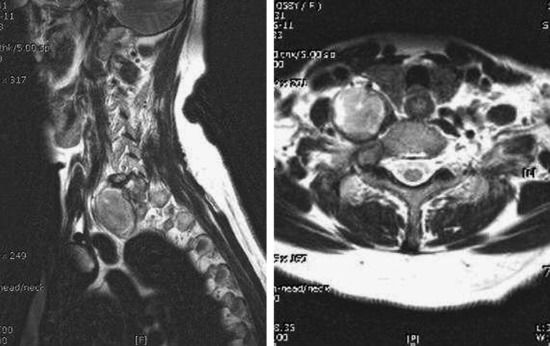
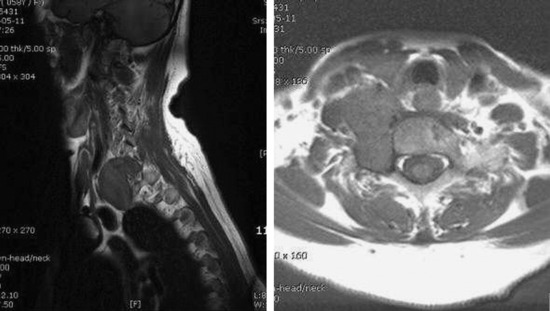
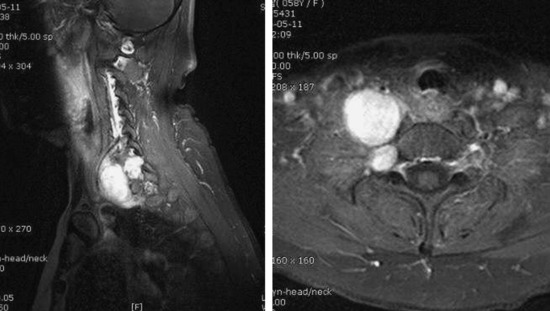
• In cases of extraspinal neurofibroma, the signal of the mass is similar to intraspinal neurofibroma (Figs. 4-21 to 4-24).
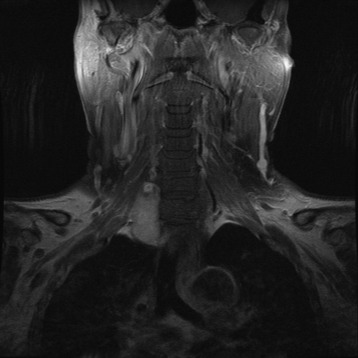
PARAGANGLIOMA
EPIDEMIOLOGY
• Pheochromocytomas are catecholamine-secreting tumors of the adrenal medulla chromaffin cells. Extra-adrenal pheochromocytomas are called paragangliomas and only rarely produce catecholamine excess.12 All but a small fraction of extra-adrenal paragangliomas arise from the carotid body and glomus jugulare.
• Paragangliomas of the spine are uncommon lesions that usually present as intradural tumors within the cauda equina.13 More than 70 cases have been reported since 1972.
DISTRIBUTION
HISTOLOGY/GRADING
• Grossly, all of the tumors located in the intradural extramedullary space are well encapsulated, red-black, very soft or friable, and bleed easily. Tumor mass is adherent to a vascular pedicle or a rich vascular network.
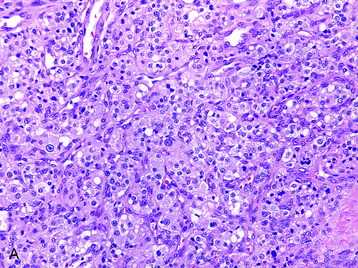
• Extra-adrenal paragangliomas are composed of neurosecretory cells, which may assume a number of architectural patterns. A nested pattern is common in which the cells are clustered into packets (“zellballen”) that are separated by numerous thin-walled vessels. Supporting, slender sustentacular cells (modified Schwann cells) surround the cell nests (Fig. 4-25, A).
• More magnified images show that the nuclei are uniformly round to oval with a regular chromatin pattern. Nucleoli are observed, and the cytoplasm is finely granular. No mitosis is observed.15
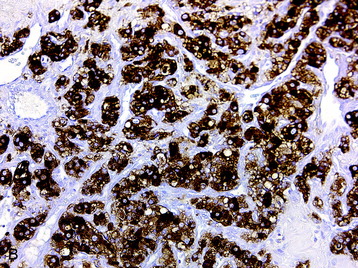
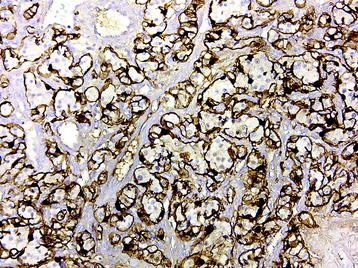
• The neurosecretory cells have a characteristic “salt and pepper” chromatin pattern and are immunoreactive for synaptophysin, chromogranin, and neuron-specific enolase (Fig. 4-25, B). The sustentacular cells demonstrate S100 protein immunoreactivity (Fig. 4-25, C).
• The histologic differential diagnosis includes ependymoma, the diagnosis initially applied to the first reported case of paraganglioma of the filum terminale. These two entities can be discerned through immunohistochemical staining with immunoreactivity for chromogranin and S100 protein (as previously discussed) in paragangliomas and glial fibrillary acidic protein immunoreactivity in ependymomas.
• Paragangliomas are grade I neoplasms, although rare circumstances of aggressive growth and malignant progression have been documented.
RADIOLOGY
• The diagnosis of pheochromocytoma or functioning paraganglioma is made by the measurements of the levels of catecholamines and their metabolites in plasma or urine.
• The sensitivity of plasma metanephrine levels for the detection of pheochromocytoma has been reported to be as high as 100%, compared with 82% for urinary and plasma catecholamine levels.16
• The m-[123I]iodobenzylguanidine scanning approach relies on nuclide uptake by the tumor and is particularly useful for locating extra-adrenal tumors.16
• The typical MR finding is a well-demarcated mass with low/intermediate signal intensity on T1WI and intermediate/high signal intensity on T2WI, in comparison with spinal cord tissue.
• On T2WI, heterogeneous signal intensity is observed. A hypointense tumor margin is commonly observed on T2WI, which indicative of paramagnetic effects from hemosiderin.14
• The hypervascular nature of paragangliomas results in punctuate areas of flow void interspersed in a matrix of increased signal intensity caused by slow flow and tumor cells. This produces a salt-and-pepper appearance on T2WI, which is considered to be a characteristic of paragangliomas (Fig. 4-26).
• Gadolinium administration produces intense heterogeneous enhancement, and high-velocity flow through the hypervascular tumors produces multiple serpiginous areas of signal void with all sequences.17
GANGLIONEUROMA
EPIDEMIOLOGY
• Ganglioneuromas belong to a group of neuronal tumor in which the neoplastic cells express a mature neuronal phenotype.18 They show a benign and slow-growing nature.
• The peripheral nervous system is much more commonly involved than the central nervous system. Peripheral ganglioneuroma arises from pluripotential neural crest cells and is closely related to the embryological development of the sympathetic nervous system.
• Common sites for peripheral ganglioneuromas include the adrenal medulla and the sympathetic nervous system in the neck, mediastinum, and retroperitoneum.19
• Central ganglioneuroma, or gangliocytoma, is less common.19 Analogous to their peripheral counterpart, both tumors represent the product of differentiation and maturation of the progeny of an initially primitive neuroblastic population.
• In contrast to the established link between peripheral ganglioneuroma and the sympathetic nervous system, the histogenesis of central ganglioneuroma remains poorly defined. Spinal ganglioneuroma encompasses the features of both the central and the peripheral types of ganglioneuroma.
• Ganglioneuromas occur most commonly in children and young adults younger than 30 years old.19 The ratio of male to females is approximately 3:2.
DISTRIBUTION
• Spinal ganglioneuromas constitute less than 10% of all ganglioneuromas. Most cases involve the paraspinal region with intraspinal extension, forming dumbbell-shaped tumors.
• Others occur within the spinal cord. Rarely, intradural extramedullary ganglioneuroma has been reported.
HISTOLOGY/GRADING
• Grossly, these tumors are usually well-circumscribed and firm in consistency. Small, focal calcifications are often seen in central ganglioneuroma. In most cases, the mass is avascular, so piecemeal removal of tumor mass is possible.
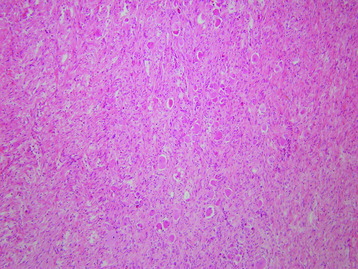
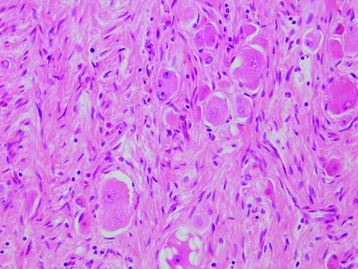
• On light microscopy, ganglioneuromas are poorly to moderately cellular tumors, with ganglion cells and spindle cells of neuronal origin with characteristic anomalies (Fig. 4-27). Mitotic figures are not seen.
• Peripheral forms usually have prominent connective tissue stroma. The stromal portion is organized into multiple fascicles, which are bundles of nerve fibers composed of Schwann cells, endoneurium connective tissue, and perineurial cells. Ganglion cells have vesicular nuclei and prominent nucleoli with abundant granular cytoplasm (Fig. 4-28).20
• The histologic evidence for neuronal origin can be obtained from the positive immunohistochemical staining for tyrosine hydroxylase, which is used for catecholamine synthesis.
• Electron microscopy studies show dense-core vesicles within neuronal cytoplasm and cell processes.21
• Ganglion cells are known not to proliferate in the ganglioneuromas with Ki-67/MIB-1 antigen study.
• Very rarely, malignant transformations arising from the stromal component of ganglioneuromas have been documented. To our knowledge, only 14 cases of malignant transformations arising from ganglioneuromas, either de novo or postirradiation, have been reported to date.22
RADIOLOGY
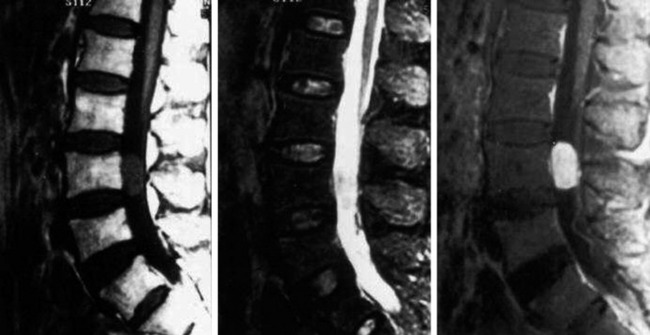
• On CT scan, ganglioneuromas appear as a homogeneous, well-circumscribed mass with heterogeneous enhancement after intravenous contrast (Fig. 4-29).23
• MRI characteristically shows low signal intensity on T1WI (Fig. 4-30) and heterogeneous high signal intensity on T2WI (Fig. 4-31).23
• With gadolinium enhancement, the mass shows bright contrast and internal focal low signal (Figs. 4-32 and 4-33).
• The differential diagnosis of an enhancin98g dumbbell-shaped mass in the spinal canal extending through the neural foramina includes neurogenic tumors, of which neurofibromas are the most common.