Chapter 27 Hypotonic (Floppy) Infant
Floppy, or hypotonic, infant is a common scenario encountered in the clinical practice of child neurology. It can present significant challenges in terms of localization and is associated with an extensive differential diagnosis (Box 27.1). As with any clinical problem in neurology, attention to certain key aspects of the history and examination allows correct localization within the neuraxis and narrows the list of possible diagnoses. Further narrowing of the differential is achievable with selected testing based on the aforementioned findings. Understanding the anatomical and etiological aspects of hypotonia in infancy necessarily begins with an understanding of the concept of tone. Tone is the resistance of muscle to stretch. Categorization of tone differs among authors, but assessment is performed with the patient at rest and all parts of the body fully supported; examination involves tonic or phasic stretching of a muscle or the effect of gravity. Tone is an involuntary function and therefore separate and distinct from strength or power, which is the maximum force generated by voluntary contraction of a muscle. Function at every level of the neuraxis influences tone, and disease processes affecting any level of the neuraxis may reduce tone. Although a comprehensive review of conditions associated with hypotonia in infancy is beyond the scope of a single chapter, this chapter considers the basic approach to evaluating the floppy infant and considers several key disorders.
Box 27.1 Differential Diagnosis of the Floppy Infant
Approach to Diagnosis
History
Several features of the history may point to a specific diagnosis or category of diagnoses leading to hypotonia, or may permit distinguishing disorders present during fetal development from disorders acquired during the perinatal period. Thoroughly investigate a family history of disorders known to be associated with neonatal hypotonia, especially in the mother or in older siblings. Certain dominantly inherited genetic disorders (e.g., myotonic dystrophy) are associated with anticipation (earlier or more severe expression of a disease in successive generations). Such disorders may be milder and therefore undiagnosed in the mother. A maternal history of spontaneous abortion, fetal demise, or other offspring who died in infancy may also provide clues to possible diagnoses. A history of reduced fetal movement is a common feature of disorders associated with hypotonia, and may indicate a peripheral cause (Vasta et al., 2005). A history of maternal fever late in pregnancy suggests in utero infection, while a history of a long and difficult delivery followed by perinatal distress suggests hypoxic-ischemic encephalopathy with or without accompanying myelopathy. Among the many potential causes of neonatal hypotonia, acquired perinatal injury is far more common than inherited disorders and is rarely overlooked. However, also consider the possibility of a motor unit disorder leading to perinatal distress and hypoxic-ischemic encephalopathy.
Physical Examination
General Features of Hypotonia
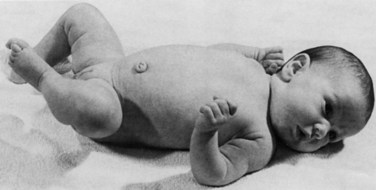
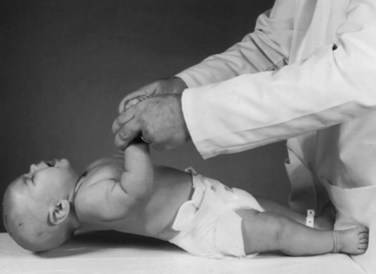
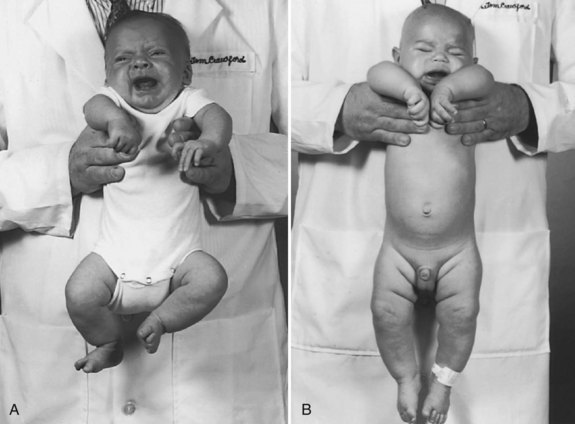
Assessing tone in an infant involves both observation of the patient at rest and application of certain examination maneuvers designed to evaluate both axial and appendicular musculature. Beginning with observation, a normal infant lying supine on an examination table will demonstrate flexion of the hips and knees so that the lower extremities are clear of the examination table, flexion of the upper extremities at the elbows, and internal rotation at the shoulders (Fig. 27.1). A hypotonic infant lies with the lower extremities in external rotation, the lateral aspects of the thighs and knees touching the examination table, and the upper extremities either extended down by the sides of the trunk or abducted with slight flexion at the elbows, also lying against the examination table. Evaluation of the traction response is done with the infant in supine position; the hands are grasped and the infant pulled toward a sitting position. A normal response includes flexion at the elbows, knees, and ankles, and movement of the head in line with the trunk after no more than a brief head lag. The head should then remain erect in the midline for at least a few seconds. An infant with axial hypotonia demonstrates excessive head lag with this maneuver (Fig. 27.2), and once upright, the head may continue to lag or may fall forward relatively quickly. Absence of flexion of the limbs may also be seen and indicates either appendicular hypotonia or weakness. The traction response is normally present after 33 weeks postconceptional age. Vertical suspension is performed by placing hands under the infant’s axillae and lifting the infant without grasping the thorax. A normal infant has enough power in the shoulder muscles to remain suspended without falling through, with the head upright in the midline and the hips and knees flexed (Fig. 27.3, A). In contrast, a hypotonic infant held in this manner slips through the examiner’s hands, often with the head falling forward and the legs extended at the knees (see Fig. 27.3, B). Infants with axial hypotonia related to brain injury may also demonstrate crossing, or scissoring, of the legs in this position, which is an early manifestation of appendicular hypertonia. In horizontal suspension, the infant is held prone with the abdomen and chest against the palm of the examiner’s hand. A normal infant maintains the head above horizontal with the limbs flexed, while a hypotonic infant drapes over the examiner’s hand with the head and limbs hanging limply. Other examination findings in hypotonic infants include various deformities of the cranium, face, limbs, and thorax. Infants with reduced tone may develop occipital flattening, or positional plagiocephaly, as the result of prolonged periods of lying supine and motionless.
Localization
The key features of disorders of cerebral function, particularly the cerebral cortex, are encephalopathy and seizures. Encephalopathy manifesting as decreased level of consciousness may be difficult to ascertain, given the large proportion of time normal infants spend sleeping. However, full-term or near-term infants with normal brain function spend at least some portion of the day awake with eyes open, particularly with feeding. Encephalopathy also manifests with excessive irritability or poor feeding, although the latter problem is rarely the sole feature of cerebral hemispheric dysfunction and may occur with disorders at more distal sites. Infants with centrally mediated hypotonia of many different etiologies frequently have relatively normal power despite a hypotonic appearance. Power may not be observable under normal circumstances because of a paucity of spontaneous movement, but it may be observable with application of a noxious stimulus such as a blood draw or placement of a peripheral intravenous catheter. Other indicators of central rather than peripheral dysfunction include fisting (trapping of the thumbs in closed hands), normal or brisk tendon reflexes, and normal or exaggerated primitive reflexes. Tendon reflexes should be tested with the infant’s head in the midline and the limbs symmetrically positioned; deviations from this technique often result in spuriously asymmetrical reflexes. Primitive reflexes are involuntary responses to certain stimuli that normally appear in late fetal development and are supplanted within the first few months of life by voluntary movements. Abnormalities of these reflexes include absent or asymmetrical responses, obligatory responses (persistence of the reflex with continued application of the stimulus), or persistence of the reflexes beyond the normal age range. Two of the most sensitive primitive reflexes are the Moro and asymmetrical tonic neck reflexes. The Moro reflex is a startle response present from 28 weeks after conception to 6 months postnatal age (Gingold et al., 1998). Quickly dropping the infant’s head below the level of the body while holding the infant supine with the head supported in one hand and the body supported in the other readily elicits this reflex. The normal response consists of initial abduction and extension of the arms with opening of the hands, followed quickly by adduction and flexion with closure of the hands. The tonic neck reflex is a vestibular response and is present from term until approximately 3 months of age. The response is elicited by rotating the head to one side while the infant is lying supine. The normal response is extension of the ipsilateral limbs while the contralateral limbs remain flexed. Central disorders resulting in hypotonia may also be associated with dysmorphism of the face or limbs, or malformations of other organs. Various defects in O-linked glycosylation of α-dystroglycan, a protein associated with the dystrophin glycoprotein complex that stabilizes the sarcolemma, result in structural defects of the brain, eye, and skeletal muscle.
Diagnostic Studies
Neuroimaging
Neuroimaging studies, in particular magnetic resonance imaging (MRI), are most useful when suspecting structural abnormalities of the CNS. T1-weighted images most readily detect congenital malformations of the brain and spinal cord, while T2-weighted images and various T2-based sequences reveal abnormalities of white matter and show evidence of ischemic injury. Specialized techniques such as MR spectroscopy may show evidence of mitochondrial disease (Matthews et al., 1993) or disorders of cerebral creatine metabolism (Frahm et al., 1994). When performing neuroimaging studies that require sedation on hypotonic infants, give particular consideration to airway management and other safety issues.
Nerve Conduction Studies and Electromyography
Nerve conduction studies and EMG are the studies of choice in a suspected motor unit disorder when other available clinical information does not suggest a specific diagnosis. The two techniques are complementary and always performed together. They allow distinction between primary disorders of muscle and peripheral nerve disorders when the two are indistinguishable on clinical grounds. Repetitive nerve stimulation (RNS) studies evaluate the integrity of the neuromuscular junction, abnormalities of which are not detectable with routine nerve conduction studies or EMG. The most commonly observed abnormality on low-rate (2-3 Hz) RNS studies of patients with various forms of myasthenia is a significant decrement, usually defined as 10% or greater, in the amplitude of the compound motor action potential (CMAP) between the first and fourth or fifth stimuli of a series. Single fiber EMG (SFEMG) is a highly specialized technique that evaluates the delay in depolarization between adjacent muscle fibers within a single motor unit, referred to as jitter. This modality is highly sensitive for neuromuscular junction abnormalities but has a low specificity and requires a cooperative patient. SFEMG with stimulation of the appropriate nerve has been described in pediatric patients (Tidwell and Pitt, 2007), but experience with this technique in infants is limited to a small number of centers. The utility of these neurophysiology studies is dependent on the skill and experience of the clinician performing the tests, as well as the precision of the question posed.
Specific Disorders Associated with Hypotonia in Infancy
Cerebral Disorders
Chromosomal Disorders
Hypotonia is a prominent feature of many disorders associated with large- or small-scale chromosomal abnormalities. Such disorders also are frequently associated with a dysmorphic appearance of the face and hands. Among the most common of these disorders is Prader-Willi syndrome, which is caused by absence of the paternal PWS/Angelman syndrome region on chromosome 15 (Butler, Meaney, and Palmer, 1986). Affected individuals often have profound hypotonia and poor feeding in infancy, suggesting a disorder of the motor unit or a combined cerebral and motor unit disorder. However, serum CK, EMG, muscle biopsy, and brain MRI are normal. The commonly recognized morphological features of almond-shaped eyes, narrow biparietal diameter, and relatively small hands and feet may not be readily apparent in early infancy. Approximately 70% of patients have a detectable small-scale deletion on chromosome 15 on high-resolution chromosomal analysis; DNA methylation studies reveal a pattern suggestive of exclusive maternal inheritance of this locus in 99%. Failure to thrive in infancy gives way in early childhood to hyperphagia and a characteristic pattern of behavioral abnormalities, intellectual disability, and hypogonadism.
Combined Cerebral and Motor Unit Disorders
Acid Maltase Deficiency
Acid maltase deficiency, an autosomal recessive deficiency of the lysosomal enzyme acid α-1,4-glucosidase, presents with a severe skeletal myopathy and cardiomyopathy and may also be associated with encephalopathy. Routine histochemical stains show accumulation of glycogen in lysosomal vacuoles and within the sarcoplasm. The diagnosis is confirmed with biochemical assay of enzyme activity in muscle or in cultured skin fibroblasts. Recombinant human enzyme is approved by the U.S. Food and Drug Administration (FDA) for replacement therapy, which can prolong survival (Kishnani et al., 2006).
Congenital Myotonic Dystrophy
Congenital myotonic dystrophy is an autosomal dominant disorder that typically presents in adolescence or early adulthood, but in some instances may be associated with profound hypotonia and weakness of the face and limbs in infancy. Approximately 25% of infants born to mothers with myotonic dystrophy are affected in this way, although the diagnosis in the mother may be unrecognized (Rakocevic-Stojanovic et al., 2005). Survivors of perinatal distress often have global developmental delay, with both intellectual impairment and motor disability throughout childhood, then develop myotonia and other characteristic symptoms of the muscular dystrophy as they approach puberty. To date, only myotonic dystrophy type 1, caused by abnormal expansion of a trinucleotide repeat within the gene, DMPK, has been associated with a congenital presentation. Genetic testing is commercially available.
Infantile Facioscapulohumeral Dystrophy
Facioscapulohumeral dystrophy (FSHD) is another dominantly inherited muscular dystrophy presenting most frequently in early adulthood, but which may have a congenital presentation. The genetic abnormality is contraction of a 3.3 kb repeat array at the D4Z4 locus. Those with the smallest integral number of repeats may have diffuse hypotonia and weakness in infancy and account for less than 5% of cases (Klinge et al., 2006). Affected infants may have cognitive impairment, epilepsy, and progressive sensorineural hearing loss. Serum CK is normal or mildly elevated. Family history may include a mildly affected parent, although cases also result from de novo mutations. Genetic testing is commercially available.
Syndromic Congenital Muscular Dystrophies
A group of congenital muscular dystrophies due to defects of O-linked glycosylation of dystroglycan, a component of the dystrophin-glycoprotein complex spanning the plasma membrane of skeletal myocytes, are associated with severe myopathy, a cerebral cortical malformation referred to as cobblestone lissencephaly, and ocular defects such as retinal dysplasia. In addition to profound hypotonia and weakness, affected infants often have intractable epilepsy. These diagnoses are suspected based on the characteristic constellation of abnormalities and have been clinically categorized as Fukuyama congenital muscular dystrophy, Walker-Warburg syndrome, and muscle-eye-brain disease. Thus far, six different causative genes have been identified (Muntoni et al., 2008), and there appears to be a far greater degree of phenotypic overlap among the different genotypes than was previously appreciated.
Congenital Disorders of Glycosylation
Congenital disorders of glycosylation are a group of recessively inherited defects in 21 different enzymes that modify N-linked oligosaccharides. Many forms present with hypotonia in infancy. The most common form, type Ia, results from a deficiency of the phosphomannomutase enzyme. In addition to hypotonia, affected infants may have hyporeflexia, global developmental delay, failure to thrive, seizures, and evidence of hepatic dysfunction, coagulopathy, and elevated thyroid-stimulating hormone (TSH). Characteristic examination findings include inverted nipples and an abnormal distribution of subcutaneous fat. Facial dysmorphism occurs but is not present in all cases. Brain MRI shows cerebellar hypoplasia. Analysis of transferrin isoforms in serum by isoelectric focusing reveals a characteristic pattern indicative of a defect in the early steps of the N-linked oligosaccharide synthetic pathway. Commercially available genetic testing identifies pathogenic sequence variants in 95% of affected individuals. Although cerebral dysfunction dominates the early clinical picture, some patients develop a demyelinating peripheral neuropathy in the first or second decade of life (Gruenwald, 2009).
Lysosomal Disorders
Certain defects of lysosomal hydrolases, in particular Krabbe disease and metachromatic leukodystrophy, result in progressive degeneration of both central and peripheral myelin (Korn-Lubetzki et al., 2003), producing both an encephalopathy and motor unit dysfunction (Cameron et al., 2004). Both disorders are associated with characteristic white matter abnormalities on brain MRI, and biochemical assays on peripheral blood of β-galactocerebrosidase in the case of Krabbe, and of arylsulfatase A in the case of metachromatic leukodystrophy confirm the diagnosis.
Infantile Neuroaxonal Dystrophy
Neuroaxonal dystrophy is a rare autosomal recessive disorder caused by mutations in the PLA2G6 gene, which encodes a calcium-independent phospholipase (Gregory et al., 2008). The classic form may present as early as 6 months of age with hypotonia, although psychomotor regression is more common, and progressive spastic tetraparesis and optic atrophy with visual impairment follow. Brain MRI shows bilateral T2 hypointensity of the globus pallidus, indicative of progressive iron accumulation, as well as thinning of the corpus callosum and cerebellar cortical hyperintensities. Nerve conduction studies show evidence of an axonal sensorimotor polyneuropathy with active denervation on EMG. The characteristic pathological finding is of enlarged and dystrophic-appearing axons on biopsy of skin, peripheral nerve, or other tissue containing peripheral nerve. Commercially available genetic testing identifies abnormalities in approximately 95% of children with early symptom onset.
Spinal Cord Disorders
Spinal Muscular Atrophy
Spinal muscular atrophy (SMA) is the most common inherited disorder of the spinal cord resulting in hypotonia in infancy, occurring with an incidence of approximately 1 in 10,000 live births per year. It is an autosomal recessive disorder in which the molecular defect leads to impaired regulation of programmed cell death in anterior horn cells and in motor nuclei of lower cranial nerves. Both populations of motor neurons are progressively lost, producing hypotonia and weakness of limb and truncal musculature, as well as bulbar dysfunction. In approximately 95% of cases, the genetic defect is homozygous deletion of the survival motor neuron 1 (SMN1) gene, which is located on the telomeric region of chromosome 5q13 (Ogino and Wilson, 2002). A virtually identical centromeric gene on 5q13, referred to as SMN2, encodes a similar but less biologically active product (Swoboda et al., 2005). While no more than two copies of SMN1 are present in the human genome, variable numbers of SMN2 copies are present. The protein product of SMN2 appears to partially rescue the SMA phenotype such that a larger SMN2 copy number generally results in a milder presentation and disease course.
Historically, SMA patients have been categorized into different phenotypes or syndromes based on age of presentation and maximum motor ability achieved. The disease results from a common genetic abnormality with a spectrum of phenotypic severity contingent upon modifying factors that include SMN2 copy number and other loci not yet identified. The classification of the most severely affected patients, with weakness and hypotonia evident at birth, is SMA type 0. These infants may have arthrogryposis multiplex congenita in addition to diffuse weakness of limb and trunk muscles, but facial weakness is usually mild if present. Perinatal respiratory failure causes death in early infancy. SMA type 1, also referred to as Werdnig-Hoffmann disease, is a designation given to infants who develop weakness within the first 6 months of life. These infants may appear normal at birth or may appear hypotonic. Facial expression is usually normal, and arthrogryposis is usually absent. Weakness is worse in proximal than in distal muscles and worse in the lower extremities, which may lead to suspicion of a congenital myopathy or muscular dystrophy. Further confounding the diagnosis is the presence of an elevated serum CK in a substantial portion of patients (Rudnick-Schoneborn et al., 1998), although CK rarely rises above 1000 U/L. In addition to limb weakness, affected infants demonstrate abdominal breathing due to relative preservation of diaphragm function as compared to abdominal and chest wall musculature. Needle EMG shows evidence of both acute and chronic denervation in the limbs and serves to distinguish this disorder from myopathies with a similar presentation.
Genetic testing is commercially available for SMN-related SMA. Among the 5% of patients without homozygous deletion of SMN1, most are compound heterozygotes with the characteristic deletion on one allele and a point mutation on the other. Parents of affected children are obligatory heterozygotes. The natural history of SMA is unique among anterior horn cell disorders in that the progression of weakness is most rapid early in the disease course and subsequently slows. Nevertheless, in the absence of supportive measures, median survival is 8 months, with death due to respiratory failure. Survivors have normal cognitive development. Several agents that act as histone deacetylase inhibitors increase the expression of SMN2 mRNA in vitro and in vivo, and among these agents, valproate, sodium phenylbutyrate, and hydroxyurea are currently or have recently been in clinical trials in SMA patients (Oskoui and Kaufmann, 2008).
Infantile Spinal Muscular Atrophy with Respiratory Distress Type 1
Infantile spinal muscular atrophy with respiratory distress type 1 (SMARD1), previously classified as a variant of SMA type 1, is a rare and distinct autosomal recessive anterior horn cell disorder. Unlike SMN-related SMA, affected infants develop early diaphragmatic paralysis and distal limb weakness that progresses to complete paralysis. Many have intrauterine growth restriction and are born with ankle contractures. Approximately one-third are born prematurely. Similar to SMN-related SMA, EMG and muscle biopsy reveal evidence of chronic active denervation. The causative gene encodes the immunoglobulin µ-binding protein 2 (IGHMBP2), for which testing is commercially available (Grohmann et al., 2001).
X-linked Spinal Muscular Atrophy
This rare X-linked anterior horn cell degenerative disorder shares a considerable degree of phenotypic overlap with SMN-related SMA. Distinctive features include polyhydramnios secondary to impaired fetal swallowing and arthrogryposis. Consider the diagnosis in any simplex case of a male infant with an SMA phenotype and normal SMN1 copy number. The only known causative gene encodes the ubiquitin-activating enzyme 1 (UBE1), for which testing is available on a research basis only (Ramser et al., 2008).
Peripheral Nerve Disorders
Polyneuropathies, both inherited and acquired, are a rare cause of infantile hypotonia. The two most common clinical designations for infantile polyneuropathies are congenital hypomyelinating neuropathy (CHN) and Dejerine-Sottas disease (DSD). In recent years, mounting evidence does not reveal that either entity is a monogenic disorder, nor are they clearly distinct from one another. Clinical features include hypotonia, distal or diffuse weakness, absent tendon reflexes, and evidence on nerve conduction studies of a demyelinating polyneuropathy. Traditionally, DSD was classified as hereditary motor and sensory neuropathy (HSMN) type III, but at least 4 genes associated with various demyelinating HMSN subtypes have been linked to the DSD and CHN phenotypes, including PMP22, MPZ, EGR2, and PRX (Plante-Bordenueve and Said, 2002). In general, patients with an infantile presentation are homozygotes or compound heterozygotes for mutations in the causative genes. The most common acquired autoimmune peripheral neuropathies, Guillain-Barré syndrome and chronic inflammatory demyelinating polyneuropathy (CIDP), occur rarely in the first year of life and typically present with weakness and hypotonia in a previously normal infant.
Neuromuscular Junction Disorders
Juvenile Myasthenia Gravis
Approximately 10% to 15% of cases of autoimmune myasthenia gravis due to endogenous production of antibodies directed against sarcolemmal nicotinic acetylcholine receptors or muscle-specific kinase occur in individuals younger than 16 years of age. The disorder is particularly rare in the first year of life (Andrews, 2004). The small number of infantile cases reported in the literature limits the conclusions drawn with respect to the occurrence of measurable antibody titers, treatment, and outcomes in this age group.
Neonatal Myasthenia
In approximately 15% of infants born to mothers with autoimmune myasthenia gravis, transitory symptoms of myasthenia occur in the neonatal period related to transfer of acetylcholine receptor antibodies across the placenta. Because the fetal nicotinic acetylcholine receptor is different from the adult form, the expression of myasthenic symptoms in newborns depends on the maternal production of antibodies against the fetal receptor. These antibodies are not active against the adult form of the receptor and therefore do not contribute to maternal symptoms. Likewise, antibodies against the fetal receptor are not detectable by commercially available assays. For these reasons, neither maternal symptom severity nor the maternal antibody titer predicts the likelihood or severity of neonatal myasthenic symptoms. As with juvenile myasthenia gravis, the predominant symptoms are ocular or bulbar, although generalized hypotonia or weakness may occur. Rarely, affected infants have arthrogryposis due to prenatal exposure to fetal antibodies, leading to prolonged immobility in utero. Affected infants may require respiratory support temporarily or may require symptomatic therapy with subcutaneous neostigmine prior to oral feeds to prevent fatigue and premature discontinuation of feeding. In a majority of cases, the symptoms resolve within the first month of life (Papazian, 1992).
Congenital Myasthenic Syndromes
Several genetic disorders of neuromuscular transmission have been identified as causing hypotonia; fluctuating or persistent weakness of ocular, bulbar, or limb muscles; or arthrogryposis in infancy. The basis of one widely used classification scheme of congenital myasthenic syndromes (CMS) is whether the abnormality occurs in the presynaptic motor nerve terminal, the synaptic cleft, or the postsynaptic sarcolemma. The cause of the presynaptic disorder is a defect in the enzyme choline acetyltransferase, which synthesizes the neurotransmitter, whereas the synaptic defect results from deficiency of the end-plate cholinesterase. The causes of the postsynaptic disorders are various abnormalities of the structure, localization, or kinetics of the acetylcholine receptor. Inheritance of most CMS is autosomal recessive, except for the slow channel syndrome, which is autosomal dominant. The clinical presentation is similar to other forms of myasthenia occurring in infancy, although deficiencies of the presynaptic enzyme, choline acetyltransferase, and of the postsynaptic acetylcholine receptor–associated protein, rapsyn, are also associated with sudden episodes of apnea (Hantai et al., 2004). Infants with CMS have negative antibody studies and demonstrate a decremental response on RNS. Specialized electrophysiological testing on fresh muscle biopsy specimens has been useful as a diagnostic tool but is not widely available. Of the 10 different genes currently known to be associated with CMS, testing is commercially available for 7, while testing of the others is available on a research basis only. Most forms of CMS are treated with cholinesterase inhibitors and/or the potassium channel inhibitor, 3,4-diaminopyridine. However, cholinesterase inhibitors may exacerbate end-plate cholinesterase deficiency and slow-channel syndrome, while the latter may respond to fluoxetine (Harper et al., 2003). The natural history of CMS is highly variable even among patients with the same genotype.
Infant Botulism
Spores of the gram-positive anaerobe Clostridium botulinum, an organism found in soil and in some cases in contaminated foods, produce an exotoxin that prevents anchoring of acetylcholine-containing vesicles to the presynaptic nerve terminal of the neuromuscular junction, disrupting neuromuscular transmission and resulting in flaccid weakness. In adults, the cause of botulism is ingestion of the preformed toxin; the organism itself cannot survive in the acidic environment of the adult digestive tract. By contrast, infants who ingest spores may be colonized and develop botulism from in situ production of the toxin. Affected infants may present any time after 2 weeks of age and may have relatively greater involvement of bulbar than appendicular muscles. The characteristic finding on RNS is an increment in the CMAP with high-rate (50 Hz) stimulation (Cornblath et al., 1983). Diagnostic confirmation is obtained by testing a stool or enema specimen with a bioassay in mice inoculated against different strains of toxin. Aside from supportive measures, early administration of botulinum immune globulin shortens the course of the disease (Arnon et al., 2006). In most cases, treatment should be initiated based on the clinical suspicion and should not be delayed while awaiting results of the bioassay.
Muscle Disorders
Congenital Myopathies
Centronuclear Myopathy
Centronuclear myopathy has X-linked, recessive, and dominant forms due to defects in three different genes, although only the first two result in congenital weakness and hypotonia. X-linked centronuclear myopathy, caused by mutations in the MTM1 gene, affects male infants. Clinical features include facial weakness, ptosis, and ophthalmoplegia in addition to severe limb weakness. Affected infants may have macrocephaly, a thin face, and long digits. Serum CK is normal or mildly elevated, and EMG shows a nonspecific myopathic pattern. The characteristic findings on muscle pathology are the presence of large, single, centrally located nuclei in more than 5% of myofibers, and predominance of hypotrophic type I fibers (Pierson et al., 2007). Mutations in the BIN1 gene result in a similar phenotype but with recessive inheritance (Nicot et al., 2007). A dominantly inherited form of centronuclear myopathy exists but presents beyond infancy.
Nemaline Myopathy
At least six different genes have been associated with this disorder, all of which encode different components of thin filaments within the sarcomere. Inheritance may be recessive or dominant, and many cases are associated with de novo mutations. Characteristic of many forms is congenital weakness involving proximal limb muscles, the face, and extraocular muscles. Muscle biopsy reveals characteristic rod-shaped sarcoplasmic inclusions best visualized on Gomori trichrome staining of frozen muscle. The most common abnormality is in the gene encoding the skeletal muscle alpha actin (ACTA1), accounting for approximately 25% of cases. Genetic testing is commercially available for this gene, as well as four of the other known causative genes (Laing, 2007).
Central Core Disease
The majority of individuals with central core disease have mild weakness, although congenital weakness with reduced fetal movement, arthrogryposis, and spinal deformities does occur. Sparing of the face and extraocular muscles is common. Histology of frozen muscle shows well-demarcated areas of absent staining by oxidative stains such as NADH-tetrazolium reductase. These areas tend to be centrally located within type I myofibers and run the entire length of the myofibers on longitudinal sections. The most common causative genetic abnormality affects the skeletal muscle ryanodine receptor 1 (RYR1), which mediates calcium release from the sarcoplasmic reticulum during excitation-contraction coupling. The disorder is allelic with susceptibility to malignant hyperthermia (Robinson et al., 2006). Some individuals have both phenotypes, others have only one of the two disorders. Both autosomal dominant and autosomal recessive inheritance of central core disease has been documented (Monnier et al., 2000).
Nonsyndromic Congenital Muscular Dystrophies
Merosin-Deficient Congenital Muscular Dystrophy
The etiology of the most common nonsyndromic congenital muscular dystrophy is a recessively inherited deficit of α-2 laminin (merosin), a component of the dystrophin-associated glycoprotein complex in skeletal muscle. Affected infants are hypotonic, with weakness of face and limb muscles and arthrogryposis. Extraocular and bulbar muscles are not usually affected. Serum CK is highly elevated, and hypomyelination of cerebral white matter is apparent on brain MRI by 6 months of age. EMG is myopathic, and some infants also have evidence of peripheral myelin dysfunction on nerve conduction studies. Muscle biopsy shows evidence of a chronic necrotizing myopathy, and endomysial lymphocytic inflammation also occurs. Immunostaining demonstrates absence of skeletal muscle merosin. Sequencing of the LAMA2 gene is commercially available. Weakness is usually static. Epilepsy occurs at a higher rate in affected infants than in the general population, although cognition is usually normal (Herrmann et al., 1996).
Ullrich Congenital Muscular Dystrophy
This autosomal recessive nonsyndromic congenital muscular dystrophy results from defects in the extracellular matrix protein, collagen VI. The presence of proximal joint contractures with striking hyperlaxity of distal joints in early life distinguishes it from other disorders in this category (Muntoni et al., 2002). Serum CK ranges from normal to 10 times the upper limit of normal. Reduced immunostaining of frozen skeletal muscle for collagen VI and production of the protein in cultured fibroblasts are diagnostic. Both assays, as well as genetic testing for abnormalities in the three different COL6A genes, are commercially available.
Andrews P.I. Autoimmune myasthenia gravis in childhood. Semin Neurol. 2004;24:101-110.
Arnon S.S., Schechter R., Maslanka S.E., et al. Human botulism immune globulin for the treatment of infant botulism. N Engl J Med. 2006;354:462-471.
Butler M.G., Meaney J.F., Palmer C.G. Clinical and cytogenetic survey of 39 individuals with Prader-Willi syndrome. Am J Med Genet. 1986;23:793-809.
Cameron C.L., Kang P.B., Burns T.M., et al. Multifocal slowing of nerve conduction in metachromatic leukodystrophy. Muscle Nerve. 2004;29:531-536.
Cornblath D.R., Sladky J.T., Sumner A.J. Clinical electrophysiology of infantile botulism. Muscle Nerve. 1983;6:448-452.
Frahm, J., Requardt, M., Helms, G., et al., 1994. Creatine deficiency in the brain: a new treatable inborn error of metabolism identified by proton and phosphorus MR spectroscopy in vivo. In: Proceedings of the Second Annual Meeting, Society of Magnetic Resonance, San Francisco, 1, 340.
Gingold M.K., Jaynes M.E., Bodensteiner J.B., et al. The rise and fall of the plantar response in infancy. J Pediatr. 1998;133:568-570.
Gregory A., Westaway S.K., Holm I., et al. Neurodegeneration associated with genetic defects in phospholipase A2. Neurology. 2008;71:1402-1409.
Grohmann K., Schuelke M., Diers A., et al. Mutations in the gene encoding immunoglobulin mu-binding protein 2 cause spinal muscular atrophy with respiratory distress type 1. Nat Genet. 2001;29:75-77.
Gruenwald S. The clinical spectrum of phosphomannomutase 2 deficiency (CDG-Ia). Biochim Biophys Acta. 2009;1792:827-834.
Hantai D., Pascale R., Koenig J., et al. Congenital myasthenic syndromes. Curr Opin Neurol. 2004;17:539-551.
Harper C.M., Fukudome T., Engel A.G. Treatment of slow channel congenital myasthenic syndrome with fluoxetine. Neurology. 2003;60:1710-1713.
Herrmann R., Straub V., Meyer K., et al. Congenital muscular dystrophy with laminin alpha 2 deficiency: Identification of a new intermediate phenotype and correlation of clinical findings to muscle immunohistochemistry. Eur J Pediatr. 1996;155:968-976.
Kishnani P.S., Nicolino M., Voit T., et al. Chinese hamster ovary cell-derived recombinant human acid alpha-glucosidase in infantile-onset Pompe disease. J Pediatr. 2006;149:89-97.
Klinge L., Eagle M., Haggerty I.D., et al. Severe phenotype in infantile facioscapulohumeral muscular dystrophy. Neuromuscul Disord. 2006;16:553-558.
Korn-Lubetzki I., Dor-Wollman T., Soffer D., et al. Early peripheral nervous system manifestations of infantile Krabbe disease. Pediatr Neurol. 2003;28:115-118.
Laing N.G. Congenital myopathies. Curr Opin Neurol. 2007;20:583-589.
Matthews P.M., Andermann F., Silver K., et al. Proton MR spectroscopic demonstration of differences in regional brain metabolic abnormalities in mitochondrial encephalomyopathies. Neurology. 1993;43:2484-2490.
Monnier N., Romero N.B., Lerale J., et al. An autosomal dominant congenital myopathy with cores and rods is associated with a neomutation in the RYR1 gene encoding the skeletal muscle ryanodine receptor. Hum Mol Genet. 2000;9:2599-2608.
Muntoni F., Bertini E., Bonnemann C., et al. 98th ENMC international workshop on congenital muscular dystrophy (CMD), 7th workshop of the MYO CLUSTER project GENRE 26-28th October, 2001, Naarden, The Netherlands. Neuromuscul Disord. 2002;12:889-896.
Muntoni F., Torelli S., Brockington M. Muscular dystrophies due to glycosylation defects. Neurotherapeutics. 2008;5:627-632.
Nicot A.S., Toussaint A., Tosch V., et al. Mutations in amphiphysin 2 (BIN1) disrupt interaction with dynamin 2 and cause autosomal recessive centronuclear myopathy. Nat Genet. 2007;39:1134-1139.
Ogino S., Wilson R.B. Genetic testing and risk assessment for spinal muscular atrophy. Hum Genet. 2002;111:477-500.
Oskoui M., Kaufmann P. Spinal muscular atrophy. Neurotherapeutics. 2008;5:499-506.
Papazian O. Transient neonatal myasthenia gravis. J Child Neurol. 1992;7:135-141.
Pierson C.R., Agrawal P.B., Blasko J., et al. Myofiber size correlates with MTM1 mutation type and outcome in X-linked myotubular myopathy. Neuromusc Disord. 2007;17:562-568.
Plante-Bordenueve V., Said G. Dejerine-Sottas disease and hereditary demyelinating polyneuropathy of infancy. Muscle Nerve. 2002;26:608-621.
Rakocevic-Stojanovic D., Savic D., Pavlovic S., et al. Intergenerational changes of CTG repeat depending on the sex of the transmitting parent in myotonic dystrophy type 1. Eur J Neurol. 2005;12:236-237.
Ramser J., Ahearn M.E., Lenski C., et al. Rare missense and synonymous variants in UBE1 are associated with X-linked infantile spinal muscular atrophy. Am Gen Hum Genet. 2008;82:188-193.
Robinson R., Carpenter D., Shaw M.A., et al. Mutations in RYR1 in malignant hyperthermia and central core disease. Hum Mutat. 2006;27:977-989.
Rudnick-Schoneborn S., Lutzenrath S., Borokowska J., et al. Analysis of creatine kinase activity in 504 patients with proximal spinal muscular atrophy types I-III from the point of view of progression and severity. Eur Neurol. 1998;39:154-162.
Swoboda K.J., Prior T.W., Scott C.B., et al. Natural history of denervation in SMA: Relation to age, SMN2 copy number, and function. Ann Neurol. 2005;57:704-712.
Tidwell T., Pitt M.C. A new analytical method to diagnose congenital myasthenia with stimulated single-fiber electromyography. Muscle Nerve. 2007;35:107-110.
Vasta I., Kinali M., Messina S., et al. Can clinical signs identify newborns with neuromuscular disorders? J Pediatr. 2005;146:73-79.