Chapter 53D Infections of the Nervous System
Prion Diseases
Prion (pronounced pree-ahn) diseases are a group of uniformly fatal neurodegenerative diseases caused by the transformation of an endogenous protein, PrP (prion-related protein), into an abnormal conformation called the prion. The term prion is derived from the term proteinaceous infectious particle and was named by Stanley Prusiner, who discovered the protein (Prusiner, 1998). For many years, prion diseases were mistakenly thought to be due to “slow viruses,” in part owing to the transmissibility of the diseases and the long incubation period between exposure and symptom onset (Brown et al., 1986b; Gajdusek, 1977). Research by Stanley Prusiner and others, however, determined that the infectious agent did not contain nucleic acid, a component of viruses. Furthermore, treating prion-contaminated material with methods that inactivated viruses and other microorganisms did not prevent these diseases from being experimentally transmitted; yet methods that denatured or destroyed proteins prevented transmission, strongly supporting the theory that the causative agent was a protein (Gajdusek et al., 1977; Prusiner, 1982). The identification of the gene encoding human PrP (Oesch et al., 1985), PRNP, and mutations of this gene in patients with familial prion disease (Goldgaber et al., 1989; Hsiao et al., 1989) further helped support the prion hypothesis. In 1997, Stanley B. Prusiner received the Nobel Prize in Physiology and Medicine for his work on identifying the prion (Prusiner, 1998). Through animal models, identification of prion gene mutations causing prion disease in humans, and in vitro production of prions with transmissibility, it essentially has been proven that the prion protein is necessary and sufficient to cause prion disease (Kim et al., 2010; Makarava et al., 2010; Mead, 2006). Although prion diseases occur in animals and humans, this chapter will focus on human prion diseases and only discuss prion diseases in animals relevant to humans.
Human Prion Diseases
Perhaps one reason many find prion diseases so fascinating is that they are unique in medicine because they can occur in three ways in humans: spontaneously (sporadic), genetically, and through transmission (acquired) (Prusiner, 1998). Approximately 85% are sporadic, 15% are genetic, and fewer than 1% are acquired (e.g., iatrogenic). Sporadic prion disease, or sporadic Jakob-Creutzfeldt disease (sCJD), is thought to occur spontaneously. Genetic prion diseases (gPrD) are due to a mutation in the PRNP gene that encodes the prion protein, PrP; gPrDs are usually classified into three clinicopathological forms: familial CJD (fCJD), Gerstmann-Sträussler-Scheinker disease (GSS), and fatal familial insomnia (FFI). Although acquired prion diseases are the least common form of human prion disease, they are perhaps the most notorious, in part owing to their occurrence through inadvertent transmission of prions from animals to humans and from human to human. Because the genetic and acquired forms of human prion disease are less common, they will be discussed in less detail in this chapter than the much more common form, sCJD.
Epidemiology
The incidence of human prion diseases is about 1 to 1.5 per million per year in most developed countries, with some variability from year to year and between countries. This means annually there are about 6000 human prion cases worldwide and about 250 to 400 in the United States. The peak age of onset of sCJD occurs around a unimodal relatively narrow peak of about 68 years (Brown et al., 1986a). Because sCJD tends to occur within a relatively narrow age range, a person’s lifetime risk of dying from sCJD is estimated to be about 1 in 9000, much higher than the incidence (which is across all age groups) of 1 in a million. It is not clear why some countries such as Switzerland have a slightly higher incidence of about 1.9 per million per year, but this might be due to ascertainment bias, where in a small country it might be easier to track all cases.
History of Creutzfeldt-Jakob Disease Nomenclature
The history of the nomenclature for CJD is quite interesting. In 1921 and 1923, Alfons Jakob published four papers describing five unusual cases of rapidly progressive dementia. He stated that his cases were nearly identical to a case described earlier by his professor Hans Creutzfeldt in 1920. This disease was referred to for many decades as Jakob’s or Jakob-Creutzfeldt disease until Clarence J. Gibbs, a prominent researcher in the field, started using the term Creutzfeldt-Jakob disease because the acronym was closer to his own initials (Gibbs, 1992). It turns out that the cases Jakob described were very different than Creutzfeldt’s case, and that only two of Jakob’s five cases actually had the disease that we now call CJD (prion disease), whereas Creutzfeldt’s case did not (Katscher, 1998). Therefore, the name for prion disease should be Jakob’s disease or possibly Jakob-Creutzfeldt disease. But because the term JC disease (JCD) might be confused with progressive multifocal leukoencephalopathy (PML) caused by the JC virus, unfortunately we continue to use the term CJD. In this chapter, we use the terms Creutzfeldt-Jakob disease and CJD. Prion diseases also have been historically called transmissible spongiform encephalopathies (TSEs) because of two common properties of prion disease; because some gPrDs might not be transmissible, and not all human prion diseases have spongiform changes (now called vacuolation due to fluid-filled vesicles in the dendrites) on pathology, the term TSE will not be used in this chapter.
What are Prions?
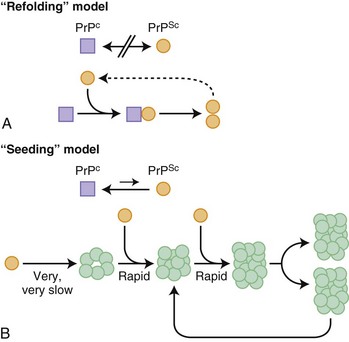
Before further discussion of various forms of prion diseases, it is important to understand what a prion is. Just as the nomenclature for human prion diseases is complicated, so is the terminology for the biology of prions. The normal prion protein (PrP) is referred to as PrPC, in which the C stands for the normal cellular form. Prion proteins, PrPC, can be transformed into prions, an abnormal infectious form of PrP called PrPSc, in which Sc refers to the abnormally shaped PrP found in scrapie, the prion disease of sheep and goats. PrPC and PrPSc essentially have identical amino acid sequences (except in genetic prion diseases; see later) but have different three-dimensional structures. Prions are characterized by their infectious properties and by the intrinsic ability of their structures to act as a template and convert the normal physiological PrPC into the pathological disease-causing form, PrPSc. Per the current prion model, when PrPC comes in contact with PrPSc, PrPC changes shape into that of PrPSc. PrPSc acts as a template for the misfolding of PrPC into PrPSc. It is believed that it is the accumulation of prions, PrPSc, in the brain that leads to nerve cell injury and death (Prusiner, 1998; Prusiner and Bosque, 2001), although some believe that it is not the accumulation of PrPSc, but rather the transformation of PrPC into PrPSc that causes neuronal injury and subsequent disease (Mallucci et al., 2007). How prions spread throughout the brain is not known. At least two models have been proposed, a refolding and a seeding model, which are not mutually exclusive (Fig. 53D.1).
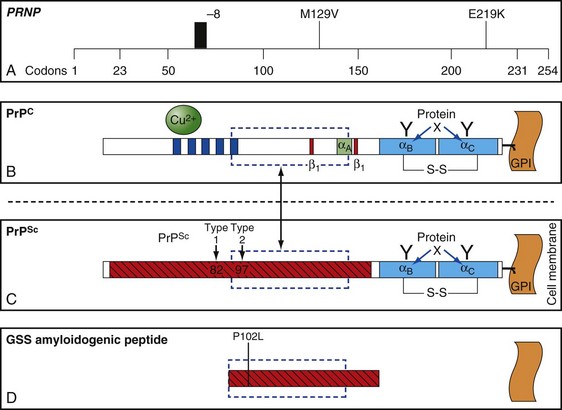
The function of PrP is still not completely known (Geschwind and Legname, 2008). It is evolutionary conserved, so it probably plays an important role in neuronal development and function (Kanaani et al., 2005). In humans, it is encoded by the PRNP gene located on the short arm of chromosome 20 (Basler et al., 1986; Oesch et al., 1985). PrPC is primarily membrane bound and resides primarily on nerve cell membranes and on other cells in the body including lymphocytes. The mature PrPC protein is attached to the outer cell membrane by a glycosylphosphatidylinositol (GPI) anchor (Borchelt et al., 1992, 1993; Taraboulos et al., 1992) (Fig. 53D.2). Mice that have had both copies of the open reading frame (ORF) of their PrP gene, Prnp, deleted (PrP−/−) have a normal lifespan and appearance (Bueler et al., 1992; Manson et al., 1994). Although clinically asymptomatic, they develop peripheral nerve demyelination (Nishida et al., 1999), have increased susceptibility to ischemic brain injury (Spudich et al., 2005; Weise et al., 2006), altered sleep and circadian rhythm (Tobler et al., 1997), altered hippocampal neuropathology and physiology including deficits in hippocampal-dependent spatial learning and hippocampal synaptic plasticity (Colling et al., 1997; Criado et al., 2005), and olfactory dysfunction (Le Pichon et al., 2008). When a slightly larger region beyond the ORF is deleted, mice develop ataxia and Purkinje cell loss later in life (Katamine et al., 1998; Rossi et al., 2001; Sakaguchi et al., 1996). Furthermore, conditional knockout mice in which the gene is not removed until after the mouse has already developed also appear normal and unaffected by gene removal.
PrPC binds to many proteins and cellular constituents. Animal and cell models have generated a variety of possible functions of PrPC including cell signaling, adhesion, proliferation, differentiation, and growth. Over the past year alone, there has been a dramatic increase in research studies postulating new roles for PrPC, including mice studies showing it mediates the toxic effects of the major protein in Alzheimer disease (AD), β-amyloid, and that it might be required for memory deficits in AD (Nygaard and Strittmatter, 2009). In fact, infusion of anti- PrPC antibodies have been used to treat cognitive deficits in AD mouse models (Chung et al., 2010; Gimbel et al., 2010; Gunther and Strittmatter, 2010). Importantly, mice devoid of PrPC can neither be infected with nor replicate prions, providing strong evidence that PrPC is necessary for prion disease (Bueler et al., 1993; Katamine et al., 1998; Prusiner et al., 1993).
Clinical Aspects of Human Prion Diseases
Sporadic Prion Disease
Sporadic CJD (sCJD) is also called classic CJD or sometimes simply CJD. It is thought to occur spontaneously, either through the spontaneous transformation of the prion protein, PrPC, into PrPSc or through a somatic mutation that results in the formation of a prion protein that is more susceptible to changing into PrPSc (see Watts et al., 2006, for a discussion on possible origins of sCJD). Nomenclature for prion diseases can be confusing. The term CJD is sometimes used to refer to all human prion diseases and sometimes to just the classic or sporadic form, sCJD. In this chapter, CJD will refer to all human prion diseases, whereas sCJD will be used to refer only to sporadic CJD.
Sporadic CJD is typically a very rapid disease with a median survival of about 7 to 8 months and mean survival of 4 months. More than 90% of patients are dead within 1 year of onset of symptoms. The mean age of onset is 68, and the median age is in the early 60s, although the range is from the 20s to 80s (Table 53D.1). Occurrence of sCJD at young (20s-40s) or old (>75) ages is uncommon (Will, 2004).
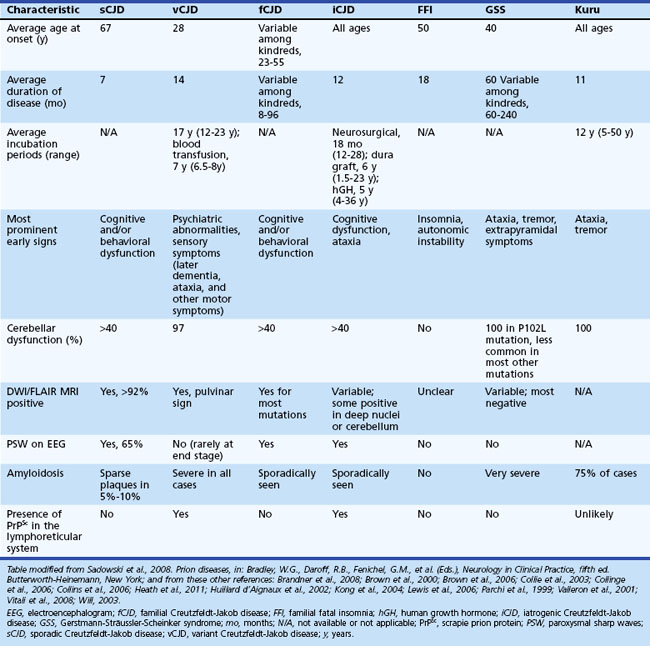
Symptoms of sCJD vary widely but typically include cognitive changes (dementia), behavioral and personality changes, difficulties with movement and coordination, visual symptoms, and constitutional symptoms (Brown et al., 1986a; Rabinovici et al., 2006). Sporadic CJD usually progresses rapidly over weeks to months from the first obvious symptoms to death. The end stage is usually an akinetic-mute state (no purposeful movement and not speaking). Most patients with prion disease die from aspiration pneumonia. Cognitive problems are often among the first symptoms in sCJD and typically include mild confusion, memory loss, and difficulty concentrating, organizing, or planning. Motor manifestations of CJD include extrapyramidal symptoms (bradykinesia, dystonia, tremor), cerebellar symptoms (gait or limb ataxia), and later in the disease, myoclonus (sudden jerking movements). Whereas the cognitive and motor symptoms are often quite obvious, other common early symptoms are often more subtle. These include behavioral or psychiatric symptoms (i.e., irritability, anxiety, depression, or other changes in personality) and constitutional symptoms (i.e., fatigue, malaise, headache, dry cough, lightheadedness, vertigo, etc.). Visual symptoms typically present as blurred or double vision, cortical blindness, or other perceptual problems; they are due to problems with processing of visual information in the brain and not due to retinal or cranial nerve abnormalities. Other symptoms such as aphasia, neglect, or apraxia (inability to do learned movements) due to cortical dysfunction might also occur and can be presenting features. Sensory symptoms such as numbness, tingling, and/or pain are less well-recognized symptoms and are probably underreported, given the magnitude of the other symptoms in sCJD (Geschwind and Jay, 2003; Lomen-Hoerth et al., 2010; Prusiner and Bosque, 2001; Will 2004).
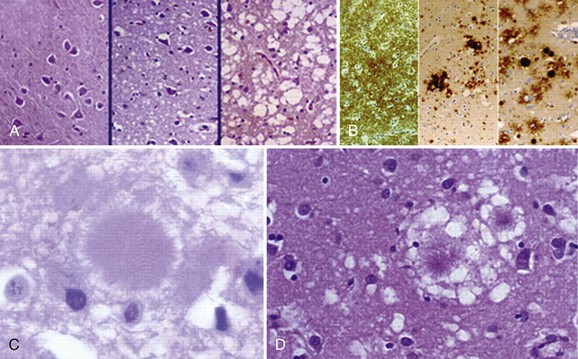
Typical neuropathological features of sCJD include neuronal loss, gliosis (proliferation of astrocytes), vacuolation (i.e., spongiform changes), and deposition of PrPSc (Fig. 53D.3). Animal models suggest the gliosis and vacuolation are early features of prion diseases (Bouzamondo-Bernstein et al., 2004). Some pathologists feel the term vacuolation is the more appropriate term to describe the spongiform changes, as these are not holes but rather fluid-filled vesicles formed in distal dendrites near synapses. Clinical and other features of several prion diseases are shown in Table 53D.1.
Molecular Classification of Sporadic Creutzfeldt-Jakob Disease
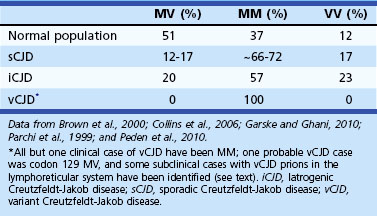
Sporadic CJD has been divided into approximately six molecular subtypes based on the genetic polymorphism at codon 129 in the prion gene (MM, MV, or VV; Table 53D.2) (also see later discussion and Figs. 53D.2 and 53D.6) and the type of protease-resistant prion (type 1 or 2). When PrPSc is extracted from brain and partially digested with proteinase, depending on which of two sites (codon 82 or 97; see Fig. 53D.2) at which PrPSc is cleaved, either a longer 21-kD (type 1) or a shorter 19-kD (type 2) peptide results when run on a Western blot. This classification to some extent separates sCJD cases based on their clinicopathological features. MM1 and MV1 are the most common forms (70%) and present as classic sCJD with rapidly progressive dementia and a duration of just a few months. VV2 (16%) starts with ataxia, later-onset dementia, and a short duration. The remaining four types—MV2 (9%), MM2-thalamic (2%), MM2-cortical (2%), and VV1 (1%)—have a duration of about 1 to 1.5 years. MV2 presents similarly to VV2 with ataxia, but have focal amyloid “kuru” plaques in the cerebellum. MM2-thalamic presents often with insomnia, followed later by ataxia and dementia, with most pathology confined to the thalamus and inferior olives and very little vacuolation; some call this form sporadic fatal insomnia (sFI), as it presents similarly to the genetic prion disease, fatal familial insomnia (FFI). MM2-cortical patients have progressive dementia with large confluent vacuoles in all cortical layers. Sporadic CJD patients with VV1 also present with progressive dementia but have severe cortical and striatal pathology with sparing of the brain stem nuclei and cerebellum. Unlike MM2-cortical, sCJD VV1 patients do not have large confluent vacuoles, but there is faint synaptic PrPSc staining (Parchi et al., 1999). Curiously, as shown in Table 53D.2, heterozygosity at codon 129 in the prion gene, PRNP, is somewhat protective against prion disease.
Diagnosis of Creutzfeldt-Jakob Disease
Several criteria exist for the diagnosis of sCJD. Unfortunately, however, most patients will only fulfill existing criteria at later stages of the disease, because most criteria are designed for epidemiological purposes to ensure that all deceased, non–autopsy-proven cases have a sufficient nonpathological diagnosis. Unfortunately, epidemiological criteria designed for CJD surveillance do not allow diagnosis at early disease stages. Most of these criteria, therefore, are not very helpful when evaluating a patient early in the disease course. Criteria generally categorize patients by level of diagnostic certainty into definite, probable, and possible. Definite criteria require pathological evidence of the presence of PrPSc in brain tissue (by biopsy or autopsy) (Budka 2003; Kretzschmar et al., 1996). Some probable diagnostic criteria are shown in Table 53D.3. The most commonly used probable criteria are the World Health Organization (WHO) Revised Criteria (1998). In the criteria, pyramidal symptoms are motor findings on exam (e.g., hyperreflexia, focal weakness, extensor response). Extrapyramidal symptoms in sCJD typically include rigidity, slowed movement (bradykinesia), tremor, or dystonia, typically due to problems in the basal ganglia or its connections. Akinetic mutism describes when patients are without purposeful movement and mute, and it occurs at the end stage of the disease. Possible CJD criteria are the same as for probable but do not require the ancillary testing (electroencephalogram [EEG] or 14-3-3 tests) (WHO, 1998). Many patients will not meet Revised WHO criteria for probable sCJD until late in the disease course. Criteria utilizing brain magnetic resonance imaging (MRI) were proposed in 2007 (Geschwind, Haman, et al., 2007; Geschwind, Josephs, et al., 2007), and in 2009, Modified European sCJD criteria allowed inclusion of brain MRI (see later discussion). Note that there are errors in these European criteria as published (Zerr et al., 2009), which it is hoped will be corrected in the future (see Table 53D.3). Regardless, there are some potential problems with the European 2009 MRI criteria (see Diagnostic Tests for Sporadic Creutzfeldt-Jakob Disease).
Table 53D.3 Several Diagnostic Criteria for Probable Sporadic Creutzfeldt-Jakob Disease
| WHO 1998 Revised Criteria (WHO, 1998) | UCSF 2007 Criteria (Geschwind et al., 2007) | European Criteria 2009† (Zerr et al., 2009) |
|---|---|---|
CSF, Cerebrospinal fluid; DWI, diffusion-weighted imaging; EEG, electroencephalogram; FLAIR, fluid-attenuated inversion recovery; MRI, magnetic resonance imaging; PSWCs, periodic sharp wave complexes; WHO, World Health Organization.
* Higher focal cortical signs include such findings or symptoms as apraxia, neglect, acalculia, aphasia, etc.
† N.B. There were errors in the table summarizing the criteria in the paper; criteria shown here are derived from the text of the paper.
Diagnostic Tests for Sporadic Creutzfeldt-Jakob Disease
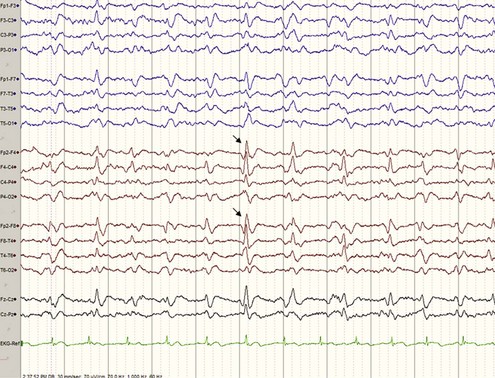
A typical EEG in sCJD has sharp or triphasic waves (periodic sharp wave complexes, or PSWCs) occurring about once every second (Fig. 53D.4); however, this EEG finding is found in only about two-thirds of sCJD patients, typically only after serial EEGs and often not until later stages of the illness (Steinhoff et al., 2004). These EEG findings are relatively specific, but PSWCs are sometimes seen in other conditions including AD, Lewy body disease, toxic-metabolic and anoxic encephalopathies, progressive multifocal leukoencephalopathy, and Hashimoto encephalopathy (Seipelt et al., 1999; Tschampa et al., 2001).
Cerebrospinal fluid (CSF) biomarkers for specific brain proteins have been used for the diagnosis of CJD since 1996, but their clinical utility is somewhat controversial, in part because of varying degrees of sensitivity and specificity around the world. The 14-3-3 protein was one of the first CSF proteins touted as a diagnostic marker for CJD, but its utility is still controversial (Chapman et al., 2000; Geschwind et al., 2003), particularly as it is elevated in many non-prion neurological conditions (Satoh et al., 1999). This test is usually a Western blot that is read subjectively, or at best semiquantitatively, as positive, negative, or inconclusive. Enzyme-linked immunosorbent assay (ELISA) tests were used for a brief period but were found to be unreliable. When first published, the 14-3-3 was reported to have 100% sensitivity and 96% specificity, but this was not a good study because of its small sample size and poor controls (Hsich et al., 1996). Since then, larger European studies have found this protein to have a sensitivity and specificity of about 85%; however, the control patients are not sufficiently characterized in some of these studies (Collins et al., 2006; Sanchez-Juan et al., 2006). Many feel that the 14-3-3 protein is merely a marker of rapid neuronal injury and has poor specificity for sCJD (Chapman et al., 2000; Geschwind et al., 2003; Satoh et al., 1999).
Other potential sCJD CSF biomarkers include total-tau (t-tau), neuron-specific enolase (NSE), and the astrocytic protein, S100β. The sensitivity and specificity of these biomarkers for sCJD varies greatly among studies. One large multicenter European study examined four biomarkers, 14-3-3, t-tau, NSE and S100β; however, not all patients had all four tests done, nor were they necessarily done in the same samples, so this study did not allow legitimate comparison of these biomarkers. Nevertheless, they found the sensitivity and specificity of the 14-3-3 to be 85% and 84%, t-tau (cutoff > 1300 pg/mL) 86% and 88%, NSE 73% and 95%, and S100β 82% and 76%, respectively (Sanchez-Juan et al., 2006). The sensitivity and specificity of these tests in other forms of prion disease such as variant CJD and gPrD are much lower than for sCJD. Some investigators have suggested the ratio of phosphorylated tau (p-tau) to t-tau (p-tau/t-tau) as a good diagnostic test. Additional potential CSF biomarkers, some with a possible functional role in prion diseases, are also being evaluated. Thus far, many feel that t-tau might be the best CSF diagnostic protein for sCJD, but it still is not close to the utility of brain MRI.
MRI has been shown to be highly sensitive and specific (91%-96%) for sCJD. Although the first MRI abnormalities reported in CJD were basal ganglia hyperintensities, particularly on T2-weighted sequences, cortical gyral hyperintensities, particularly noticeable on fluid-attenuated inversion recovery (FLAIR) and diffusion-weighted imaging (DWI) MRI sequences, are even more common. These cortical hyperintensities are commonly referred to as cortical ribboning. DWI has higher sensitivity than FLAIR. Whenever CJD is suspected, a brain MRI that includes DWI and FLAIR sequences should be obtained (Shiga et al., 2004; Vitali et al., 2008; Young et al., 2005). A typical MRI in sCJD (and some cases of genetic CJD) is shown in Fig. 53D.5. Unfortunately, many radiologists, even at academic centers, are still not familiar with the findings indicative of CJD, and a majority of sCJD MRIs are misread (Wong et al., 2009). Diagnostic criteria for sCJD that included the use of DWI and FLAIR MRI were proposed in 2007 (Geschwind, Haman, et al., 2007; Geschwind, Josephs, et al., 2007), and MRI was included in European criteria in 2009 (Zerr et al., 2009) (see Table 53D.3).
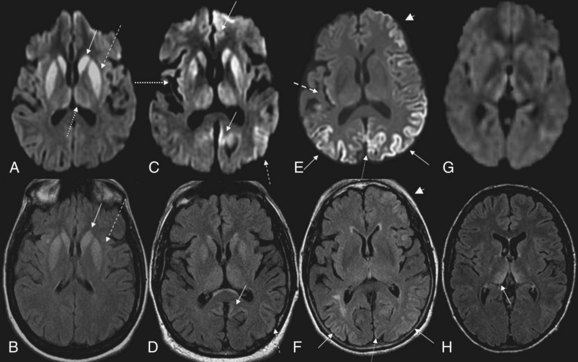
Although these European criteria are a step forward, they still have several problems. First, many patients in the study did not have DWI MRIs, and the criteria allow FLAIR without DWI MRI. This is a problem because FLAIR basal ganglia and cortical changes are less specific than DWI abnormalities for sCJD. FLAIR basal ganglia hyperintensities can be seen in many conditions other than CJD, including metabolic and autoimmune conditions (Vitali et al., 2008). Second, hyperintensities in the frontal and insular cortices, cingulate, and hippocampus were not included because of the high levels of artifact found in these regions. These artifacts, often due to air-brain interface, can be often be avoided by acquiring MRIs in different planes, such as axial and coronal. Lastly, the criteria not only did not require the use of DWI, they also did not utilize the attenuation diffusion coefficient (ADC) map to confirm the presence of restricted diffusion of water molecules. This is important because the DWI sequence is a combination of both T2 and diffusion, so a T2 shine-through effect can make certain regions appear bright on DWI despite normal diffusion. Especially when the deep gray nuclei (basal ganglia and/or thalamus) are involved in sCJD, the ADC map typically is hypointense and shows reduced levels of diffusion. Evidence suggests that the reduced diffusion on MRI in CJD is from restricted flow of water molecules inside vacuoles in the dendritic tree (Geschwind et al., 2009).
Basic laboratory studies such as complete blood cell count (CBC), chemistry, liver function tests, erythrocyte sedimentation rate (ESR), antinuclear antibody (ANA), and so forth are generally unremarkable in sCJD. CSF is typically normal with a mildly elevated protein (typically <100 mg/dL). CSF shows normal red blood cells (RBCs) and white blood cells (WBCs). Pleocytosis (>10 WBCs), an elevated immunoglobulin (Ig)G index, or the presence of oligoclonal bands is unusual in sCJD and should lead to considering other conditions, particularly infectious or autoimmune disorders. As noted earlier, an EEG that is “typical” or classic for CJD has PSWCs (see Fig. 53D.4), but often there is just slowing on EEG in sCJD.
Development of a Prion-Specific Diagnostic Test
Even MRI is not a perfect test. An ideal premortem minimally invasive test for sCJD might directly measure for the presence of prions in bodily fluids such as blood, urine, or saliva (Safar et al., 2005). One interesting technique called protein misfolding cyclic amplification (PMCA) was developed to produce large quantities of PrPSc. The methodology is conceptually similar to a PCR reaction for amplifying nucleic acid. In this assay, a very small amount of source material containing PrPSc is diluted into either fresh brain homogenate (as a source of PrPC) or recombinantly derived PrPC, then the mixture is sonicated or shaken at warm temperatures. The goal of the sonication or shaking is to break up newly formed PrPC aggregates into smaller oligomers, which then provide new seeds for the formation of PrPSc. Repeated cycles of conversion and sonication/shaking are done to amplify any PrPSc present in the sample (Watts et al., 2006; Wilham et al., 2010).
Proteinase-sensitive proteinopathy (PSPR) is a new form of sCJD. Until recently, one marker of prion diseases has been that PrPSc is resistant to proteases, enzymes that digest proteins. It has also been known since the late 1990s, however, that a portion of PrPSc is often protease sensitive (Safar et al., 1998). In the new form of sCJD that has recently been identified, the vast majority of patients’ PrPSc is protease sensitive, so standard immunohistochemical techniques that depend on identifying protease-resistant PrPSc for diagnosis are insufficient. Affected individuals had somewhat different presentations based on their codon 129 genotype (VV, MV and MV; see later discussion). Many of these cases presented with psychiatric symptoms, a frontal dementia, and most had negative ancillary tests (MRIs, EEGs, and 14-3-3). Although their mean age was commensurate with classic sCJD (late 60s), their mean disease duration was much longer at about 2.5 years (Zou et al., 2010). Such atypical features can make these cases very difficult to diagnose.
Genetic Prion Disease
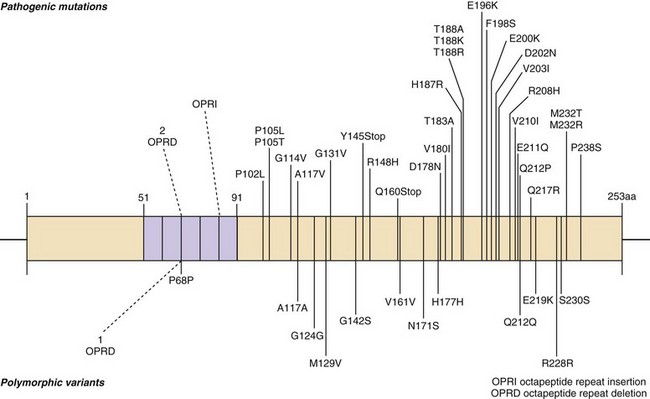
Genetic forms of prion disease (gPrD) are caused by a mutation in the prion protein (PrP) gene, PRNP. They are autosomal dominant, and most are 100% penetrant (i.e., almost everyone with a mutation develops the disease if they live a normal lifespan). More than 40 mutations, mostly point mutations but some stop codons, insertions, and deletions, have been identified. Diagnosis of gPrD is made by identification of a mutation in the PrP gene, PRNP (Kong et al., 2004) (Fig. 53D.6 and see Fig. 53D.2). It is easiest to do deoxyribonucleic acid (DNA) testing from blood while a patient is still alive; alternatively, DNA can be extracted from frozen brain autopsy tissue. Genetic prion diseases are sometimes referred to as familial, but this term can be a misnomer because more than 60% of patients with gPrD do not have a positive family history of prion disease. In retrospect, relatives were often misdiagnosed as having died of AD or Parkinson disease (PD); however, in some cases there was simply reduced penetrance of the mutation (Kovacs et al., 2005). Genetic prion diseases are often divided according to their genetic, clinical, and pathological characteristics into three forms: familial CJD (fCJD), Gerstmann-Sträussler-Scheinker disease (GSS), and fatal familial insomnia (FFI). This classification is far from perfect, because some mutations have features that blend fCJD and GSS. Compared to those with sCJD, patients with gPrD—depending on the specific PRNP mutation—usually have a younger age of onset (typically 40s-60s), have a slower course and live longer (typically a few years), and have initial symptoms of parkinsonism or ataxia, with only mild personality or cognitive changes early on. Many forms of gPrD, however, present identically to sCJD clinically, and sometimes pathologically, with rapid onset of clinical symptoms and short survival of weeks to months (Kong et al., 2004). There is often great variability in presentation and disease course of the several mutations causing gPrDs; in fact, even within the same family members carrying the same mutation, there might be great clinical variability. Several polymorphisms also have been found in PRNP, a few of which can affect the risk for getting nongenetic forms of prion disease and also affect the way genetic and nongenetic prion diseases present. The most important polymorphism is at codon 129, which can be either a methionine (M) or valine (V) (see Figs. 53D.2 and 53D.6 and Table 53D.2).
Although the precise mechanism of how disease in gPrDs occurs is unknown, it is presumed that mutations in PRNP make the normal cellular translated prion protein, PrPC, more susceptible to changing conformation into the abnormally shaped, disease-causing form, PrPSc (see later discussions for more about prion proteins, PrPC, and PrPSc) (van der Kamp and Daggett, 2010). Presumably the nascent PrPC maintains a normal conformation for most of a patient’s life and does not begin transforming shape into PrPSc until a patient gets older. Alternatively, and possibly more likely, transformation of PrPC into PrPSc occurs throughout life, with PrPSc being removed by normal cellular protein degradation pathways; but as one ages, the cellular pathways for clearing out PrPSc do not work as efficiently. The increasing accumulation of PrPSc causes transformation of nascent PrPC into PrPSc in an exponential manner, resulting in disease (Kong et al., 2004; Prusiner, 1998) (see Fig. 53D.1).
Familial Creutzfeldt-Jakob Disease
More than 15 point mutations, an insertion mutation, and octapeptide repeat insertion (OPRI) mutations with 4 or fewer 24-base-pair repeats typically present as fCJD (although often there is a great phenotypic variability in CPRI mutations). Most of these patients present similarly to sCJD, often with overlapping clinical, MRI, and EEG findings. The most common fCJD mutation is E200K. Several others are shown above the PrP gene in Fig. 53D.6.
Gerstmann-Sträussler-Scheinker Disease
At least 15 PRNP mutations have been shown to cause GSS, including about one dozen point mutations, a stop mutation, and several OPRIs (Kong et al., 2004). OPRI mutations with five or more 24-base-pair repeats typically present as GSS, whereas four or fewer OPRIs typically present as a CJD phenotype, but there are many exceptions (see Fig. 53D.6). GSS usually presents as a subacute progressive ataxic and/or parkinsonian disorder with later onset of cognitive impairment, although some mutations present with early dementia and/or behavioral abnormalities. There is considerable phenotypic variability within and between mutations and families, however (Giovagnoli et al., 2008; Kong et al., 2004; Webb et al., 2008). Because it is usually slower than sCJD or many other gPrDs, often with a duration of about 5 years (range 3 to 8 or more years), persons with GSS sometimes are mistaken to have other neurodegenerative conditions such as multiple system atrophy, spinocerebellar ataxias, idiopathic PD, AD, or Huntington disease (see Table 53D.1). Patients with GSS, however, also can present identically to sCJD with a rapidly progressive course leading to death within a few months from onset. GSS has a distinct neuropathology from most other prion diseases, with large PrPSc amyloid plaques called kuru plaques. These plaques are also seen in a minority of sCJD cases, albeit more sparsely (see Fig. 53D.3) (Parchi et al., 1999; Wadsworth et al., 2006) but differ from the florid plaques of variant CJD (Sikorska et al., 2008) (see later discussion and Fig. 53D.3). Because of the large deposits of amyloid in GSS, it might be possible to detect these pathological changes noninvasively prior to clinical onset through the use of amyloid-binding agents such as 2-(1-{6-[(2-[18F] fluoroethyl)(methyl) amino]-2-naphthyl}ethylidene) malononitrile ([18F]FDDNP) and PET scans (Kepe et al., 2010). The amyloid deposits seen in GSS contain smaller fragments of PrPSc (see Fig. 53D.2).
Fatal Familial Insomnia
FFI usually presents with progressive severe insomnia and dysautonomia, with motor and cognitive problems appearing later in the course. Autonomic symptoms often include tachycardia, hyperhidrosis, and hyperpyrexia. Progressive insomnia with disruption of circadian rhythm is eventually associated with hallucinations. Age of onset is similar to sCJD, but most FFI patients survive slightly longer, about 1.5 years. Although brain MRI is usually normal, FDG-PET imaging reveals thalamic and cingulate hypometabolism, often even before disease onset. FFI is caused by a single PRNP point mutation, D178N, with codon 129 M on the same chromosome (cis) (see Fig. 53D.6). Patients with D178N but cis codon 129 V usually present with fCJD, not FFI. Neuropathology of FFI includes profound thalamic gliosis and neuronal loss causing atrophy. Involvement of regions outside of the thalamus is greater in FFI with codon 129 MV than MM (Budka, 2003; Cortelli et al., 1997; Cortelli et al., 2006).
Acquired Creutzfeldt-Jakob Disease
Acquired forms of CJD occur because prions are transmissible. Although considered infectious, prion diseases are not as easy to transmit as many other infectious diseases such as respiratory-transmitted pathogens (e.g., certain viruses or Mycobacterium tuberculosis) or through bodily fluids (e.g., human immunodeficiency virus [HIV] and hepatitis). A relatively large amount of prions (probably several thousand proteins) are necessary to transmit prion disease. Thus, human prion diseases are not contagious; physical contact and even intimacy are not known to transmit prions between persons. Acquired prion diseases include kuru (now essentially extinct but at one time occurring among the Fore tribe in Papua New Guinea as a result of endocannibalism), iatrogenic CJD (iCJD), and the highly publicized variant CJD (vCJD), occurring primarily in the United Kingdom and France and caused by consumption of beef contaminated with bovine spongiform encephalopathy (BSE, or Mad Cow disease) (Collinge et al., 2006; Prusiner and Bosque, 2001; Will, 2003; Will et al., 2000). Kuru was one of the first identified forms of transmitted human prion disease. As noted, it is a neurodegenerative disease of the Fore ethnic group of the central highlands of Papua New Guinea. In the Fore language, kuru means “to shake or tremble.” It was transmitted through a practice in which deceased relatives were honored by ritualized endocannibalism. It is unknown how this disease began but presumably through cannibalism of persons with sCJD. Because women and children consumed the less desirable tissues, including brain and spinal cord which contained higher levels of prions, they tragically contracted a form of prion disease with severe ataxia (Gajdusek et al., 1966). The disease was essentially eliminated with the elimination of the practice of ritual cannibalism, although cases have occurred recently, suggesting an incubation period as long as 50 years or more (Collinge et al., 2006), particularly in those who are heterozygous at codon 129 in PRNP. That heterozygotes have longer incubation periods could suggest a similar phenomenon might occur with vCJD (see later discussion). Genetic risk factors, and more recently protective alleles, have been identified in the Fore population (Mead et al., 2009).
Approximately 400 cases of iatrogenic CJD (iCJD) have occurred from the use of cadaveric-derived human pituitary hormones, dura mater grafts, corneal transplants, reuse of cleaned and sterilized EEG depth electrodes implanted directly into the brain, other neurosurgical equipment, and blood transfusion (Brown et al., 2000; Brown et al., 2006; Tullo et al., 2006; Will 2003). Because the prion is a protein, not a virus or bacterium, it is not susceptible to typical decontamination methods that would inactivate such microorganisms (see Prion Decontamination) (Bellinger-Kawahara, Cleaver, et al., 1987; Bellinger-Kawahara, Diener, et al., 1987; Prusiner, 1998). This difficulty in inactivating prions has in part led to transmission of prion disease between patients. Most of the pituitary-derived (human grown hormone and gonadotrophin) cases occurred from contaminated batches in France, the United Kingdom, and the United States. Methods have since been instituted to prevent prion transmission through such hormones (Brown et al., 2006). In the United States, pituitary hormone recipient patients exposed to CJD were informed of their potential risk for developing prion disease, whereas this was not always the case in other countries. About 200 cases of iCJD have occurred from contaminated batches of dura mater (mostly Lyodura brand), with a majority of cases occurring in Japan. These cases occurred prior to 1987 when a sodium hydroxide disinfection step was added to the processing protocol (Brown et al., 2000; Brown et al., 2006). Six iCJD cases have been linked to neurosurgical procedures and at least two to corneal implants. A few other cases might have occurred through neurosurgical or other surgical procedures, but in such cases it is often very difficult to determine whether a case is iatrogenic or sporadic (Brown et al., 2006). Although it appears that the number of iCJD cases is declining, despite WHO and other recommended practices for managing potential prion-contaminated tissues, prion contamination still occurs, leaving patients at risk for iCJD.
Improved identification of prion disease should help prevent future iCJD cases, but cases will likely slip by screening, and thus strict application of efficient decontamination procedures is still critical to prevent transmission. Unfortunately, the true risk of iCJD is still unknown; many of the decontamination procedures tested were based on models using animal prions, which appear easier to decontaminate than human prions (Peretz et al., 2006). Rather than trying to decontaminate neurosurgical equipment used during surgery on potential or suspected prion subjects, some medical centers dispose of all such equipment through incineration rather than take the risk of reusing it. The most recently identified form of iCJD has been the transmission of vCJD through blood products (see later discussion).
Variant CJD, first identified in 1995, is currently the most notorious form of acquired human prion disease (Will et al., 1996). As noted earlier, it is caused by inadvertent ingestion of beef contaminated with BSE (Mad Cow disease) or, in a few cases, blood or blood product transfusion from asymptomatic patients who are carriers of vCJD (Zou et al., 2008). It is believed that BSE occurred through the practice of feeding scrapie-infected sheep products to cattle (Bruce et al., 1997; Scott et al., 1999). In general, vCJD differs from sCJD in several ways. Patients with vCJD generally are much younger, with a median age of around 27 (range 12-74), including patients in their teens and 20s. Almost all cases have occurred in persons younger than age 50. The mean disease duration is longer, about 14.5 months, versus about 7 months for sCJD. Although psychiatric symptoms often occur early in sCJD (Rabinovici et al., 2006; Wall et al., 2005), in vCJD, profound psychiatric symptoms are often the initial symptoms for several months before obvious neurological symptoms begin. A relatively unique symptom in vCJD is persistent painful paresthesias in various parts of the body. The EEG only rarely shows the classic PSWCs and, if so, then only at the end stage of disease (Binelli et al., 2006). Brain MRI usually shows the “pulvinar sign,” in which the pulvinar (posterior thalamus) is brighter than the anterior putamen on T2-weighted or DWI MRI (Collie et al., 2003) (see Fig. 53D.5); this finding is extremely rare in other human prion diseases (Petzold et al., 2004). Diagnostic criteria for probable vCJD are shown in Table 53D.4 (UK National CJD Surveillance Unit, 2010). Although several features of vCJD overlap those of sCJD, the younger age of onset, MRI findings, prominent early psychiatric features, persistent painful sensory symptoms, and chorea might help differentiate these conditions.
Table 53D.4 Current Diagnostic Criteria for Variant Creutzfeldt-Jakob Disease
EEG, Electroencephalography; MRI, magnetic resonance imaging; TSE, transmissible spongiform encephalopathy; vCJD, variant Jakob-Creutzfeldt disease.
a Spongiform change and extensive prion protein deposition with florid plaques throughout the cerebrum and cerebellum.
b Depression, anxiety, apathy, withdrawal, delusions.
c Includes frank pain and/or dysesthesias.
d The typical appearance of the EEG in sporadic CJD consists of generalized triphasic periodic complexes at approximately 1 per second. These may occasionally be seen in the late stages of vCJD.
e Tonsil biopsy is not recommended routinely nor in cases with EEG appearances typical of sporadic CJD but may be useful in suspect cases in which the clinical features are compatible with vCJD and MRI does not show bilateral pulvinar high signal.
Modified from Heath, C.A., Cooper, S.A., Murray, K., et al., 2010. Validation of diagnostic criteria for variant Creutzfeldt-Jakob disease. Ann Neurol 67, 761-770.
Definitive diagnosis of vCJD is based on pathological evidence of the variant form of PrPSc in brain biopsy or autopsy. Because vCJD is typically acquired peripherally, PrPSc can be found in the lymphoreticular system, including tonsillar tissue (Will, 2004). Brain pathology of vCJD shows abundant PrPSc deposition, in particular multiple fibrillary PrP plaques surrounded by a halo of spongiform vacuoles (so-called florid plaques) and other PrP plaques, and amorphous pericellular and perivascular PrP deposits, especially prominent in the cerebellar molecular layer. The pathognomic plaques in vCJD are called florid because they have the appearance of a flower with a dense center and surrounding ring of vacuoles (Budka, 2003) (see Fig. 53D.3). The Western blot characteristics of vCJD PrPSc also are different from those seen in other forms of prion disease (Will, 2004; Will et al., 2000).
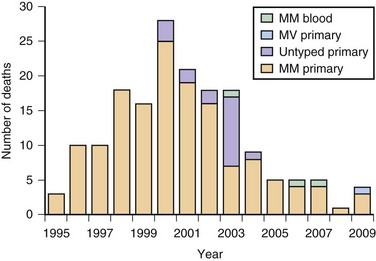
As of December 2010, approximately 218 probable or definite cases of vCJD have been documented, almost all in the United Kingdom, and no cases of vCJD have been acquired in the Western Hemisphere, including the United States or Canada (three patients in the United States and one in Canada died from vCJD but are thought to have acquired it elsewhere) (CDC, 2006; UK National CJD Surveillance Unit, 2010). Following the United Kingdom, France has the second highest number of vCJD cases, which probably have the same origin as those in the United Kingdom (Brandel et al., 2009). The peak of the vCJD epidemic was in 2000, although it is not known whether other peaks will occur, particularly in persons with different genetic susceptibility to vCJD or iatrogenically through blood products (Andrews, 2010). Two studies assessed the presence of latent vCJD in the U.K. population and found it to be much higher than expected. In one study, researchers found vCJD prions by immunostaining in 3 of 11,246 appendix samples collected from 1995-2000. Another similar study, the National Anonymous Tonsil Archive, found 1 positive sample among a subset of 10,000 tested (Garske and Ghani, 2010). Thus, there are asymptomatic persons in the U.K. population with vCJD prions in their lymphoreticular system (subclinically infected); it is not clear what their risk is of developing vCJD (becoming a clinical case) or being infectious and passing it on to others through medical/surgical procedures or blood products. As of December 2010, four patients have acquired vCJD through non-leukodepleted (white blood cells removed) blood transfusions received before 1999; three patients (all codon 129 MM) died from definite vCJD, with incubation periods of about 6 to 10 years. A fourth patient (heterozygous, MV, at codon 129) died from non-neurological causes 5 years after receiving a contaminated blood transfusion but at autopsy was found to have prions in his lymphoreticular system (Health Protection Agency, 2007). It is not known whether this latter patient would have ever developed vCJD through spread to the brain or would have simply survived as a carrier and possible reservoir for vCJD spread. One probable vCJD patient (no pathology) was found to be MV at codon 129 (Garske and Ghani, 2010). A graph of presumed vCJD cases in the United Kingdom is shown in Fig. 53D.7. Although the number of cases appears to have been stable over the past several years at five or fewer per year, there is great concern that future cases of vCJD might occur iatrogenically through transfusion of blood products or that many exposed persons, particularly with codon 129 MV or VV polymorphism, have longer incubation times. There is now evidence that vCJD also might be spread through plasma products such as factor VIII (Peden et al., 2010). These asymptomatic carriers of vCJD might pose the greatest risk for spread of vCJD through transfusion of blood products or invasive procedures. Of great concern for the risk of transmission is that these infected asymptomatic vCJD donors transmitted the disease 1.5 to 6 years before they became symptomatic (Health Protection Agency, 2007).
Prion Decontamination
Decontamination of prions requires methods that will denature proteins, as prions resist normal inactivation methods used to kill viruses and bacteria. Typical methods for reducing the load of or inactivating prions include very high temperatures for prolonged periods and autoclaving at higher-than-normal temperatures, pressure, and time, with or without denaturing agents (many of which are caustic). Recommended methods for prion decontamination that include very high temperatures with steam and caustic denaturing agents often damage equipment and instrumentation. WHO guidelines state the preferred method is steam sterilization for at least 30 minutes at 132°C in a gravity-displacement sterilizer. If a pre-vacuum sterilizer is used, they note 18 minutes at 134°C also is effective. Another option is 1M sodium hydroxide or 2% sodium hypochlorite for 1 hour, with 134°C autoclaving for at least 18 minutes. Non-fragile items may be immersed in 1N sodium hydroxide, a caustic solution, for 1 hour at room temperature and then steam sterilized for 30 minutes at a temperature of 121°C (WHO, 2003; WHO, 2006). Unfortunately, most of the literature on prion infection control is based on nonhuman prions; human prions are much more difficult to denature (Peretz et al., 2006). As already discussed, because of the risk of transmission to subsequent patients, when feasible, many hospitals dispose of neurosurgical and other surgical equipment potentially exposed to prions by incineration rather than attempting to decontaminate it for future patient use. Research into improved methods of decontamination of prions is ongoing.
Animal Prion Diseases
Unfortunately, in the mid-1980s an outbreak of BSE occurred because scrapie-contaminated material was being fed to cattle. Cattle with BSE develop an ataxic illness, weight loss, behavioral changes, and other neurological symptoms progressing to death. More than 280,000 cattle suffered from BSE. When cases were identified, entire herds were killed to prevent the disease from spreading further. Fortunately, through proper epidemiological control including feed bans and cessation of various feeding practices, the incidence of BSE has dropped dramatically, now with only a few cases occurring each year. Tragically, even though scrapie prions do not pass directly to humans, they were able to overcome the species barrier with humans by being passed through cattle. Ingestion of BSE prions unfortunately led to vCJD in the United Kingdom and several other countries (see earlier discussion). The temporal relationship between the BSE epidemic and the rise of vCJD, coupled with mouse data showing the similarity between these conditions, are strong evidence for the link between these two diseases (Bruce et al., 1997; Scott et al., 1999). As of February 2010, 21 isolated cases of BSE have been identified in North America, including three in the United States (Fig. 53D.8). Thus, it does not appear that there has been another outbreak, although it is possible that some cases are still getting into the food supply, as not all cattle are being inspected.
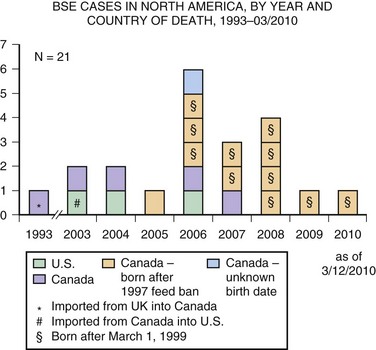
Fig. 53D.8 Bovine spongiform encephalopathy (BSE) in North America. Through February 2010, BSE surveillance has identified 21 cases in North America: 3 in the United States and 18 in Canada. Of the 3 cases identified in the United States, one was born in Canada; of the 18 cases identified in Canada, one was imported from the United Kingdom (see Fig. 53D.7). Since March 2006, each of the 14 cattle reported with BSE in North America were born in Canada and identified through the Canadian BSE surveillance system.
(Courtesy Centers for Disease Control and Prevention. Available at: http://www.cdc.gov/ncidod/dvrd/bse/index.htm.)
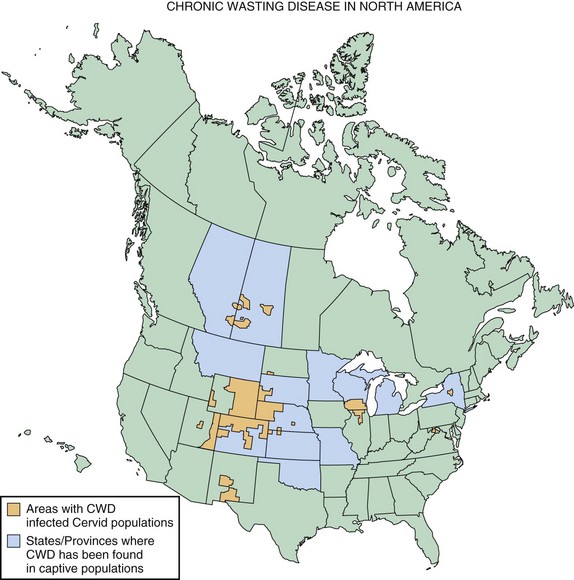
Chronic wasting disease (CWD) is a prion disease of mule deer, white-tailed deer, elk, and more recently, identified in moose. The first clinical cases were recognized in the late 1960s in North America. Clinical features of the disease include weight loss, behavioral changes such as depression, and isolation from the herd. Some other features might also include hypersalivation, polydipsia/polyuria, and ataxia. The disease primarily has been reported in the United States and Canada, with the highest concentrations occurring in the Central Mountain region of the United States, especially Colorado and Montana, as well as the Canadian provinces of Saskatchewan and Alberta. A map showing the distribution of CWD in North America is in Fig. 53D.9. A most concerning aspect of CWD is its ease of horizontal transmission between cervids. This might be due in part to the fact that CWD appears to be transmissible through blood, urine, and saliva (Haley et al., 2009). This feature makes it very difficult to prevent spread of the disease in free-ranging cervid populations (Williams, 2005). It still is not clear whether CWD can spread to humans or whether there is a species barrier, but there has been no reported increase in human prion cases in states where CWD rates have been highest (Sigurdson et al., 2009).
Treatments of Human Prion Diseases
Currently, there is no known cure for human prion diseases. All cases are uniformly fatal. Some potential mechanisms for treating prion diseases are shown in Fig. 53D.10 and include removing or reducing the endogenous substrate, PrPC, blocking the interaction of PrPC with PrPSc, removal of PrPSc, or blocking its toxicity (Korth and Peters, 2006). Several medicines have been used to treat human prion disease, but only flupirtine and quinacrine, both given orally, have been tested in randomized double-blinded placebo-controlled trials. Neither was effective in prolonging survival (Geschwind et al., manuscript in preparation; Korth and Peters, 2006). Intraventricular pentosan polysulfate has been used on a compassionate basis in the United Kingdom, Japan, and a few other countries, but observational data suggest that it does not affect survival. As of the writing of this chapter, treatment trials for human prion disease are currently underway in Italy (http://www.agenziafarmaco.it/en) and one or two other European countries, using the oral antibiotic, doxycycline, and possibly other drugs (Stewart et al., 2008). Several laboratories around the world are screening drug libraries and using medicinal chemistry to identify and develop antiprion therapies. In the absence of any curative treatments, management of prion diseases involves treating symptoms as they arise and comfort care. Insofar as there are no approved drugs in any countries for treatment of prion disease, all medications are used off-label for symptomatic treatment. There are no data supporting the use of any AD medications. At our center, we have managed hundreds of patients with prion disease and have empirically found certain medications to be helpful. For example, we commonly use SSRIs to treat depression and agitation, atypical antipsychotics (particularly quetiapine) to treat agitation and psychoses, and clonazepam to treat severe myoclonus or agitation. Valproic acid can also be useful for myoclonus, although one study suggested it increases PrPC and PrPSc in vitro but had no effect in an in vivo mouse model (Shaked et al., 2002).
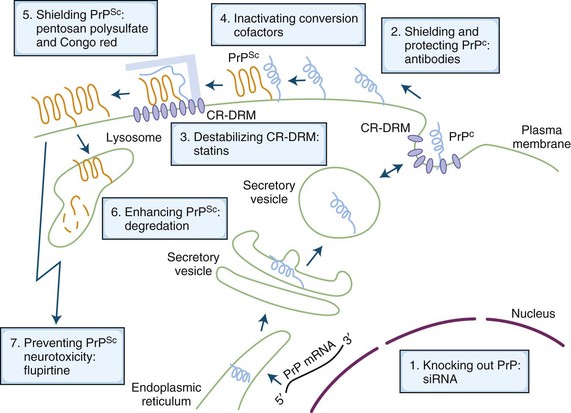
Fig. 53D.10 Schematic drawing of the life of the prion protein (PrP) inside the cell, its conversion to infectious prions, and cell biology–based possibilities of treatment intervention. The cellular PrP (PrPC) is light blue, and the scrapie PrP (PrPSc) is orange. Blue-filled ovals within the plasma membrane are cholesterol-rich, detergent-resistant membrane domains (CR-DRMs). The light blue bar represents a hypothesized conversion-assisting cofactor, sometimes referred to as protein X. For a more detailed explanation, see Korth and Peters, 2006. mRNA, Messenger ribonucleic acid; siRNA, small interfering ribonucleic acid.
(Modified from Korth, C., Peters, P.J., 2006. Emerging pharmacotherapies for Creutzfeldt-Jakob disease. Arch Neurol 63, 497-501.)
Differential Diagnosis
Other conditions such as AD, Lewy body disease, Hashimoto encephalopathy, or hepatic encephalopathy rarely also may have a “typical” EEG (Geschwind and Jay, 2003). Other rapidly progressive disorders to consider that might present similarly to CJD include Hashimoto encephalopathy (Chong and Rowland 2006; Seipelt et al., 1999), paraneoplastic or other autoimmune limbic encephalopathies (Geschwind et al., 2008; Vernino et al., 2007), cancers (particularly lymphoma, either within or outside the nervous system), central nervous system vasculitis, metabolic or toxic disorders (e.g., bismuth intoxication, hepatic encephalopathy, electrolyte imbalance, etc.), and atypical presentations of more common neurodegenerative conditions such as AD, Lewy body disease, and corticobasal degeneration (Geschwind et al., 2008).
Andrews, N.J., 2010. Incidence of variant Creutzfeldt-Jakob disease diagnoses and deaths in the UK January 1994-December 2009. London, Statistics Unit, Centre for Infections, Health Protection Agency.
Basler K., Oesch B., et al. Scrapie and cellular PrP isoforms are encoded by the same chromosomal gene. Cell. 1986;46(3):417-428.
Bellinger-Kawahara C., Cleaver J.E., et al. Purified scrapie prions resist inactivation by UV irradiation. J Virol. 1987;61(1):159-166.
Bellinger-Kawahara C., Diener T.O., et al. Purified scrapie prions resist inactivation by procedures that hydrolyze, modify, or shear nucleic acids. Virology. 1987;160(1):271-274.
Binelli S., Agazzi P., et al. Periodic electroencephalogram complexes in a patient with variant Creutzfeldt-Jakob disease. Ann Neurol. 2006;59(2):423-427.
Borchelt D.R., Rogers M., et al. Release of the cellular prion protein from cultured cells after loss of its glycoinositol phospholipid anchor. Glycobiology. 1993;3(4):319-329.
Borchelt D.R., Taraboulos A., et al. Evidence for synthesis of scrapie prion proteins in the endocytic pathway. J Biol Chem. 1992;267(23):16188-16199.
Bouzamondo-Bernstein E., Hopkins S.D., et al. The neurodegeneration sequence in prion diseases: evidence from functional, morphological and ultrastructural studies of the GABAergic system. J Neuropathol Exp Neurol. 2004;63(8):882-899.
Brandel J.P., Heath C.A., et al. Variant Creutzfeldt-Jakob disease in France and the United Kingdom: Evidence for the same agent strain. Ann Neurol. 2009;65(3):249-256.
Brandner S., Whitfield J., et al. Central and peripheral pathology of kuru: pathological analysis of a recent case and comparison with other forms of human prion disease. Philos Trans R Soc Lond B Biol Sci. 2008;363(1510):3755-3763.
Brown P., Brandel J.P., et al. Iatrogenic Creutzfeldt-Jakob disease: the waning of an era. Neurology. 2006;67(3):389-393.
Brown P., Cathala F., et al. Creutzfeldt-Jakob disease: clinical analysis of a consecutive series of 230 neuropathologically verified cases. Ann Neurol. 1986;20(5):597-602.
Brown P., Preece M., et al. Iatrogenic Creutzfeldt-Jakob disease at the millennium. Neurology. 2000;55(8):1075-1081.
Brown P., Rohwer R.G., et al. Newer data on the inactivation of scrapie virus or Creutzfeldt-Jakob disease virus in brain tissue. J Infect Dis. 1986;153(6):1145-1148.
Bruce M.E., Will R.G., et al. Transmissions to mice indicate that ‘new variant’ CJD is caused by the BSE agent. Nature. 1997;389(6650):498-501.
Budka H. Neuropathology of prion diseases. Br Med Bull. 2003;66:121-130.
Bueler H., Aguzzi A., et al. Mice devoid of PrP are resistant to scrapie. Cell. 1993;73(7):1339-1347.
Bueler H., Fischer M., et al. Normal development and behaviour of mice lacking the neuronal cell- surface PrP protein. Nature. 1992;356(6370):577-582.
CDC, 2006. vCJD (Variant Creutzfeldt-Jakob Disease). Centers for Disease Control and Prevention, Department of Health and Human Services, Atlanta.
Chapman T., McKeel D.W.Jr., et al. Misleading results with the 14-3-3 assay for the diagnosis of Creutzfeldt-Jakob disease. Neurology. 2000;55(9):1396-1397.
Chong J.Y., Rowland L.P. What’s in a NAIM? Hashimoto encephalopathy, steroid-responsive encephalopathy associated with autoimmune thyroiditis, or nonvasculitic autoimmune meningoencephalitis? Arch Neurol. 2006;63(2):175-176.
Chung E., Ji Y., et al. Anti-PrPC monoclonal antibody infusion as a novel treatment for cognitive deficits in an Alzheimer’s disease model mouse. BMC Neurosci. 2010;11:130.
Collie D.A., Summers D.M., et al. Diagnosing variant Creutzfeldt-Jakob disease with the pulvinar sign: MR imaging findings in 86 neuropathologically confirmed cases. AJNR Am J Neuroradiol. 2003;24(8):1560-1569.
Colling S.B., Khana M., et al. Mossy fibre reorganization in the hippocampus of prion protein null mice. Brain Res. 1997;755(1):28-35.
Collinge J., Whitfield J., et al. Kuru in the 21st century—an acquired human prion disease with very long incubation periods. Lancet. 2006;367(9528):2068-2074.
Collins S.J., Sanchez-Juan P., et al. Determinants of diagnostic investigation sensitivities across the clinical spectrum of sporadic Creutzfeldt-Jakob disease. Brain. 2006;129(Pt 9):2278-2287.
Cortelli P., Perani D., et al. Pre-symptomatic diagnosis in fatal familial insomnia: serial neurophysiological and 18FDG-PET studies. Brain. 2006;129(Pt 3):668-675.
Cortelli P., Perani D., et al. Cerebral metabolism in fatal familial insomnia: relation to duration, neuropathology, and distribution of protease-resistant prion protein. Neurology. 1997;49(1):126-133.
Criado J.R., Sanchez-Alavez M., et al. Mice devoid of prion protein have cognitive deficits that are rescued by reconstitution of PrP in neurons. Neurobiol Dis. 2005;19(1-2):255-265.
Gajdusek D.C. Unconventional viruses and the origin and disappearance of kuru. Science. 1977;197(4307):943-960.
Gajdusek D.C., Gibbs C.J., et al. Experimental transmission of a kuru-like syndrome to chimpanzees. Nature. 1966;209(25):794-796.
Gajdusek D.C., Gibbs C.J.Jr., et al. Precautions in medical care of, and in handling materials from, patients with transmissible virus dementia (Creutzfeldt-Jakob disease). N Engl J Med. 1977;297(23):1253-1258.
Garske T., Ghani A.C. Uncertainty in the Tail of the Variant Creutzfeldt-Jakob Disease Epidemic in the UK. PLoS One. 2010;5(12):e15626.
Geschwind M.D., Haman A., Miller B.L. Rapidly progressive dementia. Neurol Clin. 2007;25(3):783-807.
Geschwind M.D., Jay C. Assessment of rapidly progressive dementias. Concise review related to Chapter 362: Alzheimer’s disease and other primary dementias. In: Harrison’s Textbook of Internal Medicine. New York: McGraw Hill; 2003.
Geschwind M.D., Josephs K.A., Paresi J.E., et al. A 54-year-old man with slowness of movement and confusion. Neurology. 2007;69(19):1881-1887.
Geschwind M.D., Legname G. Transmissible spongiform encephalopathies. In: Smith H.J., Simons C., Sewell R.D. Protein Misfolding in Neurodegenerative Diseases: Mechanisms and Therapeutic Strategies. Boca Raton, FL: CRC Press, Taylor & Francis Group; 2008:518-532.
Geschwind M.D., Martindale J., et al. Challenging the clinical utility of the 14-3-3 protein for the diagnosis of sporadic Creutzfeldt-Jakob disease. Arch Neurol. 2003;60(6):813-816.
Geschwind M.D., Potter C.A., et al. Correlating DWI MRI with pathologic and other features of Jakob-Creutzfeldt disease. Alzheimer Dis Assoc Disord. 2009;23(1):82-87.
Geschwind M.D., Shu H., et al. Rapidly progressive dementia. Ann Neurol. 2008;64(1):97-108.
Gibbs C.J.Jr. Spongiform encephalopathies: slow, latent, and temperate virus infections–in retrospect. In: Prusiner S.B., Collinge J., Powell J., Anderton B. Prion Diseases of Humans and Animals. London: Ellis Horwood; 1992:53-62.
Gimbel D.A., Nygaard H.B., et al. Memory impairment in transgenic Alzheimer mice requires cellular prion protein. J Neurosci. 2010;30(18):6367-6374.
Giovagnoli A.R., Di Fede G., et al. Atypical frontotemporal dementia as a new clinical phenotype of Gerstmann-Sträussler-Scheinker disease with the PrP-P102L mutation. Description of a previously unreported Italian family. Neurol Sci. 2008;29(6):405-410.
Goldgaber D., Goldfarb L.G., et al. Mutations in familial Creutzfeldt-Jakob disease and Gerstmann-Sträussler-Scheinker’s syndrome. Exp Neurol. 1989;106(2):204-206.
Gunther E.C., Strittmatter S.M. Beta-amyloid oligomers and cellular prion protein in Alzheimer’s disease. J Mol Med. 2010;88(4):331-338.
Haley N.J., Seelig D.M., et al. Detection of CWD prions in urine and saliva of deer by transgenic mouse bioassay. PLoS One. 2009;4(3):e4848.
Health Protection Agency. Fourth case of transfusion-associated variant-CJD infection. Health Protection Report: weekly report. 2007;1(3):2-3.
Heath C.A., Cooper S.A., et al. Diagnosing variant Creutzfeldt-Jakob disease: a retrospective analysis of the first 150 cases in the UK. J Neurol Neurosurg Psychiatry. 2011;82(6):646-651.
Heath C.A., Cooper S.A., et al. Validation of diagnostic criteria for variant Creutzfeldt-Jakob disease. Ann Neurol. 2010;67(6):761-770.
Hsiao K., Baker H.F., et al. Linkage of a prion protein missense variant to Gerstmann-Sträussler syndrome. Nature. 1989;338(6213):342-345.
Hsich G., Kenney K., et al. The 14-3-3 brain protein in cerebrospinal fluid as a marker for transmissible spongiform encephalopathies. N Engl J Med. 1996;335(13):924-930.
Huillard d’Aignaux J.N., Cousens S.N., et al. The incubation period of kuru. Epidemiology. 2002;13(4):402-408.
Kanaani J., Prusiner S.B., et al. Recombinant prion protein induces rapid polarization and development of synapses in embryonic rat hippocampal neurons in vitro. J Neurochem. 2005;95(5):1373-1386.
Katamine S., Nishida N., et al. Impaired motor coordination in mice lacking prion protein. Cell Mol Neurobiol. 1998;18(6):731-742.
Katscher F. It’s Jakob’s disease, not Creutzfeldt’s. Nature. 1998;393(6680):11.
Kepe V., Ghetti B., et al. PET of brain prion protein amyloid in Gerstmann-Sträussler-Scheinker disease. Brain Pathol. 2010;20(2):419-430.
Kim J.I., Cali I., et al. Mammalian prions generated from bacterially expressed prion protein in the absence of any mammalian cofactors. J Biol Chem. 2010;285(19):14083-14087.
Kong Q.K., Surewicz W.K., et al. Inherited prion diseases. Biology Prion, Disease S., Prusiner B.. Cold Spring Harbor: Cold Spring Harbor Laboratory Press. 2004:673-776.
Korth C., Peters P.J. Emerging pharmacotherapies for Creutzfeldt-Jakob disease. Arch Neurol. 2006;63(4):497-501.
Kovacs G.G., Puopolo M., et al. Genetic prion disease: the EUROCJD experience. Hum Genet. 2005;118(2):166-174.
Kretzschmar H.A., Ironside J.W., et al. Diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Arch Neurol. 1996;53(9):913-920.
Le Pichon C.E., Valley M.T., et al. Olfactory behavior and physiology are disrupted in prion protein knockout mice. Nat Neurosci.. 2008.
Lewis A.M., Yu M., et al. Human growth hormone-related iatrogenic Creutzfeldt-Jakob disease with abnormal imaging. Arch Neurol. 2006;63(2):288-290.
Lomen-Hoerth C.W., Kuo A., Haman A., et al. Frequency of sensory symptoms and abnormal nerve conduction studies in a sCJD cohort. Neurology. 2010;74(Suppl. 2):A138.
Makarava N., Kovacs G.G., et al. Recombinant prion protein induces a new transmissible prion disease in wild-type animals. Acta Neuropathol. 2010;119(2):177-187.
Mallucci G.R., White M.D., et al. Targeting cellular prion protein reverses early cognitive deficits and neurophysiological dysfunction in prion-infected mice. Neuron. 2007;53(3):325-335.
Manson J.C., Clarke A.R., et al. 129/Ola mice carrying a null mutation in PrP that abolishes mRNA production are developmentally normal. Mol Neurobiol. 1994;8(2-3):121-127.
Mead S. Prion disease genetics. Eur J Hum Genet. 2006;14(3):273-281.
Mead S., Poulter M., et al. Genetic risk factors for variant Creutzfeldt-Jakob disease: a genome-wide association study. Lancet Neurol. 2009;8(1):57-66.
Nishida N., Tremblay P., et al. A mouse prion protein transgene rescues mice deficient for the prion protein gene from Purkinje cell degeneration and demyelination. Lab Invest. 1999;79(6):689-697.
Nygaard H.B., Strittmatter S.M. Cellular prion protein mediates the toxicity of beta-amyloid oligomers: implications for Alzheimer disease. Arch Neurol. 2009;66(11):1325-1328.
Oesch B., Westaway D., et al. A cellular gene encodes scrapie PrP 27-30 protein. Cell. 1985;40(4):735-746.
Parchi P., Giese A., et al. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol. 1999;46(2):224-233.
Peden A., McCardle L., et al. Variant CJD infection in the spleen of a neurologically asymptomatic UK adult patient with haemophilia. Haemophilia. 2010;16(2):296-304.
Peretz D., Supattapone S., et al. Inactivation of prions by acidic sodium dodecyl sulfate. J Virol. 2006;80(1):322-331.
Petzold G.C., Westner I., et al. False-positive pulvinar sign on MRI in sporadic Creutzfeldt-Jakob disease. Neurology. 2004;62(7):1235-1236.
Prusiner S.B. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216(4542):136-144.
Prusiner S.B. Prions. Proc Natl Acad Sci U S A. 1998;95(23):13363-13383.
Prusiner S.B., Bosque P.J. Prion diseases. In: Braunwald E., editor. Harrison’s Principles of Internal Medicine. New York: McGraw Hill, 2001. Chapter 375
Prusiner S.B., Groth D., et al. Ablation of the prion protein (PrP) gene in mice prevents scrapie and facilitates production of anti-PrP antibodies. Proc Natl Acad Sci U S A. 1993;90(22):10608-10612.
Rabinovici G.D., Wang P.N., et al. First symptom in sporadic Creutzfeldt-Jakob disease. Neurology. 2006;66(2):286-287.
Rossi D., Cozzio A., et al. Onset of ataxia and Purkinje cell loss in PrP null mice inversely correlated with Dpl level in brain. Embo J. 2001;20(4):694-702.
Safar J., Wille H., et al. Eight prion strains have PrP(Sc) molecules with different conformations. Nat Med. 1998;4(10):1157-1165.
Safar J.G., Geschwind M.D., et al. Diagnosis of human prion disease. Proc Natl Acad Sci U S A. 2005;102(9):3501-3506.
Sakaguchi S., Katamine S., et al. Loss of cerebellar Purkinje cells in aged mice homozygous for a disrupted PrP gene. Nature. 1996;380(6574):528-531.
Sanchez-Juan P., Green A., et al. CSF tests in the differential diagnosis of Creutzfeldt-Jakob disease. Neurology. 2006;67(4):637-643.
Satoh J., Kurohara K., et al. The 14-3-3 protein detectable in the cerebrospinal fluid of patients with prion-unrelated neurological diseases is expressed constitutively in neurons and glial cells in culture. Eur Neurol. 1999;41:216-225.
Scott M.R., Will R., et al. Compelling transgenetic evidence for transmission of bovine spongiform encephalopathy prions to humans. Proc Natl Acad Sci U S A. 1999;96(26):15137-15142.
Seipelt M., Zerr I., et al. Hashimoto’s encephalitis as a differential diagnosis of Creutzfeldt- Jakob disease. J Neurol Neurosurg Psychiatry. 1999;66(2):172-176.
Shaked G.M., Engelstein R., et al. Valproic acid treatment results in increased accumulation of prion proteins. Ann Neurol. 2002;52(4):416-420.
Shiga Y., Miyazawa K., et al. Diffusion-weighted MRI abnormalities as an early diagnostic marker for Creutzfeldt-Jakob disease. Neurology. 2004;I63:443-449.
Sigurdson C.J., Nilsson K.P., et al. De novo generation of a transmissible spongiform encephalopathy by mouse transgenesis. Proc Natl Acad Sci U S A. 2009;106(1):304-309.
Sikorska B., Liberski P.P., et al. Ultrastructural study of florid plaques in variant Creutzfeldt-Jakob disease: a comparison with amyloid plaques in kuru, sporadic Creutzfeldt-Jakob disease and Gerstmann-Sträussler-Scheinker disease. Neuropathol Appl Neurobiol.. 2008.
Spudich A., Frigg R., et al. Aggravation of ischemic brain injury by prion protein deficiency: role of ERK-1/-2 and STAT-1. Neurobiol Dis. 2005;20(2):442-449.
Steinhoff B.J., Zerr I., et al. Diagnostic value of periodic complexes in Creutzfeldt-Jakob disease. Ann Neurol. 2004;56(5):702-708.
Stewart L.A., Rydzewska L.H., et al. Systematic review of therapeutic interventions in human prion disease. Neurology. 2008;70(15):1272-1281.
Taraboulos A., Jendroska K., et al. Regional mapping of prion proteins in brain. Proc Natl Acad Sci U S A. 1992;89(16):7620-7624.
Tobler I., Deboer T., et al. Sleep and sleep regulation in normal and prion protein-deficient mice. J Neurosci. 1997;17(5):1869-1879.
Tschampa H.J., Neumann M., et al. Patients with Alzheimer’s disease and dementia with Lewy bodies mistaken for Creutzfeldt-Jakob disease. J Neurol Neurosurg Psychiatry. 2001;71(1):33-39.
Tullo A.B., Buckley R.J., et al. Transplantation of ocular tissue from a donor with sporadic Creutzfeldt-Jakob disease. Clin Experiment Ophthalmol. 2006;34(7):645-649.
UK National CJD Surveillance Unit, 2010. The National Creutzfeldt-Jakob Disease Surveillance Unit (NCJDSU). Western General Hospital, Edinburgh.
Valleron A.J., Boelle P.Y., et al. Estimation of epidemic size and incubation time based on age characteristics of vCJD in the United Kingdom. Science. 2001;294(5547):1726-1728.
van der Kamp M.W., Daggett V. Pathogenic mutations in the hydrophobic core of the human prion protein can promote structural instability and misfolding. J Mol Biol. 2010;404(4):732-748.
Vernino S., Geschwind M.D., et al. Autoimmune encephalopathies. The Neurologist. 2007;13(3):140-147.
Vitali P., Migliaccio R., et al. Neuroimaging in dementia. Semin Neurol. 2008;28(4):467-483.
Wadsworth J.D., Joiner S., et al. Phenotypic heterogeneity in inherited prion disease (P102L) is associated with differential propagation of protease-resistant wild-type and mutant prion protein. Brain. 2006;129(Pt 6):1557-1569.
Wall C.A., Rummans T.A., et al. Psychiatric manifestations of Creutzfeldt-Jakob disease: a 25-year analysis. J Neuropsychiatry Clin Neurosci. 2005;17(4):489-495.
Watts J.C., Balachandran A., et al. The expanding universe of prion diseases. PLoS Pathog. 2006;2(3):e26.
Webb T.E., Poulter M., et al. Phenotypic heterogeneity and genetic modification of P102L inherited prion disease in an international series. Brain. 2008;131(Pt 10):2632-2646.
Weise J., Sandau R., et al. Deletion of cellular prion protein results in reduced Akt activation, enhanced postischemic caspase-3 activation, and exacerbation of ischemic brain injury. Stroke. 2006;37(5):1296-1300.
WHO. Global Surveillance, Diagnosis and Therapy of Human Transmissible Spongiform Encephalopathies: Report of a WHO Consultation. Emerging and Other Communicable Diseases, Surveillance and Control. Geneva, Switzerland: World Health Organization; 1998.
WHO. Practical Guidelines for Infection Control in Health Care Facilities. Geneva, Switzerland: World Health Organization; 2003.
WHO. WHO Guidelines on Tissue Infectivity Distribution in Transmissible Spongiform Encephalopathies, Quality and Safety of Plasma Derivatives and Related Substances. Department of Medicines Policy and Standards, Health Technology and Pharmaceuticals Cluster. Geneva, Switzerland: World Health Organization; 2006.
Wilham J.M., Orru C.D., et al. Rapid end-point quantitation of prion seeding activity with sensitivity comparable to bioassays. PLoS Pathog. 2010;6(12):e1001217.
Will R.G. Acquired prion disease: iatrogenic CJD, variant CJD, kuru. Br Med Bull. 2003;66:255-265.
Will R.G. Variant Creutzfeldt-Jakob disease. Folia Neuropathol. 2004;42(Suppl. A)):77-83.
Will R.G., Ironside J.W., et al. A new variant of Creutzfeldt-Jakob disease in the UK. Lancet. 1996;347(9006):921-925.
Will R.G., Zeidler M., et al. Diagnosis of new variant Creutzfeldt-Jakob disease. Ann Neurol. 2000;47(5):575-582.
Williams E.S. Chronic wasting disease. Vet Pathol. 2005;42(5):530-549.
Wong K., Cattaruzza T., et al. Brain MRI in sporadic Jakob-Creutzfeldt disease is often misread. Neurology. 2009.
Young G.S., Geschwind M.D., et al. Diffusion-weighted and fluid-attenuated inversion recovery imaging in Creutzfeldt-Jakob disease: high sensitivity and specificity for diagnosis. AJNR Am J Neuroradiol. 2005;26(6):1551-1562.
Zerr I., Kallenberg K., et al. Updated clinical diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Brain. 2009;132(Pt 10):2659-2668.
Zou S., Fang C.T., et al. Transfusion transmission of human prion diseases. Transfus Med Rev. 2008;22(1):58-69.
Zou W.Q., Puoti G., et al. Variably protease-sensitive prionopathy: a new sporadic disease of the prion protein. Ann Neurol. 2010;68(2):162-172.