Insulin-Like Growth Factor-1 and Its Binding Proteins
IGF-1 Gene and Protein Structures
Receptor-Mediated Signal Transduction
IGF-2 Mannose-6 Phosphate Receptor
Specific Properties of Each Form of IGFBP
Control of IGF-1 Concentrations in Serum
Control of IGFBP Concentrations in Blood and Extracellular Fluids
Effects of IGF-1 on the Proliferation of Different Types of Cells
Effects on Cellular Differentiation
Control of IGF-1 Actions in Cells and Tissues by IGFBPs
Modulation of in-vivo Actions by IGFBPs
Transgenic Animal and Gene-Targeting Studies
IGF-1 was originally discovered because of its ability to stimulate sulfation of cartilage proteoglycans.1 The administration of GH to hypophysectomized animals resulted in induction of a substance in serum that was a potent stimulant of cartilage sulfation. In contrast, when GH was added to cartilage in vitro, it had minimal bioactivity. This suggested that a separate growth factor was induced in the serum of these animals. Purification of this substance showed that its amino acid sequence was similar to insulin and led to studies which showed that it could stimulate growth in vivo.2
IGF-1 Gene and Protein Structures
The insulin-like growth factor-1 gene is a complex, multi-component gene with 6 exons. The gene structure is shown in Fig. 17-1. The first and second exons encode the 5′ untranslated and pre-propeptide regions of IGF-1. Exon 3 encodes the distal propeptide sequence and the regions of the mature peptide that are homologous to the B chain of insulin, the region homologous to the C peptide and to the A chain region. Exon 4 encodes a D extension peptide. The fifth and sixth exons are shuffled and can encode one of two sequences termed IGF-1A and IGF-1B. This alternative splicing occurs in multiple tissues, and both IGF-1A and IGF-1B have been found to be secreted by specific cell types in culture.
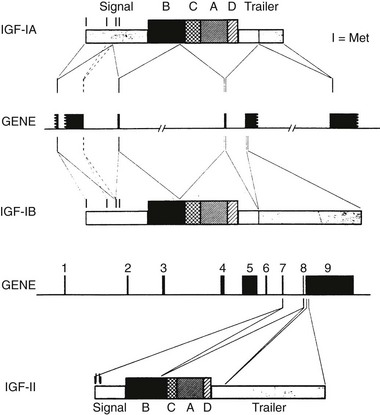
FIGURE 17-1 Structure of the human insulin-like growth factor 1 (IGF1) gene and the precursor proteins it encodes. The black boxes that are shown represent exons. The portions of each exon that encode parts of the precursor protein are shown by lines. The IGF-1A and IGF-1B precursor forms are represented by boxes. The B, C, A, and D domains of the mature peptides are noted.
Several forms of IGF-1 mRNA are transcribed, and at least four specific transcripts have been detected in tissues.3 The most abundant IGF-1 transcript (6 kb) contains multiple polyadenylation sites and a long 3′ untranslated sequence. The abundance of this transcript is regulated by GH. GH increases transcription of IGF-1 by inducing STAT5b which binds to an intronic region between exons 2 and 3 and initiates transcription. Several different fetal and tissue-specific promoters of IGF-1 have been identified, and they account for distinct transcript patterns in various tissues and the appearance of various forms at specific periods in development.4 Other abundant transcripts include a 3.2 kb transcript, a 2.7 kb transcript, and a 0.9 kb transcript. Stimuli other than GH have been shown to influence the abundance of these transcripts in various tissues.5 The small, 0.9-kb transcript is one source of the mature 70 amino acid IGF-1 peptide. This transcript is present in the liver and is an important source of the peptide that is present in the systemic circulation. Alternative processing of IGF-1 mRNA following its transcription has been shown to occur in multiple tissues and may be physiologically relevant in specific situations, such as muscle repair after injury.6 Variable polyadenylation sites and regulation of processing of the 3′ untranslated RNA extensions have been demonstrated and can result in different-length transcripts.7
The polypeptide structures of three members of the IGF gene family are shown in Fig. 17-2. Mature IGF-1 and IGF-2 contain 70 and 67 amino acids, respectively. Proinsulin has a longer C peptide region compared to IGF-1 or IGF-2. The sequence in this region is not conserved. The A chain and B chain peptide regions are of similar length. The sequences in this region are 41% and 43% homologous with proinsulin. IGF-1 and 2 contain D-domain extensions of 8 and 6 amino acids, respectively. Unlike proinsulin, IGF-1 and 2 are not cleaved into two-chain polypeptides during intracellular processing, but rather they are secreted as intact single-chain proteins. Forms of IGF-1 have been isolated from serum and from cell culture supernatants that contain the E peptide extensions (e.g., both A and B), but the relative abundance of these forms in most tissues is unknown. The frequency of processing of the E peptide domains is unclear, since longer forms of IGF-1A or IGF-1B have been shown to be secreted by cells in culture. However, some cells do not secrete IGF-1 with the E peptide extension.
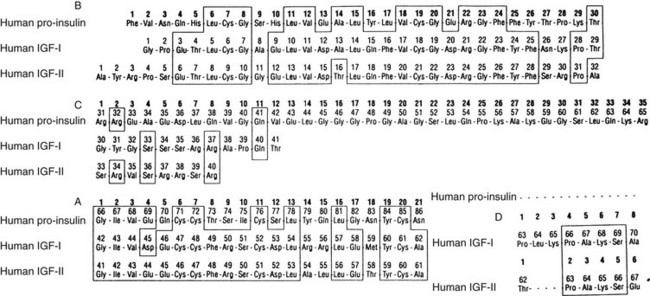
FIGURE 17-2 The sequences of proinsulin, insulin-like growth factor 1 (IGF-1), and IGF-2. The sequences are divided into the B, C, A, and D domains.
Specific amino acids within the IGF-1 molecule have been shown by site-directed mutagenesis to account for receptor and/or binding protein association (Table 17-1). Specifically, tyrosine 24, tyrosine 60, and to some extent tyrosine 31 are required for IGF-1 receptor recognition.8 The tyrosines at positions 24 and 60 are conserved within IGF-2, but tyrosine 31 is not present. The residue within the proinsulin sequence that is homologous to Tyr24 (e.g., Phe25) is important for insulin binding to its receptor. Tyrosine 60 appears to be necessary for IGF-1 to maintain a stable conformation. Studies using mutant forms of IGF-1 with large deletions indicate that the region between residues 24 and 37 contains the primary receptor binding site.9 Mutations in this region have very little effect on binding protein affinity. More recent crystallographic10 and NMR studies11 have confirmed the importance of these residues for receptor binding. These studies highlighted the importance of Phe16 and Leu54 for ligand-induced activation and suggest they are required by full activation of the receptor kinase. These studies have also shown the importance of specific residues in the C domain, particularly Arg35 and Arg36.12 Alanine scanning mutagenesis has confirmed the importance of Phe23 and 25, Tyr31, Arg36, Arg 37, Val44, and Tyr60, the residues that compose the binding site. These studies also identified a secondary site composed of Glu9, Asp12, Phe16, Leu54, and Glu58. Substitution for these residues resulted in a 33- to 100-fold reduction in receptor affinity.13
Table 17-1
Specific Amino Acids in Insulin-Like Growth Factor 1 (IGF-1) That Mediate Binding Protein and Receptor Association
| Region of IGF-1 | Ligand Interaction |
| B chain Glu3, Thr4, Gln15, Phe16 |
Required for binding to IGF-binding proteins (IGFBPs) 1-6 |
| A chain Phe 49, Arg50, Ser51 |
Required for optimal binding to IGFBP-1, 2, 4, 5 |
| Tyr24, Tyr31, Tyr60, | IGF-1 receptor |
| Tyr24-Arg37 | Contains the primary receptor binding site |
| Tyr60 | Necessary for a stable conformation |
IGF-1 and IGF-2 contain 4 amino acids that are the primary determinants of their high affinity for IGF-1 binding proteins. These include the amino acids at positions 3, 4, 15, and 16 of the B chain region of IGF-1 and the homologous residues 6, 7, 18, and 19 in IGF-2.14 These residues are critical for recognition by all six forms of IGF-binding proteins (IGFBPs). Mutant forms of IGF-1 that contain substitutions of proinsulin residues in these four positions have a nearly total loss of binding protein activity. In addition, residues at amino acids 49, 50, and 51 in the A chain are important for recognition by four of the six high-affinity binding proteins. The major exception is IGFBP-3, wherein only the B chain residues appear to be important. Recent x-ray crystallographic studies of the IGF-1/IGFBP-4 complex have confirmed the importance of these residues. They confirmed that they are the primary sites within IGF-1 that interact with IGFBP-4 and explained how the interaction between IGF-1 and IGFBP-4 interferes with receptor binding.15 Studies of the tertiary structures of IGF-1 and IGF-2 have shown that these residues are surface exposed. A recent NMR study showed that two of three helices that are present in IGF-1 form a surface-exposed hydrophobic patch that contains these A- and B-chain residues. A peptide that bound to this patch inhibited IGF-1/IGFBP binding. The C-peptide regions in each of the three proteins are divergent, and this accounts for most of the heterogeneity of sequence between IGF-1 and IGF-2. The three disulfide linkages are conserved in all three peptides.
The structure of IGF-1 is highly conserved across species. Bovine IGF-1 is identical to human, and rat differs by only three amino acids. IGF-1-like molecules have been detected in all vertebrates that have been analyzed, and even species as low on the phylogenetic tree as Caenorhabditis elegans contain IGF-1-like molecules. Computer modeling studies have indicated that the three-dimensional structure of IGF-1 is probably similar to that of insulin, which (except for the C-peptide) has been analyzed by x-ray crystallography.10 Except for residues contained in the C-peptide, many of the IGF-1 residues that bind IGF-1 receptor are also present in insulin. The high affinity of IGF-1 for the IGF-1 receptor as compared to insulin is explained by the presence of the C-peptide.12,13,14 In contrast, the higher affinity of the insulin receptor for insulin is accounted for by residues 4, 15, 49, and 51. If these insulin residues are substituted for those of IGF-1, this IGF-1 mutant has an affinity for the insulin receptor that is equal to insulin.16 Different forms of IGF-1 have been found to be present in human serum and tissues. The most extensively studied form is des-1-3 IGF-1, which occurs in brain and in serum. This IGF variant has much lower affinity for IGFBPs and therefore is more biologically active.
IGF-1 Receptor
The IGF-1 receptor is ubiquitously present and has been shown to be present in cell types derived from all three embryonic lineages. When animal tissues are analyzed, the receptor is detected uniformly, thus accounting for IGF-1’s ability to stimulate growth of all tissues. The receptor number per cell is tightly controlled and maintained in a narrow range of 20,000 to 35,000. This may be an important regulatory function, since cellular transformation in response to IGF-1 usually requires >1,000,000 receptors per cell,17 whereas cells that have <100,000 receptors per cell rarely induce tumors in experimental animal models. Thus the variables that regulate IGF-1 receptor number may be important in terms of the genesis of neoplasia.
Hormones such as GH, FSH, LH, progesterone, estradiol, and thyroxine have been shown to increase IGF-1 receptor expression.18 Similarly, PDGF, EGF, FGF, and angiotensin II up-regulate receptor expression in specific cell types.19 Following hormone binding, there is down-regulation of receptor number with internalization of receptors. However, possibly due to IGF binding proteins, the rate of internalization of IGF-1 receptors is substantially slower than that of other growth factors such as epidermal growth factor or insulin.
The biochemical structure of the receptor is similar to other polypeptide growth factor receptors (Fig. 17-3). The receptor is a heterotetrameric glycoprotein composed of two ligand binding subunits, termed alpha subunits, that contain 706 amino acids and two beta subunits that contain 627 amino acids. Only the beta subunits have a transmembrane domain (see Fig. 17-3). In man, the protein is translated from a single mRNA transcript derived from a gene that contains 21 exons located on chromosome 15, Q25-Q26.20 The prepropeptide is 1367 amino acids, and the signal peptide is removed cotranslationally. The precursor is cleaved between Lys708/Arg709 to generate the alpha and beta subunits. These are linked together by disulfide bonds to form the heterotetrameric receptor. Amino acid sequence comparison with the insulin receptor reveals 46% amino acid identity.20
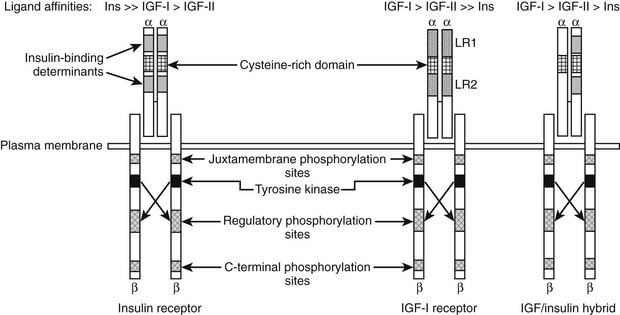
FIGURE 17-3 Structural characteristics of the insulin, insulin-like growth factor 1 (IGF-1), and hybrid receptors.
The alpha subunit contains three domains that are essential for ligand binding. These have been termed leucine rich (LR), cysteine rich (CR), and carboxy terminal (CT) domains. The receptor binds IGF-1 with a mean equilibrium dissociation constant (KD) of 10−9 M. IGF-2 binds with sixfold lower affinity, and insulin with a 200- to 300-fold lower affinity.21 The composition of crystal structure of the first three domains of the alpha subunit of both the IGF-1 and insulin receptors shows that there are two important differences, one in LR1 and one in the CR1, that account for these differences in ligand binding.22 Mutagenesis studies have shown that the residues Asp8, Tyr28, His30, Leu33, Phe58, Tyr79, and Phe90 within the LR1 domain are important for binding.23,24 The CR region contains four residues that are essential to maintaining high affinity. A short C-terminal region (692-702) is also very important, since changes in 7 of these 10 residues reduced binding affinity 10- to 30-fold.24,25 Following site one (LR-CR-CT) contact, the ligand becomes immobilized then cross-links through its second binding domain to a distinct site on the second monomer, thus resulting in high-affinity binding.
The beta subunit of the receptor is composed of an insert domain followed by two fibronectin repeat domains, then a transmembrane domain between positions 906 and 929 that is followed by an intracytoplasmic domain. This region contains intrinsic tyrosine kinase activity and critical sites of tyrosine and serine phosphorylation. The tyrosine kinase (TK) domain is 84% homologous with the insulin-receptor TK domain. The catalytic domain contains an ATP binding motif and a catalytic lysine at position 1003. Substitution for this lysine abolishes IGF-1-stimulated biological actions. Ligand binding to the alpha subunit triggers a conformational change that leads to autoactivation. This in turn leads to trans subunit autophosphorylation wherein a specific tyrosine 1135 on one beta subunit is transphosphorylated by the TK activity located on the paired beta subunit. This nonphosphorylated tyrosine is autoinhibitory, and its phosphorylation leads to kinase activation and transphosphorylation of the paired tyrosine 1135 on the corresponding subunit, followed by sustained TK activation.26
There are at least six important tyrosines contained within the cytoplasmic domain that are phosphorylated by the intrinsic tyrosine kinase. The most important is a triple tyrosine motif at positions 1131, 1135, and 1136. Substitutions for these tyrosines abolish IGF-1 signaling.24,25 Crystal structure analysis has shown that phosphorylation of all three tyrosines is required to obtain the optimal conformation.27 Following activation of the intrinsic tyrosine kinase activity, the enzyme autophosphorylates tyrosine 950 in the beta subunit, which forms a binding site for two important intracellular substrates, insulin receptor substrate 1 (IRS-1) and insulin receptor substrate 2 (IRS-2).28 Substitution for this residue attenuates IRS-1 phosphorylation. Following IRS-1 binding to Tyr950, the IGF-1R kinase phosphorylates specific sites on IRS-1 that provide binding sites for adaptor proteins, such as Grb-2, which in turn leads to Ras activation. Other kinases, such as phosphotidylinositol-3 (PI-3) kinase are activated by binding to phosphorylated IRS-1. Mutation of tyrosine 1316 in the beta subunit abrogates the ability of IGF-1R to activate PI-3 kinase. The receptor can also directly phosphorylate other substrates, including Shc, Crk, and Grb-10.23 Phosphorylation of beta subunit residues 1280 and 1283 is necessary for binding to 14-3-3, an additional signaling intermediate, and for mediating IGF-1’s anti-apoptotic activity. The NPXY motif located near the transmembrane domain is required for internalization.
Chimeric receptors that contain heterodimers of the IGF-1 and insulin receptor have been described.29 These dimers are disulfide linked. Receptor hybrids have been detected in several tissues and cell types. It is possible that they exist in all cells in which both IGF-1 and insulin receptors are expressed. The ligand specificity and affinity properties of hybrid receptors are much closer to those of the IGF-1 receptor as compared to the insulin receptor. Hybrid receptor activation has been shown to lead to stimulation of signal transduction in vitro30; however, the biological significance of hybrid receptor activation in tissues in whole animals has not been determined. Following IGF-1 activation of the receptor, it undergoes endocytosis. This is regulated in part by the adaptor protein 2 complex.31 Following its recruitment to endosomes, the receptor is cleaved by a cysteine protease, and ligand is released.32 Ubiquitination also regulates this process, and two E3 ligases, Nedd4 and MDM2, have been shown to play a role. Nedd4 binds to IGF-1R through Grb-10 and MDM2 through beta arrestin; thus these molecules also play a role in IGF-1R degradation.
The IGF-1 receptor has been overexpressed in several types of cells in culture. Receptor overexpression enhances growth in soft agar and tumor formation in nude mice.17 Studies using antisense oligonucleotides to lower IGF-1 receptor number have confirmed its importance for growth and transforming activity of human tumor cells.33 Importantly, deletion of specific tyrosines, such as tyrosines 1280 and 1281, results in a marked diminution in the transforming property of the IGF-1 receptor, although mitogenesis in vitro is still preserved.34 Additionally, the receptor is important for IGF-1’s ability to modulate the effects of other growth factors. Mouse fibroblasts containing deficient numbers of IGF-1 receptors do not undergo DNA synthesis in response to the addition of epidermal growth factor. Similarly, overexpression of the EGF and PDGF receptors does not lead to proliferation of fibroblasts in soft agar in the absence of IGF-1 receptors,35 and reexpression of the IGF-1 receptor allows proliferation to occur. Large T-antigen induction by the cellular transforming virus SV-40 requires expression of the IGF-1 receptor, and wild-type Ras activation has less of an effect on cellular transformation if the IGF-1 receptor is absent.36 Likewise, Src oncogene expression results in transforming activity only in the presence of an IGF-1 receptor.
The IGF-1 receptor has an important role in normal development and normal fetal growth. Animals that have had the IGF-1 receptor deleted by homologous recombination are born 40% of normal size.37 These animals are not viable at birth, due to hypoplasia of diaphragmatic muscle. Defects in the development of the nervous system, skin, and bones have been noted. These developmental abnormalities apparently occur relatively late in gestation. Fibroblasts obtained from these embryos have a markedly attenuated growth response compared to fibroblasts from normal embryos.
The receptor is also important for prevention of apoptosis. IGF-1 and its receptor support the viability of nonproliferating cells in culture, such as neurons. The extent of apoptosis that can be induced in neurons by osmotic hyperglycemia, ischemia, or potassium shock is dependent upon normal IGF-1 receptor expression, suggesting that it is neuroprotective.38 Hematopoietic cells that undergo apoptosis if IL-3 is withdrawn are protected by IGF-1 exposure if IGF-1 receptors are present. Plating tumor cells on a surface that does not allow ligand binding to integrins results in susceptibility to apoptosis, and this susceptibility can be reversed by incubation with IGF-1.39 In contrast to the IGF-1 receptor, overexpression of the insulin receptor is nontransforming. Likewise, overexpression of a chimeric receptor bearing the beta subunit of the insulin receptor is nontransforming, but if the IGF-1 receptor beta subunit is expressed with the insulin receptor alpha subunit, then mitogenic activity of insulin is detected at much lower ligand concentrations, and this receptor construct allows transformation to occur.40
Receptor-Mediated Signal Transduction
Following activation of the intrinsic tyrosine kinase activity and phosphorylation of tyrosine 950, the docking protein IRS-1 binds directly to the receptor (Fig. 17-4). The functionally similar protein IRS-2 has been shown to bind by a similar mechanism.41 Following binding, IRS-1 is tyrosine phosphorylated by the receptor at multiple sites, creating docking motifs that are critical for binding of intracellular proteins that contain Src homology-2 (SH-2) domains. These domains contain approximately 100 amino acids that share sequence similarity to cellular oncogene Src. Six of the tyrosines in IRS-1 occur within YXXM sequences, a recognition motif for some SH-2 domains. IRS1 gene deletion in mice results in a major decrease in body weight, with proportionate reduction in liver, heart, and spleen.42 Activation of signaling pathways that lead to enhanced IRS-1 degradation result in attenuation of IGF-1 signaling.
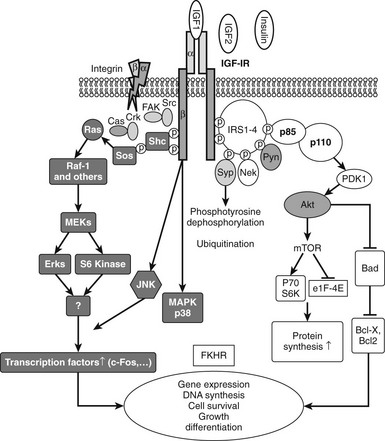
FIGURE 17-4 The two major signaling pathways that are used by the insulin-like growth factor 1 (IGF-1) receptor. These include the MAP kinase (shaded) and PI-3 kinase (open) pathways. P-110 and P-85 represent the major subunits of phosphatidylinositol-3′-kinase.
Signaling proteins that bind directly to the phosphorylated tyrosines on IRS-1 include the adaptor proteins Grb-2 and p85. Grb-2 forms a complex with the Ras-activating protein Son of Sevenless (SOS), and this complex leads to subsequent p21 Ras activation, which activates Raf and downstream components of the MAP kinase pathway.43 Activation of this pathway is important for the mitogenic function of IGF-1.
IRS-1 phosphorylation also results in binding of the p85 regulatory subunit of PI-3 kinase, and this leads to binding of the catalytic subunit p110 and its activation. This results in generation of inositol triphosphate and activation of protein tyrosine kinase B.44 This kinase activates mTOR and P70 S6 kinase, which leads to activation of protein translation. This pathway is also important for IGF-1-induced increases in cell motility and for inhibition of apoptosis. AKT also phosphorylates GSK-3 beta, leading to its inactivation, which is important for several responses that include stimulation of glucose transport.
The IGF-1 receptor can directly phosphorylate Shc, and this leads to association of Grb-2, which activates Ras and MAPK independently of IRS-1. Although Shc can be directly phosphorylated by the IGF-1 receptor, in certain situations such as following glucose-induced oxidative stress, Shc activation proceeds by a different mechanism. In vascular endothelial or smooth muscle cells, hyperglycemic stress leads to increased secretion of ligands for the αVβ3 integrin. Oxidative stress also leads to activation of c-Src. αVβ3 activation results in translocation of activated c-Src to a plasma membrane–associated scaffolding protein, SHPS-1. The IGF-1 receptor phosphorylates SHPS-1, which results in recruitment of activated Src, which recruits Shc and phosphorylates it.45 Since IRS-1 signaling is markedly down-regulated by hyperglycemia, this mechanism allows full MAP kinase activation in response to IGF-1 even in the absence of IRS-1 activation. An additional signaling molecule that is activated by the receptor is Crk, a Grb-2-like protein, with SH-2 and SH-3 domains. Crk then activates Grb-2 and SOS after it is phosphorylated by the IGF-1 receptor.21 Other signaling pathways that have been shown to be activated by IGF-1 include protein kinase-C, phospholipase-C, and direct stimulation of calcium-permeable ion channels. Activation of these proteins leads to activation of downstream signaling cascades, including G-protein activation. Additional signaling molecules that have been shown to interact with the IGF-1 receptor include RACK-146 and Grb-10.47
Since there is specificity between insulin and IGF-1 in terms of their metabolic and growth-promoting actions, it was presumed that major differences would be detected in the signal transduction pathways that each hormone utilized. However, IGF-1 and insulin-receptor kinase domains are 84% identical, and similar residues are autophosphorylated. Presumably, during normal growth or stimulation of glucose transport, either distinct domains are activated in the IRS-1 and IRS-2, or separate combinations of signaling pathways are activated. However, in pathophysiologic states such as hyperglycemia, IGF-1 receptor activation of MAP and PI-3 kinase is enhanced in some cells, whereas insulin-receptor signaling is inhibited. Other differences in signaling have also been reported. Activation of Crk-2 is specific for the IGF-1 receptor.21 Since Crk-2 has transforming activity, its activation may partially account for the ability of overexpression of IGF-1 receptors to be transforming. Insulin and IGF-1 receptors have been shown to utilize different G-protein signaling components.48 Activation of Src kinase results in phosphorylation of the IGF-1 receptor but not the insulin receptor. In summary, multiple signaling events are activated in response to IGF-1 receptor stimulation. The best characterized are those that lead to MAP or PI-3 kinase activation, but other pathways may be important in specific physiologic or pathophysiologic situations.
Blocking specific functions of intracellular signaling pathways has been shown to attenuate specific IGF-1 actions. The PI 3–kinase pathway appears important for glucose transport and for cell migration, and specific inhibitors of PI-3 kinase have been shown to inhibit these IGF-1-stimulated effects.44,49 Similarly, the MAP kinase pathway appears to be the predominant pathway for mitogenesis and rescue from apoptosis.39 Protein kinase C also appears to be essential for IGF-1-stimulated cell migration and stimulation of the transcription of specific genes. However, the requirement of stimulation of a specific pathway for a specific function is not absolute, since the results generated using specific inhibitors of each pathway support the conclusion that there are overlapping functions. In addition to interactions between the IGF-1 and insulin receptor–linked signaling pathways, several other signaling pathways have been shown to influence IGF-1-stimulated signaling events. Several hormones and growth factors such as EGF, angiotensin II, aldosterone, and estrogen have been shown to modulate IGF-1 receptor–linked signaling events.50–52 Conversely, cellular activation by IGF-1 has been shown to result in transactivation of the androgen receptor EGFR, VEGFR, and the chemokine receptor CXR4.53,54 In addition, postreceptor signaling pathway cross-talk has been demonstrated for the GH receptor, estrogen receptor, progesterone receptor, glucocorticoid receptor, and multiple cytokine pathways such as TNFα,55 which induces tissue refractoriness to IGF-1 in states of cachexia.
IGF-2 Mannose-6 Phosphate Receptor
The IGF-2/cation-independent mannose-6 phosphate receptor is a single-chain, membrane-spanning glycoprotein that contains 2451 amino acids. It binds mannose-6 phosphate residues on lysosomal enzymes as well as IGF-2. There is a large extracellular domain, a 23 amino acid transmembrane domain, and a 164 residue carboxy terminal intracytoplasmic domain. The extracellular domain is composed of 15 repeating motifs. Motifs 7 to 9 bind mannose-6 phosphate, and motif 11 contains the IGF-2 binding region.56 Analysis of this region shows that Tyr1542, Glu1544, Phe1567, Thr1520, and Ile1572 come in close contact with IGF-2, and mutagenesis studies have confirmed its importance for binding.57 Intracellularly, this receptor functions to translocate newly synthesized lysosomal enzymes into endosomes. On the cell surface, it binds to mannose-6 phosphate–containing extracellular glycoproteins, which are endocytosed into endosomes. The receptors are then recycled back to the cell surface. Proteins other than lysosomal enzymes shown to bind to this receptor include proliferin, thyroglobulin, and latent transforming growth factor beta (TGF-β). Binding of latent TGF-β has been shown to result in cleavage of the inactive form into active TGF-β. In adipocytes, it has been shown that insulin is a potent stimulant of redistribution of mannose-6 phosphate receptors from intracellular locations to the plasma membrane. The receptor binds IGF-2 with an affinity in the range of KD 1 to 3 nM. The affinity for IGF-1 is 80-fold lower, and the receptor does not bind insulin. Mannose-6 phosphate–containing proteins bind to a site that is distinct from IGF-1 or IGF-2, and the receptor can bind both types of ligands simultaneously. Once IGF-2 is bound, it is internalized and degraded. The extracellular portion of the receptor can be proteolytically cleaved in certain cell types, and the cleavage product is released. This soluble form has been detected in plasma; however, the physiologic significance of its release into plasma has not been determined.
The role of this receptor in IGF physiology is incompletely understood. Deletion of the receptor or mutations that result in loss of IGF-2 binding result in death of fetal mice.58 The receptor is subject to parental imprinting, such that only the maternal allele of the IGF-2 receptor and the paternal allele of IGF-2 are expressed. Therefore, mice that inherit a receptor allele containing a mutation from the mother have functionally altered IGF-2 receptors. These mice develop severe edema in utero prior to death.58 They are also larger than fetuses of comparable developmental age. If IGF-2 is deleted concomitantly, 50% of the fetuses survive birth; however, postnatal survival is poor. The hypothesis has been that these mice lack the putative scavenging function of the IGF-2 receptor and accumulate toxic levels of IGF-2. Although the scavenging function of the receptor is well accepted, it is clear that this receptor does not mediate the actions mediated by the IGF-1 receptor, such as growth stimulation. In most systems, inhibition of the IGF-1 receptor is sufficient to completely block the mitogenic response to IGF-1 or IGF-2 stimulation. Increases in calcium flux have been shown to occur following stimulation of 3T3 cells by IGF-2 binding to this receptor. Additionally, the receptor has been shown to activate GTP binding proteins, but the exact functional significance of these effects is undetermined. The cytoplasmic portion of the receptor encodes regions that are necessary for specific subcellular localization and endocytosis, as well as binding to GTP binding proteins.59 Partitioning of the receptor following internalization can be hormonally regulated. Treatment with insulin was found to cause a rise in the fraction of surface receptors without a change in total number. Mannose-6 phosphate stimulates a similar increase, and this can be blocked by pretreatment with pertussis toxin, implying both stimulatory and inhibitory GTP binding protein regulation.
IGF-Binding Proteins
A characteristic of IGF-1 and IGF-2 that distinguishes them from proinsulin is the ability to bind to high-affinity IGF binding proteins (IGFBPs). The IGFBPs are a family of six proteins that each have high affinity for IGF-1 and IGF-2.60 In each case, this affinity is greater than the affinity of the type 1 IGF receptor for IGF-1. One or more members of this family is present in all extracellular fluids. Therefore, they control the ability of IGF-1 and IGF-2 to bind to receptors. In addition to this property, the major functions of the IGFBPs include: (1) transporting the IGFs in the vasculature, (2) controlling their access to the extravascular space, (3) controlling tissue localization and distribution, and (4) controlling access to receptors and thereby modulating the biological responses of cells to IGF-1.
The gene structure of the IGFBPs shows that each of the six forms contains four exons.61 The mRNA species range in size from 1.4 kb (IGFBP-2) to 6 kb (IGFBP-5). Their protein structures show great similarity. Of the 18 cysteines, all are conserved in 5 of the 6 binding proteins. IGFBP-4 has 2 additional cysteines, and IGFBP-6 has only 16 cysteines. If the cysteine structure is disrupted, IGF-1 binding is markedly attenuated. All are secreted proteins and contain a hydrophobic leader sequence. The affinity of each protein for IGF-1 and IGF-2 is shown in Table 17-2. The greatest difference is in IGFBP-6, which has a 40-fold higher affinity for IGF-2.
Table 17-2
Affinities of Insulin-Like Binding Proteins (IGFBPs) for Insulin-Like Growth Factor 1 (IGF-1) and IGF-2
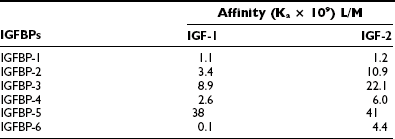
There is a high degree of sequence homology in both the N-terminal and C-terminal domains of each protein.61 Similarly, the sequences in these regions are highly conserved across species. In contrast, the middle third sequence diverges completely. This is important functionally because this is the major site of proteolytic cleavage for IGFBPs. Two of the proteins are N-glycosylated, and glycosylation sites occur in the middle third of the sequence, thereby providing specificity for this property among the different proteins. Recent structural studies have yielded a great deal of information regarding the IGF binding sites and the specific residues that are responsible for IGF binding. Two-dimensional NMR studies of IGFBPs showed that a hydrophobic pocket in the amino terminals (residues 49 to 74) contained six amino acids that form the binding pocket R49, V50, K68, L70, L72, L74.11 Mutagenesis studies confirmed the significance of this region for IGFBP-5 binding and showed that similar residues in IGFBP-3 had a similar function.62 Similarly, mutagenesis of the residues in IGFBP-2 that are comparable to Leu70, 73, 74 in IGFBP-5 results in a major decrease in IGF binding. A specific domain in the C-terminus of these proteins also contributes to IGF binding. The C-terminal binding site contribution to net affinity of the entire protein is greater for IGFBP-1 and 2.63 Recent studies have suggested there is strong cooperativity between these domains which contributes to high-affinity binding of the full-length proteins, and that covalent linkage between the N and C terminus is necessary for maximal affinity.64 The residues in IGF-2 that bind to the C-terminal domain binding site in IGFBP-6 are similar to those that bind the IGF receptor, and this probably accounts for the ability of the IGFBPs to inhibit IGF-1 binding to its receptor.
Specific Properties of Each Form of IGFBP
IGFBP-1 contains 235 amino acids and is not glycosylated. It contains an Arg-Gly-Asp near its carboxy terminus which mediates binding to the α5β1 integrin.65 IGFBP-1 has been detected in multiple types of extracellular fluids. The affinities of IGFBP-1 for IGF-1 and IGF-2 are nearly equal.
IGFBP-2 contains 289 amino acids and is not glycosylated. Its sequence is highly conserved across species, especially in the C-terminus.61 It has an Arg-Gly-Asp sequence near its carboxy terminus, and it has been shown to bind to cell surfaces. Following cleavage, its affinity for IGF-1 and 2 is greatly reduced.
IGFBP-3 contains 266 amino acids and is variably N-glycosylated.81 This accounts for its varying molecular weight estimates between 43 and 56 kD. There are three potential N-linked glycosylation sites. Digestion within N-glycanase reduces the estimated molecular mass to 34 kD. Glycosylation does not alter the affinity of this protein. IGFBP-3 contains a highly basic region between residues 216 and 244 (in which 10 of 18 amino acids are basic). This region accounts for its heparin-binding activity and its ability to adhere to glycosaminoglycans.66
IGFBP-4 contains 237 amino acids. It is N-glycosylated and therefore has a mass estimate of 28 kD in the glycosylated form and 24 kD in the nonglycosylated form. Glycosylation does not affect the affinity for IGF-1 or 2. IGFBP-4 is cleaved in most physiologic fluids to 16- and 14-kD fragments that have a reduced affinity for IGF-1 and 2.67
IGFBP-5 has 252 amino acids and is the most highly conserved form of IGF binding protein, with 97% homology in sequence between the mouse and human forms.68 It is most closely related in sequence to IGFBP-3 (e.g., 50% homology in the amino and carboxy terminal ends). IGFBP-5 contains the same heparin-binding domain as IGFBP-3 between amino acids 201 and 218.69 This sequence mediates its binding to extracellular matrix, and some specific ECM proteins that bind IGFBP-5 have been defined.69 IGFBP-5 is O-glycosylated and has size estimates between 31 and 34 kD. This protein has a high affinity for IGF-1 and IGF-2. It is proteolytically cleaved into a 22 kD fragment in physiologic fluids that has a much lower affinity for these ligands.
IGFBP-6 has 216 amino acids, and the human form has 16 cysteines. The protein is O-glycosylated. It has a high affinity for IGF-2 compared to IGF-1, but the physiologic significance of this difference has not been ascertained.70 IGFBP-6 is proteolytically cleaved in physiologic fluids.
Control of IGF-1 Concentrations in Serum
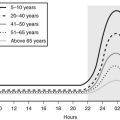
Age is an important determinant of the normal serum IGF-1 concentrations. Plasma concentrations rise from very low levels (20 to 60 ng/mL) at birth to peak values between 212 and 638 ng/mL at puberty.71 The concentrations then fall rapidly in the second decade, reaching a mean value of 284 ng/mL by age 20 and then decline more slowly over each decade (Fig. 17-5). They are reduced to <50% of the 20-year-old value by age 60 years. A portion of this change is due to age-dependent changes in GH secretion. Although the change in GH may account for much of the decline that occurs during adulthood, it does not account for all of the major increase that occurs during childhood.
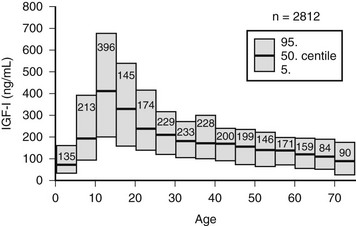
FIGURE 17-5 Serum concentrations of insulin-like growth factor 1 (IGF-1) in healthy subjects, aged birth to 75 years. The means are shown as solid lines, and the 95% confidence intervals are shown as rectangles.
There are important genetic determinants of plasma IGF-1 concentrations. Studies in twins have shown that approximately 40% of each individual’s IGF-1 variability can be accounted for on the basis of undefined genetic factors which are linked to height.72 There is a very close correlation between IGF-1 concentrations and statural height in many different types of populations that have been studied, and these appear to be due, at least in part, to this genetic factor. This genetic determinant is independent of intrinsic GH secretion. Recently a polymorphism in the IGF1 gene that occurs in 12% of Caucasians has been shown to be associated with a lower mean serum IGF-1 concentration (∼30% reduction) and a decreased final adult height (e.g., ∼2 cm). The presence of this polymorphism in individuals >60 years was associated with a twofold increase in the prevalence of type 2 diabetes and an increased incidence of heart attacks and strokes.73
The major hormonal determinant of plasma IGF-1 concentrations is growth hormone. Children with definitive evidence of growth hormone deficiency (GHD) usually have IGF-1 values that are below the 95% confidence interval.74,75 Because values vary so much throughout childhood, however, age-adjusted normative data are required to interpret low plasma IGF-1 values (see Fig. 17-5). Consideration of developmental stage (skeletal age) is also important for interpreting low values.76 In children, normal IGF-1 value is strong evidence that GH deficiency is not present. Conversely, a low IGF-1 is very suggestive of GHD, but it does not definitively prove that GHD is present.77 Other causes of growth retardation can be associated with a low IGF-1, although causes such as constitutional growth delay are usually associated with normal levels. Administration of GH to patients with GHD results in a substantial rise in IGF-1, and this occurs during the 4 to 6 hours following an injection. The values peak at 24 hours and then begin to attenuate. Because GH also increases the plasma concentrations of IGFBP-3 and a third protein termed acid labile subunit (ALS), which binds both IGFBP-3 and the IGFs, the formation of this ternary complex accounts for the extended duration of the change in serum IGF-1. The IGF-1 response of a short child to GH administration has not proven to be a useful diagnostic test of GH deficiency.78,79 In spite of these problems in interpreting low values, basal IGF-1 measurements have proven very useful for as a screening test for selecting individuals who should undergo stimulation testing to assess their GH secretory response.74,75
In states of GH excess, IGF-1 values are invariably increased. The mean IGF-1 for patients with acromegaly is seven times the normal age-adjusted control value.80 The sensitivity and specificity of a single IGF-1 measurement for accurately diagnosing acromegaly in patients older than 20 years is >97%.81 The severity of the IGF-1 abnormality appears to correlate with disease activity, and values correlate with measurement of soft tissue growth, such as heel pad thickness.80 IGF-1 measurements are useful in monitoring the response to therapy and correlate well with residual GH secretion in these patients.82 Generally, if 24-hour mean GH values are less than 1.6 ng/mL, then IGF-1 will be within the age-adjusted 95% confidence interval. IGF-1 values are also elevated during the last trimester of pregnancy, presumably due to increases in placental GH secretion.
Another hormonal variable that controls IGF-1 concentrations is thyroxine. Plasma IGF-1 concentrations are low in severe thyroxine deficiency and rise with thyroid hormone replacement.83 Serum IGF-1 values are not suppressed in Turner’s syndrome, and estrogen replacement results in little change. Prolactin has a weak, stimulatory effect on plasma IGF-1. In subjects who are severely growth hormone deficient, prolactin concentrations of 200 ng/mL or greater can maintain IGF-1 in the normal range.83
Nutritional status is an important determinant of plasma IGF-1 concentrations. Adequate caloric and protein intake have to be maintained in order to maintain an adequate serum IGF-1, both in children and adults.84 Fasting for 3 days results in substantial reduction in total serum IGF-1 and a blunted response to the administration of GH.85 Ten days of fasting results in a 70% decrease in plasma IGF-1. Following a 5-day fast, values decline by 53%, and subjects must be refed for at least 8 days for values to return to normal. During fasting and refeeding, the changes in IGF-1 correlate with changes in nitrogen balance.85 These changes are due to both energy and protein deficiency. An energy intake of 20 Kcal/kg is required to maintain a normal IGF-1, whereas an intake of 0.6 gm/kg of protein is required. The energy must be supplied as at least 100 gm of carbohydrate. Similarly, the quality of the protein intake (e.g., the amount of essential amino acids) is an important determinant of IGF-1 if the protein intake is below 0.5 g/kg/day. Children with severe protein-calorie malnutrition have low IGF-1 values that respond to treatment.85 Other catabolic conditions, such as hepatic failure, inflammatory bowel diseases, or renal failure, are associated with low serum IGF-1 concentrations.86,87 Insulin is an important determinant of IGF-1 concentrations. Although it is difficult to differentiate between nutritional regulation and insulin action, insulin perfusion of the liver in diabetic animals results in a substantial increase in plasma IGF-1. Patients with poorly controlled type 1 diabetes mellitus have low normal IGF-1s that rise into the normal range with adequate insulin treatment.88 Furthermore, in poorly controlled type 1 diabetes, there is a correlation between hemoglobin A1C values and IGF-1. Similarly, patients with severe insulin resistance have low IGF-1 values.89
Control of IGFBP Concentrations in Blood and Extracellular Fluids
IGFBP-3 is the most abundant form of IGFBP in plasma. It has the highest affinity for IGF-1 and IGF-2. It also binds to ALS, and the ternary complex that is formed has a long half-life. These characteristics explain why IGFBP-3 accounts for most of the binding protein activity in blood.90 The IGFBPs in plasma perform three functions. The first is to act as transport proteins for the IGFs. The second is to regulate their half-lives, and the third is to provide a specific means for transcapillary transport into extravascular fluid compartments.
The plasma concentrations of IGFBP-3 are regulated by GH. IGFBP-3 concentrations are low in patients with GHD and increase as a function of GH secretion.91 This increase is partially due to a direct effect of GH on IGFBP-3 synthesis; however, it is also because the half-life of IGFBP-3 is prolonged by binding to the two other proteins to form a ternary complex (consisting of IGF-1 or IGF-2, IGFBP-3, and ALS). ALS is an 88-kD glycoprotein containing several leucine-rich domains that are known to facilitate protein/protein interactions, and it is this domain structure that accounts for its binding to IGFBP-3.92 Since IGF-1 and ALS synthesis are also increased by GH, all three components are increased, and this acts to prolong the half-life of each component. The binding of IGF-1 to this complex in plasma functions to prolong its half-life from 6 minutes in the free form, which is similar to that of insulin, to 16 hours. The prolongation of the half-life of ALS-associated IGF-1/IGFBP-3 complexes is also due to the fact that this macromolecular complex (150 kD) cannot freely cross capillary barriers, and therefore it is not excreted by the kidney. If sufficient IGF-1 and IGFBP-3 are infused to exceed the binding capacity of ALS, then their half-lives are shortened substantially, indicating that it is the ternary complex that maintains the stability and prolongs their half-lives. The molar concentration of IGFBP-3 in serum is generally equal to the sum of IGF-1 and IGF-2, and therefore it is usually saturated. The affinity of IGFBP-3 for IGF-1 and IGF-2 is not lowered by binding to ALS, and its high affinity and its long half-life account for the fact that 75% of the IGF-1 and 2 in plasma is carried in this complex. The exact function of this large storage pool of IGF-1 and 2 in serum is unknown. However, it is clear that changes in the IGF-1 concentrations within this large complex correlate with the anabolic response to GH administration. Plasma IGFBP-3 levels are elevated in patients with acromegaly and low in patients with GH deficiency, as are ALS levels.91,93,94 Age is an important determinant of IGFBP-3 concentrations, and serum IGFBP-3 varies with age in a manner similar to IGF-1.76
Hormones other than GH can influence the synthesis of IGFBP-3 and therefore its plasma concentration. IGFBP-3 is low in prepubertal males and increases following testosterone administration. It decreases 40% following menopause and can be increased in postmenopausal females with physiologic estrogen replacement.95 IGFBP-3 concentrations are low in patients with hypothyroidism and increase 55% following administration of thyroxine.96
Insulin enhances the IGFBP-3 synthesis response to GH, but it does not appear to have a direct effect. Insulin also stimulates ALS secretion, and severe diabetes results in reduced ALS levels and reduced ternary complex formation. Although GH directly stimulates IGFBP-3 and ALS synthesis, infusion of IGF-1, while increasing serum IGFBP-3 transiently, acts to suppress its concentrations over time by suppressing GH release from the pituitary and thereby lowering ALS synthesis.97
IGFBP-3 abundance in serum is also regulated by protease activity.98 Several proteases that degrade IGFBP-3 have been described, including PSA and plasmin, but the exact identity of the serum protease has not been determined. Protease concentrations are abundant in human pregnancy serum99 and are also present in GH-resistant states such as diabetes.100 Proteolytic cleavage reduces the affinity of IGFBP-3 greatly, and the IGF-1 that is released binds to unsaturated IGFBP-1, 2, and 4, wherein it can equilibrate much more readily with the interstitial fluids. Therefore, an important function of proteases that cleave IGFBP-3 may be to liberate IGF-1 and IGF-2 from the IGFBP-3/ALS complex and allow them to bind to lower-affinity forms of IGFBPs that can cross capillary barriers, thus facilitating a more favorable equilibrium with the extravascular space.
The next most abundant IGFBP in plasma is IGFBP-2. The affinity of IGFBP-2 for IGF-1 is less than IGFBP-3, and its plasma concentrations are substantially lower. IGFBP-2 concentrations are inversely regulated by GH; that is, they are high in GHD, suppressed with administration of GH, and reduced in acromegaly.101 Unlike IGFBP-3, IGFBP-2 does not bind to ALS, and there is no ternary complex in plasma; therefore, its half-life when bound to IGF-1 is approximately 90 minutes. It is not saturated, and excess binding capacity exists. Intact IGFBP-2 crosses the capillary barriers. Hepatocytes appear to be the major source of serum IGFBP-2, and the abundance of its mRNA in liver is regulated in parallel with its plasma concentrations.102 Hypophysectomy in experimental animals results in a major increase in hepatic IGFBP-2 mRNA expression. GH administration to normal or GH-deficient humans results in substantial lowering of plasma IGFBP-2.60 IGF-1 is a major regulator of IGFBP-2 concentrations in serum. Following IGF-1 administration to GH-deficient humans or patients with diabetes, there is a three- to fourfold increase in IGFBP-2.60 Plasma IGFBP-2 concentrations are also increased by IGF-2, and they are elevated in patients with retroperitoneal tumors that produce IGF-2.103 Hepatic IGFBP-2 mRNA expression is significantly increased in diabetic rats and suppressed with insulin administration. Severely limiting nutrient intake in humans results in increases in plasma IGFBP-2, as does poorly controlled type 1 diabetes.104 The response to nutrient restriction is dependent upon protein intake, since it can be mimicked with low-protein diets that contain a normal caloric content, and IGFBP-2 expression in animals is increased during protein restriction.105,106 Since the half-life of the IGF-1 bound to IGFBP-2 is considerably less than IGF-1 bound to IGFBP-3, it has been assumed that IGF-1 that is bound to IGFBP-2 is in more rapid equilibrium with IGF-1 in the extravascular space.
The third most abundant protein in serum is IGFBP-1. IGFBP-1 also circulates in binary complexes with IGF-1 and IGF-2. Its affinity for the two growth factors is coequal (see Table 17-2). IGFBP-1 is acutely regulated by insulin.107 Insulin-deficient states such as fasting or type 1 diabetes are associated with very high concentrations of IGFBP-1, whereas administration of insulin or ingestion of a meal results in marked suppression.108 Major sites of synthesis of IGFBP-1 are highly restricted, and the liver is the principal site of synthesis, although kidney, maternal placenta, and uterus are other sources of this peptide. Plasma concentrations are controlled primarily by hepatic synthesis and release. Hepatic synthesis is primarily under the control of insulin.107 IGFBP-1 in blood is unsaturated, and therefore IGFBP-1 is proposed to be a major modulator of free IGF-1 levels, particularly in response to food intake. Postprandially, changes in serum insulin result in a four- to fivefold decrease in IGFBP-1. This is due to direct suppression of hepatic synthesis. Insulin directly affects IGFBP1 gene transcription, and there is an insulin-response element in the 5′ flanking region of the IGFBP1 gene.109 IGFBP-1 crosses intact capillary beds, and the amount that crosses in a fixed time period is dependent upon ambient insulin concentrations.110
Because IGFBP-1 can bind free IGF-1, it has been proposed to have a gluco-regulatory function, that is, since IGF-1 enhances insulin sensitivity, factors that lead to excessive IGFBP-1 could lead to reduced insulin sensitivity. In states of significant insulin resistance, there is enhanced phosphorylation of IGFBP-1, which increases its affinity for IGF-1 and therefore results in further attenuation of IGF-1’s ability to enhance insulin sensitivity.111 Both fasting and diabetes have been shown to cause disproportionate increases in serum IGFBP-1 concentrations.104,108 In addition, administration of glucocorticoid increases IGFBP-1 through a direct effect on IGFBP1 gene transcription.109 Administration of large concentrations of IGFBP-1 to hypophysectomized rats results in slight increases in glucose concentrations, suggesting that IGFBP-1 may have some role in regulating the insulin-like actions of IGF-1.112
The exact roles of IGFBP-1 and IGFBP-2 in controlling the distribution of the IGFs has not been determined. In catabolic states, such as nutritional deprivation, GH deficiency, or renal failure, IGFBP-1 and IGFBP-2 levels are increased. Similarly, in these conditions, the amount of IGF-1 that is bound to IGFBP-3 is decreased.113 Therefore, they can become the major serum binding component.
IGFBP-4 concentrations in serum have been shown to correlate with changes in bone physiology. Specifically, in states of low bone turnover and in states associated with low parathyroid hormone concentrations, serum IGFBP-4 concentrations are increased. There is a correlation between sunlight exposure and IGFBP-4, suggesting that vitamin D or one of its active metabolites regulates IGFBP-4.114
IGFBP-5 exists in serum mostly as proteolytic fragments, and intact IGFBP-5 is present at very low concentrations. The fragments that are present have very low affinity for IGF-1 and IGF-2, and therefore their plasma concentrations are unlikely to be major regulators of IGF-1 action. IGFBP-5 in plasma binds to ALS, and its concentrations are regulated by growth hormone and IGF-1. Both intact IGFBP-5 and its major fragment increase substantially when GH is administered to GH-deficient patients.115
Circulating IGFBP-6 levels are lower in females than males, but estrogen does not change its concentration.116 IGFBP-6 increases with physical stress, and serum concentrations are elevated in patients with critical illnesses.117 Similarly, they are increased in renal failure.118
Control of IGF-1 Synthesis in Tissues
While it is beyond the scope of this chapter to discuss the expression of IGF-1 in all tissues that have been studied, some general principles are important for a fundamental understanding of the autocrine/paracrine mediated actions of this growth factor. Connective tissue cells within a given tissue or organ are often the origin of IGF-1 transcripts. In-situ hybridization studies have shown that fibroblasts and other cells of mesenchymal origin are the primary extrahepatic source of IGF-1 in vivo.119 Importantly, the abundance of this transcript in connective tissue cells is increased in response to GH, and its synthesis is also regulated by factors that are released in response to injury, such as PDGF.120
Cartilage and Bone
In cartilage, both GH and fibroblast growth factor have been shown to be potent stimuli of IGF-1 synthesis by prechondrocytes.121 Its synthesis is most abundant in those cells that are actively differentiating; when chondrocytes reach the hypertrophic state, IGF-1 synthesis is diminished. Fetal chondrocytes, during development, have been shown to be an abundant source of IGF-1 mRNA.
Similar to cartilage, bone osteoblasts are a source of IGF-1 peptide, and it is synthesized in fetal calvarial tissue.122 GH also increases IGF-1 synthesis by osteoblasts. IGF-1 synthesis rates correlate with changes in osteoblast DNA synthesis, type I collagen synthesis, and synthesis of other components of bone extracellular matrix.123 Several bone trophic factors, such as bone morphogenic proteins, stimulate the synthesis of IGF-1.124 In bone, IGF-1 mRNA expression is down-regulated by glucocorticoids. In contrast, estrogen stimulates the expression of IGF-1 in osteoblasts.125 PTH also stimulates IGF1 gene transcription, and its effect is mediated through cAMP induction, which enhances IGF1 gene transcription.126 In contrast, the bone growth factors, FGF, PDGF, and TGF-β, down-regulate IGF-1 expression. IGF-1 appears to be an important factor for erythropoiesis. Red cell mass is decreased in IGF-1-deficient humans and is restored to normal with IGF-1 administration.127 Erythroid precursor cells synthesize IGF-1, and its synthesis can be stimulated in these cells both by GH and erythropoietin. Similarly, granulocyte precursor cells synthesize IGF-1 mRNA, and this is stimulated by granulocyte/macrophage colony-stimulating factor.
Reproductive Tract
IGF-1 expression is decreased in the ovary of the hypophysectomized rat, and ovarian expression increases in response to GH. Estrogen can increase ovarian IGF-1 expression, and this has been localized primarily to the granuloma cells of the early follicle.128 IGF-1 receptors are also present in these follicular cells, indicating the possibility for an autocrine loop. Follicular fluid contains IGF-1 and IGF-2 peptides, and their concentrations are increased following FSH administration. Several studies suggest that the effects of IGF-2 predominate over IGF-1 in the ovary, and much more IGF-2 is produced in that organ. In the oviduct, IGF-1 and IGF-2 have been shown to be present in oviductal fluid. Oviductal cells express mRNAs encoding both IGF-1 and IGF-2, as well as IGF-1 receptors. Endometrium normally expresses IGF-1 mRNA, and in rats, a 20-fold increase can be induced with estradiol administration.129 Estrogen induces IGF-1 expression primarily in the epithelium, whereas progesterone induces it in the endometrial stroma. In the late proliferative phase, IGF-1 mRNA is present almost exclusively in the stroma. Similarly, IGF-1 receptor mRNA is up-regulated during the secretory phase of the menstrual cycle. The testes express IGF-1 mRNA, and the source of origin is the Leydig cell. IGF-1 expression by Leydig cells is down-regulated by interleukin 1 and stimulated by LH.
Neural Tissue
Circulating plasma IGF-1 crosses the blood-brain barrier. However, much of the IGF-1 that is present in CSF is believed to arise from IGF-1 synthesis within the CNS. The major sites of IGF-1 mRNA are the Purkinje cells of the cerebellum, the olfactory bulb, and the hippocampus.130 The retina is also a site of postnatal expression. Astroglial cells in the cerebellum are also an important site of IGF-1 synthesis. Immunohistochemical staining has shown that IGF-1 is transported along axons and dendrites and that IGF-1 peptide is present in the choroid plexus. Factors that regulate IGF-1 synthesis in peripheral tissues such as nutrition, thyroid hormone, and estrogen also regulate CNS IGF-1 expression.131 TNFα down-regulates IGF-1 expression.
Skeletal Muscle
IGF-1 mRNA is expressed in the satellite cells and myoblasts of skeletal muscle.132 Following an ischemic or toxic injury, there is a major increase in IGF-1 mRNA expression.133 The wave of increase of expression after skeletal muscle injury coincides with the appearance of regenerating tissue and rapid cell division. Work-induced hypertrophy in muscle can also lead to an increase in expression of IGF-1 and IGF-2, indicating that this change is GH independent.134 The IGF-1B transcript is selectively increased. Cardiac muscle is also a site of IGF-1 synthesis, and it is increased in models of cardiac hypertrophy that have been induced either by pressure or volume overload.135 Blood vessels are also an important site of IGF-1 synthesis. Both endothelial and smooth muscle cells contain IGF-1 mRNA. Pressure overload, oxidative stress, and angiotensin II increase IGF-1 expression.136 Following mechanical injury to blood vessels, there is an increase in IGF-1 expression by smooth muscle cells.137
Liver
IGF-1 expression in liver correlates extremely well with changes in plasma GH concentrations. Expression in hepatic tissue is low in hypophysectomized animals and increases after administration of GH.138 The effect of GH has been shown to be mediated through the transcription factor STAT 5B. Likewise, nutritional deprivation results in a major decrease in IGF-1 mRNA abundance, and this can be restored with refeeding.139 A part of this change is due to a change in transcription, and part is due to a decrease in mRNA stability.
The liver is a major site of insulin action, and insulin regulates the ability of the liver to respond to GH with IGF-1 mRNA expression.140 Similarly, the effect of thyroxine on serum IGF-1 is mediated through its effect on hepatic IGF-1 expression.
Development
IGF-1 transcripts are easily detected in developing rats in intestine, liver, lung, and brain. Expression is present in as early as 11-day embryos, and IGF-1 mRNA abundance increases 8.6-fold by day 13.141 In early embryos, IGF-1 is detected in yolk sac, hepatic bud, and dermal myotomes, sclerotomes, and brachial-arch mesoderm. In late fetal development, IGF-1 content is increased in muscle, precartilaginous mesenchymal condensations, perichondrium, and the immature chondrocyte periosteum, as well as ossification centers. In human fetal embryos, IGF-1 mRNA levels are relatively low at 16 weeks, and the highest levels are found in placenta and stomach. At 20 weeks, fetal kidney, lung, brain, cartilage, liver, and the placenta have detectible transcripts. The perisinusoidal cells of the liver and the perichondrium appear to be foci of intense expression in 20-week fetuses, and the cells of origin appear to be fibroblast-like. Postnatally, IGF-1 expression increases markedly in skin, nerve, and muscle.
IGF-1 Expression in Kidney
IGF-1 is expressed at low levels in the fetal kidney; however, in the adult kidney in rats, IGF-1 mRNA is abundant.142 Immunohistochemical staining shows moderate amounts of IGF-1 in both the proximal and distal tubules of human fetuses. In adult rats, IGF-1 is localized primarily over the collecting ducts. Overexpression of IGF-1 in transgenic animal kidneys has been shown to result in renal growth, and GH administration to GH-deficient rats results in increased expression of IGF-1 in the kidney. Unilateral nephrectomy in rats results in compensatory growth of the contralateral kidney and in increased mRNA expression 24 hours after nephrectomy.143 This increase in compensatory synthesis is partly dependent on GH, since it is reduced in hypophysectomized animals. After ischemic injury, there is increased IGF-1 immunoreactivity in the regenerating cells of the proximal tubules.
Control of IGFBP Concentrations in Tissues
Since IGF-1 and IGF-2 function not only as endocrine hormones but also as paracrine regulators of growth and differentiation in tissues, the primary role of the IGFBPs in tissues may be to control the amount of locally produced IGF that is accessible to receptors. The exact regulation of each of the six binding proteins in each tissue in which they are expressed is beyond the scope of this chapter. The reader is referred to a review that comprehensively discusses this subject.60
Actions of the IGFs
Cell Cycle Progression
One of the most commonly studied effects of IGF-1 in vitro is its ability to stimulate DNA synthesis. IGF-1 appears to act principally by stimulating entry into DNA synthesis from the latter part of the G1 phase of the cell cycle.144 In some systems, its presence is required for progression through all 12 hours of G1. Compared to other growth factors, such as PDGF or FGF, IGF-1 is not as potent in stimulating quiescent cells to enter G1, but once cells have entered the cycle, it is often sufficient to stimulate progression through to “S” phase. In some cell types, it is possible to alter this requirement by overexpressing EGF, the c-myb proto-oncogene, or SV40 T antigen.145 Generally, these manipulations cause cells to secrete more autocrine-produced IGF-1 and thereby stimulate the IGF-1 receptor. Support for the hypothesis that constitutively synthesized IGF-1 is still required in such systems derives from studies in which antibodies that inhibit IGF-1 binding to its receptor block DNA synthesis, and cells that have had the IGF-1 receptor deleted grow poorly in response to stimulation by other growth factors.146 Similarly, in some systems, enhanced expression of the IGF-1 receptor will abrogate the need for PDGF or FGF.
Other growth factors have been shown to work cooperatively with IGF system components. PDGF and FGF increase the number of IGF-1 binding sites, and FGF, EGF, angiotensin II, and aldosterone can transactivate the IGF-1 receptor tyrosine kinase.50–52 IGF-1 is a mitogen for essentially every type of cell that possesses IGF-1 receptors. These include all mesenchymal cell types, most types of epithelial cells, including neuronal epithelium, and multiple endodermally derived cell types. Cell lines in culture that have been shown to have an increased number of IGF-1 receptors are more sensitive to IGF-1’s growth-promoting actions. A factor complicating the interpretation of all of the studies that analyze IGF-1 effects on growth in vitro is the autocrine secretion of IGF-1. This autocrine-synthesized IGF-1 is capable of binding to receptors and potentiating IGF-1 action through the IGF-1 receptor.147 Therefore, analysis of the effects of IGF-1 added to cells in culture often must take into account this confounding variable. In many of the studies in which synergism between IGF-1 and other growth factors has been analyzed, the end result is often influenced by autocrine-secreted IGF-1. Hormones such as TSH and FSH and growth factors such as PDGF and EGF may exert part of their proliferative effects by stimulating autocrine secretion of IGF-1.148
Effects of IGF-1 On The Proliferation of Different Types of Cells
Many of the growth-promoting actions of GH on skeletal growth are believed to be due to the local production of IGF-1 by prechondrocytes or early differentiating chondrocytes within the epiphyseal growth plate. In vitro, IGF-1 stimulates cartilage cell division and size, as well as proteoglycan synthesis, which contributes to enhanced extracellular matrix synthesis.149 IGF-1 also inhibits apoptosis in these cells.150 Transplantation of articular chondrocytes that had been transfected with IGF-1 cDNA showed increased cell growth and matrix synthesis.151
Bone
IGF-1 stimulates several anabolic effects on bone cells in culture. Exposure of pre-osteoblast cells to IGF-1 results in stimulation of type I collagen synthesis, DNA and RNA synthesis, as well as total protein synthesis.152 In addition, skeletal tissue is a rich source of stored IGF-1. Osteoblasts themselves can synthesize IGF-1, and several of the IGFBPs that bind to bone extracellular matrix can act as storage reservoirs.124 IGF-1 expression has been shown to be stimulated by a number of hormones and cytokines that are potent trophic growth factors for bone, implying that many of their effects may be mediated locally through IGF-1 production. Genetic models in which components of the IGF system have been altered have confirmed the importance of locally synthesized IGF-1.153 Targeted overexpression of IGF-1 in bone is associated with increased bone mineral density,154 and targeted deletion of the IGF-1 receptor is associated with poor responsiveness to parathyroid hormone.155 Targeted deletion of hepatic IGF1 gene expression, which reduces serum IGF-1, results in decreased cortical bone thickness.156
Skeletal Muscle
Several types of myoblasts in culture have been shown to respond to IGF-1 addition. Both IGF-1 and IGF-2 stimulate muscle-cell protein synthesis, as well as DNA synthesis.132 Their effects are complex, because they both stimulate differentiation in these cells (see following discussion). IGF-1 is synthesized by the satellite cells, which are pre-myoblast precursors, and its synthesis in satellite cells is controlled by the need to maximize the proliferative pool. Following stimulation of myoblast proliferation, prolonged exposure to higher concentrations of IGF-1 results in terminal differentiation. This effect is linked to the ability of IGF-1 to enhance the expression of the myogenic differentiation protein, myogenin. Muscle-specific deletion of the IGF-1 receptor results in muscle hypoplasia at birth, and IGF-1 overexpression enhances DNA synthesis during regeneration after injury.157 Increased expression also increases muscle DNA synthesis and cell number in normal animals.158 Cardiac muscle IGF-1 overexpression has been shown to reduce ventricular dilatation in models of cardiomyopathy.159
Smooth Muscle
Targeted overexpression of IGF-1 results in enhanced smooth muscle cell growth in response to balloon injury.160 The expression of contractile proteins such as myosin heavy-chain is increased in these animals, leading to enhanced contractility. Similarly, IGF-1 overexpression in intestinal smooth muscle leads to increased growth of the muscularis.
Nervous System
The major nervous-system cell types that grow in response to IGF-1 are astrocytes and glial cell precursors.161 In end-terminally differentiated neurons, IGF-1 has been shown to stimulate neurite outgrowth and myelin synthesis. Cells derived from the sympathetic nervous system, such as adrenal chromaffin cells, are stimulated to divide by IGF-1. IGF-1 is also a stimulant of neurite outgrowth in axons damaged by denervation.162 In animals, IGF-1 is required for normal growth of the olfactory bulb.163 Deletion of IGF-1 or IGF-1R results in brain growth retardation, and conversely, a localized increase in cerebellar expression was associated with increased cerebellar size.164 Detailed analysis has shown that some of these changes are due to changes in cell number. Following injury, animals that had had IGF-1 receptor expression deleted in brain showed decreased proliferation of oligodendrocytes and reduced myelin synthesis.165
Other Cell Types
Other cell types that have been shown to be IGF-1 responsive include mammary epithelial cells, vascular smooth muscle cells, endothelial cells, mesangial cells, erythroid progenitor cells, oocytes, adrenal fasciculata cells, granulosa cells, promyelocytic cells, granulocyte colony-forming cells, fetal hepatocytes, pancreatic islet cells, oligodendrocytes, Sertoli cells, and spermatogonia.148
Effects On Cell Death
In many systems, IGF-1 has been shown to be a potent inhibitor of programmed cell death. The systems that have been the best characterized are hematopoietic and neuronal cell precursors. In hematopoietic cells, erythroid progenitor cells can be induced to undergo apoptosis with serum or erythropoietin deprivation, and this effect is suppressed by IGF-1.166 IGF-1 inhibits apoptosis in myeloid precursors that occurs following the withdrawal of stimulatory cytokines, such as interleukin 3.167 In tumor cell types, transfection with a dominant negative form of the IGF-1 receptor (a form of IGF receptor that has a tyrosine kinase–defective subunit) results in enhancement of the apoptotic effect that is induced by cytotoxic agents. During ovarian follicle development, IGF-1 stimulation by gonadotrophins may prevent apoptosis of the developing follicular cells. IGF-1 has been shown to inhibit the apoptosis that occurs during development in myoblasts, neurons, and oligodendrocytes.
Effects On Cellular Differentiation
In cultured myoblasts, IGF-1 induces the expression of myogenin, a specific myoblast differentiation factor, and myogenin induction can be blocked with antisense oligonucleotides that inhibit the synthesis of autocrine-stimulated IGF-1.168 Autocrine-produced IGF-2 may have similar effects. The programmed events that occur during differentiation in response to IGF-1 are time specific since, in L-6 myoblasts, cellular exposure to high concentrations of IGF-1 early in the differentiation program acts to inhibit differentiation, but at later time points, it is accelerated.169 IGF-2 can inhibit apoptosis that occurs during transition from proliferation to differentiation in myoblastic cell lines. Differentiation markers have also been shown to be preferentially stimulated in response to IGF-1 or 2 in osteoclasts, chondrocytes, and neural cells. The addition of IGF-1 to different types of cultured neurons has been shown to enhance neuronal differentiation. Maintenance of neuroepithelial cultures in several model systems has been shown to be enhanced by IGF-1, probably by inhibiting apoptosis.
Effects On Specific Cellular Functions
Production of steroids by ovarian granulosa cells and thecal cells has been shown to be stimulated by IGF-1 and IGF-2, and their effects are synergistic with FSH.170 IGF-1 also stimulates steroid hormone secretion by ACTH-responsive, adrenal cortical cells.171 IGF-1 stimulates testosterone secretion from Leydig cells and acts synergistically with LH to increase the response. Similarly, thyroglobulin production by thyroid follicular cells is synergistically enhanced with TSH plus IGF-1. GH secretion by pituitary cells is inhibited by IGF-1.172 IGF-1 inhibits glutamate-stimulated release of gamma amino butyric acid from Purkinje cells. IGF-1 is a specific stimulant of IGFBP-5 transcription by muscle cells and fibroblasts.173 Other proteins whose transcription is stimulated by IGF-1 include elastin by smooth muscle cells, crystallin by lens epithelial cells, and cholesterol side cleavage enzyme by adrenal cortical cells. Some proteins whose expression is increased following IGF-1 have been shown to result in specific functional changes in that cell type (e.g., the increased α actin in skeletal muscle174 and the increased myelin in neuronal cells).164 Microarray studies have shown that IGF-1 selectively up-regulates the expression of several genes and some, such as heparin-binding EGF and twist, may have important implications for cellular growth.175,176
Several metabolic processes that are stimulated by IGF-1 in a variety of cell types have been analyzed. These include glucose uptake, glycolysis, glycogen synthesis, and glucose oxidation in skeletal muscle cells.177 These metabolic effects can be mediated by the insulin receptor if sufficient IGF-1 is added in vitro (e.g., concentrations > 10−8 M); however, antibody-blocking studies have indicated that IGF-1 can have direct effects on this process through its own receptor in some cell types. Similarly, the hybrid IGF-1/insulin receptor may play a role in mediating these effects in some cell types. Total protein synthesis, extracellular matrix protein synthesis, cell migration, and the synthesis of proteoglycans and collagen, in particular, have been analyzed extensively in connective tissue cells. IGF-1 often acts in concert with other growth factors to stimulate connective tissue cell protein synthesis. IGF-1 is a potent stimulant of cell migration and stimulates this process by both chemotaxis and chemokinesis.178 IGF-1 is not directly angiogenic, but it can stimulate the synthesis of angiogenic peptides such as vascular endothelial cell growth factor.
Role of IGF-1 in Malignant Tumors
Because IGF-1 is a potent inhibitor of apoptosis, it has been proposed that it may function to enhance tumor formation in several experimental animal models. The presence of an intact IGF-1 receptor is required for propagation of several types of tumors.179 In the absence of IGF-1 receptors, C6 glioma cells do not form tumors, and they undergo apoptosis. Often the presence of a normal IGF-1 receptor number is inadequate for tumor formation, and the IGF-1 receptors need to be overexpressed.17 However, several processes that are necessary for tumor formation can be facilitated by IGF-1, even in the absence of enhanced receptor number, such as prevention of cell death. Deletion of the receptor results in inability of cells that would normally be tumorigenic in nude mice to form tumors, and mutation of specific tyrosine residues on the receptor and expression of these mutated receptors results in lack of tumor formation.180 In human tumors, a direct causal role for the receptor in tumor pathogenesis has been difficult to prove. All of the data that exist are correlative. In Wilms tumor, small cell lung carcinoma, uterine cancer, and some colorectal cancers, IGF-1 receptor number is increased.179 No mutations of the receptor have been identified as a cause of human tumors.
Several cell types that form tumors in animals have been shown to overproduce IGF-1 or IGF-2. However, in these systems, antisense IGF-1 often does not inhibit tumor formation or induce apoptosis. In contrast to the effects that are induced by blocking receptor-binding ovarian carcinomas that overexpress IGF-2 have a higher rate of metastasis.181 Precancerous liver nodules that occur in virally-induced models of hepatic cancers overexpress IGF-2. Pancreatic tumor cells that have been transformed with SV-40 T antigen require IGF-2 for continued growth. Certain fetal tumors, such as Wilms tumor and neuroblastoma, are accompanied by loss of imprinting of the IGF-2 gene, and overproduction of IGF-2 accompanies tumor formation.182 The IGF-2 receptor has also been implicated as a tumor suppressor in hepatocellular carcinomas, possibly through its role in the clearance and degradation of IGF-2. The only paraneoplastic syndrome that is known to be definitively linked to IGF-2 overproduction occurs with retroperitoneal sarcomas. Overproduction of IGF-2 by the tumor results in hypoglycemia.183 The mechanism that has been proposed is that IGF-2 forms binary complexes with specific forms of IGFBPs in plasma that do not bind ALS (such as IGFBP-2), and this allows accelerated equilibration of IGF-1 and IGF-2 with extravascular fluids, thus leading to increased IGF-1 in interstitial fluids and to hypoglycemia.
Studies in mice have shown that IGF-1 overexpression is associated with mammary intraepithelial neoplasia; conversely, expression of dominant negative forms of the IGF-1 receptor is associated with decreased tumor progression.184 Similarly, animals with low serum IGF-1 due to gene targeting of hepatic IGF-1 have delayed onset and reduced severity of many types of tumors.180 Transgenic overexpression of IGF-1 in mouse prostate also leads to a higher prevalence of tumors at younger ages compared to control animals.185 Recent studies have documented the important role of IGF-1/IGF receptor in immunocompromised animals having brain tumor xenografts. These studies have shown that anti-IGF-1 receptor antibody and tyrosine kinase inhibitors have potent effects in inhibiting tumor cell propagation, and they prolong mouse survival.186,187 In addition, studies have shown that antibodies can alter the metastatic potential of the primary tumor,188 suggesting that IGF-1/IGF-1R may play a role in tumor cell dissemination.188
Control of IGF-1 Actions in Cells and Tissues by IGFBPs
Variables That Regulate IGFBP Affinity
Proteolysis
Proteolysis of IGFBP-3 by serum proteases has been shown to result in marked reduction in affinity for IGF-1 and a significant but less intense change in affinity for IGF-2. The principle fragment that is retained, the 32-kD fragment, has at least a 20-fold reduction in affinity for IGF-1. This protease activity is increased in pregnancy, diabetes, and nutritional deprivation. Matrix metalloproteases, such as MMP-1, MMP-2, and MMP-9, degrade several forms of IGFBPs, including IGFBP-3, and constitute part of the protease activity of pregnancy.189 Several well-defined proteases have been shown to degrade IGFBP-3, including plasmin, cathepsin-D, and prostate-specific antigen. IGFBP-3 proteolytic activity has been noted in lymph, follicular fluid, peritoneal fluid, and amniotic fluid. The IGFBP-2 protease is also a cation-dependent serine protease and cleaves IGFBP-2 into multiple fragments. The rate of IGFBP-2 cleavage is increased by IGF-1 binding. IGFBP-5 is cleaved by proteases in a variety of physiologic fluids, including serum, and by the complement C1s that is present in cell-culture supernatants from fibroblasts, osteoblasts, and smooth muscle cells. IGFBP-5, like IGFBP-3, is also cleaved by MMP-2, MMP-9, and PAPP-A. Blocking proteolytic cleavage by incubating IGF-1 with a mutated, protease-resistant form of IGFBP-5 was shown to result in inhibition of IGF-1-stimulated cell growth. IGFBP-4 proteases are also present in several physiologic fluids. PPAP-A, a metalloprotease, has been shown to cleave IGFBP-4.190 Like IGFBP-2, IGFBP-4 proteolytic activity is enhanced by IGF binding to IGFBP-4. There is correlative data suggesting that degradation of IGFBP-4 results in relief of inhibition of IGF-1 actions in vivo.191
IGFBP Phosphorylation
Three of the six forms of IGFBPs have been shown to be phosphorylated: IGFBP-1, IGFBP-3, and IGFBP-5. IGFBP-1 is phosphorylated on serine residues at positions 101, 119, and 169. Casein kinase 2 phosphorylates IGFBP-1, which increases its affinity for IGF-1 by sixfold. The form of IGFBP-1 that is increased during poorly controlled diabetes is a very highly phosphorylated form.111 IGFBP-3 is phosphorylated at positions 111 and 113, and IGF-1 stimulates its phosphorylation. Casein kinase 2 phosphorylates IGFBP-3.
Adherence to Cell Surface, Extracellular Matrix, and Glycosaminoglycans
Both IGFBP-3 and IGFBP-5 have been shown to adhere to cell surfaces. Proteoglycans may be important cell surface–binding components for both proteins. Specific receptors have been postulated to exist for IGFBP-3. The type-V TGF-β receptor is a cell-surface protein that binds IGFBP-3.192 IGF-1 that is bound to ECM or cell-associated IGFBP-3 is in more favorable equilibrium with receptors, since IGFBP-3 binding to cells lowers its affinity. IGFBP-5 binding to ECM or to proteoglycans causes an eightfold reduction in its affinity. In addition to proteoglycans, other types of extracellular matrix proteins bind to IGFBP-5 (e.g., plasminogen activator inhibitor-1, osteopontin, and thrombospondin).193 Localization of IGFBP-5 within the extracellular matrix may provide an important means for focally concentrating IGF-1 or IGF-2 in the pericellular environment.194
EFFECTS OF SPECIFIC FORMS OF IGFBPs ON IGF-1 ACTIONS
When present in concentrations that are greater than IGF-1, IGFBP-1 inhibits IGF-1 actions. If high-affinity forms of IGFBP-1 are added in a 4 : 1 molar excess over IGF-1, they inhibit DNA synthesis, glucose incorporation, and glucose transport.195 IGFBP-1 has been shown to block IGF-1 binding to receptors on human endometrial membranes, and it inhibits the mitogenic response of endometrial stromal cells to EGF.60 IGFBP-1 can also enhance the cellular response to IGF-1. If the dephosphorylated form of IGFBP-1 is utilized and added in an equimolar ratio or less with IGF-1, IGFBP-1 can potentiate the in-vitro response of smooth muscle cells, keratinocytes, and fibroblasts to IGF-1.60 It also enhances the effect of IGF-1 on adrenal steroidgenesis.196 IGFBP-1 has been shown to directly stimulate migration of CHO cells, fibroblasts, and trophoblasts by binding to the α5β1 integrin receptor through its RGD sequence.65 This effect does not require IGF-1 binding to IGFBP-1. In general, IGFBP-1 is induced by stresses such as insulin deficiency,107 hypoxia,197 and endoplasmic-reticulum stress.198 This suggests that it functions to coordinate the IGF-1 response in these pathophysiologic conditions.
IGFBP-2
IGFBP-2 has also been shown to be inhibitory in most in-vitro experiments. Using purified IGFBP-2, it was shown to inhibit IGF-1-stimulated thymidine incorporation into chick embryo fibroblasts and rat astroglial cells, as well as a human lung carcinoma cell line. IGFBP-2 inhibited IGF-1- or IGF-2-stimulated protein synthesis in MDBK cells, and des IGF-1, a form which does not bind to IGFBP-2, was stimulatory.60 Overexpression in renal epithelial cells in vitro resulted in inhibition of IGF-1 actions. IGFBP-2 was shown to mediate the inhibitory effect of TGFβ on lung epithelial cell growth.199 Conversely, IGFBP-2 has been shown to stimulate IGF-1-stimulated glucose incorporation and AIB transport in microvascular endothelial cells and DNA synthesis in smooth muscle cells.200 IGFBP-2 contains a heparin-binding domain which has been shown to stimulate neuroblastoma bone cell proliferation; carboxy terminal fragments that contain this domain enhance chondrocyte proliferation.201,202 IGFBP-2 enhances glioblastoma invasion and stimulates the growth of prostate cancer cells, but whether these effects are direct or mediated by enhancing IGF-1 activity has not been determined.203 IGFBP-2 adheres to ECM through its heparin-binding domain, and the matrix-associated protein enhanced the effect of IGF-2 on osteoblast growth.204 These effects require IGFBP-2 association with ECM through its heparin-binding domain. Recently IGFBP-2 has been shown to stimulate growth of hematopoietic stem cells, implying a direct role in precursor-cell compartmental expansion.205 Like IGFBP-1, IGFBP-2 is up-regulated in response to injury, particularly in the CNS, and this is believed to be an important mechanism for targeting IGFs to sites of injury.206
IGFBP-3
IGFBP-3, if added in molar excess, inhibits glucose incorporation into fat cells, as well as IGF-1-stimulated DNA synthesis in human fibroblasts.60 Maximum inhibition was noted at a 5:1 molar ratio. IGFBP-3 inhibits IGF-1-stimulated glucose incorporation. If IGFBP-3 is pre-incubated with muscle cells then removed from the medium, it can potentiate their AIB transport response to IGF-1.60 Using this experimental paradigm, IGFBP-3 was also shown to enhance the IGF-1-stimulated DNA synthesis response of human fibroblasts, but co-incubation with IGFBP-3 was inhibitory. IGFBP-3 inhibited IGF-1-stimulated cyclic AMP generation by rat granulosa cells and inhibited IGF-1-stimulated collagen synthesis by osteoblasts. Addition of IGFBP-3 to breast epithelial cells results in growth inhibition.207 It inhibits the growth of breast cancer cells in part by stimulating the activity of a phosphatase that down-regulates IGF-1 signaling.208 Like IGFBP-5, IGFBP-3 has been shown to adhere to fibroblast extracellular matrix, and matrix-associated IGFBP-3 enhanced the ability of IGF-1 to stimulate MAP kinase activation.209
IGF-Independent Effects of IGFBP-3: IGFBP-3 has been shown to bind to the type-V TGF-β receptor. Direct addition of IGFBP-3 has been shown to attenuate the effects of several growth factors, including FGF, on cell growth. TGF-β is believed to cause part of its growth-inhibitory effect in breast carcinoma cells through induction of IGFBP-3. Increasing the expression of IGFBP-3 has been shown to inhibit the proliferative actions of several growth factors.210 IGFBP-3 inhibited TGF-β-stimulated chrondrocyte proliferation by inducing STAT-1 phosphorylation and increasing the expression of the CDK inhibitor p21.211 IGFBP-3 can inhibit growth of fibroblasts that do not possess IGF-1 receptors, indicating that IGFBP-3 has growth-suppressive effects that are independent of IGF-1 binding.212 IGFBP-3 has also been shown to stimulate apoptosis in certain cell lines, including cells that do not possess IGF-1 receptors. In addition to its ability to bind to the type-V TGF-β receptor, IGFBP-3 has been shown to bind the RXRα receptor and to inhibit retinoic acid signaling.213 This response requires nuclear translocation of IGFBP-3. The delineation of the specific amino acids that mediate IGF binding to IGFBP-3 has made it possible to prepare mutant forms that do not bind IGF-1 or IGF-2. These studies have shown that the non-IGF-binding mutants enhance cytokine-induced apoptosis. Similarly, expression of one of these mutants inhibited proliferative tumor growth in mice.214 In contrast, the mutant accelerated the growth of esophageal cancer xenografts, whereas the wild-type protein was inhibitory, implying that it maintained its function as an inhibitor solely by inhibiting the effects of IGF-1.215 This mutant also inhibited insulin-stimulated glucose transport in adipocytes.216 Therefore, it appears that IGFBP-3 can have IGF-dependent and IGF-independent inhibitory actions. In general, the IGF-independent actions have been shown to be inhibitory, but when the IGF-dependent effects are analyzed, IGFBP-3 can enhance or inhibit IGF-1 actions, depending upon the cellular context.
IGFBP-4
IGFBP-4 has been consistently shown in in-vitro experiments to inhibit the actions of IGF-1 on cartilage and bone growth.217 IGFBP-4 that is synthesized constitutively by intestinal carcinoma cells inhibits their growth. Several differentiated functions of IGF-1 have been shown to be inhibited by IGFBP-4, including the generation of cyclic AMP by osteoblasts, protein synthesis by prostatic cells, and glycogen synthesis by osteosarcoma cells, as well as the steroidogenic response of granulosa cells to FSH. IGFBP-4 potently inhibits smooth muscle cell replication and AIB transport.218 A protease-resistant mutant of IGFBP-4 inhibited osteoblast proliferation. Cultured myoblasts overexpressing IGFBP-4 showed impaired proliferation and differentiation. The proteolytic cleavage of IGFBP-4 by PAPP-A has been proposed as a mechanism for releasing IGFs to bind to receptors and thus enhancing IGF actions.219
IGFBP-5
IGFBP-5 has been shown to potentiate the effects of IGF-1 in stimulating protein synthesis and DNA synthesis in skeletal tissue, including myoblasts, smooth muscle cells, fibroblasts, osteoblasts, and chondrocytes.69 The potentiation of IGF-1-stimulated fibroblast and smooth muscle cell growth is believed to occur by association of IGFBP-5 with ECM.220 ECM binding requires a specific region of basic amino acids that are located between positions 201 and 218, and mutation of specific basic residues within this motif results in the loss of ECM association and an inability of IGFBP-5 to potentiate IGF’s effects.220 IGFBP-5 binds to several specific ECM proteins, and association with these proteins has been shown to facilitate its capacity to enhance IGF-1 actions.60 IGFBP-5 can also potentiate the effect of IGF-2 on mouse osteoblast, DNA, and protein synthesis. IGFBP-5 has been shown to have effects that are independent of IGF-1. A fragment of IGFBP-5 that does not bind IGF-1 has been shown to potentiate the effect of IGF-1 on osteoblast DNA synthesis and to stimulate mesangial cell migration. Direct injection of IGFBP-5 into bone of IGF-1-deficient mice resulted in enhanced osteoblast growth.221 Overexpression of IGFBP-5 has been shown to activate MAP kinase independently of IGF-1.222 Deletion of IGFBP-5 expression by osteosarcoma cells showed that it was important for cell survival. Schwann-cell differentiation that is stimulated by IGF-1 has been shown to be potentiated by IGFBP-5. In contrast, some studies have demonstrated that IGFBP-5 overexpression results in growth inhibition.223 In mammary gland, its expression is induced during involution; inhibition of its expression reduces epithelial cell apoptosis.224 Additionally, IGFBP-5 enhanced the proapoptotic effect of TNFα on breast carcinoma cell proliferation.225 In skeletal myoblasts, IGFBP-5 has been shown to inhibit this proliferation and thereby facilitate differentiation.226
IGFBP-6
IGFBP-6 appears to preferentially inhibit the effects of IGF-2 in several tissues and cell types.227 Addition of IGFBP-6 inhibited cartilage growth, and its overexpression in rhabdomyosarcoma or bronchial epithelial cells resulted in growth inhibition.228 It also has been shown to inhibit cancer cell adhesion and migration.229 It facilitated apoptosis in oligodendrocytes and inhibited the antiapoptotic effect of IGF-1. Cellular toxins have been shown to induce IGFBP-6, and blocking its induction has been associated with a reduction in toxin-induced cell death.230
Actions of IGF-1 in vivo
IGF-1 was initially termed somatomedin because it mediated the growth-promoting actions of GH, and it was presumed to be a growth stimulant for all tissues. Several correlative types of experiments were conducted to support this hypothesis. These included hypophysectomy, which lowered serum IGF-1 and reduced growth or implantation of GH-secreting tumors, which raised serum IGF-1 and stimulated growth. Additionally, serum IGF-1 concentrations were shown to correlate with changes in GH secretion and growth rates. The initial hypothesis stated that GH-stimulated IGF-1 synthesis in the liver resulted in increased plasma IGF-1, which was transported to skeletal tissues where it acted to stimulate growth.1 The development of cDNA probes for IGF-1 has allowed new types of experiments that led to refinement of this hypothesis. IGF-1 synthesis was demonstrated in multiple extrahepatic tissues, and paracrine-synthesized IGF-1 stimulated growth.148 This raised the question as to what percentage of the generalized growth-promoting actions of IGF-1 is mediated by this autocrine/paracrine secretion, and what percentage is mediated by its endocrine effects.
Administration of IGF-1 to whole animals results in balanced growth, and if the animal has been hypophysectomized, the effect is enhanced.3 A rate-limiting factor is the amount of IGF-1 that can be infused, since very high concentrations will induce hypoglycemia. IGF-1 also feeds back on the pituitary and suppresses GH. This results in a reduction in total serum IGF-1 concentrations due to suppression of ALS and IGFBP-3. If animals are made catabolic, either by nutritional deprivation231 or administration of glucocorticoids,232 administration of IGF-1 results in a partial reversal of catabolism. Likewise, systemic administration of IGF-1 has been shown to improve wound healing, enhance recovery of renal function after kidney injury, and stimulate whole-body protein accretion.233 When IGF-1 is given to nutritionally compromised animal models, the increase in the weight of organs such as spleen and kidney appears to be enhanced preferentially compared to changes in skeletal growth.233,234 In contrast, in well-nourished, hypophysectomized rats and mice, there is proportionate body growth in response to IGF-1, with skeletal tissue being stimulated in a manner nearly identical to nonskeletal tissue.234 IGF-1 stimulates an increase in glomerular filtration rate and has a direct trophic effect on gut epithelial proliferation. Infusion of IGF-1 tends to lower IGFBP-3 and raise IGFBP-2, changes which are similar to those that occur in GH deficiency. Infusion of IGF-1 to insulin-deficient, diabetic rats results in improved growth and improvement in glucose utilization. Similarly, peripheral glucose uptake and glycerol synthesis are stimulated. Infusion of IGF-1 into the insulin-deficient BB rat results in suppression of hepatic glucose output, possibly due to a suppressive effect on glucagon and GH, and these actions led to enhanced sensitivity to insulin.235 Diabetic animals that receive IGF-1 have less increase in body fat compared to animals that are treated with insulin.
Modulation of in-vivo Actions by IGFBPs
In-vivo studies have been performed wherein specific forms of IGFBPs have been administered with IGF-1. Administration of an equimolar amount of IGFBP-1 with IGF-1 reduced the growth response of hypophysectomized rats compared to IGF-1 alone.236 Administration of a large, single dose of IGFBP-1 without IGF-1 resulted in a modest increase (6%) in plasma glucose concentrations. Acute increases in plasma IGFBP-1 result in decreased protein synthesis basally and in response to IGF-1. In contrast, administration of IGFBP-1 with IGF-1 (1:4 molar ratio) to wounds results in enhanced wound healing, including increases in re-epithelialization and formation of granulation tissue.237 Similarly targeted deletion of IGFBP-1 in liver decreases hepatic regeneration after injury.238 In addition, overexpression of IGFBP-1 in pancreas in vivo was shown to have a trophic effect on islet cells. These findings indicate that in some specialized circumstances, increased tissue expression of IGFBP-1 may enhance IGF-1 actions as compared to global inhibition that occurs when IGFBP-1 is administered systemically. Subcutaneous administration of IGFBP-2 together with IGF-2 has been shown to stimulate bone formation and inhibit the development of disuse osteoporosis in mice.204 Administration of a complex of IGFBP-2 and IGF-2 stimulated osteoblast differentiation.239
Because of its role in carrying IGFs in serum, animal studies in which IGF-1 and IGFBP-3 are infused together have been important for defining the endocrine actions of IGF-1. In-vivo administration of a combination of IGF-1 and IGFBP-3 has been shown to consistently enhance of IGF-1’s trophic effects.240 Administration of an equimolar concentration of IGF-1/IGFBP-3 to hypophysectomized rats showed increased bone mineralization and increased growth rates compared to IGF-1 alone. Administration of equimolar concentrations of IGF-1/IGFBP-3 to estrogen-deficient rats resulted in 30% improvement in bone mineral density. Muscle mass was also increased in these animals. A recent study showed that IGFBP-3 induced insulin resistance when administered to rats without IGF-1241; however, when administered with IGF-1, it protected mice against the development of diabetes.242 When IGFBP-3 is given without IGF-1, it can induce apoptosis.243,244 Administration of IGFBP-4 with IGF-1 to mice resulted in increased serum IGF-1 and increased rates of bone formation.245 This effect was dependent upon ongoing IGFBP-4 proteolysis. IGFBP-5 administration with IGF-1 to ovariectomized mice resulted in enhanced bone formation.246
Transgenic Animal and Gene-Targeting Studies
Several transgenic animal models of IGF-1 action have been utilized in which IGF-1 has been overexpressed. To determine if IGF-1 could substitute for GH and stimulate generalized somatic growth, GH secretion was attenuated by cytotoxic destruction of somatotrophs, and then IGF-1 replacement was affected by expressing IGF-1 mRNA in several tissues.247 These animals grew normally, although there was some disproportionate growth of the kidneys, liver, pancreas, and spleen. Additionally, small-bowel length and mass are greater, as is villus height and crypt depth. Likewise, brain size appeared to be particularly sensitive to IGF-1 transgene overexpression. If IGF-1 is overexpressed on a background of no-growth hormone deficiency, more modest increases in somatic growth compared to control animals are noted; however, total body size can be increased by 30%. Brain size is increased disproportionately by 50%. The effect is due in part to inhibition of apoptosis.248 Whether suppression of GH results in the inability to attain greater growth rates following IGF-1 overexpression is unknown. Interestingly, the GH-deficient mice have a somewhat hypoplastic liver, and this effect is not totally reversed by IGF-1 transgene overexpression.247 The major conclusion from these studies was that most but not all of the growth-promoting effects of GH are mediated by IGF-1 using both autocrine/paracrine, as well as endocrine, mechanisms and that local expression of IGF-1 in tissues such as brain results in disproportionate increases in growth.
Attempts to determine the effects of IGFBPs have also utilized transgenic animals. IGFBP-1 transgenic animals show variable phenotypes, depending upon which organs express the transgene. Mice that had expression predominantly in pancreas, kidney, and brain had normal organ sizes except in brain, which was decreased in size.249 Since IGFBP-1 is not constitutively expressed in brain, it presumably bound to IGF-1 or IGF-2, and the animals had a reduction in brain growth. In contrast, in mice with abundant hepatic expression, there was a slight growth retardation at birth and a 10% to 15% reduction in postnatal growth.250 Hepatic overexpression during fetal life also results in growth retardation (e.g., 18% reduction in birth weight).251 If the level of expression of IGFBP-1 in the liver is increased to a very high level, this results in more severe growth retardation and delayed skeletal maturation.252
Overexpression of IGFBP-2 resulted in fetal and postnatal growth retardation.253 This effect is present even in the face of GH and IGF-1 excess. In contrast, deletion of IGFBP-2 expression showed a reduction in bone turnover and reduced bone mineral content at age 16 weeks in mice.254 Histologic evaluation confirmed decreased bone turnover rate.254
In IGFBP-3 transgenic animals, there is modest (10%) fetal and postnatal reduction in growth despite a 2.8-fold increase in total serum IGF-1 concentrations. Analysis of bone showed that resorption was increased and formation was decreased.255 Overexpression of the non-IGF binding mutant form of IGFBP-3 resulted in an increase in GH and IGF-1 levels in serum but no evidence of growth retardation.256 Deletion of IGFBP-3 alone has not been reported, but generalized knockout of steroid receptor coactivator 3, which regulates IGFBP-3 synthesis, was associated with a 20% decrease in postnatal growth.257 Because there are six forms of IGFBPs, following deletion of a single form of IGFBP, compensatory changes which offset the effects of the single gene deletion can occur. Mice were prepared that had deletion of IGFBP-3, 4, and 5, and this resulted in a significant reduction (55%) in serum IGF-1 and a 38% reduction in postnatal growth rate.258
Targeted overexpression of IGFBP-4 in smooth muscle or in bone has been shown to attenuate IGF-1 actions. Cancellous bone formation was reduced, and this was associated with impaired growth.259 When IGFBP-4 is overexpressed in smooth muscle, several organs (e.g., bladder and uterus) show disproportionate growth impairment. In contrast, generalized deletion of IGFBP-4 resulted in a 10% reduction in body weight.260 Overexpression of IGFBP-5 (fourfold increase in serum concentrations) resulted in fetal growth retardation and a significant (17% to 23%) reduction in body size in the early postnatal period.261 Overexpression of ALS resulted in modest postnatal growth restriction (e.g., 5.3% to 8.1%),262 and deletion of ALS also resulted in modest growth retardation.263 Taken together these studies have demonstrated that low levels of expression of the IGFBPs 2, 3, 4, and 5, as well as ALS, are necessary for normal growth. However, overexpression in which there is an imbalance between the concentration of a specific form of IGFBP and IGF-1 often results in growth retardation. This finding suggests that it is both the balance between free and bound IGF-1 and the ability of IGFBPs to prolong IGF-1’s half-life and deliver the optimal amount to tissue receptors that determine how they modulate the growth response.
A great deal of information regarding the fetal and postnatal growth-promoting effects of IGF-1 has been obtained by homologous recombination experiments. In experiments in which the IGF-1 gene was deleted, the fetuses were born alive and were 60% of normal birth length and weight.264 Homozygous animals had extremely high juvenile mortality rates, and only approximately 10% to 20% of these animals survived to adulthood. This appears to be due somewhat to the gene dosage effects, since animals that had only a partial reduction in IGF-1 expression survived into adulthood. The animals that do survive to adulthood are disproportionately short and have an abnormally slow growth rate during the juvenile period. They reach 50% of normal adult size. They also have poor Leydig cell development and small brain sizes. Skeletal abnormalities were also noted. The cause for the increased premature death is unknown. No apparent abnormalities of differentiation have been noted. Fetal growth retardation begins at day 13.5 in utero, and body size is reduced progressively at each stage up to birth.
Deletion of the IGF-1 receptor results in a much more severe phenotype. The animals are 45% of the normal size at birth.264 All have a hypoplastic diaphragm and die at birth. Likewise, there are multiple skeletal and skin defects, indicating that the receptor is necessary for normal muscle, skin, and bone development in utero. Haploinsufficiency of the IGF-1 receptor results in survival and modest growth retardation (e.g., 8% reduction in adult size).265 These animals tolerate oxidative stress better than controls and have a 16% to 33% increase in lifespan. Deletion of IGF-1 receptor expression in endothelium resulted in some protection against the development of neovascularization.266
IGF-2 gene deletion gives a very different phenotype. The animals are approximately 60% of normal size at birth, but unlike the IGF-1 mice, they grow normally postnatally and do not die in excessive numbers.267 No differentiation defects or structural tissue defects are noted. Deletion of both IGF-1 and IGF-2 resulted in extremely small mice approximately 30% of normal size. This manipulation is lethal, since the mice cannot generate a normal inspiration. They are phenotypically similar to the mice lacking the IGF-1 receptor.
Autocrine/Paracrine Regulation of IGF-1-Mediated Growth
An example of local control of IGF-1 is the response to injury that occurs following several types of injury models, such as freezing ear cartilage or thermal burns.148 Fibroblast or chondrocyte precursor cells surrounding the damaged area immediately begin to synthesize IGF-1, and the peak of synthesis usually occurs between 3 and 7 days after injury. Following balloon denudation of blood vessels, the increase in IGF-1 mRNA expression coincides with an increase in the number of precursor cells that are entering the proliferative pool. Therefore, it has been assumed that local regulation of growth, particularly in response to injury but also to other stimuli such as unilateral nephrectomy, wherein the contralateral kidney makes more IGF-1 and enlarges, may be more responsive to local IGF-1 regulation.
Transgenic animals that overexpress IGF-1 in tissues (other than liver) showed normal growth rates if a high level of IGF-1 expression is maintained.247 Another type of experiment that has reinforced the importance of tissue expression is analysis of brain growth. The blood-brain barrier provides some partitioning between blood IGF-1 and locally produced IGF-1. Transgenic animals in which there is intense expression of IGF-1 within the CNS show larger brains than animals that do not have this intense expression, indicating a paracrine regulation of growth that is probably partially independent of blood IGF-1 concentrations.248
A recent experimental animal model that has helped to further understanding of the relative components of autocrine/paracrine–produced IGF-1 as compared to blood-transported IGF-1 is the mouse in which hepatic IGF-1 expression has been selectively targeted.268 This results in an 80% reduction in plasma IGF-1 concentrations. In contrast to global IGF-1 knockout animals, in which the expression of IGF-1 in peripheral tissues as well as liver is eliminated, all other tissues in these animals synthesized IGF-1 normally. These animals were normal size at birth and postnatal growth was very minimally retarded (e.g., 6%). This indicates that deletion of IGF-1 expression in the liver results in a major reduction in endocrine-produced IGF-1 and that autocrine/paracrine IGF-1 in these experimental mice is adequate to allow normal statural growth. Since peripheral tissue IGF-1 expression is also under the control of GH, this type of experiment does not distinguish between how much of the locally produced IGF-1 is regulated by factors other than GH and how much is under GH control. It does eliminate the possibility that in order to grow normally, one has to have a completely normal blood IGF-1 concentration. It also proves definitively that the major source of blood IGF-1 is the liver. Although it might not be surprising that fetal growth was normal in these animals, since IGF-2 is an important fetal growth factor, it is striking that there was no juvenile growth retardation, in spite of these low plasma IGF-1 concentrations. These studies have been extended by simultaneously deleting liver ALS expression. This dual inhibition results in a 16% reduction in growth and a more severe decrease in serum IGF-1.269 These animals also have reduced bone mineral density. In contrast, deletion of ALS alone resulted in only mild growth retardation.270 These findings indicate that a normal serum IGF-1 is necessary for normal growth but also that tissue IGF-1 is making a very significant contribution. In more recent studies, an animal model in which IGF-1 expression is deleted in peripheral tissues but transgenic overexpression of IGF-1 is induced in liver has been developed. These animals have a sixfold increase in serum IGF-1 which results in maintenance of normal growth. Therefore, supraphysiologic concentrations of IGF-1 in serum alone are sufficient for normal growth; however, these animals show a disproportionate increase in growth of the spleen, thymus, and kidneys.271 In summary, these findings support the conclusion that both hepatic and extrahepatic IGF-1 synthesis are necessary for normal growth. They further support the conclusion that normal GH secretion is required for balanced organ growth and for coordination between the affects of blood-transported and locally synthesized IGF-1.
Effects of IGF-1 in Humans
Administration of IGF-1 to normal humans results in changes that are comparable to those noted previously in animal studies. A large bolus of rapidly administered IGF-1 (e.g., 100 mcg/kg) results in hypoglycemia.272 When analyzed on a molar basis, IGF-1 is one twelfth as potent as insulin in reducing glucose. A continuous infusion of 24 mcg/kg/hr of IGF-1 to normal humans results in a 50% reduction in C-peptide but maintenance of euglycemia. Peripheral glucose uptake is increased at these infusion rates, and hepatic glucose production and free fatty acid levels are suppressed. Protein breakdown is also decreased. However, using lower infusion rates (5 mcg/kg/hr), which do not necessitate supplemental glucose to avoid hypoglycemia, there is no effect on protein breakdown. Insulin sensitivity is also enhanced, as assessed by insulin-to-glucose ratios measured during the IGF-1 infusion. IGF-1 has consistently suppressed insulin levels and resulted in more efficient glucose responsiveness to insulin.273 Since GH is also suppressed, inhibition of several of the known insulin-antagonist actions of GH may contribute to this change. IGF-1 also suppresses glucagon, and such suppression probably contributes to the enhanced insulin sensitivity that is observed during IGF-1 infusions.273
Administration of exogenous IGF-1 to catabolic subjects results in improvement in nitrogen balance. The degree of improvement is comparable to that achieved with GH administration.274 A study that used the same design (i.e., 6 days of a 50% caloric restriction) showed that concomitant administration of GH with a 12 mcg/kg/hr infusion of IGF-1 resulted in further enhancement of nitrogen retention compared to either treatment alone.97 GH inhibited the development of symptomatic hypoglycemia. Infusion of IGF-1 alone resulted in suppression of IGFBP-3 concentrations and suppression of acid-labile subunit, but administration of concomitant GH resulted in maintenance of normal levels of IGFBP-3 and ALS in plasma.97 This high level of the IGF-1/IGFBP-3 probably contributed to improved nitrogen balance. Other changes in IGFBPs also occur. IGF-1 alone increases IGFBP-2 concentrations threefold, suggesting that a larger fraction of the IGF-1 is bound to IGFBP-2 under these conditions, and thus it has a shorter half-life. A reduced anabolic response to IGF-1 alone may occur as a consequence of these changes in IGFBP profiles. Several other studies have suggested that maintenance of ternary-complex activity results in a better anabolic response. Administration of the IGF-1/IGFBP-3 complex to patients with severe burns resulted in increased protein synthesis rates.275 The mechanism of improvement may be multifactorial, since administration of the complex to thermally injured rats results in preservation of normal gut mucosa and improved nutrient absorption.276 Administration of IGF-1/IGFBP-3 to osteoporotic patients for 4 weeks following hip fracture showed that it was anabolic and improved bone density.277
Cholesterol is also lowered in response to IGF-1 infusion, as is potassium. Renal function improves, with an approximately 25% increase in glomerular filtration rate and renal blood flow. The fractional excretion of phosphate is decreased, which probably contributes to the antiphosphaturic effect noted in acromegaly. In addition to improvement in renal function, there is an improvement in the anemia that accompanies renal failure.278
GH selectively stimulates whole-body protein synthesis and has a lesser effect on inhibiting proteolysis. IGF-1 infusions at relatively high concentrations inhibit proteolysis but have no effect on protein synthesis. With prolonged administration of IGF-1 (e.g., 5 to 7 days given as a subcutaneous injection), there is no effect on proteolysis but a marked increase in protein synthesis, and the effects are indistinguishable from GH.279 Therefore, the mode of administration and the actual dose of IGF-1 given are important determinants of whether IGF-1 has an acute insulin-like effect on protein synthesis (e.g., inhibiting proteolysis) or a chronic GH-like effect in preferentially stimulating protein synthesis. The combination of GH plus IGF-1 has a greater effect on decreasing protein oxidation in GH-deficient subjects compared to either substance given alone.280 When catabolism is induced by administering high doses of glucocorticoids, IGF-1 has a significant effect on attenuating proteolysis and a small effect on increasing protein synthesis.281 These effects are less dramatic than those with GH.280 The effect of IGF-1 in enhancing insulin sensitivity appears to be preserved even in dexamethasone-treated patients.
Bone Metabolism
Short-term IGF-1 administration to normal subjects increases bone turnover, with a preferential effect on bone formation.282 Young women with anorexia nervosa and severe osteopenia also respond by increasing bone turnover, and there is a short-term anabolic effect.283 IGF-1 is also an effective stimulant of bone formation in men with osteoporosis.284 Patients with growth hormone deficiency also respond to IGF-1 with increased bone turnover.285 IGF-1 has been given to elderly subjects with osteoporosis and results in increased markers of bone resorption, such as pyridinoline crosslinks in the urine. However, there are also increases in markers of bone formation, indicating that bone turnover is stimulated. Serum IGF-1 concentrations correlated with the presence of vertebral factures in diabetic postmenopausal women and predicted low bone mass in premenopausal women.286 However, the net effect of long-term administration of IGF-1 on bone mineral content is unknown. Studies in rats have shown that administration of IGF-1 in combination with IGFBP-3 may be a potent stimulant of cortical bone formation, and a 4-month course of treatment in humans with osteoporosis with IGF-1/IGFBP-3 supported this conclusion.277 These enhanced effects of IGF-1/IGFBP-3 compared to IGF-1 alone may be due to the inability of IGF-1 administration alone to sustain high plasma IGF-1 concentrations over prolonged time periods.
Other Effects of IGF-1
IGF-1 in Diabetes
Studies in mice have shown that overexpression of IGFBP-3 results in reduced insulin action in muscle and fat. Similarly, deletion of IGF-1 expression in liver results in increased GH secretion and reduced insulin sensitivity.287 In contrast, overexpression of IGFBP-2 results in enhanced insulin sensitivity. This effect is probably direct, since the animals are refractory to the development of glucose intolerance even when fed a high-fat diet.288 When insulin sensitivity is assessed formally with the euglycemic hyperinsulinemic clamp method, IGF-1 administration to type 2 diabetics results in a substantial improvement in sensitivity to insulin.289 This also occurs in insulin-deficient diabetics and patients with extreme insulin resistance syndromes, including those involving mutations of the insulin receptor. IGF-1 infusion into type 1 diabetics lowers hepatic glucose output and increases peripheral glucose utlization.290 Preliminary studies have indicated that administration of IGF-1 to patients with severe insulin resistance results in long-term lowering of glucose and improved insulin sensitivity.291 Adolescents with type 1 diabetes who were treated for 4 weeks with subcutaneously administered IGF-1 had reduced insulin requirements and improved their metabolic control. These effects were attributed to suppression of the “dawn phenomenon.”292 Administration of IGF-1 to patients with type A extreme insulin resistance has resulted in improved metabolic control. Some studies, however, have not seen the same degree of improvement in patients with type A insulin resistance. Administration of IGF-1 to subjects with type 2 diabetes shows that it results in a 3.4-fold improvement in insulin resistance as assessed by direct measurement. More importantly, IGF-1 lowers hemoglobin A1C by 1.7% and improves glucose tolerance.293 Insulin concentrations are lowered in these patients, suggesting that a change in insulin sensitivity is the primary mechanism accounting for this improvement. Similarly, in type 1 diabetics, the requirement for exogenous insulin can be lowered by IGF-1 while maintaining good glycemic control.291–294 In a large (n = 208) group of type 2 diabetics who were treated with 4 different doses of IGF-1 for 3 months, the groups that received the two highest doses had a 1.6% reduction in hemoglobin A1C, indicating that long-term improvement in diabetic control is achievable with IGF-1.
Side effects have been noted both in normal subjects and in diabetics who have received high concentrations of IGF-1 for several weeks. These include parotid gland tenderness, subcutaneous edema, and a 10% increase in heart rate. In rare subjects, there is edema of the retina, and occasionally pseudotumor cerebri has been noted. Other unusual side effects include Bell’s palsy and severe myalgias. All of these side effects have been noted to be reversible and remit after stopping IGF-1.293–296 Administration of the combination of IGFBP-3 and IGF-1 to type 1 diabetics for 2 weeks resulted in a 48% reduction in insulin dosage and a 23% reduction in blood glucose, indicating improvement in insulin sensitivity.295 This combination also improved control in patients with type 2 diabetes and was associated with a reduction in side effects.296
Growth Hormone Insensitivity Syndromes
The types of genetic defects that lead to the growth hormone sensitivity syndrome have been expanded to include GH receptor mutations, Stat 5b mutations, ALS mutations, and mutations in the IGF-1 gene.297 Administration of IGF-1 to patients with GH insensitivity syndrome who had mutations of the GH receptor resulted in improvement in growth rates. Analysis of growth rates in nine such subjects who were treated for 1 year showed that they grew 7.5 cm in the first year, as compared to pretreatment growth rates of 4 cm/yr. Longer-term studies administering IGF-1 at 50 mcg/kg twice a day subcutaneously to patients with GH insensitivity syndrome have shown that the first-year growth velocity cannot be maintained in the second year, and the growth rates are reduced to 6 cm per year.298–300 This growth rate has been maintained for periods as long as 4 years in subjects who received IGF-1, so there appears to be a growth benefit which, if projected to adulthood, would result in significant improvement in final adult stature. However, the growth rates during the second through fifth years are not as robust as those in GH-deficient subjects who received GH during a similar interval. Hypoglycemia occurs occasionally in these patients but is usually avoidable by a dosage adjustment. Other side effects that have been noted with acute, high-dose administration of IGF-1 to adults have not been observed in these children. One child with a GH insensitivity syndrome did develop pseudotumor cerebri, which resolved while treatment was continued. Another troublesome feature that has been noted, however, is a coarsening of the facial features, particularly in those subjects who are receiving the treatment during initiation of adolescence. This effect appears to be more significant than that noted with GH administration during puberty. Evaluation of these patients 1 to 2 years after stopping IGF-1 shows that their coarse facial features resolve. Whether the suboptimal growth rates and coarsening of facial features are due to stimulation by IGF-1 in the absence of the direct actions of GH that are mediated through the GH receptor is unknown. Similar growth responses have been demonstrated using the IGF-1/IGFBP-3 combination.301
A single patient has been described in whom there was a mutation resulting in deletion of a major portion of the IGF1 gene.302 This resulted in severe growth retardation at birth that persisted into adulthood. Head circumference was also reduced. Administration of IGF-1 prior to epiphyseal fusion resulted in growth acceleration. It also resulted in improvement in insulin sensitivity. Four patients have been described with either a single allele or point mutation in the IGF-1 receptor.303–305 All cases resulted in growth retardation. One child with two point mutations responded to high doses of GH with an increase in growth.305
References
1. Salmon, WD, Jr., Daughaday, WH. A hormonally controlled serum factor which stimulates sulfate incorporation by cartilage in vitro. J Lab Clin Med. 1957;49:825.
2. Schoenle, E, Zapf, J, Humbel, RE, et al. Insulin-like growth factor I stimulates growth in hypophysectomized rats. Nature. 1982;296:252.
3. Murphy, LJ, Freisen, HG. Differential effects of estrogen and growth hormone on uterine and hepatic insulin like growth factor-I gene expression in ovariectomized and hypophysectomized rat. Endocrinology. 1988;122:325.
4. Holt, EC, Van Wyk, JJ, Lund, PK. Tissue and development specific regulation of a complex family of insulin-like growth factor I messenger ribonucleic acids. Mol Endocrinol. 1988;2:1077.
5. Carlsson, B, Carlsson, L, Billig, H. Estrus cycle dependent covariation of the insulin-like growth factor I (IGF-I) messenger ribonucleic acid and protein in rat ovary. Mol Cell Endocrinol. 1989;64:271.
6. Rotwein, P, Bichell, DP, Kikuchi, K. Multifactorial regulation of IGF-I gene expression. Mol Reprod Dev. 1993;35:358.
7. Hepler, JE, VanWyk, JJ, Lund, PK. Different half-lives of insulin-like growth factor-I mRNA that differ in length of 3′ untranslated sequence. Endocrinology. 1990;127:155.
8. Bayne, ML, Applebaum, J, Chicchi, GG, et al. The roles of tyrosines 24, 31, and 60 in the high-affinity binding of insulin-like growth factor-I to the type 1 insulin-like growth factor receptor. J Biol Chem. 1990;265:15648.
9. Bayne, ML, Applebaum, J, Underwood, D, et al. The C region of human insulin-like growth factor (IGF) I is required for high-affinity binding to the type I IGF receptor. J Biol Chem. 1989;264:11004.
10. Vajdos, FF, Ultsch, M, Schaffer, ML, et al. Crystal structure of human insulin-like growth factor-1: detergent binding inhibits binding protein interactions. Biochem. 2001;40:11022.
11. Zeslawski, W, Beisel, HG, Kamionka, M, et al. The interaction of insulin-like growth factor-I with the N-terminal domain of IGFBP-5. EMBO J. 2001;20:3638.
12. Shaffer, ML, Deshayes, K, Nakamura, G, et al. Complex with a phage display-derived peptide provides insight into the function of insulin-like growth factor I. Biochem. 2003;42:9324.
13. Gauguin, L, Delaine, Ca, Alvino, CL, et al. Alanine scanning of a putative receptor binding surface of insulin-like growth factor-I. J Biol Chem. 2008;283:20821.
14. Clemmons, DR, Dehoff, MH, Busby, WH, et al. Competition for binding to insulin-like growth factor (IGF) binding protein-2, 3, 4, and 5 by the IGFs and IGF analogs. Endocrinology. 1992;131:890.
15. Siwanowicz, I, Popowicz, GM, Wisniewska, M, et al. Structural basis for the regulation of insulin-like growth factors by IGF binding proteins. Structure (Camb). 2005;103:155.
16. Gauguin, L, Klaproth, B, Sajid, W, et al. Structural basis for the lower affinity of the insulin-like growth factors for the insulin receptor. J Biol Chem. 2008;283:2604.
17. Kaleko, M, Rutter, WJ, Miller, D. Overexpression of the human insulin-like growth factor-I receptor promotes ligand dependent neoplastic transformation. Mol Cell Biol. 1990;10:464.
18. Werner, H, Roberts, CT, Jr. The IGFI receptor gene: a molecular target for disrupted transcription factors. Genes Chrom Can. 2003;36:113.
19. Du, J, Meng, XP, Delafontaine, P. Transcriptional regulation of the insulin-like growth factor-I receptor gene: Evidence for protein kinase C dependent and independent pathways. Endocrinology. 1996;138:1378.
20. Abbott, AM, Bueno, R, Pedrin, MT, et al. Insulin-like growth factor-I receptor gene structure. J Biol Chem. 1992;267:10759.
21. LeRoith, D, Werner, H, Beitner-Johnson, D, et al. Molecular and cellular aspects of the insulin-like growth factor-I receptor. Endocrine Rev. 1995;16:143.
22. Lou, M, Garrett, TP, McKern, NM, et al. The first three domains of the insulin receptor differ structurally from the insulin-like growth factor 1 receptor in the regions governing ligand specificity. Proc Natl Acad Sci U S A. 2006;103:12429.
23. Keyhanfar, M, Booker, GW, Whittaker, J, et al. Precise mapping of an IGF-I binding site on the IGF-1R. Biochem J. 2007;401:269.
24. Lawrence, MC, McKern, NM, Ward, CW. Insulin receptor structure and its implications for the IGF-1 receptor. Curr Opin Struct Biol. 2007;17:699.
25. Whittaker, J, Groth, AV, Mynarcik, DC, et al. Alanine scanning mutagenesis of a type I insulin-like growth factor receptor ligand binding site. J Biol Chem. 2001;276:43980.
26. Wu, J, Li, W, Craddock, BP, et al. Small-molecule inhibition and activation-loop trans-phosphorylation of the IGF1 receptor. EMBO J. 2008;27:1985.
27. Pautsch, A, Zoephel, A, Ahorn, H, et al. Crystal structure of bisphosphorylated IGF-1 receptor kinase: insight into domain movements upon kinase activation. Structure. 2001;9:955.
28. Yamasaki, H, Pager, D, Gebremedhin, S, et al. Human insulin-like growth factor-I receptor 950 tyrosine is required for somatotroph growth factor signal transduction. J Biol Chem. 1992;267:20953.
29. Soos, MA, Field, CE, Siddle, K. Purified hybrid insulin/insulin-like growth factor-I receptors bind insulin-like growth factor-I but not insulin with high affinity. Biochem J. 1993;290:419.
30. Pandini, G, Frasca, F, Mineo, R, et al. Insulin/insulin-like growth factor I hybrid receptors have different biological characteristics depending on the insulin receptor isoform involved. J Biol Chem. 2002;277:39684.
31. Rotem-Yehudar, R, Galperin, E, Horowitz, M. Association of insulin-like growth factor 1 receptor with EHD1 and SNAP29. J Biol Chem. 2001;276:33054.
32. Navab, R, Chevet, E, Authier, F, et al. Inhibition of endosomal insulin-like growth factor-I processing by cysteine proteinase inhibitors blocks receptor-mediated functions. J Biol Chem. 2001;276:13644.
33. Bohula, EA, Salibury, AJ, Sohail, M, et al. The efficacy of small interfering RNAs targeted to the type 1 insulin-like growth factor receptor (IGF1R) is influenced by secondary structure in the IGF1R transcript. J Biol Chem. 2003;278:15991.
34. Li, S, Resnicoff, M, Boneya, R. Effect of mutations at serines 1280–1281 on the mitogenic and transforming activities of the IGF-I receptor. J Biol Chem. 1996;271:12254.
35. DeAngelis, T, Ferber, A, Baserga, R. The insulin-like growth factor I receptor is a requirement for the mitogenic and transforming activities of the platelet derived growth factor receptor. J Cell Physiol. 1995;164:214.
36. Sell, C, Rubini, R, Rubin, R, et al. Simian virus 40 large tumor antigen is unable to transform mouse embryo fibroblasts lacking type I insulin like growth factor receptor. Proc Natl Acad Sci U S A. 1993;90:11217.
37. Liu, JP, Baker, J, Perkins, AS, et al. Mice carrying null mutations of the genes encoding insulin like growth factor I (IGF-I) and type 1 IGF receptor (IGF/r). Cell. 1993;75:73.
38. D’Mello, SR, Galli, C, Ciott, T, et al. Induction of apoptosis in cerebellar granule neurons by low potassium: inhibition of death by insulin-like growth factor-I and cAMP. Proc Natl Acad Sci U S A. 1993;90:10989.
39. Rubin, R, Baserga, R. Biology of disease: Insulin-like growth factor-I receptor: its role in cell proliferation, apoptosis, and tumorigenicity. Lab Invest. 1995;13:311.
40. Faria, TN, Blakesley, VA, Kato, H, et al. Role of the carboxy-terminal domain of the insulin and insulin-like growth factor-I receptors in receptor function. J Biol Chem. 1994;269:13922.
41. Sun, XJ, Wang, LM, Zhang, Y, et al. Role of IRS-2 in insulin and cytokine signaling. Nature. 1995;377:173.
42. Pete, G, Fuller, Cr, Oldham, JM, et al. Postnatal growth responses to insulin-like growth factor I in insulin receptor substrate-1-deficient mice. Endocrinol. 1999;140:5478.
43. Myers, MJ, White, MF. The IRS-1 signaling system. Trends in Biological Sciences. 1994;19:289.
44. Myers, MG, Sun, XJ, Cheatham, B, et al. IRS-I is a common element in insulin and insulin-like growth factor I signalling to the phosphatidylinositol 5′-kinase. Endocrinology. 1993;132:1421.
45. Clemmons, DR, Maile, LA. Interaction between insulin-like growth factor-I receptor and alphaVbeta3 integrin linked signaling pathways: cellular responses to change sin multiple signaling inputs. Mol Endocrinol. 2004;19:1.
46. Kiely, PA, Sant, A, O’Connor, R. RACK1 is an insulin-like growth factor 1 (IGF-1) receptor-interacting protein that can regulate IGF-I mediated Akt activation and protection from cell death. J Biol Chem. 2002;277:22581.
47. Giovannone, B, Lee, E, Laviola, L, et al. Two novel proteins that are linked to insulin-like growth factor (IGF-I) receptors by the Grb10 adapter and modulate IGF-I signaling. J Biol Chem. 2003;278:31564.
48. Dalle, S, Ricketts, W, Imamura, T, et al. Insulin and insulin-like growth factor I receptors utilize different G protein signaling components. J Biol Chem. 2001;276:15688.
49. Myers, MJ, White, MF. Insulin signal transduction and the IRS proteins. Ann Rev Pharmacol. 1996;36:615.
50. Lauzier, MC, Page, EL, Michaud, MD, et al. Differentiation regulation of hypoxia-inducible factor-1 through receptor tyrosine kinase transactivation in vascular smooth muscle cells. Endocrinol. 2007;148:4023–4031.
51. Holzman, JL, Liu, L, Duke, BJ, et al. Transactivation of the IGF-1R aldosterone. Am J Physiol Renal Physiol. 2006;292:F1219.
52. Song, RX, Zhang, Z, Chen, Y, et al. Estrogen signaling via a linear pathway involving insulin-like growth factor I receptor, matrix metalloproteinases, and epidermal growth factor receptor to activate mitogen-activated protein kinase in MCF-7 breast cancer cells. Endocrinol. 2007;148:4091.
53. Verras, M, Sun, Z. 2005 Beta-Catenin is involved in insulin-like growth factor 1-mediated transactivation of the androgen receptor. Mol Endocrinol. 2005;19:391.
54. El-Shewy, HM, Kelly, FL, Barki-Harrington, L, et al. Ectodomain shedding-dependent transactivation of epidermal growth factor receptors in response to insulin-like growth factor type 1. Mol Endocrinol. 2004;18:2727.
55. Venters, HD, Tang, Q, Liu, Q, et al. A new mechanism of neurodegeneration: a proinflammatory cytokine inhibits receptor signaling by a survival peptide. Proc Natl Acad Sci U S A. 1999;96:9879.
56. Brown, J, Delaine, C, Zaccheo, OJ, et al. Structure and functional analysis of the IGF-II/IGF2R interaction. EMBO J. 2008;27:265.
57. Williams, C, Rezgui, D, Prince, SN, et al. Structural insights into the interaction of insulin-like growth factor 2 with IGF2R domain 11. Structure. 2007;15:1065.
58. Lau, MM, Stewart, CHE, Liu, Z, et al. Loss of imprinted IGF-II cation independent mannose 6 phosphate receptor results in fetal overgrowth and perinatal lethality. Gene Dev. 1994;8:2953.
59. McKinnon, T, Chakraborty, C, Gleeson, L, et al. Stimulation of human extravillous trophoblast migration by IGF-II is mediated by IGF type 2 receptor involving inhibitory G protein(s) and phosphorylation. J Clin Endocrinol Metab. 2001;86:3665.
60. Jones, JI, Clemmons, DR. Insulin like growth factor and their binding proteins: biologic actions. Endocrine Rev. 1995;16:3.
61. Rechler, MM. Insulin like growth factor binding proteins. Vitamins and Hormones. 1993;47:1.
62. Imai, Y, Moralez, A, Andag, C, et al. Substitutions for hydrophobic amino acids in the N-terminal domains of IGFBP-3 and -5 markedly reduce IGF-I binding and alter their biologic actions. J Biol Chem. 2000;275:18188.
63. Bach, LA, Headley, SJ, Norton, RS. IGF-binding proteins-the pieces are falling into place. Trends Endocrinol Metab. 2005;16:228.
64. Kuang, Z, Yao, S, McNeil, KA, et al. Cooperativity of the N- and C-terminal domains of insulin-like growth factor (IGF) binding protein 2 in IGF binding. Biochem. 2007;46:13720.
65. Jones, JL, Gockerman, A, Busby, WH, Jr., et al. Insulin-like growth factor binding protein 1 stimulates cell migration and binds to the α5β1 integrin by means of its Arg-Gly-Asp sequence. Proc Natl Acad Sci U S A. 1993;90:10553.
66. Booth, BA, Boes, M, Dake, BL, et al. Structure function relationships in the heparin binding C-terminal region of insulin-like growth factor binding protein-3. Growth Regul. 1996;6:206.
67. Durham, SK, Keifer, MR, Riggs, BL, et al. Regulation of insulin like growth factor binding protein-4 by a specific insulin like growth factor binding protein-4 protease in normal human osteoblast like cells. Implications on human cell physiology. J Bone Mineral Res. 1994;9:111.
68. James, PL, Jones, SB, Busby, WH, et al. IGF binding protein-5 is expressed in myoblast differentiation and is highly conserved. J Biol Chem. 1993;268:22305.
69. Clemmons, DR. Insulin-like growth factor binding proteins and their role in controlling IGF actions. Cytokine Growth Factor Rev. 1997;8:45.
70. Martin, JL, Willetts, KE, Baxter, RC. Purification and properties of a novel insulin-like growth factor-II binding protein from transformed human fibroblasts. J Biol Chem. 1990;265:4124.
71. Aimaretti, G, Boschetti, M, Corneli, G, et al. Normal age-dependent values of serum insulin growth factor-1: results from a healthy Italian population. J Endocrinol Invest. 2008;31:455.
72. Hong, Y, Pedesen, NL, Brismar, K, et al. Quantitative genetic analyses of insulin like growth factor I (IGF-I), IGF binding protein-1 and insulin levels in middle-aged and elderly twins. J Clin Endocrinol Metab. 1996;81:1791.
73. Vaessen, N, Heutink, P, Janssen, JA, et al. A polymorphism in the gene for IGF-I: functional properties and risk for type 2 diabetes and myocardial infarction. Diabetes. 2001;50:637.
74. Juul, A, Skakkebaek, NE. Prediction of the outcome of growth hormone provocative testing in short children by measurement of serum levels of insulin-like growth factor I and insulin-like growth factor binding protein 3. J Pediatr. 1997;130:197–204.
75. Ranke, MB, Schweizer, R, Elmlinger, MW, et al. Significance of basal IGF-I, IGFBP-3 and IGFBP-2 measurements in the diagnostics of short stature in children. Horm Res. 2000;54:60.
76. Juul, A, Dalgaard, P, Blum, WF, et al. Serum levels of insulin-like growth factor (IGF) binding protein-3 (IGFBP-3) in healthy infants, children, and adolescents: the relation to IGF-I, IGF-II, IGFBP-1, IGFBP-2, age, sex, body mass index, and pubertal maturation. J Clin Endocrinol Metab. 1995;80:2534.
77. Mitchell, H, Dattani, MT, Naduri, V, et al. Failure of IGF-I and IGFBP-3 to diagnose growth hormone insufficiency. Arch Dis Child. 1999;80:443.
78. Jorge, AA, Souza, SC, Arnhold, IJ, et al. Poor reproducibility of IGF-I and IGF binding protein-3 generation test in children with short stature and normal coding region of the GH receptor gene. J Clin Endocrinol Metab. 2002;87:469.
79. Jorgensen, JO, Hansen, TK, Conceicaso, FL, et al. Short-term tools to measure responsiveness to growth hormone replacement. Horm Res. 2001;55(suppl 2):40.
80. Clemmons, DR, Underwood, LE, Ridgeway, EC. Evaluation of acromegaly by radioimmunoassay of somatomedin-C. N Engl J Med. 1979;301:1138.
81. Melmed, S. Acromegaly. N Engl J Med. 1990;322:966.
82. Stoffel-Wagner, B, Springer, W, Bidlingmaier, F, et al. A comparison of different methods for diagnosing acromegaly. Clin Endocrinol. 1997;46:531.
83. Clemmons, DR. Clinical utility of measurements of insulin-like growth factor 1. Nat Clin Pract Endocrinol Metab. 2006;2:436.
84. Noel, M, Chevenne, D, Porquet, D. Utility of insulin-like growth factor-I and its binding protein assays. Curr Opin Clin Nutr Metab Care. 2001;4:399.
85. Thissen, JP, Underwood, LE, Ketelslegers, JM. Regulation of insulin-like growth factor-I in starvation and injury. Nutr Rev. 1999;57:167.
86. Tonshoff, B, Kiepe, D, Ciarmatori, S. Growth hormone/insulin-like growth factor system in children with chronic renal failure. Pediatr Nephrol. 2005;20:279.
87. De Palo, EF, Bassanello, M, Lancerin, F, et al. GH/IGF system, cirrhosis and liver transplantation. Clin Chim Acta. 2001;310:31.
88. Bereket, A, Lang, CH, Blethen, SL, et al. Effect of insulin on the insulin-like growth factor system in children with new onset insulin dependent diabetes. J Clin Endocrinol Metab. 1995;80:1312.
89. Morrow, LA, O’Brien, MB, Moller, DE, et al. Recombinant human insulin-like growth factor-I therapy improves glycemic control and insulin action in the type A syndrome of severe insulin resistance. J Clin Endocrinol Metab. 1994;79:205.
90. Baxter, RC, Martin, JL. Radioimmunoassay of growth hormone dependent insulin-like growth factor binding protein in human plasma. J Clin Invest. 1986;78:1504.
91. Blum, WF, Albertsson-Wikland, K, Rosberg, S, et al. Serum levels of insulin-like growth factor I (IGF-I) and IGF binding protein 3 reflect spontaneous growth hormone secretion. J Clin Endocrinol Metab. 1993;76:1610.
92. Leogney, SR, Baxter, RC, Carrerato, T, et al. Structure and functional expression of acid-labile subunit of the insulin-like growth factor binding protein complex. Mol Endocrinol. 1992;6:870.
93. Grinspoon, S, Clemmons, DR, Swearingen, B, et al. Serum insulin-like growth factor-binding protein-3 levels in the diagnosis of acromegaly. J Clin Endocrinol Metab. 1995;80:927.
94. Laursen, T, Flyvbjerg, A, Jorgensen, JO, et al. Stimulation of the 150-kilodalton insulin-like growth factor binding protein-3 ternary complex by continuous and pulsatile patterns of growth hormone (GH) administration in GH-deficient patients. J Clin Endocrinol Metab. 2000;85:4310.
95. Pfeilschifter, J, Scheidt-Nave, C, Leidig-Bruckner, G, et al. Relationship between circulating insulin-like growth factor components and sex hormones in a population based sample of 50- to 80-year-old men and women. J Clin Endocrinol Metab. 1996;81:2534.
96. Miell, JP, Taylor, AM, Zini, M, et al. Effects of hypothyroidism and hyperthyroidism on insulin-like growth factors (IGFs) and growth hormone- and IGF binding proteins. J Clin Endocrinol Metab. 1993;76:950.
97. Kupfer, SR, Underwood, LE, Baxter, RC, et al. Enhancement of the anabolic effects of growth hormone and insulin like growth factor-I by the use of both agents simultaneously. J Clin Invest. 1993;91:391.
98. Maile, LA, Holly, JM. Insulin-like growth factor binding protein (IGFBP) proteolysis: occurrence, identification, role and regulation. Growth Horm IGF Res. 1999;9:85–95.
99. Olausson, H, Lof, M, Brismar, K, et al. Longitudinal study of the maternal insulin-like growth factor system before, during and after pregnancy in relation to fetal and infant weight. Horm Res. 2008;69:99.
100. Clemmons, DR, Sleevi, M, Busby, WH, Jr. Recombinant, nonglycosylated human insulin-like growth factor-binding protein-3 (IGFBP-3) is degraded preferentially after administration to type II diabetics, resulting in increased endogenous glycosylated IGFBP-3. J Clin Endocrinol Metab. 2005;90:6561.
101. Blum, WF, Brier, BH. Radioimmunoassays for IGFs and IGFBPs. Growth Regulation. 1994;4:11.
102. Ooi, GT, Orlowski, CC, Brown, AL, et al. Different tissue distribution and hormonal regulation of messenger RNAs encoding rat insulin-like growth factor binding proteins-1 and 2. Mol Endocrinol. 1990;4:321.
103. Daughaday, WH, Trivedi, B, Baxter, RC. Serum “big” insulin-like growth factor-II from patients with tumor hypoglycemia lacks normal E-domain O-linked glycosylation, a possible determinant of normal propeptide processing. Proc Natl Acad Sci U S A. 1993;90:5283.
104. Bereket, A, Lang, CH, Wilson, TA. Alterations in the growth hormone-insulin-like growth factor axis in insulin dependent diabetes mellitus. Horm Metab Res. 1999;31:172–181.
105. Smith, WJ, Underwood, LE, Clemmons, DR. Effects of Caloric or Protein Restriction on Insulin-Like Growth Factor-I (IGF-I) and IGF-Binding Proteins in Children and Adults. J Clin Endocrinol Metab. 1995;80:443.
106. Straus, DS, Takemoto, CD. Effect of dietary protein deprivation insulin-like growth factor IGF-I and II, IGF binding protein-2 and serum albumin gene expression in the rat. Endocrinology. 1990;127:1849.
107. Ooi, GT, Tseng, LY, Tran, MQ, et al. Insulin rapidly decreases insulin-like growth factor-binding protein-1 gene transcription in streptozotocin-diabetic rats. Mol Endocrinol. 1992;6:2219.
108. Busby, WH, Snyder, DK, Clemmons, DR. Radioimmunoassay of a 26,000 dalton plasma insulin like growth factor binding protein: control by nutritional variables. J Clin Endocrinol Metab. 1988;67:1225.
109. Gan, L, Pan, H, Unterman, TG. Insulin response sequence-dependent and independent mechanisms mediate effects of insulin on glucocorticoid-stimulated insulin-like growth factor binding protein-1 promoter activity. Endocrinol. 2005;146:4274.
110. Bar, RS, Boes, M, Clemmons, DR, et al. Insulin differentially alters transcapillary movement of intravascular IGFBP-1, IGFBP-2 and endothelial cell IGF binding proteins in rat heart. Endocrinology. 1990;127:497.
111. Westwood, M, Gibson, JM, Williams, AC, et al. Hormonal regulation of circulating insulin-like growth factor-binding protein-1 phosphorylation status. J Clin Endocrinol Metab. 1995;80:3520.
112. Lewitt, MS, Denyer, GS, Cooney, GJ, et al. Insulin-like growth factor binding protein-1 modulates blood glucose levels. Endocrinol. 1991;129:2254.
113. Davies, SC, Wass, JAH, Ross, RJM, et al. Induction of a specific protease for insulin-like growth factor binding protein-3 in the circulation during severe illness. J Endocrinol. 1991;130:469.
114. Scharla, SH, Strong, DD, Rosen, C, et al. 1,25-Dihydroxyvitamin D3 increases secretion of insulin-like growth factor binding protein-4 (IGFBP-4) by human osteoblast-like cells in vitro and elevates IGFBP-4 serum levels in vivo. J Clin Endocrinol Metab. 1993;77:1190–1197.
115. Ehrnborg, C, Ohisson, C, Mohan, S, et al. Increased serum concentration of IGFBP-4 and IGFBP-5 in healthy adults during one month’s treatment with supraphysiological doses of growth hormone. Growth Horm IGF Res. 2007;17:234.
116. Rooman, RP, De Beeck, LO, Martin, M, et al. IGF-I, IGF-II, free IGF-I and IGF-binding proteins-2 to -6 during high dose estrogen treatment in constitutionally tall girls. Eur J Endocrinol. 2002;146:823.
117. Baxter, RC. Changes in the IGF-IGFBP axis in critical illness. Best Pract Res Clin Endocrinol Metab. 2001;15:421.
118. Houang, M, Cabrol, S, Perin, L, et al. Insulin-like growth factor-I (IGF-I), insulin-like growth factor binding proteins (IGFBP) and insulin-like growth factor type I receptor in children with various status of chronic renal failure. Growth Horm IGF Res. 2000;10:332.
119. Han, VKM, D’Ercole, AJ, Lund, PK. Cellular location of somatomedin (insulin-like growth factor) messenger RNA in the human fetus. Science. 1987;236:193.
120. Roberts, CT, Lasky, SR, Lowe, WL, et al. Molecular coding of rat insulin-like growth factor-I complementary DNA: Differential messenger RNA processing of regulation by growth hormone in extrahepatic tissue. Mol Endocrinol. 1987;1:243.
121. Isgaard, J, Nilsson, A, Vikma, A, et al. Growth hormone regulates the level of IGF-I mRNA in rat growth plate. Endocrinology. 1988;122:1515.
122. Grundberg, E, Brandstrom, H, Lam, KC, et al. Systemic assessment of the human osteoblast transcriptome in resting and induced primary cells. Physiol Genomics. 2008;33:301–311.
123. Silver, DM, Fudo, H, Halperin, D, et al. Differential expression of insulin like growth factor I (IGF-I) and IGF-II messenger ribonucleic acid in growing rat bone. Endocrinology. 1993;132:1158.
124. McCarthy, TL, Ji, C, Centrella, M, et al. Links among growth factors. Hormones and nuclear factors with essential roles in bone formation. Crit Rev Oral Biol Med. 2000;11:409.
125. Mendez-Davila, C, Garcia-Moreno, C, Trubi, C, et al. Effects of 17beta-estradiol, tamoxifen and raloxifene on the protein and mRNA expression of interleukin-6, transforming growth factor-beta1 and insulin-like growth factor-1 in primary human osteoblast cultures. J Endocrinol Invest. 2004;27:904.
126. Rosen, CJ. The cellular and clinical parameters of anabolic therapy for osteoporosis. Crit Rev Eukaryot Gene Expr. 2003;13:25.
127. Sivan, B, Lilos, P, Laron, Z. Effects of insulin-like growth factor-I deficiency and replacement therapy on the hematopoietic system in patients with Laron syndrome (primary growth hormone insensitivity). J Pediatr Endocrinol Metab. 2003;16:509.
128. Davidson, Tr, Chamberlain, CS, Bridges, TS, et al. Effect of follicle size on in-vitro production of steroids and insulin-like growth factor (IGF-I), IGF-II, and the IGF binding proteins by equine ovarian granulosa cells. Biol Reprod. 2002;66:1640.
129. Moyano, P, Rotwein, P. Mini-review: estrogen action in the uterus and insulin-like growth factor-I. Growth Horm IGF Res. 2004;14:431.
130. D’Ercole, AJ, Ye, P. Expanding the mind: IGF-I and brain development. Endocrinol epub. July 31, 2008.
131. Cardona-Gomez, GP, Mendez, P, DonCarlos, LL, et al. Interactions of estrogen and insulin-like growth factor-I in the brain: molecular mechanisms and functional implications. J Steroid Biochem Mol Biol. 2002;83:211.
132. Adamo, ML, Farrar, RP. Resistance training, and IGF involvement in the maintenance of muscle mass during the aging process. Ageing Res Rev. 2006;5:310.
133. Edwall, D, Schalling, M, Jennische, E, et al. Induction of insulin-like growth factor I messenger ribonucleic acid during regeneration of rat skeletal muscle. Endocrinol. 1989;124:820.
134. Paul, AC, Rosenthal, N. Different modes of hypertrophy in skeletal muscle fibers. J Cell Biol. 2002;156:751.
135. McMullen, JR, Izumo, S. Role of the insulin-like growth factor I (IGFI)/phosphoinositide-3-kinase (PI3K) pathway mediating physiological cardiac hypertrophy. Novartis Found Symp. 2006;274:90.
136. Cooper, SA, Whaley-Connell, A, Habibi, J, et al. Renin-angiotensin-aldosterone system and oxidative stress in cardiovascular insulin resistance. Am J Physiol Heart Circ Physiol. 2007;293:2009.
137. Cercek, B, Fishbein, MC, Forrester, JS, et al. Induction of insulin-like growth factor I messenger RNA in rat aorta after balloon denudation. Circ Res. 1990;66:1755.
138. Eleswarapu, S, Gu, Z, Jiang, H. Growth hormone regulation of insulin-like growth factor-I gene expression may be mediated by multiple distal signal transducer and activator of transcription 5 binding sites. Endocrinol. 2008;249:2230.
139. Lowe, WL, Adamo, M, Werner, H, et al. Regulation by fasting of insulin-like growth factor I and its receptor: Effects on gene expression and binding. J Clin Invest. 1989;84:619.
140. Butler, ST, Marr, AL, Pelton, Sh, et al. Insulin restores GH responsiveness during lactation-induced negative energy balance in dairy cattle: effects on expression of IGF-I and GH receptor. J Endocrinol. 2003;176:205.
141. Rotwein, P, Pollack, KM, Watson, M, et al. Insulin-like growth factor gene expression during rat embryonic development. Endocrinology. 1987;121:2141.
142. Hirschberg, R. The physiology and pathophysiology of IGF-I in the kidney. Adv Exp Med Biol. 1993;343:345.
143. Flyvbjerg, A, Bennett, WF, Rasch, R, et al. Compensatory renal growth in uninephrectomized adult mice is growth hormone dependent. Kidney Int. 1999;56:2048.
144. Dupont, J, Pierre, A, Froment, P, et al. The insulin-like growth factor axis in cell cycle progression. Horm Metab Res. 2003;35:740.
145. Travali, S, Reiss, K, Ferber, A, et al. Constitutively expressed c-myb abrogates the requirement for insulin-like growth factor-I in 3T3 fibroblasts. Mol Cell Biol. 1991;11:731.
146. Coppola, D, Ferber, A, Miura, A, et al. A functional insulin-like growth factor I receptor is required for the mitogenic and transforming activities of the epidermal growth factor receptor. Mol Cell Biol. 1994;14:4588.
147. Pietrzkowski, Z, Lammers, R, Carpenter, G, et al. Constitutive expression of insulin-like growth factor-I and insulin-like growth factor-I receptor abrogates all requirements for exogenous growth factors. Cell Growth and Differentiation. 1992;3:199.
148. Lowe, WL. Biologic actions of the insulin-like growth factors. In: LeRoith D, ed. Insulin-like Growth Factors: Molecular and Cellular Aspects. Boca Raton: CRC Press; 1991:49.
149. Nilsson, O, Marino, R, De Luca, F, et al. Endocrine regulation of the growth plate. Horm Res. 2005;64:157.
150. Loeser, RF, Shanker, G. Autocrine stimulation by insulin-like growth factor 1 and insulin like growth factor 2 mediates chondrocyte survival in vitro. Arthritis Rheum. 2000;43:1552.
151. Madry, H, Zurakowski, D, Trippel, SB. Overexpression of human insulin-like growth factor-I promotes new tissue formation in an ex vivo model of articular chrondrocyte transplantation. Gene Ther. 2001;8:1443.
152. Yakar, S, Rosen, C. From mouse to man: redefining the role of insulin-like growth factor-I in the acquisition of bone mass. Pro Soc Expo Biol Med. 1998;228:245.
153. Zhang, M, Xuan, S, Bouxsein, ML, et al. Osteoblast-specific knockout of the insulin-like growth factor (IGF) receptor gene reveals an essential role of IGF signaling in bone matrix mineralization. J Biol Chem. 2002;277:44005.
154. Zhao, G, Monier-Faugere, MC, Langub, MC, et al. Targeted overexpression of insulin-like growth factor I in osteoblasts of transgenic mice: increased trabecular bone volume without increased osteoblast proliferation. Endocrinol. 2000;141:2674.
155. Wang, Y, Nishida, S, Boudignon, BM, et al. IGF-I receptor is required for the anabolic actions of parathyroid hormone on bone. J Bone Miner Res. 2007;22:1329.
156. Sjogren, K, Sheng, M, Moverare, S, et al. Effects of liver-derived insulin-like growth factor I on bone metabolism in mice. J Bone Miner Res. 2002;17:1977.
157. Takahashi, T, Ishida, K, Itoh, K, et al. IGF-I gene transfer by electroporation promotes regeneration in a muscle injury model. Gene Ther. 2003;10:612.
158. Banu, J, Wang, L, Kalu, DN. Effects of increased muscle mass on bone in male mice overexpressing IGF-I in skeletal muscles. Calcif Tissue Int. 2003;73:196.
159. Welsh, S, Plank, D, Witt, S, et al. Cardiac-specific IGF-1 expression attenuates dilated cardiomyopathy in tropomodulin-overexpressing transgenic mice. Circ Res. 2002;90:641.
160. Zhu, B, Zhao, G, Witte, DP, et al. Targeted overexpression of IGF-I in smooth muscle cells of transgenic mice enhances neointimal formation through increased proliferation and cell migration after intraarterial injury. Endocrinol. 2001;142:3598.
161. Cao, Y, Gunn, AJ, Bennet, L, et al. Insulin-like growth factor (IGF)-1 suppresses oligodendrocyte caspase-3 activation and increases glial proliferation after ischemia in near-term fetal sheep. J Cereb Blood Flow Metab. 2003;23:739.
162. Homma, K, Koriyama, Y, Mawatari, K, et al. Early down-regulation of IGF-I decides the fate of rat retinal ganglion cells after optic nerve injury. Neurochem Int. 2007;50:741.
163. Vicario-Abejon, C, Yusta-Boyo, MJ, Fernandez-Moreno, C, et al. Locally born olfactory bulb stem cells proliferate in response to insulin-related factors and require endogenous insulin-like growth factor-I for differentiation into neurons and glia. J Neurosci. 2003;23:895.
164. Ye, P, Xing, YZ, Dai, ZH, et al. In vivo actions of insulin-like growth factor-I (IGF-I) on cerebellum development in transgenic mice: Evidence that IGF-I increases proliferation of granule cell progenitors. Dev Brain Res. 1996;95:44.
165. Bateman, JM, McNeill, H. Insulin/IGF signaling in neurogenesis. Cell Mol Life Sci. 2006;63:1701.
166. Muta, K, Krontes, B. Apoptosis of human erythroid colony forming cells is decreased by stem cell factor and insulin-like growth factor-I as well as erythropoietin. J Cell Physiol. 1993;156:264.
167. Rodriguez-Tarduchy, G, Collins, MKL, Garcia, I, et al. Insulin-like growth factor I inhibits apoptosis in IL-3 dependent hematopoietic cells. J Immunol. 1992;149:535.
168. Florini, JR, Ewton, DZ, Root, SL. IGF-I stimulates terminal myogenic differentiation by induction of myogenin gene expression. Mol Endocrinol. 1991;5:718.
169. Palmer, S, Myerson, G, Lindgren, E, et al. Insulin-like growth factor-I shifts from promoting cell division to potentiating maturation during normal differentiation. Proc Natl Acad Sci USA. 1991;88:9994.
170. Khamsi, F, Roberge, S, Yavas, Y, et al. Recent discoveries in physiology of insulin-like growth factor-1 and its interaction with gonadotropins in folliculogenesis. Endocrine. 2001;16:151–165.
171. Mesiano, S, Jaffe, RB. Role of growth factors in the developmental regulation of the human fetal adrenal cortex. Steroids. 1997;62:62.
172. Yamasaki, H, Prager, D, Gebremedhin, S, et al. Insulin-like growth factor-I (IGF-I) attenuation of growth hormone is enhanced by overexpression of pituitary IGF-I receptors. Mol Endocrinol. 1991;5:890.
173. Duan, C, Hawes, S, Prevette, T, et al. Insulin like growth factor-I (IGF-I) stimulates IGF binding protein-5 synthesis through transcriptional activation of the gene in aortic smooth muscle cells. J Biol Chem. 1996;271:4280.
174. Spangenburg, EE, Bowles, DK, Booth, FW. IGF-I induced transcriptional activity of the skeletal {alpha} action gene is regulated by signaling mechanisms linked to voltage-gated calcium channels during myoblast differentiation. Endocrinol. 2003;145:2054.
175. Dupont, J, Fernandez, AM, Glackin, CA, et al. Insulin-like growth factor 1 (IGF-1) induced twist expression is involved in the anti-apoptotic effects of the IGF-I receptor. J Biol Chem. 2001;276:26699.
176. Mulligan, C, Rochford, J, Denyer, G, et al. Microarray analysis of insulin and insulin-like growth factor-1 (IGF-1) receptor signaling reveals the selective up-regulation of the mitogen heparin-binding EGF-like growth factor by IGF-1. J Biol Chem. 2002;277:42480.
177. Grohmann, M, Foulstone, E, Welsh, G, et al. Isolation and validation of human prepubertal skeletal muscle cells: maturation and metabolic effects of IGF-I, IGFBP-3 and TNFalpha. J Physiol. 2005;568:229.
178. Zheng, B, Clemmons, DR. Blocking ligand occupancy of the αVβ3 integrin inhibits IGF-I signaling in vascular smooth muscle cells. Proc Natl Acad Sci U S A. 1998;95:11217.
179. Samani, AA, Yakar, S, LeRoith, D, et al. The role of the IGF system in cancer growth and metastasis: overview and recent insights. Endocr Rev. 2007;28:20.
180. Yakar, S, LeRoith, D, Brodt, P. The role of the growth hormone/insulin-like growth factor axis in tumor growth and progression: lessons from animal models. Cytokine and Growth Fact Rev. 2005;16:407.
181. Sayer, RA, Lancaster, JM, Pittman, J, et al. High insulin-like growth factor-2 (IGF-2) gene expression is an independent predictor of poor survival for patients with advanced stage serous epithelial ovarian cancer. Gynecol Oncol. 2005;96:355.
182. Haruta, M, Arai, Y, Sugawara, W, et al. Duplication of paternal IGF2 or loss of maternal IGF2 imprinting occurs in half of Wilms tumors with various structural WT1 abnormalities. Genes Chrom Can. 2008;47:712.
183. Baxter, RC, Daughaday, WH. Impaired function of the ternary insulin like growth factor binding complex in patients with hypoglycemia due to non-islet cell tumors. J Clin Endocrinol Metab. 1991;73:696.
184. Sachdev, D, Hartell, JS, Lee, AV, et al. A dominant negative type I insulin-like growth factor receptor inhibits metastasis of human cancer cells. J Biol Chem. 2004;279:5017.
185. DiGiovanni, J, Kiguchi, K, Frijhoff, A, et al. Deregulated expression of insulin-like growth factor 1 in prostate epithelium leads to neoplasia in transgenic mice. Proc Natl Acad Sci U S A. 2000;97:3455.
186. Wu, KD, Zhou, L, Burtrum, D, et al. Antibody targeting of the insulin-like growth factor I receptor enhances the anti-tumor response of multiple myeloma to chemotherapy through inhibition of tumor proliferation and angiogenesis. Cancer Immunol Immunother. 2007;56:343.
187. Wu, JD, et al. In-vivo effects of the human type I insulin-like growth factor receptor antibody A12 on androgen-dependent and androgen-independent human prostate tumors. Clin Cancer Res. 2005;11:3065.
188. Miyamoto, S, et al. Blockade of paracrine supply of insulin-like growth factors using neutralizing antibodies suppresses the liver metastasis of human colorectal cancers. Clin Cancer Res. 2005;11:3494.
189. Fowlkes, J, Enghild, JJ, Suzukik, N, et al. Matrix metalloproteases degrade insulin like growth factor binding protein-3 in dermal fibroblast cultures. J Biol Chem. 1994;269:25742.
190. Lawrence, JB, Oxvig, C, Overgaard, MT, et al. The insulin-like growth factor (IGF-I)-dependent IGF binding protein-4 protease secreted by human fibroblasts is pregnancy-associated plasma protein-A. Proc Natl Acad Sci USA. 1999;196:3149.
191. Nichols, TC, Busby, WH, Jr., Merricks, E, et al. Protease-resistant insulin-like growth factor (IGF) binding protein-4 inhibits IGF-I actions and neointimal expansion in a porcine model of neointimal hyperplasia. Endocrinol. 2007;148:5002.
192. Leal, SM, Liu, Q, Huang, GS, et al. The type V transforming growth factor beta receptor is a putative insulin like growth factor binding protein 3 receptor. J Biol Chem. 1997;272:20572.
193. Clemmons, DR. Use of mutagenesis to probe IGF binding protein structure/function relationships. Endocr Rev. 2001;22:800.
194. Rechler, MM, Clemmons, DR. Regulatory actions in insulin-like growth factor binding proteins. Trends Endocrinol Metab. 1998;9:176.
195. Burch, WW, Correa, J, Shaveley, JE, et al. The 25 k Dalton insulin-like growth factor (IGF) binding protein inhibits both basal and IGF mediated growth in chick embryonic pelvic cartilage in vitro. J Clin Endocrinol Metab. 1990;70:173.
196. Fottner, C, Spottl, G, Weber, MM. The divergent effect of insulin-like growth factor binding protein (IGFBP)-1 on IGF induced steroidogenesis in bovine adrenocortical cells is not due to its phosphorylation status. Exp Clin Endocrinol Diabetes. 2007;115:232.
197. Kajimura, S, Aida, K, Duan, C. Understanding hypoxia-induced gene expression in early development: in vitro and in vivo analysis of hypoxia-inducible factor-1 regulated zebra fish insulin-like growth factor binding protein gene expression. Mol Cell Biol. 2006;26:1142.
198. Marchand, A, Tomkiewicz, C, Magne, L, et al. Endoplasmic reticulum stress induction of insulin-like growth factor-binding protein-1 involves ATF4. J Biol Chem. 2006;281:19124.
199. Wolf, E, Lahm, H, Wu, M, et al. Effects of IGFBP-2 overexpression in vitro and in vivo. Pediatr Nephrol. 2000;14:572.
200. Bar, RS, Booth, BA, Bowes, M, et al. Insulin-like growth factor binding proteins from cultured endothelial cells: purification, characterization, and intrinsic biologic activities. Endocrinology. 1989;125:1910.
201. Russo, VC, Schutt, BS, Andaloro, E, et al. Insulin-like growth factor binding protein-2 binding to extracellular matrix plays a critical role in neuroblastoma cell proliferation, migration and invasion. Endocrinol. 2005;146:4445.
202. Kiepe, D, Van der Pas, A, Ciarmatori, S, et al. Defined carboxy-terminal fragments of insulin-like growth factor binding protein-2 exert similar mitogenic activity on cultures rat growth plate chondrocytes as IGF-I. Endocrinol. 2008;149:4901–4911.
203. Wang, H, Wang, H, Shen, W, et al. Insulin-like growth factor binding protein 2 enhances glioblastoma invasion by activating invasion-enhancing genes. Cancer Res. 2003;63:4315.
204. Conover, CA, Johnstone, EW, Turner, RT, et al. Subcutaneous administration of insulin-like growth factor (IGF)-II/IGF binding protein-2 complex stimulates bone formation and prevents loss of bone mineral density in a rat model of disuse osteoporosis. Growth Horm IGF Res. 2002;12:178.
205. Zhang, CC, Kaba, M, Iizuka, S, et al. Angiopoietin-like 5 and IGFBP2 stimulate ex-vivo expansion of human cord blood hematopoietic stem cells as assayed by NOD/SCID transplantation. Blood. 2008;111:3415.
206. Chesik, D, De Keyser, J, Wilczak, N. Insulin-like growth factor binding protein-2 as a regulator of IGF actions in CNS: implications in multiple sclerosis. Cytokine & Growth Factor Reviews. 2007;18:267.
207. Strange, KS, Wilkinson, D, Emerman, JT. Mitogenic properties of insulin-like growth factors I and II, insulin-like growth factor binding protein-3 and epidermal growth factor on human breast carcinoma cells. Breast Cancer Res Treat. 2002;75:203.
208. Ricort, JM, Binoux, M. Insulin-like growth factor binding protein-3 activates a phosphotyrosine phosphatase. Effects on the insulin-like growth factor signaling pathway. J Biol Chem. 2002;277:19448.
209. Martin, JL, Jambazov, S. Insulin-like growth factor binding protein-3 in extracellular matrix stimulates adhesion of breast epithelial cells and activation of p44/42 mitogen-activated protein kinase. Endocrinol. 2006;147:4400.
210. Huynh, H, Yang, XF, Pollak, M. Estradiol and antiestrogens regulate a growth-inhibitory insulin-like growth factor binding protein 3 autocrine loop in human breast cancer cells. J Biol Chem. 1996;271:1016.
211. O’Rear, L, Longobardi, L, Torello, M, et al. Signaling cross talk between IGF-binding protein-3 and transforming growth factor (beta) in mesenchymal chondroprogenitor cell growth. J Mol Endocrinol. 2005;34:723.
212. Silha, JV, Sheppard, PC, Mishra, S, et al. Insulin-like growth factor (IGF) binding protein-3 attenuates prostate tumor growth by IGF-dependent and IGF-independent mechanisms. Endocrinol. 2006;147:2112.
213. Liu, B, Lee, HY, Weinzimer, SA. Direct functional interactions between insulin-like growth factor-binding protein-3 and retinoid X receptor- regulate transcriptional signaling and apoptosis. J Biol Chem. 2000;275:33607.
214. Schedlich, LJ, Young, TF, Fifth, SM, et al. Insulin-like growth factor-binding protein (IGFBP)-3 and IGFBP-5 share a common nuclear transport pathway in T47D human breast carcinoma cells. J Biol Chem. 1998;273:18347.
215. Takaoka, M, Kim, SH, Okawa, T, et al. IGFBP-3 regulated esophageal tumor growth through IGF-dependent and independent mechanisms. Cancer Biol Ther. 2007;6:534.
216. Chan, SS, Twigg, SM, Firth, SM, et al. Insulin-like growth factor binding protein-3 leads to insulin resistance in adipocytes. J Clin Endocrinol Metab. 2005;90:6588.
217. Mohan, S, Bautista, CM, Wergedal, J, et al. Isolation of inhibitory insulin-like growth factor (IGF) binding protein from bone cell conditioned medium: a potential local regulator of IGF action. Proc Natl Acad Sci U S A. 1989;86:8338.
218. Conover, CA, Durham, SK, Zapf, J, et al. Cleavage analysis of insulin-like growth factor (IGF)-dependent IGF-binding protein-4 proteolysis and expression of protease-resistant IGF-binding protein-4 mutants. J Biol Chem. 1995;270:4395.
219. Spicer, LJ. Proteolytic degradation of insulin-like growth factor binding proteins by ovarian follicles. Biol Reprod. 2003;70:1223.
220. Parker, A, Rees, C, Clarke, JB, et al. Binding of insulin-like growth factor binding protein-5 to smooth muscle cell extracellular matrix is a major determinant of the cellular response to IGF-I. Mol Biol Cell. 1998;9:2383.
221. Miyakoshi, N, Richman, C, Kasukawa, Y, et al. Evidence that IGF-binding protein-5 functions as a growth factor. J Clin Invest. 2001;107:71.
222. Kuemmerle, JF, Zhou, H. Insulin-like growth factor-binding protein-5 (IGFBP-5) stimulates growth and IGF-I secretion in human intestinal smooth muscle by Ras-dependent activation of p38 MAP kinase and Erk1/2 pathways. J Biol Chem. 2002;277:20563.
223. Butt, AJ, Dickson, KA, McDougall, F, et al. Insulin-like growth factor-binding protein-5 inhibits the growth of human breast cancer cells in vitro and in vivo. J Biol Chem. 2003;278:29676.
224. Marshman, E, Green, KA, Flint, DJ, et al. Insulin-like growth factor binding protein-5 and apoptosis in mammary epithelial cells. J Cell Sci. 2003;116:675.
225. Butt, AJ, Dickson, KA, Jambazov, S, et al. Enhancement of tumor necrosis factor-alpha-induced growth inhibition by insulin-like growth factor-binding protein-5 (IGFBP-5), but not IGFBP-3 in human breast cancer cells. Endocrinol. 2005;146:3113.
226. Mukherjee, A, Wilson, EM, Rotwein, P. Insulin-like growth factor (IGF) binding protein-5 blocks skeletal muscle differentiation by inhibiting IGF actions. Mol Endocrinol. 2008;22:206.
227. Bach, LA. IGFBP-6 five years on; not so “forgotten”? Growth Horm IGF Res. 2005;15:185.
228. Kiepe, D, Ulinski, T, Powell, DR, et al. Differential effects of insulin-like growth factor binding proteins 1, -2, -3 and -6 on cultured growth plate chondrocytes. Kidney Int. 2002;62:1591.
229. Fu, P, Thomson, JA, Bach, LA. Promotion of cancer cell migration: an insulin-like growth factor (IGF) independent action of IGF-binding protein-6. J Biol Chem. 2007;282:22298.
230. Guo, L, Zhao, YY, Zhao, YY, et al. Toxic effects of ICDD on osteogenesis through altering IGFBP-6 gene expression in osteoblasts. Biol Pharm Bull. 2007;30:2018.
231. Douglas, RG, Gluckman, PD, Ball, B, et al. The effects of infusion of insulin-like growth factor I (IGF-I), IGF-II and insulin on glucose and protein metabolism in fasted lambs. J Clin Invest. 1991;88:614.
232. Chrysis, D, Zhang, J, Underwood, LE. Divergent regulation of proteasomes by insulin-like growth factor I and growth hormone in skeletal muscle of rats made catabolic with dexamethasone. Growth Horm IGF Res. 2002;12:434.
233. Lang, CH, Frost, RA. Role of growth hormone, insulin-like growth factor-I and insulin-like growth factor binding proteins in the catabolic response to injury and infection. Curr Opin Nutr Metab Care. 2002;5:271.
234. Thissen, JP, Underwood, LE, Maiter, DM, et al. Failure of IGF-I infusion to promote growth in protein-restricted rats despite normalization of serum IGF-I concentrations. Endocrinology. 1991;128:885.
235. Jacob, RJ, Sherwin, RS, Bowen, L, et al. Metabolic effects of IGF-I and insulin in spontaneously diabetic BB/w rats. Am J Physiol. 1991;260:E262.
236. Cox, GN, McDermott, MJ, Merkel, E, et al. Recombinant human insulin-like growth factor binding protein-1 inhibits growth stimulated by IGF-I and growth hormone in hypophysectomized rats. Endocrinol. 1994;35:1913.
237. Galiano, RD, Zhao, L, Clemmons, DR, et al. Interaction between the insulin-like growth factor family and the integrin receptor family in tissue repair processes. J Clin Invest. 1996;98:2462.
238. Leu, JI, Crissey, MA, Taub, R. Massive hepatic apoptosis associated with TGF-beta1 activation after Fas ligand treatment of IGF binding protein-1-deficient mice. J Clin Invest. 2003;111:129.
239. Palermo, C, Manduca, P, Gazzerro, E, et al. Potentiating role of insulin-like growth factor binding protein (IGFBP)-2 on IGF-II-stimulated alkaline phosphatase activity in differentiating osteoblasts. Am J Physiol Endocrinol Metab. 2004;286:E648.
240. Bagi, CM, Brommage, R, Adams, SO, et al. Benefit of systemically administered rh IGF-I and rh IGF-I/IGBP-3 on cancellous bone in oophorectomized rats. J Bone Mineral Res. 1994;9:1301.
241. Kim, HS, Ali, O, Shim, M, et al. Insulin-like growth factor binding protein-3 induces insulin resistance in adipocytes in vitro and in rats in vivo. Pediatr Res. 2007;61:159.
242. Chen, W, Salojin, KV, Mi, QS, et al. Insulin-like growth factor (IGF)-I/IGF-binding protein-3 complex: therapeutic efficacy and mechanism of protection against type 1 diabetes. Endocrinol. 2004;145:627.
243. Zappala, G, Elbi, C, Edwards, J, et al. Induction of apoptosis in human prostate cancer cells by insulin-like growth factor binding protein-3 does not require binding to retinoid X receptor-alpha. Endocrinol. 2007;149:1802.
244. Fang, P, Hwa, V, Little, BM, et al. IGFBP-3 sensitizes prostate cancer cells to interferon-gamma-induced apoptosis. Growth Horm IGF Res. 2007;18:38.
245. Miyakoshi, N, Qin, X, Kasukawa, Y, et al. Systemic administration of insulin-like growth factor (IGF) binding protein-4 (IGFBP-4) increases bone formation parameters in mice by increasing IGF bioavailability via an IGFBP-4 protease-dependent mechanism. Endocrinol. 2001;142:2641.
246. Andress, DL. IGF-binding protein-5 stimulates osteoblast activity and bone accretion in ovariectomized mice. Am J Physiol Endocrinol Metab. 2001;281:283.
247. Behringer, RR, Lewin, TM, Quaife, CJ, et al. Expression of insulin-like growth factor I stimulates normal somatic growth in growth hormone deficient transgenic mice. Endocrinology. 1990;127:1033.
248. Chrysis, D, Calikoglu, AS, Ye, P, et al. Insulin-like growth factor-I overexpression attenuates cerebellar apoptosis by altering the expression of Bcl family proteins in a developmentally specific manner. J Neurosci. 2001;21:1481.
249. Dai, A, Xing, Y, Boney, CM, et al. Human insulin-like growth factor binding protein-1 (hIGFBP-1) transgenic mice: characterization and insights into the regulation of IGFBP-1 expression. Endocrinology. 1994;135:1316.
250. Rajkumar, K, Barron, D, Lewitt, M, et al. Growth retardation and hyperglycemia in insulin-like growth factor binding protein-1 transgenic mice. Endocrinology. 1995;136:4029.
251. Watson, CS, Bialek, P, Anzo, M, et al. Elevated circulating insulin-like growth factor binding protein-1 is sufficient to cause fetal growth restriction. Endocrinol. 2006;147:1175.
252. Ben Lagha, N, Menuelle, P, Seurin, P, et al. Bone formation in the context of growth retardation induced by hIGFBP-1 overexpression in transgenic mice. Connect Tissue Res. 2002;43:515.
253. Eckstein, F, Pavicic, T, Nedbal, S, et al. Insulin-like growth factor binding protein-2 (IGFBP-2) overexpression negatively regulates bone size and mass, but not density, in the absence and presence of growth hormone/IGF-I excess in transgenic mice. Anat and Embryology. 2003;206:139.
254. DeMambro, VE, Clemmons, DR, Horton, LG, et al. Gender-specific changes in bone turnover and skeletal architecture in IGFBP-2 null mice. Endocrinol. 2008;149:2051.
255. Silha, JV, Mishra, S, Rosen, CJ, et al. Perturbations in bone formation and resorption in insulin-like growth factor binding protein-3 transgenic mice. J Bone Miner Res. 2003;18:1834.
256. Silha, JV, Gui, Y, Mishra, S, et al. Overexpression of gly56/gly80/gly81-mutant insulin-like growth factor binding protein-3 in transgenic mice. Endocrinol. 2004;146:1523.
257. Liao, L, Chen, X, Wang, S, et al. Steroid receptor coactivator 3 maintains circulating insulin-like growth factor I (IGF-I) by controlling IGF-binding protein 3 expression. Mol Cell Biol. 2008;28:2460.
258. Ning, Y, Schuller, AG, Bradshaw, S, et al. Diminished growth and enhanced glucose metabolism in triple knockout mice containing mutations of insulin-like growth factor binding protein-3, -4, and -5. Mol Endocrinol. 2006;20:2173.
259. Zhang, M, Faugere, MC, Malluche, H, et al. Paracrine overexpression of IGFBP-4 in osteoblasts of transgenic mice decreases bone turnover and causes global growth retardation. J Bone Miner Res. 2003;18:836.
260. Ning, Y, Schuller, AG, Conover, CA, et al. Insulin-like growth factor (IGF) binding protein-4 is both a positive and negative regulator of IGF activity in vivo. Mol Endocrinol. 2008;22:1213.
261. Salih, Da, Tripathi, G, Holding, C, et al. Insulin-like growth factor binding protein 5 (IGFBP-5) compromises survival, growth, muscle development, and fertility in mice. Proc Natl Acad Sci U S A. 2004;101:4314.
262. Silha, JV, Gui, Y, Modric, T, et al. Overexpression of the acid-labile subunit of the IGF ternary complex in transgenic mice. Endocrinol. 2001;142:4305.
263. Domene, HM, Bengolea, SV, Jasper, HG, et al. Acid-labile subunit deficiency: phenotypic similarities and differences between human and mouse. J Endocrinol Invest. 2005;28:43.
264. Baker, J, Liu, JP, Robertson, EJ, et al. Role of insulin like growth factors in embryonic and postnatal growth. Cell. 1993;75:83.
265. Holzenberger, M, Leneuve, P, Hamard, G, et al. A targeted partial invalidation of the insulin-like growth factor receptor gene in mice causes a postnatal growth deficit. Endocrinol. 2000;141:2557.
266. Kondo, T, Vicent, D, Suzuma, K, et al. Knockout of insulin and IGF-I receptors on vascular endothelial cells protects against retinal neovascularization. J Clin Invest. 2003;111:1835.
267. DeChiara, RM, Efstratiadis, A, Robertson, EJ. A growth deficiency phenotype in heterozygous mice carrying an insulin-like growth factor II gene disruption. Nature. 1990;345:78.
268. Sjogren, K, Liu, JL, Blad, K, et al. Liver-derived insulin-like growth factor I (IGF-I) is the principal source of IGF-I in blood but is not required for postnatal body growth in mice. Proc Natl Acad Sci U S A. 1999;96:7088.
269. Yakar, S, Rosen, CJ, Beamer, WG, et al. Circulating levels of IGF-I directly regulate bone growth and density. J Clin Invest. 2002;110:771.
270. Ueki, I, Ooi, GT, Tremblay, ML, et al. Inactivation of the acid labile subunit gene in the mice results in mild retardation of postnatal growth despite profound disruptions in the circulating insulin-like growth factor system. Proc Natl Acad Sci U S A. 2000;97:6868.
271. Wu, Y, Sun, H, Yakar, S, et al. Elevated levels of IGF-1 in serum rescue the severe growth retardation of IGF-1 null mice. Endocrinology. 2009, Epub ahead of print.
272. Guler, H-P, Zapf, J, Froesch, ER. Short-term metabolic effects of recombinant human insulin-like growth factor-I in healthy adults. N Engl J Med. 1987;317:137.
273. Kerr, D, Tamborlane, V, Rife, F, et al. Effect of insulin-like growth factor-I on the responses to and recognition of hypoglycemia in humans. A comparison with insulin. J Clin Invest. 1993;91:141.
274. Clemmons, DR, Smith-Banks, A, Celniker, AC, et al. Reversal of diet-induced catabolism by infusion of recombinant insulin-like growth factor-I (IGF-I) in humans. J Clin Endocrinol Metab. 1992;75:234.
275. Herndon, DN, Ramzy, PI, DebRoy, MA, et al. Muscle protein catabolism after severe burn: effects of IGF-1/IGFBP-3 treatment. Ann Surg. 1999;229:713.
276. Jeschke, MG, Bolder, U, Chung, DH, et al. Gut mucosal homeostasis and cellular mediators after severe thermal trauma and the effect of insulin-like growth factor-1 in combination with insulin-like growth factor binding protein-3. Endocrinol. 2007;148:354.
277. Boonen, S, Rosen, C, Bouillon, R, et al. Musculoskeletal effects of the recombinant human IGF-I/IGF binding protein-3 complex in osteoporotic patients with proximal femoral fracture: a double-blind, placebo-controlled pilot study. J Clin Endocrinol Metab. 2002;87:1593.
278. Deicher, R, Horl, WH. Hormonal adjuvants for the treatment of renal anaemia. Eur J Clin Invest. 2005;35(suppl 3):75.
279. Mauras, N, Martha, PM, Jr., Quarmby, V, et al. rhIGF-I administration in humans: metabolic effects of bolus vs continuous subcutaneous delivery. Am J Physiol. 1997;272:E628.
280. Mauras, N, Haymond, MW. Are the metabolic effects of GH and IGF-I separable? Growth Horm IGF Res. 2005;15:19.
281. Mauras, N, Beaufree, B. Recombinant human insulin like growth factor I enhances whole-body protein anabolism and significantly diminishes the protein catabolic effects of prednisone in humans without a diabetogenic effect. J Clin Endocrinol Metab. 1995;80:869.
282. Ebling, PR, Jones, JD, O’Fallon, WM, et al. Short term effects of recombinant human insulin like growth factor I on bone turnover in normal women. J Clin Endocrinol Metab. 1993;77:1384.
283. Grinspoon, S, Baum, HBA, Lee, K, et al. Effects of short term rhIGF-I on bone turnover in osteopenic women with anorexia nervosa. J Clin Endocrinol Metab. 1996;81:3364.
284. Johansson, AG, Lindh, E, Blum, WF, et al. Effects of growth hormone and insulin-like growth factor-I in men with osteoporosis. J Clin Endocrinol Metab. 1996;81:44.
285. Biandi, T, Glatz, Y, Bouillon, R, et al. Effects of short term insulin-like growth factor-I (IGF-I) or growth hormone treatment on bone metabolism and on production of 1,25 dihydroxycholecalciferol in GH deficient adults. J Clin Endocrinol Metab. 1998;83:81.
286. Liu, JM, Zhao, HY, Ning, G, et al. IGF-1 as an early marker for low bone mass or osteoporosis in premenopausal and postmenopausal women. J Bone Miner Metab. 2008;26:159.
287. Haluzik, M, Yakar, S, Gavrilova, O, et al. Insulin resistance in the liver-specific IGF-1 gene-deleted mouse is abrogated by deletion of the acid-labile subunit of the IGF-binding protein-3 complex: relative roles of growth hormone and IGF-1 in insulin resistance. Diabetes. 2003;52:2483.
288. Ezzat, VA, Duncan, ER, Wheatcroft, SB, et al. The role of IGF-I and its binding proteins in the development of type 2 diabetes and cardiovascular disease. Diabetes Obesity and Metab. 2008;10:198.
289. Pratipanawatr, T, Pratipanawatr, W, Rosen, C, et al. Effect of IGF-I on FFA and glucose metabolism in control and type 2 diabetic subjects. Am J Physiol Endocrinol Metab. 2002;282:E1360.
290. Simpson, HL, Jackson, NC, Shojaee-Moradie, F, et al. Insulin-like growth factor I has a direct effect on glucose and protein metabolism, but not effect on lipid metabolism in type 1 diabetes. J Clin Endocrinol Metab. 2004;89:425.
291. Kuzuya, H, Matsuura, N, Sakamoto, M, et al. Trial of insulin-like growth factor-I therapy for patients with extreme insulin resistance syndromes. Diabetes. 1993;42:696.
292. Cheetham, TD, Jones, J, Taylor, AM, et al. The effects of recombinant insulin-like growth factor I administration or growth hormone levels and insulin requirements in adolescents with type 1 insulin-dependent diabetes mellitus. Diabetologia. 1993;36:678.
293. Moses, AC, Young, SCJ, Morrow, LA, et al. Recombinant human insulin-like growth factor I increases insulin sensitivity and improves glycemic control in Type II diabetes. Diabetes. 1996;45:95.
294. Quattrin, T, Thrailkill, K, Baler, L, et al. Dual hormonal replacement with insulin and recombinant insulin like growth factor I in insulin dependent diabetes mellitus: effects on glycemic control, IGF-I levels, and safety profiles. Diabetes Care. 1997;20:374.
295. Clemmons, DR, Moses, AC, McKay, MJ, et al. The combination of insulin-like growth factor I and insulin-like growth factor-binding protein-3 reduces insulin requirements in insulin-dependent type 1 diabetes: evidence for in vivo biological activity. J Clin Endocrinol Metab. 2000;85:1518.
296. Clemmons, DR, Sleevi, M, Allan, G, et al. Effects of combined recombinant insulin-like growth factor (IGF)-I and IGF binding protein-3 in type 2 diabetic patients on glycemic control and distribution of IGF-I and IGF-II among serum binding protein complexes. J Clin Endocrinol Metab. 2007;92:2652.
297. Rosenfeld, RG, Belgorosky, A, Camacho-Hubner, C, et al. Defects in growth hormone receptor signaling. Trends Endocrinol Metab. 2007;4:1043.
298. Guevara-Aguirre, J, Vasconez, O, Martinez, V, et al. A randomized, double-blind, placebo-controlled trial of safety and efficacy of recombinant human insulin-like growth factor I in children with growth hormone receptor deficiency. J Clin Endocrinol Metab. 1995;80:1393.
299. Backeljaw, PF, Underwood, LE. Prolonged treatment with recombinant insulin-like growth factor I in children with growth hormone insensitivity syndrome: a clinical research center study. J Clin Endocrinol Metab. 1996;81:3312.
300. Chernausek, SD, Backeljauw, PF, Frane, J, et al. Long-term treatment with recombinant insulin-like growth factor (IGF)-I in children with severe IGF-I deficiency due to growth hormone insensitivity. J Clin Endocrinol Metab. 2007;92:902.
301. Keating, GM. Mecasermin. BioDrugs. 2008;22:177.
302. Camacho-Hubner, C, Woods, KA, Miraki-Moud, F, et al. Effects of recombinant human insulin-like growth factor I (IGF-I) therapy on the growth hormone IGF system of a patient with partial IGF-I gene deletion. J Clin Endocrinol Metab. 1999;84:1611.
303. de Lacerda, L, Carvalho, JA, Stannard, B, et al. In vitro and in vivo responses to short-term recombinant human insulin-like growth factor-1 (IGF-I) in a severely growth-retarded girl with ring chromosome 15 and deletion of a single allele for the type 1 IGF receptor gene. Clin Endocrinol. 1999;51:541.
304. Okubo, Y, Siddle, K, Firth, H, et al. Cell proliferation activities on skin fibroblasts from a short child with absence of one copy of the type 1 insulin-like growth factor receptor (IGF1R) gene and a tall child with three copies of the IGF1R gene. J Clin Endocrinol Metab. 2003;88:5981.
305. Abuzzahab, MJ, Schneider, A, Goddard, A, et al. IGF-I receptor mutations resulting in intrauterine and postnatal growth retardation. N Engl J Med. 2003;349:2211.