[level-membership-for-basic-science-category]
Immunochemical Methods Used for Organism Detection
1. List the reasons a laboratory or clinician would use immunochemical tests to diagnose disease.
2. Define a polyclonal antibody and a monoclonal antibody and explain the difference between the two.
3. Explain how monoclonal antibodies are produced. How has their development affected immunochemical testing?
4. Define the four types of immunochemical testing—precipitation, particle agglutination, immunofluorescent assays, and enzyme immunoassays—and provide a clinical application for each.
5. Explain the difference between a direct fluorescent antibody (DFA) test and an indirect fluorescent antibody (IFA) test and explain how each is used in the clinical laboratory.
6. Explain the function of the hypoxanthine, aminopterin, and thymidine (HAT) medium in hybridoma production.
The diagnosis of an infectious disease by culture and biochemical techniques can be hindered by several factors. These factors include the inability to cultivate an organism on artificial media, such as Treponema pallidum, the agent that causes syphilis, or the fragility of an organism and its subsequent failure to survive transport to the laboratory, such as with respiratory syncytial virus and varicella-zoster virus. Another factor, the fastidious nature of some organisms (e.g., Leptospira or Bartonella spp.) can result in long incubation periods before growth is evident. In addition, administration of antimicrobial therapy before specimen collection, such as with a patient who has received partial treatment, can impede diagnosis. In these cases, detecting a specific product of the infectious agent in clinical specimens is very important, because this product would not be present in the specimen in the absence of the agent. This chapter discusses the direct detection of microorganisms in patient specimens using immunochemical methods and the identification of microorganisms by these methods once they have been isolated on laboratory media. Chapter 10 discusses the diagnosis of infectious diseases using serological methods.
Production of Antibodies for Use in Laboratory Testing
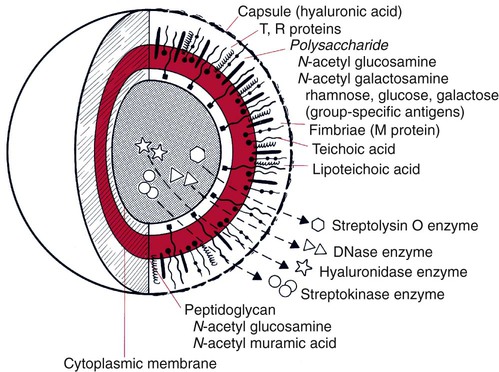
Immunochemical methods use antigens and antibodies as tools to detect microorganisms. Antigens are substances recognized as “foreign” in the human body. Antigens are usually high-molecular-weight proteins or carbohydrates that elicit the production of other proteins, called antibodies, in a human or animal host (see Chapter 3). Antibodies attach to the antigens and aid the host in removing the infectious agent (see Chapters 3 and 10). Antigens may be part of the physical structure of the pathogen, such as the bacterial cell wall, or they may be a chemical produced and released by the pathogen, such as an enzyme or a toxin. Each antigen contains a region that is recognized by the immune system. These regions are referred to as antigenic determinants or epitopes. Figure 9-1 shows the multiple molecules within group A Streptococcus (Streptococcus pyogenes) that are recognized by the immune system as antigenic.
Monoclonal Antibodies
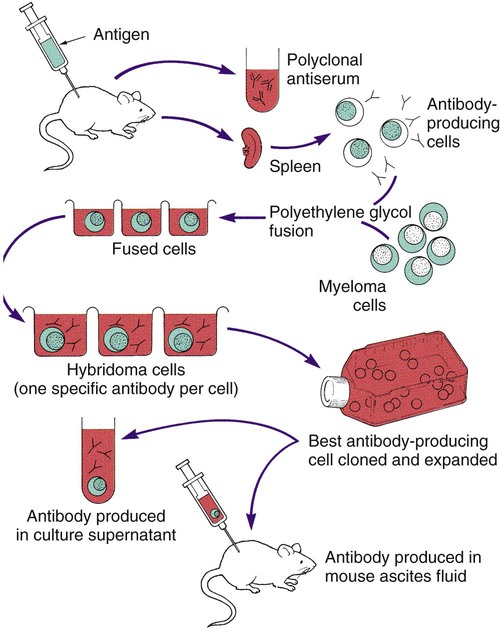
Monoclonal antibodies are antibodies that are completely characterized and highly specific. The ability to create an immortal cell line that produces large quantities of a monoclonal antibody has revolutionized immunologic testing. Monoclonal antibodies are produced by the fusion of a malignant single antibody-producing myeloma cell with an antibody-producing plasma B cell, forming a hybridoma cell. Clones of the hybridoma cells continuously produce specific monoclonal antibodies. One technique for the production of a clone of cells is illustrated in Figure 9-2.
Principles of Immunochemical Methods Used for Organism Detection
Precipitation Tests
Double Immunodiffusion
In the double immunodiffusion method, small circular wells are cut in an agarose gel, a gelatin-like matrix derived from agar, which is a chemical purified from the cell walls of brown algae. The agarose forms a porous material through which molecules can readily diffuse. The patient specimen containing antigen is placed in a well, and antibody directed against the antigen is placed in the adjacent well. Over 18 to 24 hours, the antigen and antibody diffuse toward each other, producing a visible precipitin band (a lattice structure or visible band) at the point in the gel where the antigen and antibody are in equal proportion (zone of equivalence). If the concentration of antibody is significantly higher than that of the antigen, no lattice forms and no precipitation reaction occurs; this is known as prozone effect. Conversely, if excess antigen prevents lattice formation, resulting in no band formation, the effect is termed postzone. Immunodiffusion is currently used to detect exoantigens produced by the systemic fungi to confirm their presence in culture (Figure 9-3). However, the technique is extremely time-consuming and is no longer used regularly in the clinical laboratory for antigen detection in patient specimens.
Particle Agglutination
Latex Agglutination
Antibody molecules can be bound in random alignment to the surface of latex (polystyrene) beads (Figure 9-4). The number of antibody molecules bound to each latex particle is large, resulting in a high number of exposed potential antigen binding sites. Antigen present in a specimen binds to the combining sites of the antibody exposed on the surfaces of the latex beads, forming cross-linked aggregates of latex beads and antigen. The size of the latex bead (0.8 µm or larger) enhances the ease with which the agglutination reaction is visualized. Levels of bacterial polysaccharides detected by latex agglutination have been shown to be as low as 0.1 ng/mL.
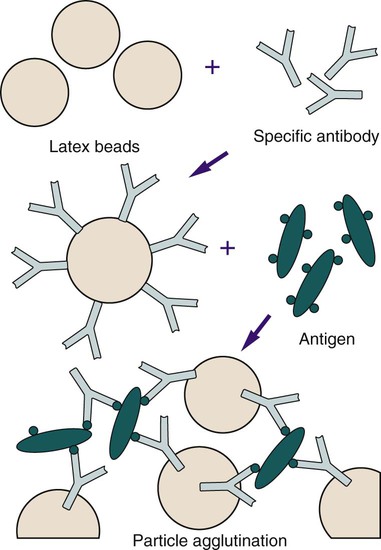
Latex tests are very popular in clinical laboratories for detecting antigen to Cryptococcus neoformans in cerebrospinal fluid or serum (Figure 9-5) and to confirm the presence of beta-hemolytic Streptococcus from culture plates (Figure 9-6). Latex tests are continually being developed for a variety of organisms. Some examples of additional latex tests are available for the detection of Clostridium difficile toxins A and B, rotavirus, and Escherichia coli 0157:H7 from suspect colonies of E coli.


Coagglutination
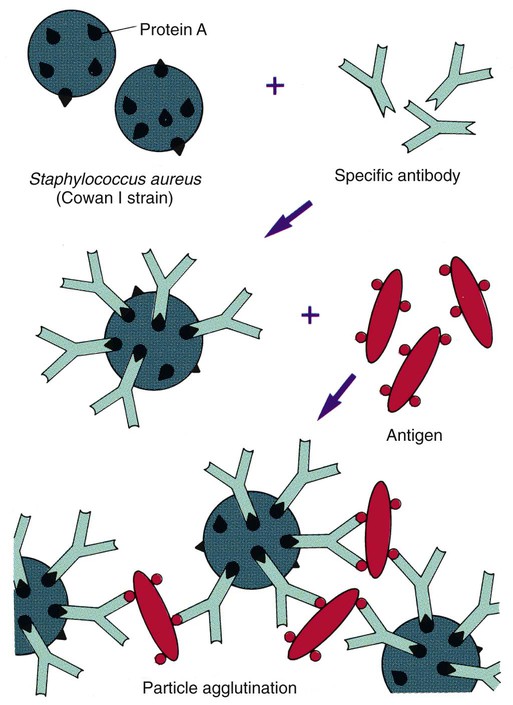
Similar to latex agglutination, coagglutination uses antibody bound to a particle to enhance the visibility of the agglutination reaction between antigen and antibody. In this case the particles are killed and treated S. aureus organisms (Cowan I strain), which contain a large amount of an antibody-binding protein, protein A, in their cell walls. In contrast to latex particles, these staphylococci bind only the base of the heavy chain portion of the antibody, leaving both antigen-binding ends free to form complexes with specific antigen (Figure 9-7). Several commercial suppliers have prepared coagglutination reagents for identification of streptococci, including Lancefield groups A, B, C, D, F, G, and N; Streptococcus pneumoniae; Neisseria meningitidis; and Haemophilus influenzae types A to F grown in culture. The coagglutination reaction is highly specific and demonstrates reduced sensitivity in comparison to commercially prepared latex agglutination systems. Therefore, coagglutination is not usually used for direct antigen detection.
Immunofluorescent Assays
Immunofluorescent assays are frequently used for detecting bacterial and viral antigens in clinical laboratories. In these tests, antigens in the patient specimens are immobilized and fixed onto glass slides with formalin, methanol, ethanol, or acetone. Monoclonal or polyclonal antibodies conjugated (attached) to fluorescent dyes are applied to the specimen. After appropriate incubation, washing, and counterstaining (staining of the background with a nonspecific fluorescent stain such as rhodamine or Evan’s blue), the slide is viewed using a microscope equipped with a high-intensity light source (usually halogen) and filters to excite the fluorescent tag. Most kits used in clinical microbiology laboratories use fluorescein isothiocyanate (FITC) as the fluorescent dye. FITC fluoresces a bright apple-green (Figure 9-8).
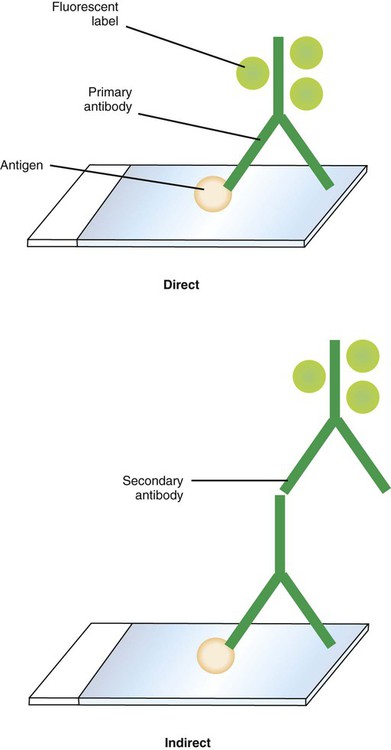
Fluorescent antibody tests are performed using either a direct fluorescent antibody (DFA) or and indirect fluorescent antibody (IFA) technique (Figure 9-9). In the DFA technique, FITC is conjugated directly to the specific antibody. In the IFA technique, the antigen-specific antibody is unlabeled, and a second antibody (usually raised against the animal species from which the antigen-specific antibody was harvested) is conjugated to the FITC. The IFA is a two-step, or sandwich, technique. The IFA technique is more sensitive than the DFA method, although the DFA method is faster because it involves a single incubation.
Enzyme Immunoassays
Solid-Phase Immunoassay
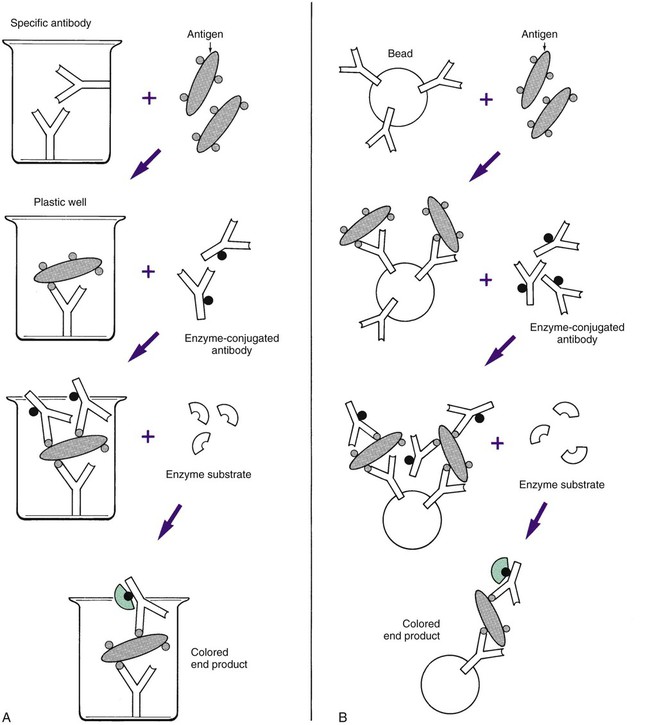
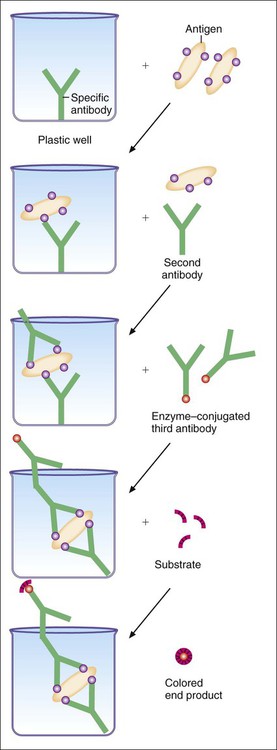
Most ELISA systems developed to detect infectious agents consist of antibody firmly fixed to a solid matrix, either the inside of the wells of a microdilution tray or the outside of a spherical plastic or metal bead or some other solid matrix (Figure 9-10). Such systems are called solid-phase immunosorbent assays (SPIA). If antigen is present in the specimen, stable antigen-antibody complexes form when the sample is added to the matrix. Unbound antigen is thoroughly removed by washing, and a second antibody against the antigen is then added to the system. This antibody has been complexed to an enzyme such as alkaline phosphatase or horseradish peroxidase. If the antigen is present on the solid matrix, it binds the second antibody, forming a sandwich with antigen in the middle. After washing has removed unbound, labeled antibody, the addition and hydrolysis of the enzyme substrate causes the color change and completes the reaction. The visually detectable end point appears wherever the enzyme is present (Figure 9-11). Because of the expanding nature of the reaction, even minute amounts of antigen (greater than 1 ng/mL) can be detected. These systems require a specific enzyme-labeled antibody for each antigen tested. However, it is simpler to use an indirect assay in which a second, unlabeled antibody is used to bind to the antigen-antibody complex on the matrix. A third antibody, labeled with enzyme and directed against the nonvariable Fc portion of the unlabeled second antibody, can then be used as the detection marker for many different antigen-antibody complexes (Figure 9-12). ELISA systems are important diagnostic tools for hepatitis Bs (surface) and hepatitis Be (early) antigens and HIV p24 protein, all indicators of early, active, acute infection.
Membrane-Bound SPIA
The flow-through and large surface area characteristics of nitrocellulose, nylon, and other membranes have been exploited to enhance the speed and sensitivity of ELISA reactions. An absorbent material below the membrane pulls the liquid reactants through the membrane and helps to separate nonreacted components from the antigen-antibody complexes bound to the membrane; washing steps are also simplified. Membrane-bound SPIA systems are available for several viruses (Figure 9-13), group A beta-hemolytic streptococci antigen directly from throat swabs, and group B streptococcal antigen in vaginal secretions. In addition to their use in clinical laboratories, these assays are expected to become more prevalent for home testing systems.
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
Immunochemical Methods Used for Organism Detection
1. List the reasons a laboratory or clinician would use immunochemical tests to diagnose disease.
2. Define a polyclonal antibody and a monoclonal antibody and explain the difference between the two.
3. Explain how monoclonal antibodies are produced. How has their development affected immunochemical testing?
4. Define the four types of immunochemical testing—precipitation, particle agglutination, immunofluorescent assays, and enzyme immunoassays—and provide a clinical application for each.
5. Explain the difference between a direct fluorescent antibody (DFA) test and an indirect fluorescent antibody (IFA) test and explain how each is used in the clinical laboratory.
6. Explain the function of the hypoxanthine, aminopterin, and thymidine (HAT) medium in hybridoma production.
The diagnosis of an infectious disease by culture and biochemical techniques can be hindered by several factors. These factors include the inability to cultivate an organism on artificial media, such as Treponema pallidum, the agent that causes syphilis, or the fragility of an organism and its subsequent failure to survive transport to the laboratory, such as with respiratory syncytial virus and varicella-zoster virus. Another factor, the fastidious nature of some organisms (e.g., Leptospira or Bartonella spp.) can result in long incubation periods before growth is evident. In addition, administration of antimicrobial therapy before specimen collection, such as with a patient who has received partial treatment, can impede diagnosis. In these cases, detecting a specific product of the infectious agent in clinical specimens is very important, because this product would not be present in the specimen in the absence of the agent. This chapter discusses the direct detection of microorganisms in patient specimens using immunochemical methods and the identification of microorganisms by these methods once they have been isolated on laboratory media. Chapter 10 discusses the diagnosis of infectious diseases using serological methods.
Production of Antibodies for Use in Laboratory Testing
Immunochemical methods use antigens and antibodies as tools to detect microorganisms. Antigens are substances recognized as “foreign” in the human body. Antigens are usually high-molecular-weight proteins or carbohydrates that elicit the production of other proteins, called antibodies, in a human or animal host (see Chapter 3). Antibodies attach to the antigens and aid the host in removing the infectious agent (see Chapters 3 and 10). Antigens may be part of the physical structure of the pathogen, such as the bacterial cell wall, or they may be a chemical produced and released by the pathogen, such as an enzyme or a toxin. Each antigen contains a region that is recognized by the immune system. These regions are referred to as antigenic determinants or epitopes. Figure 9-1 shows the multiple molecules within group A Streptococcus (Streptococcus pyogenes) that are recognized by the immune system as antigenic.
Monoclonal Antibodies
Monoclonal antibodies are antibodies that are completely characterized and highly specific. The ability to create an immortal cell line that produces large quantities of a monoclonal antibody has revolutionized immunologic testing. Monoclonal antibodies are produced by the fusion of a malignant single antibody-producing myeloma cell with an antibody-producing plasma B cell, forming a hybridoma cell. Clones of the hybridoma cells continuously produce specific monoclonal antibodies. One technique for the production of a clone of cells is illustrated in Figure 9-2.
Principles of Immunochemical Methods Used for Organism Detection
Precipitation Tests
Double Immunodiffusion
In the double immunodiffusion method, small circular wells are cut in an agarose gel, a gelatin-like matrix derived from agar, which is a chemical purified from the cell walls of brown algae. The agarose forms a porous material through which molecules can readily diffuse. The patient specimen containing antigen is placed in a well, and antibody directed against the antigen is placed in the adjacent well. Over 18 to 24 hours, the antigen and antibody diffuse toward each other, producing a visible precipitin band (a lattice structure or visible band) at the point in the gel where the antigen and antibody are in equal proportion (zone of equivalence). If the concentration of antibody is significantly higher than that of the antigen, no lattice forms and no precipitation reaction occurs; this is known as prozone effect. Conversely, if excess antigen prevents lattice formation, resulting in no band formation, the effect is termed postzone. Immunodiffusion is currently used to detect exoantigens produced by the systemic fungi to confirm their presence in culture (Figure 9-3). However, the technique is extremely time-consuming and is no longer used regularly in the clinical laboratory for antigen detection in patient specimens.

[/not-level-membership-for-basic-science-category]
