Chapter 47 Inhibitors of Bacterial Ribosomal Actions
| Abbreviations | |
|---|---|
| AIDS | Acquired immunodeficiency syndrome |
| CSF | Cerebrospinal fluid |
| GI | Gastrointestinal |
| IM | Intramuscular |
| IV | Intravenous |
| MIC | Minimal inhibitory concentration |
| MLSB | Macrolide-lincosamide-streptogramin B |
| mRNA | Messenger ribonucleic acid |
| MRSA | Methicillin-resistant Staphylococcus aureus |
| tRNA | Transfer ribonucleic acid |
| VRE | Vancomycin-resistant enterococci |
Therapeutic Overview
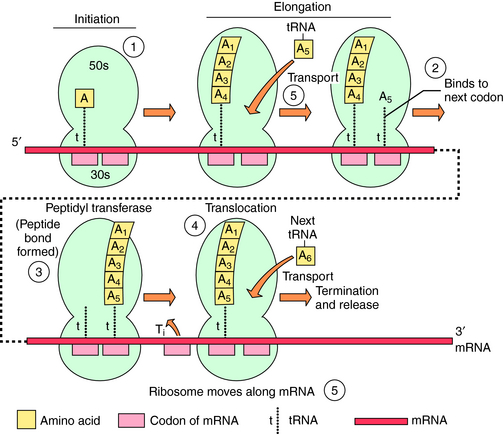
The ribosome-binding sites for macrolides such as erythromycin, azithromycin, clarithromycin, and clindamycin are on the same 50S subunit (S represents the sedimentation parameter), but the structures of the drugs and the spectrum of activities differ considerably. Erythromycin, one of the first macrolides developed, is relatively safe and widely used, especially for the treatment of infections in children (Box 47-1). Because of the success of macrolides in the treatment of pulmonary infections, these drugs continue to be used in the treatment of respiratory tract infections in adults. The primary differences among erythromycin, clarithromycin, and azithromycin are related to relative activities against certain bacterial species such as Mycobacterium, gastrointestinal (GI) tolerability, and pharmacokinetics. Clindamycin displays antimicrobial activity somewhat similar to that
of erythromycin. However, the two differ structurally, and clindamycin displays extensive anaerobic activity while having no activity for atypical respiratory pathogens.
The tetracyclines and synthetic glycylcycline analogs bind to the 30S ribosomal subunit and are effective against aerobic and anaerobic gram-positive and gram-negative organisms. Given their wide spectrum of activity, these agents remain widely used for treatment of bacterial, chlamydial, rickettsial, and mycoplasmal infections, although the development of bacterial resistance has reduced their efficacy against some pathogens (Box 47-2).
The streptogramins and oxazolidinones are newer classes of antibiotics that were developed primarily for the treatment of gram-positive organisms and often have activity against organisms that are resistant to β-lactams and glycopeptides. Both classes inhibit protein synthesis, but their structures and mechanisms of action differ.
| Therapeutic Overview |
|---|
| Aminoglycosides |
| Inhibit gram-negative aerobes |
| Narrow therapeutic index |
| Renal and otic toxicities can be serious |
| Pharmacokinetics are important considerations |
| Plasmid-mediated resistance is a problem |
| Macrolides |
| Inhibit Mycoplasma, Chlamydia, Legionella |
| Inhibit gram-positive organisms |
| Clindamycin |
| Inhibits gram-positive cocci and anaerobic species |
| Active against clostridium difficile-associated diarrhea and colitis |
| Chloramphenicol |
| Kills major meningitis pathogens |
| Serious toxicity |
| Ketolides |
| Inhibit respiratory pathogens |
| Active against penicillin and macrolide-resistant S. pneumoniae |
| Tetracyclines |
| Inhibit broad spectrum of organisms |
| Streptogramins |
| Inhibit gram-positive organisms |
| Active against VRE |
| Oxazolidinones |
| Inhibit gram-positive organisms |
| Active against VRE |
These drugs represent important agents for the treatment of multidrug-resistant gram-positive infections, but prudent use will be important to prevent the development of resistance to these agents.
Mechanisms of Action
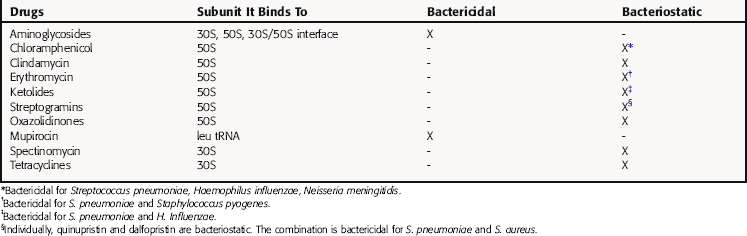
The bacterial ribosomal subunit to which each of these drugs binds and the bactericidal or bacteriostatic response of susceptible bacteria to the drugs are in Table 47-1. The principal steps in bacterial ribosomal synthesis of proteins, as carried out by the 70S ribosomes and relevant RNAs, and the points at which the drugs act, are depicted in Figure 47-1.
Aminoglycosides consist of amino sugars linked through glycosidic bonds to an aminocyclitol. The structures of streptomycin, gentamicin, and other clinically important aminoglycosides are shown in Figure 47-2. The particular amino sugars and specific locations of the amino groups distinguish the compounds and are important for their antimicrobial effects and toxicity. Gentamicin consists of a mixture of three species with little differences in activities.

The aminoglycosides can cross the complex cell membrane structure of gram-negative bacteria (see Fig. 46-3) and are more effective against aerobic gram-negative than gram-positive bacteria.
Binding to the ribosome leads to inhibition of protein synthesis. This takes place on the ribosomes, where messenger RNA (mRNA) acts as a template for the addition of activated amino acids attached to tRNAs. The 70S ribosomal particles move along the mRNA template, adding the appropriate amino acid (see Fig. 47-1). Aminoglycosides bind to several ribosomal sites (see Table 47-1) of the 30S and 50S subunits of the bacterial ribosome. Streptomycin, the most thoroughly studied, binds to the 30S subunit, although this can be altered by mutation of particular amino acids. Binding of aminoglycosides interferes with protein synthesis in two ways:
Bacterial resistance to aminoglycosides results from:
Clinically, enzymatic modification of the aminoglycosides is the most important mechanism of resistance. Although resistance resulting from altered ribosomes does occur in enterococci, it is rarely seen in gram-negative bacteria and is relatively uncommon. Resistance as a consequence of the inadequate transport of drug across the cytoplasmic membrane is uncommon in aerobic or facultative species but is seen in strict anaerobes. Mutants with alterations in the electron transfer chain and in adenosine triphosphatase activity have been identified, but they are very rare. The resistance of some Pseudomonas species to aminoglycosides may be related to failure of the drug to distort the lipopolysaccharide of the outer membrane, thus not allowing drug to enter the bacterial cell.
Macrolides, Chloramphenicol, and Clindamycin
The macrolides (erythromycin, clarithromycin, azithromycin, and dirithromycin), chloramphenicol, and clindamycin are discussed as a group because these agents bind to the same site or sites on the ribosomal 50S subunit. They bind to bacterial 70S ribosomes but not to the 80S ribosomes of mammalian cells. Their structures are shown in Figure 47-3. Bacterial resistance is observed for all of these agents.
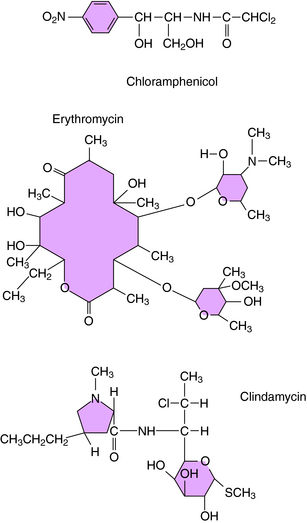
Erythromycin and the newer macrolides reversibly bind to 50S ribosomal subunits, causing dissociation of peptidyl-tRNA from the ribosome and interference with peptide elongation. Erythromycin inhibits the binding of chloramphenicol to 50S ribosomes, but chloramphenicol does not inhibit erythromycin binding. The activity of macrolides is primarily bacteriostatic; however, bactericidal activity is observed for certain organisms (see Table 47-1).
Bacterial resistance to macrolides occurs by several mechanisms, some of which also confer resistance to clindamycin and streptogramin type B. The most problematic forms of resistance arise either from alteration of ribosomal binding sites or drug efflux. Alteration of ribosomal binding sites occurs via a plasmid-encoded enzyme that methylates the 50S ribosomal subunit. Methylation likely causes a conformational change of the ribosomal target and decreased binding. This type of resistance is associated with the erm (erythromycin ribosome methylation) gene and is referred to as the macrolide-lincosamide-streptogramin B (MLSB) phenotype, because it confers resistance to macrolides, clindamycin, and streptogramin B. Both erythromycin and clindamycin induce this enzyme, but erythromycin has greater activity. This is clinically important, because an organism resistant to erythromycin and susceptible to clindamycin can become resistant to both drugs during therapy. This applies to the treatment of infections caused by methicillin-resistant Staphylococcus aureus (MRSA), which is often resistant to erythromycin but may appear susceptible to clindamycin. This form of resistance is present on plasmids that can pass from enterococci to streptococci and is encountered in strains of macrolide-resistant Streptococcus pneumoniae and Streptococcus pyogenes.
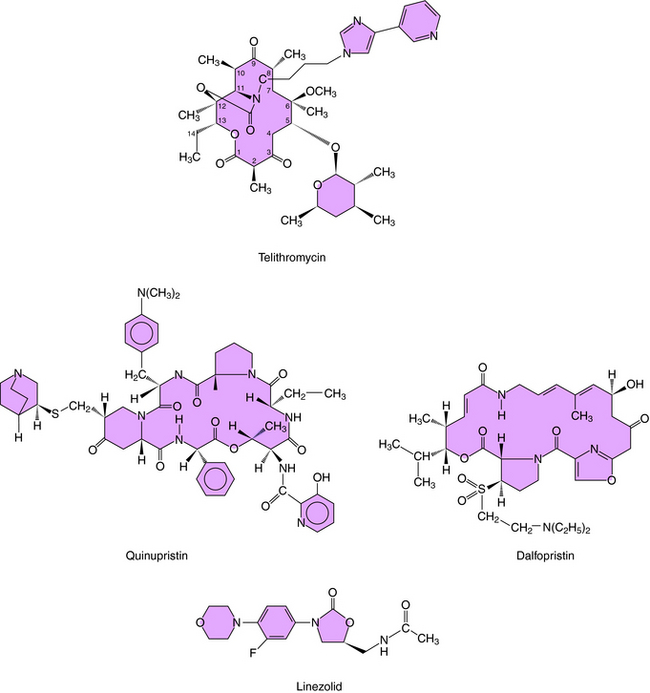
The ketolides are semisynthetic derivatives of erythromycin and are part of the MLSB family of antimicrobials. The currently approved ketolide is telithromycin; its structure is shown in Figure 47-4. Similar to the macrolides, ketolides inhibit protein synthesis at the 50S ribosomal subunit. However, ketolides demonstrate a higher binding affinity. Ketolides are primarily bacteriostatic but demonstrate bactericidal activity against some pathogens, including S. pneumoniae and H. influenzae.
Tetracyclines and Glycylcyclines
The structures of the tetracyclines are shown in Figure 47-5. These compounds bind to 30S ribosomes, thereby preventing attachment of the aminoacyl-tRNA to its acceptor site and preventing the addition of amino acids to the peptide chain being synthesized. Differences in the activities of individual tetracyclines are related to their solubility in lipid membranes of the bacteria. These drugs enter the cytoplasm of gram-positive bacteria by an energy-dependent process, but in gram-negative organisms, they pass through the outer membrane by diffusion through porins. Because minocycline and doxycycline are more lipophilic, they can enter gram-negative cells through the outer lipid membrane and through the porins. Once in the periplasmic space, the tetracyclines are transported across the inner cytoplasmic membrane by a protein-carrier system.
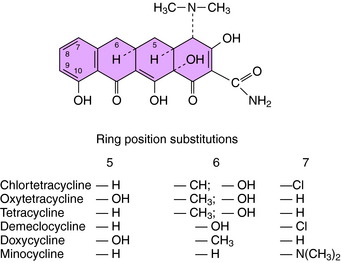
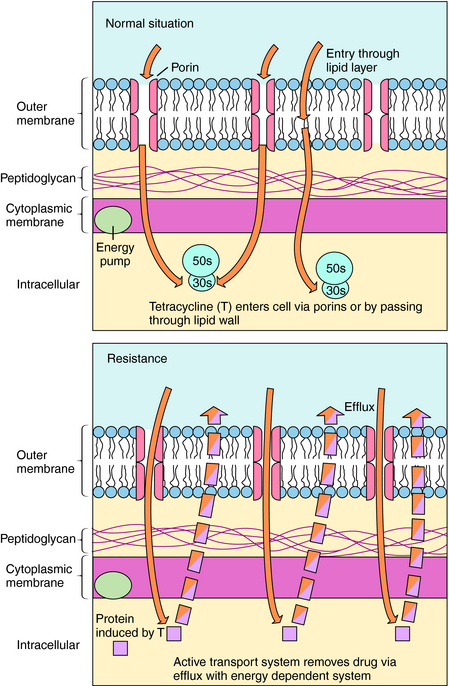
There are several mechanisms of resistance to tetracyclines. The most common, found in both gram-positive and gram-negative bacteria, is plasmid or transposon mediated and involves decreased intracellular accumulation and increased transport of the drug out of the bacterial cell (Fig. 47-6). Drug efflux occurs as a result of the action of a new protein, likely induced by the drug. A second mechanism involves alteration of outer membrane proteins resulting from mutations in chromosomal genes. In a third mechanism the ribosomal binding site is protected as a result of the presence of a plasmid-generated protein that binds to the ribosome. Resistance to one tetracycline usually implies resistance to all tetracyclines. However, some staphylococci and some Bacteroides species are resistant to tetracycline but susceptible to minocycline and doxycycline because of the lipophilicity of these latter agents.
Streptogramins are a family of compounds derived from Streptomyces pristinaespiralis whose members are classified into two groups based upon structure. For clinical use, two streptogramins, quinupristin and dalfopristin, whose structures are shown in Figure 47-4, are combined as quinupristin-dalfopristin in a 30:70 mixture. The sequential binding of each component to the 50S ribosomal subunit leaves a stable drug-ribosome complex that interferes with peptide chain elongation and peptidyl transferase. The activity of each component is bacteriostatic, but the combination is bactericidal against some organisms. The spectrum of activity of quinupristin-dalfopristin encompasses many gram-positive organisms, whereas activity against gram-negative organisms is limited.
Oxazolidinones inhibit protein synthesis by binding to the 50S ribosomal subunit and preventing formation of the initiation complex. The oxazolidinone available for use in the United States is linezolid (see Fig. 47-4). Linezolid is bacteriostatic, with activity directed primarily against gram-positive organisms, including those resistant to other antibiotics.
Mupirocin is a topical agent, previously known as pseudomonic acid, which inhibits gram-positive and some gram-negative bacteria by binding to isoleucyl-tRNA synthetase, preventing isoleucine incorporation into bacterial proteins. Mupirocin has been shown to eliminate the nasal carriage of methicillin-resistant staphylococci.
Pharmacokinetics
The pharmacokinetic parameters for the antibiotics that inhibit bacterial protein synthesis are given in Table 47-2.
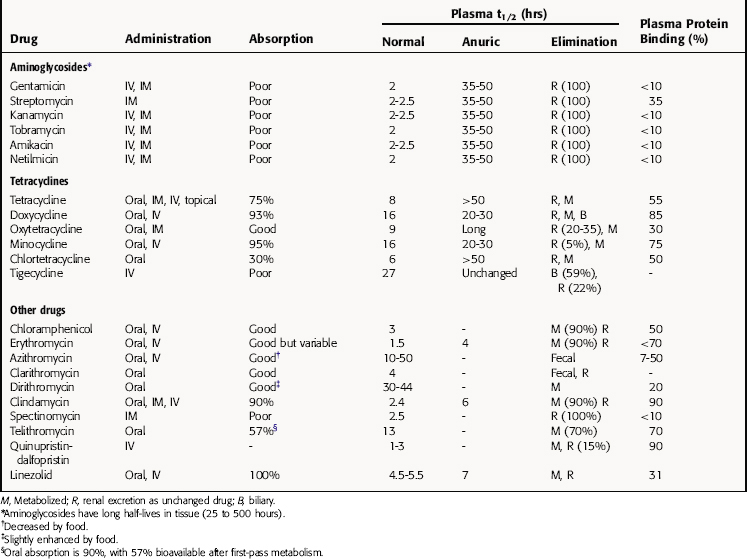
Because of their high polarity, aminoglycosides do not enter phagocytic or other cells, the brain, or the eye. They are distributed into interstitial fluid, with a volume of distribution essentially equal to that of the extracellular fluid. The highest concentrations of aminoglycosides occur in the kidney, where they concentrate in proximal tubular cells. Urine concentrations are generally 20 to 100 times greater than those in plasma and remain so for 24 hours after a single dose. These drugs enter peritoneal, pleural, and synovial fluids relatively slowly but eventually achieve concentrations that are only slightly less than those in plasma.
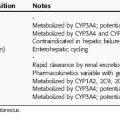
High concentrations of free drug may accumulate in infants deficient in forming glucuronides. In addition, hydrolysis of the succinate may be depressed in newborns and infants and requires careful monitoring of serum concentrations when this drug is used in these groups. Because of toxicity, bacterial resistance, and the availability of other antibiotics, chloramphenicol is rarely used.
Metabolism of linezolid occurs by nonenzymatic oxidation throughout the body, with most of the unchanged linezolid and its primary metabolites eliminated by urinary excretion. Dosage adjustment is not required for either mild or moderate hepatic impairment or for renal insufficiency. Accumulation of metabolites does occur in renal insufficiency, but the clinical significance is unknown.
Relationship of Mechanisms of Action to Clinical Response
The initial treatment of suspected sepsis has consisted of an aminoglycoside such as gentamicin or tobramycin in combination with a penicillin or cephalosporin, but newer cephalosporins and other β-lactams such as aztreonam or imipenem have lower toxicity and are being used with increased frequency in this setting (see Chapter 46). Local antimicrobial resistance patterns should be used to help define empiric therapies for sepsis.
Streptomycin is used primarily to treat uncommon infections such as those caused by Francisella tularensis, Brucella species, Yersinia pestis, and resistant tuberculosis strains or infections in patients allergic to the usual antituberculosis drugs (see Chapter 49). Amikacin is also used to treat multidrug resistant tuberculosis.
The macrolides, erythromycin, azithromycin, clarithromycin, and dirithromycin, are active primarily against gram-positive species such as staphylococci and streptococci but also inhibit some gram-positive bacilli (see Box 47-1). Chlamydiae, M. pneumoniae, Ureaplasma urealyticum, Legionella, Corynebacterium diphtheriae, Bordetella species, Campylobacter jejuni, and most oral anaerobic species are inhibited. Most aerobic gram-negative bacilli are resistant, although azithromycin inhibits Salmonella.
Erythromycin and other macrolides are used as an alternative to penicillin, particularly in children and especially in those with streptococcal pharyngitis, erysipelas, scarlet fever, cutaneous streptococcal infections, and pneumococcal pneumonia. However, levels of macrolide resistance in S. pneumoniae and S. pyogenes continue to increase; thus therapy with macrolides is increasingly problematic. Despite increasing resistance, macrolides continue to be used widely in combination with β-lactams for the treatment of community-acquired pneumonia because of their excellent activity against atypical respiratory pathogens. Although macrolides can cure S. aureus infections, the high frequency of resistance does not make them an initial choice for therapy. Azithromycin is useful for treating sexually transmitted diseases, including ones caused by Chlamydiae (potential for treatment with a single 1-g dose that significantly increases compliance), and erythromycin can be used to treat chlamydial pneumonia of the newborn. Trachoma can be treated effectively with a single dose of azithromycin. Recent data also support the use of azithromycin for the treatment of traveler’s diarrhea. Erythromycin is also useful for eradicating the carrier state of diphtheria and may shorten the course of pertussis if administered early. Clarithromycin and azithromycin are useful in both preventing and treating M. avium complex infections in patients with acquired immunodeficiency syndrome (AIDS). Bacillary angiomatosis in patients with AIDS has also been successfully treated with erythromycin. Erythromycin can be used to prevent bacterial endocarditis in penicillin-allergic patients with rheumatic fever.
Tetracyclines are broad-spectrum agents that inhibit a wide variety of aerobic and anaerobic gram-positive and gram-negative bacteria and other microorganisms such as rickettsiae, Ehrlichia, mycoplasmas, chlamydiae, and some mycobacterial species. The tetracyclines have many clinical uses, but because of increasing bacterial resistance and development of other drugs, they are no longer as widely used. For example, some S. pneumoniae, S. pyogenes, and staphylococci are now resistant to most tetracyclines. Among the Enterobacteriaceae, resistance has increased greatly in recent years, such that many E. coli and Shigella species and virtually all P. aeruginosa are resistant. However, tetracyclines do inhibit Pasteurella multocida, F. tularensis, Yersinia pestis, Vibrio species, and Brucella organisms. Doxycycline inhibits Bacteroides fragilis, but most Bacteroides species are resistant to the other tetracyclines. Other anaerobic species such as Fusobacterium and Actinomyces are inhibited, as are Borrelia burgdorferi (the cause of Lyme disease) and others. Mycobacterium marinum is inhibited, and some activity is observed against Plasmodium species.
Tetracyclines are the preferred agents for the treatment of rickettsial diseases such as Rocky Mountain spotted fever, typhus, scrub typhus, rickettsial pox, and Q fever (see Box 47-2), with doxycycline being the preferred agent from this class. Doxycycline is also the drug of choice for the treatment of ehrlichiosis and is used to treat Lyme disease and relapsing fever caused by Borrelia species. Atypical respiratory pathogens including Mycoplasma pneumoniae, Chlamydia pneumoniae, and Chlamydia psittaci respond to tetracyclines, which may be better tolerated by adults than erythromycin. Systemic Vibrio infections and peptic ulcer disease associated with Helicobacter pylori may also be treated with tetracyclines. Chlamydial infections of a sexual origin, such as nongonococcal urethritis, salpingitis, cervicitis, and lymphogranuloma venereum, are effectively treated with doxycycline. Tetracyclines are also effective for the treatment of inclusion conjunctivitis and trachoma caused by chlamydiae. For penicillin-allergic patients, tetracycline or doxycycline represent important alternative treatments for some forms of syphilis. In addition, doxycycline is a recommended treatment for granuloma inguinale.
Pharmacovigilance: Side Effects, Clinical Problems, and Toxicity
Reversible renal impairment develops in 5% to 25% of patients receiving an aminoglycoside for more than 3 days. The impairment can progress to severe renal insufficiency in a small number of patients, but it is usually reversible. In the renal cortex, aminoglycosides are transported across the luminal brush border and accumulate in proximal tubular cells. The initial decrease in glomerular filtration that occurs in response to aminoglycosides may result from inhibition of vasodilatory prostaglandins. Aminoglycosides also inhibit several enzymes and alter mitochondria and ribosomes in proximal tubular cells.
Erythromycin also inhibits the cytochrome P450 system, which can lead to significant drug-drug interactions. Erythromycin prolongs the half-life of theophylline and can lead to theophylline toxicity. It also inhibits the metabolism of carbamazepine, cyclosporine, corticosteroids, warfarin, and digoxin.
Quinupristin and dalfopristin inhibit cytochrome P450 enzymes, creating the potential for significant drug-drug interactions. Concomitant use of quinupristin-dalfopristin and drugs known to prolong the QTc interval should be avoided.
Linezolid has been known to interact with serotonergic agents, resulting in an increased risk of serotonin syndrome (see Chapter 30). Weak and reversible inhibition of monoamine oxidase occurs with linezolid use. Therefore patients taking linezolid should avoid eating large quantities of food with high tyramine content.
Ackermann G, Rodloff AC. Drugs of the 21st century: Telithromycin (HMR 3647)—the first ketolide. J Antimicrob Chemother. 2003;51:497-511.
Anonymous. Choice of antibacterial drugs. Treat Guidel Med Lett. 2007;5:33-50.
Anonymous. Tigecycline (tygacil). Med Lett. 2005;47:73-74.
Eliopoulos GM. Quinupristin-dalfopristin and linezolid. Evidence and opinion. Clin Infect Dis. 2003;36:473-481.
Hancock RE. Mechanisms of action of newer antibiotics for gram-positive pathogens. Lancet Infect Dis. 2005;5:209-218.
Noskin GA. Tigecycline: A new glycylcycline for treatment of serious infections. Clin Infect Dis. 2005;41(Suppl):S303-S314.