Chapter 25 Inflammatory Response and Mediators in Retinal Injury
Retinal injury
Ischemia–hypoxia
The retina consumes oxygen more rapidly than any other tissue in the body in part to fuel the constant renewal and replacement of photoreceptor outer segments.1 The presence of abundant mitochondria in the photoreceptor inner segments is evidence of this high energy demand.2 In addition, the choroid receives a high rate of blood flow to supply oxygen and glucose to the retinal pigment epithelium (RPE).3 Inadequate blood supply decreases oxygen delivery to the retina (hypoxia) and may result in ischemic injury. The subsequent ischemia can induce neuronal apoptosis and neovascularization. Classical examples of the retinal diseases associated with hypoxia and ischemia are diabetic retinopathy and retinal vein occlusion.
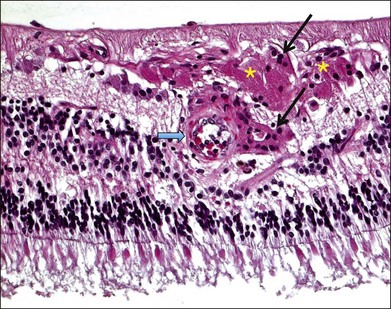
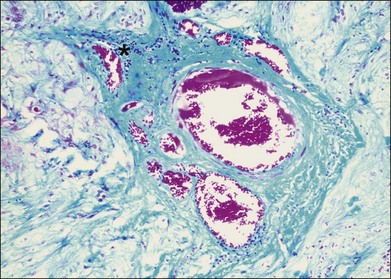
Inflammation often occurs in hypoxic-ischemic conditions. Diabetic retinopathy is characterized by retinal vascular nonperfusion and leakage. In an experimental rat model of diabetes mellitus (DM) induced by streptozotocin, retinovascular leukostasis develops within 1 week, followed by a progressive breakdown of the blood–retinal barrier. After 1 week, areas of nonperfusion and reperfusion develop, and extravascular leukocytes are observed in the retina.4 On a microscopic level in human diabetic retinopathy, the inflammation is demonstrated by vascular dilatation, exudation and transudation of fluids, and leukocyte (macrophages, neutrophils, and T lymphocytes) infiltration in the retina (Fig. 25.1).5–7 Retinal vein occlusion is another common condition associated with hypoxia and ischemia. In a histopathological study of 29 cases with central vein occlusion, lymphocytes and macrophages were observed at the occlusion site in 47% of cases (Fig. 25.2).8
Further evidence of the involvement of inflammatory processes in diabetic retinopathy and retinal vein occlusion is based on the upregulation of several inflammatory genes and the detection of inflammatory cytokines and chemokines under hypoxic-ischemic conditions. Tumor necrosis factor (TNF)-α, interleukin (IL)-1β, monocyte chemotactic protein 1 (MCP-1/CCL2), and macrophage inflammatory protein (MIP)/CCL3 transcripts have been detected in the ischemic-hypoxic retina. These proinflammatory cytokines, particularly TNF-α and IL-1β, are thought to play a major role in the breakdown of the blood–retinal barrier and the degeneration of retinal capillaries.9 CCL2 and RANTES (regulated upon activation, normal T-cell expressed and secreted)/CCL5 are significantly elevated in sera of patients with severe nonproliferative diabetic retinopathy compared with those who have less severe retinopathy.7 Increased C-reactive protein (CRP), IL-6, and TNF-α, and especially intercellular adhesion molecule (ICAM)-1, vascular cell adhesion molecule-1, and E-selectin are associated with nephropathy, retinopathy, and cardiovascular disease in both type 1 and type 2 diabetes.10 In proliferative diabetic retinopathy, vitreous cytokine levels of IL-6, IL-8, and CCL2 are strongly correlated with elevated VEGF.11 Patients with central retinal vein occlusion have significantly higher levels of aqueous and vitreous VEGF and IL-6 versus control subjects with nonischemic ocular disease.12,13
Type 1 DM is now considered a common autoimmune disorder with multiple genetic susceptibility loci as well as environmental risk factors. Recently, a genomewide association study of type 1 DM was combined in a meta-analysis with two other studies to examine a total of 7514 cases and 9045 reference samples. Several genetic regions, including those containing the immunoregulatory cytokine genes IL-10, IL-19, and IL-20, showed significant associations with type 1 DM, suggesting possible functional relevance of these genes to the disease pathogenesis.14 The Wellcome Trust Case Consortium genomewide association study of type 1 diabetic families identified a genetic susceptibility region which is close to the inflammatory genes Toll-like receptor (TLR)7 and TLR8.15 In another study, a custom panel of 1536 single-nucleotide polymorphisms (SNPs) located in 193 candidate genes was used to genotype 437 African Americans with type 1 DM, 128 with and 309 without severe diabetic retinopathy. The results indicated an association between genes involved in inflammation, such as ANGPT1 (angiopoietin 1), FLT1 (fms-related tyrosine kinase 1), PLA2G4A (cytosolic phospholipase A2), OLR1 (oxidized low-density lipoprotein lectin-like receptor 1), and PPARG (peroxisome proliferator-activated receptor-γ), and the progression of diabetic retinopathy.16 In contrast, a study of 315 patients and 335 age- and gender-matched control subjects showed no significant genetic association between central retinal vein occlusion and variants in many inflammation-related genes, including TNF-α, IL-1β, IL-6, IL-8, IL-10, and CCL2.17
The concept that diabetic retinopathy is a persistent low-grade inflammatory response to ischemia-induced retinal neovascularization and damage has led to the clinical application of anti-inflammatory treatments such as corticosteroids and anti-TNF-α agents.5,18 In addition to being a potent inflammatory molecule, TNF-α can also promote apoptosis and loss of retinal vascular pericytes and endothelial cells,19,20 increase retinal endothelial permeability,21 and activate microglia22 in experimental diabetic rats. Treatments that target factors related to retinal–blood barrier breakdown have also been used in retinal vein occlusion.
As corticosteroids have anti-inflammatory, antiangiogenic, and antivascular permeability properties, the administration of intravitreal corticosteroids (triamcinolone) has shown promising results for diabetic macular edema in a number of randomized clinical trials.9,23 However, in a carefully designed, prospective, randomized trial, the Diabetic Retinopathy Clinical Research Network showed that focal or grid laser photocoagulation was more effective and had fewer side-effects than intravitreal triamcinolone treatment of diabetic macular edema.24 Corticosteroids have also been used to treat both central and branch retinal vein occlusions.25 Case series have suggested that intravitreal injection of triamcinolone may be useful for the treatment of macular edema in patients with branch retinal vein occlusion.26 Nonetheless, due to a high rate of adverse events, the use of this treatment was not supported by the Standard care versus Corticosteroid for Retinal vein occlusion (SCORE) study, a randomized trial which included 411 patients with branch retinal vein occlusion and vision loss from macular edema.27 In another study of 437 patients with central retinal vein occlusion and 830 with branch vein occlusion, the longer-term delivery of intravitreal corticosteroids through a dexamethasone implant was reported to be associated with a shorter time to achieve a gain in visual acuity.28 Elevation of intraocular pressure and cataract progression are among the potential significant adverse effects for patients receiving intravitreal corticosteroids. Further development of noncorticosteroid agents that target the inflammatory components underlying retinovascular disease pathogenesis may provide future therapeutic options.
Oxidative stress
Oxidative processes occur through the removal of electrons from molecules. In biologic systems, energy is released when lipids, proteins, and carbohydrates are oxidized to form carbon dioxide and water. Oxidative reactions may also result in the production of reactive oxygen intermediates (ROIs), such as free radicals, hydrogen peroxide, and singlet oxygen. ROIs can damage other molecules and increase under conditions of irradiation, aging, reperfusion, inflammation, increased partial pressure of oxygen, cigarette smoke, and air pollution.29 The biologic mechanisms to prevent the detrimental effects of ROIs include cellular compartmentalization, repair of DNA and proteins, and neutralization by antioxidant compounds. The retina is an ideal environment for the generation of ROIs for several reasons: (1) high oxygen consumption; (2) high levels of cumulative irradiation; (3) RPE phagocytosis, which is an oxidative stress that produces ROIs; (4) high levels of polyunsaturated fatty acids in the photoreceptor outer-segment membranes; and (5) abundant photosensitizers in the neurosensory retina and RPE.29
Oxidative stress results when there is an imbalance between pro-oxidants and antioxidants, leading to molecular damage and/or a disruption of redox signaling.30 Inflammation and oxidative stress are closely interrelated: inflammatory cells can generate ROIs, and oxidative stress can induce inflammation through NF-κβ-mediated inflammatory gene expression.30 Oxidative stress plays a role in the pathogenesis of several retinal disorders, including AMD and retinopathy of prematurity (ROP).
Multiple changes are associated with the aging eye, including thickening of Bruch’s membrane, accumulation of lipofuscin in the RPE, and loss of parafoveal rods. In a model outlined by Curcio et al.31 and others, the RPE and Bruch’s membrane are modified or damaged by oxidative stress and enzymatic processes over time. The resultant damage slows the normal flow of materials such as lipids, protein, and lipofuscin from the outer retina and RPE through Bruch’s membrane. However, simple accumulation of material does not lead to AMD. The retained materials, including lipoproteins, may be modified by oxidative stress and become stimuli for inflammation.31 This model of Bruch’s membrane lipoprotein accumulation and modification in a chronic proinflammatory environment is similar to the current understanding of the pathogenesis of atherosclerosis.
Oxidative stress and inflammation in AMD
Several studies have provided evidence that oxidative stress is involved in AMD pathogenesis. Crabb et al. performed a proteomic analysis of drusen from normal and AMD donor eyes.32 They identified multiple proteins modified by oxidation, including cross-linked molecules of carboxyethyl pyrrole (CEP) protein adducts, tissue metalloproteinase inhibitor 3, and vitronectin. Carboxymethyl lysine, an advanced glycation end product produced through oxidation of carbohydrate, was also isolated. These oxidation-related products were identified more frequently in drusen from AMD eyes. In addition, drusen in AMD donor eyes were also more likely to contain crystallins, which are proteins upregulated during cellular stress such as oxidation. In another proteomics study, Yuan et al. isolated the Bruch’s membrane–choroid complex from postmortem AMD and control eyes.33 They found elevated levels of retinoid-processing proteins in the early and midstage AMD eyes. These retinoids are highly susceptible to oxidative damage, can accumulate in RPE lipofuscin granules, and activate complement. They also found significant elevation of an oxidation product-related receptor in eyes with advanced dry AMD. In the study by Yuan et al.,33 approximately 60% of the elevated proteins were involved in immune response or host defense, including complement factors C5 and C7, α-crystallin A, α-crystallin B, and major histocompatibility complex (MHC) class II molecule DRα. In contrast, several proteins classified as damage-associated molecular patterns (DAMPs) were present at similar levels in AMD and control eyes. Damaged cells release DAMPs which can bind receptors in the complement system, Toll-like receptors, and receptors for advanced glycation end products. These data suggest that both normal and AMD eyes express proteins in response to aging-associated damage; however, in AMD eyes the response to injury may evolve into a chronic proinflammatory environment.
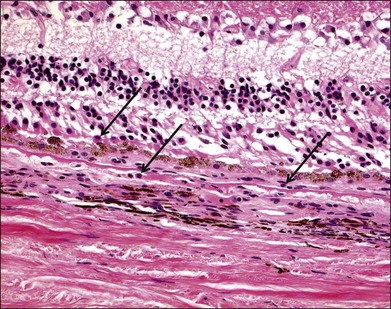
Numerous histopathologic studies of AMD eyes have documented the presence of macrophages and multinucleated giant cells, mainly associated with vascular channels and breaks in Bruch’s membrane (Fig. 25.3).34–39 It is unclear whether the macrophages contribute to the development of AMD lesions or if they accumulate as an adaptive response to injury associated with AMD. Some evidence suggests the relationship between oxidative stress and inflammation in AMD may be mediated in part by macrophages similar to the pathogenesis of atherosclerotic plaques. Oxidation of lipoproteins exposes phosphorylcholine (PC), which is also present on the membranes of apoptotic cells. PC can be bound by CRP, an opsonic molecule that activates the classical complement pathway and enhances phagocytosis via phagocytic complement receptors.40,41 In this manner, oxidative damage to the retina may lead to macrophage infiltration and activation via the innate immune response. An immunohistochemical study of surgically excised AMD-associated choroidal neovascular (CNV) membranes provides in vivo evidence to support this hypothesis.42 Oxidized lipoproteins were detected in RPE cells as well as on macrophages associated with the CNV membranes. Scavenger receptors for oxidized lipoproteins were also present on RPE cells, macrophages, and some endothelial cells in the CNV membranes, although the type and amount of scavenger receptor differed for each cell type. These data suggest macrophages accumulate in AMD lesions in order to phagocytize oxidized lipoproteins via scavenger receptors.
Hollyfield et al. have tied together earlier in vitro experiments and in vivo observations by producing AMD-like lesions in mice exposed to an oxidative product.43,44 They immunized mice with CEP adducts formed by the covalent interaction with an oxidation fragment of docosahexanoic acid (DHA), and found higher levels of antibodies against CEP in these mice. The authors hypothesized that the mouse immune system responds to the CEP antibodies by depositing complement below the RPE. In support of this idea, they detected complement C3d in Bruch’s membrane and on RPE cells in the CEP-immunized mice but not controls. Furthermore, there was a close relationship between the level of CEP-specific antibodies and the severity of RPE pathology. An intact immune response was required for CEP-induced RPE changes since Rag-deficient mice which lack mature T and B cells did not develop RPE pathology after CEP immunization. Monocytes and macrophages were observed in the interphotoreceptor matrix. Hollyfield et al. suggested the macrophages probably did not initiate the pathology since many lesions were observed without associated macrophages. They hypothesized that macrophages could be attracted by the release of cytokines from lysed cells and participate in debris removal, as supported by the presence of melanin-laden macrophages in the RPE lesions.
Microglia and AMD
Microglia are specialized tissue macrophages which constitute the main resident immune cells in the neuroretina. In concert with the RPE cells, choroidal macrophages, and dendritic cells, the microglia play an important role in maintaining normal retinal immune function as well as supporting the surrounding cells composing the neuroretina. In their quiescent state, microglia are inconspicuous stellate cells that produce anti-inflammatory IL-10 in response to RPE-derived cytokines. Microglia can rapidly transform into enlarged, activated microglia in response to microenvironment signals of infection, neuronal injury, ischemia, or oxidative stress.45–49 Quiescent and activated microglia secrete polypeptide neurotrophic factors, including brain-derived neurotrophic factor, ciliary neurotrophic factor (CNTF), nerve growth factor, neurotrophin-3, and basic fibroblast growth factor which influence neuron physiology and survival.47
The sub-RPE space is immunologically protected by the outer blood–retinal barrier and normally devoid of immune cells. It is hypothesized that as lipofuscin and oxidized material accumulate in the RPE and Bruch’s membrane of the aging eye, the RPE may be stimulated to produce inflammatory cytokines and chemokines which activate microglia and stimulate sub-RPE migration.48,50 Under conditions of chronic inflammation and activation, advanced glycation end products and oxidized lipids bind microglia and promote further inflammatory response. The microglia may have a detrimental effect in the neuroretina as they perpetuate the inflammation and injury cycle by secreting molecules that kill neighboring cells. Ma et al. cocultured activated retinal microglia with primary cultures of mouse RPE cells and measured lower levels of junctional proteins and RPE-65 as well as prominent structural disorganization and increased proliferation of the RPE cells in comparison to controls.51 They also found increased RPE expression and secretion of multiple proinflammatory cytokines, including IL-1β, TNF-α, IL-6, and granulocyte–macrophage colony-stimulating factor; the anti-inflammatory cytokine IL-10 was not markedly changed. Coculture with activated microglia also fostered a proangiogenic environment with increased mRNA levels of the matrix metalloproteinases MMP1 and MMP9 as well as protein levels of MMP1, MMP2, MMP9, and VEGF. Finally, Ma et al. evaluated the in vivo effects of subretinal activated microglia by transplanting them into the subretinal space of adult wild-type mice. When the animals were euthanized and examined 4 days after the subretinal injections, there were large and prominent choroidal neovascular membranes in these mice.
Both human and mouse in vivo studies provide evidence of microglial migration in conditions of aging or stress. Gupta et al. examined three AMD eyes with geographic atrophy and two normal donor eyes.52 They found quiescent microglia in the inner layers of the normal retinas; in contrast, the microglia were balloon-shaped and present in the inner and outer nuclear layers, often in association with degenerate rods in the AMD eyes. The activated microglia were also observed in the subretinal space of the AMD eyes; their rhodopsin-positive cytoplasmic inclusions were evidence of their role in phagocytizing photoreceptor debris. The findings suggest microglial migration occurs in AMD.
Xu et al. studied the mouse retina and observed that the numbers of subretinal microglia increased with age.50 They also noted an age-dependent accumulation of lipofuscin in subretinal microglia, possibly related to phagocytosis of photoreceptor outer segments. Shen et al. reported that naloxone, a potent microglial inhibitor, ameliorated AMD-like retinal lesions in Ccl2/Cx3cr1 double deficient with rd8 background mice via suppression of proinflammatory molecules of nitric oxide (NO), TNF-α, and IL-β.53 Under experimentally induced photo-oxidative stress in rats exposed to blue light, Rutar et al. used microarray expression analyses to show that light-induced injury induced differential expression of complement-related genes, including upregulation of complement activators and receptors.54 They also detected complement C3-expressing microglia in the outer nuclear layer and in the subretinal spaces adjacent to areas of injury.
Other inflammatory-related molecules and pathways
The inflammatory response to oxidative stress involves many other molecules and pathways which may represent future therapeutic targests. Neuroprotectin D1 (NPD1) is an endogenous anti-inflammatory mediator which is derived from the essential omega-3 polyunsaturated fatty acid, DHA.55,56 Multiple studies have shown the RPE generates NPD1 in response to oxidative stress.57–60 NPD1 promotes RPE survival by upregulating antiapoptotic genes and downregulating antiapoptotic genes57,58,61 and also inhibits cytokine-mediated proinflammatory gene induction.57,62
The PPARs are transcription factors which are activated by fatty acids. They exist in three forms, PPAR-γ, -α, and -δ (also known as -β, NUC-1, or FAAR), which differ in tissue distribution and ligand specificity.63 PPAR-γ is expressed by normal human RPE and upregulated in response to oxidative stress, including in AMD lesions.64 PPAR-γ activation may protect against oxidative injury through upregulation of antioxidative enzymes,65,66 downregulation of chemokines such as CXCR4 and CCL2,67,68 and modulation of microglial activation.69 Pharmacologic agonists for both PPAR-α (fibrates) and -γ (thiazolidinediones) have been investigated as potential treatments for ocular neovascularization through inhibition of the VEGF pathway63,70–72; however, due to the overlap between many components of the pathways of angiogenesis and inflammation, further clinical studies are warranted.73
Nuclear erythroid-2-related factor (NRF2) is another transcription factor which plays an important role in the cellular response to oxidative stress and may be regulated by PPAR-γ.68 NRF2 regulates antioxidant gene expression and complement activation to prevent further inflammation-mediated oxidative stress.74 To date, most studies have focused on the role of NRF2 in lung disease (modulation of macrophage activation in response to cigarette smoke)75 and neuroinflammation (downmodulation of activated microglia).76,77
Heat shock proteins (Hsps) prevent harmful protein aggregation by serving as molecular chaperones that bind damaged intracellular proteins and assist in the processes of repair or removal and degradation.78 Hsps participate in chaperone-mediated autophagy which is an oxidative stress-activated lysosomal pathway that results in degradation of cytoplasmic material and organelles. Some Hsps also have cytoprotective effects through modulation of apoptosis and stabilization of cytoskeletal proteins.79,80 Age-related decreases in Hsp activity and autophagy may contribute to the accumulation of aggregations of oxidized proteins and lipids (lipofuscin) in RPE cells.81–83 Oxidative stress is known to increase the expression of many of the Hsps, especially Hsp27 (also known as HspB1) and Hsp 70.80,84,85 Several studies have measured higher levels of Hsps, including Hsp27, αA- and αB-crystallin in drusen, Bruch’s membrane, and choroid in AMD eyes compared to normal controls.86,87 It is hypothesized that AMD eyes have higher levels of Hsps as an attempt to adapt to increased oxidative stress. However, the Hsp αB-crystallin may actually contribute to angiogenesis through modulation of VEGF-A, as suggested by the work of Kase et al.88
Genes and inflammation in AMD
Multiple genes related to inflammation have been linked to AMD risk and may provide evidence for the roles of oxidative stress and inflammation.89 Variations in the complement factor H gene (CFH) were the first genetic changes associated with AMD.90–93 CFH is a regulatory protein which suppresses formation of the C3 cleavage enzyme and inhibits the alternative pathway of complement activation. Variations in CFH may limit the protein’s ability to inhibit complement activation and contribute to the development of a chronic inflammatory state. A SNP in the DNA repair gene ERCC6 has a modest association with AMD risk; however, when present in combination with a SNP in the CFH intron there is a strong correlation with AMD.94 Polymorphisms in other complement genes, including factors C3 and I, have also been linked with increased risk of AMD.95–97 Conversely, variants associated with factors B, C2, and C7, as well as SERPING1, which encodes complement component 1 inhibitor, may be protective against AMD.98–100
Multiple chemokine and cytokine gene variants have also been associated with increased risk of AMD. CX3CR1 is a chemokine receptor found on macrophages, microglia, T cells, and astrocytes which mediates leukocyte migration and activation.101,102 SNPs in CX3CR1 have been associated with AMD risk. Combadière et al. showed that monocytes from AMD patients who were homozygous for a SNP in CX3CR1 had impaired chemotaxis and suggested a possible mechanism for the subretinal accumulation of microglia frequently observed in AMD.103 Other inflammation-related genes that may be linked to AMD risk include IL-8, TLR3, and TLR4.104–106
Finally, data from several studies suggest variants in two additional genes in an AMD susceptibility locus on chromosome 10q26 may play a role in AMD risk. ARMS2 has been localized to mitochondria which are a site of ROI generation.107 Multiple studies have shown correlations between an ARMS2 variant and advanced AMD.89,108,109 Of note, Wang et al. demonstrated an especially strong risk in AMD patients with elevated serum levels of the inflammatory markers CRP, IL-6, and soluble ICAM-1.110 Another gene on chromosome 10q26, HtrA1, is a stress-inducible member of the family of heat shock serine proteases. Multiple AMD-associated SNPs have been described, although there is some controversy whether HtrA1 or ARMS2 is the true gene associated with AMD.89,111
Oxidative stress and inflammation in retinopathy of prematurity
Oxidative stress also plays a role in ROP. Development of the human retinal vasculature continues through the 40th week of gestation; therefore, premature infants are born with an incomplete retinal blood supply. Inability to autoregulate the blood flow to the retina and choroid combined with external oxygen supplementation leads to exaggerated retinal oxygenation and increased oxidative stress.112,113 Oxidative injury causes endothelial cell apoptosis and death with resultant obliteration of retinal vessels.114 In addition to the inherent pro-oxidative characteristics of the retina discussed earlier in this section, relative to the adult, the neonatal retina contains more free iron to catalyze oxidizing reactions and fewer antioxidants. In the neonatal period, the cytochrome C oxidase and NO synthase inflammatory pathways are highly active, leading to production of ROIs and prostaglandins. ROIs react with NO to produce reactive nitrogen species which also contribute to retinal vascular degeneration.115–118 Lipid peroxidation of endothelial cell membranes and polyunsaturated fatty acids in the retina results in further damage. Peroxidation of arachidonic acid produces isoprostanes that have indirect cytotoxic effects on the retinal vascular endothelium.119 Lipid peroxidation also produces several proinflammatory mediators including lysophosphatidic acid, platelet-activated factor, and the eicosanoids derived from omega-3 and omega-6 long-chain polyunsaturated fatty acids.120 Oxidative stress and ROIs may also play an important role in neovascularization, one of the major complications of ROP, by triggering angiogenesis through VEGF and other signaling pathways, including Janus kinase-signal transducer and activator of transcription (JAK/STAT).121–124
Anti-inflammatory and antioxidant therapies in AMD and ROP
As research studies continue to clarify and expand our knowledge of the roles of oxidative stress and inflammation in the pathogenesis of AMD and ROP, the rationale for therapies targeting these pathways grows. A large randomized and controlled clinical trial, the Age-Related Eye Disease Study (AREDS), demonstrated the benefit of vitamin and mineral supplementation in preventing progression from early to advanced AMD.125 Although the exact mechanism is unknown, it is hypothesized that the AREDS vitamin formulation counteracts oxidative stresses in the retina. Several studies suggested supplementation with vitamin E, a potent scavenger of ROIs, suppressed the development of severe ROP disease in premature infants; however, subsequent trials have not reproduced these results.112 Dietary enrichment with anti-inflammatory lipids is another potential direction for disease prevention in both AMD and ROP.112,126 In AMD, multiple areas of evidence support the use of systemic and local anti-inflammatory therapies for disease prevention or in combination with anti-VEGF medications as treatment for neovascular disease. Several AMD clinical trials have demonstrated the benefits of immunomodulatory medications such as methotrexate, sirolimus, and daclizumab; ongoing studies are now investigating complement component inhibitors.127 In the future, treatment and prevention strategies for retinal diseases should be multifactorial in order to address the complex and interacting pathways associated with aging, oxidative stress, and inflammation.
Trauma
Inflammation also plays a role in physical retinal injury such as rhegmatogenous retinal detachment (RRD) and intraocular foreign body. Retinal vasculitis may develop after RRD (Fig. 25.4). Inflammatory cells, including foreign-body giant cells, are often present surrounding an intraocular foreign body. Separation of the neuroretina from the RPE disrupts the normal homeostasis of the retina. Studies of experimental RRD animal models and human surgical specimens provide insight into the cellular and molecular processes that occur in the detached neuroretina. In the earliest phase after detachment, the photoreceptor outer segments become disorganized and slough off in the subretinal space where they are phagocytized and removed by infiltrating macrophages and RPE cells.128–130 Subsequently, some photoreceptor cells become apoptotic131–133 and are phagocytized by macrophages and activated microglia infiltrating the subretinal space.128,134 Other changes associated with RRD include proliferation of Müller cells, RPE, astrocytes, microglia, pericytes, and capillary endothelial cells,135 as well as Müller cell hypertrophy and retinal gliosis. RPE cells frequently undergo hypertrophy, hyperplasia, hypotrophy or atrophy, and migrate to the subretinal space.130,136,137 Over time, unless the retina is reattached, further photoreceptor atrophy accompanies glial scarring, subretinal fibrosis, proliferative vitreoretinopathy (PVR), and epiretinal membrane formation.138,139
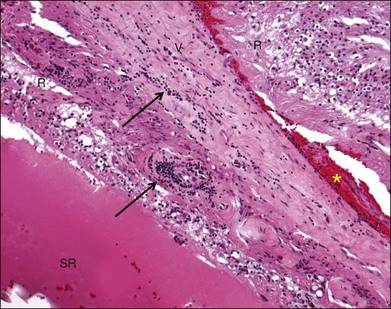
Several studies provide evidence of an early inflammatory response to RRD. As early as 1 hour after experimental RRD, Nakazawa et al. measured increased mRNA and protein levels of TNF-α, IL-1β, and CCL2.140 Hollborn et al. analyzed mRNA levels 24 hours after experimental RRD and identified upregulation of genes related to inflammation and immune response, antioxidants and metal homeostasis, intracellular proteolysis, and blood coagulation/fibrinolysis.141 The inflammation-related genes included CCL2, MHC class I and II molecules, interferon-regulatory factors, TLR-2, TLR-4, and IL-18. In another gene expression study 7 days after experimental RRD, Zacks reported multiple upregulated genes, including those involved in inflammation (i.e., interferon pathway-related), immune complexes (i.e., CFH), antigen presentation, acute stress response (i.e., crystallins), and metabolism.129
High levels of CCL2 are produced by Müller cells after RRD.140,142 CCL2 mediates photoreceptor apoptosis and the macrophage/microglial response to RRD. Blocking or deleting expression of CCL2 in an experimental model of RRD nearly eliminates photoreceptor apoptosis perhaps by preventing macrophage/microglia generation of ROIs.142 In another experiment, Nakazawa et al. administered intraperitoneal dexamethasone and suppressed upregulation of TNF-α and photoreceptor degeneration after experimental RRD.143
Photoreceptor loss can lead to irreversible vision loss even after retinal reattachment; however, significant numbers of photoreceptors may survive for days to weeks after RRD. In neural systems, IL-6 could actually enhance cell survival by preventing apoptosis and therefore reducing photoreceptor death after RRD. Zacks et al. showed that genes in the IL-6 pathway are upregulated within 24 hours of experimental RRD.144 Transcript and protein levels of IL-6 as well as CNTF, a member of the IL-6 family with neuroprotective effects, were increased in experimentally detached retina.144 They also measured increased transcription of two pathways that might have beneficial effects on photoreceptor survival, transforming growth factor-β (antiapoptosis) and aryl hydrocarbon receptor (antioxidative stress).144 In another study, Chong et al. showed that absence of IL-6 function resulted in increased photoreceptor apoptosis after experimental RRD in both IL-6 double-knockout as well as in wild-type mice treated with anti-IL-6 antibody.145 Subretinal injection of IL-6 immediately or up to 2 weeks after creation of the experimental RD in wild-type Norway rats resulted in preservation of photoreceptors 4 weeks later, but this effect had disappeared at 8 weeks. Finally, intraperitoneal injections of minocycline, a semisynthetic tetracycline antibiotic with antiapoptotic and anti-inflammatory effects, resulted in preservation of the photoreceptor inner and outer segments, decreased photoreceptor apoptosis, and decreased glial activation.146
Among the complications of RRD, PVR is associated with an increased risk of recurrent detachment and worse visual outcome. Chronic or poorly regulated inflammation may be correlated with the development of PVR. Multiple studies of vitreous and subretinal fluid specimens from human RRD patients have demonstrated increased levels of inflammatory cytokines and related molecules (i.e., IL-1, IL-6, TNF-α, interferon-γ, ICAM-1) and growth factors (i.e., VEGF, fibroblast growth factor, and hepatocyte growth factor) in association with PVR.147–159 Examination of PVR membranes also has revealed macrophages, lymphocytes, MHC class II positive cells, as well as deposits of immunoglobulins, complement factors, and cellular adhesion molecules.160 In an attempt to decrease the incidence of PVR, multiple adjunctive treatments have been investigated. Two antiproliferative agents, 5-fluorouracil and daunorubicin, were administered intravitreally during or at the completion of vitrectomy for PVR with varied success depending on the outcomes measured.160,161 Despite several clinical trials, neither adjunctive agent is currently in wide use.
An intraocular foreign body may occur as a result of penetrating ocular trauma or through an iatrogenic mechanism such as the use of a silicone oil tamponade for repair of a retinal detachment. Although some intraocular foreign bodies such as acrylic plastic may be tolerated with minimal inflammation, others such as organic material may elicit immediate and intense inflammation. Surgical specimens are rarely available for pathologic examination; however, a few case reports in the literature have shed light on the intraocular inflammatory responses to foreign bodies. Lee et al. described a focal posttraumatic choroidal granulomatous inflammation that presented as a subretinal mass 19 weeks after the eye sustained a penetrating injury with a metal projectile.162 The surgically excised mass consisted of fibrovascular tissue with reactive vascularization, histiocytes, lymphocytes, and occasional giant cells. Several cases were previously described in the literature; all were associated with penetrating trauma and several had identifiable foreign material.163 They differ from sympathetic ophthalmia due to the absence of inflammation in the noninjured eye.
Wickham et al. examined enucleated globes that had a history of retinal detachment surgery with silicone oil.164 They reported the presence of silicone oil in the iris, ciliary body, retina, optic nerve, trabecular meshwork, and epiretinal membranes. Macrophages were observed in close association with the silicone oil, and the number of macrophages generally reflected the degree of silicone oil distribution. The macrophages often contained phagocytized silicone oil. Focal areas of retinal infiltration by macrophages occurred at retinectomy sites and were associated with subretinal silicone oil. T and B lymphocytes were located only in the inflammatory membranes (i.e., cyclitic, PVR, epiretinal, and glaucomatous) and among the perivascular inflammation. In general, the distribution and number of lymphocytes did not differ from the control eyes with a history of RRD or trauma. The authors concluded that intraocular silicone oil elicits a localized inflammatory response mediated by macrophages.
Conclusion
Acute and chronic inflammatory processes are intricately involved in ischemia, oxidative stress, and trauma in the retina. The inflammatory response may represent a double-edged sword with both harmful and beneficial effects.165 Acute inflammation is often an essential and helpful response to maintain normal function; for example, macrophages can remove necrotic or apoptotic cells and promote wound healing. However, prolonged or dysregulated chronic inflammation can magnify and perpetuate damage of the retina, causing atrophic chorioretinal scarring or PVR. Since inflammation is only one component of most retinal diseases, current anti-inflammatory therapies merely provide palliative treatment. As we better understand the inflammatory pathways and mechanisms, we will develop more effective prevention and treatment strategies to manage all retinal diseases.
1 Cohen LH, Noell WK. Relationships between visual function and metabolism. In: Graymore CN, ed. Biochemistry of the retina. New York: Academic Press, 1965.
2 Young RW, Bok D. Participation of the retinal pigment epithelium in the rod outer segment renewal process. J Cell Biol. 1969;42:392–403.
3 Wilson TM, Strang R, Wallace J, et al. The measurement of the choroidal blood flow in the rabbit using 85-krypton. Exp Eye Res. 1973;16:421–425.
4 Miyamoto K, Khosrof S, Bursell SE, et al. Prevention of leukostasis and vascular leakage in streptozotocin-induced diabetic retinopathy via intercellular adhesion molecule-1 inhibition. Proc Natl Acad Sci U S A. 1999;96:10836–10841.
5 Adamis AP, Berman AJ. Immunological mechanisms in the pathogenesis of diabetic retinopathy. Semin Immunopathol. 2008;30:65–84.
6 Cusick M, Chew EY, Chan CC, et al. Histopathology and regression of retinal hard exudates in diabetic retinopathy after reduction of elevated serum lipid levels. Ophthalmology. 2003;110:2126–2133.
7 Meleth AD, Agron E, Chan CC, et al. Serum inflammatory markers in diabetic retinopathy. Invest Ophthalmol Vis Sci. 2005;46:4295–4301.
8 Green WR, Chan CC, Hutchins GM, et al. Central retinal vein occlusion: a prospective histopathologic study of 29 eyes in 28 cases. Retina. 1981;1:27–55.
9 Kaur C, Foulds WS, Ling EA. Blood–retinal barrier in hypoxic ischaemic conditions: basic concepts, clinical features and management. Prog Retin Eye Res. 2008;27:622–647.
10 Goldberg RB. Cytokine and cytokine-like inflammation markers, endothelial dysfunction, and imbalanced coagulation in development of diabetes and its complications. J Clin Endocrinol Metab. 2009;94:3171–3182.
11 Yoshimura T, Sonoda KH, Sugahara M, et al. Comprehensive analysis of inflammatory immune mediators in vitreoretinal diseases. PLoS ONE. 2009;4:e8158.
12 Noma H, Funatsu H, Mimura T, et al. Vitreous levels of interleukin-6 and vascular endothelial growth factor in macular edema with central retinal vein occlusion. Ophthalmology. 2009;116:87–93.
13 Noma H, Funatsu H, Mimura T, et al. Aqueous humor levels of vasoactive molecules correlate with vitreous levels and macular edema in central retinal vein occlusion. Eur J Ophthalmol. 2010;20:402–409.
14 Barrett JC, Clayton DG, Concannon P, et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet. 2009;41:703–707.
15 Cooper JD, Walker NM, Smyth DJ, et al. Follow-up of 1715 SNPs from the Wellcome Trust Case Control Consortium genome-wide association study in type 1 diabetes families. Genes Immun. 2009;10(Suppl 1):S85–S94.
16 Roy MS, Hallman DM, Fu YP, et al. Assessment of 193 candidate genes for retinopathy in African Americans with type 1 diabetes. Arch Ophthalmol. 2009;127:605–612.
17 Maier R, Steinbrugger I, Haas A, et al. Role of inflammation-related gene polymorphisms in patients with central retinal vein occlusion. Ophthalmology. 2011;119:1125–1129.
18 Sfikakis PP, Grigoropoulos V, Emfietzoglou I, et al. Infliximab for diabetic macular edema refractory to laser photocoagulation: a randomized, double-blind, placebo-controlled, crossover, 32-week study. Diabetes Care. 2010;33:1523–1528.
19 Behl Y, Krothapalli P, Desta T, et al. Diabetes-enhanced tumor necrosis factor-alpha production promotes apoptosis and the loss of retinal microvascular cells in type 1 and type 2 models of diabetic retinopathy. Am J Pathol. 2008;172:1411–1418.
20 Joussen AM, Doehmen S, Le ML, et al. TNF-alpha mediated apoptosis plays an important role in the development of early diabetic retinopathy and long-term histopathological alterations. Mol Vis. 2009;15:1418–1428.
21 Aveleira CA, Lin CM, Abcouwer SF, et al. TNF-alpha signals through PKCzeta/NF-kappaB to alter the tight junction complex and increase retinal endothelial cell permeability. Diabetes. 2010;59:2872–2882.
22 Ibrahim AS, El-Shishtawy MM, Pena A, Jr., et al. Genistein attenuates retinal inflammation associated with diabetes by targeting of microglial activation. Mol Vis. 2010;16:2033–2042.
23 Bandello F, Iacono P, Battaglia Parodi M. Treatment options for diffuse diabetic macular edema. Eur J Ophthalmol. 2010;21:45–50.
24 Diabetic Retinopathy Clinical Research Network. A randomized trial comparing intravitreal triamcinolone acetonide and focal/grid photocoagulation for diabetic macular edema. Ophthalmology. 2008;115:1447–1449. 1449. e1–10
25 Wong TY, Scott IU. Clinical practice. Retinal-vein occlusion. N Engl J Med. 2010;363:2135–2144.
26 Jonas JB, Akkoyun I, Kamppeter B, et al. Branch retinal vein occlusion treated by intravitreal triamcinolone acetonide. Eye (Lond). 2005;19:65–71.
27 Scott IU, Ip MS, VanVeldhuisen PC, et al. A randomized trial comparing the efficacy and safety of intravitreal triamcinolone with standard care to treat vision loss associated with macular edema secondary to branch retinal vein occlusion: the Standard care vs Corticosteroid for Retinal Vein occlusion (SCORE) study report 6. Arch Ophthalmol. 2009;127:1115–1128.
28 Haller JA, Bandello F, Belfort R, Jr., et al. Randomized, sham-controlled trial of dexamethasone intravitreal implant in patients with macular edema due to retinal vein occlusion. Ophthalmology. 2010;117:1134–1146. e3
29 Beatty S, Koh H, Phil M, et al. The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv Ophthalmol. 2000;45:115–134.
30 Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011;21:103–115.
31 Curcio CA, Johnson M, Huang JD, et al. Apolipoprotein B-containing lipoproteins in retinal aging and age-related macular degeneration. J Lipid Res. 2010;51:451–467.
32 Crabb JW, Miyagi M, Gu X, et al. Drusen proteome analysis: an approach to the etiology of age-related macular degeneration. Proc Natl Acad Sci U S A. 2002;99:14682–14687.
33 Yuan X, Gu X, Crabb JS, et al. Quantitative proteomics: comparison of the macular Bruch membrane/choroid complex from age-related macular degeneration and normal eyes. Mol Cell Proteomics. 2010;9:1031–1046.
34 Killingsworth MC, Sarks JP, Sarks SH. Macrophages related to Bruch’s membrane in age-related macular degeneration. Eye (Lond). 1990;4:613–621.
35 Green WR, Enger C. Age-related macular degeneration histopathologic studies. The 1992 Lorenz E. Zimmerman Lecture. Ophthalmology. 1993;100:1519–1535.
36 Dastgheib K, Green WR. Granulomatous reaction to Bruch’s membrane in age-related macular degeneration. Arch Ophthalmol. 1994;112:813–818.
37 Grossniklaus HE, Cingle KA, Yoon YD, et al. Correlation of histologic 2-dimensional reconstruction and confocal scanning laser microscopic imaging of choroidal neovascularization in eyes with age-related maculopathy. Arch Ophthalmol. 2000;118:625–629.
38 Grossniklaus HE, Miskala PH, Green WR, et al. Histopathologic and ultrastructural features of surgically excised subfoveal choroidal neovascular lesions: submacular surgery trials report no. 7. Arch Ophthalmol. 2005;123:914–921.
39 Cao X, Shen D, Patel MM, et al. Macrophage polarization in the maculae of age-related macular degeneration: a pilot study. Pathol Int. 2011;61:528–535.
40 Chang MK, Binder CJ, Torzewski M, et al. C-reactive protein binds to both oxidized LDL and apoptotic cells through recognition of a common ligand: phosphorylcholine of oxidized phospholipids. Proc Natl Acad Sci U S A. 2002;99:13043–13048.
41 Hazen SL, Chisolm GM. Oxidized phosphatidylcholines: pattern recognition ligands for multiple pathways of the innate immune response. Proc Natl Acad Sci U S A. 2002;99:12515–12517.
42 Kamei M, Yoneda K, Kume N, et al. Scavenger receptors for oxidized lipoprotein in age-related macular degeneration. Invest Ophthalmol Vis Sci. 2007;48:1801–1807.
43 Hollyfield JG, Bonilha VL, Rayborn ME, et al. Oxidative damage-induced inflammation initiates age-related macular degeneration. Nat Med. 2008;14:194–198.
44 Hollyfield JG, Perez VL, Salomon RG. A hapten generated from an oxidation fragment of docosahexaenoic acid is sufficient to initiate age-related macular degeneration. Mol Neurobiol. 2010;41:290–298.
45 Ng TF, Streilein JW. Light-induced migration of retinal microglia into the subretinal space. Invest Ophthalmol Vis Sci. 2001;42:3301–3310.
46 Chen L, Yang P, Kijlstra A. Distribution, markers, and functions of retinal microglia. Ocul Immunol Inflamm. 2002;10:27–39.
47 Langmann T. Microglia activation in retinal degeneration. J Leukoc Biol. 2007;81:1345–1351.
48 Xu H, Chen M, Forrester JV. Para-inflammation in the aging retina. Prog Retin Eye Res. 2009;28:348–368.
49 Karlstetter M, Ebert S, Langmann T. Microglia in the healthy and degenerating retina: insights from novel mouse models. Immunobiology. 2010;215:685–691.
50 Xu H, Chen M, Manivannan A, et al. Age-dependent accumulation of lipofuscin in perivascular and subretinal microglia in experimental mice. Aging Cell. 2008;7:58–68.
51 Ma W, Zhao L, Fontainhas AM, et al. Microglia in the mouse retina alter the structure and function of retinal pigmented epithelial cells: a potential cellular interaction relevant to AMD. PLoS ONE. 2009;4:e7945.
52 Gupta N, Brown KE, Milam AH. Activated microglia in human retinitis pigmentosa, late-onset retinal degeneration, and age-related macular degeneration. Exp Eye Res. 2003;76:463–471.
53 Shen D, Cao X, Zhao L, et al. Naloxone ameliorates retinal lesions in Ccl2/Cx3cr1 double-deficient mice via modulation of microglia. Invest Ophthalmol Vis Sci. 2011;52:2897–2904.
54 Rutar M, Natoli R, Kozulin P, et al. Analysis of complement expression in light-induced retinal degeneration: Synthesis and deposition of C3 by microglia/macrophages is associated with focal photoreceptor degeneration. Invest Ophthalmol Vis Sci. 2011;52:5347–5358.
55 Bazan NG. Homeostatic regulation of photoreceptor cell integrity: significance of the potent mediator neuroprotectin D1 biosynthesized from docosahexaenoic acid: the Proctor Lecture. Invest Ophthalmol Vis Sci. 2007;48:4866–4881.
56 Serhan CN, Yacoubian S, Yang R. Anti-inflammatory and proresolving lipid mediators. Annu Rev Pathol. 2008;3:279–312.
57 Mukherjee PK, Marcheselli VL, Serhan CN, et al. Neuroprotectin D1: a docosahexaenoic acid-derived docosatriene protects human retinal pigment epithelial cells from oxidative stress. Proc Natl Acad Sci U S A. 2004;101:8491–8496.
58 Mukherjee PK, Marcheselli VL, Barreiro S, et al. Neurotrophins enhance retinal pigment epithelial cell survival through neuroprotectin D1 signaling. Proc Natl Acad Sci U S A. 2007;104:13152–13157.
59 Mukherjee PK, Marcheselli VL, de Rivero Vaccari JC, et al. Photoreceptor outer segment phagocytosis attenuates oxidative stress-induced apoptosis with concomitant neuroprotectin D1 synthesis. Proc Natl Acad Sci U S A. 2007;104:13158–13163.
60 Bazan NG. Neurotrophins induce neuroprotective signaling in the retinal pigment epithelial cell by activating the synthesis of the anti-inflammatory and anti-apoptotic neuroprotectin D1. Adv Exp Med Biol. 2008;613:39–44.
61 Antony R, Lukiw WJ, Bazan NG. Neuroprotectin D1 induces dephosphorylation of Bcl-xL in a PP2A-dependent manner during oxidative stress and promotes retinal pigment epithelial cell survival. J Biol Chem. 2010;285:18301–18308.
62 Marcheselli VL, Hong S, Lukiw WJ, et al. Novel docosanoids inhibit brain ischemia-reperfusion-mediated leukocyte infiltration and pro-inflammatory gene expression. J Biol Chem. 2003;278:43807–43817.
63 Malchiodi-Albedi F, Matteucci A, Bernardo A, et al. PPAR-gamma, microglial cells, and ocular inflammation: new venues for potential therapeutic approaches. PPAR Res. 2008;2008:295784.
64 Herzlich AA, Ding X, Shen D, et al. Peroxisome proliferator-activated receptor expression in murine models and humans with age-related macular degeneration. Open Biol J. 2009;2:141–148.
65 Garg TK, Chang JY. Oxidative stress causes ERK phosphorylation and cell death in cultured retinal pigment epithelium: prevention of cell death by AG126 and 15-deoxy-delta 12, 14-PGJ2. BMC Ophthalmol. 2003;3:5.
66 Qin S, McLaughlin AP, De Vries GW. Protection of RPE cells from oxidative injury by 15-deoxy-delta12,14-prostaglandin J2 by augmenting GSH and activating MAPK. Invest Ophthalmol Vis Sci. 2006;47:5098–5105.
67 Kostadinova R, Wahli W, Michalik L. PPARs in diseases: control mechanisms of inflammation. Curr Med Chem. 2005;12:2995–3009.
68 Rodrigues GA, Maurier-Mahé F, Shurland DL, et al. Differential effects of PPARgamma ligands on oxidative stress-induced death of retinal pigmented epithelial cells. Invest Ophthalmol Vis Sci. 2011;52:890–903.
69 Bernardo A, Minghetti L. PPAR-gamma agonists as regulators of microglial activation and brain inflammation. Curr Pharm Des. 2006;12:93–109.
70 Murata T, He S, Hangai M, et al. Peroxisome proliferator-activated receptor-gamma ligands inhibit choroidal neovascularization. Invest Ophthalmol Vis Sci. 2000;41:2309–2317.
71 Del V, Cano M, Gehlbach PL. PPAR-alpha ligands as potential therapeutic agents for wet age-related macular degeneration. PPAR Res. 2008;2008:821592.
72 Pershadsingh HA, Moore DM. PPARgamma agonists: potential as therapeutics for neovascular retinopathies. PPAR Res. 2008;2008:164273.
73 Herzlich AA, Tuo J, Chan CC. Peroxisome proliferator-activated receptor and age-related macular degeneration. PPAR Res. 2008;2008:389507.
74 Cano M, Thimmalappula R, Fujihara M, et al. Cigarette smoking, oxidative stress, the anti-oxidant response through Nrf2 signaling, and age-related macular degeneration. Vision Res. 2010;50:652–664.
75 Rangasamy T, Cho CY, Thimmulappa RK, et al. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J Clin Invest. 2004;114:1248–1259.
76 Innamorato NG, Rojo AI, García-Yagüe AJ, et al. The transcription factor Nrf2 is a therapeutic target against brain inflammation. J Immunol. 2008;181:680–689.
77 Rojo AI, Innamorato NG, Martín-Moreno AM, et al. Nrf2 regulates microglial dynamics and neuroinflammation in experimental Parkinson’s disease. Glia. 2010;58:588–598.
78 Kaarniranta K, Salminen A, Eskelinen EL, et al. Heat shock proteins as gatekeepers of proteolytic pathways – implications for age-related macular degeneration (AMD). Ageing Res Rev. 2009;8:128–139.
79 Alge CS, Priglinger SG, Neubauer AS, et al. Retinal pigment epithelium is protected against apoptosis by alphaB-crystallin. Invest Ophthalmol Vis Sci. 2002;43:3575–3582.
80 O’Reilly AM, Currie RW, Clarke DB. HspB1 (Hsp 27) expression and neuroprotection in the retina. Mol Neurobiol. 2010;42:124–132.
81 Louie JL, Kapphahn RJ, Ferrington DA. Proteasome function and protein oxidation in the aged retina. Exp Eye Res. 2002;75:271–284.
82 Kapphahn RJ, Bigelow EJ, Ferrington DA. Age-dependent inhibition of proteasome chymotrypsin-like activity in the retina. Exp Eye Res. 2007;84:646–654.
83 Jung T, Bader N, Grune T. Lipofuscin: formation, distribution, and metabolic consequences. Ann N Y Acad Sci. 2007;1119:97–111.
84 Strunnikova N, Baffi J, Gonzalez A, et al. Regulated heat shock protein 27 expression in human retinal pigment epithelium. Invest Ophthalmol Vis Sci. 2001;42:2130–2138.
85 Yaung J, Jin M, Barron E, et al. alpha-Crystallin distribution in retinal pigment epithelium and effect of gene knockouts on sensitivity to oxidative stress. Mol Vis. 2007;13:566–577.
86 Nakata K, Crabb JW, Hollyfield JG. Crystallin distribution in Bruch’s membrane–choroid complex from AMD and age-matched donor eyes. Exp Eye Res. 2005;80(6):821–826.
87 Pons M, Cousins SW, Csaky KG, et al. Cigarette smoke-related hydroquinone induces filamentous actin reorganization and heat shock protein 27 phosphorylation through p38 and extracellular signal-regulated kinase 1/2 in retinal pigment epithelium: implications for age-related macular degeneration. Am J Pathol. 2010;177:1198–1213.
88 Kase S, He S, Sonoda S, et al. alphaB-crystallin regulation of angiogenesis by modulation of VEGF. Blood. 2010;115:3398–3406.
89 Ding X, Patel M, Chan CC. Molecular pathology of age-related macular degeneration. Prog Retin Eye Res. 2009;28:1–18.
90 Hageman GS, Anderson DH, Johnson LV, et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci U S A. 2005;102:7227–7232.
91 Edwards AO, Ritter R, 3rd., Abel KG, et al. Complement factor H polymorphism and age-related macular degeneration. Science. 2005;308:421–424.
92 Haines JL, Hauser MA, Schmidt S, et al. Complement factor H variant increases the risk of age-related macular degeneration. Science. 2005;308:419–421.
93 Klein RJ, Zeiss C, Chew EY, et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308:385–389.
94 Tuo J, Ning B, Bojanowski CM, et al. Synergic effect of polymorphisms in ERCC6 5’ flanking region and complement factor H on age-related macular degeneration predisposition. Proc Natl Acad Sci U S A. 2006;103:9256–9261.
95 Yates JR, Sepp T, Matharu BK, et al. Complement C3 variant and the risk of age-related macular degeneration. N Engl J Med. 2007;357:553–561.
96 Fagerness JA, Maller JB, Neale BM, et al. Variation near complement factor I is associated with risk of advanced AMD. Eur J Hum Genet. 2009;17:100–104.
97 Thakkinstian A, McKay GJ, McEvoy M, et al. Systematic review and meta-analysis of the association between complement component 3 and age-related macular degeneration: a huge review and meta-analysis. Am J Epidemiol. 2011;173:1365–1379.
98 Gold B, Merriam JE, Zernant J, et al. Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat Genet. 2006;38:458–462.
99 Dinu V, Miller PL, Zhao H. Evidence for association between multiple complement pathway genes and AMD. Genet Epidemiol. 2007;31:224–237.
100 Ennis S, Jomary C, Mullins R, et al. Association between the SERPING1 gene and age-related macular degeneration: a two-stage case-control study. Lancet. 2008;372:1828–1834.
101 Chan CC, Tuo J, Bojanowski CM, et al. Detection of CX3CR1 single nucleotide polymorphism and expression on archived eyes with age-related macular degeneration. Histol Histopathol. 2005;20:857–863.
102 Tuo J, Smith BC, Bojanowski CM, et al. The involvement of sequence variation and expression of CX3CR1 in the pathogenesis of age-related macular degeneration. FASEB J. 2004;18:1297–1299.
103 Combadière C, Feumi C, Raoul W, et al. CX3CR1-dependent subretinal microglia cell accumulation is associated with cardinal features of age-related macular degeneration. J Clin Invest. 2007;117:2920–2928.
104 Goverdhan SV, Ennis S, Hannan SR, et al. Interleukin-8 promoter polymorphism -251A/T is a risk factor for age-related macular degeneration. Br J Ophthalmol. 2008;92:537–540.
105 Zareparsi S, Buraczynska M, Branham KE, et al. Toll-like receptor 4 variant D299G is associated with susceptibility to age-related macular degeneration. Hum Mol Genet. 2005;14:1449–1455.
106 Yang Z, Stratton C, Francis PJ, et al. Toll-like receptor 3 and geographic atrophy in age-related macular degeneration. N Engl J Med. 2008;359:1456–1463.
107 Fritsche LG, Loenhardt T, Janssen A, et al. Age-related macular degeneration is associated with an unstable ARMS2 (LOC387715) mRNA. Nat Genet. 2008;40:892–896.
108 Jakobsdottir J, Conley YP, Weeks DE, et al. Susceptibility genes for age-related maculopathy on chromosome 10q26. Am J Hum Genet. 2005;77:389–407.
109 Schmidt S, Hauser MA, Scott WK, et al. Cigarette smoking strongly modifies the association of LOC387715 and age-related macular degeneration. Am J Hum Genet. 2006;78:852–864.
110 Wang JJ, Ross RJ, Tuo J, et al. The LOC387715 polymorphism, inflammatory markers, smoking, and age-related macular degeneration. A population-based case-control study. Ophthalmology. 2008;115:693–699.
111 Friedrich U, Myers CA, Fritsche LG, et al. Risk- and non-risk-associated variants at the 10q26 AMD locus influence ARMS2 mRNA expression but exclude pathogenic effects due to protein deficiency. Hum Mol Genet. 2011;20:1387–1399.
112 Sapieha P, Joyal JS, Rivera JC, et al. Retinopathy of prematurity: understanding ischemic retinal vasculopathies at an extreme of life. J Clin Invest. 2010;120:3022–3032.
113 Hartnett ME. The effects of oxygen stresses on the development of features of severe retinopathy of prematurity: knowledge from the 50/10 OIR model. Doc Ophthalmol. 2010;120:25–39.
114 Saito Y, Geisen P, Uppal A, et al. Inhibition of NAD(P)H oxidase reduces apoptosis and avascular retina in an animal model of retinopathy of prematurity. Mol Vis. 2007;13:840–853.
115 Hardy P, Dumont I, Bhattacharya M, et al. Oxidants, nitric oxide and prostanoids in the developing ocular vasculature: a basis for ischemic retinopathy. Cardiovasc Res. 2000;47:489–509.
116 Beauchamp MH, Sennlaub F, Speranza G, et al. Redox-dependent effects of nitric oxide on microvascular integrity in oxygen-induced retinopathy. Free Radic Biol Med. 2004;37:1885–1894.
117 Gu X, El-Remessy AB, Brooks SE, et al. Hyperoxia induces retinal vascular endothelial cell apoptosis through formation of peroxynitrite. Am J Physiol Cell Physiol. 2003;285:C546–C554.
118 Balazy M. Trans-arachidonic acids: new mediators of inflammation. J Physiol Pharmacol. 2000;51:597–607.
119 Brault S, Martinez-Bermudez AK, Marrache AM, et al. Selective neuromicrovascular endothelial cell death by 8-Iso-prostaglandin F2alpha: possible role in ischemic brain injury. Stroke. 2003;34:776–782.
120 Hardy P, Beauchamp M, Sennlaub F, et al. New insights into the retinal circulation: inflammatory lipid mediators in ischemic retinopathy. Prostaglandins Leukot Essent Fatty Acids. 2005;72:301–325.
121 Ushio-Fukai M. VEGF signaling through NADPH oxidase-derived ROS. Antioxid Redox Signal. 2007;9:731–739.
122 Saito Y, Uppal A, Byfield G, et al. Activated NAD(P)H oxidase from supplemental oxygen induces neovascularization independent of VEGF in retinopathy of prematurity model. Invest Ophthalmol Vis Sci. 2008;49:1591–1598.
123 Liu T, Castro S, Brasier AR, et al. Reactive oxygen species mediate virus-induced STAT activation: role of tyrosine phosphatases. J Biol Chem. 2004;279:2461–2469.
124 Al-Shabrawey M, Bartoli M, El-Remessy AB, et al. Inhibition of NAD(P)H oxidase activity blocks vascular endothelial growth factor overexpression and neovascularization during ischemic retinopathy. Am J Pathol. 2005;167:599–607.
125 Krishnadev N, Meleth AD, Chew EY. Nutritional supplements for age-related macular degeneration. Curr Opin Ophthalmol. 2010;21:184–189.
126 Stahl A, Krohne TU, Sapieha P, et al. Lipid metabolites in the pathogenesis and treatment of neovascular eye disease. Br J Ophthalmol. 2011;95:1496–1501.
127 Wang Y, Wang VM, Chan CC. The role of anti-inflammatory agents in age-related macular degeneration (AMD) treatment. Eye (Lond). 2011;25:127–139.
128 Lewis GP, Sethi CS, Carter KM, et al. Microglial cell activation following retinal detachment: a comparison between species. Mol Vis. 2005;11:491–500.
129 Zacks DN. Gene transcription profile of the detached retina (an AOS thesis). Trans Am Ophthalmol Soc. 2009;107:343–382.
130 Wickham L, Charteris DG. Glial cell changes of the human retina in proliferative vitreoretinopathy. Dev Ophthalmol. 2009;44:37–45.
131 Chang CJ, Lai WW, Edward DP, et al. Apoptotic photoreceptor cell death after traumatic retinal detachment in humans. Arch Ophthalmol. 1995;113:880–886.
132 Cook B, Lewis GP, Fisher SK, et al. Apoptotic photoreceptor degeneration in experimental retinal detachment. Invest Ophthalmol Vis Sci. 1995;36:990–996.
133 Arroyo JG, Yang L, Bula D, et al. Photoreceptor apoptosis in human retinal detachment. Am J Ophthalmol. 2005;139:605–610.
134 Hisatomi T, Sakamoto T, Sonoda KH, et al. Clearance of apoptotic photoreceptors: elimination of apoptotic debris into the subretinal space and macrophage-mediated phagocytosis via phosphatidylserine receptor and integrin alphavbeta3. Am J Pathol. 2003;162:1869–1879.
135 Geller SF, Lewis GP, Anderson DH, et al. Use of the MIB-1 antibody for detecting proliferating cells in the retina. Invest Ophthalmol Vis Sci. 1995;36:737–744.
136 Charteris DG, Sethi CS, Lewis GP, et al. Proliferative vitreoretinopathy-developments in adjunctive treatment and retinal pathology. Eye (Lond). 2002;16:369–374.
137 Iandiev I, Uckermann O, Pannicke T, et al. Glial cell reactivity in a porcine model of retinal detachment. Invest Ophthalmol Vis Sci. 2006;47:2161–2171.
138 Wickham L, Sethi CS, Lewis GP, et al. Glial and neural response in short-term human retinal detachment. Arch Ophthalmol. 2006;124:1779–1782.
139 Lewis GP, Chapin EA, Luna G, et al. The fate of Müller’s glia following experimental retinal detachment: nuclear migration, cell division, and subretinal glial scar formation. Mol Vis. 2010;16:1361–1372.
140 Nakazawa T, Matsubara A, Noda K, et al. Characterization of cytokine responses to retinal detachment in rats. Mol Vis. 2006;12:867–878.
141 Hollborn M, Francke M, Iandiev I, et al. Early activation of inflammation- and immune response-related genes after experimental detachment of the porcine retina. Invest Ophthalmol Vis Sci. 2008;49:1262–1273.
142 Nakazawa T, Hisatomi T, Nakazawa C, et al. Monocyte chemoattractant protein 1 mediates retinal detachment-induced photoreceptor apoptosis. Proc Natl Acad Sci U S A. 2007;104:2425–2430.
143 Nakazawa T, Kayama M, Ryu M, et al. Tumor necrosis factor-alpha mediates photoreceptor death in a rodent model of retinal detachment. Invest Ophthalmol Vis Sci. 2011;52:1384–1391.
144 Zacks DN, Han Y, Zeng Y, et al. Activation of signaling pathways and stress-response genes in an experimental model of retinal detachment. Invest Ophthalmol Vis Sci. 2006;47:1691–1695.
145 Chong DY, Boehlke CS, Zheng QD, et al. Interleukin-6 as a photoreceptor neuroprotectant in an experimental model of retinal detachment. Invest Ophthalmol Vis Sci. 2008;49:3193–3200.
146 Yang L, Kim JH, Kovacs KD, et al. Minocycline inhibition of photoreceptor degeneration. Arch Ophthalmol. 2009;127:1475–1480.
147 Kauffmann DJ, van Meurs JC, Mertens DA, et al. Cytokines in vitreous humor: interleukin-6 is elevated in proliferative vitreoretinopathy. Invest Ophthalmol Vis Sci. 1994;35:900–906.
148 Kon CH, Occleston NL, Aylward GW, et al. Expression of vitreous cytokines in proliferative vitreoretinopathy: a prospective study. Invest Ophthalmol Vis Sci. 1999;40:705–712.
149 Limb GA, Chignell AH. Vitreous levels of intercellular adhesion molecule 1 (ICAM-1) as a risk indicator of proliferative vitreoretinopathy. Br J Ophthalmol. 1999;83:953–956.
150 Mitamura Y, Takeuchi S, Matsuda A, et al. Hepatocyte growth factor levels in the vitreous of patients with proliferative vitreoretinopathy. Am J Ophthalmol. 2000;129:678–680.
151 El-Ghrably IA, Dua HS, Orr GM, et al. Intravitreal invading cells contribute to vitreal cytokine milieu in proliferative vitreoretinopathy. Br J Ophthalmol. 2001;85:461–470.
152 La Heij EC, van de Waarenburg MP, Blaauwgeers HG, et al. Basic fibroblast growth factor, glutamine synthetase, and interleukin-6 in vitreous fluid from eyes with retinal detachment complicated by proliferative vitreoretinopathy. Am J Ophthalmol. 2002;134:367–375.
153 Ogata N, Nishikawa M, Nishimura T, et al. Inverse levels of pigment epithelium-derived factor and vascular endothelial growth factor in the vitreous of eyes with rhegmatogenous retinal detachment and proliferative vitreoretinopathy. Am J Ophthalmol. 2002;133:851–852.
154 Dieudonné SC, La Heij EC, Diederen R, et al. High TGF-beta2 levels during primary retinal detachment may protect against proliferative vitreoretinopathy. Invest Ophthalmol Vis Sci. 2004;45:4113–4118.
155 Banerjee S, Savant V, Scott RA, et al. Multiplex bead analysis of vitreous humor of patients with vitreoretinal disorders. Invest Ophthalmol Vis Sci. 2007;48:2203–2207.
156 Dieudonné SC, La Heij EC, Diederen RM, et al. Balance of vascular endothelial growth factor and pigment epithelial growth factor prior to development of proliferative vitreoretinopathy. Ophthalmic Res. 2007;39:148–154.
157 Yoshimura T, Sonoda KH, Sugahara M, et al. Comprehensive analysis of inflammatory immune mediators in vitreoretinal diseases. PLoS ONE. 2009;4:e8158.
158 Rasier R, Gormus U, Artunay O, et al. Vitreous levels of VEGF, IL-8, and TNF-alpha in retinal detachment. Curr Eye Res. 2010;35:505–509.
159 Ricker LJ, Kijlstra A, Kessels AG, et al. Interleukin and growth factor levels in subretinal fluid in rhegmatogenous retinal detachment: a case-control study. PLoS ONE. 2011;6:e19141.
160 Charteris DG. Proliferative vitreoretinopathy: pathobiology, surgical management, and adjunctive treatment. Br J Ophthalmol. 1995;79:953–960.
161 Charteris DG, Sethi CS, Lewis GP, et al. Proliferative vitreoretinopathy-developments in adjunctive treatment and retinal pathology. Eye (Lond). 2002;16:369–374.
162 Lee SJ, Aaberg TM, Sr., Grossniklaus HE. Surgically excised focal posttraumatic choroidal granulomatous inflammation. Retina. 2003;23:719–720.
163 Wilson MW, Grossniklaus HE, Heathcote JG. Focal posttraumatic choroidal granulomatous inflammation. Am J Ophthalmol. 1996;121:397–404.
164 Wickham L, Asaria RH, Alexander R, et al. Immunopathology of intraocular silicone oil: enucleated eyes. Br J Ophthalmol. 2007;91:253–257.
165 Gronert K. Resolution, the grail for healthy ocular inflammation. Exp Eye Res. 2010;91:478–485.