Chapter 33 Inborn Errors of Urea Synthesis
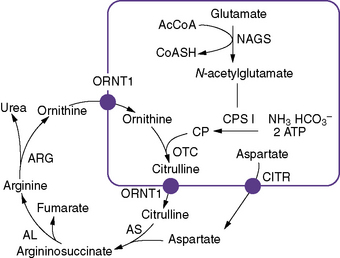
Inherited urea cycle disorders represent a devastating group of inborn errors of metabolism that are associated with hyperammonemic encephalopathy and high mortality and morbidity rates. They comprise deficiencies in any of the six enzymes and two amino acid transporters involved in urea synthesis (Figure 33-1). Accordingly, these disorders are named as follows (estimated prevalence rates are given) [Brusilow and Maestri, 1996; Tuchman, 1992; Yamanouchi et al., 2002]:








These disorders are inherited as autosomal-recessive traits, except for ornithine transcarbamylase deficiency, which is X-linked. Because of the absence of mass newborn screening for these disorders, the true incidence of urea cycle disorders is unknown. Based on case reports and questionnaires about referred patients, the combined prevalence of all urea cycle disorders is estimated to be 1 per 8200 [Brusilow and Maestri, 1996].
Other than in arginase deficiency, infants with a complete deficiency of a urea cycle enzyme (N-acetylglutamate synthase, CPS I, ornithine transcarbamylase, argininosuccinate synthetase, or argininosuccinate lyase) commonly present in the newborn period with hyperammonemic coma. Despite aggressive treatment that relies primarily on hemodialysis, the mortality rate in infancy has been reported to approximate 50 percent [Maestri et al., 1999], and as demonstrated by our previous studies, virtually all of the survivors are left with developmental disabilities [Msall et al., 1984; Krivitzky et al., 2009]. Patients with late-onset disease (those with partial enzyme deficiencies, including ornithine transcarbamylase-deficient female heterozygotes) may present at any age with hyperammonemic crises that carry a 10 percent mortality rate and a significant risk of intellectual disabilities [Batshaw et al., 1986]. Even asymptomatic ornithine transcarbamylase-deficient heterozygotes have been shown to have mild cognitive deficits [Batshaw et al., 1980; Gyato et al., 2004].
The Urea Cycle
Dietary protein, on average, contains approximately 16 percent nitrogen. More than 90 percent of the nitrogen that is not used for anabolic processes normally is metabolized and excreted as urea. Therefore, substantial urea synthesis capacity (approximately 16 g per day in adults) is required [Linder, 1985]. With a deficiency of one of the urea cycle enzymes, an insufficient amount of urea will be formed, and nitrogen in the form of ammonia will accumulate. Accumulation in brain causes altered mental status and encephalopathy.
The urea cycle was proposed by Hans Krebs and Kurt Henseleit in 1932, and was the first cyclic pathway elucidated. Six enzymes, one co-factor, and two transporters are necessary for optimal urea cycle activity (see Figure 33-1). The clinically most important co-factor is N-acetylglutamate, which is formed from acetyl coenzyme A (acetyl-CoA) and glutamate in a reaction catalyzed by N-acetylglutamate synthase. N-acetylglutamate activates the first enzyme of the urea cycle, carbamyl phosphate synthetase I, which uses adenosine triphosphate, bicarbonate, and glutamine or ammonia to synthesize carbamyl phosphate, contributing the first atom of waste nitrogen to the cycle. Carbamyl phosphate synthetase I is expressed in periportal hepatocytes and intestinal mucosa epithelial cells [Ryall et al., 1985]. It is the most abundant protein in liver mitochondria, accounting for 20 percent of the mitochondrial matrix protein [Lusty, 1978]. The enzyme consists of a single polypeptide with a molecular weight of 165,000 and approximately 1500 amino acid residues [Haraguchi et al., 1991].
Clinical Description of Urea Cycle Disorders
N-Acetylglutamate Synthase Deficiency
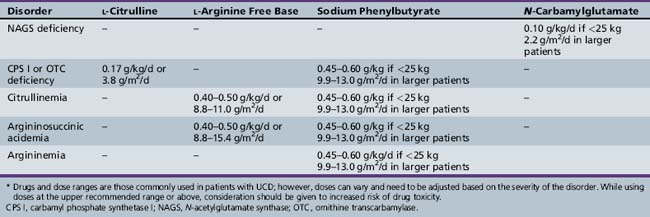
Inherited N-acetylglutamate synthase (NAGS) deficiency leads to hyperammonemia by causing a secondary deficiency of CPS I. This disorder has been reported in approximately two dozen patients to date [Bachmann et al., 1982; Elpeleg et al., 2002; Caldovic et al. 2007], but is likely to be underdiagnosed because of the lack of specific biochemical markers and the physiologically low abundance of NAGS in the liver. NAGS deficiency is inherited as an autosomal-recessive disorder and has a phenotype that is similar to that found in CPS I deficiency. It is characterized by hyperammonemia in the newborn period or later in life, and can be fatal or lead to mental retardation and other developmental disabilities. Plasma amino acid analysis usually demonstrates an increased level of glutamine and reduced or absent levels of citrulline. Urinary orotic acid level is normal or low. NAGS activity in the liver has a variable degree of deficiency (ranging from undetectable to normal residual activity) that is unresponsive to l-arginine. Because enzyme analysis requires large amounts of liver tissue and may not be entirely reliable, analysis of genomic DNA for mutations in the N-acetylglutamate synthase gene is the best diagnostic method [Morizono et al., 2004]. Treatment of NAGS deficiency used to consist of a low-protein diet and use of ammonia-scavenging drugs (Table 33-1). A more specific therapy using N-carbamyl-l-glutamate is now possible because N-carbamyl-l-glutamate has been approved for the treatment of NAGS deficiency. N-carbamyl-l-glutamate is a stable structural analog of N-acetylglutamate and can substitute for it in the activation of CPS I.
Carbamyl Phosphate Synthetase I Deficiency
CPS I deficiency was first reported in 1969 [Hommes et al., 1969]. This mitochondrial urea cycle enzyme should be distinguished from a similar cytosolic enzyme, carbamyl phosphate synthetase 2, which is involved in de novo pyridine synthesis. This disorder can manifest with hyperammonemic coma in the newborn period or later in childhood. Biochemically, the principal findings are hyperammonemia, an increased level of plasma glutamine, and reduced level or absence of citrulline on plasma amino acid analysis. Urinary orotic acid level is normal or low. Patients with neonatal-onset disease generally demonstrate less than 5 percent normal CPS I activity in liver, whereas those with late-onset disease have higher residual activity [Qureshi et al., 1986]. Like most of the other urea cycle disorders, CPS I deficiency does not arise from a common mutation [Funghini et al., 2003; Summar, 1998]. Therapy consists of dialysis and/or intravenous alternate pathway therapy during severe hyperammonemic episodes, and low-protein diet and oral alternate pathway therapy for chronic treatment (see Table 33-1). Patients with the neonatal-onset form who survive the initial crisis generally require liver transplantation for long-term survival.
Ornithine Transcarbamylase Deficiency
Ornithine transcarbamylase deficiency was first reported in 1962 in two girls, aged 20 months and 6 years, who were found to have hyperammonemia associated with episodic vomiting, delirium, stupor, failure to thrive, and mental retardation [Russell et al., 1962]. The mothers of the children were sisters, and both demonstrated evidence of a similar but less marked metabolic disorder. Both girls died before the age of 8 years.
Several years later, this disorder also was identified in males. The delayed recognition in males was attributable to the almost total lack of enzyme activity and rare survival of affected males beyond the newborn period. The reason for this difference between males and females subsequently was determined to be related to its X-linked inheritance pattern, with the occurrence of symptomatic females explained by skewed X chromosome inactivation [Ricciuti et al., 1976]. The classic presentation of OTC deficiency in hemizygous males is as a catastrophic illness in the first week of life. In symptomatic female heterozygotes and in males with partial OTC deficiency, symptoms rarely present in the newborn period. Only one case of lethal OTC deficiency in a female neonate has been reported [Klosowski et al., 1998]. In partial deficiencies, age at presentation after the newborn period covers a wide spectrum, with development of hyperammonemic episodes in infancy in some patients, in later childhood in others, and not until adulthood in still others [Ahrens et al., 1996; Ausems et al., 1997; McCullough et al., 2000]. These patients generally have 5–30 percent of normal OTC activity in liver on in vitro measurement. Biochemically, the principal findings are hyperammonemia, hyperglutaminemia, reduced levels or complete absence of citrulline in plasma, and increased urinary orotic acid level.
More than 340 different point mutations and polymorphisms have been found in OTC-deficient patients, and no mutations are prevalent [McCullough et al., 2000; Tuchman et al., 2002]. In about 80 percent of affected families, prenatal diagnosis using DNA techniques is possible [Grompe et al., 1991]. When the mutation has been identified, carrier testing and prenatal diagnosis can be offered to the family. With severe OTC deficiency being an X-linked lethal disease, the calculated probability for the mother of an affected male to be a carrier is 2/3 or 66 percent, and the probability that the patient has a de novo mutation is 1/3 or 33 percent. The identification of common intragenic polymorphisms allows tracking of the mutant allele, even when the deleterious mutation is unknown [Plante and Tuchman, 1998]. Although most families have point mutations, 8 percent of families have large deletions of one or more exons, 10 percent have small deletions or insertions of a few basepairs, 18 percent have splice site mutations, and in about 20 percent no mutation can be found. In about half of those where no mutation could be found, large deletions involving the OTC locus were detected using microarray technology [Shchelochkov et al., 2009]. There are several disease genes very close to the OTC locus, including the Duchenne muscular dystrophy gene and the chronic granulomatous disease gene. Patients with these large deletions may thus have other severe genetic diseases at the same time, making them extremely difficult to manage [Deardorff et al., 2008]. Amongst those patients with point mutations, mutations causing neonatal disease affect amino acid residues that are in the interior of the enzyme, especially around the active site, whereas those associated with late-onset and milder phenotypes tend to be located on the surface of the protein [Tuchman et al., 1998].
Heterozygote detection of OTC deficiency is important both to identify at-risk family members and to offer prenatal diagnosis. It can be accomplished by either molecular studies or, in cases where no mutation was found, by provocation testing. The best provocation testing is an allopurinol load [Hauser et al., 1990]. This test has replaced the protein loading test, which can precipitate a hyperammonemic episode. The allopurinol load (300 mg given orally to adults) leads to increased excretion of orotic acid, reaching 10–20 times control values in 90 percent of OTC heterozygotes [Hauser et al., 1990]. Approximately 15 percent of OTC-deficient heterozygous females will become symptomatic during their lifetime [Batshaw et al., 1986]. (Heterozygotes for other urea cycle disorders are asymptomatic.) Therapy for the neonatal-onset form of OTC deficiency consists of dialysis and administration of intravenous ammonia scavenger drugs, followed by maintenance on a low-protein diet and long-term alternative pathway therapy (see Table 33-1). Patients with the neonatal-onset form who survive the initial crisis generally require liver transplantation.
Citrullinemia
Citrullinemia was first reported in 1962 [McMurray et al., 1962]. Its name derives from the marked elevation of citrulline in blood of affected persons. This disorder also has been called citrullinuria because of the increased excretion of citrulline in urine, and argininosuccinic acid (argininosuccinate) synthetase deficiency to denote its enzyme deficit. Heterogeneity is seen clinically, biochemically, and at the molecular level. Two distinct forms have been reported: neonatal/childhood-onset citrullinemia (type I; with diminished levels of argininosuccinate synthetase in all organs) and citrullinemia type II or citrin deficiency, an adult-onset citrullinemia that is in some but not all cases preceeded by neonatal cholestasis and decreased synthetic function of the liver (caused by a defect in citrin) [Saheki et al., 1987].
Biochemically, the principal findings are hyperammonemia, citrullinemia, and citrullinuria. Citrulline levels generally are elevated 50–100-fold [normal levels less than 50 μmol/L] [Batshaw et al., 1981]. Urinary orotic acid levels also may be increased but less so than in OTC deficiency or argininemia. Citrullinemia is inherited as an autosomal-recessive trait. The gene has been localized to the q34 region of chromosome 9, and the nucleotide coding sequence and deduced amino acid sequence for the enzyme are known [Gao et al., 2003]. To date, more than 50 mutations have been identified; some involve single base changes in the coding sequence, and others involve skipping of an exon in the messenger RNA (mRNA) due to abnormal splicing. Most patients appear to be compound heterozygotes of two different mutations. In neonatal-onset cases, argininosuccinate synthetase activity in liver is less than 5 percent of normal, whereas in childhood-onset cases, 10–25 percent residual activity is seen [Brusilow and Horwich, 2001].
Therapy for citrullinemia (type I) consists of dialysis during severe hyperammonemic crises, followed by low-protein diet and long-term alternative pathway therapy (see Table 33-1). After the initial crisis, patients are in general more stable and easier to manage than patients with more proximal defects (CPS I and OTC deficiencies).
Citrullinemia Type II or Citrin Deficiency
Citrullinemia type II was identified first in Japan but the mutation has been traced back to China. Citrin deficiency is caused by mutations in a gene encoding a previously unknown calcium-dependent mitochondrial membrane protein named citrin (SLC25A13) [Kobayashi et al., 2003]. This inner mitochondrial membrane carrier enables the exchange of matrix aspartate for cytosolic glutamate across the inner mitochondrial membrane [Palmieri et al., 2001]. Plasma ammonia levels are less severely elevated during acute episodes than in other urea cycle disorders, and citrulline levels are elevated up to 20-fold [Kobayashi et al., 1993]. It has been noted that serum pancreatic secretory trypsin inhibitor also is increased and may be useful as a diagnostic marker for the disorder [Kobayashi et al., 1993].
Citrullinemia type II, citrin deficiency, manifests in adulthood with cyclical bizarre behavior patterns (aggression, irritability, hyperactivity), dysarthria, seizures, motor weakness, and coma. Dementia and hepatomegaly eventually develop. Cases of hepatocellular carcinoma also have been reported in affected persons [Hagiwara et al., 2003]. In retrospect, many of the patients have had symptoms since childhood that suggested hyperammonemia, including recurrent episodes of vomiting, lethargy, and irritability [Okeda et al., 1989]. Treatment generally relies on alternate pathway therapy (arginine and phenylbutyrate) [Imamura et al., 2003]; however, liver transplantation is becoming a more common practice in the treatment of this disorder because of the possible liver complications [Ikeda et al., 2001; Yazaki et al., 2004].
More recently, a neonatal-onset form of citrin deficiency has been identified; this form is associated with intrahepatic cholestasis [Tamamori et al., 2002; Tazawa et al., 2001]. Affected infants have multiple metabolic abnormalities, including aminoacidemia, galactosemia, hypoproteinemia, hypoglycemia, and cholestasis. Treatment usually is by high-protein/low-carbohydrate diet, and symptoms often disappear within a year [Saheki et al., 2004]. A few children, however, have a severe form of the disorder with liver damage and tyrosinemia that necessitate liver transplantation. Hyperammonemia is not a major component of this disorder.
Argininosuccinicacidemia
Argininosuccinicacidemia was first described in 1958 by Allan et al. [1958]. Its name derives from the marked elevation of argininosuccinic acid in blood of affected persons. This disorder also has been called argininosuccinicaciduria (ASA) because of the increased excretion of argininosuccinic acid in urine, and argininosuccinate lyase deficiency to denote the underlying enzyme deficiency. In addition to hyperammonemic coma in the newborn period and recurrent hyperammonemic episodes later in childhood, a specific abnormality of the hair termed trichorrhexis nodosa develops in affected children. Nodules appear on the hair shaft, and the hair is friable. A generalized erythematous maculopapular skin rash also may appear in this disorder. Both conditions are associated with arginine deficiency and respond to arginine supplementation [Brusilow and Horwich, 2001].
Chronic marked hepatomegaly has been reported in patients managed with protein restriction and in those receiving arginine supplementation, but this finding is not universal. Pathologic examination reveals modest fatty infiltration and fibrosis. Results of liver function tests frequently are abnormal, especially during hyperammonemic crises, and patients may develop cirrhosis [Zimmermann et al., 1986]. Why some patients with ASA develop cirrhosis and others do not is not understood.
The gene for ASL has been localized to the long arm of chromosome 7 [Kleijer et al., 2002]. The deficient enzyme, a homotetramer of 50-kilodalton (kDa) subunits, is expressed in multiple tissues, including the brain. Evidence of multiple allelic mutations and intragenic complementation, indicating extensive genetic heterogeneity, is characteristic of this disorder [Yu et al., 2001]. The frequency of some mutations, however, is higher than that of others; such mutations may occur at “hot spots” with higher susceptibility for alteration [Linnebank et al., 2002]. The multiple mutations may account for some of the observed heterogeneity in this disease at the clinical level.
Biochemically, the principal findings are elevated citrulline level, hyperammonemia, argininosuccinicacidemia, and argininosuccinicaciduria. The plasma argininosuccinic acid peak is large (normally it is undetectable) [Batshaw et al., 1981]. Of note, this peak may be missed, because it can overlie the peak for leucine or isoleucine. The presence of the two anhydrides of argininosuccinic acid, however, in areas of the chromatogram where homocystine and gamma-aminobutyric acid (GABA) normally are found, aids in the diagnosis. Markedly increased levels of argininosuccinic acid also are readily identifiable in the urine. Additionally, citrulline levels are increased 3–10-fold (to 100 to 300 μmol/L).
The diagnosis can be confirmed by measuring ASL in erythrocytes or fibroblasts, although this is rarely needed. In cerebrospinal fluid, elevated concentrations of argininosuccinic acid and its anhydrides, as well as pyrimidines (pseudouridine and uridine), have been found [Gerrits et al., 1993]. After the initial hyperammonemic crisis and establishment of the diagnosis, recommended treatment for argininosuccinicacidemia consists of a low-protein diet and l-arginine supplementation (see Table 33-1). Many metabolic physicians are also using ammonia scavengers in their treatment regimen.
Argininemia
Argininemia, or hyperargininemia, was first described in 1969 by Terheggen and colleagues [Terheggen et al., 1969]; it is caused by a deficiency of arginase 1. Its name derives from the marked elevation of arginine in the blood of affected persons. Argininemia presents differently from all of the other congenital urea cycle disorders. It usually appears as a progressive neurologic disorder, rather than as an acute encephalopathy [Cederbaum et al., 1979; Prasad et al., 1997].
Clinical symptoms do not classically start with hyperammonemic coma in infancy. In one report, however, a 2-month-old child with recurrent vomiting, persistent jaundice, and hepatomegaly (with associated cirrhosis) was diagnosed as having hyperargininemia [Braga et al., 1997]. Another patient presented with cerebral edema and growth retardation [Harrington et al., 2000]. More commonly, development for the first few years of life appears to be normal, although a detailed history often reveals evidence of protein aversion (often with anorexia, vomiting, and irritability) and some developmental delay. The disease runs a chronic course, but with acute episodes of ataxia, behavioral disturbances, vomiting, lethargy, and seizures. Such episodes often are precipitated by intercurrent viral illnesses [Grody et al., 1993]. Associated biochemical abnormalities usually include moderately elevated plasma ammonia levels of 3–4 times normal and plasma arginine levels of greater than 5 times normal, often exceeding 1000 μmol/L (normal is less than 120 μmol/L).
The unique feature of this disorder is the development of progressive muscle weakness, tremor, and spasticity (diplegia or quadriplegia) [Prasad et al., 1997; Scheuerle et al., 1993]. Mental retardation and growth failure also commonly are evident by childhood, and glaucoma has been reported [Sacca et al., 1996]. Affected children generally do not succumb to hyperammonemic coma and therefore have a longer life span than those affected by proximal urea cycle disorders.
Arginase 1 is found in liver and red blood cells. By contrast, renal arginase (arginase 2) is found in the mitochondrial matrix and differs from the liver type of enzyme in biochemical, molecular, and antigenic properties [Cederbaum et al., 2004]. Arginase 2 also is found in the small intestine and the brain, and its levels have been found to be elevated in patients with argininemia [Iyer et al., 1998]. It is possible that the presence of arginase 2 in hyperargininemia provides some degree of protection from nitrogen accumulation, resulting in less severe hyperammonemic episodes than in other urea cycle disorders. The gene for liver arginase has been localized to chromosome band 6q23 [Sparkes et al., 1986]. Available evidence suggests multiple point mutations and microdeletions in this disorder, indicating extensive genetic heterogeneity [Vockley et al., 1996], and many affected persons are compound heterozygotes. Correlation between the severity of the mutation and the degree of clinical symptoms has been shown [Uchino et al., 1998].
The principal biochemical finding is markedly elevated plasma levels of arginine. Arginine plus ornithine, aspartate, threonine, glycine, and methionine levels are elevated in cerebrospinal fluid [Cederbaum et al., 1982]. In addition, a generalized dibasic aminoaciduria (argininuria, lysinuria, cystinuria, ornithinuria) is present. Urinary excretion of orotic acid and guanidine compounds also is markedly increased [Marescau et al., 1990]. The diagnosis can be confirmed by measuring arginase 1 activity in erythrocytes.
The mechanism responsible for the spasticity and cognitive deficits in argininemia is unknown but is unlikely to be the result of the generally moderate hyperammonemia. Arginine, its guanidine metabolites, and altered biogenic amines are candidate neurotoxins [Marescau et al., 1990]. Arginine is the substrate for nitric oxide synthetase, so overproduction of nitric oxide may play a role in neuropathology [Iyer et al., 1998]. It is theoretically possible that pharmacologic inhibitors of nitric oxide synthetase may be beneficial. Treatment with alternate pathway therapy appears to halt the progression of the spasticity, and botulinum toxin (Botox) and surgical tendon release may improve function.
Hyperornithinemia-Hyperammonemia-Homocitrullinuria Syndrome
HHH syndrome was first described in 1969 [Shih et al., 1969], and only about 50 cases have been reported in the literature, most being in the French-Canadian population in Quebec [Gjessing et al., 1986]. Clinical symptoms are similar to those in other urea cycle disorders but rarely develop in infancy [Zammarchi et al., 1997]. Spastic paraparesis also has been noted and occasionally coagulation disorders occur [Gallagher et al., 2001; Lemay et al., 1992; Salvi et al., 2001a, b]. Plasma ornithine concentrations are elevated, ranging from 400 to 600 mmol/L. Plasma lysine level typically is low, and urinary excretion of homocitrulline is increased. The high blood levels and urinary excretion of homocitrulline likely are the result of conversion of lysine to homocitrulline by ornithine transcarbamylase, in the absence of ornithine in mitochondria. The etiology of HHH syndrome begins with a mutation in the ornithine transporter gene, ORNT1, also called SLC25A15, whose product is a member of the solute mitrochondrial carrier protein family [Camacho et al., 1999]. This leads to decreased ornithine levels in mitochondria and secondary impairment of urea synthesis. The expression of ORNT2, an intronless gene or processed pseudogene, encoding a protein about 90 percent identical to OTNT 1, may explain the milder clinical signs and symptoms compared with those in CPS I and OTC deficiencies [Camacho et al., 2003]. Treatment of HHH syndrome involves protein restriction, phenylbutyrate, and citrulline supplementation.
Common Clinical Presentations of Urea Cycle Disorders
The classic presentation of a complete defect in the urea cycle (other than arginase) is as a catastrophic illness in the first week of life. Clinical manifestations appear between 24 and 72 hours of age, starting as a poor suck, hypotonia, vomiting, lethargy, and hyperventilation with rapid progression to coma and seizures. The electroencephalographic (EEG) pattern during hyperammonemic coma is one of low voltage with slow waves and asymmetric delta and theta waves. The tracing may demonstrate a burst suppression pattern, and the duration of the interburst interval may correlate with the height of ammonia levels [Clancy and Chung, 1991]. Neuroimaging studies reveal cerebral edema with small ventricles, flattening of cerebral gyri, and diffuse low density of white matter; evidence of intracranial hemorrhage also may be seen [Kendall et al., 1983].
Partial urea cycle enzyme deficiencies have a spectrum of presentations, with hyperammonemic episodes developing in infancy in some patients, in later childhood in others, and not until adulthood in still others. Symptoms may be delayed in onset with a mild deficiency or by dietary self-restriction – specifically, avoidance of meats, fish, eggs, milk, and other high-protein foods. Signs and symptoms in childhood include anorexia, ataxia, and behavioral abnormalities such as episodes of erratic behavior, acting out of character, irritability, cloudedness to frank mental status change, nocturnal restlessness, and attention-deficit and hyperactivity [Rowe et al., 1986]. In adults, signs and symptoms may mimic those of psychiatric or neurologic disorders, and include migraine-like headache, nausea, dysarthria, ataxia, confusion, hallucinations, and visual impairment (blurred vision, scotomas, lost vision) [Arn et al., 1990]. In a case report of late-onset OTC deficiency, the patient was a heterozygote who presented with a syndrome mimicking complex partial status epilepticus [Bogdanovic et al., 2000]. Neurologic findings may include increased deep tendon reflexes, papilledema, and decorticate/decerebrate posturing. Seizures generally are a late complication; the seizure episode typically is preceded by alteration in consciousness. There are indications that patients with urea cycle disorders may also be prone to seizures outside of hyperammonemic episodes, as seizures seemed to be observed more often in this patient group than in the general population in one clinic [Zecavati et al., 2008]. Analysis of data collected longitudinally from patients with urea cycle disorders [Tuchman et al., 2008; Seminara et al., 2010] will allow further investigation of this impression. A possible explanation for increased frequency of seizures in hyperammonemic coma and chronic hyperammonemia in urea cycle disorder patients could be increased extracellular potassium levels [Lichter-Konecki et al., 2008a; Lichter-Konecki, 2008b], which lower the seizure threshold.
In affected persons, hyperammonemic episodes have been precipitated by high-protein meals, infection, medication, trauma, surgery, and delivery/postpartum period. It is not uncommon for the initial hyperammonemic episode in a child with a partial deficiency to occur after weaning, when low-protein breast milk is replaced by formula or cow’s milk with a higher protein content. This is also the time when protein avoidance/self-restriction may first be observed. The puerperium in OTC-deficient heterozygotes and valproate therapy in partial CPS, OTC, and ASL deficiencies also has been associated with hyperammonemic crises [Arn et al., 1990; Batshaw et al., 1981; Honeycutt et al., 1992; Morgan et al., 1987]. The posited mechanism of valproate-induced hyperammonemia is through accumulation of the branched short-chain fatty acyl-CoA metabolites of valproate, which inhibit N-acetylglutamate formation, the co-factor of the first enzyme in the urea cycle, CPS I. This also has been the proposed explanation for hyperammonemia in the organic acidemias where propionyl-CoA was found to competitively inhibit N-acetylglutamate synthase. In a case report of haloperidol-induced hyperammonemia in a patient with partial OTC deficiency, a similar mechanism is likely [Rubenstein et al., 1990].
Associated Medical Conditions
A number of medical conditions have been associated with urea cycle disorders. For example, it is recognized that chronic liver dysfunction develops in patients with argininosuccinicacidemia. Other examples are anecdotal observations of a propensity for hypertension, electrolyte abnormalities (hypokalemia and hypomagnesemia), gastrointestinal abnormalities, including anorexia, stunted growth, microcytic anemia, and nutritional deficiencies [Brusilow and Horwich, 2001]. Pancreatitis has also been described, although the etiology is unclear [Anadiotis et al., 2001]. In addition, metabolic stroke-like episodes have been described in CPS and OTC deficiencies involving the caudate and putamen, with resultant extrapyramidal syndromes [Keegan et al., 2003].
Histopathologic Features of Urea Cycle Disorders
Histopathologic examination of liver in urea cycle disorders may be normal but often demonstrates diffuse microvesicular steatosis, marked increased glycogen in periportal cells, and variable portal fibrosis [Badizadegan and Perez-Atayde, 1997]. Cirrhosis has been identified in some patients with argininosuccinicaciduria (ASA), citrullinemia type II, and arginase deficiency [Zimmermann et al., 1986]. Why some patients with ASA develop cirrhosis is not known at this point but nitric oxide metabolism may play a role [Brunetti-Pierri et al., 2009].
Neuropathologic findings in urea cycle disorders are similar to those following hepatic encephalopathy and hypoxic-ischemic insults. They depend both on the duration of hyperammonemic coma, and on the interval between coma and death. Neonates dying in hyperammonemic coma have prominent cerebral edema and generalized neuronal cell loss on postmortem examination [Crome and France, 1971]. Histologically, astrocyte swelling was one of the first observations made in an animal model for hepatic encephalopathy [Norenberg, 1977], leading to the hypothesis that swollen astrocytes may be the cellular correlate of the brain edema in acute hyperammonemia. The central role of astrocytes in hyperammonemic encephalopathy was felt to be underscored by the finding of Alzheimer type II astrocytes in the brains of patients in whom coma had taken an irreversible course. In survivors of prolonged coma, changes observed on neuroimaging studies obtained months later include ventriculomegaly with increased sulcal markings, bilateral symmetric low-density white matter defects, cystic degeneration, injury to the bilateral lentiform nuclei, and diffuse atrophy with sparing of the cerebellum [Kendall et al., 1983; Majoie et al., 2004; Takanashi et al., 2003; Yamanouchi et al., 2002]. Neuropathologic findings in those children who subsequently died were consistent with the neuroimaging findings and included ulegyria, cortical atrophy with ventriculomegaly, prominent cortical neuronal loss, gliosis (often with Alzheimer type II astrocytes), and spongiform changes at the gray–white matter interface and in the basal ganglia and thalamus [Dolman et al., 1988].
Mechanism of Neuropathology
The mechanism of the ammonia-induced neuropathology remains unclear [Bachmann et al., 2004]. Ammonia normally is detoxified in astrocytes by glutamate dehydrogenase and glutamine synthase. Accumulation of ammonia and glutamine in brain has a number of potentially toxic effects.
Downregulation of Astrocytic Glutamate Transporters
Astrocytic glutamate transporters were shown to be downregulated [Desjardin et al., 2001] and extracellular glutamate levels to be elevated [Felipo and Butterworth, 2002] in hyperammonemia. This led to the hypothesis that the brain damage in hyperammonemic coma is caused by overstimulation of neurons by elevated extracellular glutamate levels, a mechanism of brain damage called excitotoxicity. However, only a limited amount of neuronal cell death and little indication of excitotoxicity were observed in the animal models studied [Butterworth, 2007].
Elevated Glutamine Levels
A negative correlation between the height of brain glutamine levels and brain myoinositol levels has been observed, suggesting depletion of the osmolyte myoinositol by high glutamine levels [Takanashi et al 2002; Gropman et al., 2008]. High brain glutamine levels may have an osmotic effect and cause brain swelling. Inhibition of glutamine synthase by methionine sulfoximine was reported to prevent astrocyte swelling in experimental hyperammonemia [Willard-Mack et al., 1996].
Altered Water Transport Through Aquaporin 4 Water Channels
Water transport at blood–brain and brain–cerebrospinal fluid interfaces is facilitated by channel proteins called aquaporins. Aquaporin 4 (Aqp4) is the main astrocytic water channel and is expressed in astrocytic endfeet at the brain vasculature. While upregulation of Aqp4 expression in astrocytes exposed to ammonia in culture was reported [Rama Rao et al., 2003b], downregulation of this channel was found in the brains of hyperammonemic animals. The regulation of Aqp4 expression was shown to depend on the type of brain injury and type of edema.
Altered Glucose Metabolism/Disturbed Energy Metabolism
Ammonia stimulates phosphofructokinase, the key enzyme of glycolysis. Ammonium may play a significant role in regulating glycolysis in astrocytes under physiological conditions in vivo [Tsacopoulos and Magistretti, 1996]. Increased ammonia levels, on the other hand, inhibit α-ketoglutarate dehydrogenase in brain [Lai and Cooper, 1986]. Increased α-ketoglutarate levels were measured in the brain of hyperammonemic mice [Ratnakumari et al., 1994]. Stimulation of phosphofructokinase, as well as inhibition of α-ketoglutarate dehydrogenase, the key enzyme of the tricarboxylic cycle, will cause increased formation of lactate in brain. A block in the tricarboxylic cycle will lead to compromised brain energy metabolism.
Interference with the Normal Flux of Potassium Ions
The consideration that ammonia could interfere with potassium homeostasis is due to the fact that NH4+ has properties similar to those of the potassium ion. An important astrocytic function is the maintenance of potassium homeostasis. NH4+ can cross cell membranes through ion channels or membrane transporters, and can replace K+ on different transporters. In cultured mouse astrocytes, ammonium was shown to enter the cell through inward rectifying K+ channels [Marcaggi and Coles, 2001]. Ammonia may thus interfere with potassium channel or potassium transporter function. An elevated extracellular K+ concentration was measured in the parietal cortex of healthy rats infused with ammonium acetate to cause hyperammonemia [Sugimoto et al., 1997]. Lichter-Konecki et al. [2008a] found downregulation of the major astrocytic water, gap-junction, and potassium channels in hyperammonemia in an animal model for urea cycle disorders, and concluded a possible disturbance of potassium and water homeostasis in brain during hyperammonemia.
Oxidative and Nitrosative Stress
An increase in free radical production and nitric oxide synthesis causes oxidative/nitrosative (O/N) stress. Ammonia causes free radical production and a decrease in antioxidant enzyme activity, while antioxidants prevented astrocyte swelling after ammonia exposure. In addition to oxidative stress, there is also nitrosative stress in hyperammonemic encephalopathy, as an upregulation of nitric oxide synthase (NOS) activity and expression was detected in animal models for hepatic encephalopathy [Rao et al., 1997]. Increased amounts of nitric oxide were found in the brains of such animals, and NOS inhibitors prevented astrocyte swelling in vivo and in vitro. How O/N stress would cause astrocyte swelling is not known but a mechanism under consideration is induction of mitochondrial permeability transition (MPT). In this scenario, oxidative stress would cause opening of the permeability transition pore (PTP) of the inner mitochondrial membrane and ions would flow, changing the membrane potential. This would lead to mitochondrial dysfunction and more free radical production, causing cell damage.
Besides the amino acid glutamine, tryptophan and serotonin also are increased in the brain of the spf/Y mouse, an animal model for OTC deficiency, and in cerebrospinal fluid of children with urea cycle disorders [Bachmann et al., 1982; Batshaw et al., 1993; Inoue et al., 1987]. This finding led to the hypothesis that high glutamine levels also may cause other amino acid imbalances in the brain. Increased tryptophan levels result in increased levels of its metabolite, quinolinate, [Robinson et al., 1995], an excitotoxin at the N-methyl-d-aspartate receptor.
Differential Diagnosis
In the newborn period, hyperammonemia is similar in presentation to a number of acquired conditions, including sepsis, intracranial hemorrhage, and cardiorespiratory disorders. The measurement of plasma ammonia levels is critical to distinguish between these conditions. The acquired disorders generally are not associated with significant elevations in ammonia levels, whereas the inborn errors are. As a consequence, plasma ammonia levels should be measured as part of the routine evaluation for serious illness in the newborn period. Apart from urea cycle disorders, a number of other inborn errors of metabolism can cause hyperammonemia. These include organic acidemias, nonketotic hyperglycinemia, congenital lactic acidoses, lysinuric protein intolerance, and defects in fatty acid oxidation. An algorithm for distinguishing between these disorders is presented in Figure 33-2.
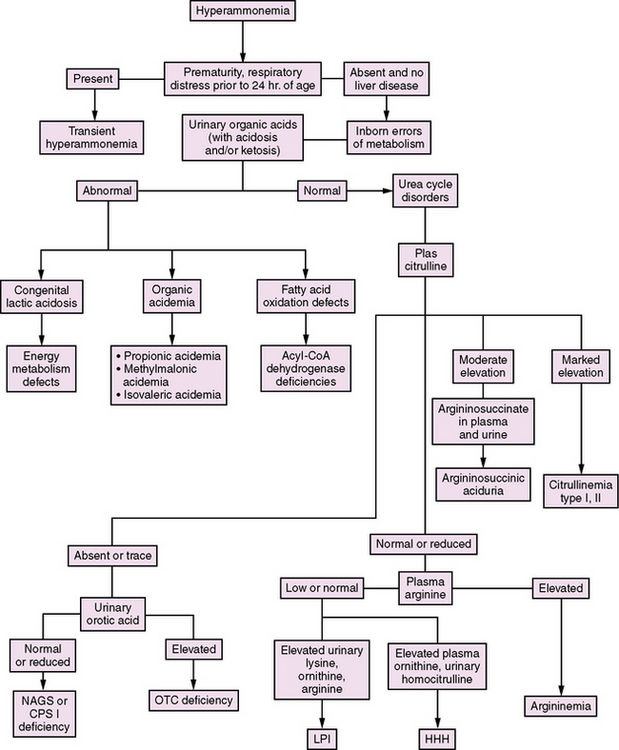
In the organic acidemias (e.g., methylmalonicacidemia, propionicacidemia, isovalericacidemia, glutaricacidemia type II, multiple carboxylase deficiency), the usual findings include a marked metabolic acidosis, ketosis, and increased anion gap. Hypoglycemia and pancytopenia also may be present. Mass spectrometry of urine organic acids or acylcarnitine analysis in blood will make the specific diagnosis. In propionic acidemia/organic acidurias, plasma (but not cerebrospinal fluid) will have an elevated glycine level – whence came the initial name, ketotic hyperglycinemia. By contrast, with nonketotic hyperglycinemia, ketosis is absent, glycine levels in both plasma and cerebrospinal fluid are markedly elevated, and cerebrospinal fluid glycine to plasma glycine ratio is increased [Tada and Hayasaka, 1987].
Fatty acid oxidation defects, caused by different-chain-length acyl-CoA dehydrogenase deficiencies, manifest in the neonatal period with hypoglycemia and decreased ketones, as well as liver and heart disease. Analysis of urinary organic acids shows a dicarboxylic aciduria; plasma carnitine levels may be low; and abnormal acylcarnitine species are found [Roe and Ding, 2001].
Congenital lactic acidosis can be the result of a primary deficiency of pyruvate carboxylase or pyruvate dehydrogenase, or secondary to a defect within the mitochondrial respiratory chain [McCormick et al., 1985]. The principal biochemical finding is lactic acidosis. In pyruvate dehydrogenase deficiency and mild pyruvate carboxylase deficiency, the ratio of lactate to pyruvate remains normal. In severe pyruvate carboxylase deficiency, mitochondrial respiratory chain defects, and secondary lactic acidosis, however, this ratio is significantly increased [Zeviani et al., 1989]. In oxidative phosphorylation defects and other mitochondrial diseases the 3-hydroxybutyrate to acetoacetate ratio also is elevated.These findings are in contrast with those in inborn errors of urea synthesis, in which the urinary organic acid profile is normal and plasma lactate level is normal or mildly increased. Although peak plasma ammonia levels generally are in the range of 150 to 500 μM (normal 15 to 40 μM) in these disorders, they are likely to be above 500 μM in neonatal-onset urea cycle disorders.
Plasma amino acid patterns also are distinct in urea cycle disorders, with low levels of arginine and ornithine and either low or high levels of citrulline. Citrulline is the product of CPS I and OTC, and the substrate for ASS and ASL. Thus, it either is absent, or is present only in trace amounts, in NAGS, CPS I, and OTC deficiencies, and levels are markedly elevated in ASS and ASL deficiencies [Hudak et al., 1985]. Differentiation of ASS from ASL deficiencies depends on finding argininosuccinic acid in plasma and urine in ASL deficiency. In HHH syndrome, biochemical findings include accumulation of ornithine in blood and of homocitrulline in the urine.
In older children and adults, identification of partial-defect urea cycle disorders often has been delayed by misdiagnoses that have included migraine, cyclical vomiting, viral encephalitis, stroke, Reye’s syndrome, drug toxicity, child abuse, psychosis, postpartum depression, seizure disorder, and cerebral palsy [Christodoulou et al., 1993; de Bruijn et al., 1976; DiMagno et al., 1986; Drogari and Leonard, 1988; Finkelstein et al., 1990]. Studies have found a mean delay of 8–16 months between the onset of symptoms and diagnosis of late-onset urea cycle disorders in children [Rowe et al., 1986]. During this time, progressive abnormalities in growth and cognitive/motor development may be noted.
Diagnosis of late-onset urea cycle disorders may be less clear-cut than in neonatal cases. Plasma ammonia levels may be in the range of 150–250 μM during symptomatic episodes and normal when the patient is clinically stable. Serum (blood) urea nitrogen, which clearly is subnormal in neonatal-onset deficiencies, may be normal. Plasma citrulline levels often are low to normal in partial OTC and CPS I deficiencies, rather than absent or trace, as in complete defects. Yet, as noted previously, oroticaciduria can be provoked by an allopurinol load to detect partial OTC deficiencies, and marked elevations of argininosuccinic acid in blood and urine are characteristic in ASL deficiency (up to 1500 μM in plasma, normally undetected), of citrulline in ASS deficiency (range 1000–5000 μM in plasma, normal 6–50 μM), and of arginine in arginase deficiency (up to 1500 μM, normal less than 100 μM) [Batshaw et al., 1981]. In HHH syndrome, marked elevations of homocitrulline in urine and of ornithine in blood are seen.
Apart from urea cycle disorders, other inborn errors of metabolism that manifest with symptomatic hyperammonemia in later childhood/adulthood include the organic acidemias and lysinuric protein intolerance [Simell et al., 1975]. These disorders can be distinguished by specific patterns of urinary organic acids, urinary amino acids, plasma amino acids, and plasma carnitine/acylcarnitine levels. A number of acquired disorders also can manifest with hyperammonemia, including hepatic encephalopathy, Reye’s syndrome, drug toxicity, and hepatotoxins. The medical history, prothrombin time, a urinary toxin screen, and analysis of plasma amino acid pattern (low branched-chain amino acids, high aromatic amino acids in chronic liver disease, elevated lysine in Reye’s syndrome) should help differentiate these disorders.
Treatment
In the newborn period, treatment of urea cycle disorders may be reactive or anticipatory. In families who have had a previously affected child or in OTC-deficient kindreds, the birth of an at-risk or prenatally diagnosed infant provides the opportunity for prospective management. Within hours of birth, the child can be placed on oral alternate pathway therapy before the development of hyperammonemia (see Table 33-1). Of 15 infants with urea cycle disorders treated in this fashion, all but 3 survived the newborn period without hyperammonemic coma [Maestri et al., 1991].
Hemodialysis
In infants who are diagnosed during hyperammonemic coma, hemodialysis should be started immediately [McBryde et al., 2004; Tuchman, 1992]. In addition, l-arginine HCl (except in hyperargininemia), sodium benzoate, and sodium phenylacetate (Ammonul, Ucyclyd Pharma) should be given intravenously at the same doses used to treat intercurrent hyperammonemia.
Liver Transplantation
Long-term treatment of urea cycle disorders depends on the severity of the disease. For neonatal-onset CPS I and OTC deficiencies, current clinical guidelines recommend that liver transplantation be considered as a form of enzyme replacement therapy [Leonard and McKiernan, 2004]. This procedure usually is performed at 6–12 months of age, with use of alternate pathway therapy before surgery [Leonard and McKiernan, 2004; Saudubray et al., 1999]. More recently, and as a result of favorable outcome with liver transplantation, other neonatal-onset urea cycle and poorly controlled late-onset disorders also have been considered for this treatment.
The Studies in Pediatric Liver Transplantation (SPLIT) reported that 114 children with urea cycle disorders (UCDs) received a liver transplant between December of 1995 and June 2008 [Arnon et al., 2010]. The 1-year survival rate for patients with UCDs was 95.2 percent and the 1-year survival rate of their grafts was 91.8 percent. The 5-year patient survival rate was 88.7 percent, and the 5-year graft survival rate 83.7 percent. Living donors provided the liver in 7.9 percent of cases, and some form of cadaveric liver (whole, split, reduced) was used in 87.8 percent of cases. SPLIT is a cooperative research consortium that was established in 1995 to assess transplanted patients, their grafts, and management practices. SPLIT registers about 60 percent of liver transplants performed in children in the United States.
A number of case reports have been published concerning the results of liver transplantation with regard to correction of the metabolic defects in urea cycle disorders [Leonard and McKiernan, 2004; Saudubray et al., 1999]. These reports suggest that transplantation corrects most of the metabolic abnormalities (although citrulline levels remain low in OTC and CPS I deficiencies) and prevents future hyperammonemic episodes. In terms of morbidity, concerns have included neurodevelopmental lags, and frequent and prolonged hospitalizations for treatment of infections and regulation of immunosuppressing drugs. Overall, however, the reported quality of life has been much improved with normalization of diet and a decreased frequency of hospital admissions [Leonard and McKiernan, 2004].
Protein Restriction
In all urea cycle disorders other than citrullinemia type II, a protein-restricted diet should be combined with alternate pathway therapy unless liver transplantation has been performed. In general, using the minimum daily protein requirement for the child’s age is recommended. For neonatal-onset disease, this generally is given half as an essential amino acid supplement (e.g., Cyclinex, Ross Pharmeuticals) and half as natural protein. For mild late-onset disease, protein restriction alone may be sufficient. In children receiving sodium phenylbutyrate, monitoring branched-chain amino acid levels and supplementing these as needed also has been suggested [Scaglia et al., 2004]. Additional calories can be provided via nonprotein formulas, such as Prophree or MJ80056. The diet should be supplemented with vitamins, minerals, and trace elements. Also recommended is routine monitoring of weight, growth, hair, skin, nails, and biochemical indices of nutritional status.
Alternative Pathway Therapy
For late-onset CPS I and OTC deficiencies, as well as for the other urea cycle disorders, long-term therapy generally involves nitrogen restriction combined with alternative pathway therapy (see Table 33-1) [Batshaw et al., 2001; Berry and Steiner, 2001]. Approaches to stimulating alternative pathways of waste nitrogen excretion vary with the site of the enzymatic block. In the case of ASS and ASL deficiencies (including citrullinemia type II), arginine can stimulate waste nitrogen excretion through enhanced production and excretion of citrulline and argininosuccinic acid [Imamura et al., 2003]. In CPS I and OTC deficiencies, sodium phenylbutyrate has been used to provide an alternative pathway. It initially is converted to sodium phenylacetate, which then conjugates with glutamine to form phenylacetylglutamine, which is excreted by the kidney [Burlina et al., 2001; MacArthur et al., 2004]. Arginase deficiency has been managed with an arginine-restricted diet supplemented with sodium phenylbutyrate.
N-Carbamyl-l-Glutamate
In NAGS deficiency, administration of N-carbamyl-l-glutamate (Carbaglu, Orphan Europe, Paris, France) may be an effective therapy. N-carbamyl-l-glutamate is a stable structural analog of N-acetylglutamate and theoretically could substitute for it in the activation of CPS I [Hall et al., 1958]. Although the affinity of N-carbamyl-l-glutamate for CPS I is lower than that of N-acetylglutamate (KM = 2 and 0.15 μM, respectively) [Rubio and Grisolia, 1981], it is much more resistant to degradation by aminoacylase [Kim et al., 1972]. N-carbamyl-l-glutamate is therefore attractive for therapy of N-acetylglutamate synthase deficiency. Moreover, unlike N-acetylglutamate, N-carbamyl-l-glutamate can cross the mitochondrial membrane into the mitochondrion, where N-acetylglutamate synthase resides [Kim et al., 1972]. Several patients with N-acetylglutamate synthase deficiency have been reported to respond clinically to treatment with N-carbamyl-l-glutamate [Guffon et al., 1995; Hinnie et al., 1997; Morris et al., 1998; Schubiger et al., 1991]. One homozygous affected subject underwent a 15N-tracer study of ureagenesis rate before and after 3-day treatment with oral N-carbamyl-l-glutamate, it completely restored normal urea cycle function [Caldovic et al., 2003].
Management of Intercurrent Hyperammonemic Crises
If vomiting occurs or if lethargy becomes evident, and ammonia levels are above 3–5 times normal, more aggressive treatment is needed. This approach involves hospitalization, with temporary complete elimination of protein and institution of intravenous administration of sodium benzoate, sodium phenylacetate, and l-arginine. (Caution: arginine HCl may cause metabolic acidosis, and extravasation may lead to tissue necrosis.) Although these medications are not associated with severe adverse events when they are given at therapeutic doses, severe toxicity and death have resulted from dosing errors [Praphanphoj et al., 2000]. Therefore, double-checking of drug administration orders is essential, and monitoring plasma levels is recommended.
In the event that ammonia levels do not respond to this conservative management approach and biochemical abnormalities or clinical signs and symptoms worsen, hemodialysis should be initiated. The relative effectiveness of peritoneal dialysis, exchange transfusion, hemodialysis, and continuous arteriovenous hemofiltration has been the subject of some controversy. Yet nitrogen balance studies clearly demonstrate the advantage of hemodialysis, with continuous arteriovenous hemodiafiltration being second best if hemodialysis is unavailable [McBryde et al., 2004]. Hemodialysis should be continued until ammonia levels fall to less than five times normal; peritoneal dialysis or continuous hemofiltration may then be initiated if needed. Hemodialysis using an extracorporeal membrane oxygenation pump has been reported to be effective for rapidly removing ammonia [Summar et al., 1996].
Outcome
Before the development of alternative pathway therapy, few children with a complete urea cycle defect survived infancy [Shih, 1976]. Most died in the newborn period, and the remainder succumbed to intercurrent hyperammonemic episodes or protein malnutrition later in infancy or early childhood. Long-term survival, however, has improved with the introduction of alternative pathway therapy and liver transplantation [Brusilow and Horwich, 2001]. The mortality and morbidity rates currently cited for urea cycle disorders were obtained in the 1980s, when the index of suspicion for these disorders was low, leading to significant delays in diagnosis. In addition, no standardized treatment regimen was in use. At that time, the 5-year survival rate for a small prospective cohort of patients with neonatal-onset disease was reported to be approximately 25 percent, and the morbidity (e.g., mental retardation, seizure disorders, cerebral palsy) was found to be virtually universal for infants rescued from hyperammonemic coma [Batshaw et al., 1982; Msall et al., 1984]. In a retrospective study of mortality among infants with neonatal-onset urea cycle disorders referred to one major metabolic center during that period [Maestri et al., 1999], 34 of 74 infants (46 percent) died during their neonatal hyperammonemic episode, and among the 40 infants surviving the initial hyperammonemic episode, the median survival period was 3.8 years. More recently, with the general availability of plasma ammonia assays at most hospitals, an increased index of suspicion for metabolic disorders, and standardized alternative pathway therapy, it is likely that the outcome has improved.
Although mortality has decreased, morbidity remains high among survivors of neonatal hyperammonemic coma. More than three-quarters of these children have mental retardation, and frequent comorbid conditions include cerebral palsy, seizure disorder, and visual deficits [Msall et al., 1984]. A correlation between intellectual function and duration of hyperammonemic coma has been noted. Children in coma for less than 3 days had a far better outcome than those in coma for longer periods [Msall et al., 1984]. Cognitive outcome also was better in infants with complete defects who received prospective treatment and in children with partial defects. Of interest, even OTC-deficient women with subtle clinical signs or symptoms (e.g., protein aversion) have been found to have mild cognitive impairments suggestive of a nonverbal learning disability [Gropman and Batshaw, 2004; Maestri et al., 1991; Nagata et al., 1991; Widhalm et al., 1992].
References
 The complete list of references for this chapter is available online at www.expertconsult.com.
The complete list of references for this chapter is available online at www.expertconsult.com.
Ahrens M.J., Berry S.A., Whitley C.B., et al. Clinical and biochemical heterogeneity in females of a large pedigree with ornithine transcarbamylase deficiency due to the R141Q mutation. Am J Med Genet. 1996;66:311-315.
Allan J.D., Cusworth D.C., Dent C.E., et al. A disease, probably hereditary characterised by severe mental deficiency and a constant gross abnormality of aminoacid metabolism. Lancet. 1958;1:182.
Anadiotis G., Ierardi-Curto L., Kaplan P.B., et al. Ornithine transcarbamylase deficiency and pancreatitis. J Pediatr. 2001;138:123.
Arn P.H., Hauser E.R., Thomas G.H., et al. Hyperammonemia in women with a mutation at the ornithine carbamoyltransferase locus. A cause of postpartum coma. N Engl J Med. 1990;322:1652.
Arnon R., Kerkar N., Davis M.K., et al. Liver transplantation in children with metabolic diseases: The studies of pediatric liver transplantation experience. Pediatr Transplant. 2010. Jun 17. [Epub ahead of print] PMID: 20557477 [PubMed – as supplied by publisher]
Arnon R., Kerkar N., Davis M.K., SPLIT Research Group, et al. Liver transplantation in children with metabolic diseases: the studies of pediatric liver transplantation experience. Pediatr Transplant. 2010;14(6):796-805. Sep 1. Epub 2010 Jun 27
Ausems M.G., Bakker E., Berger R., et al. Asymptomatic and late-onset ornithine transcarbamylase deficiency caused by a A208T mutation: Clinical, biochemical and DNA analyses in a four-generation family. Am J Med Genet. 1997;68:236.
Bachmann C., Colombo J.P., Jaggi K. N-acetylglutamate synthetase (NAGS) deficiency: Diagnosis, clinical observations and treatment. Adv Exp Med Biol. 1982;153:39.
Bachmann C., Braissant O., Villard A.M., et al. Ammonia toxicity to the brain and creatine. Mol Genet Metab. 2004;81(Suppl 1):S52.
Badizadegan K., Perez-Atayde A.R. Focal glycogenosis of the liver in disorders of ureagenesis: Its occurrence and diagnostic significance. Hepatology. 1997;26:365.
Batshaw M.L., Roan Y., Jung A.L., et al. Cerebral dysfunction in asymptomatic carriers of ornithine transcarbamylase deficiency. N Engl J Med. 1980;302:482.
Batshaw M.L., Thomas G.H., Brusilow S.W. New approaches to the diagnosis and treatment of inborn errors of urea synthesis. Pediatrics. 1981;68:290.
Batshaw M.L., Brusilow S., Waber L., et al. Treatment of inborn errors of urea synthesis: Activation of alternative pathways of waste nitrogen synthesis and excretion. N Engl J Med. 1982;306:1387.
Batshaw M.L., Msall M., Beaudet A.L., et al. Risk of serious illness in heterozygotes for ornithine transcarbamylase deficiency. J Pediatr. 1986;108:236.
Batshaw M.L., Robinson M.B., Hyland K., et al. Quinolinic acid in children with congenital hyperammonemia. Ann Neurol. 1993;34:676.
Batshaw M.L., MacArthur R.B., Tuchman M. Alternative pathway therapy for urea cycle disorders: Twenty years later. J Pediatr. 2001;138:S464. discussion S54
Berry G.T., Steiner R.D. Long-term management of patients with urea cycle disorders. J Pediatr. 2001;138:S56. discussion S60
Bogdanovic M.D., Kidd D., Briddon A., et al. Late onset heterozygous ornithine transcarbamylase deficiency mimicking complex partial status epilepticus. J Neurol Neurosurg Psychiatry. 2000;69:813.
Braga A.C., Vilarinho L., Ferreira E., et al. Hyperargininemia presenting as persistent neonatal jaundice and hepatic cirrhosis. J Pediatr Gastroenterol Nutr. 1997;24:218.
Brunetti-Pierri N., Erez A., Shchelochkov O., et al. 4.Systemic hypertension in two patients with ASL deficiency: a result of nitric oxide deficiency? Mol Genet Metab. 2009;98(1–2):195-197. Epub 2009 Jun 13
Brusilow S., Horwich A.L. Urea cycle enzymes. In: Beaudet A.L., Sly W.S., Valle D., editors. The metabolic and molecular basis of inherited disease. ed 8. New York: McGraw-Hill; 2001:1990.
Brusilow S.W., Maestri N.E. Urea cycle disorders: Diagnosis, pathophysiology, and therapy. Adv Pediatr. 1996;43:127.
Burlina A.B., Ogier H., Korall H., et al. Long-term treatment with sodium phenylbutyrate in ornithine transcarbamylase–deficient patients. Mol Genet Metab. 2001;72:351.
Butterworth R.F. Neuronal cell death in hepatic encephalopathy. Metab Brain Dis. 2007;22:309-320.
Caldovic L., Morizono H., Panglao M.G., et al. Null mutations in the N-acetylglutamate synthase gene associated with acute neonatal disease and hyperammonemia. Hum Genet. 2003;112:364.
Caldovic L., Morizono H., Tuchman M. Mutations and polymorphisms in the human N-acetylglutamate synthase (NAGS) gene. Hum Mutat. 2007;28(8):754-759.
Camacho J.A., Obie C., Biery B., et al. Hyperornithinaemia-hyperammonaemia-homocitrullinuria syndrome is caused by mutations in a gene encoding a mitochondrial ornithine transporter. Nat Genet. 1999;22:151.
Camacho J.A., Rioseco-Camacho N., Andrade D., et al. Cloning and characterization of human ORNT2: A second mitochondrial ornithine transporter that can rescue a defective ORNT1 in patients with the hyperornithinemia-hyperammonemia-homocitrullinuria syndrome, a urea cycle disorder. Mol Genet Metab. 2003;79:257.
Cederbaum S.D., Shaw K.N., Spector E.B., et al. Hyperargininemia with arginase deficiency. Pediatr Res. 1979;13:827.
Cederbaum S.D., Moedjono S.J., Shaw K.N., et al. Treatment of hyperargininaemia due to arginase deficiency with a chemically defined diet. J Inherit Metab Dis. 1982;5:95.
Cederbaum S.D., Yu H., Grody W.W., et al. Arginases I and II: Do their functions overlap? Mol Genet Metab. 2004;81(Suppl 1):S38.
Christodoulou J., Qureshi I.A., McInnes R.R., et al. Ornithine transcarbamylase deficiency presenting with strokelike episodes. J Pediatr. 1993;122:423.
Clancy R.R., Chung H.J. EEG changes during recovery from acute severe neonatal citrullinemia. Electroencephalogr Clin Neurophysiol. 1991;78:222.
Crome L., France N.E. The pathological findings in a case of argininosuccinic aciduria. J Ment Defic Res. 1971;15:266.
de Bruijn J.G., Bruyn G.W., Klawans H.L.Jr. Further observation on the possible relationship between migraine and serum ammonia levels. Clin Neurol Neurosurg. 1976;79:151.
Deardorff M.A., Gaddipati H., Kaplan P., et al. Complex management of a patient with a contiguous Xp11.4 gene deletion involving ornithine transcarbamylase: A role for detailed molecular analysis in complex presentations of classical diseases. Mol Genet Metab. 2008;94(4):498-502. Epub 2008 Jun 3
Desjardins P., Belanger M., Butterworth R.F. Alterations in expression of genes coding for key astrocytic proteins in acute liver failure. J Neurosci Res. 2001;66:967-971.
DiMagno E.P., Lowe J.E., Snodgrass P.J., et al. Ornithine transcarbamylase deficiency –a cause of bizarre behavior in a man. N Engl J Med. 1986;315:744.
Dolman C.L., Clasen R.A., Dorovini-Zis K. Severe cerebral damage in ornithine transcarbamylase deficiency. Clin Neuropathol. 1988;7:10.
Drogari E., Leonard J.V. Late onset ornithine carbamoyl transferase deficiency in males. Arch Dis Child. 1988;63:1363.
Elpeleg O., Shaag A., Ben-Shalom E., et al. N-acetylglutamate synthase deficiency and the treatment of hyperammonemic encephalopathy. Ann Neurol. 2002;52:845.
Felipo V., Butterworth R.F. Neurobiology of ammonia. Prog Neurobiol. 2002;67:259.
Finkelstein J.E., Hauser E.R., Leonard C.O., et al. Late-onset ornithine transcarbamylase deficiency in male patients. J Pediatr. 1990;117:897.
Funghini S., Donati M.A., Pasquini E., et al. Structural organization of the human carbamyl phosphate synthetase I gene (CPS1) and identification of two novel genetic lesions. Hum Mutat. 2003;22:340.
Gallagher A.C., Pike M., Standing S. HHH syndrome associated with callosal agenesis and disordered neuronal migration. Dev Med Child Neurol. 2001;43:430.
Gao H.Z., Kobayashi K., Tabata A., et al. Identification of 16 novel mutations in the argininosuccinate synthetase gene and genotype-phenotype correlation in 38 classical citrullinemia patients. Hum Mutat. 2003;22:24.
Gerrits G.P., Gabreels F.J., Monnens L.A., et al. Argininosuccinic aciduria: Clinical and biochemical findings in three children with the late onset form, with special emphasis on cerebrospinal fluid findings of amino acids and pyrimidines. Neuropediatrics. 1993;24:15.
Gjessing L.R., Lunde H.A., Undrum T., et al. A new patient with hyperornithinaemia, hyperammonaemia and homocitrullinuria treated early with low protein diet. J Inherit Metab Dis. 1986;9:186.
Grody W.W., Kern R.M., Klein D., et al. Arginase deficiency manifesting delayed clinical sequelae and induction of a kidney arginase isozyme. Hum Genet. 1993;91:1.
Grompe M., Caskey C.T., Fenwick R.G. Improved molecular diagnostics for ornithine transcarbamylase deficiency. Am J Hum Genet. 1991;48:212.
Gropman A.L., Batshaw M.L. Cognitive outcome in urea cycle disorders. Mol Genet Metab. 2004;81(Suppl 1):S58.
Gropman A.L., Fricke S.T., Seltzer R.R., et al. 1H MRS identifies symptomatic and asymptomatic subjects with partial ornithine transcarbamylase deficiency. Mol Genet Metab. 2008;95(1–2):21-30. Epub 2008 Jul 26
Guffon N., Vianey-Saban C., Bourgeois J., et al. A new neonatal case of N-acetylglutamate synthase deficiency treated by carbamylglutamate. J Inherit Metab Dis. 1995;18:61.
Gyato K., Wray J., Huang Z.J., et al. Metabolic and neuropsychological phenotype in women heterozygous for ornithine transcarbamylase deficiency. Ann Neurol. 2004;55:80.
Hagiwara N., Sekijima Y., Takei Y., et al. Hepatocellular carcinoma in a case of adult-onset type II citrullinemia. Intern Med. 2003;42:978.
Hall L.M., Metzenberg R.L., Cohen P.P. Isolation and characterization of a naturally occurring cofactor of carbamyl phosphate biosynthesis. J Biol Chem. 1958;230:1013.
Haraguchi Y., Uchino T., Takiguchi M., et al. Cloning and sequence of a cdna encoding human carbamyl phosphate synthetase I: Molecular analysis of hyperammonemia. Gene. 1991;107:335.
Harrington J.W., Stiefel M., Gianos E. Arginase deficiency presenting with cerebral oedema and failure to thrive. J Inherit Metab Dis. 2000;23:517.
Hauser E.R., Finkelstein J.E., Valle D., et al. Allopurinol-induced orotidinuria. A test for mutations at the ornithine carbamoyltransferase locus in women. N Engl J Med. 1990;322:1641.
Hinnie J., Colombo J.P., Wermuth B., et al. N-acetylglutamate synthetase deficiency responding to carbamylglutamate. J Inherit Metab Dis. 1997;20:839.
Hommes F.A., De Groot C.J., Wilmink C.W., et al. Carbamylphosphate synthetase deficiency in an infant with severe cerebral damage. Arch Dis Child. 1969;44:688.
Honeycutt D., Callahan K., Rutledge L., et al. Heterozygote ornithine transcarbamylase deficiency presenting as symptomatic hyperammonemia during initiation of valproate therapy. Neurology. 1992;42:666.
Hudak M.L., Jones M.D.Jr, Brusilow S.W. Differentiation of transient hyperammonemia of the newborn and urea cycle enzyme defects by clinical presentation. J Pediatr. 1985;107:712.
Ikeda S., Yazaki M., Takei Y., et al. Type II (adult onset) citrullinaemia: Clinical pictures and the therapeutic effect of liver transplantation. J Neurol Neurosurg Psychiatry. 2001;71:663.
Imamura Y., Kobayashi K., Shibatou T., et al. Effectiveness of carbohydrate-restricted diet and arginine granules therapy for adult-onset type II citrullinemia: A case report of siblings showing homozygous SLC25A13 mutation with and without the disease. Hepatol Res. 2003;26:68.
Inoue I., Gushiken T., Kobayashi K., et al. Accumulation of large neutral amino acids in the brain of sparse-fur mice at hyperammonemic state. Biochem Med Metab Biol. 1987;38:378.
Iyer R., Jenkinson C.P., Vockley J.G., et al. The human arginases and arginase deficiency. J Inherit Metab Dis. 1998;21(Suppl 1):86.
Keegan C.E., Martin D.M., Quint D.J., et al. Acute extrapyramidal syndrome in mild ornithine transcarbamylase deficiency: Metabolic stroke involving the caudate and putamen without metabolic decompensation. Eur J Pediatr. 2003;162:259.
Kendall B.E., Kingsley D.P., Leonard J.V., et al. Neurological features and computed tomography of the brain in children with ornithine carbamoyl transferase deficiency. J Neurol Neurosurg Psychiatry. 1983;46:28.
Kim S., Paik W.K., Cohen P.P. Ammonia intoxication in rats: Protection by N-carbamoyl-L-glutamate plus L-arginine. Proc Natl Acad Sci USA. 1972;69:3530.
Kleijer W.J., Garritsen V.H., Linnebank M., et al. Clinical, enzymatic, and molecular genetic characterization of a biochemical variant type of argininosuccinic aciduria: Prenatal and postnatal diagnosis in five unrelated families. J Inherit Metab Dis. 2002;25:399.
Klosowski S., Largilliere C., Storme L., et al. Lethal ornithine transcarbamylase deficiency in a female neonate: A new case. Acta Paediatr. 1998;87:227.
Kobayashi K., Shaheen N., Kumashiro R., et al. A search for the primary abnormality in adult-onset type II citrullinemia. Am J Hum Genet. 1993;53:1024.
Kobayashi K., Bang Lu Y., Xian Li M., et al. Screening of nine SLC25A13 mutations: Their frequency in patients with citrin deficiency and high carrier rates in Asian populations. Mol Genet Metab. 2003;80:356.
Krivitzky L., Babikian T., Lee H.S., et al. Intellectual, adaptive, and behavioral functioning in children with urea cycle disorders. Pediatr Res. 2009;66(1):96-101.
Lai J.C., Cooper A.J. Brain alpha-ketoglutarate dehydrogenase complex: kinetic properties, regional distribution, and effects of inhibitors. J Neurochem. 1986;47:1376-1386.
Lemay J.F., Lambert M.A., Mitchell G.A., et al. Hyperammonemia-hyperornithinemia-homocitrullinuria syndrome: Neurologic, ophthalmologic, and neuropsychologic examination of six patients. J Pediatr. 1992;121:725.
Leonard J.V., McKiernan P.J. The role of liver transplantation in urea cycle disorders. Mol Genet Metab. 2004;81(Suppl 1):S74.
Lichter-Konecki U., Mangin J.M., Gordish-Dressman H., et al. Gene expression profiling of astrocytes from hyperammonemic mice reveals altered pathways for water and potassium homeostasis in vivo. Glia. 2008;56(4):365-377.
Lichter-Konecki U. Profiling of Astrocyte Properties in the Hyperammonemic Brain: Shedding New Light on the Pathophysiology of the Brain Damage in Hyperammonemia. J Inherit Metab Dis. 2008;31:492-502. Epub 2008 Aug 9
Linder M.. Nutrition and metabolism of proteins. Nutritional biochemistry and metabolism with clinical applications. 1985:51.
Linnebank M., Tschiedel E., Haberle J., et al. Argininosuccinate lyase (ASL) deficiency: Mutation analysis in 27 patients and a completed structure of the human ASL gene. Hum Genet. 2002;111:350.
Lusty C.J. Carbamyl phosphate synthetase. Bicarbonate-dependent hydrolysis of ATP and potassium activation. J Biol Chem. 1978;253:4270.
MacArthur R.B., Altincatal A., Tuchman M. Pharmacokinetics of sodium phenylacetate and sodium benzoate following intravenous administration as both a bolus and continuous infusion to healthy adult volunteers. Mol Genet Metab. 2004;81(Suppl 1):S67.
Maestri N.E., Hauser E.R., Bartholomew D., et al. Prospective treatment of urea cycle disorders. J Pediatr. 1991;119:923.
Maestri N.E., Clissold D., Brusilow S.W. Neonatal onset ornithine transcarbamylase deficiency: A retrospective analysis. J Pediatr. 1999;134:268.
Majoie C.B., Mourmans J.M., Akkerman E.M., et al. Neonatal citrullinemia: Comparison of conventional MR, diffusion-weighted, and diffusion tensor findings. Am J Neuroradiol. 2004;25:32.
Marcaggi P., Coles J.A. Ammonium in nervous tissue transport across cell membranes, fluxes from neurons to glial cells, and role in signalling. Prog Neurobiol. 2001;64:157.
Marescau B., De Deyn P.P., Lowenthal A., et al. Guanidino compound analysis as a complementary diagnostic parameter for hyperargininemia: Follow-up of guanidino compound levels during therapy. Pediatr Res. 1990;27:297.
McBryde K.D., Kudelka T.L., Kershaw D.B., et al. Clearance of amino acids by hemodialysis in argininosuccinate synthetase deficiency. J Pediatr. 2004;144:536.
McCormick K., Viscardi R.M., Robinson B., et al. Partial pyruvate decarboxylase deficiency with profound lactic acidosis and hyperammonemia: Responses to dichloroacetate and benzoate. Am J Med Genet. 1985;22:291.
McCullough B.A., Yudkoff M., Batshaw M.L., et al. Genotype spectrum of ornithine transcarbamylase deficiency: Correlation with the clinical and biochemical phenotype. Am J Med Genet. 2000;93:313.
McMurray W., Mohyuddin F., Rossiter RJ., et al. Citrullinuria: A new aminoaciduria associated with mental retardation. Lancet. 1962;1:138.
Morgan H.B., Swaiman K.F., Johnson B.D. Diagnosis of argininosuccinic aciduria after valproic acid–induced hyperammonemia. Neurology. 1987;37:886.
Morizono H., Caldovic L., Shi D., et al. Mammalian N-acetylglutamate synthase. Mol Genet Metab. 2004;81(Suppl 1):S4.
Morris A.A., Richmond S.W., Oddie S.J., et al. N-acetylglutamate synthetase deficiency: Favourable experience with carbamylglutamate. J Inherit Metab Dis. 1998;21:867.
Msall M., Batshaw M.L., Suss R., et al. Neurologic outcome in children with inborn errors of urea synthesis. Outcome of urea-cycle enzymopathies. N Engl J Med. 1984;310:1500.
Nagata N., Matsuda I., Matsuura T., et al. Retrospective survey of urea cycle disorders: Part 2. Neurological outcome in forty-nine Japanese patients with urea cycle enzymopathies. Am J Med Genet. 1991;40:477.
Norenberg M.D. A light and electron microscopic study of experimental portal-systemic (ammonia) encephalopathy. Progression and reversal of the disorder. Lab Invest. 1977;36:618.
Okeda R., Tanaka M., Kawahara Y., et al. Adult-type citrullinemia. Acta Neuropathol (Berl). 1989;78:96.
Palmieri L., Pardo B., Lasorsa F.M., et al. Citrin and aralar1 are Ca2+-stimulated aspartate/glutamate transporters in mitochondria. Embo J. 2001;20:5060.
Plante R.J., Tuchman M. Polymorphisms in the human ornithine transcarbamylase gene useful for allele tracking. Mutations in brief no. 193. Online. Hum Mutat. 1998;12:289.
Praphanphoj V., Boyadjiev S.A., Waber L.J., et al. Three cases of intravenous sodium benzoate and sodium phenylacetate toxicity occurring in the treatment of acute hyperammonaemia. J Inherit Metab Dis. 2000;23:129.
Prasad A.N., Breen J.C., Ampola M.G., et al. Argininemia: A treatable genetic cause of progressive spastic diplegia simulating cerebral palsy: Case reports and literature review. J Child Neurol. 1997;12:301.
Qureshi I.A., Letarte J., Ouellet R., et al. Kinetic abnormalities of carbamyl phosphate synthetase-I in a case of congenital hyperammonaemia. J Inherit Metab Dis. 1986;9:253.
Rama Rao K.V., Chen M., Simard J.M., et al. Increased aquaporin-4 expression in ammonia-treated cultured astrocytes. Neuroreport. 2003;14:2379-2382.
Rao V.L.R., Audet R.M., Butterworth R.F. Increased neuronal nitric oxides synthase expression in brain following portacaval anastomosis. Brain Res. 1997;765:169-172.
Ratnakumari L., Qureshi I.A., Butterworth R.F. Regional amino acid Neurotransmitter changes in brains of spf/Y mice with congenital ornithine transcarbamylase dificiency. Metab Brain Dis. 1994;9:43-51.
Ricciuti F.C., Gelehrter T.D., Rosenberg L.E. X-chromosome inactivation in human liver: Confirmation of X-linkage of ornithine transcarbamylase. Am J Hum Genet. 1976;28:332.
Robinson M.B., Hopkins K., Barshaw M.L., et al. Evidence of excitotoxicity in the brain of the ornithine carbamoyltransferase deficient sparse fur mouse. Brain Res Dev Brain Res. 1995;90:35.
Roe C., Ding J. Mitochondrial fatty acid oxidation disorders. In: Scriver C., Beauder A., Sly W., et al, editors. The metabolic and molecular bases of inherited diseases. ed 8. New York: McGraw-Hill; 2001:2297.
Rowe P.C., Newman S.L., Brusilow S.W. Natural history of symptomatic partial ornithine transcarbamylase deficiency. N Engl J Med. 1986;314:541.
Rubenstein J.L., Johnston K., Elliott G.R., et al. Haloperidol-induced hyperammonaemia in a child with citrullinaemia. J Inherit Metab Dis. 1990;13:754.
Rubio V., Grisolia S. Treating urea cycle defects. Nature. 1981;292:496.
Russell A., Levin B., Oberholzer V.G., et al. Hyperammonaemia. A new instance of an inborn enzymatic defect of the biosynthesis of urea. Lancet. 1962;2:699.
Ryall J., Nguyen M., Bendayan M., et al. Expression of nuclear genes encoding the urea cycle enzymes, carbamoyl-phosphate synthetase I and ornithine carbamoyl transferase, in rat liver and intestinal mucosa. Eur J Biochem. 1985;152:287.
Sacca S.C., Campagna P., Ciurlo G. Congenital glaucoma associated with an arginase deficit: A case report. Eur J Ophthalmol. 1996;6:421.
Saheki T., Kobayashi K., Ichiki H., et al. Molecular basis of enzyme abnormalities in urea cycle disorders. With special reference to citrullinemia and argininosuccinic aciduria. Enzyme. 1987;38:227.
Saheki T., Kobayashi K., Iijima M., et al. Adult-onset type II citrullinemia and idiopathic neonatal hepatitis caused by citrin deficiency: Involvement of the aspartate glutamate carrier for urea synthesis and maintenance of the urea cycle. Mol Genet Metab. 2004;81(Suppl 1):S20.
Salvi S., Dionisi-Vici C., Bertini E., et al. Seven novel mutations in the ORNT1 gene (SLC25A15) in patients with hyperornithinemia, hyperammonemia, and homocitrullinuria syndrome. Hum Mutat. 2001;18:460.
Salvi S., Santorelli F.M., Bertini E., et al. Clinical and molecular findings in hyperornithinemia-hyperammonemia-homocitrullinuria syndrome. Neurology. 2001;57:911.
Saudubray J.M., Touati G., Delonlay P., et al. Liver transplantation in urea cycle disorders. Eur J Pediatr. 1999;158(Suppl 2):S55.
Scaglia F., Carter S., O’Brien W.E., et al. Effect of alternative pathway therapy on branched chain amino acid metabolism in urea cycle disorder patients. Mol Genet Metab. 2004;81(Suppl 1):S79.
Scheuerle A.E., McVie R., Beaudet A.L., et al. Arginase deficiency presenting as cerebral palsy. Pediatrics. 1993;91:995.
Schubiger G., Bachmann C., Barben P., et al. N-acetylglutamate synthetase deficiency: Diagnosis, management and follow-up of a rare disorder of ammonia detoxication. Eur J Pediatr. 1991;150:353.
Seminara J., Tuchman M., Krivitzky L., et al. Establishing a consortium for the study of rare diseases: The Urea Cycle Disorders Consortium. Mol Genet Metab. 2010;100(Suppl 1):S97-S105. Epub 2010 Feb 10
Shchelochkov O.A., Li F.Y., Geraghty M.T., et al. High-frequency detection of deletions and variable rearrangements at the ornithine transcarbamylase (OTC) locus by oligonucleotide array CGH. Mol Genet Metab. 2009;96(3):97-105. Epub 2009 Jan 12
Shih V. Hereditary urea-cycle disorders. In: Grisolia S., Baguena R., Mayor F., editors. The urea cycle. New York: John Wiley & Sons; 1976:367.
Shih V.E., Efron M.L., Moser H.W. Hyperornithinemia, hyperammonemia, and homocitrullinuria. A new disorder of amino acid metabolism associated with myoclonic seizures and mental retardation. Am J Dis Child. 1969;117:83.
Simell O., Perheentupa J., Rapola J., et al. Lysinuric protein intolerance. Am J Med. 1975;59:229.
Sparkes R.S., Dizikes G.J., Klisak I., et al. The gene for human liver arginase (ARG1) is assigned to chromosome band 6q23. Am J Hum Genet. 1986;39:186.
Sugimoto H., Koehler R.C., Wilson D.A., et al. Methionine sulfoximine, a glutamine synthetase inhibitor, attenuates increased extracellular potassium activity during acute hyperammonemia. J Cereb Blood Flow Metab. 1997;17:44-49.
Summar M.L. Molecular genetic research into carbamoyl-phosphate synthase I: Molecular defects and linkage markers. J Inherit Metab Dis. 1998;21(Suppl 1):30.
Summar M., Pietsch J., Deshpande J., et al. Effective hemodialysis and hemofiltration driven by an extracorporeal membrane oxygenation pump in infants with hyperammonemia. J Pediatr. 1996;128:379.
Tada K., Hayasaka K. Non-ketotic hyperglycinaemia: Clinical and biochemical aspects. Eur J Pediatr. 1987;146:221.
Takanashi J., Kurihara A., Tomita M., et al. Distinctly abnormal brain metabolism in late-onset ornithine transcarbamylase deficiency. Neurology. 2002;59:210.
Takanashi J., Barkovich A.J., Cheng S.F., et al. Brain MR imaging in acute hyperammonemic encephalopathy arising from late-onset ornithine transcarbamylase deficiency. Am J Neuroradiol. 2003;24:390.
Tamamori A., Okano Y., Ozaki H., et al. Neonatal intrahepatic cholestasis caused by citrin deficiency: Severe hepatic dysfunction in an infant requiring liver transplantation. Eur J Pediatr. 2002;161:609.
Tazawa Y., Kobayashi K., Ohura T., et al. Infantile cholestatic jaundice associated with adult-onset type II citrullinemia. J Pediatr. 2001;138:735.
Terheggen H., Lowenthal A., Lavinha F. Arginemia with arginase deficiency. Lancet. 1969;2:748.
Tsacopoulos M., Magistretti P.J. Metabolic coupling between glia and neurons. J Neurosci. 1996;16:877-885.
Tuchman M. The clinical, biochemical, and molecular spectrum of ornithine transcarbamylase deficiency. J Lab Clin Med. 1992;120:836.
Tuchman M., Morizono H., Rajagopal B.S., et al. The biochemical and molecular spectrum of ornithine transcarbamylase deficiency. J Inherit Metab Dis. 1998;21(Suppl 1):40.
Tuchman M., Jaleel N., Morizono H., et al. Mutations and polymorphisms in the human ornithine transcarbamylase gene. Hum Mutat. 2002;19:93.
Tuchman M., Lee B., Lichter-Konecki U., et al. Additional members of Urea Cycle Disorders Consortium of the Rare Diseases Clinical Research Network (2008) Cross-sectional multicenter study of patients with urea cycle disorders in the United States. Mol Genet Metab. 2008;94(4):397-402. Epub 2008 Jun 17
Uchino T., Endo F., Matsuda I. Neurodevelopmental outcome of long-term therapy of urea cycle disorders in Japan. J Inherit Metab Dis. 1998;21(Suppl 1):151.
Vockley J.G., Jenkinson C.P., Shukla H., et al. Cloning and characterization of the human type II arginase gene. Genomics. 1996;38:118.
Widhalm K., Koch S., Scheibenreiter S., et al. Long-term follow-up of 12 patients with the late-onset variant of argininosuccinic acid lyase deficiency: No impairment of intellectual and psychomotor development during therapy. Pediatrics. 1992;89:1182.
Willard-Mack C.L., Koehler R.C., Hirata T., et al. Inhibition of glutamine synthetase reduces ammonia-induced astrocyte swelling in rat. Neuroscience. 1996;71:589.
Yamanouchi H., Yokoo H., Yuhara Y., et al. An autopsy case of ornithine transcarbamylase deficiency. Brain Dev. 2002;24:91.
Yazaki M., Hashikura Y., Takei Y., et al. Feasibility of auxiliary partial orthotopic liver transplantation from living donors for patients with adult-onset type II citrullinemia. Liver Transpl. 2004;10:550.
Yu B., Thompson G.D., Yip P., et al. Mechanisms for intragenic complementation at the human argininosuccinate lyase locus. Biochemistry. 2001;40:15581.
Zammarchi E., Ciani F., Pasquini E., et al. Neonatal onset of hyperornithinemia-hyperammonemia-homocitrullinuria syndrome with favorable outcome. J Pediatr. 1997;131:440.
Zecavati N., Lichter-Konecki U., Singh R., et al. Seizures in Urea Cycle Disorders (UCDs): An under-recognized symptom in patients outside of the acute metabolic phase. American Society of Human Genetics 58th Annual Meeting. 2008. Philadelphia
Zeviani M., Bonilla E., DeVivo D.C., et al. Mitochondrial diseases. Neurol Clin. 1989;7:123.
Zimmermann A., Bachmann C., Baumgartner R. Severe liver fibrosis in argininosuccinic aciduria. Arch Pathol Lab Med. 1986;110:136.