Chapter 76 Hypoxic-Ischemic Encephalopathy in Infants and Older Children
Cardiac arrest in childhood is often a catastrophic event that is associated with mortality or poor neurologic outcome in the in-hospital or out-of-hospital setting [Moler et al., 2009]. Because of the dismal neurologic outcomes associated with cardiac arrest, especially in the out-of-hospital setting, interventions to ameliorate hypoxic-ischemic encephalopathy following cardiac arrest have been sought for decades. This chapter reviews the pathophysiologic and clinical aspects of hypoxic-ischemic encephalopathy, primarily as it applies to older children. Chapter 17 discusses hypoxic-ischemic injury in newborns.
Cardiac Arrest: Etiology, Survival, and Neurological Outcome
Recent reports from the American Heart Association National Registry of Cardiopulmonary Resuscitation and from the Pediatric Emergency Care Applied Research Network have provided extensive information concerning clinical characteristics, hospital courses, and outcomes for children who experience in-hospital cardiac arrests [Meert et al., 2009; Nadkarni et al., 2006]. A multicenter study of patients who had a sustained return of circulation after in-hospital cardiac arrest showed a survival to hospital discharge of 48.7 percent [Meert et al., 2009]. Of those who survived to discharge, 76.7 percent had a good neurologic outcome.
In contrast, a recent large prospective pediatric cohort study revealed that overall survival from an out-of-hospital cardiac arrest was only 6.4 percent [Atkins et al., 2009]. Similarly, Berg et al. [2008] reported only a 10 percent survival to discharge following an out-of-hospital cardiac arrest. Young et al. [2004] also reported a low survival to hospital discharge of pediatric out-of-hospital arrest patients (8.6 percent), and demonstrated that only one-third survivors had good neurologic outcomes.
For the first time, a multicenter cohort study was conducted in children with cardiac arrest and return of circulation that compares and contrasts major clinical features of in-hospital and out-of-hospital cardiac arrest [Moler et al., 2009]. This study reported pre-arrest characteristics, post-arrest interventions, mortality, and neurological outcome, and concluded that these represent discrete entities with respect to cause, first monitored rhythm recorded, mortality during resuscitation and in the post-cardiac arrest period, and neurological outcome. Differences in outcome between in-hospital and out-of-hospital cardiac arrest may simply represent a shorter duration of no flow and higher-quality cardiopulmonary resuscitation (CPR) for patients after in-hospital than after out-of-hospital cardiac arrest [Manole et al., 2009]. Thus, when evaluating different therapies, these groups of patients should be analyzed separately, as they represent different degrees of disease severity. A comparison of these two groups is presented in Table 76-1 and Table 76-2.
Table 76-1 Comparison of the Etiology of Cardiac Arrest for In-Hospital and Out-of-Hospital Cohorts*†
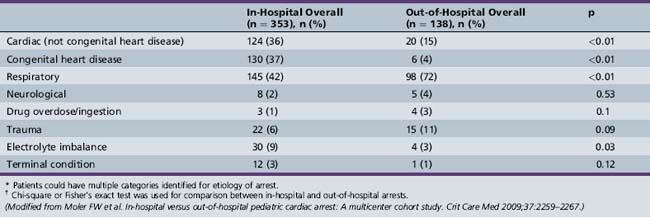
Table 76-2 Characteristics of Successfully Resuscitated Pediatric Patients with In-Hospital or Out-of-Hospital Cardiac Arrest
| Out-of-Hospital Cardiac Arrest | In-Hospital Cardiac Arrest | |
|---|---|---|
| Initial rhythm (%) | Asystolic (46) | Bradycardia (49) |
| Bradycardia (10) | Asystole (16) | |
| VF/VT (7) | VF/VT (10) | |
| CPR duration (median, min) | 31 | 9 |
| Cause (%) | Respiratory (72) | Cardiac (73) |
| Pre-existing chronic condition | 49 | 88 |
| Good neurologic outcome | 24 | 47 |
| Mortality rate (%) | 62 | 51 |
| Neurologic injury as the cause of mortality | 69 | 20 |
CPR, cardiopulmonary resuscitation; VF, ventricular fibrillation; VT, ventricular tachycardia.
(Modified from Moler FW et al. In-hospital versus out-of-hospital pediatric cardiac arrest: A multicenter cohort study. Crit Care Med 2009;37:2259–2267.)
Favorable neurological outcome is more often achieved in patients with in-hospital cardiac arrest as compared with out-of-hospital cardiac arrest (47 versus 24 percent), in spite of patients with in-hospital cardiac arrest having a higher frequency of abnormal pre-arrest neurological status (34 versus 17 percent) and chronic pre-existing conditions (88 versus 49 percent) (see Table 76-2). Also, neurologic injury was the main cause of death in the out-of-hospital cohort, with mortality being significantly greater in this group (69 versus 20 percent), whereas neurologic injury as the cause of death was uncommon in the in-hospital cohort.
Cardiac arrest is not a rare event during childhood for either in-hospital or out-of-hospital patients [Berg et al., 2008]. The incidence of cardiac arrests in infants approaches that of adults and is higher as compared with children and adolescents [Manole et al., 2009; Atkins et al., 2009]. Despite still being poor, pediatric cardiac arrest outcomes have been improving mainly because of increased understanding of its pathophysiology, expansion of resuscitation training programs, and improvement in resuscitation techniques [Fioretto, 2009; Berg et al., 2008; Fuchs, 2008]. Also, post-arrest care has evolved and substantial attention has been focused on this period, as neurologic injury is one of the most important complications [Fioretto, 2009; Manole et al., 2009].
Post-Cardiac Arrest Syndrome
Pediatric cardiac arrest results from asphyxia in the majority of cases [Manole et al., 2009; Nadkarni et al., 2006]. It is a distinct entity from adult cardiac arrest, which is predominantly due to ventricular fibrillation. The morphological pattern of brain damage is different in asphyxial and ventricular fibrillation cardiac arrest due to the differences in patterns of cerebral blood flow [Vaagenes et al., 1997].
Cardiac arrests in children generally result from progressive tissue hypoxia and acidosis due to respiratory failure, circulatory shock, or both [Nadkarni et al., 2006; Young et al., 2004]. Electrocardiographic rhythms of cardiac arrests in children usually progress through bradyarrhythmias to asystole or pulseless electrical activity rather than to ventricular fibrillation. Although the outcomes from cardiac arrhythmias or shock in children are generally good, the outcomes from pulseless cardiac arrests in children are poor [Nadkarni et al., 2006; Young et al., 2004].
Post-cardiac arrest syndrome includes brain injury and myocardial dysfunction, as well as systemic ischemia and reperfusion [Manole et al., 2009; Adrie et al., 2002]. The four phases of resuscitation in cardiac arrest are: pre-arrest (events leading to cardiac arrest, i.e., hypoxia, bradycardia, hypovolemia, arrhythmia), arrest (no flow), resuscitation (chest compressions: low or no flow, and assisted ventilation), and recovery after resuscitation [Topjian et al., 2008; Yannopoulos et al., 2010].
Therapy targeting each of these phases has a defined goal; interventions in the early phases focus on limiting further injury and vital support, whereas those in the later phases focus on prognostication and finally rehabilitation [Nolan et al., 2008].
Response to Inadequate Oxygen Delivery: Mechanisms of Brain Injury
Asphyxia produces a state of hypoxemic hypotensive perfusion before cardiac arrest. Pediatric and neonatal models of hypoxia-ischemia and asphyxia exist so that therapies, brain tissue monitoring, cerebral blood flow determinations, and outcomes can be studied [Manole et al., 2009; Fink et al., 2004]. Data related to mechanisms of brain damage from children after cardiac arrest are limited. During ischemia, energy stores of the brain are depleted, and toxic metabolites accumulate (lactate and hydrogen ion). Upon reperfusion, injury to the brain is secondary to excitotoxicity, calcium accumulation, protease activation, and formation of reactive oxygen and nitrogen species. The mechanism of neuronal damage after cardiac arrest is a combination of necrosis, apoptosis, and autophagy, along with inflammation. Necrosis is a process characterized by immediate mitochondrial and energy failure, leading to cellular swelling, loss of cell membrane integrity, and a prominent inflammatory response in surrounding tissue. Apoptosis is an energy-requiring process that generally needs new protein synthesis. Enzymatic degradation of cytoskeletal proteins results in cell somal and nuclear shrinkage, and DNA is characteristically fragmented via endonucleases. In contrast to necrosis, apoptosis produces minimal inflammation and autophagic stress, and also results in cell death. Autophagy is an adaptive response to starvation, and results in autodigestion of cellular proteins and organelles to feed the cell. Triggering of autophagy after acute insults could potentially be beneficial or detrimental, likely depending upon the degree or duration of injury [Fink et al., 2008]. The role of autophagy after cerebral ischemia is currently under investigation [Hotchkiss et al., 2009].
Brain Energy Failure
The brain depends on large amounts of exogenous substrate (oxygen and glucose) because of its high metabolic demands, limited energy stores, and reliance on oxidative metabolism. Cellular energy depletion is postulated to be the triggering event that initiates the many cascades of injury that occur during ischemia [Krause et al., 1988]. Adenosine 5-triphosphate (ATP) is the energy source that drives all cellular physiologic processes [Bellamy et al., 1996; Sweeney et al., 1995]. One of the most vital processes performed is the preservation of ATP to maintain membrane ionic gradients [Ackerman and Clapham, 1997]. Under normal conditions, maintenance of ionic gradients across the cell membrane accounts for nearly 50 percent of total cellular expenditure [Erecinska and Silver, 1989]. Interruption of cerebral blood flow results in loss of consciousness and reductions in electroencephalographic (EEG) activity within seconds. Within 5–7 minutes, complete energy failure occurs, accompanied by disturbances of neuronal and glial ion homeostasis, including sodium and water influx and efflux of potassium (Figure 76-1). When the extracellular potassium concentration reaches a critical threshold, voltage-gated channels undergo depolarization, precipitating extracellular calcium influx [Sweeney et al., 1995]. If flow remains inadequate and energy failure persists, calcium-mediated events, such as phospholipase and protease activation, can lead to irreversible cellular injury and necrosis, as well as cerebral acidosis with elevated lactate levels and decreased hydrogen ion concentration (pH). If blood flow is restored, recovery of basal cellular metabolism (ATP levels, protein synthesis, oxygen consumption) and normal pH occurs if the ischemic injury is of limited duration [Ljunggren et al., 1974].
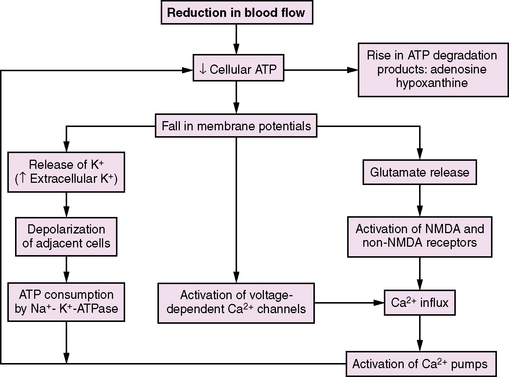
Certain neurons have long been known to be especially vulnerable to global hypoxic-ischemic insults, particularly the hippocampal CA1 and CA4 zones, and cortical layers III and V [Krause et al., 1988; White et al., 1996]. Five minutes of complete global brain ischemia produces cell death in these regions within 48–72 hours. This time interval between ischemia and cell death consists of an early postischemic period when ATP and lactate levels recover, followed, after 24 hours, by a decrease in ATP levels and lactate re-accumulation [Kochanek, 1993; Siesjo et al., 1995].
After anoxia or ischemia, restoration of aerobic metabolism is essential but not sufficient for recovery. Although the restoration of brain perfusion (as occurs with successful CPR) will re-establish energy stores, cell injury and death continue in a process known as reperfusion injury [Hoesch et al., 2008]. Mechanisms of reperfusion injury include lipid peroxidation and other damage caused by oxygen free radicals that accumulate after reintroduction of oxygen to the ischemic region, and neuronal damage mediated by inflammatory cells. Reperfusion is also characterized by continued activation of extracellular glutamate and intracellular calcium-dependent enzyme systems, calcium-dependent gene expression, caspase activation and immune-mediated damage by microglia [Hoesch et al., 2008].
Despite global metabolic recovery, certain neurons progress to cell death. Restoration of blood flow and oxidative metabolism after energy failure in the brain leads to specific cellular and molecular dysfunction because of two related processes – cell necrosis and apoptosis – both of which evolve during and after resuscitation [Hockenbery, 1995; Schnaper, 1994]. These processes, coupled with the need to restore highly integrated functions, explain the unfortunate clinical scenario of the vegetative state despite restoration of normal function in other organ systems. The relation of these two forms of cell death to selective vulnerability of neurons in the brain is beginning to emerge.
Cell necrosis, which is characterized by denaturation and coagulation of cellular proteins, is the basic pattern of pathological cell death and results from progressive reduction in cellular ATP content [Buja et al., 1993; Sweeney et al., 1995]. Necrosis involves progressive derangements in energy and substrate metabolism that are followed by a series of morphologic alterations, including swelling of cells and organelles, development of subsurface cellular blebs, amorphous deposits in mitochondria, condensation of nuclear chromatin, and breaks in plasma and cell organelle membranes [Brierley et al., 1973]. It was traditionally assumed that an ischemic cell death occurred through this process and this selective vulnerability represented a specific predilection for the development of necrosis in certain neurons after transient ischemic insults.
It has been demonstrated, however, that cell death after hypoxic-ischemic insults can occur by a second pathway, apoptosis. Programmed cell death is important during development, in normal physiology, and in the pathogenesis of disease it is called apoptosis [Schnaper, 1994]. Apoptosis has been described as directed cell suicide [Hockenbery, 1995]. Its unique feature is that, although external events may trigger apoptosis, all of the machinery for the process is contained within the cell. Activation of this machinery may result from toxicity of the extracellular milieu, binding to a specific cell receptor, or removal of a factor that prevents apoptosis from occurring. Apoptosis is thus clearly distinct from necrosis. Necrosis occurs despite the cell’s effort to survive. The development of apoptosis involves new protein synthesis and the activation of endonucleases with a resultant characteristic cleavage of DNA at linkage regions between nucleosomes to form fragments of double-stranded DNA [Mackey et al., 1997].
The stimuli triggering apoptosis are not clearly defined, although protease activation or oxidant injury to DNA has been proposed. Reports indicate that cell death in selectively vulnerable brain regions, such as the CA1 region of the hippocampus, after transient global brain ischemia, occurs by an apoptotic mechanism. After a global ischemic insult, DNA fragmentation is most pronounced in neurons of the CA1 region of the hippocampus, which suggests that apoptosis may play a role in both selective neuronal necrosis and delayed neuronal death [Nitatori et al., 1995]. The concept of delayed neuronal death is important because it implies that there is a window of opportunity for treatment after global ischemia.
Apoptosis in the post-ischemic brain is not limited to scattered neuronal death in what has traditionally been deemed selectively vulnerable regions; it is also seen in penumbral regions around evolving cerebral infarctions [Li et al., 1995]. The severity of the ischemic insult and other local factors likely determine whether an injured neuron recovers, undergoes programmed cell death, or dies a necrotic death. It is quite possible, although only a speculative notion, that after cardiopulmonary arrest and resuscitation, a continuum exists in neurons from recovery to necrosis that depends on the duration of the insult, the local milieu, and the given brain region. None the less, in any given brain region, whether neuronal death is produced by necrosis, apoptosis, or both, a highly complex series of events is involved during the arrest and after restoration of spontaneous circulation. Because apoptosis occurs in stages, there exist several potential strategies for reducing cell death [Kochanek et al., 2001]. See chapters 13–15 for additional discussion of these mechanisms.
Calcium-Mediated Injury
Calcium plays a strategic role in regulation of many cellular metabolic processes; therefore, the concentration of cytosolic free calcium is tightly controlled. Hypoxic-ischemic injury interrupts intracellular calcium homeostasis, which results in massive increases in the intracellular concentration of calcium. This calcium accumulation is believed to promote irreversible cellular injury [Bellamy et al., 1996; Lipton and Rosenberg, 1994] (Figure 76-2).
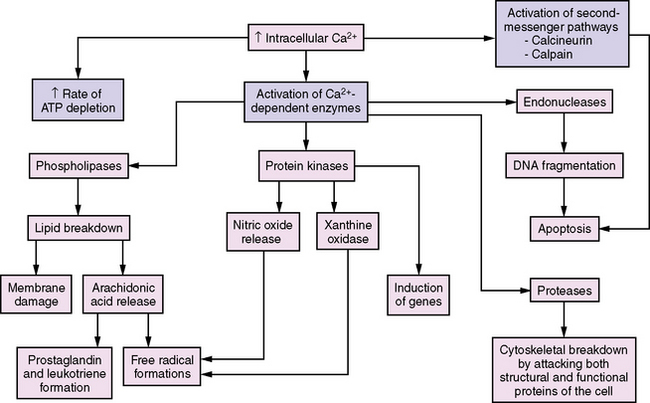
Fig. 76-2 Increases in intracellular Ca2+ mediate several enzymatic pathways that cause cell injury.
Transient calcium accumulation occurs in all cells during ischemia, but secondary irreversible accumulation occurs in the selectively vulnerable zones many hours later. Electrophysiologic studies demonstrate that delayed neuronal death is preceded by neuronal hyperactivity. It is hypothesized that ischemic and early post-ischemic calcium accumulation leads to complex series of derangements in cellular metabolism [Morgenstern and Pettigrew, 1997; Sweeney et al., 1995]. The intracellular accumulation of calcium:
Excitotoxic Injury
During hypoxic-ischemic damage, pathologically prolonged membrane depolarization occurs and, in certain neuronal populations, leads to excessive release of neurotransmitters into the synaptic cleft [Lipton and Rosenberg, 1994; Rogers and Kirsch, 1989]. The effect of these neurotransmitters is prolonged by failure of the ATP-dependent presynaptic reuptake mechanisms. Glutamate and aspartate are the major excitatory amino acid neurotransmitters in the mammalian central nervous system (CNS); both also have neurotoxic properties [Rogers and Kirsch, 1989]. Hypoxia-induced neuronal death is mediated by synaptic activity. Inhibition of synaptic glutamate release or blockade of glutamate receptors may prevent hypoxic neuronal injury. Glutamate is the major neurotransmitter in the selectively vulnerable zones and accumulates extracellularly in these regions after hypoxic or ischemic insults. The mechanisms by which glutamate may harm neurons during ischemia and reperfusion are becoming more clearly defined [Kalia et al., 2008].
Glutamate is released at the presynaptic terminal in response to neuronal stimulation and acts by binding to postsynaptic dendritic receptors. Two main classes of excitatory neurotransmitter receptors have been identified. One class consists of the ligand-gated ion channels (ionotropic receptors) and includes N-methyl-d-aspartate (NMDA); alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA), or quisqualate; and kainate receptor types [Rogers and Kirsch, 1989; Sweeney et al., 1995]. Toxicity caused by NMDA receptor activation is usually rapid, whereas AMPA or kainate receptor-mediated cell death is slower to develop [Dugan and Choi, 1994]. The current understanding of the glutamate receptor subtypes is that the NMDA receptor is more complex and contains more modulatory sites than does the kainate receptor [Lipton and Rosenberg, 1994]. The other class of excitatory neurotransmitter receptors includes the metabotropic receptors that are coupled with G proteins and modulate intracellular second messengers such as calcium, cyclic nucleosides, and inositol triphosphate. Seven subtypes of the metabotropic glutamate receptor have been identified [Nakanishi, 1992].
When activated, the ionotropic glutamate receptors open sodium channels and may also have an important role in initiation and propagation of membrane depolarization and spreading depression. With inotropic receptor activation, rapid excitatory amino acid-mediated calcium accumulation occurs [Sweeney et al., 1995]. In the presence of ischemia, this calcium accumulation is exacerbated by cellular energy failure, which disables the Na+/K+-ATPase membrane pump and results in further calcium accumulation. Altered calcium homeostasis leads to activation of many deleterious processes, including activation of phospholipases, proteases, endonucleases, protein kinases, and calmodulin-regulated enzymes such as nitric oxide synthase. There is evidence that nitric oxide is an important mediator of glutamate neurotoxicity [Dawson, 1994]. Re-establishment of the energy supply can reverse these changes. Delayed glutamate-related neuronal injury is most likely caused by activation of inotropic receptors and subsequent calcium influx.
Intracellular free calcium may increase during ischemia either by influx of extracellular calcium or by release of bound or sequestered intracellular calcium [Morgenstern and Pettigrew, 1997]. Cerebral ischemia is accompanied by a significant decrease in extracellular calcium concentration, which is consistent with an intracellular shift of calcium. Energy failure during ischemia results in membrane depolarization, which allows calcium influx via voltage-sensitive calcium channels and causes glutamate release. Glutamate can promote calcium influx by three different mechanisms [Haun et al., 1996]. The first and most obvious mechanism is by opening the NMDA receptor-gated calcium channel. Second, stimulation of non-NMDA receptors opens sodium channels and results in massive influx of sodium and subsequent membrane depolarization, which then allows calcium entry via voltage-sensitive calcium channels. Third, the ion channel gated by non-NMDA receptors allows direct influx of calcium. Three additional possibilities exist for calcium entry during ischemia:
Massive influx of sodium during ischemia creates conditions that inhibit or even reverse sodium/calcium exchange, leading to further calcium influx. It is also possible that calcium enters through areas of the cell membrane that have been damaged by the ischemic insult. Release of bound or sequestered calcium likely occurs through two different mechanisms. First, hydrogen ion, which accumulates during ischemia, can directly displace bound calcium. Second, sequestered intracellular calcium may be mobilized by inositol-1,4,5-triphosphate, which is produced by glutamate-stimulated hydrolysis of polyphosphoinositides, mediated by the metabotropic receptor [Berridge and Irvine, 1984; Sugiyama et al., 1987].
Although most work has centered on the role of calcium influx, some investigations have focused on the importance of the release of sequestered calcium. Mitani et al. [1993] demonstrated that two-thirds of the increase in the intracellular concentration of calcium seen in cultures of hippocampal neurons subjected to glucose and oxygen deprivation is caused by release of sequestered calcium, whereas only one-third is caused by extracellular calcium influx. Although the relative contribution of influx and release from internal stores remains unclear, there is accumulating evidence that release from internal stores does play a role.
Activation of Intracellular Enzymes
The marked increase in intracellular calcium concentration activates at least four classes of enzymes [Morgenstern and Pettigrew, 1997; Sweeney et al., 1995]. Phospholipases break down the lipid cellular membrane, releasing arachidonic acid that generates prostaglandins and free radicals. Protein kinases activate enzymes in an unordered manner, including nitric oxide synthase and xanthine oxidase, which are also generators of free radicals. Proteases begin the uninhibited breakdown of the cytoskeleton, and endonucleases initiate DNA fragmentation.
Protease activation may play a central role in mediating both necrosis and programmed cell death. With regard to necrosis, numerous calcium-dependent enzymes become activated during ischemia and produce important neuronal structural injury. One class of calcium-dependent proteases, calpains, has received the greatest amount of attention. Calpains are cytosolic thiol proteases that degrade numerous cytoskeletal proteins such as neurotubules and neurofilaments, as well as activate protein kinase C and phospholipases. Inhibition of calpain activation has produced marked reduction in ischemic brain injury, particularly after focal ischemia [Bartus et al., 1994]. Proteases may also play a pivotal role in the initiation of programmed cell death.
There is growing evidence that some neuronal death after ischemia is mediated by the activity of cysteine-requiring aspartate-directed proteases (caspases), the protease responsible for apoptosis in mammals [Hotchkiss et al., 2009].
Phospholipase Release of Free Fatty Acids
Metabolism of membrane phospholipids through activation of phospholipases is postulated to play a key role in the pathophysiology of ischemic brain injury (Figure 76-3).
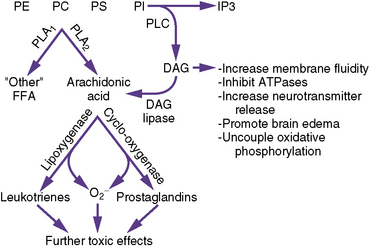
Free fatty acids are released from neuronal membranes during ischemia; the amount of release is proportional to the duration of ischemia. Free fatty acid release is the only known cerebral metabolic indicator that continues to increase in proportion to the duration of ischemia after completion of energy failure [Shin et al., 1983]. Free fatty acids are released by two distinct but related processes. First, phosphatidylinositol is hydrolyzed by phospholipase C with the production of diacylglycerol and inositol phosphates [Abe et al., 1987]. Phospholipase C-mediated hydrolysis begins during the initial moments of ischemia and is related to the magnitude of neurotransmitter receptor stimulation. Diacylglycerol is then hydrolyzed by lipases to free fatty acids, predominantly arachidonic acid and stearic acid. Second, other brain glycerophospholipids are hydrolyzed by phospholipase A2, which is activated by increases in intracellular calcium concentration. The process of free fatty acid release and metabolism is not a generalized process in the neuronal membrane but is concentrated in synaptic regions and is thus related to excitotoxicity.
The free fatty acids released have potential detrimental effects through at least three mechanisms. First, free fatty acid metabolism via the cyclo-oxygenase pathway contributes to oxygen radical production during reperfusion [Kontos, 1987]. Second, free fatty acid and diacylglycerol directly increase membrane fluidity, inhibit ATPases, increase neurotransmitter release, promote brain edema, and uncouple oxidative phosphorylation. Third, enzymatic oxidation of arachidonic acid during reperfusion by cyclo-oxygenase, lipoxygenase, or cytochrome P-450 produces a large number of bioactive lipids (prostaglandins, thromboxanes, leukotrienes, and hydroxyl acids), many of which have detrimental effects. Ischemia induces a pro-inflammatory state that increases tissue vulnerability to further injury on reperfusion [Collard and Gelman, 2001].
Activation of Nitric Oxide Synthesis
Nitric oxide plays a multifaceted role in the brain as a neurotransmitter and a regulator of cerebral blood flow [Bhardwaj et al., 1997; Dawson and Dawson, 1995; Dawson et al., 1992]. Sites of nitric oxide production include neurons, vascular endothelium, perivascular neurons, and astrocytes [Bhardwaj et al., 1997]. If present in abnormally high concentrations, nitric oxide may exert neurotoxic effects [Dawson, 1994; Moncada and Higgs, 1993].
Nitric oxide is produced in a reaction catalyzed by nitric oxide synthase, in which oxygen and l-arginine are converted into l-citrulline and nitric oxide. Three isoforms of nitric oxide synthase have been described [Samdani et al., 1997] (Table 76-3). There are two constitutive isoforms: neuronal and endothelial. An inducible isoform has also been described and has been demonstrated in macrophages, microglia, and astrocytes. The constitutive enzymes are calcium-calmodulin-dependent enzymes. Inducible nitric oxide synthase is calcium-independent, is induced by endotoxin and cytokines, and, when stimulated, produces large amounts of nitric oxide in a sustained manner [Clark et al., 1996]. Neurons containing nitric oxide synthase are present throughout the brain, with the highest concentration in the cerebellum and the lowest concentration in the medulla.
| Type I: Neuronal NOS | Type II: Inducible NOS | Type III: Endothelial NOS |
|---|---|---|
| Activity depends on elevated Ca2+ | Activity is independent of Ca2+ | Activity depends on elevated Ca2+ |
| First identified in neurons | First identified in macrophages | First identified in endothelial cells |
| Constitutively expressed, but inducible | Inducible under pathologic conditions | Constitutively expressed, but inducible under pathologic conditions |
| Plays a prominent role in the early stage of neuronal injury after cerebral ischemia | Plays a role in the later stages of neuronal injury after cerebral ischemia | Plays a protective role in cerebral ischemia by maintaining cerebral blood flow |
| Protein and catalytic activity are upregulated within 10 minutes and peak 3 hours after cerebral ischemia | Protein and catalytic activity are upregulated within 12 hours and peak 48 hours after cerebral ischemia | Protein and catalytic activity are upregulated within 1 hour and peak 24 hours after cerebral ischemia |
In some neurons, nitric oxide synthase activity is regulated by the NMDA receptor. NMDA receptor stimulation results in calcium influx and activation of nitric oxide synthase. Nitric oxide is produced and then diffuses to target cells and stimulates guanylate cyclase, which leads to the production of cyclic guanosine monophosphate. Cyclic guanosine monophosphate then produces the physiologic effect (e.g., vasorelaxation, cell signaling). Inducible nitric oxide synthase is expressed by several cell types (including macrophages, microglia, and astrocytes) in response to stimulation by cytokines. Nitric oxide produced by this nitric oxide synthase isoform is involved in cell-mediated cytotoxicity [Samdani et al., 1997]. The mechanism of this cytotoxicity is not yet fully understood but may involve inhibition of key enzymes necessary for DNA replication and mitochondrial energy production.
As part of the excitotoxic cascade of injury, glutamate release activates both endothelial and neuronal nitric oxide synthase, which increase nitric oxide production during focal and global ischemia [Bari et al., 1997; Samdani et al., 1997]. During ischemia, endothelial nitric oxide synthase production may be protective by increasing cerebral blood flow, whereas during ischemia, and more so during reperfusion, neuronal nitric oxide release synthase production may cause additional neurotoxicity. Nitric oxide produced under conditions of ischemia contributes to the cytotoxicity of glutamate, presumably through hydroxyl radical production (Figure 76-4). Nitric oxide contributes to hydroxyl radical production through its reaction with the superoxide radical. Nitric oxide and superoxide radical react to form peroxynitrite, which then decomposes to yield nitrogen dioxide and the toxic hydroxyl radicals. Several days after focal ischemia, activation of inducible nitric oxide synthase from phagocytic cells may contribute to delayed injury. The majority of evidence suggesting a role for nitric oxide has been demonstrated in models of focal ischemia. Less convincing but definite evidence exists for a potential neurotoxic role during global ischemia. Although one of the major effects of nitric oxide is to increase cyclic guanosine monophosphate, multiple mechanisms have been proposed to define its neurotoxic effects.
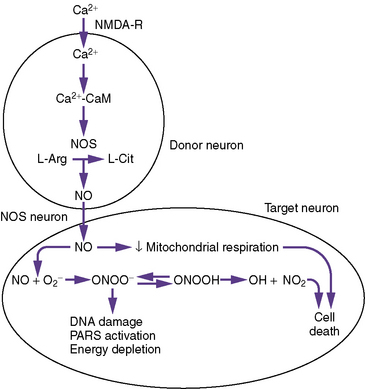
An established pathway of nitric oxide-mediated neuronal cell death is nitric oxide activation of the nuclear enzyme poly-adenosine diphosphate (ADP)-ribose-synthetase (PARS) [Dawson and Dawson, 1995; Samdani et al., 1997]. Nitric oxide activates PARS by damaging DNA. PARS participates in DNA repair by catalyzing the transfer of ADP-ribose units from nicotinamide adenine dinucleotide (NAD) to nuclear proteins. For every mole of ADP-ribose transferred, 1 mol of NAD is consumed, and four free energy equivalents of ATP are necessary to regenerate NAD. Therefore, over-activation of PARS can rapidly deplete cellular energy stores.
Depending on its source, nitric oxide may be toxic or protective in the brain under ischemic conditions. Overproduction of nitric oxide from either neuronal or inducible nitric oxide synthase leads to neurotoxicity; however, nitric oxide production from endothelial nitric oxide synthase protects brain tissue by maintaining regional cerebral blood flow. Studies emphasize the necessity for developing truly selective inhibitors for neuronal and inducible nitric oxide synthase to protect the brain adequately from ischemic injury caused by overproduction of nitric oxide and simultaneously to maintain or enhance regional cerebral blood flow [Samdani et al., 1997].
Formation of Oxygen Radicals
Toxic oxygen radical species, produced during post-ischemic reperfusion, have been implicated as important contributors to reperfusion injury and delayed cell death [Siesjo et al., 1989]. Oxygen free radicals are not one specific compound. They are a group of substances that are formed during ischemia, when oxygen becomes unavailable as the terminal electron acceptor in the electron transport chain. A free radical is any molecule that has an unpaired electron in its outermost orbit; such molecules are short-lived and highly reactive. They include nitric oxide, superoxide anion, hydrogen peroxide (not itself a free radical), hydroxyl radical, and peroxynitrate. The lone electron results in molecular instability and the tendency to initiate and propagate chain reactions.
Oxygen free radicals are generated during arachidonic acid metabolism to prostaglandins, as a byproduct of xanthine oxidase-catalyzed production of uric acid through catecholamine oxidation, through a mitochondrial leak during the oxidation of hemoglobin, and through the action of nitric oxide synthase [Kontos, 1987; Siesjo et al., 1995; Traystman et al., 1991] (Figure 76-5). Free radicals are quite destructive to cellular components such as membrane lipids, especially in the presence of iron. Iron is normally transported in the blood, tightly bound to transferrin and stored inside the cell bound to ferritin. In ischemic conditions with accompanying acidosis, iron may be displaced from its normal binding sites and can catalyze reactions that promote oxygen radical formation [Komara et al., 1986]. Most commonly implicated is the Haber–Weiss/Fenton reaction, whereby potent hydroxyl radicals are produced from hydrogen peroxide in the presence of free iron.
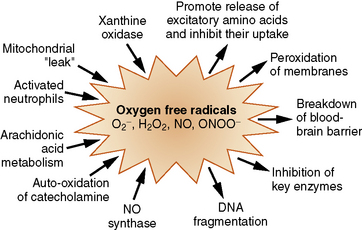
The impact of the liver and the intestines in producing dysfunction in other organ systems needs further clarification. The liver and intestines have a great deal of xanthine dehydrogenase, which during ischemia can be converted to xanthine oxidase [Bellamy et al., 1996; Nielsen et al., 1997]. The latter could enter the systemic circulation, generate free radicals, and cause additional dysfunction and injury at distant sites.
The brain may be particularly vulnerable to free radical injury for several reasons. One is the high concentration of polyunsaturated fatty acids, especially arachidonic acid. As noted previously, free fatty acids are released throughout ischemia. On exposure to oxygen radical species, these free fatty acids are vulnerable to lipid peroxidation [Krause et al., 1988]. Cerebrospinal fluid has low concentrations of iron-binding proteins; therefore, iron released from injured neurons or glia is likely to contribute to these peroxidation reactions. Byproducts of these reactions – for example, malondialdehyde and conjugated dienes – have been used as markers of the extent of lipid peroxidation after brain injury [Schmidley, 1990]. Lipid peroxides accumulate in the selectively vulnerable zones during reperfusion after transient forebrain ischemia [Bromont et al., 1989; Komara et al., 1986]. The peroxides do not accumulate during the ischemic period itself or in areas that are not reperfused, and thus are implicated in reperfusion injury [Oliver et al., 1990].
Lipid membranes are a natural target of free radicals, especially in the brain, because they are abundant, and their polyunsaturated nature makes them easy to oxidize [Schmidley, 1990]. The effects of free radicals on membranes include changes in membrane fluidity and alteration of ion channels and transport proteins. In addition to disruption of the cell membrane, fragmentation of the mitochondrial membrane results in a decrease in cellular energy production.
The ability of the cell to defend itself against free radicals is limited, and free radical scavenging enzymes, such as superoxide dismutase and catalase, may be overwhelmed after an ischemic area is reperfused [Vannucci and Perlman, 1997]. Surprisingly, restoration of blood flow to hypoxic-ischemic areas is potentially detrimental because the influx of oxygen can be used as a source of oxygen free radicals through the processes noted previously. Stimulated by high tissue carbon dioxide tension, low oxygen tension, and low pH, restored blood flow to an ischemic area is often increased above normal (luxury perfusion). In experimental preparations, oxygen free radicals are detected primarily during reperfusion. After luxury perfusion, there is prolonged hypoperfusion and an associated decrease in cerebral metabolism. In animal experiments, these decreases are not seen with pretreatment with oxygen free radical scavengers.
Inflammatory Reaction
There is growing evidence that acute inflammation contributes to neuronal damage after ischemia and reperfusion [Hoesch et al., 2008; Adrie et al., 2002]. The mechanisms for inflammatory injury can be divided into three broad categories: adverse effects on blood rheology; vasculature injury; and cytotoxic injury to neurons [Doherty and Hutchison, 2009]. They are also discussed in more detail in Chapter 15.
Genetic Damage and Regulation
Studies have demonstrated that ischemia and reperfusion can induce gene damage in the CNS. Reactive oxygen species generated by cerebral oxidative stress interact with nucleic acids and cause the formation of oxidative DNA and RNA lesions that result from DNA base modifications or single-stranded breaks in neurons or astrocytes [Liu and Arora, 2002; Liu et al., 2001]. Oxidative DNA lesions cause a change in coding properties during DNA and RNA synthesis (replication and transcription), or may terminate chain elongation during transcription and translation. Either process can affect protein synthesis.
The development of microarray systems for gene expression profiling since the mid-1990s has provided insights into the mechanisms of the brain’s response to global hypoxic injury [Jin et al., 2001; Papadopoulos et al., 2000; Tang et al., 2002]. Future studies should not only increase the understanding of the molecular basis of ischemic injury, but also suggest potential therapies. Because investigators in such studies frequently use several thousand gene transcripts, using different animal models with varying degrees of severity and duration of global hypoxia-ischemia, as well as varying time periods of reperfusion, it will be some time before a unified or general understanding of the underlying mechanisms of injury are more clearly appreciated.
Early during hypoxia-ischemia, overall gene expression in the brain is reduced or ceases to maintain energy metabolism to support essential cell activities. Immediate early genes undergo rapid induction after global ischemia [Kogure and Kato, 1993]. Some of the immediate early gene products that have been identified include c-fos, c-jun, and the zinc finger family of proteins; heat shock proteins; and amyloid precursor protein. Immediate early genes are transcription factors that bind to target genes and alter their expression. These stress response proteins may directly affect neuronal death and survival after ischemia. However, despite much research into their expression, it remains unclear which ones contribute to the outcome of hypoxic-ischemic injury [Papadopoulos et al., 2000]. This confusion occurs partly because some are upregulated, whereas others are downregulated, and their respective neurotoxic or neuroprotective roles remain to be defined.
The heat shock proteins are an early expressed group; they are molecular chaperones, which bind intracellular proteins and prevent their inappropriate folding [Papadopoulos et al., 2000]. Hsp-70 is markedly induced by hypoxia and is neuroprotective. Hsp-32 is another heat shock protein that may be neuroprotective after hypoxia. It encodes heme-oxygenase-1, which, after induction, has been found to protect neurons in models of transient forebrain ischemia, possibly by producing bile pigments that are strong antioxidants, as well as carbon monoxide, which can cause vasodilatation and inhibits platelet aggregation.
Growth factors are also released within hours after ischemia and may also serve a neuroprotective role. They are polypeptides and the major types of growth factors include nerve growth factor; brain-derived neurotropic factor; neurotrophin-3, 4, 5; and basic fibroblast growth factor [Papadopoulos et al., 2000]. Nerve growth factor, brain-derived neurotropic factor, and neurotrophin-3 bind receptors on the cell surface that have tyrosine kinase activity, and activation of tyrosine kinase is associated with neuronal survival.
Hours later, cytokines, adhesion molecules, isoforms of nitric oxide synthase, and gene products involved in apoptosis are upregulated. Some of these genes protect the cells from dying; some promote the death of injured cells; and others have no known effects on cell survival. Cytokines are produced by activated lymphocytes, macrophages, and astrocytes, and include tumor necrosis factor-α, transforming growth factor-β, and interleukin-1β. Cytokines participate in the inflammatory response and accelerate entry of inflammatory cells from the circulation into the brain. Certain cytokines (e.g., tumor necrosis factor-α and interleukin-1β) may also induce the expression of glycoproteins known as adhesion molecules on endothelial cells, which also facilitate recruitment of inflammatory cells into the ischemic brain [Papadopoulos et al., 2000].
Neuronal degeneration can also be promoted by induction of apoptosis genes or genes that cause a stress to the cells, such as those related to free radical production or to the production of various nitric oxide synthase isoforms [Koistinaho and Hokfelt, 1997]. Cells that undergo apoptosis shrink down and ultimately are phagocytosed by neighboring cells. Genes that accelerate apoptosis include members of the bax and bad families, p53, and Fas; those that inhibit apoptosis are associated with bcl-2, bcl-x (L), and DAD1 families [Papadopoulos et al., 2000]. When apoptosis is initiated, cytochrome c is released from damaged mitochondria and, in concert with apoptotic protease-activating factor, induces members of the caspase family, including interleukin-1-converting enzyme. As apoptosis proceeds, nuclear components and the cell skeleton are destroyed, detach other cell constituents, and are phagocytosed.
Two studies demonstrated both the elegance of the application of microarray technology and the dilemmas in synthesizing the results. In one study, 15 minutes of global cerebral ischemia in rats was associated with 1.7-fold or greater increased expression of 57 genes and 1.7-fold or greater decreased expression of 34 genes studied up to 72 hours after hypoxia [Jin et al., 2001]. Induced genes included those involved in protein synthesis, pro-apoptotic genes, anti-apoptotic genes, injury-response genes, receptors, ion channels, and enzymes. Transcriptional induction of several genes was also reported, as was co-induction of several groups of related genes (e.g., vascular endothelial growth factor and its receptor, neuropilin-1). A second study examined 8000 transcripts in rat brains 24 hours after exposure to 6 hours of hypoxia (8 percent oxygen) and found very little gene induction (15 upregulated genes and 11 downregulated genes) [Tang et al., 2002]. In contrast, the same investigators found that, in a model of permanent focal ischemia, 415 genes were upregulated and 158 were downregulated. Presumably, the differences between global hypoxia and ischemia were attributable to greater tissue injury in the ischemic model, but there is no apparent explanation of the differences in the response to global hypoxia in these two groups of studies.
Clinical Pathophysiology
Cerebral Blood Flow and Metabolism After Resuscitation
The pioneering studies, in which global cerebral blood flow and cerebral metabolic rate for oxygen were measured in animal models of global ischemia or cardiac arrest, focused on the early post-resuscitation period. In their classic study, Snyder et al. [1975] demonstrated that, after 15 minutes of global brain ischemia in dogs, cerebral blood flow transiently increased to levels well above baseline. After 15–30 minutes, cerebral blood flow progressively decreased to a level below normal for the rest of the monitoring period (90 minutes). This pattern of early transient post-ischemic hyperemia and subsequent delayed post-ischemic hypoperfusion has been observed almost universally in global cerebral ischemia models, including asphyxia-induced cardiac arrest. The level of hyperemia and subsequent hypoperfusion varies in relation to the duration of the insult. Although these phases of increased and decreased cerebral blood flow characterize the net global effect, regional cerebral blood flow is often heterogeneous, particularly during post-ischemic hypoperfusion, when areas of decreased and increased perfusion may coexist. Metabolism, as assessed by cerebral metabolic rate for oxygen, is reduced during the early post-ischemic period and then progressively recovers to a level that varies, depending on the model used and the duration of ischemia. In some models, including ventricular fibrillation in dogs, significant recovery of cerebral metabolic rate for oxygen may occur during the first few hours, despite persistent post-ischemic hypoperfusion, creating the potential for a secondary ischemic insult during reperfusion. Whether this increase in cerebral metabolic rate for oxygen represents appropriate synaptic activity, seizures, or changes in the basal metabolic rate is uncertain. In other models, global cerebral blood flow and cerebral metabolic rate for oxygen are matched during the first few hours after ischemia. In such cases, post-ischemic cerebral blood flow appears to be determined by the metabolic needs of the brain, and delayed hypoperfusion may not be a significant cause of neuronal injury. Thus, delayed hypoperfusion is likely the result of brain injury rather than the cause [Michenfelder and Milde, 1990].
Of historic interest is the concept of “no reflow,” which was suggested in the 1960s by Ames et al. [1968]. These investigators noted that, after a period of global cerebral ischemia, reflow could not be re-established. At a microscopic level, areas of “no reflow” are interspersed with areas of restored blood flow and microinfarcts [Fink et al., 2008]. Brain regions that are selectively vulnerable include the thalamus, amygdala, hippocampus, and striatum. It is hypothesized that vasospasm, perivascular edema, and increased blood viscosity play a role in the development of this “no reflow” phenomenon.
The second phase of cerebral blood flow is characterized by increased global cerebral blood flow, and is often referred to as the “hyperemic” phase. Some believe that this hyperemic phase is essential for neuronal functional recovery [Fink et al., 2008]. This opinion is based on studies that show that hypertensive reperfusion improves neurologic outcome, whereas post-arrest hypotension worsens neurologic outcome. In addition to hypertensive reperfusion, hemodilatation and thrombolysis in this phase have been shown to improve outcome in animal models [Fink et al., 2008].
Drugs such as nimodipine can increase cerebral blood flow during the early post-ischemic hypoperfusion phase after global cerebral ischemia. Cerebral metabolic rate for oxygen recovery generally is not increased by treatment, even though nimodipine improves neurologic outcome after global ischemia in some models. Safar et al. [1996] reported that a multifaceted flow-promotion treatment strategy to increase cerebral blood flow and reduce cerebral metabolic rate for oxygen early after ventricular fibrillation in dogs improved outcome. This improvement was accomplished with cardiopulmonary bypass, mild hypothermia, hemodilution, and transient hypertension.
Studies in which investigators measured cerebral blood flow and cerebral metabolic rate for oxygen in humans after cardiac arrest shed interesting light on these data from animals. Whereas cerebral blood flow and cerebral metabolic rate for oxygen typically are measured early (0–2 hours after arrest) in experimental animal studies, such measurements in humans are obtained beginning 6–12 hours after arrest. Beckstead et al. [1978] measured global cerebral blood flow and cerebral metabolic rate for oxygen in 25 adults after cardiac arrest. In all, 21 of these patients had a vegetative outcome. In agreement with the experimental animal studies, the researchers observed hypoperfusion and hypometabolism with coupling of cerebral blood flow and cerebral metabolic rate for oxygen. During the subsequent 2 days, however, global cerebral blood flow increased to levels equal to or greater than normal, whereas metabolism failed to recover. Thus, in humans with poor neurologic outcome after cardiac arrest, absolute or relative delayed hyperemia followed the hypoperfusion phase. Results in two more studies in adults have confirmed these findings. Using xenon-133 washout, Cohan et al. [1989] observed that patients regaining consciousness had increased cerebral blood flow (hyperemia) within 24 hours. Similarly, Love et al. [1989] used stable xenon-enhanced computed tomography (CT) and discovered that the combination of diffuse hyperemia and loss of reactivity of cerebral blood flow to changes in PaCO2 was seen 100 hours after arrest in adults who never regained consciousness.
Thus, immediately after cardiac arrest accompanied by restoration of systemic hemodynamic stability, transient global brain hyperemia occurs and is followed by a period of patchy hypoperfusion. The magnitude and duration of these alterations in flow appear to be related to the duration of the insult. In patients with good outcomes, global cerebral blood flow recovers over a subsequent 24–72 hours, and CO2 reactivity remains intact. In patients who do not regain consciousness or progress to brain death, absolute or relative cerebral blood flow hyperemia develops, with impaired CO2 reactivity [Prough and Zornow, 1997]. It must be recognized that the measurement of metabolism in these studies has traditionally been cerebral metabolic rate for oxygen. Bergsneider et al. [1995] used positron emission tomography (PET) in humans after traumatic brain injury and reported the occurrence of delayed hyperglycolysis in some comatose patients. Studies of cerebral glucose utilization with PET after cardiac arrest are needed.
Results from clinical studies of asphyxia-induced cardiac arrest in children are scarce and somewhat conflicting with regard to the prognostic implications. Normal values of post-cardiac arrest cerebral blood flow with loss of responsivity to PaCO2 were observed in children with poor outcome at 24 hours after asphyxia-induced cardiac arrest by strangulation, as studied with stable-xenon CT [Ashwal et al., 1991]. Similarly, in studies of subjects between 24 and 48 hours after near-drowning, Ashwal et al. [1990] observed low cerebral blood flow and no relation between cerebral blood flow and PaCO2 in the seven nonsurvivors, which again suggests loss of cerebral blood flow reactivity to changes in PaCO2. In that study hyperemia was not routinely observed in either vegetative survivors or children who died, but only a single cerebral blood flow measurement was made. Beyda [1987] obtained serial measurements of post-cardiac arrest cerebral blood flow with xenon-enhanced CT in a series of children who had suffered asphyxia-induced cardiac arrest from submersion accidents. Children with good neurologic outcomes had slightly decreased cerebral blood flow values at 12 hours, which increased to normal during the subsequent 24–60 hours. In these children, cerebral blood flow reactivity to CO2 was intact. Children with an eventual vegetative outcome or brain death exhibited hyperemia with loss or attenuation of CO2 reactivity. This hyperemia progressed to low or normal flow over the next 12–72 hours in children with vegetative outcome, and progressed to low and then no flow, with the development of brain death.
Major Disorders Causing Cardiac Arrest
Table 76-4 lists common causes, by category, of pediatric cardiorespiratory arrest taken from major published pediatric cardiac arrest series. Common specific disease entities resulting in cardiac arrest are sudden infant death syndrome, drowning, strangulation, nonaccidental trauma, and lightning/electrical injury.
| Etiology | Patients (%) |
|---|---|
| Submersion | 27 |
| Sudden infant death syndrome | 20 |
| Trauma | 15 |
| Respiratory | 9 |
| Sepsis | 9 |
| Heart disease | 6 |
| Other/unknown | 14 |
(Data from Hickey RW, Cohen DM, Strausbaugh S, et al: Pediatric patients requiring CPR in the pre-hospital setting, Ann Emerg Med 25:495–501, 1995, and Schindler MB, Bohn D, Cox PN, et al: Outcome of out-of-hospital cardiac or respiratory arrest in children, N Engl J Med 335:1473–1479, 1996.)
Sudden Infant Death Syndrome
Sudden infant death syndrome (SIDS) is defined as the sudden death of an infant younger than 1 year that remains unexplained after a thorough case investigation, including performance of a complete postmortem examination, examination of the death scene, and review of the medical history [Willinger et al., 1991]. The addition of requirements for examining the death scene and reviewing the history give greater precision to the diagnosis, but it remains one of exclusion, based on the absence of any specific findings on routine postmortem examination. In many unresolved cases, the history, investigation, or postmortem examination reveals information that excludes the diagnosis of SIDS but does not explain the cause of death; suspected abuse, neglect, or accidental suffocation; vomiting or diarrhea without evidence of infection; unreliable information; or findings such as mild bronchopneumonia, supraglottitis, or maternal drug use are not sufficient to explain death [Gilbert-Barness and Barness, 1995]. Even though almost all cases of sudden unexpected death in infants continue to be ascribed to SIDS, the uncritical certification of the syndrome as a diagnosis, without a postmortem examination, death scene investigation, and careful history, prevents accurate diagnosis of the cause of death [Bass et al., 1986; Valdes-Dapena, 1995].
Epidemiology
Incidence and age at death
SIDS is the leading cause of postneonatal infant death in the United States and is the third leading cause of infant mortality overall [Kinney and Thach, 2009]. Although SIDS may occur during the first month of life, most neonatal deaths can be accounted for by specific perinatal factors, including prematurity, infection, and congenital diseases.
The incidence of SIDS peaks between 2 and 4 months of age; 95 percent of all deaths from the syndrome occur by 6 months of age [Hunt, 1992]. SIDS, therefore, is primarily a disorder that strikes children between 1 and 6 months. It is so rare outside those age limits that the diagnosis in such cases should be made with caution, and only after all possible causes, including metabolic diseases, are excluded [Gilbert-Barness and Barness, 1995]. The age-at-death distribution of SIDS is the most consistent, unique, and provocative characteristic identified [Peterson, 1988].
Identification of risk factors
In recent years, SIDS has been substantially demystified by major advances in our understanding of its relation to sleep and homeostasis, environmental and genetic risk factors, and biochemical and molecular abnormalities [Kinney and Thach, 2009]. The most important advance has been the discovery that the prone (on the stomach) sleep position more than triples the risk of SIDS, which in the early 1990s led to national and international campaigns advocating a supine sleep position for infants [Willinger et al., 1994].
Considerable investigation has been carried out to identify other factors that may contribute to SIDS. Unfortunately, no epidemiological differences have been of sufficient sensitivity and specificity to permit prospective identification of future SIDS victims [Hoffman and Hillman, 1992]. One way of conceptualizing the emerging multidisciplinary data is with the Triple-Risk Model, first proposed in 1994 [Filiano and Kinney, 1994]. Like other models it emphasizes the interaction of multiple factors in the pathogenesis of SIDS [Kinney and Thach, 2009]. According to this model, SIDS occurs when three factors present simultaneously. The first factor is an underlying vulnerability in the infant; the second is a critical developmental period; and third, an exogenous stressor (Box 76-1).
Box 76-1 Components of the Triple-Risk Model of Sudden Infant Death Syndrome











(Modified from Kinney HC, Thach BT. The sudden infant death syndrome. N Engl J Med 2009;361:795–785.)
Risk factors for SIDS can be divided into extrinsic and intrinsic categories [Paterson et al., 2006]. Extrinsic risk factors are physical stressors that would place a vulnerable infant at risk for asphyxia or other homeostatic derangement [Kinney and Thach, 2009]. Such extrinsic factors include prone and side-sleeping positions, bed clothes that cover the head, sleeping on sofas or other soft furniture in which the infant could become wedged, a high ambient temperature in the sleeping environment, soft bedding, and bed sharing [Kinney and Thach, 2009].
Sleeping in the prone position by infants has been one of the most consistent risk factors for SIDS; an avoidance of prone sleeping has been the focus of several national campaigns to prevent the syndrome. The evidence that prone sleeping is a risk factor for SIDS has been so compelling that the American Academy of Pediatrics Task Force on Infant Positioning and SIDS [1992] recommended the supine sleeping position for infants. The fear that aspiration would occur if an infant vomited or regurgitated while sleeping in the supine position appears to be unwarranted; in fact, the incidence of death from aspiration has actually decreased among infants sleeping in the supine position [Gilbert-Barness and Barness, 1995]. Although the lateral position for sleeping has also been recommended, positioning infants on their sides may not be as safe, because the infant may roll over into the prone position.
The recommendation for sleep in a nonprone or supine position is for healthy infants only. Gastroesophageal reflux and certain upper airway anomalies that predispose to airway obstruction and perhaps some other illness may be indications for a prone sleeping position [AAP, 1996].
Although the incidence of prone sleep position is currently 20 percent or less, 30–50 percent of infants with SIDS are still found in the prone position [Hauck and Tanabe, 2008; Mitchell et al., 2008].
Scientific studies have demonstrated that bed sharing by mother and infant can alter and synchronize sleep patterns of the two. These studies have led to speculation in the lay press that bed sharing, sometimes referred to as co-sleeping, may also reduce the risk of SIDS. Although bed sharing may have certain benefits (such as encouraging breastfeeding), no scientific studies demonstrate that bed sharing reduces the incidence of SIDS. Conversely, there are studies suggesting that bed sharing, under certain conditions, may actually increase the risk of the syndrome [AAP, 1997].
In addition to extrinsic factors related to external events around the time of death, intrinsic factors are postulated to affect the underlying vulnerability of the infant and thus increase the risk of SIDS. Intrinsic risk factors can be subdivided into developmental factors, such as prematurity, and putative genetic factors, such as familial SIDS, male sex (by 2:1 ratio), and race or other ethnic group [Kinney and Thach, 2009].
Certain genetic polymorphisms have been associated with SIDS [Kinney and Thach, 2009]. Polymorphisms associated with SIDS have been reported in a variety of genes involved in autonomic function, neurotransmission, energy metabolism, and response to infection [Hunt, 2005; Weese-Mayer et al., 2007]. In addition, the vulnerable infant’s response to environmental factors may actually reflect aberrant intrinsic responses. For that reason, events and environmental conditions extrinsic to the infant, such as poverty, adverse prenatal response to certain substances, and postnatal exposure to cigarette smoke may trigger intrinsic responses in the vulnerable infant [Kinney and Thach, 2009].
For example, prenatal exposure to alcohol and cigarette smoke may have a direct effect on neurotransmitter systems that are critical to homeostatic control in the developing human brain [Duncan et al., 2008]. A number of epidemiologic studies have documented that smoking during pregnancy and after birth are two major and independent risk factors for SIDS [Perkin, 2004]. With the reduction in the incidence of infants being put to sleep prone, maternal smoking has become the major modifiable risk factor for SIDS [Horne et al., 2002].
Pathophysiology
The most compelling hypothesis to explain SIDS is that of a brainstem abnormality in cardiorespiratory control [Hunt, 1992]. Both the pathologic data derived from postmortem examination studies and the clinical data obtained in infants at increased epidemiologic risk for SIDS support this hypothesis. The postmortem findings, although subtle, are consistent with a maturational or developmental abnormality associated with the brainstem areas relevant to autonomic control of cardiorespiratory regulation. The clinical data to support the cardiorespiratory control hypothesis were initially obtained from assessments of patients with apnea of infancy and of asymptomatic infants at increased epidemiologic risk for SIDS.
Infants at increased risk for SIDS have diminished ventilator responsiveness to hypercarbia and hypoxia [Hunt, 1992]. The extent of overlap between control infants and at-risk infants, however, precludes the use of hypercarbic or hypoxic ventilator responsiveness, or both, as a test to predict risk for the syndrome. Because of overlapping values, an individual infant cannot be classified as normal or abnormal. In addition, these tests are too time-consuming and costly to be useful for large-scale application.
Arousal from sleep that is triggered by abnormal levels of carbon dioxide and oxygen is essential for the initiation of protective airway responses; indeed, head turning and escape to fresh air are critical for survival from an asphyxial micro-environment [Kinney and Thach, 2009]. Arousal involves a progressive activation of specific subcortical to cortical brain structures and consists of ascending and descending components that mediate cortical and subcortical arousal, respectively, with feedback loops between them. Cortical arousal involves noradrenergic, serotonergic (5-hydroxytryptamine), dopaminergic, cholinergic, and histaminergic neurons in the brainstem, basal forebrain, and hypothalamus, which excite the cerebral cortex and cause cortical activation [Sinton and McCarley, 2004]. Subcortical arousal, on the other hand, is mediated mainly by brainstem pathways that increase the heart rate, blood pressure, respiration, and postural tone without changes in cortical activity. Prospective studies of infants who have subsequently died of SIDS have supported the presence of arousal deficits in infants with SIDS [Kinney and Thach, 2009].
In severe hypoxia or ischemia, normal breathing fails and is replaced by gasping. Gasping increases the volume of air in the lungs, followed by oxygen transport to the heart, increased cardiac output, and finally brain perfusion and re-oxygenation. The act of gasping involves neurons within the rostral medulla that demonstrate a “bursting” discharge due to pacemaker activity and that are modulated by 5-hydroxytryptamine and norepinephrine, which, in combination, sustain gasping and restore rhythmic respiratory activity [Kinney and Thach, 2009]. Tracings from infants who subsequently died of SIDS have indicated that their gasping was ineffectual, with large-amplitude breaths, abnormally complex gasps, and an inability to increase the heart rate.
Some infants with acute life-threatening events (characterized by apnea and unresponsiveness that is interrupted by resuscitation, with no underlying disorder diagnosed) may represent potential SIDS cases in which the failure in gasping is averted by successful intervention. The incidence of acute, life-threatening events is significantly increased in infants with SIDS [Platt et al., 2000].
A strong history of SIDS, apparent life-threatening events, and obstructive sleep apnea in family members predisposes an infant to obstructive sleep apnea during the first year of life [McNamara and Sullivan, 2000]. The potential link among SIDS, apparent life-threatening events, and obstructive sleep apnea in infants is unclear; however, an increased incidence of obstructive sleep apnea in subsequent siblings of infants who died of SIDS and infants with apparent life-threatening events has been described. In addition, studies have documented obstructive events during sleep in infants who subsequently became victims of SIDS [Kato et al., 2001]. These findings suggest that SIDS and apparent life-threatening events are related to obstructive sleep apnea. Sleep studies should be obtained in young infants with multiple family histories of SIDS, apparent life-threatening events, and obstructive sleep apnea.
Cardiorespiratory recordings from infants who subsequently died of SIDS have shown episodes of tachycardia and bradycardia hours or days before death, suggesting a primary failure of autonomic mechanisms [Kinney and Thach, 2009]. Thus, one pathway for SIDS may involve inadequate compensatory autonomic responses to a hypotensive challenge that results from a cardiac arrhythmia, a “shocklike” sequence, or respiratory perturbation with secondary hypotension.
Compared with QTc intervals in matched control infants, QTc intervals are increased in future SIDS victims [Franco et al., 2008]. Such a prolongation could be related to the autonomic dysfunction already reported in these patients.
The biologic role of SIDS risk factors becomes comprehensible in light of the above-mentioned pathways, since many risk factors can trigger asphyxia or other homeostatic stressors and exacerbate the underlying vulnerability. An increased risk of SIDS in the first 6 months of life probably reflects a convergence of immature homeostatic systems [Kinney and Thach, 2009]. Premature infants may be at greater risk than term infants because they appear to have shorter arousal episodes and immature cardiorespiratory patterning. The prone sleep position increases the likelihood of rebreathing in the face-down position, upper airway obstruction, and hyperthermia. Developmental motor mechanisms also may underlie the risk associated with a prone sleep position, since all newborns obviously lack experience in sleeping in this position. Furthermore, infants have not fully learned the efficient protective strategies of head lifting and turning. Conceivably, developmental deficits in neural pathways that support reflexive motor learning may increase the risk associated with sleeping in the prone position. In addition, the prone sleep position is associated with impaired arousal from sleep, reduced vasomotor tone, and diminished laryngeal chemoreflexes and baroreceptor reflexes [Kinney and Thach, 2009].
The search for underlying vulnerability in SIDS infants has led to intense analysis of peripheral and central sites critical to protective responses to asphyxia and hypoxia. The major focus of SIDS research has been on the brainstem because it contains critical neural networks that mediate respiration, chemosensitivity, autonomic function, sleep, and arousal. Abnormalities in various neurotransmitters or their receptors have now been reported in relevant brainstem regions in infants with SIDS. To date, the most robust evidence for a neurochemical abnormality comes from research on the medullary 5-hydroxytryptamine system, in that approximately 50–75 percent of infants with SIDS appear to have abnormalities in this system [Paterson et al., 2006; Kinney and Thach, 2009]. The medullary 5-hydroxytryptamine system, which is considered critical for the modulation and integration of diverse homeostatic functions, according to the level of arousal, is involved in ventilation and gasping, thermoregulation, autonomic control, responses to carbon dioxide and oxygen, arousal from sleep, and hypoxia-induced plasticity [Kinney and Thach, 2009]. Abnormalities in 5-hydroxytryptamine neuronal number and differentiation receptors, or transporter, have been reported in the medulla of infants with SIDS, as compared with control infants, in four independent data sets. Given the wide array of homeostatic functions modulated by the medullary 5-hydroxytryptamine system, sudden death may result from a convergence of defects in protective responses to homeostatic stressors during sleep that are modulated by 5-hydroxytryptamine, probably in conjunction with related neurotransmitters.
A recent report demonstrated that levels of serotonin (5-hydroxytryptamine) were low in infants dying from SIDS, pointing to a deficiency, as opposed to an excess, of 5-hydroxytryptamine in the pathogenesis of the disorder [Duncan et al., 2010].
Drowning (Submersion Injury)
Submersion accidents are one of the most common causes of cerebral hypoxic-ischemic injury in children [Hasibeder, 2003]. A number of imprecise terms have been used in the literature that relate to submersion events. Terms such as drowning versus near-drowning, primary versus secondary, wet versus dry, cold versus warm, active versus passive, and fresh versus salt-water drowning have been used [Habib and Perkin, 2008; Papa et al., 2005]. These terms and definitions have made submersion data difficult to compare and they have obscured the interpretation of many issues related to treatment and prognosis. To reach accurate and meaningful conclusions regarding prevention, treatment, and prognosis of submersion events, it is necessary that uniform definitions of submersion events and outcomes are accepted. The World Congress on Drowning held in 2002 recommended that nearly all terms other than “drowning” be eliminated [Papa et al., 2005]. Specifically, the terms near-drowning and secondary drowning, which implied an outcome of survival beyond 24 hours after submersion, should be abandoned. Drowning is defined as the process resulting in respiratory impairment from submersion or immersion in a liquid medium [Papa et al., 2005]. Water rescue refers to a person who is alert but experiences some distress while swimming, displays transient or minimal symptoms, and does not require further emergency department evaluation [American Heart Association, 2000]. Immersion hypothermia refers to prolonged exposure to extremely cold water (<10°C) with core body temperature ≤32°C (89°F) [Stewart, 2006]. Immersion syndrome is a form of drowning caused by sudden exposure to very cold water (<10°C) that precipitates a lethal arrhythmia such as ventricular fibrillation [Stewart, 2006]. Death from drowning refers to any cause of death, at any time, if there is a clear chain of causality related to a submersion event [Idris et al., 2003].
Epidemiology
In the United States, submersion accidents remain the second most common cause of injury and death in children 1–4 years of age [Habib and Perkin, 2008]. Motor vehicle collisions are the leading cause of unintentional injury of children; however, swimming pools are nearly 14 times more likely than a motor vehicle to be involved in the death of a child [Habib and Perkin, 2008]. Children younger than 5 years have the highest drowning mortality rates [Rivara, 2009; Habib and Perkin, 2008]. Annually, about 1800 drowning deaths occur in the United States in children younger than 19 years [Ibsen and Koch, 2002; Burford et al., 2005]. Approximately 75 percent of these victims are children of the ages of 1–4 years. Just over one-half of these drowning deaths occur in residential pools [Burford et al., 2005]. These drowning events are more frequent in the first 6 months of pool ownership and are typically described by caretakers as silent, with few reports of crying or splashing [Habib and Perkin, 2008; Wintemute et al., 1991]. Toddler drowning tends to occur because of a lapse in parental or adult supervision. A responsible supervising adult can be identified in 84 percent of all toddler drownings, but only 18 percent are witnessed [Quan et al., 1989; Wintemute, 1990]. Infant drowning is reported to occur most often in bathtubs and accounts for about 13 percent of all submersion events [Habib and Perkin, 2008]. Although the majority of infant submersions are due to a lapse in supervision, up to one-third of cases may result from intentional abuse [Hasibeder, 2003; Quan et al., 1989; Lavelle et al., 1995; Gillenwater et al., 1996]. Other unique types of vessels in which infants or toddlers have drowned include toilets, 3–5-gallon buckets, hot tubs, and ice chests [Burford et al., 2005].
The distribution of submersion accidents by age follows a well-defined bimodal pattern [Habib and Perkin, 2008]. Children younger than age 5 years represent the first peak, which is followed by a second peak at ages 16–24 years. In all pediatric age groups, the rate of male submersions exceeds that of females [Burford et al., 2005]. Older children and teenagers are more prone to drown in bodies of fresh water such as lakes, rivers, and canals [Hasibeder, 2003]. Between 15 and 20 percent of submersion injuries involve adolescents, and these accidents primarily are related to water-sport activities, use of illicit drugs, alcohol, and risk-taking behaviors [Habib and Perkin, 2008]. The history associated with these drowning events often involves high-energy impacts, such as diving, jet-skis, or boat collisions [Hwang et al., 2003]. Overall, fewer than 5 percent of all submersions are associated with major trauma, and cervical spine injuries are rare, occurring in fewer than 0.5 percent of drowning accidents [Ibsen and Koch, 2002; Hwang et al., 2003].
Certain medical conditions are associated with an increased risk of drowning. Individuals with epilepsy are at four times higher risk for submersion accidents, regardless of age [Orlowski, 1979]. This threat extends to bathtubs as well, and some investigators have suggested that children with seizure disorders not bathe without supervision, or that they shower instead. Less obvious, and often overlooked, conditions include familial cardiac arrhythmias, such as long QT syndrome and polymorphic ventricular tachycardia [Ackerman and Porter 1998]. These inherited arrhythmias have been associated with drowning accidents and can be triggered by strenuous swimming or immersion in very cold water [Burford et al., 2005].
Pathophysiology
Neurological
The initial phase of a drowning event is marked by intense fright, panic, and a struggle to maintain the airway above water; young nonswimmers are incapable of doing this. Once this effort becomes unsuccessful, the victim submerges and voluntary breath holding occurs until the stimulus of air hunger is overwhelming. Fluid is swallowed first into the stomach, but will eventually be vomited and aspirated into the lungs by active inhalation [Hasibeder, 2003]. As alveoli are flooded and hypoxia intensifies, there is loss of airway reflexes and further pulmonary aspiration of water occurs [Idris et al., 2003]. Active aspiration of fluid into the lungs occurs in at least 90 percent of all severe submersion events. The final steps are cardiovascular collapse with bradycardia/asystole, leading to onset of global cerebral hypoxic-ischemic injury.
The remarkable changes in blood gas values in normothermic animals during a submersion event have been studied [Habib and Perkin, 2008]. Within moments of drowning, there is rapid onset of respiratory acidosis. Carbon dioxide tension doubles by 10 minutes and the pH falls below 7.0. There is a precipitous decline in blood oxygen tension to as low as 4 mmHg within 5 minutes of the submersion event. Within 2–4 minutes, there is sufficient hypoxemia to cause loss of consciousness. Cardiac arrest occurs within 5–10 minutes of total alveolar flooding or complete airway obstruction (laryngospasm). Following cessation of brain circulation, residual oxygen stores are depleted in about 10 seconds, and exhaustion of high-energy phosphates occurs within 5 minutes. It is widely accepted that the onset of irreversible neuronal damage begins soon thereafter.
Respiratory system
Following a submersion event, most children survive, and due to laryngospasm, present with relatively dry lungs and little respiratory distress. Only with active aspiration of gastric contents or a complete cardiac arrest leading to passive aspiration does the drowning victim present with significant pulmonary abnormalities. Fresh or salt-water aspiration into the lungs can lead to rapid changes in the normal function of surfactant. Aspiration of fresh water dilutes and washes out surfactant, rendering alveoli unstable [Habib and Perkin, 2008]. Salt water, with a tonicity similar to 3 percent saline, generates an influx of protein-rich plasma from the pulmonary capillaries, resulting in secondary surfactant dysfunction. The net physiologic effect of either type of fluid is the rapid increase of alveolar surface tension and production of large areas of atelectasis in the lungs. The resulting loss in pulmonary compliance creates widespread areas of ventilation/perfusion (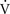 /
/ ) abnormalities and intrapulmonary shunt. Pulmonary aspiration also may trigger an inflammatory response in the alveolar capillary membranes and progress to acute respiratory distress syndrome (ARDS). These changes in pulmonary function often result in refractory hypoxemia. This was previously referred to as secondary drowning. Finally, alveolar rupture and free air in the thorax have been observed after drowning. This may result from airway bronchospasm or forced expiration against bronchi that are filled with fluid and debris.
) abnormalities and intrapulmonary shunt. Pulmonary aspiration also may trigger an inflammatory response in the alveolar capillary membranes and progress to acute respiratory distress syndrome (ARDS). These changes in pulmonary function often result in refractory hypoxemia. This was previously referred to as secondary drowning. Finally, alveolar rupture and free air in the thorax have been observed after drowning. This may result from airway bronchospasm or forced expiration against bronchi that are filled with fluid and debris.
Cardiovascular system
Hypoxic cardiac injury is first manifest by severe bradycardia, followed by mechanical asystole with pulseless electrical activity and eventual electrical asystole. In the majority of submersion events in which cardiopulmonary arrest has occurred, permanent cardiac dysfunction is rare [Ibsen and Koch, 2002]. After return of spontaneous circulation (ROSC), the cardiovascular system often is characterized by a transient state of low cardiac output. There also is elevated systemic vascular resistance caused by intense vasoconstriction from release of endogenous catecholamines and hypothermia. These hemodynamic changes mimic those of cardiogenic shock, and high after-load in the presence of sustained myocardial ischemia may produce cardiac failure with high ventricular filling pressures. After cardiopulmonary arrest, the two most common dysrhythmias encountered at the scene or during warming in children are sinus bradycardia and cardiac asystole. However, 20–30 percent of drowning victims present with ventricular tachycardia or fibrillation [Ibsen and Koch, 2002]. The presence of ventricular dysrhythmias may reflect a shorter period of hypoxia-ischemia and offer a narrow opportunity for improved cerebral salvage if rapidly converted to a pulsatile rhythm [Young and Seidel, 1999]. There are several case reports of submersion events associated with congenital forms of long QT syndrome. Typically, these children present with wide QRS complex tachydysrhythmias, like torsades de pointes.
Electrolytes
Clinically significant changes in blood volume or electrolyte concentrations occur only if large volumes of aspirated fluid (11–22 mL/kg) are absorbed into the systemic circulation (Hasibeder, 2003]. Typically, drowning victims aspirate small volumes (3–4 mL/kg), causing only mild to moderate changes in electrolyte concentrations. In most reports of submersion accidents, electrolytes are measured in the normal physiologic range [Habib and Perkin, 2008]. There is little evidence to demonstrate a significant electrolyte difference between fresh- and salt-water emersion. Aspirated fresh water may enter the circulation and cause hypervolemia and mild hemodilution with an increased risk of red cell hemolysis and hyperkalemia. Aspirated salt water rarely may cause clinically significant hypovolemia and hemoconcentration.
Hypothermia
Next to immediate resuscitation, hypothermia is the only known effective measure to reduce hypoxic brain injury. The definition of hypothermia is a core body temperature of less than 35°C (95°F). The current record for longest cold-water submersion with an intact neurological recovery is 66 minutes in a 2½-year-old female with core body temperatures as low as 13.7°C (57°F) [Suominen et al., 1997]. The key to hypoxic cerebral survival may be associated with cerebral hypothermia. Brain metabolism and cerebral oxygen demand is reduced by roughly 7 percent per degree Celsius drop in body temperature. At 30°C, cerebral metabolic rate is reduced by nearly 50 percent and the brain can tolerate no blood flow for about 20 minutes [Suominen et al., 2002]. Under controlled conditions in the operating room, induction of deep hypothermia (18–25°C) can protect the brain from hypoxic injury for as long as 70–110 minutes [Suominen et al., 2002]. In cold-water drowning, cerebral preservation occurs naturally when the brain becomes hypothermic before asphyxial cardiac arrest. The critical factor for cerebral outcome is the rate of cerebral cooling and the overall temperature of the brain before arterial hypoxia has fallen to injurious levels [Suominen et al., 2002].
In warm-water drowning, hypoxia and asphyxial cardiac arrest usually develop before or during cerebral cooling, which does not assure protection from anoxic brain damage. Under these conditions, the presence, in the emergency department, of a rectal temperature consistent with severe hypothermia is thought to reflect prolonged exposure to anoxia, and equilibrium with ambient water temperatures, and usually correlates with extensive neurological injury [Graf et al., 1995]. Even in warm-water environments (>20°C), it is not uncommon for a child who is under water for approximately 5 minutes to have a core body temperature in the range of 32–34°C when removed from the water [Habib and Perkin, 2008]. Other environmental and clinical factors that affect initial temperature in the emergency department include the ambient water and air temperatures, duration of exposure, and the time elapsed during rescue and transport to the emergency department. Therefore, relative hypothermia is common in warm-water drowning, and historical information related to the circumstances of the submersion event (water temperature, duration of submersion, and time elapsed during resuscitation and transport) must be considered when deciding issues of resuscitation.
Outcomes and Prognosis
Outcomes
In the United States, nearly three-quarters of all submersion victims will survive neurologically intact [Habib and Perkin, 2008]. Between 25 and 30 percent of children will have a poor outcome, defined as either death (17 percent), or survival in a persistent vegetative state (12 percent). Asphyxia leads to cardiac arrest in 20–30 percent of drowning accidents and prognosis for these victims is much worse [Christensen et al., 1997]. Approximately 40–50 percent of these victims will have ROSC and one-quarter will survive to hospital discharge, but only 6 percent will survive neurologically intact. Methods devised to identify those submersion victims who will have a good versus a poor outcome are not reliable. The use of prognostic variables to guide aggressive resuscitation or withdrawal of support is weakened by reports of patients who have intact recovery despite many unfavorable prognostic indicators [Lavelle and Shaw, 1993].
Prognosis
Historical, clinical, and laboratory variables have been unable to predict reliably, with 100 percent specificity, those victims who will have a poor neurological outcome. The most dependable predictors of outcome following warm-water submersion are factors that reflect the duration and extent of hypoxia and ischemia at the scene or in the field [Quan and Kinder, 1992]. Those victims who have prolonged submersion times or require CPR for cardiac arrest will have the poorest outcomes. If the duration of submersion exceeds 25 minutes and the victim requires CPR for cardiac arrest, the intact survival rate approaches zero. Likewise, neurological outcome after more than 25 minutes of effective CPR is uniformly poor. However, most drowning events are unwitnessed, and the length of submersion or the onset of resuscitation care (e.g., CPR), as reported by first responders, is generally regarded as an approximate guess [Christensen et al., 1997]. Confusion and emotional issues often make the history of the submersion event unreliable.
The physiologic status of the drowning victim on arrival at the emergency department is an important indicator of prognosis [Habib et al., 1996]. The absence of a palpable rhythm on arrival at the emergency department is a poor prognostic factor. Similarly, the need to continue CPR in the emergency department has been shown to correlate with a poor neurological outcome [Christensen et al., 1997].
More recently, the Pediatric Risk of Mortality Score system (PRISM) was applied in the emergency department to determine death and neurological impairment from submersion accidents in children [Zuckerman et al., 1998]. Fifty victims were evaluated. A PRISM score of more than 25 consistently identified all children that subsequently died or sustained severe neurologic impairment. A PRISM score is based on 14 physiologic parameters, is real-time, and is calculated on data from the initial presentation before interventions in the emergency department affect physiologic variables.
The neurological status of the submersion victim on arrival at the emergency department has been correlated with outcome. On arrival, fixed and dilated pupils are a sign of significant brainstem injury, yet are not consistently associated with poor outcome [Habib and Perkin, 2008]. The Glasgow Coma Scale (GCS) is an index of the level of consciousness and is used as a surrogate for neurological function. More than 80 percent of submersion victims with a GCS score in the emergency department of less than 5 will have a poor outcome [Dean and Kaufman, 1981]. The GCS assessment is dependent upon on-going clinical treatments, such as ventilation, oxygenation, CPR, administration of neurotropic agents, and the elapsed interval of time from submersion to ROSC. Interestingly, a large number (44 percent) of comatose victims in the emergency department will improve their neurological status and GCS during transport from the emergency department to the pediatric intensive care unit [Habib and Perkin, 2008]. Allman’s data showed that 25 percent of comatose victims (16 of 66) initially assessed with a GCS of 3 (post-CPR) in the emergency department continued to improve and survived neurologically intact [Allman et al., 1986]. Except for those victims who have suffered lengthy submersions, or required prolonged CPR, the neurological function on arrival to the emergency department is not static and not reliable enough to make critical decisions about resuscitation.
In general, on arrival at the emergency department, factors strongly associated with unfavorable outcomes include an unwitnessed warm-water submersion, duration of submersion longer than 25 minutes, delayed resuscitation efforts for more than 10 minutes, epinephrine usage at the scene or during transport, a nonperfusing rhythm with on-going CPR in the emergency department, persistent apnea, or coma, unresponsive pupils, initial pH <7.1, and glucose >200 mg/dL [Habib and Perkin, 2008]. Despite these many ominous variables, prognostic formulas are not 100 percent specific (zero false-positive rate) for poor outcomes in the emergency department, or, like the PRISM method, require future prospective validation. On the other hand, good outcomes are very likely when the submersion event lasts less than 5 minutes; duration of CPR is less than 10 minutes; and the victim arrives at the emergency department breathing spontaneously, with a pulsatile rhythm and reactive pupils, and is not comatose.
After a severe drowning event, individual outcomes cannot be reliably predicted in the emergency department; therefore, aggressive cardiopulmonary and cerebral resuscitative efforts should be performed [Christensen et al., 1997]. Neurological outcomes are positively affected by interventions at the scene or in the emergency department, and mental status often will change during this time. About one-third of submersion victims initially will present with clinical findings consistent with moderate degrees of hypoxic injury but whose outcomes are uncertain. This group of children appears to have potential for improved outcomes and may benefit from aggressive resuscitation therapies.
In the pediatric critical care unit, the prognosis for neurologic outcome has also proved difficult to evaluate. The use of brainstem auditory-evoked potentials, cerebral blood flow, and metabolic rate, in combination with initial blood glucose values, have all demonstrated some promise but are not available for widespread use [Ashwal et al., 1990; Fisher et al., 1992].
Studies involving xenon cerebral blood flow measurements revealed that total frontal gray- and white-matter flows, and temporal and parietal gray-matter flows, were significantly decreased in patients who ultimately died, in comparison with those who were normal or survived in a vegetative state [Ashwal et al., 1990]. Cerebral blood flow measurements were not useful in discriminating the quality of life in those who survived.
Proton magnetic resonance spectroscopy and magnetic resonance imaging (MRI) are useful in predicting outcome in near-drowning victims [Auld et al., 1995; Dubowitz et al., 1998; Holshouser et al., 1997; Kreis et al., 1996]. When performed sequentially in occipital gray matter, proton MR spectroscopy provides useful objective information that can significantly enhance the ability to establish prognosis after near-drowning. Patients with poor outcomes appear to have increased lactate and lower metabolic ratios, specifically the N-acetylaspartate (NAA)/creatine and NAA/choline ratios [Holshouser et al., 1997].
It seems that the clinical examination, particularly after a stabilization period of 12–24 hours, provides the best estimate of long-term outcome. In children who manifest no neurologic improvement within the first 3–6 hours after admission to the intensive care unit (i.e., remain comatose, flaccid, and without pupillary response), survival is extremely unlikely. Continuation of aggressive care beyond this point should be considered on a case-by-case basis. Evaluation at 12–24 hours has revealed that failure of return of spontaneous respiration after resuscitation is associated with a 100 percent rate of mortality [Jacobsen et al., 1983]. In addition, abnormal posturing or seizures persisting beyond 12 hours may also indicate a poor prognosis.
In retrospective investigation of 44 children, Bratton et al. [1994] found that that neurologic examination 24 hours after warm-water near-drowning distinguished satisfactory outcome from unsatisfactory outcome (death or dependence on total custodial care). All survivors who were normal or had only mild deficits were awake and initiated spontaneous, purposeful movements within 24 hours after submersion. All children who survived with severe deficits or who died remained comatose 24 hours after submersion.
Christensen et al. [1997], in a retrospective review of 274 near-drowning patients, demonstrated that an observation period of 48 hours was necessary to avoid loss of intact survivors. They suggested the following strategies to maximize the number of intact survivors while minimizing the personal, hospital, and social costs of poor outcome:



Prevention
Quan et al. [1989], in a 10-year study of drowning and near-drowning in Kings County, Washington, found that 89 percent of all victims had no supervision, regardless of whether in a bathtub, pool, or ocean. Adults should be assigned specifically as observers without additional duties or distractions. Home pools should have accessible portable telephones so adults do not have to leave the poolside. If duties call the supervising adult into the house, children should leave the pool and preferably be barred from the area until the adult returns. Most toddlers who drown gain access to the pool from inside the house through doors to the pool area, rather than being engaged in water activities. Fences are the only intervention that has proved effective, reducing pool immersion incidents by 50–70 percent [Present, 1987]. Risk of drowning or near-drowning involving unintended access to an unfenced pool is 3.76 times higher than the risk associated with a fenced pool [Pitt and Balanda, 1991]. There should be four-sided fencing immediately surrounding pools, not at the property line. This kind of fencing allows children access to their yards but isolates the pool.
CPR may produce acceptable outcomes if immersion times are less than 5 minutes and resuscitation time is less than 10 minutes; however, 42 percent of children who drowned in their own home pools were retrieved by a lay person, usually a family member, and CPR was not instituted until emergency personnel arrived [Wintemute et al., 1987]. Immediate resuscitation before the arrival of paramedical personnel is associated with a better neurologic outcome [Kyriacou et al., 1994]. Formal swimming lessons have now been associated with a lower risk of drowning for preschool children aged 1–4 years [Brenner et al., 2009]. This is the age group at greatest risk of drowning and for which the idea of swimming lessons has been most controversial [Rivara, 2009]. The American Academy of Pediatrics Committee on Injury, Violence, and Poison Prevention [2003] has produced a list of 23 recommendations to reduce the incidence of drowning in infants, children, and adolescents. Undoubtedly, the most effective and best-proven intervention to prevent swimming-pool drowning is four-sided fencing with self-closing, self-latching gates. Every coalition and municipality should work with pool contractors, public health officials, and local media to encourage isolation fencing on all new pools, particularly for families with young children. Institution of pool safety upgrades should be encouraged when older homes with pools are sold. Industries should continue to explore aesthetically acceptable fencing and effective self-closures for gates, doors, and sliding doors that lead into pool areas.
Strangulation Injury
Strangulation injuries result from external compression of the neck. Several forms have been described, according to the method of application of compression, including hanging, postural strangulation, manual strangulation, and ligature strangulation [Scott and Wiebe, 1997]. Hanging is “complete” when the suspension point is above the height of the victim and a part of the body is totally suspended in air, and is “incomplete” when the suspension point is below the height of the victim and a part of the body is supported by the floor or other object.
The cause of hanging varies with age. For children younger than 5 years, hanging is almost exclusively accidental [Sabo et al., 1996]. These incidents occur secondary to heads getting caught in crib slats, curtain cords, electric windows, Venetian blinds, or hammock cords, and when clothing or a pacifier ribbon becomes caught on an object. However, homicides in infants have been reported. From ages 5–12, hangings are both accidental and suicidal. After the age of 12, most hangings are suicidal or accidental during autoerotic actions [Krol and Wolfe, 1994; Sabo et al., 1996]. Suicide is the third leading cause of death in adolescents in North America, and hanging is the second most common method of suicide in this age group. Asphyxial games, often referred to as the “choking game” by the media and in the medical literature, are now a recognized thrill-seeking behavior among young people [Andrew et al., 2009; McClave et al., 2010; Cowell, 2009]. Recent reports indicate that knowledge of and participation in such activities is more common and that death can occur, particularly when the activity is solitary in nature. Hanging incidents prompting admission must receive, in addition to supportive care, close scrutiny for the opportunity for behavioral intervention [Andrew et al., 2009].
The mechanism of injury in strangulation and hangings is influenced by the amount of physical force used, how the force was applied, and the duration of the force. Death is a consequence of direct injury to the CNS, immediate cardiac arrest, or compression and injury to the structures of the neck. Direct injury to the CNS results from the drop force when the victim is completely suspended, as in complete hangings. The cervical spinal cord or brainstem is disrupted, and death is instantaneous. In complete or judicial hangings, the ligature knot is located centrally over the occiput with the ligature curved symmetrically around the anterior portion of the neck. This placement may result in partial or complete avulsion of the carotid arteries, laryngeal fractures, and fractures of the verbal bodies and base of the skull. Death from immediate cardiac arrest is thought to be secondary to a massive vagal reflex from bilateral carotid body or vagal sheath stimulation [Scott and Wiebe, 1997].
Compression of neck structures and its sequelae are the most common mechanisms of injury in strangulation and incomplete hangings. The venous drainage of the head and neck is through the jugular system and a series of intercommunicating veins related to the spinal column. Because of their superficial location, minimal external pressure results in occlusion of internal and external jugular veins, with obstruction of venous outflow from the head and neck. Venous obstruction results in ischemia, hypoxia, and loss of consciousness. The victim becomes flaccid, with loss of muscle tone, and compression of the carotid arteries occurs, resulting in increasing ischemia and hypoxia. Depending on the severity and duration of the insult, neuronal damage occurs, with cerebral edema and elevated intracranial pressure [Sabo et al., 1996]. As the glottis and tongue are lifted into the pharynx, loss of muscle tone results in airway occlusion. The carotid arteries above the level of the cricoid cartilage are separated from the surface of the skin by the platysma and sternocleidomastoid muscles and deep cervical fascia. These vessels are not easily occluded by external pressure, but traction injuries can occur, ranging from small intimal tears to total disruption.
The laryngeal skeleton includes the hyoid bone and thyroid and cricoid cartilages. These structures in children are semi-rigid, and calcification is not complete until the third decade. Laryngeal fractures are rare in childhood, but the prevalence of fractures increases with age, coincidentally with calcification of the laryngeal skeleton. The most common laryngeal injuries in childhood are mild and include endolaryngeal edema, hematomas, and mucosal tears. More severe injuries result in extensive edema, hematomas and lacerations, cartilage fractures, and vocal cord paralysis. These injuries may cause airway discontinuity and obstruction. Cervical fractures are also rare in children [Krol and Wolfe, 1994]. Feldman and Simms [1980] reviewed 233 childhood strangulations and did not find cervical or laryngeal fractures or evidence of airway compromise.
Cerebrovascular hemodynamic profiles have been obtained in children with hypoxic-ischemic injuries after strangulation [Ashwal et al., 1991; Hanigan et al., 1996]. In general, the profiles reveal cerebral hyperemia, loss of auto-regulation, and intact but blunted vascular response to PaCO2. Not unexpectedly, the accumulated data demonstrate that absolute values of CO2 reactivity vary considerably, and single-point determination encompassing wide epochs may not reflect the continuous changes in hemodynamics of the cerebrovascular bed to hypoxic-ischemic injury and subsequent treatment. Many patients manifest negative CO2 reactivities in various deep and cortical regions, which may represent redistribution of flow from responsive to nonresponsive vascular beds.
Exaggerated sympathetic stimulation due to strangulation can result in acute reversible myocardial dysfunction mimicking myocardial infarction [Sivanandan et al., 2009].
Abusive Head Trauma
The term abusive head trauma applies to all mechanisms of inflicted CNS injury [Christian and Block, 2009; McKinney et al., 2008]. It is also referred to using such terms as “shaken baby syndrome,” “shaken infant syndrome,” “non-accidental trauma,” and “inflicted traumatic brain injury,” to name a few. Abusive head trauma is a broadened terminology that allows one to account for the multitude of primary and secondary injuries that result from abusive head trauma, some of which contribute to the often permanent and significant brain damage sustained by abused infants and children. Although it was initially described as “shaken baby syndrome,” shaking might not be the sole etiology of brain injury in abusive head trauma. Recent studies suggest that the most severe clinical and histologic manifestations of abusive head trauma are usually related to hypoxic-ischemic encephalopathy [McKinney et al., 2008; Aaen et al., 2010]. Interestingly, a recent article suggests that, in young children (<36 months) presenting with acute head trauma, hypoxic-ischemic encephalopathy more commonly occurs in abusive head trauma than in accidental head trauma [Ichord et al., 2007].
Patients with severe traumatic head injury experience immediate alterations in cerebral and systemic physiology that substantially influence neurologic outcome [Atkinson, 2000]. Two immediate pathophysiologic events occur with the onset of severe head injury that substantially affect subsequent outcome: head injury-induced apnea and a stress-related massive sympathetic discharge. The combined effects of hypoxia, hypercarbia, and acidosis and a surge in blood pressure, as well as the direct effects of catecholamines on tissue, lead to a synergistic injury effect in the patient.
The extent of apnea and catecholamine surge after severe head injury is directly related to the amount of energy transmitted to the brainstem [Atkinson, 2000]. As a result, many patients with severe head injury live or die at the scene, depending on whether they resume breathing. In patients who resume spontaneous respiration, apnea-induced hypoxic brain and cardiac injury augmented by elevated stress hormones may result in serious morbidity.
Johnson et al. [1995] examined intentional closed-head injury (i.e., child abuse) in 28 children and found that 57 percent had a verifiable history of apnea before hospitalization. They concluded that trauma-induced apnea causes cerebral hypoxia and ischemia, and these were more important in modulating outcome than was the mechanism of primary brain injury (i.e., subdural hematoma, subarachnoid hemorrhage, diffuse axonal injury, or contusions).
Geddes et al. [2001] identified a high incidence of widespread microscopic neuronal hypoxic brain damage in a cohort of children with abusive head trauma. In a proportion of infants, they found significant microscopic changes of focal axonal damage to the craniocervical junction on the cervical cord. For infants who presented with a history of apnea and hypoxic brain damage, they proposed a mechanism of cervical hyperextension and flexion during shaking, which led to damage of the brainstem respiratory centers with a consequent bad outcome from severe hypoxic brain damage.
Similarly, Oehmichen et al. [2008] concluded that death in inflicted brain injury results from “a global cerebral ischemia secondary to subdural hematoma, focal vasospasm, and trauma-induced respiratory and/or circulatory failure.” Additional recent evidence further confirms that, in abusive head trauma, hypoxic-ischemic injury is of greater importance with regard both to signs and symptoms at presentation and to long-term outcome [Kemp et al., 2003; Newton and Vandevan, 2009; Aaen et al., 2010; Ashwal et al., 2010]. These issues are discussed in more depth in chapter 75.
Lightning and Electrical Injuries
Lightning is an uncommon form of electrical injury. Only 150–300 people die each year in the United States of injury secondary to lightning strike; the death rate is highest in adolescents [Fontanarosa, 1993; Volinsky et al., 1994]. Nearly two-thirds of people struck by lightning survive, although many have significant sequelae.
The lightning strike is direct-current electrical discharge with a high magnitude of energy and short duration of exposure. When the electrical discharge in a cloud is greater than the air resistance (30,000–50,000 V), energy is discharged to the ground. Lightning energy is estimated to be about 25,000–200,000 A, or 100 million to 2 billion V. The duration of the electrical discharge is from 0.00001 to 0.001 second. The estimated heat generated from the electrical discharge is between 8000°C and 30,000°C at the contact area [Patten, 1992]. Survival from lightning strikes is possibly related to the short duration of electrical contact. With the presence of metal objects on the body, however, the electrical contact time is increased, resulting in a deeper burn and increased rates of morbidity and mortality. Thermal forces generated by lightning are capable of producing tremendous pressure waves from the rapid expansion and cooling of the air around the lightning strike. These pressure waves are significant enough to cause serious blunt-force trauma.
Lightning strike may cause injury as a result of one of three types of energy transfer: electrical, thermal, or mechanical. The electrical injury depends on the voltage, amperage, degree of resistance, and anatomic pathway. Electrical energy follows the path of least resistance. Because tissues that have a low water and electrolyte content have a higher resistance, tissue resistance decreases in the following order: bone, fat, tendon, skin, muscle, blood vessel, and nerve [Gibbs and Keel, 2003]. The most important resistor to the flow of current is skin. When the skin is dry, the electrical path is deflected over the body because of the higher resistance of the skin. The thermal energy produced may cause a break in the skin, forming a pathway for the electrical charge to pass through to less resistant tissues, such as muscle, nerves, and blood vessels.
The primary effect of lightning results from a massive direct-current insult to the entire myocardium and CNS. This insult leads to asystole and loss of function of the medullary respiratory centers [Courtman et al., 2003]. Lightning effects on the respiratory center last longer than the cardiac effects. Cardiac rhythm returns if apnea is corrected; if apnea is not corrected, secondary hypoxic cardiac arrest ensues [Courtman et al., 2003]. The mortality from lightning is related to the effects of apnea, not asystole. Resuscitation attempts in lightning victims have resulted in higher success rates than in victims of other cardiac dysrhythmias. The direct current of lightning causes a single countershock to the entire myocardium, as opposed to alternating current, which distributes a repetitive fibrillating stimulus to the heart. The resumption of spontaneous electrical activity may be secondary to the automaticity of the myocardium. Patients with initial rhythms associated with a poor prognosis may recover completely with prompt institution of respiratory and cardiac support. Aggressive and persistent resuscitation efforts should be continued until cerebral function can be assessed.
Anoxic cardiac and cerebral injury may be reduced in lightning injuries caused by a delay in anoxic cellular destruction. Several authors have suggested that the reason for possible recovery after prolonged cardiac arrest is that lightning causes cellular metabolism to cease instantly, thereby delaying the onset of anoxic tissue destruction and reducing the potential for anoxic cardiac and cerebral injury [Fontanarosa, 1993]. However, the metabolic basis for this concept has not been verified, and available clinical evidence does not support the concept that lightning strike confers any cellular protective mechanism.
The CNS is highly sensitive to electrical injury. Injury to the CNS may result from direct electrical injury or anoxia, or may be secondary to vascular injury [Patten, 1992]. The pathophysiologic mechanism of CNS injury includes coagulation in the cortex, formation of epidural or subdural hematomas, paralysis of the respiratory center, and intraventricular hemorrhage [Cherington, 2002]. Symptoms of CNS injury include loss of consciousness, confusion, transient disorientation, seizures, restlessness, retrograde amnesia, and varying degrees of transient paralysis. Coma and symptoms of cerebral edema are not unusual. Transient paralysis may involve the lower extremities in two-thirds of victims and the upper extremities in one-third. Late sequelae include amnesia, paraplegia, neuropathy, decreased reflexes, and chronic subdural hematomas. The presence of sequelae may be related to hypoxic or blunt-force CNS injury.
Severe head trauma can occur as a consequence of falling, or being thrown to the ground by the enormous blast force of a thunderous lightning strike, or as a direct effect of the lightning strike [Cherington, 2002]. Blunt head trauma patients often present with subarachnoid and intraventricular hemorrhages, as well as mass lesions of epidural and subdural hematomas [Cherington, 2002].
Electrical Shock
More than 20 percent of all electrical injuries occur in children [Fontanarosa, 1993; Thompson and Ashwal, 1983]. Risk-taking behavior among adolescent boys, such as climbing utility poles, playing near electrical railway lines, and trespassing through transformer substations, can expose them to high-tension electrical sources. The inquisitive nature of toddlers and their habit of exploring their environment with their mouths contribute directly to the most common electrical injury in childhood, the perioral burn, which affects 4000 children per year.
Low-voltage alternating current (60 Hz) has a narrow margin of safety [Fontanarosa, 1993]. Low-intensity current at 1–4 mA is perceived as an unpleasant tingling sensation. Contact with alternating current at 10–20 mA causes repetitive stimulation of muscle fibers, produces tetanic skeletal muscle contractions, which may prevent the victim’s hand from releasing its grasp from the current source, and lengthens the duration of contact with the energized source. Thoracic muscle tetany involving the diaphragm and intercostal muscles occurs at 30–50 mA and can result in respiratory arrest. Prolonged contact leads to sustained apnea with hypoxemia and secondary cardiac arrest. Ventricular fibrillation may occur with exposure to as little as 50–100 mA. The repetitive frequency of alternating current increases the likelihood of current delivery to the myocardium during the vulnerable recovery period of the cardiac cycle and can precipitate ventricular fibrillation.
Respiratory arrest may last minutes to hours after the electrical shock terminates and may persist after restoration of spontaneous circulation. In some patients, cardiac activity may be maintained in the absence of spontaneous ventilation. If respiratory arrest is not corrected promptly by artificial ventilation and oxygenation, secondary hypoxic cardiac arrest may occur.
Sudden Cardiac Arrest in Children and Adolescents
Sudden cardiac arrest is the sudden cessation of cardiac activity, so that the victim becomes unresponsive with no normal breathing and no signs of circulation. Unless the victim receives immediate CPR and other treatment to restore normal cardiac activity, he or she will die. Although the precise incidence of sudden cadiac arrest in children is unknown, it is not a leading cause of death [Hazinski et al., 2004].
When sudden cardiac arrest does occur in children and adolescents, it may be precipitated by ventricular fibrillation or rapid ventricular tachycardia (pulseless ventricular tachycardia). Ventricular fibrillation occurs in 5–15 percent of all pediatric victims of out-of-hospital cardiac arrest, and is reported in up to 20 percent of pediatric in-hospital arrests at some point during the resuscitation [American Heart Association, 2005]. The incidence increases with age. These abnormal heart rhythms in children are typically caused by inherited or congenital cardiac conditions or by acute medical problems that cause inflammation of the heart. Examples of such conditions include long QT syndrome, hypertrophic cardiomyopathy, abnormal development of the coronary arteries, aortic dissection, myocarditis, coronary artery aneurysm associated with Kawasaki’s disease, and congenital aortic stenosis [Wu et al., 2009]. Many of these conditions are not detected during routine screening for school physical examinations or sports activities; thus, sudden cardiac arrest may be the first sign of these conditions [Maron, 2003].
Sudden cardiac arrest may also result from commotio cordis, a condition resulting from a blow to the chest that causes ventricular tachycardia or fibrillation. Each year approximately 10 cases of this condition are reported in the United States for victims of all ages [Maron et al., 2002].
If a child develops sudden cardiac arrest caused by ventricular fibrillation or pulseless ventricular tachycardia, immediate CPR by a bystander and early defibrillation are needed. Automatic external defibrillators are computerized defibrillators designed for use by lay rescuers to treat sudden cardiac arrest. The automatic external defibrillator provides voice and visual prompts to guide the rescuer. When attached to an unresponsive victim in cardiac arrest, the automatic external defibrillator analyzes the victim’s heart rhythm and determines whether a shock is needed, charges to an appropriate shock dose, and prompts the rescuer to deliver a shock. The automatic external defibrillator delivers a shock only if ventricular fibrillation or ventricular tachycardia is present [Hazinski et al., 2004].
The U.S. Food and Drug Adminstration has cleared automatic external defibrillators for use in children younger than 8 years. Two of these devices have accurately identified ventricular fibrillation and rapid ventricular tachycardia in young children [Atkinson et al., 2003]. The American Heart Association states that automatic external defibrillators may be used with CPR for treatment of prehospital cardiac arrest in children 1–8 years of age [Hazinski et al., 2004]. For children 1–8 years of age, the rescuer should use a pediatric dose-attenuator system, if one is available [American Heart Association, 2005].
Neurologic Complications After Cardiac Arrest
Delayed Posthypoxic Injury
Delayed posthypoxic injury may occure 24–48 hours after the initial recovery of consciousness after coma. In children, this injury occurs most frequently after near-drowning accidents or cardiac surgery, in which profound hypothermia and either low-flow bypass or circulatory arrest was used. MRI studies may reveal evidence suggestive of edema or demyelination, which may be postanoxic or osmotic in etiology. In addition, diffusion-weighted imaging may reveal restricted diffusion, typically in the basal ganglia, cerebellum, and cortex. These changes are seen initially in gray matter and later in white matter [Bydder and Rutherford, 2001].
Post-Ischemic Seizures
Excessive excitatory transmitter release during brain ischemia can lead to paroxysmal cortical discharges, especially in the hippocampus, beginning within minutes and lasting several hours despite cerebral reperfusion. Repetitive discharges may progress to clinically overt seizures, and suppression is then necessary to prevent further injury. Seizures, including episodes of convulsive and nonconvulsive status epilepticus, after anoxic injury are associated with poor outcome and persistent unconsciousness [Jumas and Brenner, 1990; Krumholz et al., 1988]. The large study by Krumholz et al. [1988] showed that seizures or myoclonus per se are not significantly related to outcome, but convulsive status epilepticus and myoclonic status epilepticus confer poor outcome, as judged by survival and recovery of consciousness (Table 76-5). In addition, electrographically documented status epilepticus without somatic motor manifestations has also been suggestive of poor outcome [Simon and Aminoff, 1986].
Table 76-5 Survival and Consciousness at Discharge of Comatose Patients After Cardiopulmonary Resuscitation
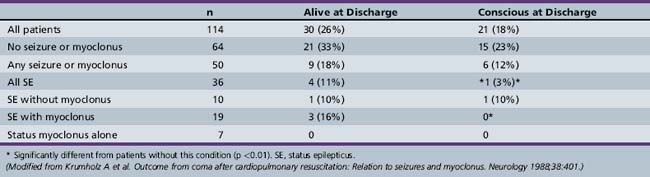
Status epilepticus can itself result in permanent anoxic brain damage and necessitates immediate attention. Clinical seizures should be treated aggressively with benzodiazepines and phenytoin. If these agents are unsuccessful, barbiturates may be added. The disadvantage of barbiturates in this setting is that they will further depress the sensorium and make prognostication difficult. Levetiracetam and valproic acid also may be considered, as several recent studies in pediatric status epilepticus demonstrate their safety and efficacy [Kirmani et al., 2009; Mehta et al., 2007; Abend et al., 2008].
Post-ischemic seizures account for 5–10 percent of cases of childhood status epilepticus [Pellock, 1994]. Children who have seizures should be treated aggressively; EEG is indicated to guarantee that seizure activity is ablated. Nonconvulsive status epilepticus may exist after apparently successful treatment of convulsive status epilepticus [Perkin et al., 1996]. A potential risk of intensive care unit therapy is failure to recognize status epilepticus in the critically ill, paralyzed patient with injury [Michelson and Ashwal, 2004; Weise and Bleck, 1997]. This clinical setting may pose significant risk for increased morbidity in the already compromised patient; continuous encephalographic monitoring may be warranted if paralysis is required for cardiorespiratory management.
Delayed Postanoxic Myoclonic Seizures
Delayed postanoxic myoclonic seizures may occur after severe anoxic brain injury. Myoclonus usually appears within the first 24–48 hours after cardiac arrest and is generalized, involving all voluntary muscles. The EEG is usually abnormal, with generalized synchronous spike discharges or burst-suppression activity. Control of the myoclonic seizure activity is difficult with phenytoin or phenobarbital, and treatment with intravenous benzodiazepines is frequently necessary. The presence of postanoxic myoclonus is usually indicative of a high risk for long-term neurologic sequelae [Krumholz et al., 1988].
In one study of comatose adult patients after cardiac resuscitation, myoclonic seizures developed in 37 percent and all of those patients died [Wijdicks et al., 1994]. The authors concluded that myoclonic status epilepticus in comatose survivors after cardiac resuscitation indicates devastating anoxic brain injury. Myoclonic status epilepticus should be considered an agonal phenomenon, and its presence should strongly influence the decision to withdraw life support.
However, the decision to withdraw supportive care in patients with postanoxic myoclonic status epilepticus should be based on more precise predictors of outcome, such as somatosensory-evoked potentials and serial neurological examination [Young, 2009]. If continued supportive management is desired because of family choice or prognostic uncertainty, and standard anticonvulsant medications have failed to ameliorate myoclonic status epilepticus, the decision to pursue pharmacological coma or neuromuscular blockade must be individualized. Because most myoclonic status abates within 48 hours, it may be practical to use anesthetic agents and neuromuscular blockade for this period of time without prolonged loss of the ability to examine the patient [Krumholz et al., 1988].
Neurologic Prognosis After Cardiac Arrest
Currently, the most powerful individual predictor of neurologic outcome after cardiorespiratory arrest is the neurologic examination. Sequential observations of a patient’s breathing pattern, eye movements, body posture, and reflexes provide essential information to help establish expectation of outcome [Trubel et al., 2003]. Levy et al. [1985] applied multivariate analysis to variables from the neurologic examination in a series of 210 comatose adults studied after hypoxic-ischemic insults. Of the 15 patients who were vegetative at 1 month, none regained independent function. The oculocephalic response and the 3-day motor response enabled the investigators to identify all patients who gained independent function. This identification was even true for 2 of the 57 patients who remained in eyes-closed coma for 3 days and then ultimately regained independent function. Edgren et al. [1987] applied the GCS scores of less than 6 at 3 days after cardiac arrest and showed that they were predictive of poor outcome at 6 months, with no false-negative results. A study involving 109 comatose children with hypoxic-ischemic encephalopathy documented that the absence of a motor response on the third day after admission was associated with an unfavorable outcome [Beca et al., 1995]. Nevertheless, in 23 percent of the children, clinical evaluation was difficult because of neuromuscular paralysis or heavy sedation. This reinforces the relevance of other early prognostic factors, such as electrophysiologic data.
Although these studies did not include patients treated with hypothermia, a small study of 37 initially comatose patients who underwent hypothermia showed recovery of awareness (on day 6) in 2 of 14 patients who had a motor response that was no better than extensor posturing on day 3 [Al Thenayan et al., 2008]. Thus, caution is warranted in relying on the motor response alone before day 6 (or possibly later) for patients who have received hypothermia therapy [Young, 2009].
The absence of a pupillary reaction to light suggests a poor prognosis but has unclear specificity when assessed early after a cardiac arrest. In prospective studies involving 491 patients, 152 of whom had absent pupillary light reflex on hospital admission, false-positive rates (i.e., outcome better than poor despite a positive test result) ranged from 0 to 31 percent. However, all 108 patients whose pupillary reactions were absent at day 3 after cardiac arrest had poor outcomes [Young, 2009].
After review of existing evidence in adults in coma, the American Academy of Neurology stated that outcome is invariably poor when there is absent pupillary light reflex, absent corneal reflex, and no or only extensor motor response to pain 3 days after cardiac arrest [Wijdicks et al., 2006].
Although a few patients are thought to “defy the textbooks,” they likely represent cases of asphyxia without cardiac arrest, coma of an origin other than asphyxia-induced cardiac arrest, unrecognized hypothermia, or an associated overdose of a cerebrally protective drug. Although the GCS score was not predictive of outcome when applied after asphyxia-induced cardiac arrest in pediatric patients, its predictive power was tested with only a single score on initial presentation [Dean and Kaufman, 1981]. Careful studies to determine which components of the neurologic examination in children after hypoxic injury correlate best with outcome have not been done [Jacinto et al., 2001; Kriel et al., 1994; Trubel et al., 2003].
Electroencephalography
The EEG also can provide prognostic information after cardiopulmonary arrest. Chen et al. [1996] retrospectively examined the relation between first postcardiac arrest EEG and clinical outcome in 408 adult cases. A five-grade classification was used to categorize EEGs. Although permanent severe neurologic damage was observed in some patients with grade I EEG, none of the 208 patients with grade IV or V (i.e., severely abnormal) EEGs had a good neurologic recovery. Adjunctive prognostic information also can be derived from cerebrospinal fluid enzyme elevations of the brain isoenzyme of creatine phosphokinase or enolase. The neurologic examination, EEG, and cerebrospinal fluid neuron specific enzyme levels, used together, could provide powerful prognostic information as early as 48–72 hours after resuscitation.
Before a study by Mandel et al. [2002], there was no prospective research on the pediatric population in which the relation between early EEG criteria and long-term outcome was analyzed. In their study, the first EEG was obtained within the first 24 hours after admission. Among their population of children who were still comatose at 24 hours, the presence of a discontinuous tracing and the absence of reactivity were indicative of an unfavorable outcome, as was also observed by Chen et al. [1996]. The presence of spike or epileptiform discharges was always associated with an unfavorable outcome. EEG findings must be interpreted cautiously because they may be altered by sedative medication. Review of all available data to date suggests that specific findings on EEG may be helpful but do not have sufficient prognostic accuracy [Haque et al., 2008].
Somatosensory and Auditory-Evoked Potentials
Evoked potential monitoring has been used in an attempt to provide early prognostic information about outcome after CPR. Evoked potentials are inexpensive and can be obtained within the intensive care unit; their advantages over EEG are that the responses are not affected by sedative administration and they are insensitive to electrical noise in the environment. Madl et al. [1996] tested somatosensory-evoked potentials in 441 adults after successful resuscitation from cardiac arrest. They reported that 20 percent of the patients had no N20 response, and observed that the presence or absence and latency of the cortical N20 peak reliability differentiated between bad and favorable outcome with 100 percent predictive ability. They also found that a preserved N20 peak was not useful for discriminating whether an individual patient would survive. Testing was performed within 48 hours of resuscitation, which is a clinically relevant time frame. Beca et al. [1995] used somatosensory-evoked potentials in children within 48 hours after different neurologic injuries (e.g., trauma, ischemia, meningitis). They demonstrated predictive power of 100 percent for good outcome in patients with normal somatosensory-evoked potentials and 100 percent prediction for poor outcome with absence of somatosensory-evoked potentials in 99 children tested, when they excluded certain cases in which there was a physical barrier that impeded cutaneous reception of the electrical impulse (e.g., subdural effusions). A systematic review of studies of somatosensory-evoked potential results in comatose patients enables estimation of outcomes with a higher degree of certainty than is available from individual studies [Robinson et al., 2003]. Adults in coma from hypoxic-ischemic injury with absence of somatosensory-evoked potential responses have a less than 1 percent chance of awakening. However, in children, exceptions to this rule occur; of the 7 percent who awaken despite absence of evoked responses, the majority have an outcome better than a severe disability [Robinson et al., 2003].
Brainstem auditory evoked testing has been used in children who experienced cardiac arrest after submersion accidents [Fisher et al., 1992]. Normal evoked responses were observed in all children who recovered neurologically intact. Children who recovered with significant handicaps demonstrated reduction in wave V amplitude over time and prolonged wave I–V interpeak latencies.
Of interest and importance, initial data suggest that somatosensory evoked potentials still provide prognostic information when hypothermia is utilized, although the velocity of conduction is slowed modestly [Bleck, 2006].
The predictive powers of the clinical examination, EEG, and somatosensory evoked potentials in determining the prognosis of patients in anoxic coma have been evaluated and compared in adult patients to identify combinations that may improve the accuracy of prediction. In one study, clinical examination correctly predicted outcome in 58 percent of patients, somatosensory evoked potentials in 59 percent, and EEG in 41 percent [Basetti et al., 1996]. The combination of clinical examination, somatosensory evoked potentials, and EEG raised the percentage of correct predictions to 82 percent. Another report of 34 adult patients with anoxic coma revealed that patients with extensor posturing to painful stimulation and a “malignant” EEG, or those with flexed posturing and bilateral absence of somatosensory evoked potentials, on day 3 or later, had poor outcomes [Chen et al., 1996]. These findings are important and suggest that evoked response measurement can provide important adjunctive information to aid in decision making after asphyxia-induced cardiac arrest (Figure 76-6). However, decisions regarding continuation or foregoing of life-sustaining therapies should be based on all of the available clinically relevant information.
The objective of establishing the most accurate information possible is essential in the discussion with the family. It is currently recommended that maximal support be given to the child for the first 24 hours. After this time, the association of the clinical factors (GCS score <5, absence of spontaneous respirations and pupillary reflexes) and the presence of electrophysiologic factors (discontinuous activity in the EEG, absence of EEG reactivity, and abolition of the N20 wave) are highly predictive of death or severe long-term disability [Mandel et al., 2002].
Biochemical Markers
Biochemical variables in the cerebrospinal fluid, such as lactate, creatine phosphokinase BB-isoenzyme, lactate dehydrogenase, and neuron specific enolase, have been investigated as early prognostic markers of neurologic outcome after cardiac arrest [Fogel et al., 1997; Garcia et al., 1994; Karkela et al., 1992]. Examination of the cerebrospinal fluid in the early phase after cardiac arrest may be hazardous, particularly if there is the possibility of an intracranial mass lesion. Moreover, serial examinations of cerebrospinal fluid might be difficult to justify. Therefore, valid biochemical markers of outcome in serum must be established.
Several proteins synthesized in astroglial cells or neurons have been proposed as serum biochemical markers of cell damage in the CNS [Qureshi, 2002]. The ideal serum markers should have high specificity and sensitivity for brain injury, be released only after irreversible destruction of brain tissue, have a rapid appearance in serum, and have temporal correlation with the onset of injury. The variability in measurement should be low, to ensure a predictable relationship between the serum concentration and the amount of brain injury. The marker should be able to provide prognostic information regarding brain injury. Furthermore, reliable assays for immediate analyses should be available [Qureshi, 2002].
With regard to reliable biochemical markers of brain injury, increased serum levels of the low-molecular-weight protein S-100B have been reported in cases of cardiac arrest and traumatic brain injury [Mussack et al., 2002; Spinella et al., 2003]. Urinary S-100B protein concentrations are used as a marker for brain damage in asphyxiated infants [Gazzolo et al., 2004]. The biologic role of S-100B protein, a calcium-binding protein found in the nervous system, is still unclear. Its half-life is about 1 hour, and it is excreted mainly in the urine [Gazzolo et al., 2004]. Increased S-100B concentrations in biologic fluids are a reliable marker of brain damage in adults, infants, and fetuses [Gazzolo et al., 2004]. The short half-life of S-100B protein makes it an interesting marker for longitudinal monitoring. Elevated S-100B levels 12 hours after cardiac arrest are suggestive of severe global cellular brain damage, which is correlated with an unfavorable outcome after 12 months [Mussack et al., 2002]. In one analysis, neuron-specific enolase concentrations in serum samples were correlated with patient outcome after cardiac arrest [Fogel et al., 1997]. A serum neuron-specific enolase concentration of 33 ng/mL, determined up to day 3 after cardiac arrest, can predict poor neurologic outcome.
Neuroimaging
Studies have indicated that normal results of head CT performed immediately after anoxic injury are not correlated with outcome [Han et al., 1990; Romano et al., 1993]. However, abnormal results obtained in the initial hours after injury are associated with a poor prognosis.
Patients with anoxic-ischemic insults may exhibit a variety of findings on CT of the brain. In a study of 53 patients (all ages) scanned 3 days after an out-of-hospital cardiac arrest, clinical differences were found between patients who had cerebral edema and those who did not [Morimoto et al., 1993]. In all, 23 of 25 patients with cerebral edema had cardiac arrest as a result of respiratory distress, whereas this finding was true in only 5 of 28 patients without cerebral edema. Initial arterial pH was significantly lower in patients with cerebral edema, and significantly more of these patients remained comatose or were diagnosed as brain-dead. Cerebral edema was apparent in 9 of these 25 patients when scanned on the first day of hospitalization. Over the next 2 days, low-density areas involving the basal ganglia, midbrain, and thalamus developed in 22 of the 25 patients. Eventually, loss of the normal gray–white matter differentiation was seen in all patients. It was suggested that brain swelling in the early postresuscitation period might be one of the predictors of a poor neurologic outcome in patients who experience an out-of-hospital cardiac arrest [Morimoto et al., 1993].
Although basal ganglia injury is a well-known cerebral effect of hypoxic-ischemic injury, documentation comes from postmortem studies, with only sporadic reports in live patients [Jacobs et al., 1990]. MRI makes it possible to detect lesions in the basal ganglia after cardiopulmonary arrest, which are correlated with the development of movement disorder. The course of the clinical signs is correlated with the presence of abnormalities on MRI [Jacobs et al., 1990; Wallays et al., 1995], as well as with the results of diffusion-weighted imaging.
Recent developments in brain MRI – in particular, diffusion-weighted MRI – provide a new and potentially powerful marker of early global ischemic brain injury. Preliminary studies have shown widespread diffusion-weighted MRI abnormalities in poor-outcome post-cardiac arrest patients during the first week [Wijman et al., 2009].
Magnetic Resonance Spectroscopy
Proton MR spectroscopy is clinically useful for identifying the biochemical state of the CNS noninvasively, as reviewed in Chapter 11. It has been used to study patients after acute nontraumatic and traumatic injuries [Ashwal et al., 1995, 1997; Ross and Michaelis, 1994]. Pediatric patients with severe closed-head injuries or strokes with severe long-term neurologic sequelae exhibit reduced NAA peaks and reduced NAA/creatine ratios [Ashwal et al., 2000, 2006]. Adults with cardiac arrest, elevated brain lactate levels on proton MR spectroscopy, and absence of N20 somatosensory-evoked responses had a poor prognosis if they were still alive 4 weeks after injury [Berek et al., 1995].
Several studies have addressed the metabolic changes in response to acute cerebral injury in children through the use of proton spectroscopy [Holshouser et al., 1997]. Proton MR spectroscopy has been used to evaluate the prognosis of pediatric near-drowning victims [Dubowitz et al., 1998; Kreis et al., 1996]. Of 16 children reported, those with excessive lactate levels universally had a poor outcome, usually surviving in a vegetative state. Lactate level was also more likely to peak by day 4 rather than immediately after injury. Holshouser et al. [1997] demonstrated the potential value of proton MR spectroscopy in predicting outcome after acute CNS injuries in 82 infants and children. In this study, changes in metabolite ratios and the presence or absence of lactate, in combination with clinical and MRI score data, predicted outcome correctly in 91 percent of CNS-injured neonates and in 100 percent of CNS-injured infants and children. Poor-outcome groups had lower GCS scores, longer periods of unconsciousness, and lower NAA/creatine and NAA/choline ratios, and lactate was more likely to be present. Several clinical examples of children with acute brain injuries and abnormal proton spectra that demonstrate the clinical utility of MR spectroscopy are depicted in Figure 76-7. MR spectroscopy has also been found useful for outcome prediction after accidental [Ashwal et al., 2000, 2006] and nonaccidental trauma in children [Aaen et al., 2010; Ashwal et al., 2010].
A, Spectra from occipital cortex in a normal 21-month-old female, displaying the increases in N-acetyl aspartate (NAA) and in creatine-phosphocreatine (Cre), with decreases in choline (Cho) and myoinositol (mI) that are seen in comparison with younger patients. (See Chapter 11 for spectra in younger children.) Glx, glutamine. B, Spectra from the occipital cortex in an 18-month-old female with severe post-hypoxic-ischemic injury on day 4 after a near-drowning injury with a normal T2-weighted MRI at the same time. The decreases in NAA and Cre, with increase in Glx, are apparent (echo time [TE] = 20 milliseconds, repetition time [TR] = 3 seconds). This child made some recovery to a moderate to severe disability. C, Spectra from occipital cortex in a 17-month-old female in a permanent vegetative state resulting from a near-drowning accident. Her spectrum discloses a low NAA peak, which was difficult to measure because of the greatly increased Glx with reversal of the Cho/Cre peaks and the presence of lactate, all of which suggest a poor prognosis (TE = 20 milliseconds, TR = 3 seconds). T2-weighted MRI demonstrates diffuse edema.
(B, Courtesy of Dr. Barbara Holshouser, Department of Radiology, Loma Linda University Medical Center.)
Treatment
As previously mentioned, there are at least four phases of cardiac arrest:
Interventions to improve outcome from pediatric cardiac arrest should be targeted to optimize therapies according to the timing, duration, intensity, and phase of resuscitation, as suggested in Table 76-6. The pre-arrest phase represents the largest potential opportunity to impact patient survival by preventing pulseless cardiopulmonary arrest. Interventions during the pre-arrest phase focus on prevention. Infant safety seats, pedestrian safety programs, and safe driving to prevent traumatic arrests, supine sleep position for SIDS prevention, water safety programs and swimming lessons to prevent drowning arrests, and medication safety caps to prevent drug poisoning arrests are well-known, highly effective efforts to prevent cardiac arrests. Because most pediatric cardiac arrests are caused by progressive respiratory failure and/or shock, the main focus of Pediatric Advanced Life Support (PALS) is the early recognition and treatment of respiratory failure and shock in children (i.e., prevention of cardiac arrest in the pre-arrest phase). Within the hospital, medical emergency teams (rapid response teams) are being formed to recognize and intervene when cardiac arrest is impending [VanVoorhis and Willis, 2009].
| Phase | Interventions |
|---|---|
| Pre-arrest (protect) | Optimize community education regarding child safety |
| Optimize patient monitoring and rapid emergency response | |
| Recognize and treat respiratory failure and/or shock to prevent cardiac arrest | |
| SIDS prevention | |
| Arrest (no-flow) phase response (preserve) | Minimize interval to BLS and ACLS (organized ) |
| Minimize interval to defibrillation when indicated | |
| Low-flow (CPR) phase (resuscitate) | Push hard, push fast |
| Allow full chest recoil | |
| Minimize interruptions in compressions | |
| Avoid over-ventilation | |
| Consider adjuncts to improve vital organ perfusion during CPR | |
| Consider ECMO if standard CPR/ALS not promptly successful | |
| Postresuscitation phase: short term | Optimize cardiac output and cerebral perfusion |
| Treat arrhythmias, if indicated | |
| Avoid hyperglycemia, hyperthermia, hyperventilation | |
| Consider mild postresuscitation systemic hypothermia | |
| Postresuscitation phase: longer-term rehabilitation (regenerate) | Early intervention with occupational and physical therapy Bioengineering and technology interface |
ACLS, Advanced cardiac life support; ALS, advanced life support; BLS, basic life support; CPR, cardiopulmonary resuscitation; ECMO, extracorporeal membrane oxygenation.
(Modified from Topjian AA et al. Pediatric cardiopulmonary resuscitation: Advances in science, techniques, and outcomes. Pediatrics 2008;122:1086–1098.)
Resuscitation
Outcomes following pediatric out-of-hospital cardiac arrest are worse than after in-hospital arrests [Moler et al., 2009]. In particular, neurologic outcomes appear to be much worse among children who are out-of-hospital arrest survivors. Two diseases have especially poor outcomes: traumatic arrests and SIDS [Nadkarni and Berg, 2009]. Traumatic cardiac arrests typically result from either airway compromise and severe, prolonged hypoxia, or exsanguination resulting in profound circulatory shock. Not surprisingly, chest compressions with an inadequately filled heart are not likely to provide adequate coronary and cerebral perfusion. SIDS patients are typically discovered a long time after cardiac arrest and resuscitation efforts are understandably unsuccessful. For other pediatric cardiac arrests in the prehospital setting, CPR and advanced life support from emergency medical service providers may be applied late, after prolonged hypoxia and hypoperfusion of vital organs. Animal and human data both indicate that CPR for children can be very effective. In addition, these data underline the fact that basic life support delivered early is more important than advanced life support delivered late. Prompt action by a citizen bystander in the prehospital setting is generally more effective than late heroic efforts in the intensive care unit [Nadkarni and Berg, 2009].
Interventions during the no-flow phase of untreated pulseless cardiac arrest focus on early recognition of cardiac arrest and the initiation of basic and advanced life support [American Heart Association, 2005]. When there is insufficient oxygen delivery to the brain or heart, CPR should be started. The goal of effective CPR is to optimize coronary perfusion pressure and blood flow to critical organs during the low-flow phase [Paradis et al., 1990]. Basic life support provided by continuous, effective chest compressions (characterized by an effort to push hard, push fast, allow full chest recoil, minimize interruptions, and do not overventilate) is the emphasis of this phase.
One of the most common precipitating events for cardiac arrest in children is respiratory insufficiency. Adequate oxygen delivery to meet metabolic demand and removal of carbon dioxide is the goal of initial assisted ventilation during advanced life support. Effective bag-mask ventilation remains the cornerstone of providing effective emergency ventilation. During CPR, cardiac output and pulmonary blood flow are approximately 10–25 percent of what they are during normal sinus rhythm. Consequently, much less ventilation is necessary for adequate gas exchange from the blood transversing the pulmonary circulation during CPR. Overventilation during CPR is common and can substantially compromise venous return and cardiac output [Aufderheide and Lurie, 2004].
During CPR, epinephrine’s alpha-adrenergic effect on vascular tone is most important. The alpha-adrenergic action increases systemic vascular resistance, thereby increasing diastolic blood pressure, which in turn increases the likelihood of return of spontaneous circulation. High-dose epinephrine (0.05–0.2 mg/kg) improves myocardial and cerebral blood flow during CPR more than standard-dose epinephrine (0.01–0.02 mg/kg), and may increase the incidence of initial return of spontaneous circulation. However, studies indicate that the use of high-dose epinephrine in children does not improve survival and may be associated with a worse neurologic outcome [Gueugniaud et al., 1998; Lister and Fontan, 2004].
Vasopressin is a long-acting endogenous hormone that acts at specific receptors to mediate systemic vasoconstriction (V1 receptor) and reabsorption of water in the renal tubule (V2 receptor). In experimental models of cardiac arrest, vasopressin increases blood flow to the heart and brain and improves long-term survival compared with epinephrine. Vasopressin may decrease splanchnic blood flow during or following CPR. Wenzel et al. [2004] demonstrated the success of vasopressin alone and vasopressin followed by epinephrine in refractory asystolic cardiac arrest – an important breakthrough in the science of resuscitation. Vasopressin has also been reported to be useful in low cardiac output states associated with sepsis syndrome and organ recovery in children. While vasopressin will not likely replace epinephrine as a first-line agent in pediatric cardiac arrest, there are preliminary data to suggest that its use in conjunction with epinephrine in pediatric cardiac arrest deserves further investigation [Aung and Htay, 2005].
Postresuscitation Interventions
The underlying foundation of neurointensive care is excellent supportive care. State-of-the-art care for a patient who has suffered a hypoxic-ischemic injury consists of maintenance of adequate blood pressure; oxygenation; ventilation; electrolyte homeostasis; nutritional supplementation; and prevention of hyperglycemia, hyperthermia, and seizures [Gisvold et al., 1996]. Several features of supportive post-ischemic intensive care warrant further discussion. These include intracranial pressure monitoring and control, glucose homeostasis, cardiovascular support, temperature regulation, and reduction of metabolic rate.
Intracranial Pressure Monitoring and Control
Results of clinical studies suggest that intracranial pressure elevation is more common after asphyxia-induced cardiac arrest than after isolated global brain ischemia or ventricular fibrillation-induced cardiac arrest [Scollo-Lavizzari and Bassetti, 1987]. This finding is not surprising in view of the severe histopathology of the asphyxia-induced cardiac arrest and the long insult times from which the myocardium can recover in children. Sustained elevation of intracranial pressure (>20 mmHg) was a predictor of poor outcome in a series of pediatric submersion accidents [Dean and McComb, 1981; Nussbaum and Galant, 1983; Sarnaik et al., 1985]. Unfortunately, as in ventricular fibrillation-induced cardiac arrest, the threshold for poor outcome from asphyxia-induced cardiac arrest appears to be below the threshold for the occurrence of intracranial hypertension because many patients experience poor outcome despite normal intracranial pressure. Although there are anecdotal cases of asphyxia-induced cardiac arrest in patients with intact neurologic outcome despite elevated intracranial pressure, the ability to control intracranial pressure elevation does not result in meaningful survival. Routine monitoring and treatment of intracranial pressure are not currently recommended after asphyxia-induced cardiac arrest.
Hyperventilation has been advocated after cerebral injury as a means of decreasing intracranial pressure. However, the use of hyperventilation, even after traumatic brain injury, has been re-evaluated [Muizelaar et al., 1991; Skippen et al., 1997; Yundt and Diringer, 1997]. There is no evidence of any sustained beneficial effect of hyperventilation after hypoxic-ischemic brain injury, and its use should be avoided [Hickey and Zuckerbraun, 2003]. The failure of intracranial pressure-directed therapy to improve outcome after cardiopulmonary arrest and the commonly observed period of hypoperfusion in the first hours to days after the arrest raises serious questions about the blind application of an intervention with the potential to reduce cerebral blood flow further. Thus, hyperventilation should be reserved for patients with signs of cerebral herniation or suspected pulmonary hypertension [Hickey and Zuckerbraun, 2003].
The traditional approach to cerebral resuscitation has also recommended the use of mannitol in the post-cardiac arrest period. Again, studies in the setting of cardiopulmonary arrest are lacking. In dogs subjected to 6 minutes of global ischemia, mannitol (2 g/kg) further reduced cerebral blood flow during the post-ischemic hypoperfusion phase [Arai et al., 1986]. This unwanted effect of mannitol likely represents a result of dehydration. A theoretically beneficial oxygen radical-scavenging effect of mannitol has also been proposed, but if significant oxygen radical-induced brain injury occurs, significantly more potent oxygen radical scavengers are available.
Glucose Homeostasis
Observations in animals and humans have suggested that an elevated serum glucose level at or about the time of an ischemic insult is associated with enhanced post-ischemic damage of cerebral tissue [Kushner et al., 1990]. Pre-existing hyperglycemia worsens the outcome from global hypoxic-ischemic injury in adult and juvenile animals [Combs et al., 1990; Kalimo et al., 1981; Lanier et al., 1987; Pulsinelli et al., 1982]. Outcome from out-of-hospital cardiac arrest is worse in adults with elevated glucose concentrations at admission [Longstreth, 1987] and in hyperglycemic children after submersion injury [Ashwal et al., 1990]. Hyperglycemia has also been described as increasing brain damage when traumatic brain injury is complicated by secondary ischemia [Cherian et al., 1997]. As a mechanism for the adverse effect of hyperglycemia, most attention has focused on the neuronal effects of increased tissue lactate production and lowered tissue pH [Cherian et al., 1997; Kawai et al., 1997].
Several studies have demonstrated that:
In addition, researchers have demonstrated that, when subjected to the same ischemic insult, hyperglycemic animals have a more pronounced histopathologic injury than do normoglycemic animals [Kalimo et al., 1981]. In children, increased brain lactate level, as detected by MR spectroscopy, has been associated with poorer long-term neurologic outcomes [Ashwal et al., 1997]. The mechanisms by which lactate and acidosis may augment cellular injury during ischemia remain unclear [Kim et al., 1996; Steingrub and Mundt, 1996]. Direct cytotoxicity, cellular swelling, oxygen radical-mediated damage, and alterations in intracellular calcium homeostasis have been proposed as mechanisms of acidosis-induced injury [Kim et al., 1996; Siesjo et al., 1993]. Lactate induces alterations in calcium homeostasis and may contribute to synaptic dysfunction.
Other mechanisms by which hyperglycemia may enhance cerebral ischemic injury include effects on cerebral vasculature and cerebral blood flow [Cipolla et al., 1997; Kawai et al., 1997], alteration in the modulation of protein synthesis in the post-ischemic period [Lin et al., 1997], decreased adenosine production [Hsu et al., 1991], blood–brain barrier breakdown [Dietrich et al., 1993], and hyperosmolality [Gisselsson et al., 1992]. On the basis of these findings, it has also been suggested that, in the presence of impaired oxygen delivery, on-going delivery of glucose may be detrimental. The administration of glucose, once performed in every comatose patient, is questioned, and protocols involving glucose-free solutions are suggested [Sieber and Traystman, 1991].
In summary, the optimal level of blood glucose after resuscitation from either a global or focal hypoxic-ischemic event has not been determined. From a practical point, it seems wise to avoid hypoglycemia in a neonate with hypoxic-ischemic injury and to avoid hyperglycemia in older patients. There is evidence that hyperglycemia is common in acutely ill children and that time of onset, duration, and intensity of hyperglycemia are associated with mortality [Srinivasan et al., 2004]. Many pediatic intensive care practitioners have begun tentatively to control blood glucose in their patients. Reports do suggest that this control may be achieved safely and effectively [Clark et al., 2008; Verhoeven et al., 2009].
Cardiovascular Support
Although the optimal cerebral perfusion pattern for neuronal recovery remains to be defined, blood pressure fluctuations, both high and low, adversely affect outcome. In several important reviews, investigators have suggested a benefit of transient hypertension during the immediate postresuscitation period, hypothesizing that this improves flow in areas with microvascular sludging. Safar et al. [1996] suggested that transient hypertension was beneficial after cardiac arrest in dogs. However, it was used as part of a multifaceted treatment protocol, and its specific benefit is controversial.
The phase of therapy following return of spontaneous circulation has recently emerged as a critical window of opportunity for impact on survival from cardiac arrest [Trzeciak et al., 2009]. However, the particular factors associated with survival in the post-cardiac arrest syndrome have been insufficiently studied and remain poorly understood. Persistent circulatory insufficiency is common in the post-return of spontaneous circulation state. Specifically, arterial hypotension can result from a combination of factors, including myocardial stunning, neurally mediated cardiac dysfunction, relative adrenal insufficiency, and a profound proinflammatory response related to global ischemia and reperfusion injury that induces a “sepsis-like” state [Trzeciak et al., 2009; Adrie et al., 2002]. Because of the multiple influences on cardiac output, cardiac support can be difficult to manage. Careful use of fluids, inotropic agents, and vasoactive drugs is indicated. Vasodilators may result in profound reduction in blood pressure. In a retrospective human study, postresuscitative hypertension was associated with a better neurologic outcome [Morris and Nadkarni, 2003].
Extracorporeal Membrane Oxygenation-Cardiopulmonary Resuscitation
Perhaps the ultimate technology to control postresuscitation temperature and hemodynamic parameters is extracorporeal membrane oxygenation (ECMO). In addition, the concomitant administration of heparin may optimize microcirculatory flow. The use of venoarterial ECMO to re-establish circulation and provide controlled reperfusion following cardiac arrest has been published, but prospective, controlled studies are lacking [Chen et al., 2003; Masetti et al., 2005; Morris et al., 2004). Nevertheless, these series have reported extraordinary results with the use of ECMO as a rescue therapy for pediatric cardiac arrests, especially from potentially reversible acute postoperative myocardial dysfunction or arrhythmias. In one study, 11 children who suffered cardiac arrest in the pediatric intensive care unit after cardiac surgery were placed on ECMO during CPR after 20–110 minutes of CPR [del Nido et al., 1992]. Prolonged CPR was continued until ECMO cannulas, circuits, and personnel were available. Six of these 11 children were long-term survivors without apparent neurologic sequelae. Increasingly improved survival rates have been reported for pediatric cardiac patients provided with mechanical cardiopulmonary support within 20 minutes of the initiation of CPR. Despite these promising results, CPR and ECMO are not curative treatments; rather, they are simply cardiopulmonary supportive measures that may allow tissue perfusion and viability until recovery from the precipitating disease process can occur. Most remarkably, Morris et al. reported 66 children who, over 7 years, were placed on ECMO during CPR at the Children’s Hospital of Philadelphia [Morris et al., 2004]. The median duration of CPR before establishment of ECMO was 50 minutes, and 35 percent (23 of 66) of these children survived to hospital discharge. It is important to emphasize that these children had brief periods of “no flow,” excellent CPR during the “low-flow” period, and a well-controlled postresuscitation phase. Potential advantages of ECMO come from its ability to maintain tight control of physiologic parameters after resuscitation. Parameters including blood-flow rates, oxygenation, ventilation, anticoagulation, and body temperature can all be manipulated precisely through the ECMO circuit. As we learn more about the process of secondary injury following cardiac arrest, ECMO might enable controlled perfusion and temperature management to minimize reperfusion injury and maximize cell recovery.
Temperature Control
The effectiveness of hypothermia as a cerebral protective intervention (i.e., before cardiopulmonary arrest) is unquestioned. Hypothermia has been successfully applied during cerebrovascular and cardiovascular surgery for decades [Marion et al., 1996]. The beneficial effects of hypothermia applied immediately before and during cardiopulmonary arrest are clearly demonstrated by the clinical experience with victims of cold-water submersion accidents and with victims of accidental deep hypothermia [Walpoth et al., 1997].
When considering hypothermia (defined as a core temperature <35°C), the clinician should distinguish between uncontrolled (i.e., spontaneous, accidental) and controlled hypothermia induced by artificial cooling, which is used to prevent or attenuate various forms of neurologic injury [Polderman, 2004]. There is growing evidence that induced hypothermia can have neuroprotective effects in some patients with neurologic injury [Hickey and Nadkarni, 2008; Manole et al., 2009]. Class I evidence (from animal experiments, two randomized controlled trials, and various nonrandomized trials) supports the use of hypothermia in selected categories of patients after cardiopulmonary resuscitation [Bernard and Buist, 2003; Bernard et al., 2002; Hypothermia after Cardiac Arrest Group, 2002; Sessler, 1997]. Cooling should be initiated as soon as possible, but should not be withheld, even if delays of up to 8 hours occur [Polderman, 2004]. Treatment should be continued for at least 24 hours at brain temperatures of 32–34°C [Manole et al., 2009]. Patients should be rewarmed slowly, because there is evidence that quick rewarming may have deleterious effects [Hickey et al., 2003; Safar and Kochanek, 2002].
The effect of hypothermia on the injured brain is complex. For each 1°C decrease in temperature, the cerebral metabolic rate for oxygen decreases by 6–7 percent [Bernard and Buist, 2003]. However, hypothermia’s main beneficial effect is to provide protection against numerous deleterious biochemical mechanisms [Safar and Kochanek, 2002]. Over a period of days after the restoration of spontaneous circulation, these mechanisms – which include calcium shifts, excitotoxicity, lipid peroxidation and other free radical reactions, DNA damage, mitochondrial injury, and inflammation – lead to death of neurons in vulnerable regions of the brain [Safar and Kochanek, 2002].
Potential mechanisms for the protective effect of hypothermia include inhibition of glutamate release, inhibition of adenine nucleotide depletion, prevention of brain acidosis, and preservation of protein kinase activity [Cardell et al., 1991; Chen et al., 1992; Duhaime and Ross, 1990; Sutton et al., 1991]. Hypothermia also may inhibit free radical mechanisms by decreasing free fatty acid release, preserving endogenous antioxidants, and antagonizing free radical insults [du Plessis and Johnston, 1997]. The marked amelioration of delayed energy failure and specific inhibition of apoptosis (demonstrated in a piglet model) suggest that hypothermia may delay apoptotic commitment, allowing endogenous protective mechanisms to restore cellular integrity [Thoresen and Wyatt, 1997].
Given that therapeutic hypothermia in the range of 32–34°C appears to be safe and potentially improves outcome, it should be considered in children with cardiac arrest [Manole et al., 2009; Hutchinson et al., 2008; International Liaison Committee on Resuscitation, 2006]. Reviews of therapeutic hypothermia in children, along with methods of cooling, have been recently published [Hutchinson et al., 2008; Sloniewsky, 2011; Statler and Bratton, 2010]. The potential benefits and harms of hypothermia therapy are summarized in Box 76-2.
Box 76-2 Benefits and Harms of Hypothermia Therapy



Potential Harms





(Modified from Hutchinson JS et al. Hypothermia therapy for cardiac arrest in pediatric patients. Pediatr Clin N Am 2008;55:529–544.)
Conversely, hyperthermia is common after cardiac arrest in children and is associated with poor neurologic outcome [Hickey et al., 2003]. This may represent either noxious effects of hyperthermia on the injured brain or a worse insult overall with multiorgan system failure [Laptook et al., 2008]. As little as 1° of hyperthermia appears to be important [Manole et al., 2009]. Therefore, post-cardiac arrest pyrexia should be actively prevented and treated.
Post-Cardiac Arrest Brain Injury – Potential Therapies
Post-cardiac arrest brain injury is a common cause of morbidity and mortality. The unique vulnerability of the brain is attributed to its limited tolerance of ischemia and its unique response to reperfusion. As previously described, the mechanism of brain injury triggered by cardiac arrest and resuscitation are complex and include excitotoxicity, disrupted calcium homeostasis, free radical formation, pathological protease cascades, and activation of cell-death signaling pathways [Neumar et al., 2008]. Many of these pathways are executed over periods of hours to days and possibly weeks after return of spontaneous circulation. Histologically, selectively vulnerable neuronal subpopulations in the hippocampus, cortex, cerebellum, corpus striatum, and thalamus degenerate over period of hours to days. Both neuronal necrosis and apoptosis have been reported after cardiac arrest. The relative contribution of each cell-death pathway remains controversial, however, and is dependent in part on patient age and the neuronal subpopulation under examination. The relatively protracted duration of injury cascades and histological change suggests a therapeutic window for neuroprotective strategies after cardiac arrest [Neumar et al., 2008].
Hypothermia and ECMO are the only treatment modalities available for improving brain recovery in the post-cardiac arrest syndrome [Manole et al., 2009]. Many potential therapies have been proposed for cardiac arrest. Over the past three decades, investigators have used animal models of global cerebral ischemia to study numerous neuroprotective modalities, including anesthetics, anticonvulsants, calcium and sodium channel antagonists, immunosuppressants, growth factors, protease inhibitors, magnesium, and gamma-aminobutyric acid (GABA) agonists. Many of these targeted, pharmacological, neuroprotective strategies that focus on specific injury mechanisms have shown benefit in preclinical studies. Yet, none of the interventions tested thus far in prospective clinical trails has improved outcomes after out-of-hospital cardiac arrest [Neumar et al., 2008]. Some of these potential therapies are presented below.
Modulation of Endogenous Defenses
The potential mechanisms involved in cell death after cardiac arrest and restoration of circulation are complex. An evolving understanding of these mechanisms may allow clinicians to take advantage of these endogenous neuroprotectants as future therapies to improve neuronal survival after cardiac arrest. Several events are essential if neurons are to recover. First and foremost, ATP levels must be restored and maintained. Reperfusion is essential, although not the sole requirement for neuronal rescue, in as much as the cells require energy sources and the means to eliminate their waste. Therefore, a second critical factor is that the cells must not be subject to overwhelming cytosolic calcium burdens; therefore, not only must ATP levels be maintained, but also excitotoxicity should be minimized [Sweeney et al., 1995]. Other factors thought to be important include avoiding excessive formation of free radicals and metabolism of free fatty acids. In response to the complex sequence of pathophysiologic events that evolve after brain injury, several biochemical and molecular mediators that may have neuroprotective roles are present. These endogenous neuroprotectants are produced, induced, or activated after ischemia, and their postulated or proven functions improve cell (specifically neuronal) survival in in vivo, in vitro, or both models.
Heat shock proteins
Heat shock proteins are one family of candidate neuroprotectants that are highly conserved among biologic species and are induced in cells after a variety of stimuli. Heat stress is the classic example; however, any insult that damages protein structure, including ischemic and traumatic brain injury, can produce a heat shock protein response [Raghupathi et al., 1995; Simon et al., 1991]. Heat shock proteins have generated major interest as potential neuroprotectants because their prior induction by a sublethal stress can afford protection from subsequent injury. Transient entire-body hyperthermia reduces subsequent ischemic brain injury in rats [Chopp, 1989].
Heat shock proteins likely play an important role in cellular defense against environmental stresses [Kogure and Kato, 1993]. The HSP70 gene is the heat shock gene that has been best characterized after cerebral ischemia. Numerous studies have demonstrated increased transcription of HSP70 messenger RNA and increased synthesis of HSP70 and HSP72 after global cerebral ischemia [Gonzalez et al., 1991; Nowak et al., 1993; Vass et al., 1988]. This gene appears to be expressed in proportion to the duration of ischemia. Increasing durations of ischemia lead to HSP70 expression, first in neurons from selectively vulnerable regions, next in relatively resistant neurons, then in glia, and, finally, in endothelial cells [Sharp et al., 1993; Simon et al., 1991]. Although a direct cause-and-effect relation between heat shock proteins and neuroprotection in vivo has not been established, novel techniques specifically to increase heat shock proteins in neurons after injury should help in answering this question.
Adenosine
An endogenous biochemical mediator that may serve a protective role after cerebral ischemia, particularly early after injury, is adenosine. Adenosine levels are increased in brain tissue after experimental ischemia and in response to hypoxia, hypotension, and hypoglycemia [Laudignon et al., 1991; Morii et al., 1987; Ruth et al., 1993]. The bulk of the extracellular adenosine derives from the intracellular catabolism of ATP. The release of adenosine after ischemia could afford neuroprotection by a combination of several mechanisms. When bound to A2 receptors, adenosine is a cerebrovasodilator [Sweeney et al., 1995]. When bound to A1 receptors, adenosine reduces neuronal metabolism and excitatory amino acid release, stabilizes postsynaptic membranes, and inhibits platelet activation and neutrophil function [Miller and Hsu, 1992]. Thus, the beneficial effects of adenosine after cerebral ischemia include improved regional blood flow, reduced local oxygen demand, attenuation of excitotoxicity, increases in intracellular calcium, and anti-inflammatory and rheologic effects.
The effects of adenosine and synthetic adenosine derivatives on ischemic, hypoxic, or traumatic CNS damage, when given before or after the insult, protect against neuronal damage [Rudolphi and Schubert, 1996]. The therapeutic value of the agonist approach is questionable because most adenosine derivatives have prohibitive CNS and cardiovascular side effects, such as strong sedation, hypotension, cardiodepression, and renal vasoconstriction.
Targeting Delayed Neuronal Death
Although a portion of neuronal death occurs immediately after acute brain injury, it is clear that some portion of neurons die in a delayed manner by apoptosis [Kochanek et al., 2001]. Because apoptosis occurs in stages, there exist several potential strategies for reducing cell death. Targeting upstream regulators of the programmed cell death cascade, such as BCL-2 family proteins and cell death receptors is one such strategy [Kochanek et al., 2001]. Regulation of cell death may depend on the proportion of pro-apoptotic versus anti-apoptotic BCL-2 proteins in the cell after injury.
Another potential anti-apoptotic strategy is to target the final common pathway of the apoptotic cell death cascade. Activation of caspase-8, caspase-9, and other caspases is the key committed step in apoptosis [Youle and Strasser, 2008; Clark et al., 2000]. Caspase-3 activates endonucleases and other proteases, inactivates enzymes important in DNA repair, and cleaves cytoskeletal proteins, culminating in morphologic features of apoptosis [Kochanek et al., 2001]. Pan-caspase and relatively selective peptide inhibitors of many caspases are available and undergoing experimental trials.
Several contemporary strategies used for ischemic injury may reduce apoptosis. Hypothermia, antioxidants, and anti-excitotoxic strategies may prevent apoptosis and are discussed elsewhere in this chapter and Chapter 14.
Reduction of Excitotoxicity
Competitive antagonists at NMDA binding sites have been disappointing in animal studies because of the large doses (and hence systemic adverse effects) necessary to penetrate the blood–brain barrier and reduce the high ischemic levels of glutamate at the NMDA receptor. These factors limit the therapeutic index and clinical utility of competitive antagonists [du Plessis and Johnston, 1997].
MK-801 is a noncompetitive NMDA antagonist; that is, it does not compete with glutamate for binding to the NMDA receptor but instead blocks influx of extracellular calcium by binding to a site within the NMDA receptor-gated calcium channel. Results of numerous studies suggest that this agent is neuroprotective in the setting of focal ischemia [McCulloch, 1992; Pulsinelli et al., 1993]; however, results of MK-801 treatment in the setting of global brain ischemia are discouraging.
Blockade of non-NMDA receptors (i.e., AMPA and kainate receptors) with quinoxalinedione compounds has most consistently prevented delayed neuronal cell loss in the hippocampal CA1 region in animal models of global cerebral ischemia. NBQX (2,3-dihydroxy-6-nitro-7-sulfamoyl-benzoquinoxaline) is a non-NMDA receptor antagonist that appears to provide histologic protection in the cortex, hippocampus, or both in rat models of global ischemia [Buchan et al., 1991; Diemer et al., 1993; Nellgard and Wicloch, 1992]. Of interest, delayed treatment with NBQX appears to be effective. Treatment 12 hours after global ischemia in the rat [Li and Buchan, 1993] and 24 hours after global ischemia in the gerbil [Sherdown et al., 1993] resulted in significantly decreased delayed neuronal death in the CA1 region of the hippocampus.
The protective efficacy of AMPA-receptor antagonists against delayed neuronal death may be explained in the following ways. First, in the post-ischemic phase, AMPA receptors may become upregulated and more responsive [du Plessis and Johnston, 1997]. Furthermore, NMDA antagonists increase and AMPA antagonists decrease glucose metabolism. Consequently, during the period of disturbed metabolism after ischemia, a decrease in glucose metabolism, as occurs with AMPA antagonists, promotes neuronal survival [du Plessis and Johnston, 1997].
The divalent cation Mg2+ acts as a glutamate receptor antagonist to the extent that it blocks the neuronal influx of Ca2+ within the ion channel. In this regard, magnesium sulfate has reduced the severity of hypoxic-ischemic brain damage in immature rats [Vannucci and Perlman, 1997]. Clinical trials have revealed variable effects [Gisvold et al., 1996].
Inhibition of Calcium Accumulation
Calcium accumulation in neurons and release of stored intracellular calcium are pivotal events leading to irreversible cellular damage during the reperfusion phase after an ischemic insult. Calcium accumulation occurs through receptor-operated channels, such as the NMDA receptor-activated channels, voltage-operated calcium channels, and nonspecific membrane channels, and through release of intracellular calcium via the inositol second-messenger pathway. Conflicting results regarding the ability of the voltage-operated calcium channel antagonists (lidoflazine, nimodipine, nicardipine, flunarizine) to attenuate calcium accumulation in brain cells after ischemia have been obtained [Grotta et al., 1990]. The contribution of the voltage-gated channels to post-ischemic calcium accumulation is apparently less than that of the NMDA-operated channels in selectively vulnerable zones. Nevertheless, clinical experience with the voltage-operated calcium channel antagonists and their cerebral vasodilatory effects prompted laboratory and clinical evaluations.
Calcium channel blockers have been studied in a variety of animal models of global ischemia and have been subjected to clinical trials. Despite some encouraging findings in animal models, the clinical trials have failed to yield positive results [Morgenstern and Pettigrew, 1997].
Nimodipine, a dihydropyridine compound that selectively blocks the L-type calcium channel, has been studied in animal models of global cerebral ischemia and in human survivors of cardiac arrest. Using a canine model, Steen et al. [1983] demonstrated that pre-ischemic treatment with nimodipine resulted in increased cerebral blood flow and improved neurologic outcome after 10 minutes of global ischemia. However, when the drug was administered after ischemia, the results were intermediate; cerebral blood flow was improved, but outcome was unchanged [Steen et al., 1984]. Subsequently, Milde et al. [1986] studied the effect of post-ischemic treatment with nimodipine on cerebral blood flow in the early reperfusion period. Nimodipine-treated dogs had significantly higher cerebral blood flow, but nimodipine treatment had no effect on cerebral metabolism. Again, this raises the question of whether improving blood flow in the early reperfusion period has any effect on neurologic outcome. These results culminated in a clinical trial of nimodipine treatment in survivors of cardiac arrest (ventricular fibrillation) [Roine et al., 1990]. There was no difference in neurologic outcome or survival at 3 or 12 months after cardiac arrest.
Lidoflazine is a nonselective voltage-sensitive calcium channel antagonist that, in early animal studies, appeared to ameliorate hypoxic-ischemic brain injury. However, later studies, including a large clinical trial, failed to substantiate the results of the early experiments [Brain Resuscitation Clinical Trial II Study Group, 1991].
Antioxidants
There are two distinct types of superoxide dismutases in mammalian cells: copper-zinc and manganese [Tainer et al., 1983]. Copper-zinc superoxide dismutase is located in the cytosol, whereas manganese superoxide dismutase is located in the mitochondria. Superoxide dismutases are widely distributed throughout the body, but plasma levels are very low as a result of efficient renal clearance [Marklund, 1984].
Copper-zinc superoxide dismutase has been proposed as a therapeutic agent for reperfusion injury because of its ability to scavenge oxygen radicals. Superoxide dismutase has two drawbacks as a therapeutic agent. First, it is cleared rapidly by the kidney and has a circulatory half-life of approximately 8 minutes [Turrens et al., 1984]. Second, and more important, copper-zinc superoxide dismutase is a large, water-soluble molecule and therefore cannot readily penetrate cell membranes or cross the blood–brain barrier in significant quantities after intravenous administration [Beckman et al., 1988; Freeman et al., 1983]. Thus, if access to intracellular compartments is required for therapeutic efficacy, it appears unlikely that copper-zinc superoxide dismutase would provide a protective effect.
Investigators have tried two different modifications in the delivery of superoxide dismutase in an effort to increase the circulatory half-life and intracellular access of the intravenously administered enzyme: liposome-entrapped superoxide dismutase and polyethylene glycol-conjugated superoxide dismutase. Superoxide dismutase delivered in positively charged liposomes has a circulatory half-life of approximately 4 hours and has dramatically increased access into cultured endothelial cells [Freeman et al., 1983; Turrens et al., 1984]. Administration of liposome-entrapped superoxide dismutase produces both increased brain superoxide dismutase activity and reduced extent of infarctions in a rat model of global ischemia [Imaizumi et al., 1990]. Conjugation of polyethylene glycol monomers to superoxide dismutase increases its circulatory half-life to approximately 37 hours and increases its uptake into cultured endothelial cells [Beckman et al., 1988]. However, polyethylene glycol-conjugated superoxide dismutase does not appear to increase brain superoxide dismutase activity [Haun et al., 1991].
The amount of oxygen administered during resuscitation also appears to influence the severity of the free radical damage, and the practice of using 100 percent oxygen during cardiac arrest needs to be re-examined [Lipinski et al., 1999; Liu et al., 1998; Zwemer et al., 1994]. Although it is important to ensure that patients are not hypoxemic, a growing body of preclinical evidence suggests that hyperoxia during the early stages of reperfusion harms post-ischemic neurons by causing excessive oxidative stess [Neumar et al., 2008]. On the basis of preclinical evidence alone, unnecessary arterial hyperoxia should be avoided, especially during the initial post-cardiac arrest period. This can be achieved by adjusting the supplemental oxygen to produce an arterial oxygen saturation of 94–96 percent [Balan et al., 2006; Koch et al., 2008].
Interventions directed at pathways indirectly related to free radical generation may be beneficial for reducing oxidative stress in the brain. Hypothermia reduces lipid peroxidation and maintains endogenous antioxidant activity during reperfusion from global brain ischemia [Baiping et al., 1994]. Acidosis during ischemia increases free radical production and may inactivate endogenous antioxidants [Links, 1988]; however, the role of buffer therapy is controversial.
Reduction of Lipid Peroxidation
Lipid-soluble agents rapidly cross the blood–brain barrier, a beneficial property for cerebral resuscitation. Administration of alpha-tocopherol after cerebral ischemia reduces ischemic neuronal damage in gerbils [Calapai et al., 1993].
Lipid-soluble compounds termed lazaroids have been developed to reduce iron-dependent lipid peroxidation after cerebral ischemia. The 21-aminosteroid lazaroids are potent inhibitors of lipid peroxidation but lack classic steroidal activities [Braughler et al., 1987]. However, they have multiple antioxidant actions:
The most extensively studied 21-aminosteroid, U74006F (tirilazad mesylate), appears to possess multiple antioxidant properties; however, studies of the use of this agent in global cerebral ischemia have yielded mixed results [Sweeney et al., 1995]. An explanation for the limited effect in global ischemia is that tirilazad is lipophilic and therefore accumulates in cell membranes with a high affinity for vascular endothelium. Tirilazad may protect the vascular endothelium from peroxidative damage but is not expected to cross the blood–brain barrier [Schmid-Elsaesser et al., 1997]. Recent experimental study has demonstrated that the lazaroid compound U74389G, administered during cardiac arrest, mitigated postresuscitation myocardial dysfunction and improved survival rates [Wang et al., 2004].
It has been proposed that free radical injury of the brain mainly involves the microvasculature, although both brain parenchyma and vascular endothelium have the potential to generate free radicals [Grammas et al., 1993; Siesjo et al., 1995]. One newly discovered group of antioxidants, the pyrrolopyrimidines, have a significantly greater potential to enter the brain parenchyma. Results of early investigations suggest that these compounds, which are able to cross the blood–brain barrier, may have a greater potential to treat cerebral ischemia [Schmid-Elsaesser et al., 1997].
Nitric Oxide Inhibition
As described earlier, nitric oxide could be both beneficial and detrimental to the brain during ischemia and reperfusion. Possible beneficial effects include vasodilatation, inhibition of platelet aggregation, and downregulation of NMDA receptors, whereas possible detrimental effects include production of hydroxyl radicals, increased inflammatory effects mediated through inducible nitric oxide synthase, and inhibition of key enzymes necessary for DNA replication and mitochondrial energy production [Choi, 1993]. It is therefore not surprising that animal studies in which nitric oxide synthase inhibitors were used for treatment of cerebral ischemia have yielded both positive and negative results. Nitric oxide synthase inhibitors are analogs of l-arginine, the substrate for nitric oxide synthase. These analogs competitively inhibit the conversion of l-arginine to l-citrulline and nitric oxide.
In animal models, inhibitors of nitric oxide synthesis, including desmethyl tirilazad [Fernandez et al., 1997], 7-nitroindazole [O’Neill et al., 1996], and N-omega-nitro-l-arginine [Kohno et al., 1997], have all reduced the effects of global hypoxic-ischemic injury. However, because the competing neurotoxic versus neuroprotective vasodilator effects of nitric oxide remain poorly understood in focal and global ischemic injury, extensive animal and subsequent human clinical research is needed.
Modulation/Protein Synthesis
Protein synthesis is essential for cell survival. Proteins are involved in nearly every process in the cell (e.g., enzymatic reactions, transport, storage, intracellular signaling, intercellular signaling, and cytoskeletal support). Obviously, intact protein synthesis is important for cellular recovery after hypoxic-ischemic injury. However, total protein synthesis is impaired after cerebral ischemia [Kleihues and Hossmann, 1971; Nowak et al., 1985]. Inhibition of protein synthesis is slow to recover, requiring hours to days to return to normal levels. The inhibition of protein synthesis is not regionally homogeneous; inhibition is more severe and prolonged in the cortex, hippocampus, and caudate [Bodsch et al., 1986; Widmann et al., 1991]. Failure of protein synthesis to recover after ischemia is the biochemical correlate of cell death. Protein synthesis in neurons of the vulnerable CA1 region of the hippocampus fails to recover after even short periods of ischemia. The inhibition of protein synthesis, which appears to be reperfusion-specific, prevents cells from responding to free radical damage by synthesizing the enzymes necessary to detoxify radicals and repair membrane damage [White et al., 1996].
Although total protein synthesis is decreased after cerebral ischemia, the synthesis of some proteins is actually increased. It is enticing to postulate that these proteins may play an important role in reparative processes after cerebral ischemia; however, it is equally plausible that the selective expression of some genes may contribute to delayed neuronal death, possibly through apoptotic mechanisms [Akins et al., 1996]. Ischemia appears to cause dramatic changes in gene expression [Kogure and Kato, 1993; Sharp et al., 1993]. Genes that have received the most attention in models of cerebral ischemia include the immediate early genes (c-fos and c-jun), heat shock protein genes, and trophic factor genes. Immediate early genes code for proteins (fos, jun, and others) that function as transcription factors and control the expression of many genes in the CNS [Nowak et al., 1993]. Increased expression of c-fos and c-jun has been demonstrated after global cerebral ischemia [Akins et al., 1996].
Trophic factors appear to be produced in the CNS after ischemic brain injury and may contribute to the repair of damaged cells [Kogure and Kato, 1993]. Fibroblast growth factor and nerve growth factor are two growth factors elaborated in the CNS that appear to be important for the survival and development of neurons. Takami et al. [1992] demonstrated increased production of fibroblast growth factor messenger RNA and fibroblast growth factor after global ischemia in the rat. Nerve growth factor levels have also been demonstrated to increase after cerebral ischemia. Both fibroblast growth factor and nerve growth factor, administered by injection into the lateral ventricle, decrease neuronal injury after cerebral ischemia in gerbils [Shigeno et al., 1991; Wen et al., 1995]. Thus, increased expression of growth factors after ischemia appears to be an attempt by the injured brain to prevent further injury and promote repair. Use of neurotrophic factors or novel brain-penetrating neurotrophic factor receptor agonists to prevent neuronal death or to promote regeneration and rewiring of injured brain represents an exciting area of study.
Insulin is a member of an important group of growth factors that includes insulin-like growth factor-1 and nerve growth factor. There is evidence that members of the insulin growth factor family, when delivered to the brain in the 2 hours after reperfusion, can salvage a percentage of the neurons that would otherwise die [White et al., 1996].
Gene expression after cerebral ischemia is incompletely understood but exciting, in that it may provide a mechanism to improve outcome. In animal studies in which gene regulation was examined in a model of hypoxic conditioning in the newborn, preliminary data revealed the potential for manipulating gene regulation after hypoxic insults [Bernaudin et al., 2002].
Inhibition of Arachidonic Acid Metabolism
Although many avenues exist for manipulation of phospholipid-derived mediator formation after global cerebral ischemia or cardiorespiratory arrest, most studies have focused on the arachidonic acid cascade. Pretreatment with cyclo-oxygenase inhibitors (indomethacin, piroxicam, diclofenac, or ibuprofen) before 5 minutes of global brain ischemia in gerbils attenuated selectively vulnerable cell death [Nakagomi et al., 1989; Sasaki et al., 1988]. In contrast, inhibition of the lipoxygenase pathway did not prevent selectively vulnerable cell death. Prostaglandins potentiate the effects of excitatory amino acids, and inhibition of this effect may explain the protection afforded by these agents. Similarly, inhibition of superoxide anion synthesis during the metabolism of arachidonic acid also may be an important mechanism for this effect. Alternatively, cyclo-oxygenase metabolites of arachidonic acid are important regulators of normal cerebral blood flow, especially in the immature brain, and cyclo-oxygenase inhibitors increase cerebral blood flow after global ischemia. However, indomethacin administration after ischemia does not increase cerebral blood flow, and the effect of resuscitation with cyclo-oxygenase inhibitors on cerebral blood flow after cardiorespiratory arrest has not been studied. A limit to the clinical effectiveness of cyclo-oxygenase inhibitors may be that a burst of cyclo-oxygenase product formation occurs within seconds of reperfusion.
Glucocorticoids
Glucocorticoids inhibit phospholipase A2 activity and have a history of use in cerebral resuscitation. The use of corticosteroids as a therapy after cardiorespiratory arrest, however, is unsupported. Pretreatment with dexamethasone in global ischemia models worsened neuronal survival, perhaps because of steroid-induced hyperglycemia. Corticosteroid treatment after global cerebral ischemia in animal models has produced effects ranging from very detrimental to minimally protective. Jastremski et al. [1989] retrospectively assessed the impact of steroid use in the 262 patients who did not receive thiopental in the Brain Resuscitation Clinical Trial I. Four groups (receiving no, low-dose, medium-dose, or high-dose steroid) were compared. No effect on mortality or neurologic outcome was seen.
Hyperbaric Oxygen Treatment
Hyperbaric oxygen treatment has been demonstrated to prolong survival [Krakovsky et al., 1998] and have some neuroprotective properties when used in acute settings in models of global ischemia [Kapp et al., 1982; Takahashi et al., 1992; Zhou et al., 2003]. However, clinical reviews of patients with traumatic brain injury [Longhi and Stocchetti, 2004] or stroke [Rusyniak et al., 2003] have suggested that it is unlikely to be of benefit, but clinical trials examining the potential benefits versus risk of hyperbaric oxygen treatment have not yet been performed.