[level-membership-for-pathology-category]
Chapter 8 Ischaemia, infarction and shock
Blood suffers the various pathological processes that occur in all tissues (neoplasia, nutritional defects, etc., see Ch. 23) but because blood is a tissue that circulates there is also a specific set of pathologies related to defects in flow.
Thrombo-embolic events are major causes of morbidity and mortality in the UK and other developed countries. The majority of readers (and authors) of this book will die from events somewhere along the sequence described in this chapter. Common and serious disorders in which thrombo-embolic mechanisms participate include:
However, no organ is exempt from the harmful consequences of an impaired blood supply, although some (e.g. brain, myocardium), because of their nutritional requirements and inability to regenerate, are more vulnerable than others.
Ischaemia is the result of impaired vascular perfusion, depriving the affected tissue of vital nutrients, especially oxygen. The effects on the tissue can be reversible, but this depends on:
Infarction is death (necrosis) of tissue as a result of ischaemia. Infarction is irreversible, but tissues vary in their ability to repair and replace the loss. Infarction usually results from thrombo-embolic phenomena completely occluding the artery supplying the affected tissue.
Shock (pathophysiological rather than psychological surprise) is a state of circulatory collapse resulting in impaired tissue perfusion. Ischaemia, infarction and shock are, therefore, interrelated phenomena. The cellular responses to impaired nutrition are summarised in Chapter 6.
Although the most common causes of ischaemia and infarction are thrombo-embolic phenomena, vascular insufficiency can also result from other causes (Fig. 8.1).
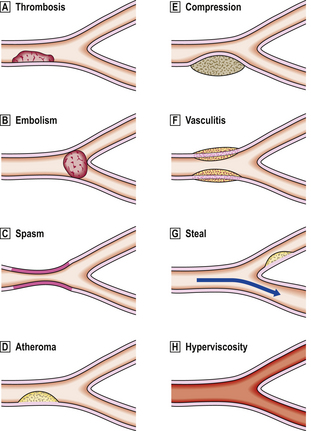
Fig. 8.1 Vascular lesions causing ischaemia.  Thrombosis: initiated by either abnormal flow (e.g. stasis, turbulence), damage to vessel wall (e.g. denudation of endothelial lining) or abnormal blood constituents.
Thrombosis: initiated by either abnormal flow (e.g. stasis, turbulence), damage to vessel wall (e.g. denudation of endothelial lining) or abnormal blood constituents.  Embolism.
Embolism.  Spasm: due to contraction of smooth muscle in media of vessel, for example due to lack of nitric oxide from endothelium.
Spasm: due to contraction of smooth muscle in media of vessel, for example due to lack of nitric oxide from endothelium.  Atheroma: occurs only in arteries and may in turn be complicated by thrombosis and embolism.
Atheroma: occurs only in arteries and may in turn be complicated by thrombosis and embolism.  Compression: veins are more susceptible because of their thinner walls and lower intraluminal pressure.
Compression: veins are more susceptible because of their thinner walls and lower intraluminal pressure.  Vasculitis: inflammation of vessel wall narrows lumen and may be complicated by superimposed thrombosis.
Vasculitis: inflammation of vessel wall narrows lumen and may be complicated by superimposed thrombosis.  Vascular steal: for example, an artery may be narrowed by atheroma but flow is still sufficient to maintain viability of perfused territory; however, flow may be compromised by increased demands of a neighbouring territory.
Vascular steal: for example, an artery may be narrowed by atheroma but flow is still sufficient to maintain viability of perfused territory; however, flow may be compromised by increased demands of a neighbouring territory.  Hyperviscosity: increased viscosity, for example in hypergammaglobulinaemia resulting from myeloma, causes impaired flow and predisposes to thrombosis.
Hyperviscosity: increased viscosity, for example in hypergammaglobulinaemia resulting from myeloma, causes impaired flow and predisposes to thrombosis.
NON-THROMBO-EMBOLIC VASCULAR INSUFFICIENCY
Vascular flow can be impeded by abnormalities other than thrombo-embolic phenomena.
In arteries, the commonest lesion is atheroma, which in turn may be complicated by thrombo-embolism. In medium-sized arteries, atheromatous plaques (Ch. 13) often narrow the lumen, causing ischaemia and sometimes atrophy of tissues in the hypoperfused territory. Serious consequences include the symptom of angina due to myocardial ischaemia, often heralding the development of irreversible infarction, and hypertension due to renal artery narrowing and hypoperfusion of a kidney, which responds physiologically by increased renin secretion.
Transient arterial narrowing can result from spasm of the smooth muscle in the vessel wall. This can be due to a decrease in nitric oxide (endothelium-derived relaxing factor) production by the vascular endothelium due to cellular injury or loss. Spasm of coronary arteries can lead to angina and both may be relieved by glyceryl trinitrate. Arterial spasm is also responsible for the transient ischaemia of the fingers in Raynaud’s phenomenon.
Blood vessels can be partially or totally occluded by external compression. This is done intentionally during surgery by ligation to prevent haemorrhage from severed vessels, although the results can be disastrous if, accidentally, the wrong vessel is tied off! Because of their thin walls and low intraluminal pressure, veins are more susceptible to occlusion by external compression. This occurs commonly in strangulated hernias, testicular torsion and torsion of ovaries containing cysts or tumours.
‘Steal’ syndromes occur when blood is diverted (‘stolen’) from a vital territory. This results when, proximal to an area of atheromatous narrowing insufficient on its own to produce ischaemia, the arterial stream is diverted along another branch vessel to meet the increased demands of a competing territory or lesion; the territory supplied by the atheromatous vessel then becomes ischaemic. This is a relatively uncommon cause of ischaemia, but often the most challenging diagnostically.
Ischaemia at the arteriolar, capillary and venular level can result from increased whole blood viscosity. Viscosity effects contribute relatively little to the flow characteristics of blood in vessels of large calibre, but in small vessels they are a major factor. Hyperviscosity of blood can occur in myeloma, a tumour of plasma cells, as a result of the abnormally high concentration of gammaglobulin in the plasma and rouleaux formation by red cells.
THROMBO-EMBOLIC VASCULAR OCCLUSION
Clot
When blood stagnates due to the cessation of the pumping action of the heart, or if blood is allowed to stand in a bottle or test tube, then the clotting process is set in motion. A complex series of enzymatic steps (Ch. 23) is activated, resulting in the formation of a fibrin meshwork that entraps the cells into a solid but elastic clot. When this process occurs in the body after death, the red cells tend to settle out before the clot forms, so that these postmortem clots have two layers: a lower, deep red layer (resembling redcurrant jelly) and an upper, clearer layer (resembling chicken fat), with platelets evenly distributed throughout. Since these clots have formed within the body and represent the blood content of the vessel during life, they are moulded to the shape of the vessels in which they have formed. Some time after death, the various blood cells and the cells of the vessel wall begin to release their hydrolytic enzymes and the clot is dissolved.
The sequence of enzymatic reactions involved in the clotting cascade and abnormalities of this system are discussed in Chapter 23.
Thrombosis





Thrombus is a solidification of blood contents that forms within the vascular system during life and is therefore different in concept from a clot. Its mode of formation, its structure and its appearance are all different from those of a clot and the two should never be confused.
Role of platelets
The mechanism for closing small gaps in vessel walls brought about by trauma involves the platelets. Platelets are smaller than red blood cells, rather angular in appearance, and have no nucleus. They are derived from large, multinucleated cells in the bone marrow called megakaryocytes. Although platelets have no nucleus, they are highly structured internally and contain a variety of organelles, some of which are specific to them. As well as mitochondria and the various cytoskeletal elements found in most cells, platelets also contain alpha granules and dense granules. The alpha granules contain several substances involved in the process of platelet adhesion to damaged vessel walls (fibrinogen, fibronectin, platelet growth factor and an antiheparin as well as less well defined substances), and the dense granules contain substances such as adenosine diphosphate (ADP) that cause platelets to aggregate.
Platelets are activated and the contents of their granules are released when the platelets come into contact with collagen, as may be found in damaged vessel walls, or with polymerising fibrin. The platelets change shape and extend pseudopodia; their granules release their contents and the platelets form a mass that covers the vessel wall defect until the endothelial cells have regenerated and repaired the vessel permanently. However, if this process is activated within an intact vessel, it results in a thrombus.
Thrombus formation
There are three predisposing situations that may result in thrombus formation. These were described originally by Virchow and are known as Virchow’s triad. The three factors are:
Not all three are needed for thrombosis to occur, but any one of them may result in thrombosis in a particular case.
If we consider the sequence of events involved in the formation of thrombus on the basis of an atheromatous plaque (Ch. 13), this will serve as a very good example of the factors listed by Virchow.
Arterial thrombosis
In its earliest phase the atheromatous plaque may consist of a slightly raised fatty streak on the intimal surface of any artery, such as the aorta (Fig. 8.2). With time, the plaque enlarges and becomes sufficiently raised to protrude into the lumen and cause a degree of turbulence in the blood flow. This turbulence eventually causes loss of intimal cells, and the denuded plaque surface is presented to the blood cells, including the platelets. The turbulence itself will predispose to fibrin deposition and to platelet clumping; the bare luminal surface of the vessel will have collagen exposed and platelets will settle on this surface. Thus we have two of the factors described in Virchow’s triad operating in an atheromatous plaque. If this plaque exists in the aorta of a smoker or someone with a high cholesterol level and a high level of low density lipoprotein—common risk factors for atheroma—then the third of Virchow’s factors is introduced, since these changes in blood constituents are well known to predispose to thrombus formation. This process, once begun, may be self-perpetuating, as it has been shown that platelet-derived growth factor, which is contained in the alpha granules, causes proliferation of arterial smooth muscle cells, which are an important constituent of the atheromatous plaque.
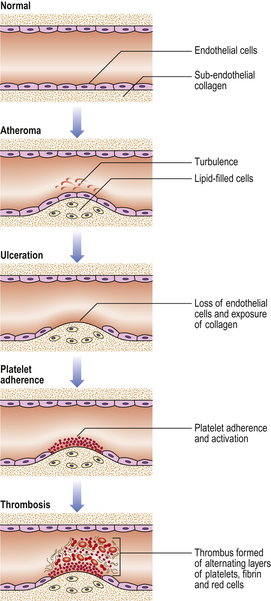
Fig. 8.2 Thrombosis. Thrombosis is exemplified by its occurrence on an atheromatous plaque, a particularly common event. Important steps in this sequence include: loss of endothelial cells and exposure of the underlying collagen; platelet adherence and activation; partial or complete arterial occlusion by the multilayered thrombus.
Thus, the first layer of the thrombus is a platelet layer. Formation of this layer in turn causes the precipitation of a fibrin meshwork, in which red cells are trapped, and a layer of this meshwork is developed on top of the platelet layer. The alternating bands of white platelets and red blood cells in thrombi were first described by Zahn and are called the lines of Zahn. This complex structure now protrudes even further into the lumen, causing more turbulence and forming the basis for further platelet deposition. The normal flow of blood within the vessels is laminar; the cells move in the swifter central lane and the plasma runs along the walls. Therefore, the greatest degree of turbulence occurs at the downstream side of arterial thrombi, as the blood passes over the thrombus, and on the upstream side of venous thrombi for the same reason. Thrombi will therefore grow in the direction of blood flow; this process is known as propagation.
Venous thrombosis
In veins, however, the blood pressure is lower than in arteries and atheroma does not occur; so what initiates venous thrombus formation? Most venous thrombi seem to begin at valves. Valves naturally produce a degree of turbulence because they protrude into the vessel lumen and they may be damaged by trauma, stasis and occlusion. However, thrombi can also form in veins of young, active individuals with no predisposing factors that can be identified. Once they begin, the thrombi grow by successive deposition in the manner described previously and this process may produce a highly patterned, coralline growth (Fig. 8.3). Since normal flow within the vessels is laminar, most of the blood cells are kept away from diseased walls or from damaged vein valves. However, if the blood pressure is allowed to fall during surgery or following a myocardial infarction, then flow is slower through the vessels and thrombosis becomes a likely event. Similarly, the venous return from the legs is very reliant upon calf muscle contraction and relaxation, which massages the veins and, because of the valves, tends to return the blood heartwards. So, if elderly subjects are immobilised for any reason, they are at great risk from the formation of deep leg vein thromboses. The frequency with which deep vein thrombosis is found to occur following surgery is directly related to the enthusiasm with which it is sought (e.g. by the pathologist at postmortem examination) and the sensitivity of the methods used to demonstrate it. Postmortem studies on unselected medical and surgical patients show significant deep vein thrombosis in 34% of the former and 60% of the latter, regardless of the cause of death.
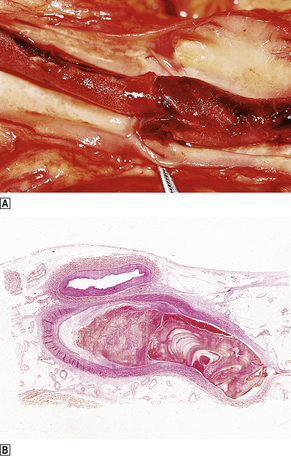
Fig. 8.3 Venous thrombus.  Femoral vein opened at autopsy to reveal a thrombus.
Femoral vein opened at autopsy to reveal a thrombus.  Histological section showing the characteristic laminated or coralline structure of a thrombus.
Histological section showing the characteristic laminated or coralline structure of a thrombus.
When a vein becomes thrombosed it evokes an inflam-matory reaction, a phenomenon known as thrombophlebitis; the opposite process also occurs—a vein that is inflamed will often thrombose and this is known as phlebothrombosis. The end effect is the same, a thrombosed and inflamed vein, but clearly if there is a predisposing cause then the cause needs to be recognised and treated.
Clinical effects
The effects of thrombosis are apparent only if the thrombus is sufficiently large to affect the flow of blood significantly. Arterial thrombosis results in loss of pulses distal to the thrombus and all the signs of impaired blood supply: the area becomes cold, pale and painful, and eventually the tissue dies and gangrene results. In venous thromboses, 95% of which occur in leg veins, the area becomes tender, swollen and reddened, as blood is still carried to the site by the arteries but cannot be drained away by the veins. The tenderness is due to developing ischaemia in the vein wall initially, but there is also general ischaemic pain as the circulation worsens. The more specific clinical effects of thrombosis depend on the tissue that is affected.
Myocardial infarction is often associated with thrombus formation in coronary arteries and is responsible for numerous sudden deaths (Ch. 13).
Strokes may be due to the formation of thrombus in a cerebral vessel, although they may be also the result of haemorrhage or embolism (Ch. 26).
Thrombophlebitis migrans occurs in previously healthy veins in any area of the body. The thromboses appear and disappear, changing site all the time, and the condition may persist for months or even years. It is extremely ominous and usually indicates the presence of visceral cancer, commonly of the pancreas. The mechanism remains obscure.
Fate of thrombi
Various fates await the newly formed thrombus (Fig. 8.4). In the best scenario it may resolve; the various degradative processes available to the body may dissolve it and clear it away completely. It is not known what proportion of thrombi follow this course, but the total number is likely to be large. A second possibility is that the thrombus may become organised into a scar by the invasion of macrophages, which clear away the thrombus, and fibroblasts, which replace it with collagen, occasionally leaving a mural nodule or web that narrows the vessel lumen. A third possibility is that the intimal cells of the vessel in which the thrombus lies may proliferate, and small sprouts of capillaries may grow into the thrombus and later fuse to form larger vessels. In this way the original occlusion may become recanalisedand the vessel patent again. Another common result is that the thrombus affects some vital centre and causes death before either the body or the clinicians can make an effective response; this event is very common. Finally, fragments of the thrombus may break off into the circulation, a process known as embolism.
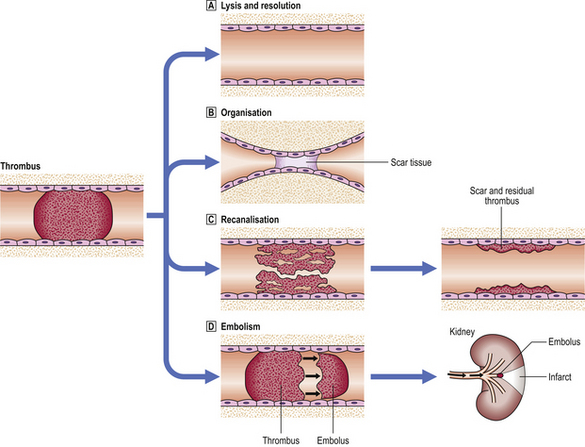
Fig. 8.4 Consequences of thrombosis.  Lysis of the thrombus and complete restitution of normal structure usually can occur only when the thrombus is relatively small and is dependent upon fibrinolytic activity (e.g. plasmin).
Lysis of the thrombus and complete restitution of normal structure usually can occur only when the thrombus is relatively small and is dependent upon fibrinolytic activity (e.g. plasmin).  The thrombus may be replaced by scar tissue which contracts and obliterates the lumen; the blood bypasses the occluded vessel through collateral channels.
The thrombus may be replaced by scar tissue which contracts and obliterates the lumen; the blood bypasses the occluded vessel through collateral channels.  Recanalisation occurs by the ingrowth of new vessels which eventually join up to restore blood flow, at least partially.
Recanalisation occurs by the ingrowth of new vessels which eventually join up to restore blood flow, at least partially.  Embolism caused by fragmentation of the thrombus and resulting in infarction at a distant site.
Embolism caused by fragmentation of the thrombus and resulting in infarction at a distant site.
Embolism
 An embolus is a mass of material in the vascular system able to become lodged within a vessel and block its lumen
An embolus is a mass of material in the vascular system able to become lodged within a vessel and block its lumen


An embolus is a mass of material in the vascular system able to lodge in a vessel and block its lumen. The material may have arisen within the body or have been introduced from outside. The material may be solid, liquid or gaseous. The end results of embolism are more dependent upon the final resting place of the embolic material than on its nature. Emboli travel in the circulation, passing through the vascular tree until they reach a vessel whose diameter is small enough to prevent their further passage. The clinical effects will therefore depend upon the territory supplied by that vessel and the presence or absence of an alternative (collateral) circulation to that area. The most frequent source of embolic material is a thrombus formed in any area of the circulatory system, but other sources of embolic material should not be disregarded. Over 90% of major emboli are derived from thrombi, so we shall first consider the principal clinical syndromes associated with this situation and then briefly mention other forms of embolism.
Pulmonary embolism
Around 95% of venous thrombosis occurs in leg veins; the majority of the rest occur in pelvic veins, and a very few occur in the intracranial venous sinuses. Therefore, most emboli from such thrombi will arrive in the pulmonary circulation—pulmonary embolism. The only possibility for such emboli to arrive in the arterial side of the circulation is if there is an arterial–venous communication, such as a perforated septum in the heart (paradoxical embolus), but this event is exceptionally rare.
The effects of pulmonary emboli depend upon their size. Small emboli may occur unnoticed and be lysed within the lung or they may become organised and cause some permanent, though small, respiratory deficiency. Such a respiratory deficiency may only come to light with the eventual accumulation of damage from many such tiny embolic events. The accumulation of such damage over a long period may be the cause of so-called ‘idiopathic’ pulmonary hypertension (Ch. 14).
A second class of pulmonary emboli may be large enough to cause acute respiratory and cardiac problems that may resolve slowly with or without treatment. The main symptoms are chest pain and shortness of breath due to the effective loss of the area of lung supplied by the occluded vessel; the area may even become infarcted. This occlusion puts some strain on the heart, which is evident on the ECG as right heart strain with deep S waves in lead 1 and the presence of Q waves and inverted T waves in lead 3 (S1, Q3, T3). This ECG pattern is one of the few that a pathologist will look for in the case notes before performing an autopsy on a patient suspected of having had a pulmonary embolus in life; without these findings the embolus is unlikely to have caused death. Although many patients recover from such episodes, their lung function is impaired and, of course, they are at risk from further emboli from the same source. Consequently, they require symptomatic therapy for the embolus as well as treatment for the causative thrombus.
The third class of pulmonary emboli are massive and result in sudden death. These are usually long thrombi derived from leg veins and have the shape of the vessels in which they arose, rather than that of the vessels in which they are found at postmortem examination. They are often impacted across the bifurcation of one of the major pulmonary arteries as a ‘saddle embolus’, a descriptive term for their appearance. As with all thrombi, even if they have undergone embolisation they retain the appearance of a thrombus, with lines of Zahn and a granular, friable consistency. This consistency is distinct from the elastic, ‘chicken fat and redcurrant jelly’ appearance of postmortem clots.
Systemic embolism
Systemic emboli arise in the arterial system and again their effects are due to their size and to the vessel in which they finally lodge. The thrombi from which they come generally form in the heart or on atheromatous plaque (Fig. 8.5). In the heart, thrombi may form on areas of cardiac muscle that has died as a result of myocardial infarction, as these areas will have lost their normal endothelial lining and will expose the underlying collagen to the circulating platelets. These areas of dead myocardium will also be adynamic and will disrupt the normal blood flow within the heart, creating turbulence and predisposing to thrombus formation at that site.
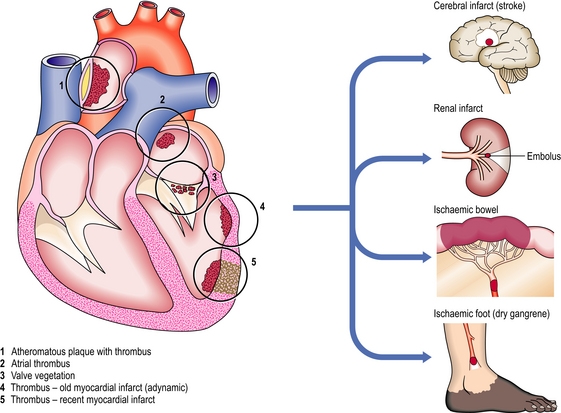
Fig. 8.5 Origins and effects of systemic arterial emboli. Systemic arterial emboli almost invariably originate from the left side of the heart or from major arteries. Infarction or gangrene is the usual consequence.
Another common cause of thrombosis within the heart is the presence of atrial fibrillation. This ineffectual movement of the atria causes blood to stagnate in the atrial appendages and thrombosis to occur. When the normal heart rhythm is re-established the atrial thrombus may be fragmented and emboli broken off.
Emboli from the heart are usually derived from thrombi on the left side of the circulation. These emboli may travel to the brain, causing cerebrovascular incidents such as transient ischaemic attacks or strokes, or they may travel to any of the viscera, or to the limbs.
Large emboli may lodge at the bifurcation of the aorta as a saddle embolus cutting off the blood supply to the lower limbs, a situation that requires rapid diagnosis if the embolus is to be removed before the changes in the limbs become irreversible. Smaller emboli may lodge in smaller vessels nearer the periphery and cause gangrene of the digits. Small emboli may travel into the kidneys or spleen and be relatively asymptomatic, even when they cause the death of the area of tissue distal to their site of impaction; ischaemic scars are not uncommon findings at autopsy with no clinical history to lead one to suspect that such events had been occurring.
More dramatic consequences develop as a result of emboli travelling to the intestine, often passing down the superior mesenteric artery; this impaction can cause death of whole sections of small bowel, which, unlike kidneys or spleen, depends upon the whole organ to be intact in order to function. The death of even a small area of bowel means perforation and peritonitis, whereas the death of a small area of kidney or spleen means only a small scar.
Vegetations on the heart valves are an important source of emboli. Most seriously, in infective endocarditis the vegetations consist of micro-organisms, usually bacteria, and are extremely friable. Marantic vegetations, consisting of platelets and fibrin, occur on the heart valves of patients who are severely debilitated, for example by cancer; these vegetations are often firmly adherent and are less likely to embolise.
Embolic atheroma
Fragments of atheromatous plaque may embolise and these are frequently seen in the lower limbs of arteriopathic patients. The precise cause of such ischaemic toes is rarely investigated thoroughly enough to be diagnosed. The embolic fragments may be recognised in histological preparations by the cigar-shaped clefts left behind when the cholesterol crystals dissolve out during histological processing.
Platelet emboli
Since the early stages of atheroma involve mainly platelet deposition, emboli from early lesions may be composed solely of platelets. In general these are very tiny emboli and do not present with severe clinical signs. The exception is in the brain, where even small emboli manifest with striking clinical symptoms and signs. A stroke that lasts for less than 24 hours and that is associated with complete clinical recovery is termed a transient ischaemic attack (TIA); although these show complete resolution, they are risk markers for subsequent major strokes. While many TIAs are thought to be due to platelet emboli, there is some evidence that in some cases they may also be caused by arterial spasm.
Infective emboli
Infected lesions within the blood stream, in particular the vegetations on diseased heart valves (Ch. 13), may break off and lodge in small vessels in the usual way. But here, the usual effects of emboli are compounded by the infective agent present and this agent may weaken the wall of the vessel, causing the development of a ‘mycotic’ aneurysm. (Mycotic is a misnomer because the infective agent is usually bacterial, not fungal.)
Fat embolism
Fat embolism usually arises following some severe trauma with fracture to long bones. Fat from the bone marrow is released into the circulation and comes to lodge in various organs. A similar situation arises in severe burns and in extensive soft tissue injury. Much of the circulating fat enters the lungs and this indicates that it must travel by way of the venous system. However, fat globules are fluid and so small that they may cross the pulmonary vascular bed and, therefore, many also enter the systemic arterial circulation, causing confusion or coma, renal impairment and skin petechiae. It has also been suggested that systemic effects of trauma, particularly burns, can cause changes in the stability of fat held in micellar suspension, resulting in free fat appearing in the circulation. This fat could then travel in the circulation in exactly the same way as a true fat embolism. Some degree of fat embolism probably occurs in most cases of long bone injury but it is generally asymptomatic.
Gas embolism
There are various causes of embolic events involving gas; several are iatrogenic. The classic form is caisson disease, experienced by divers when they are transferred too rapidly from high to low pressure environments. At high pressure, increased volumes of gas dissolve in the blood and during rapid decompression these come out as bubbles. In the case of air, the oxygen and carbon dioxide redissolve but the nitrogen bubbles remain and enter bones and joints, causing the pains of the ‘bends’, or they lodge in the lungs, causing the respiratory problems of the ‘chokes’.
The other causes of gas embolism are mainly surgical when some vessel is opened to the air. This also occurs in suicide attempts when the neck veins are cut, or accidentally when patients are disconnected from intravenous lines and air enters. The ‘secret murders’ by air injection so favoured by thriller writers are rare, as the volume of air needed to cause death in this fashion is around 100 ml.
The pathological signs of this condition at autopsy include visible bubbles in the vessels, such as those of the meninges, and sometimes a frothy ball of fibrin and air in the right side of the heart, occluding one of the valves.
Amniotic embolism
With the vastly increased pressures in the uterus during labour, amniotic fluid may be forced into the maternal uterine veins. These amniotic fluid emboli travel in the circulation and lodge in the lungs, causing respiratory distress like other pulmonary emboli. They can be recognised histologically because they contain the shed skin cells of the infant.
Tumour embolism
Tumour emboli are mainly small and break off as tumours that penetrate vessels (Ch. 11). They do not usually cause immediate physical problems in the way that other emboli do, but this mechanism is a major route of dissemination of malignancies through the body (metastasis).
Embolism of foreign matter
Particles of foreign matter may contaminate fluids injected intravenously. This is rare when such fluids are injected for medical reasons, but talc, etc. is a common contaminant of fluids injected by intravenous drug abusers. The foreign particles elicit a granulomatous reaction in the organs in which they lodge.
INFARCTION




Infarction is ischaemic death of tissue within the living body. This means that death of tissue from other causes, such as toxins or trauma, is not infarction but is simply necrosis, which is the general term for death of tissue within the living organism. Only death of tissue due to restricted blood supply is infarction. The word infarction means ‘stuffed full’ and reflects the fact that the first types of infarction that were recognised were those in which the blockage was venous and the arterial supply continued to pump blood into the organ when the outlet was blocked. A similar effect may be seen in those cases where a second blood supply is present and, although the arterial inflow is blocked, blood still enters the organ from this second supply; a good example of this is the lung, which has both pulmonary and bronchial arterial supplies. In the past, pathologists classically divided infarcts into grey and red infarcts, but these merely represent stages in the same process; not all infarcts in the same organ go through these stages, so the division is pointless.
Reperfusion injury
Many of the tissue effects of ischaemic injury paradoxically seem to occur not during the ischaemic episode but when perfusion is re-established. This is not as illogical as it may seem because much of this damage is oxygen dependent and the only way for oxygen to get to the site is by blood flow. When the blood flow returns to an area of tissue that has been ischaemic, it encounters tissue where transport mechanisms across the cell membrane have been disrupted to a variable extent and, in particular, where calcium transport out of the cell and from organelles such as mitochondria is impaired. This appears to be the trigger for the activation of oxygen-dependent free radical systems that begin the clearing away of dead cells which we recognise as a part of reperfusion injury. At the same time polymorphs and macrophages enter the area and begin to clear away debris and themselves import their own intrinsic oxygen free radicals into the area.
Experimental studies reveal that cell death in reperfusion injury and, in some instances, in pure ischaemia involves the intracellular mechanisms of apoptotic cell death. This may result from disruption of the cell–matrix interactions on which cells depend for survival.
Experimentally, reperfusion injury can be prevented with anti-oxidants but this has only a small effect on the ultimate amount of tissue loss. What we see here is another example of an adaptive process (clearing up of dead and damaged cells) that produces deleterious effects (scarring) and that can be marginally modified pharmacologically.
Morphology of infarcts
Infarcted areas vary in appearance depending on the time that has elapsed between the infarct occurring and the lesion coming to the attention of the pathologist (Fig. 8.6). If the tissue is examined within 24 hours of the infarct there will be no direct evidence of the event. Between 24 and 48 hours the dead tissue is beginning to evoke a response from the surrounding living tissues and inflammatory cells can be seen moving into the infarcted area. In routine histological sections stained with haematoxylin and eosin the cytoplasm contains proteins, which stain pink, and RNA, which stains blue. In normal tissue, the cytoplasm therefore has a slightly purple tinge and in areas that have been dead a few hours the RNA is broken down and the cytoplasm becomes bright pink. It should be borne in mind that all histological sections of tissue are ‘dead’ and that what we are looking at is the consequence of the time that has elapsed between the infarcted tissue dying and the rest of the tissue being killed by being dropped into formalin or some other fixative.
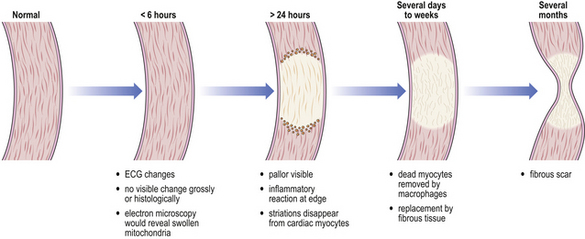
Fig. 8.6 Evolution of an infarct. This is typified by a myocardial infarct in which naked-eye abnormalities are rarely evident until many hours have elapsed. The dead tissue elicits an inflammatory reaction, characteristically neutrophil polymorphs and, later, macrophages. Unless complications intervene, the infarct heals by fibrosis.
If the infarcted tissue has stayed in the living patient for some days before being removed (by biopsy, surgery or autopsy), the degradative processes of the body in the form of macrophages and polymorphs will have begun to clear away the dead tissue, which will consequently have an amorphous, acellular appearance apart from the numerous inflammatory cells. At this stage, the tissue is at its weakest; subjects with myocardial infarction who have survived the acute episode 10 days previously may suddenly die with rupture of the healing infarct and consequent haemopericardium and cardiac tamponade (Ch. 13). The gross appearance of the tissue at this time is very variable; if small blood vessels in the vicinity have also become ischaemic they may die and blood will escape into the infarct, giving it a patchy or confluent red appearance. On the other hand, there may be no bleeding into the area, in which case it remains pale with a red hyperaemic rim, and grows progressively paler as healing takes place.
If the patients survive this danger period the damaged tissue either regenerates or repairs itself with the formation of a scar (Ch. 6). Such a scar is apparent as a grey, contracted area consisting of collagenous fibrous tissue. A scar solves the tissue deficit in the sense that the organ is intact and the hole is mended, but the scarred area is no longer functional; a healed myocardial infarct is adynamic and can be the site of further problems for the patient. In the heart, this may take the form of an aneurysm as the scar is subjected to cyclic pressure loads and becomes stretched without any ability to contract again.
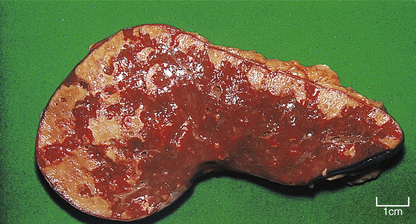
The overall shape of infarcts depends upon the territory of perfusion of the occluded blood supply; some classic appearances are the wedge-shaped infarcts seen in the lung and the triangular infarcts (conical in three dimensions) seen in the kidneys at autopsy. Other scarred infarcts, such as those in the spleen (Fig. 8.7), are less predictable because the blood supply is less regular and marked overlaps of vascular territories occur, and because the soft tissue distorts as the scar contracts. In the brain the dead tissue is cleared away so efficiently that a fluid-filled cyst is often all that remains (Ch. 26).
Gangrene
When whole areas of a limb or a region of the gut have their arterial supply cut off and large areas of mixed tissues die in bulk, such a process is termed gangrene. Two types of gangrene are recognised:
Another mechanism that results in gangrene is torsion: the gut may twist on a lax mesentery, or an ovary or testis may twist on its pedicle, occluding the venous return. The organ swells and the oedema further compresses the drainage. The arteries continue to pump blood into the organ, but ischaemia supervenes and infarction develops.
Gas gangrene results from infection of ischaemic tissue by gas-producing anaerobic bacteria such as Clostridium perfringens.
Capillary ischaemia
In frostbite, the capillaries are damaged in exposed areas and contract so severely that the area they normally supply becomes ischaemic and dies. Exposure to cold without freezing causes capillary contraction followed by a fixed dilatation; this is the mechanism of damage in ‘trench foot’ and related conditions.
Capillaries may also be blocked by parasites, by abnormal cells in sickle cell disease or by abnormal proteins that precipitate in the cold (cryoglobulinaemia) and these phenomena also lead to local ischaemia and infarction.
The balance of thrombotic and thrombolytic mechanisms is delicate; this balance may be secondarily disturbed by several different disease processes and, unfortunately, by some therapeutic interventions. In such cases, thrombosis may become activated without effective counterbalance, with the result that minute thrombi may form throughout the body and, consequently, bleeding may occur at multiple sites due to consumption of clotting factors; this phenomenon is called disseminated intravascular coagulation or DIC (Fig. 8.8). It occurs as a complication of many disease states such as cancer or infection (Ch. 23).
Susceptibility to ischaemia
Different tissues show differing susceptibility to ischaemia for a variety of reasons. Some tissues have only one arterial supply and if this is blocked there is no possibility of collateral supplies taking over; one such ‘end artery’ situation is the retinal artery and thrombosis of this artery leads inevitably to blindness. Tissues also vary in the degree of ischaemia that they can tolerate, commonly as a function of differing metabolic needs; even within a tissue, different areas have different susceptibilities. Within the heart the sub-endocardial zone is at a watershed between the coronary supply from the outside and the diffusion zone from blood within the chambers. If the coronary arteries are narrowed by the presence of atheroma, these patients are at great risk of developing sub-endocardial infarctions if their systemic blood pressure drops for any reason. Consequently, such patients may develop the complications of sub-endocardial infarction following trauma, surgery or toxic shock from infections.
Low-flow infarction
In some tissues, infarction may be due to impaired blood flow (or oxygenation) rather than an absolute cessation of flow. Tissues that are especially vulnerable to low-flow infarction include:
‘Watershed’ areas
Tissue at the interface between the adjacent territories of two arteries is prone to infarction if there is an impairment of blood or oxygen supply. The tissue is normally situated precariously on the fringes of the territories perfused by the arteries, with no collateral circulation to provide blood from alternative vessels.
Patients who are severely shocked and hypotensive may develop ischaemic lesions in these sites.
Portal vasculature
Some tissues are perfused by blood that has already passed through one set of capillaries; this vascular arrangement is described as portal. Therefore, there is normally a drop in intravascular pressure across the first set of capillaries and a reduction in oxygen saturation, rendering the tissue perfused by the second set of capillaries vulnerable to ischaemic injury.
These patterns of vascular microanatomy account for the pituitary infarction, renal tubular necrosis and acute pancreatitis that may occur in severely shocked patients.
Arterial stenoses
Atheromatous narrowing or stenosis of arteries may be of insufficient severity to cause infarction distally in normotensive individuals. However, if the blood pressure and, therefore, blood flow falls, the tissue distal to the arterial stenosis may become infarcted. Thus, patients who become severely shocked may develop ischaemic changes in various organs without there being any sign of total vascular occlusion.
Transient arterial spasm can also cause infarction in vulnerable tissues such as the brain and heart.
Infarction and metabolic activity
Cells with large metabolic requirements are exceptionally vulnerable to ischaemic damage and infarction. Cerebral neurones are the most at risk; irreversible damage occurs within a few minutes of cessation of blood flow and oxygenation. Cardiac myocytes also have a considerable requirement for oxygen and other nutrients; they may be irreversibly damaged if the coronary arteries, which may be narrowed by atheroma, cannot supply these requirements during tachycardia associated with exertion.
Low flow without infarction (erectile impotence)
Male sexual function (and arousal in both sexes) depends upon effective vascular responsiveness of the sexual apparatus to produce penile erection. This may be impaired in many clinical situations (diabetes, drug treatment of systemic hypertension, surgery, etc.). Recently a new drug (sildenafil, Viagra) has been developed which specifically targets this vascular bed with consequent improvement in erectile responsiveness.
SHOCK




The word ‘shock’ has different meanings in different contexts. In the emotional context, which almost all of us experience to varying degrees, it means a severe psychological reaction to an event for which we were unprepared. Another meaning is the unpleasant and often painful sensation experienced when high voltage electricity flows through the body. These meanings of the word ‘shock’ are not relevant here, but they can be a source of great confusion when talking to patients.
Shock as a pathological process is characterised by profound circulatory failure resulting in life-threatening hypoperfusion of vital organs. Compensatory mechanisms maintain blood pressure until they too are defeated, resulting in hypotension. Shock may be classified as:
In the early stages the arterial networks in many vital organs can compensate to some extent. For example, by a process of autoregulation, the cerebral arteries dilate when blood pressure is reduced, so that the cerebral vascular resistance falls and a normal rate of flow is maintained. In other tissues, with less vital functions, there is compensatory vasoconstriction in order to increase peripheral vascular resistance and thus maintain the effective blood pressure supplying vital organs; this increased vascular tone is mediated by adrenergic mechanisms and by the effects of angiotension.
If the compensatory mechanisms fail and hypotension ensues, various tissues will be vulnerable to ischaemic injury (Fig. 8.9). This is an extremely serious clinical problem which, depending on the primary cause, is frequently fatal. The clinicopathological consequences include:
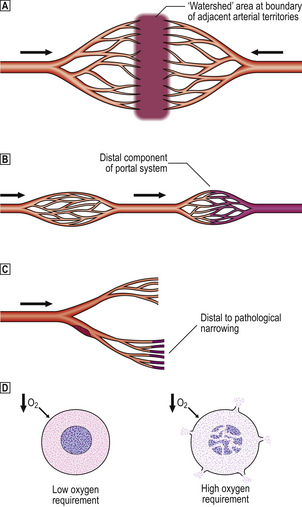
Fig. 8.9 Determinants of the vulnerability of tissues to ischaemic damage in hypotensive shock.  Ischaemic damage may occur at the boundary (‘watershed’ area) between adjacent arterial territories (e.g. splenic flexure of the colon between superior and inferior mesenteric arterial supplies, and regions of the brain at the border of cerebral arterial territories).
Ischaemic damage may occur at the boundary (‘watershed’ area) between adjacent arterial territories (e.g. splenic flexure of the colon between superior and inferior mesenteric arterial supplies, and regions of the brain at the border of cerebral arterial territories).  Tissues supplied by the distal components of portal vascular systems (e.g. anterior pituitary, renal tubules, some pancreatic acini) may suffer ischaemia due to critical fall in perfusion pressure across the proximal capillary bed.
Tissues supplied by the distal components of portal vascular systems (e.g. anterior pituitary, renal tubules, some pancreatic acini) may suffer ischaemia due to critical fall in perfusion pressure across the proximal capillary bed.  Hypotension may precipitate ischaemic damage distal to a region of arterial narrowing (e.g. by atheroma).
Hypotension may precipitate ischaemic damage distal to a region of arterial narrowing (e.g. by atheroma).  Cells with high oxygen requirements (e.g. cerebral neurones) may die from hypoxia, whereas neighbouring cells with lower oxygen demands survive.
Cells with high oxygen requirements (e.g. cerebral neurones) may die from hypoxia, whereas neighbouring cells with lower oxygen demands survive.
Cardiogenic shock
The commonest cause of cardiogenic shock is acute myocardial infarction. Death of part of the left ventricular myocardium reduces the heart’s functional capacity and, even at rest, the left ventricular stroke volume is reduced. If shock proceeds to the hypotensive phase, it should be corrected only with extreme caution; by artificially increasing the blood pressure, additional strain will be placed on the myocardium, with the risk of catastrophic failure.
Hypovolaemic shock
Hypovolaemic shock is characterised by loss of effective circulating blood volume. This may be due to:
The pathogenesis of shock resulting from haemorrhage is logical. Internal or external bleeding, for example due to traumatic rupture of an internal organ or accidental severing of a major vessel, causes a reduction in the normal blood volume. Blood pressure is initially maintained by compensatory mechanisms, some of which enable the condition to be suspected clinically; symptoms and signs include a cold, clammy skin and a high pulse rate.
Shock due to generalised increased vascular permeability and/or dilatation can occur as a result of:
In these situations there are varying degrees of vasodilatation and increased vascular permeability. Blood pools in the dilated vessels and, if there is endothelial damage (for example in
Commonly confused conditions and entities relating to ischaemia, infarction and shock
| Commonly confused | Distinction and explanation |
|---|---|
| Clot and thrombus | Both are blood solidified by coagulation, but a clot is formed in non-flowing blood whereas a thrombus is formed in flowing blood and, therefore, often has a laminated structure. |
| Phlebothrombosis andthrombophlebitis | Phlebothrombosis is thrombosisin a vein (Greek: phlebos). Thrombophlebitis is an inflammatory reaction to phlebothrombosis. |
| Necrosis and infarction | Necrosis means abnormal death of sheets of cells or tissue (normal single cell death is usually by apoptosis). Infarction is necrosis due to deprivation of blood supply. |
| Infarction and gangrene | Gangrene is infarction of mixed tissues in bulk (e.g. all layers of bowel wall, part of limb from skin to bone). |
severe burns or bacterial toxaemia), fluid leaks from the vessels into the extravascular compartment, causing a profound reduction in the effective circulating blood volume. Examples mediated by bacterial toxins include (Ch. 3):
Other vascular effects of bacterial toxaemia
The bacterial toxins not only have a hypotensive effect but also activate the complement and blood coagulation cascades. The latter may lead to disseminated intravascular coagulation (DIC) (Ch. 23). In this condition, which may also be precipitated by the release of tissue thromboplastins from necrotic tissue, fibrin is deposited on endothelial surfaces of blood vessels, thus interfering with trans-endothelial flow of nutrients. The fibrin may also form a mesh across the lumen of small blood vessels resulting in:
The fibrinolytic activity of endothelial cells can cope with small amounts of fibrin deposition; DIC results when the rate of fibrin deposition exceeds the rate of fibrinolysis. DIC is diagnosed by its multisystem clinical features, by the frequent accompaniments of haemolysis and thrombocytopenia, by the haemorrhagic tendency due to consumption of coagulation factors (consumption coagulopathy), and by finding fibrin degradation products.
Gowda R.M., Fox J.T., Khan I .A.. Cardiogenic shock: basics and clinical considerations.. International Journal of Cardiology. 2008;123:221-228.
Hovens M.M., Snoep J.D., Tamsma J.T., Huisman M .V.. Aspirin in the prevention and treatment of venous thromboembolism. Journal of Thrombosis and Haemostasis. 2006;4:1470-1475.
Kuipers S., Schreijer A .J., Cannegieter S.C., Büller H.R., Rosendaal F.R., Middeldorp S.. Travel and venous thrombosis: a systematic review. Journal of Internal Medicine. 2007;262:615-634.
Levi M.. Disseminated intravascular coagulation.. Critical Care Medicine. 2007;35:2191-2195.
Munford R.S.. Severe sepsis and septic shock: the role of Gram-negative bacteremia. Annual Review of Pathology. 2006;1:467-496.
Prandoni P. Acquired risk factors of venous thromboembolism in medical patients. Pathophysiology of Haemostasis and Thrombosis. 2006;35:128-132
Stafford I., Sheffield J.. Amniotic fluid embolism. Obstetrics and Gynecology Clinics of North America. 2007;34:545-553.
Taviloglu K., Yanar H.. Fat embolism syndrome.. Surgery Today. 2007;37:5-8.
Tissier R., Berdeaux A., Ghaleh B., et al. Making the heart resistant to infarction: how can we further decrease infarct size? Frontiers in Bioscience. 2008;13:284-301.
[/level-membership-for-pathology-category][not-level-membership-for-pathology-category]
Chapter 8 Ischaemia, infarction and shock
Blood suffers the various pathological processes that occur in all tissues (neoplasia, nutritional defects, etc., see Ch. 23) but because blood is a tissue that circulates there is also a specific set of pathologies related to defects in flow.
Thrombo-embolic events are major causes of morbidity and mortality in the UK and other developed countries. The majority of readers (and authors) of this book will die from events somewhere along the sequence described in this chapter. Common and serious disorders in which thrombo-embolic mechanisms participate include:
However, no organ is exempt from the harmful consequences of an impaired blood supply, although some (e.g. brain, myocardium), because of their nutritional requirements and inability to regenerate, are more vulnerable than others.
Ischaemia is the result of impaired vascular perfusion, depriving the affected tissue of vital nutrients, especially oxygen. The effects on the tissue can be reversible, but this depends on:
Infarction is death (necrosis) of tissue as a result of ischaemia. Infarction is irreversible, but tissues vary in their ability to repair and replace the loss. Infarction usually results from thrombo-embolic phenomena completely occluding the artery supplying the affected tissue.
Shock (pathophysiological rather than psychological surprise) is a state of circulatory collapse resulting in impaired tissue perfusion. Ischaemia, infarction and shock are, therefore, interrelated phenomena. The cellular responses to impaired nutrition are summarised in Chapter 6.
Although the most common causes of ischaemia and infarction are thrombo-embolic phenomena, vascular insufficiency can also result from other causes (Fig. 8.1).
Fig. 8.1 Vascular lesions causing ischaemia. Thrombosis: initiated by either abnormal flow (e.g. stasis, turbulence), damage to vessel wall (e.g. denudation of endothelial lining) or abnormal blood constituents. Embolism. Spasm: due to contraction of smooth muscle in media of vessel, for example due to lack of nitric oxide from endothelium. Atheroma: occurs only in arteries and may in turn be complicated by thrombosis and embolism. Compression: veins are more susceptible because of their thinner walls and lower intraluminal pressure. Vasculitis: inflammation of vessel wall narrows lumen and may be complicated by superimposed thrombosis. Vascular steal: for example, an artery may be narrowed by atheroma but flow is still sufficient to maintain viability of perfused territory; however, flow may be compromised by increased demands of a neighbouring territory. Hyperviscosity: increased viscosity, for example in hypergammaglobulinaemia resulting from myeloma, causes impaired flow and predisposes to thrombosis.
NON-THROMBO-EMBOLIC VASCULAR INSUFFICIENCY
Vascular flow can be impeded by abnormalities other than thrombo-embolic phenomena.
In arteries, the commonest lesion is atheroma, which in turn may be complicated by thrombo-embolism. In medium-sized arteries, atheromatous plaques (Ch. 13) often narrow the lumen, causing ischaemia and sometimes atrophy of tissues in the hypoperfused territory. Serious consequences include the symptom of angina due to myocardial ischaemia, often heralding the development of irreversible infarction, and hypertension due to renal artery narrowing and hypoperfusion of a kidney, which responds physiologically by increased renin secretion.
Transient arterial narrowing can result from spasm of the smooth muscle in the vessel wall. This can be due to a decrease in nitric oxide (endothelium-derived relaxing factor) production by the vascular endothelium due to cellular injury or loss. Spasm of coronary arteries can lead to angina and both may be relieved by glyceryl trinitrate. Arterial spasm is also responsible for the transient ischaemia of the fingers in Raynaud’s phenomenon.
Blood vessels can be partially or totally occluded by external compression. This is done intentionally during surgery by ligation to prevent haemorrhage from severed vessels, although the results can be disastrous if, accidentally, the wrong vessel is tied off! Because of their thin walls and low intraluminal pressure, veins are more susceptible to occlusion by external compression. This occurs commonly in strangulated hernias, testicular torsion and torsion of ovaries containing cysts or tumours.
‘Steal’ syndromes occur when blood is diverted (‘stolen’) from a vital territory. This results when, proximal to an area of atheromatous narrowing insufficient on its own to produce ischaemia, the arterial stream is diverted along another branch vessel to meet the increased demands of a competing territory or lesion; the territory supplied by the atheromatous vessel then becomes ischaemic. This is a relatively uncommon cause of ischaemia, but often the most challenging diagnostically.
Ischaemia at the arteriolar, capillary and venular level can result from increased whole blood viscosity. Viscosity effects contribute relatively little to the flow characteristics of blood in vessels of large calibre, but in small vessels they are a major factor. Hyperviscosity of blood can occur in myeloma, a tumour of plasma cells, as a result of the abnormally high concentration of gammaglobulin in the plasma and rouleaux formation by red cells.
THROMBO-EMBOLIC VASCULAR OCCLUSION
Clot
When blood stagnates due to the cessation of the pumping action of the heart, or if blood is allowed to stand in a bottle or test tube, then the clotting process is set in motion. A complex series of enzymatic steps (Ch. 23) is activated, resulting in the formation of a fibrin meshwork that entraps the cells into a solid but elastic clot. When this process occurs in the body after death, the red cells tend to settle out before the clot forms, so that these postmortem clots have two layers: a lower, deep red layer (resembling redcurrant jelly) and an upper, clearer layer (resembling chicken fat), with platelets evenly distributed throughout. Since these clots have formed within the body and represent the blood content of the vessel during life, they are moulded to the shape of the vessels in which they have formed. Some time after death, the various blood cells and the cells of the vessel wall begin to release their hydrolytic enzymes and the clot is dissolved.
The sequence of enzymatic reactions involved in the clotting cascade and abnormalities of this system are discussed in Chapter 23.
Thrombosis
Thrombus is a solidification of blood contents that forms within the vascular system during life and is therefore different in concept from a clot. Its mode of formation, its structure and its appearance are all different from those of a clot and the two should never be confused.
Role of platelets
The mechanism for closing small gaps in vessel walls brought about by trauma involves the platelets. Platelets are smaller than red blood cells, rather angular in appearance, and have no nucleus. They are derived from large, multinucleated cells in the bone marrow called megakaryocytes. Although platelets have no nucleus, they are highly structured internally and contain a variety of organelles, some of which are specific to them. As well as mitochondria and the various cytoskeletal elements found in most cells, platelets also contain alpha granules and dense granules. The alpha granules contain several substances involved in the process of platelet adhesion to damaged vessel walls (fibrinogen, fibronectin, platelet growth factor and an antiheparin as well as less well defined substances), and the dense granules contain substances such as adenosine diphosphate (ADP) that cause platelets to aggregate.
Platelets are activated and the contents of their granules are released when the platelets come into contact with collagen, as may be found in damaged vessel walls, or with polymerising fibrin. The platelets change shape and extend pseudopodia; their granules release their contents and the platelets form a mass that covers the vessel wall defect until the endothelial cells have regenerated and repaired the vessel permanently. However, if this process is activated within an intact vessel, it results in a thrombus.
Thrombus formation
There are three predisposing situations that may result in thrombus formation. These were described originally by Virchow and are known as Virchow’s triad. The three factors are:
Not all three are needed for thrombosis to occur, but any one of them may result in thrombosis in a particular case.
If we consider the sequence of events involved in the formation of thrombus on the basis of an atheromatous plaque (Ch. 13), this will serve as a very good example of the factors listed by Virchow.
Arterial thrombosis
In its earliest phase the atheromatous plaque may consist of a slightly raised fatty streak on the intimal surface of any artery, such as the aorta (Fig. 8.2). With time, the plaque enlarges and becomes sufficiently raised to protrude into the lumen and cause a degree of turbulence in the blood flow. This turbulence eventually causes loss of intimal cells, and the denuded plaque surface is presented to the blood cells, including the platelets. The turbulence itself will predispose to fibrin deposition and to platelet clumping; the bare luminal surface of the vessel will have collagen exposed and platelets will settle on this surface. Thus we have two of the factors described in Virchow’s triad operating in an atheromatous plaque. If this plaque exists in the aorta of a smoker or someone with a high cholesterol level and a high level of low density lipoprotein—common risk factors for atheroma—then the third of Virchow’s factors is introduced, since these changes in blood constituents are well known to predispose to thrombus formation. This process, once begun, may be self-perpetuating, as it has been shown that platelet-derived growth factor, which is contained in the alpha granules, causes proliferation of arterial smooth muscle cells, which are an important constituent of the atheromatous plaque.
Fig. 8.2 Thrombosis. Thrombosis is exemplified by its occurrence on an atheromatous plaque, a particularly common event. Important steps in this sequence include: loss of endothelial cells and exposure of the underlying collagen; platelet adherence and activation; partial or complete arterial occlusion by the multilayered thrombus.
Thus, the first layer of the thrombus is a platelet layer. Formation of this layer in turn causes the precipitation of a fibrin meshwork, in which red cells are trapped, and a layer of this meshwork is developed on top of the platelet layer. The alternating bands of white platelets and red blood cells in thrombi were first described by Zahn and are called the lines of Zahn. This complex structure now protrudes even further into the lumen, causing more turbulence and forming the basis for further platelet deposition. The normal flow of blood within the vessels is laminar; the cells move in the swifter central lane and the plasma runs along the walls. Therefore, the greatest degree of turbulence occurs at the downstream side of arterial thrombi, as the blood passes over the thrombus, and on the upstream side of venous thrombi for the same reason. Thrombi will therefore grow in the direction of blood flow; this process is known as propagation.
Venous thrombosis
In veins, however, the blood pressure is lower than in arteries and atheroma does not occur; so what initiates venous thrombus formation? Most venous thrombi seem to begin at valves. Valves naturally produce a degree of turbulence because they protrude into the vessel lumen and they may be damaged by trauma, stasis and occlusion. However, thrombi can also form in veins of young, active individuals with no predisposing factors that can be identified. Once they begin, the thrombi grow by successive deposition in the manner described previously and this process may produce a highly patterned, coralline growth (Fig. 8.3). Since normal flow within the vessels is laminar, most of the blood cells are kept away from diseased walls or from damaged vein valves. However, if the blood pressure is allowed to fall during surgery or following a myocardial infarction, then flow is slower through the vessels and thrombosis becomes a likely event. Similarly, the venous return from the legs is very reliant upon calf muscle contraction and relaxation, which massages the veins and, because of the valves, tends to return the blood heartwards. So, if elderly subjects are immobilised for any reason, they are at great risk from the formation of deep leg vein thromboses. The frequency with which deep vein thrombosis is found to occur following surgery is directly related to the enthusiasm with which it is sought (e.g. by the pathologist at postmortem examination) and the sensitivity of the methods used to demonstrate it. Postmortem studies on unselected medical and surgical patients show significant deep vein thrombosis in 34% of the former and 60% of the latter, regardless of the cause of death.
Fig. 8.3 Venous thrombus. Femoral vein opened at autopsy to reveal a thrombus. Histological section showing the characteristic laminated or coralline structure of a thrombus.
When a vein becomes thrombosed it evokes an inflam-matory reaction, a phenomenon known as thrombophlebitis; the opposite process also occurs—a vein that is inflamed will often thrombose and this is known as phlebothrombosis. The end effect is the same, a thrombosed and inflamed vein, but clearly if there is a predisposing cause then the cause needs to be recognised and treated.
Clinical effects
The effects of thrombosis are apparent only if the thrombus is sufficiently large to affect the flow of blood significantly. Arterial thrombosis results in loss of pulses distal to the thrombus and all the signs of impaired blood supply: the area becomes cold, pale and painful, and eventually the tissue dies and gangrene results. In venous thromboses, 95% of which occur in leg veins, the area becomes tender, swollen and reddened, as blood is still carried to the site by the arteries but cannot be drained away by the veins. The tenderness is due to developing ischaemia in the vein wall initially, but there is also general ischaemic pain as the circulation worsens. The more specific clinical effects of thrombosis depend on the tissue that is affected.
Myocardial infarction is often associated with thrombus formation in coronary arteries and is responsible for numerous sudden deaths (Ch. 13).
Strokes may be due to the formation of thrombus in a cerebral vessel, although they may be also the result of haemorrhage or embolism (Ch. 26).
Thrombophlebitis migrans occurs in previously healthy veins in any area of the body. The thromboses appear and disappear, changing site all the time, and the condition may persist for months or even years. It is extremely ominous and usually indicates the presence of visceral cancer, commonly of the pancreas. The mechanism remains obscure.
Fate of thrombi
Various fates await the newly formed thrombus (Fig. 8.4). In the best scenario it may resolve; the various degradative processes available to the body may dissolve it and clear it away completely. It is not known what proportion of thrombi follow this course, but the total number is likely to be large. A second possibility is that the thrombus may become organised
[/not-level-membership-for-pathology-category]
