[level-membership-for-pulmolory-and-respiratory-category]
Interstitial Lung Disease
After reading this chapter you will be able to:
 Organize and distinguish between the entities grouped as interstitial lung diseases (ILDs).
Organize and distinguish between the entities grouped as interstitial lung diseases (ILDs).
 Interpret symptoms, examination signs, and pulmonary function testing in ILD.
Interpret symptoms, examination signs, and pulmonary function testing in ILD.
 List emerging pathophysiologic characteristics that are associated with selected ILDs.
List emerging pathophysiologic characteristics that are associated with selected ILDs.
 Describe how to manage ILD in general and how some specific ILDs can be treated.
Describe how to manage ILD in general and how some specific ILDs can be treated.
Interstitial lung disease (ILD) comprises a broad category of lung diseases rather than a specific disease entity.1,2 This category includes various illnesses affecting the lung parenchyma with many different causes, treatments, and prognoses. These disorders are grouped together because of similarities in their clinical presentations, appearance on plain chest radiography, and physiologic features.
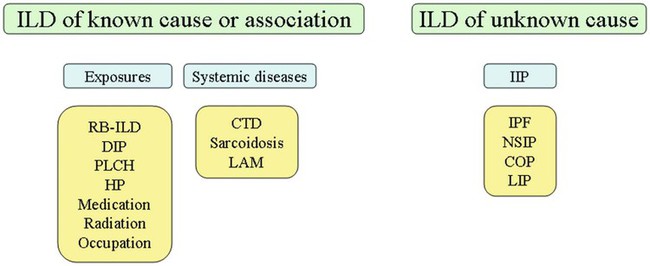
With over 100 separate disorders, it is essential to group ILDs based on etiology, disease associations, or pathology. An organizational scheme is presented in Figure 24-1. When evaluating patients with an ILD, one first must consider diseases with known causes or associations, such as diseases related to specific exposures, diseases associated with systemic conditions, and diseases with a known genetic basis. Most patients do not have an ILD with a known cause, and their disorder is classified by pathologic pattern. These groups are divided into specific disease entities. Using this organizational scheme to guide a careful and complete history, one is able to understand the disease processes and work efficiently toward an accurate diagnosis.
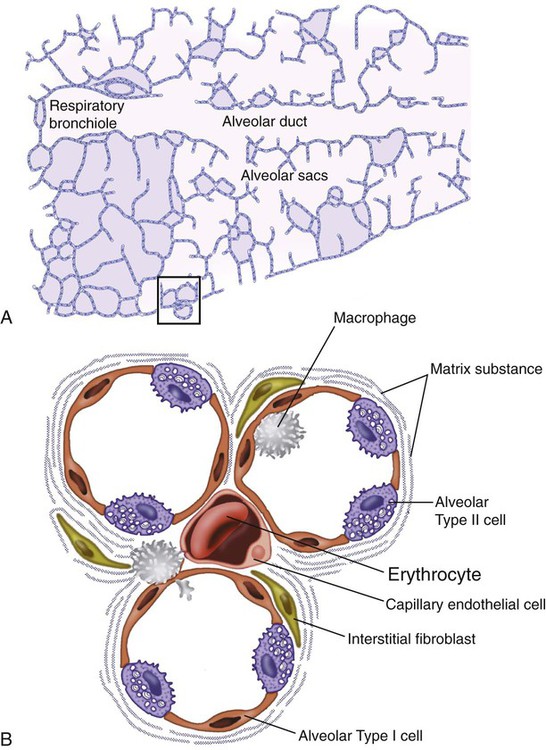
As the name ILD implies, the histologic abnormalities that characterize ILD involve the pulmonary interstitium to a greater extent than the alveolar spaces or airways, although exceptions exist. Figure 24-2 illustrates the components of the normal pulmonary parenchyma. The interstitium is the area between the capillaries and the alveolar space. As Figure 24-2 shows, in the normal state, this space allows close apposition of gas and capillaries with minimal connective tissue matrix, fibroblasts, and inflammatory cells such as macrophages. The interstitium supports the delicate relationship between the alveoli and capillaries allowing for efficient gas exchange. When responding to any injury—whether from a specific exposure (e.g., asbestos, nitrofurantoin, or moldy hay), an autoimmune-mediated inflammation from a systemic connective tissue disease (e.g., rheumatoid arthritis), or unknown injury (e.g., idiopathic pulmonary fibrosis [IPF])—the lung must respond to the damage and repair itself. If the exposure or injury persists or if the injury repair process is imperfect, the lung may be permanently damaged with increased interstitial tissue replacing the normal capillaries, alveoli, and healthy interstitium.
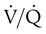 mismatching, shunt, and decreased diffusion across the abnormal interstitium. Work of breathing is markedly increased because of decreased lung compliance. Together, these physiologic impairments lead to the exercise intolerance seen in all of the ILDs. If the initiating injury or abnormal repair from injury is not halted, progressive tissue damage leading to worsening physiologic impairment and death can occur.
mismatching, shunt, and decreased diffusion across the abnormal interstitium. Work of breathing is markedly increased because of decreased lung compliance. Together, these physiologic impairments lead to the exercise intolerance seen in all of the ILDs. If the initiating injury or abnormal repair from injury is not halted, progressive tissue damage leading to worsening physiologic impairment and death can occur.Characteristics of Interstitial Lung Disease
Clinical Signs and Symptoms of Interstitial Lung Disease
Many ILDs have similar clinical features and are not easily distinguished based on history or examination. Symptoms are generally limited to the respiratory tract. However, extrapulmonary symptoms should not be ignored because they may point to an ILD associated with a systemic diagnosis. Exertional breathlessness (dyspnea) and a nonproductive cough are the most common reasons patients seek medical attention. Other respiratory symptoms, such as sputum production, hemoptysis, pneumothorax, or wheezing, can occur and be suggestive of specific diseases (Table 24-1). If the patient also has prominent extrapulmonary symptoms, such as myalgia, arthralgia, sclerodactyly, gastroesophageal reflux, or Raynaud phenomenon, ILD resulting from underlying connective tissue disease may be present (Table 24-2).
TABLE 24-1
Unusual Pulmonary Findings and Likely Diagnosis
| Pulmonary Findings | Disease | Frequency |
| Hemoptysis | LAM | Rare |
| Chyloptysis | LAM | Rare |
| Pneumothorax | LAM, BHD | Common |
| Wheeze | Sarcoidosis, LAM | Common |
| Chylous pleural effusion | LAM | Common |
| Exudative pleural effusion | RA | Common |
TABLE 24-2
Extrapulmonary Findings and Likely Diagnosis
| Extrapulmonary Findings | Disease | Frequency |
| Raynaud phenomenon | All CTD | Common |
| Arthralgia | All CTD | Common |
| Myalgia | Polymyositis | Common |
| Large muscle weakness | Polymyositis | Common |
| Sclerodactyly | Scleroderma | Common |
| Rheumatoid skin nodules | RA | Rare |
| Fingertip fissures | Antisynthetase syndrome | Common |
| Dorsal hand rash | Dermatomyositis | Common |
| Facial rash | Dermatomyositis | Common |
| Exudative pleural effusion | RA | Common |
| Shawl distribution skin nodules | TSC | Common |
| “Pencil eraser” facial skin nodules | BHD | Common |
| Central nervous system benign cortical tuber | TSC | Common |
| Abdominal angiomyolipoma | LAM | Common |
| Renal cancer | BHD | Common |
| Cardiomyopathy | Sarcoidosis, polymyositis | Rare |
| Cardiac conduction block | Sarcoidosis | Rare |
| Violaceous facial skin nodules | Sarcoidosis | Common |
| Subcutaneous nodules | Sarcoidosis | Common |
| Cranial neuropathy | Sarcoidosis | Rare |
| Small fiber neuropathy | Sarcoidosis | Rare |
Radiographic Features
ILDs manifest as abnormal lung parenchyma that cast abnormal radiographic shadows. For most ILDs, the chest radiograph reveals reduced lung volumes with bilateral reticular or reticulonodular opacities. However, the chest radiograph has limited value because the three-dimensional abnormalities are summed into a two-dimensional image with loss of spatial information. High-resolution cross-sectional imaging via computed tomography (CT) provides detailed images representing pulmonary pathology.3 High-resolution CT images allow noninvasive evaluation of ILDs and are a key element in making a confident diagnosis and managing ILD.4
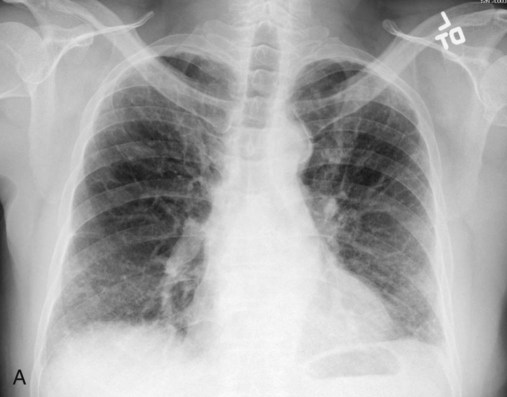
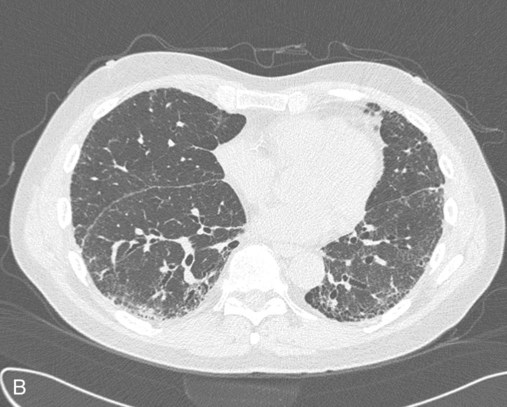
Plain chest radiographs and high-resolution CT images of usual interstitial pneumonitis (UIP) show the prototypic fibrotic injury pattern. The chest radiograph (Figure 24-3, A) and high-resolution CT image (Figure 24-3, B) in UIP typically reveal a bilateral, patchy, peripheral (subpleural), and basilar predominant disease with reticulonodular infiltrates, often with honeycomb, cystic change. The lung architecture is distorted in patients with moderate or severe disease burden, with reduced lung volume and traction bronchiectasis, especially at the lung bases. Reticulogranular (ground-glass) abnormalities, increased attenuation of the lung tissue without distortion of the underlying blood vessels or bronchi, are absent or minimal in IPF. Pleural disease, air trapping, micronodules, and significant lymphadenopathy are not seen, although two-thirds of patients with IPF can have mild mediastinal adenopathy.5
 Rule of Thumb
Rule of Thumb Rule of Thumb
Rule of ThumbPhysiologic Features
Similar to the radiographic findings, there can be considerable variability among the specific diseases in the physiologic abnormalities seen. However, a restrictive physiologic impairment is the common finding.6 Both forced expiratory volume in 1 second (FEV1) and forced vital capacity (FVC) are diminished, and the FEV1/FVC ratio is preserved or even supranormal. Lung volumes are reduced, as is the diffusing capacity of the lung for carbon monoxide (DLCO). This reduction in diffusing capacity reflects a pathologic disturbance of the alveolus-capillary interface.
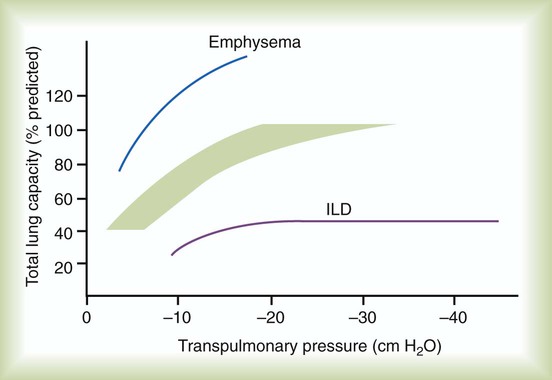
Although not commonly pursued, the compliance characteristics of the lungs can be evaluated with an esophageal balloon to measure intrathoracic pressure at various lung volumes. In almost all ILDs, the lungs have reduced compliance and require supranormal transpleural pressures to ventilate (Figure 24-4). This lack of compliance results in small lung volumes and increased work of breathing.
Less frequently, a pattern of physiologic obstruction may be seen. This obstruction can be the result of the primary disease process (e.g., LAM, PLCH, or sarcoidosis in some patients) or concomitant emphysema or asthma.7 If ILD develops in a patient with significant emphysema, the opposing physiologic effects of the two diseases may result in deceptively normal spirometry and lung volume measurements and apparently normally compliant lungs. However, because both emphysema and ILD result in impaired gas exchange, DLCO is significantly decreased.
 Rule of Thumb
Rule of ThumbSelected Specific Types of Interstitial Lung Disease and Therapies
Exposure-Related Interstitial Lung Disease
Tobacco-Associated Interstitial Lung Disease
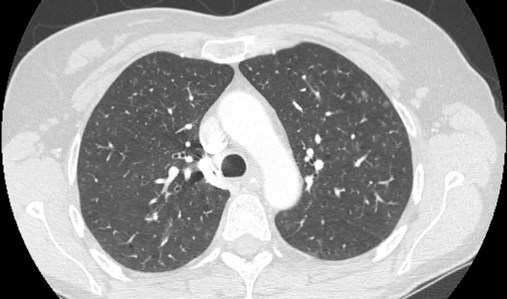
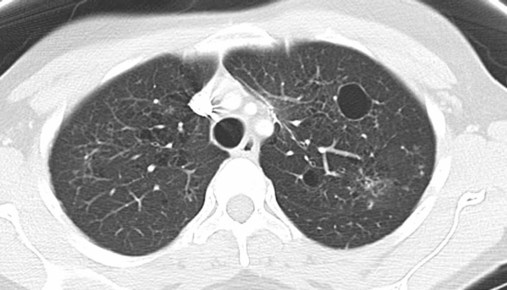
Although the association of first-hand tobacco smoke and obstructive lung disease is common and well known, tobacco smoke is also an avoidable cause of ILD. Although the association is rarer than with obstructive lung disease, first-hand tobacco smoke inhalation can lead to three types of ILD in susceptible individuals: RB-ILD, DIP, and PLCH. The first two disorders are related. Each disease consists of increased numbers of polyclonal activated macrophages. The diseases differ by the location of these overly abundant cells. In RB-ILD, macrophages accumulate in the respiratory bronchioles leading to bronchiolar remodeling and fibrosis of adjacent alveoli. As expected for a disease with combined airway and alveolar injury, pulmonary function testing reveals mixed restriction and obstruction with frequent air trapping. High-resolution CT images show this mixed pathologic location with indistinct centrilobular nodules (Figure 24-5). In DIP, the increased macrophages fill the alveoli, manifesting as restrictive impairment on pulmonary function testing and diffuse ground-glass attenuation on high-resolution CT imaging (Figure 24-6).
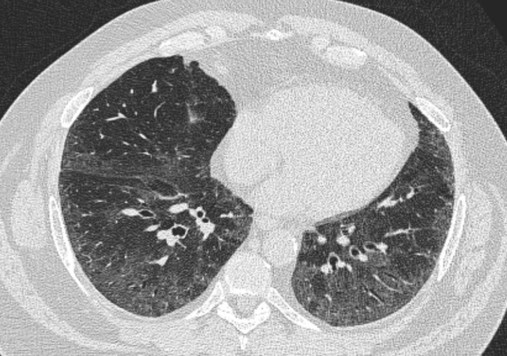
Pulmonary Langerhans cell histiocytosis (PLCH) is the third interstitial manifestation of tobacco smoke. Increased numbers of polyclonal macrophages play a prominent role. However, in PLCH, they are accompanied by fibroblasts and eosinophils in nodules concentrated around small airways. These nodules are stellate and destroy adjacent lung tissue, leading to the classic high-resolution CT image of stellate nodules associated with cysts as seen in Figure 24-7. Although adult smoking-associated PLCH is pathologically similar to childhood Langerhans cell histiocytosis, the adult form does not involve bone and has not proven to respond to chemotherapy as the childhood form does. The relationship of these two disorders has yet to be defined.
In each of these three diseases, the primary treatment is complete tobacco abstinence. With abstinence, most patients either minimally improve or remain stable,8 but a few progressively worsen and may require lung transplantation. Active treatment with prednisone or other immunosuppressive medications is discouraged because few, if any, patients improve.9
Drug-Related and Radiation-Related Interstitial Lung Disease
Many drugs have been associated with pulmonary complications of various types, including interstitial inflammation and fibrosis, bronchospasm, pulmonary edema, and pleural effusions. Drugs from many different therapeutic classes can cause ILD, most commonly chemotherapeutic agents, antibiotics, antiarrhythmic drugs, and immunosuppressive agents (Box 24-1). There are no distinct physiologic, radiographic, or pathologic patterns of drug-induced ILD, and the diagnosis is usually made when a patient is exposed to a medication known to result in lung disease, the timing of the exposure is appropriate for the development of the disease, and other causes of ILD have been eliminated. Treatment is avoidance of further exposure and systemic corticosteroids in markedly impaired or declining patients. In addition to future drug exposure, bleomycin injury is accentuated by increased fractional inspired oxygen (FiO2), even months after last drug exposure, and supplemental oxygen (O2) should be used only if absolutely necessary in these patients.10
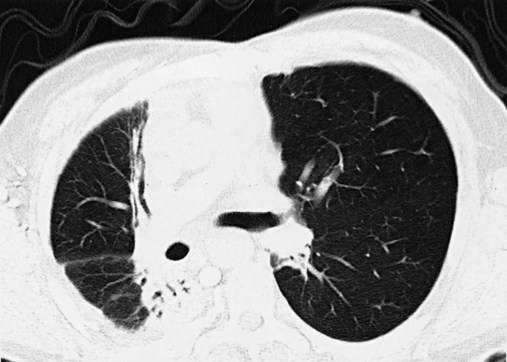
Exposure to therapeutic radiation in the management of cancer may result in ILD. Patients presenting within 6 months of radiation therapy generally have ground-glass abnormalities thought to represent acute inflammation. The ground-glass abnormalities can occur in both radiation-exposed tissue and unexposed tissue. Short-term systemic corticosteroid treatment can improve lung function. In contrast, radiographic abnormalities that develop more than 6 months after therapy typically appear as densely fibrotic tissue within the radiation port. On CT scan, a straight line indicating the margin of radiation is frequently evident as seen in Figure 24-8. These patients do not improve with corticosteroid therapy, and treatment is supportive.
Hypersensitivity Pneumonitis
Hypersensitivity pneumonitis (HP) is a cell-mediated immune reaction to inhaled antigens in susceptible persons.11 Patients must be sensitized by an initial exposure, with subsequent reexposure leading to either acute HP usually after a brief but intense exposure or chronic HP from persistent exposure. Patients with acute HP present to medical attention with a history of a few days of shortness of breath, chest pain, fever, chills, malaise, and a cough that may be productive of purulent sputum. Patients who are chronically exposed to low levels of inhaled antigens may develop subtle interstitial inflammatory reactions in the lung that do not result in noticeable symptoms for months to years and can present with severe, impairing disease, which can be very difficult to distinguish from IPF.
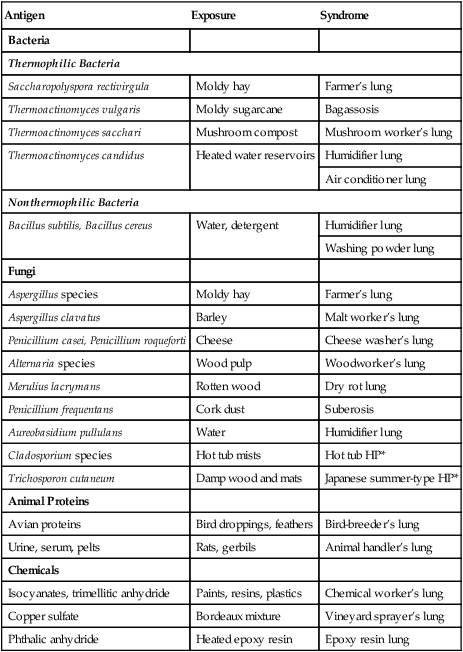
Common organic antigens known to cause HP include bacteria and fungi, which may be found in moldy hay (farmer’s lung) or in the home environment, in particular, in association with central humidification systems (humidifier lung), indoor hot tubs, and animal proteins (e.g., bird breeder’s lung). Inorganic antigens from vaporized paints and plastics can also lead to HP. Numerous established antigens are listed in Table 24-3 along with the typical source of exposure and the associated syndrome.
TABLE 24-3
Etiologies of Hypersensitivity Pneumonitis
| Antigen | Exposure | Syndrome |
| Bacteria | ||
| Thermophilic Bacteria | ||
| Saccharopolyspora rectivirgula | Moldy hay | Farmer’s lung |
| Thermoactinomyces vulgaris | Moldy sugarcane | Bagassosis |
| Thermoactinomyces sacchari | Mushroom compost | Mushroom worker’s lung |
| Thermoactinomyces candidus | Heated water reservoirs | Humidifier lung |
| Air conditioner lung | ||
| Nonthermophilic Bacteria | ||
| Bacillus subtilis, Bacillus cereus | Water, detergent | Humidifier lung |
| Washing powder lung | ||
| Fungi | ||
| Aspergillus species | Moldy hay | Farmer’s lung |
| Aspergillus clavatus | Barley | Malt worker’s lung |
| Penicillium casei, Penicillium roqueforti | Cheese | Cheese washer’s lung |
| Alternaria species | Wood pulp | Woodworker’s lung |
| Merulius lacrymans | Rotten wood | Dry rot lung |
| Penicillium frequentans | Cork dust | Suberosis |
| Aureobasidium pullulans | Water | Humidifier lung |
| Cladosporium species | Hot tub mists | Hot tub HP* |
| Trichosporon cutaneum | Damp wood and mats | Japanese summer-type HP* |
| Animal Proteins | ||
| Avian proteins | Bird droppings, feathers | Bird-breeder’s lung |
| Urine, serum, pelts | Rats, gerbils | Animal handler’s lung |
| Chemicals | ||
| Isocyanates, trimellitic anhydride | Paints, resins, plastics | Chemical worker’s lung |
| Copper sulfate | Bordeaux mixture | Vineyard sprayer’s lung |
| Phthalic anhydride | Heated epoxy resin | Epoxy resin lung |
Because the causal relationship between exposure and lung disease may not be obvious, a careful systematic occupational, environmental, and avocational history is crucial in evaluating patients with ILD. Elements that strongly suggest a diagnosis of HP are exposure to an appropriate antigen and the correct temporal relationship of symptoms to the exposure. Blood samples may be obtained to determine whether there has been an antibody response to certain antigens associated with HP (serum precipitins). However, the presence of such antibodies is insufficient to establish the diagnosis of HP because many individuals develop antibodies in the absence of disease. Likewise, the absence of detectable antibodies does not rule out the diagnosis of HP because the culprit may be an antigen that is not included in the blood analysis.12
Specific therapies for HP are strict antigen avoidance and immunosuppression with corticosteroids in patients with symptomatic or physiologically impairing disease. In acute HP, corticosteroids seem to hasten recovery but do not improve ultimate lung function.13 In chronic HP, patients with fibrosis on CT scan have a shorter survival, and it is unknown if long-term immunosuppression is beneficial.14
Occupational Interstitial Lung Disease
Predictable clinical and radiographic abnormalities occur in susceptible patients who have been exposed to asbestos.15 These abnormalities include pleural changes (plaques, fibrosis, effusions, atelectasis, and mesothelioma) and parenchymal scarring and lung cancer. Asbestos exposure alone increases the risk of lung cancer only minimally (1.5 to 3.0 times). However, asbestos exposure and cigarette smoking act synergistically to increase greatly the risk of cancer. Asbestos exposure also may result in benign asbestos pleural effusions or an entity known as rounded atelectasis. Benign asbestos pleural effusions may be asymptomatic or may be associated with acute chest pain, fever, and dyspnea. Generally, a shorter lag time exists between initial asbestos exposure and the development of benign asbestos pleural effusions (<15 years) than is seen with other manifestations of asbestos exposure. The effusions are characteristically exudative and are often bloody. In a patient with a history of asbestos exposure and a bloody pleural effusion, the major differential diagnostic concern is malignant pleural effusion from a mesothelioma. The clinical course of benign asbestos pleural effusions is characterized by spontaneous resolution, often with recurrences, and treatment is drainage to reduce symptoms. Rounded atelectasis typically manifests as a pleural-based parenchymal mass that may be mistaken for carcinoma. The characteristic CT features, such as local volume loss, pleural thickening, and the “comet tail” appearance of bronchi and vessels curving into the lesion, help distinguish rounded atelectasis from carcinoma.
The term asbestos-related pulmonary disease encompasses all of these entities, whereas asbestosis is reserved for patients who have evidence of parenchymal fibrosis. Most patients with asbestosis have had considerable airborne asbestos exposure many years before manifestation of the lung disease. Exposure frequently is associated with occupations such as shipbuilding or insulation work. Patients report very slowly progressive dyspnea on exertion16 and have crackles on lung examination. Physiologic testing shows restrictive impairment with reduced DLCO. The chest radiograph reveals bilateral lower zone reticulonodular infiltrates similar to infiltrates seen in IPF. With an appropriate exposure history, the presence of radiographic pleural plaques or rounded atelectasis indicates asbestos as the likely cause of ILD, although neither history nor radiographic findings is required for establishing the diagnosis. Differentiating indolent IPF from asbestosis can be difficult, and the presence of nonfibrotic pulmonary manifestation of asbestos exposure strongly argues for asbestosis, whereas indolent disease interrupted by rapid stair step decline usually occurs only in IPF. Surgical lung biopsy with asbestos body determination can establish a definitive diagnosis, but this is infrequently performed owing to the age and debility of these patients. No medical therapy has been shown to improve or decrease progression of asbestosis. Severe impairment typically occurs 30 to 40 years after exposure, making almost all patients ineligible for lung transplantation because of age. Management of asbestosis is supportive.
It is important to recognize the association of silicosis with lung cancer and active tuberculosis.17 Patients with silicosis are at increased risk of lung cancer, and the risk is increased when combined with exposure to tobacco smoke, diesel exhaust, or radon gas. Patients with silicosis develop active tuberculosis 2-fold to 30-fold more frequently than coworkers without silicosis. This association is especially important in societies with a high incidence of HIV infection, which markedly increases the risk of silicosis-associated active tuberculosis.
Systemic Disease–Associated Interstitial Lung Disease
Connective Tissue Disease–Associated Interstitial Lung Disease
ILD is a well-known complication of various connective tissue diseases.18 The most commonly implicated disorders are scleroderma, rheumatoid arthritis, Sjögren syndrome, polymyositis/dermatomyositis, and systemic lupus erythematosus.
High-resolution CT findings are variable and range from normal lung architecture to ground-glass abnormalities to reticular and fibrotic changes.19 The pathologic pattern of injury with these diseases is as equally diverse and correlates with the high-resolution CT findings. Inflammatory injury patterns are most commonly seen, such as nonspecific interstitial pneumonitis (NSIP) and organizing pneumonia (OP). The NSIP inflammatory injury pattern appears as ground-glass abnormalities on high-resolution CT scan, whereas OP is shown by patchy consolidated lung with air bronchograms. Both of these pathologic patterns can improve with aggressive immunosuppression. At the other end of the pathologic response spectrum is fibrotic injury, which manifests as UIP, which shows reticular fibrotic opacities and honeycomb cystic changes on high-resolution CT scan and typically does not improve with immunosuppression, although long-term controlled studies are lacking.
Specific treatment of connective tissue disease–associated ILD must be individualized. Patients with evidence of extrapulmonary inflammation, an inflammatory pathologic pattern such as NSIP or OP on high-resolution CT or biopsy, or rapidly progressive symptoms are usually treated with prolonged immunosuppressive agents such as cyclophosphamide, azathioprine, mycophenolate, or tacrolimus.20,21
More recent studies have begun to provide evidence-based therapy for these diverse patients. The Scleroderma Lung Study showed that 1 year of oral cyclophosphamide modestly improved lung function compared with modest decline in the control group.20 The patients with the highest degree of fibrosis on high-resolution CT improved most, and ground-glass abnormalities or an inflammatory pattern on bronchoalveolar lavage were not predictive of benefit. After 1 year off immunosuppressive therapy, the patients treated with cyclophosphamide worsened and were indistinguishable from the untreated patients in the control group.22 Many clinicians hypothesize that to preserve any lung function gained by cyclophosphamide, continued immunosuppression may be necessary, and mycophenolate is most often used.
Sarcoidosis
Sarcoidosis is an idiopathic multisystem inflammatory disorder that commonly involves the lung.23 It is the most common ILD in the United States. The tissue inflammation that occurs in sarcoidosis has a characteristic pattern in which the inflammatory cells collect in microscopic nodules called granulomas. In contrast to IPF, sarcoidosis is more common among young adults than among older adults. Sarcoidosis often follows a benign course of inflammation without symptoms or long-term consequences that spontaneously remits.
The most common manifestation of sarcoidosis is asymptomatic hilar adenopathy. Less frequently, the chest radiograph shows parenchymal opacities in the midlung zone that may be nodular, reticulonodular, or alveolar. When symptoms occur, cough, chest pain, dyspnea, and wheezing are most common. Pulmonary physiology may be normal, restrictive, obstructive, or mixed, all with reduced DLCO. Obstructive impairment may be related to endobronchial granulomatous inflammation or scarring.24
Corticosteroids are commonly used in the management of sarcoidosis, but treatment usually is reserved for patients with marked symptoms or physiologic impairment attributable to the disease.25 Although corticosteroids almost always reduce active sarcoid inflammation, long-term side effects should limit duration. For patients requiring long-term immunosuppression, alternative immunosuppressive agents such as methotrexate, azathioprine, leflunomide, or tumor necrosis factor-alpha inhibitors such as infliximab should be used.26 Other organs that may require corticosteroid therapy include cardiac involvement, uveitis, and peripheral or central nervous system involvement with cranial nerve abnormalities. Disease activity is difficult to ascertain in many patients. Serum angiotensin-converting enzyme levels and gallium scans are not well correlated with disease activity, and their routine use is discouraged.27
Lymphangioleiomyomatosis
Lymphangioleiomyomatosis (LAM) is a rare disorder of abnormal smooth muscle tissue proliferating around small airways leading to severe obstruction and destruction of alveoli with resultant thin-walled cyst formation.28 All patients are women, although both men and women with tuberous sclerosis complex can develop lung pathology identical to LAM that is called tuberous sclerosis complex LAM. This peculiar pathology is caused by abnormalities in the TSC-2 gene.29
Interstitial Lung Disease of Unknown Cause
Idiopathic Interstitial Pneumonias
Idiopathic Pulmonary Fibrosis
Idiopathic pulmonary fibrosis (IPF) is the most common IIP and is defined as a progressive fibrotic lung disease isolated to the lung.30 Most patients are older than 60 years, and IPF is extremely unusual in persons younger than 40. Risk factors for development of IPF include exposure to smoke, metal dust, farming dust, and hairdressing chemicals. Patients present with chronic cough and exertional dyspnea, and high-resolution CT suggests a fibrotic process.
The diagnosis is made by noting a lack of exposure or systemic disease known to cause ILD and determining UIP as the pathologic pattern of injury. The diagnosis of UIP is made when high-resolution CT shows bilateral and basilar-predominant peripheral reticular fibrosis and honeycomb cystic change with absence of significant ground-glass abnormalities, micronodules, and air trapping. Without these classic findings, a surgical lung biopsy is needed for diagnosis.31,32 Patients who do not have IPF can have UIP on surgical lung biopsy (e.g., connective tissue disease), so this pattern of injury and repair is not unique to IPF.
Most patients die as a result of progressive fibrotic lung disease within 4 years of diagnosis. Data show that approximately half of patients die with gradually progressive disease over several years.33 The other half experience stable lung function or minimal decline for months to years and then have sudden worsening over a few weeks or months leading to death.34 Baseline parameters that predict an increased risk of death include severity of dyspnea, severity of restrictive physiologic defect, reduced DLCO, pulmonary arterial hypertension, degree of fibrosis on high-resolution CT, and SaO2 desaturation on exertion.35 Serial parameters that predict poor survival include worsening dyspnea, FVC, and DLCO.
No medical therapy has proven beneficial or is recommended for IPF. Prior trials have shown no benefit with aggressive immunosuppression,36–38 interferon gamma,39 etanercept,40 bosentan, ambrisentan, sildenafil,41 or imatinib.42 Several medications are currently under investigation, including pirfenidone, azathioprine in combination with oral corticosteroids and N-acetyl cysteine, and warfarin.
More recent studies have highlighted the importance of pulmonary arterial hypertension in IPF.43 The degree of pulmonary arterial hypertension does not always correlate with the burden of fibrosis on CT scan or FVC, implying that a vascular process other than obliteration of the capillary bed from fibrosis occurs.44 Significant pulmonary arterial hypertension is suggested in patients with markedly impaired diffusion capacity but relatively preserved FVC. At the present time, medications that benefit pulmonary arterial hypertension such as bosentan and sildenafil have not proven beneficial for IPF. Treatment with other agent used for pulmonary arterial hypertension are under investigation in IPF, but their use outside of trials is not recommended.
Nonspecific Interstitial Pneumonia
NSIP is an IIP with diffuse inflammation seen on surgical lung biopsy.45 Patients are on average 7 to 10 years younger than patients with IPF, but considerable overlap exists. The degree of accompanying interstitial fibrosis is variable among patients. The combination of fibrosis and inflammation (fibrotic NSIP) is most common, whereas pure cellular NSIP is less common. Patients present with chronic or subacute cough and dyspnea. High-resolution CT shows predominant ground-glass abnormalities in cellular NSIP and both ground-glass abnormalities and fibrotic changes in fibrotic NSIP. Given that there is significant clinical and radiographic overlap between fibrotic NSIP and IPF, surgical lung biopsy is frequently required to distinguish these two entities, such as when elements of classic UIP are not present on high-resolution CT images.
The prognosis is much better for NSIP than IPF with most patients surviving 7 to 10 years. Immunosuppression with oral corticosteroids and cytotoxic immunosuppressive agents is the primary therapy. Type and duration of therapy are guided by disease activity and degree of inflammation on biopsy and ground-glass abnormalities on high-resolution CT. Pathologic NSIP is found frequently as an IIP and is the most common pattern of injury seen in connective tissue disease–associated ILD. Owing to this frequent association, many authors consider NSIP a connective tissue disease isolated to the lung.46,47
Lymphocytic Interstitial Pneumonia
Lymphocytic interstitial pneumonia is a rare disorder of polyclonal lymphocyte aggregates that accumulate diffusely in the interstitium.48 The diagnosis almost always requires surgical lung biopsy. Patients are typically younger than patients with IPF and present with subacute dyspnea and cough. Pulmonary function testing may show a mixed picture, and high-resolution CT typically shows diffuse ground-glass attenuation with variable amounts of fibrosis. Most patients respond well to oral corticosteroids with a few requiring long-term immunosuppression. Lymphocytic interstitial pneumonia is frequently associated with connective tissue diseases, especially Sjögren syndrome, and with immunodeficiency, and these possibilities should be investigated in all patients with lymphocytic interstitial pneumonia.
 Problem
ProblemNonspecific Therapies for Interstitial Lung Disease
Oxygen Therapy
We favor continuous rather than pulse delivery because the desaturation with activity seen in most patients that is not rectified with pulse therapy, and pulse units vary greatly in the amount of O2 delivered.49 For most patients, liquid O2 is the best source to provide adequate flow rates. In motivated patients, transtracheal delivery of supplemental O2 increases the efficiency of delivery and improves cosmetics. However, patients must be chosen carefully because of the need for frequent care and risk of mucus dessication and rare hemorrhage.
 Problem
ProblemPulmonary Rehabilitation and Exercise Therapy
Pulmonary rehabilitation, a mainstay of treatment for obstructive lung disease, has also proven beneficial in the management of ILD. Pulmonary rehabilitation is important in building aerobic fitness, maintaining physical activity, and improving quality of life. When pulmonary rehabilitation is stopped, the benefits wane over a few months.50 We encourage all of our patients to enroll in outpatient pulmonary rehabilitation and to continue maintenance therapy.
Transplantation
The only therapy shown to prolong life in patients with end-stage, particularly fibrotic, ILD is lung transplantation.51 Transplantation has been performed successfully in the management of most ILDs. Enthusiasm for the procedure is tempered by the significant risk of mortality at 1 year (10% to 25%) and 5 years (50% to 60%). Many patients with ILD are older than the upper age limit of “physiologic” age 65. Additionally, comorbidities such as gastroesophageal reflux disease, which is common in many ILDs, preclude lung transplantation owing to the increased risk of chronic rejection and death.
[/level-membership-for-pulmolory-and-respiratory-category][not-level-membership-for-pulmolory-and-respiratory-category]
Interstitial Lung Disease
After reading this chapter you will be able to:
Organize and distinguish between the entities grouped as interstitial lung diseases (ILDs).
Interpret symptoms, examination signs, and pulmonary function testing in ILD.
List emerging pathophysiologic characteristics that are associated with selected ILDs.
Describe how to manage ILD in general and how some specific ILDs can be treated.
Interstitial lung disease (ILD) comprises a broad category of lung diseases rather than a specific disease entity.1,2 This category includes various illnesses affecting the lung parenchyma with many different causes, treatments, and prognoses. These disorders are grouped together because of similarities in their clinical presentations, appearance on plain chest radiography, and physiologic features.
With over 100 separate disorders, it is essential to group ILDs based on etiology, disease associations, or pathology. An organizational scheme is presented in Figure 24-1. When evaluating patients with an ILD, one first must consider diseases with known causes or associations, such as diseases related to specific exposures, diseases associated with systemic conditions, and diseases with a known genetic basis. Most patients do not have an ILD with a known cause, and their disorder is classified by pathologic pattern. These groups are divided into specific disease entities. Using this organizational scheme to guide a careful and complete history, one is able to understand the disease processes and work efficiently toward an accurate diagnosis.
As the name ILD implies, the histologic abnormalities that characterize ILD involve the pulmonary interstitium to a greater extent than the alveolar spaces or airways, although exceptions exist. Figure 24-2 illustrates the components of the normal pulmonary parenchyma. The interstitium is the area between the capillaries and the alveolar space. As Figure 24-2 shows, in the normal state, this space allows close apposition of gas and capillaries with minimal connective tissue matrix, fibroblasts, and inflammatory cells such as macrophages. The interstitium supports the delicate relationship between the alveoli and capillaries allowing for efficient gas exchange. When responding to any injury—whether from a specific exposure (e.g., asbestos, nitrofurantoin, or moldy hay), an autoimmune-mediated inflammation from a systemic connective tissue disease (e.g., rheumatoid arthritis), or unknown injury (e.g., idiopathic pulmonary fibrosis [IPF])—the lung must respond to the damage and repair itself. If the exposure or injury persists or if the injury repair process is imperfect, the lung may be permanently damaged with increased interstitial tissue replacing the normal capillaries, alveoli, and healthy interstitium.
Characteristics of Interstitial Lung Disease
Clinical Signs and Symptoms of Interstitial Lung Disease
Many ILDs have similar clinical features and are not easily distinguished based on history or examination. Symptoms are generally limited to the respiratory tract. However, extrapulmonary symptoms should not be ignored because they may point to an ILD associated with a systemic diagnosis. Exertional breathlessness (dyspnea) and a nonproductive cough are the most common reasons patients seek medical attention. Other respiratory symptoms, such as sputum production, hemoptysis, pneumothorax, or wheezing, can occur and be suggestive of specific diseases (Table 24-1). If the patient also has prominent extrapulmonary symptoms, such as myalgia, arthralgia, sclerodactyly, gastroesophageal reflux, or Raynaud phenomenon, ILD resulting from underlying connective tissue disease may be present (Table 24-2).
TABLE 24-1
Unusual Pulmonary Findings and Likely Diagnosis
| Pulmonary Findings | Disease | Frequency |
| Hemoptysis | LAM | Rare |
| Chyloptysis | LAM | Rare |
| Pneumothorax | LAM, BHD | Common |
| Wheeze | Sarcoidosis, LAM | Common |
| Chylous pleural effusion | LAM | Common |
| Exudative pleural effusion | RA | Common |
TABLE 24-2
Extrapulmonary Findings and Likely Diagnosis
| Extrapulmonary Findings | Disease | Frequency |
| Raynaud phenomenon | All CTD | Common |
| Arthralgia | All CTD | Common |
| Myalgia | Polymyositis | Common |
| Large muscle weakness | Polymyositis | Common |
| Sclerodactyly | Scleroderma | Common |
| Rheumatoid skin nodules | RA | Rare |
| Fingertip fissures | Antisynthetase syndrome | Common |
| Dorsal hand rash | Dermatomyositis | Common |
| Facial rash | Dermatomyositis | Common |
| Exudative pleural effusion | RA | Common |
| Shawl distribution skin nodules | TSC | Common |
| “Pencil eraser” facial skin nodules | BHD | Common |
| Central nervous system benign cortical tuber | TSC | Common |
| Abdominal angiomyolipoma | LAM | Common |
| Renal cancer | BHD | Common |
| Cardiomyopathy | Sarcoidosis, polymyositis | Rare |
| Cardiac conduction block | Sarcoidosis | Rare |
| Violaceous facial skin nodules | Sarcoidosis | Common |
| Subcutaneous nodules | Sarcoidosis | Common |
| Cranial neuropathy | Sarcoidosis | Rare |
| Small fiber neuropathy | Sarcoidosis | Rare |
Radiographic Features
ILDs manifest as abnormal lung parenchyma that cast abnormal radiographic shadows. For most ILDs, the chest radiograph reveals reduced lung volumes with bilateral reticular or reticulonodular opacities. However, the chest radiograph has limited value because the three-dimensional abnormalities are summed into a two-dimensional image with loss of spatial information. High-resolution cross-sectional imaging via computed tomography (CT) provides detailed images representing pulmonary pathology.3 High-resolution CT images allow noninvasive evaluation of ILDs and are a key element in making a confident diagnosis and managing ILD.4
Plain chest radiographs and high-resolution CT images of usual interstitial pneumonitis (UIP) show the prototypic fibrotic injury pattern. The chest radiograph (Figure 24-3, A) and high-resolution CT image (Figure 24-3, B) in UIP typically reveal a bilateral, patchy, peripheral (subpleural), and basilar predominant disease with reticulonodular infiltrates, often with honeycomb, cystic change. The lung architecture is distorted in patients with moderate or severe disease burden, with reduced lung volume and traction bronchiectasis, especially at the lung bases. Reticulogranular (ground-glass) abnormalities, increased attenuation of the lung tissue without distortion of the underlying blood vessels or bronchi, are absent or minimal in IPF. Pleural disease, air trapping, micronodules, and significant lymphadenopathy are not seen, although two-thirds of patients with IPF can have mild mediastinal adenopathy.5
Physiologic Features
Similar to the radiographic findings, there can be considerable variability among the specific diseases in the physiologic abnormalities seen. However, a restrictive physiologic impairment is the common finding.6 Both forced expiratory volume in 1 second (FEV1) and forced vital capacity (FVC) are diminished, and the FEV1/FVC ratio is preserved or even supranormal. Lung volumes are reduced, as is the diffusing capacity of the lung for carbon monoxide (DLCO). This reduction in diffusing capacity reflects a pathologic disturbance of the alveolus-capillary interface.
Although not commonly pursued, the compliance characteristics of the lungs can be evaluated with an esophageal balloon to measure intrathoracic pressure at various lung volumes. In almost all ILDs, the lungs have reduced compliance and require supranormal transpleural pressures to ventilate (Figure 24-4). This lack of compliance results in small lung volumes and increased work of breathing.
Less frequently, a pattern of physiologic obstruction may be seen. This obstruction can be the result of the primary disease process (e.g., LAM, PLCH, or sarcoidosis in some patients) or concomitant emphysema or asthma.7 If ILD develops in a patient with significant emphysema, the opposing physiologic effects of the two diseases may result in deceptively normal spirometry and lung volume measurements and apparently normally compliant lungs. However, because both emphysema and ILD result in impaired gas exchange, DLCO is significantly decreased.
Selected Specific Types of Interstitial Lung Disease and Therapies
Exposure-Related Interstitial Lung Disease
Tobacco-Associated Interstitial Lung Disease
Although the association of first-hand tobacco smoke and obstructive lung disease is common and well known, tobacco smoke is also an avoidable cause of ILD. Although the association is rarer than with obstructive lung disease, first-hand tobacco smoke inhalation can lead to three types of ILD in susceptible individuals: RB-ILD, DIP, and PLCH. The first two disorders are related. Each disease consists of increased numbers of polyclonal activated macrophages. The diseases differ by the location of these overly abundant cells. In RB-ILD, macrophages accumulate in the respiratory bronchioles leading to bronchiolar remodeling and fibrosis of adjacent alveoli. As expected for a disease with combined airway and alveolar injury, pulmonary function testing reveals mixed restriction and obstruction with frequent air trapping. High-resolution CT images show this mixed pathologic location with indistinct centrilobular nodules (Figure 24-5). In DIP, the increased macrophages fill the alveoli, manifesting as restrictive impairment on pulmonary function testing and diffuse ground-glass attenuation on high-resolution CT imaging (Figure 24-6).
Pulmonary Langerhans cell histiocytosis (PLCH) is the third interstitial manifestation of tobacco smoke. Increased numbers of polyclonal macrophages play a prominent role. However, in PLCH, they are accompanied by fibroblasts and eosinophils in nodules concentrated around small airways. These nodules are stellate and destroy adjacent lung tissue, leading to the classic high-resolution CT image of stellate nodules associated with cysts as seen in Figure 24-7. Although adult smoking-associated PLCH is pathologically similar to childhood Langerhans cell histiocytosis, the adult form does not involve bone and has not proven to respond to chemotherapy as the childhood form does. The relationship of these two disorders has yet to be defined.
In each of these three diseases, the primary treatment is complete tobacco abstinence. With abstinence, most patients either minimally improve or remain stable,8 but a few progressively worsen and may require lung transplantation. Active treatment with prednisone or other immunosuppressive medications is discouraged because few, if any, patients improve.9
Drug-Related and Radiation-Related Interstitial Lung Disease
Many drugs have been associated with pulmonary complications of various types, including interstitial inflammation and fibrosis, bronchospasm, pulmonary edema, and pleural effusions. Drugs from many different therapeutic classes can cause ILD, most commonly chemotherapeutic agents, antibiotics, antiarrhythmic drugs, and immunosuppressive agents (Box 24-1). There are no distinct physiologic, radiographic, or pathologic patterns of drug-induced ILD, and the diagnosis is usually made when a patient is exposed to a medication known to result in lung disease, the timing of the exposure is appropriate for the development of the disease, and other causes of ILD have been eliminated. Treatment is avoidance of further exposure and systemic corticosteroids in markedly impaired or declining patients. In addition to future drug exposure, bleomycin injury is accentuated by increased fractional inspired oxygen (FiO2), even months after last drug exposure, and supplemental oxygen (O2) should be used only if absolutely necessary in these patients.10
