Chapter 551 Hypopituitarism
Hypopituitarism denotes underproduction of growth hormone (GH) alone or in combination with deficiencies of other pituitary hormones. Affected children have postnatal growth impairment that is specifically corrected by replacement of GH. The incidence of congenital hypopituitarism is thought to be between 1 in 4,000 and 1 in 10,000 live births. With expanding knowledge of the genes that direct pituitary development or hormone production, an increasing proportion of cases can be attributed to specific genetic disorders. Mutations in 7 candidate genes account for 13% of isolated growth hormone deficiency (IGHD) and 20% of multiple pituitary hormone deficiency (MPHD) cases. The likelihood of finding mutations is increased by positive family histories and decreased in cases with adrenocorticotropin hormone (ACTH) deficiency. The genes, hormonal phenotypes, associated abnormalities and modes of transmission for such established genetic disorders are shown in Tables 551-1 and 551-2. Acquired hypopituitarism usually has a later onset and different causes (Table 551-3).
Table 551-1 ETIOLOGIC CLASSIFICATION OF MULTIPLE PITUITARY HORMONE DEFICIENCY
| GENE OR LOCATION | PHENOTYPE | INHERITANCE |
|---|---|---|
| GENETIC FORMS | ||
| POU1F1 (PIT1) | GH, TSH, PRL | R, D |
| PROP1 | GH, TSH, PRL, LH, FSH, ±ACTH, variable AP | R |
| LHX3 | GH, TSH, PRL, LH, FSH, variable AP, ±short neck | R |
| LHX4 | GH, TSH, ACTH, small AP, EPP, ±Arnold Chiari | D |
| TPIT | ACTH, severe neonatal form | R |
| HESX1 | GH, variable for others, small AP, EPP | R, D |
| SOX3 | Variable deficiencies, ±MR, EPP, small AP and stalk | XL |
| PTX2 | Rieger syndrome | D |
| GLI2 | Holoprosencephaly, midline defects | D |
| GLI3 | Hall-Pallister syndrome | D |
| SHH (Sonic hedgehog) | GH deficiency with single central incisor | D |
| ACQUIRED FORMS | ||
| Idiopathic | ||
| Irradiation | GH deficiency precedes other deficiencies | |
| Inflammation | Histiocytosis, sarcoidosis | |
| Autoimmune | Hypophysitis | |
| Post-surgical | Stalk section, vascular compromise | |
| Tumor | Craniopharyngioma, glioma, pinealoma | |
| Trauma | Battering, shaken baby, vehicular | |
| UNCERTAIN ETIOLOGY | ||
| Idiopathic | ||
| Congenital absence of pituitary | ||
| Septo-optic dysplasia | ||
| Birth trauma | ||
ACTH, adrenocorticotropic hormine; AP, anterior pituitary; D, dominant; EPP, ectopic posterior pituitary; FSH, follicle-stimulating hormone; GH, growth hormone; LH, luteinizing hormone; MR, mental retardation; PRL, prolactin; R, recessive; TSH, thyroid-stimulating hormone; XL, X-linked.
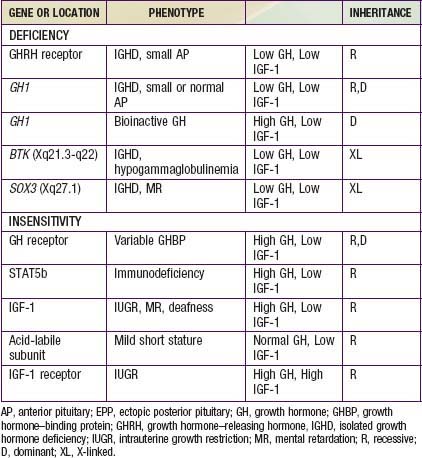
Table 551-3 CAUSES OF ACQUIRED HYPOPITUITARISM
BRAIN DAMAGE*
PITUITARY TUMORS*
NON-PITUITARY TUMORS
INFECTION
INFARCTION
AUTOIMMUNE DISORDER
OTHER
* Pituitary tumors are classically the most common cause of hypopituitarism. However, new findings imply that causes related to brain damage might outnumber pituitary adenomas in causing hypopituitarism.
From Schneider HJ, Aimaretti G, Kreitschmann-Andermahr I, et al: Hypopituitarism, Lancet 369:1461–1470, 2007.
Multiple Pituitary Hormone Deficiency
Genetic Forms
HESX1
The HESX1 gene is expressed in precursors of all 5 cell types of the anterior pituitary early in embryologic development. Mutations result in a complex phenotype with defects in development of the optic nerve. Heterozygotes for loss-of-function mutations show the combinations of isolated GH deficiency and optic nerve hypoplasia. Homozygotes can have full expression of septo-optic dysplasia (SOD). This condition combines incomplete development of the septum pellucidum with optic nerve hypoplasia and other midline abnormalities. Clinical observation of nystagmus and visual impairment in infancy leads to the discovery of optic nerve and brain abnormalities. SOD is associated with anterior and/or posterior pituitary hormone deficiencies in about 25% of the cases. These patients often show the triad of a small anterior pituitary gland, an attenuated pituitary stalk, and an ectopic posterior pituitary bright spot. The great majority of patients with SOD do not have HESX1 mutations. The etiology might involve mutations in another gene or a nongenetic explanation (Chapters 585 and 623).
Other Congenital Forms
Pituitary hypoplasia can occur as an isolated phenomenon or in association with more extensive developmental abnormalities such as anencephaly or holoprosencephaly. Midfacial anomalies (cleft lip, palate; Chapter 302) or the finding of a solitary maxillary central incisor indicate a high likelihood of GH or other anterior or posterior hormone deficiency. At least 12 genes have been implicated in the complex genetic etiology of holoprosencephaly (Chapter 585.7). In the Hall-Pallister syndrome, absence of the pituitary gland is associated with hypothalamic hamartoblastoma, postaxial polydactyly, nail dysplasia, bifid epiglottis, imperforate anus, and anomalies of the heart, lungs, and kidneys. The combination of anophthalmia and hypopituitarism has been associated with mutations in the SIX6, SOX2, and OTX2 genes.
Acquired Forms
Any lesion that damages the hypothalamus, pituitary stalk, or anterior pituitary can cause pituitary hormone deficiency (see Table 551-3). Because such lesions are not selective, multiple hormonal deficiencies are usually observed. The most common lesion is the craniopharyngioma (Chapter 491). Central nervous system germinoma, eosinophilic granuloma (histiocytosis), tuberculosis, sarcoidosis, toxoplasmosis, meningitis, and aneurysms can also cause hypothalamic-hypophyseal destruction. Trauma, including shaken child syndrome (Chapter 37), motor vehicle accidents, traction at delivery, anoxia, and hemorrhagic infarction, can also damage the pituitary, its stalk, or the hypothalamus.
Isolated Growth Hormone Deficiency and Insensitivity
Genetic Forms of Growth Hormone Deficiency
Growth Hormone Insensitivity
Clinical Manifestations
Acquired Hypopituitarism
The child is normal initially, and manifestations similar to those seen in idiopathic pituitary growth failure gradually appear and progress. When complete or almost complete destruction of the pituitary gland occurs, signs of pituitary insufficiency are present. Atrophy of the adrenal cortex, thyroid, and gonads results in loss of weight, asthenia, sensitivity to cold, mental torpor, and absence of sweating. Sexual maturation fails to take place or regresses if already present. There may be atrophy of the gonads and genital tract with amenorrhea and loss of pubic and axillary hair. There is a tendency to hypoglycemia. Growth slows dramatically. Diabetes insipidus (Chapter 552) may be present early but tends to improve spontaneously as the anterior pituitary is progressively destroyed.
If the lesion is an expanding tumor, symptoms such as headache, vomiting, visual disturbances, pathologic sleep patterns, decreased school performance, seizures, polyuria, and growth failure can occur (Chapter 491). Slowing of growth can antedate neurologic signs and symptoms, especially with craniopharyngiomas, but symptoms of hormonal deficit account for only 10% of presenting complaints. Evidence of pituitary insufficiency might first appear after surgical intervention. In children with craniopharyngiomas, visual field defects, optic atrophy, papilledema, and cranial nerve palsy are common.
Differential Diagnosis
Constitutional Growth Delay
Constitutional growth delay is one of the variants of normal growth commonly encountered by the pediatrician. Length and weight measurements of affected children are normal at birth, and growth is normal for the 1st 4-12 mo of life. Height is sustained at a lower percentile during childhood. The pubertal growth spurt is delayed, so their growth rates continue to decline after their classmates have begun to accelerate. Detailed questioning often reveals other family members (often one or both parents) with histories of short stature in childhood, delayed puberty, and eventual normal stature. IGF-1 levels tend to be low for chronological age but within the normal range for bone age. GH responses to provocative testing tend to be lower than in children with a more typical timing of puberty. The prognosis for these children to achieve normal adult height is guarded. Predictions based on height and bone age tend to overestimate eventual height to a greater extent in boys than in girls. Boys with >2 yr of pubertal delay can benefit from a short course of testosterone therapy to hasten puberty after 14 yr of age. The cause of this variant of normal growth is thought to be persistence of the relatively hypogonadotropic state of childhood (Chapter 13).
Primary Hypothyroidism
Primary hypothyroidism (Chapter 559) is more common than GH deficiency. Low total or free T4 and elevated TSH levels establish the diagnosis. Responses to GH provocative tests may be subnormal and the sella may be enlarged. Pituitary hyperplasia recedes during treatment with thyroid hormone. Because thyroid hormone is a necessary prerequisite for normal GH synthesis, it must always be assessed before GH evaluation.
Psychosocial Causes
Emotional deprivation is an important cause of retardation of growth and mimics hypopituitarism. The condition is known as psychosocial dwarfism, maternal deprivation dwarfism, or hyperphagic short stature. The mechanisms by which sensory and emotional deprivation interfere with growth are not fully understood. Functional hypopituitarism is indicated by low levels of IGF-1 and by inadequate responses of GH to provocative stimuli. Puberty may be normal or even premature. Appropriate history and careful observations reveal disturbed mother-child or family relations and provide clues to the diagnosis (Chapter 37). Proof may be difficult to establish because the parents or caregivers often hide the true family situation from professionals, and the children rarely divulge their plight. Emotionally deprived children often have perverted or voracious appetites, enuresis, encopresis, insomnia, crying spasms, and sudden tantrums. The subgroup of children with hyperphagia and a normal body mass index tends to show catch-up growth when placed in a less stressful environment.
Abuzzahabab MJ, Schneider A, Goddard A, et al. IGF-1 receptor mutations resulting in intrauterine and postnatal growth retardation. New Eng J Med. 2003;349:2211-2222.
Albertsson-Wikland K. Growth hormone in children with idiopathic short stature. BMJ. 2011;342:d1248.
Amundson E, Wide-Boman U, Barrenäs ML, et al. Impact of growth hormone therapy on quality of life in adults with Turner syndrome. J Clin Endocrinol Metab. 2010;95:1355-1359.
Badaru A, Wilson DM. Alternatives to growth hormone stimulation testing in children. Trends Endocrinol Metab. 2004;15:252-258.
Besson A, Salemi S, Deladoey J, et al. Short stature caused by a biologically inactive mutant growth hormone. J Clin Endocr Metab. 2005;90:2493-2499.
Carel JC, Ecosse E, Nicolino M, et al. Adult height after long term treatment with recombinant growth hormone for idiopathic isolated growth hormone deficiency: observational follow up of the French population based registry. BMJ. 2002;25:70-73.
Choc NA, Choh SA, Jehangir J, et al. Posterior pituitary ectopia with absent pituitary stalk—a rare cause of hypopituitarism. J Pediatr Endocrin Metab. 2009;22:407-408.
Dattani MT. Growth hormone deficiency and combined pituitary hormone deficiency: does the genotype matter? Clin Endocrinol. 2005;63:121-130.
Deodati A, Cianfarani S. Impact of growth hormone therapy on adult height of children with idiopathic short stature: systematic review. BMJ. 2011;342:c7157.
Domene HM, Bengolea SV, Martinez AS, et al. Deficiency of the circulating insulin-like growth factor system associated with inactivation of the acid-labile subunit gene. New Eng J Med. 2004;350:570-577.
Drake AJ, Kelnar CJH. The evaluation of growth and the identification of growth hormone deficiency. Arch Dis Child Educ Pract Ed. 2006;91:ep61-ep67.
Finkelstein BS, Imperiale TF, Speroff T, et al. Effect of growth hormone therapy on height in children with idiopathic short stature. Arch Pediatr Adolesc Med. 2002;156:230-240.
Grimberg A, Feemster KA, Pati S, et al. Medically underserved girls receive less evaluation for short stature. Pediatrics. 2011;127:696-702.
Growth Hormone Research Society. Consensus guidelines for the diagnosis and treatment of growth hormone (GH) deficiency in childhood and adolescence: summary statement of the GH Research Society. J Clin Endocrinol Metab. 2000;85:3998.
Hughes IP, Choong CS, Cotterill A, et al. Gender bias in children receiving growth hormone treatment. J Clin Endocrinol Metab. 2010;95:1191-1198.
Hwa V, Little B, Adiyaman P, et al. Severe growth hormone insensitivity resulting from total absence of signal transducer and activator of transcription 5b. J Clin Endocr Metab. 2005;90:4260-4266.
Kerr JM, Wierman ME. Pituitary apoplexy. BMJ. 2011;342:d1270.
Lee MM. Idiopathic short stature. N Engl J Med. 2006;354:2576-2582.
Leschek EW, Rose SR, Yanovski JA, et al. Effect of growth hormone treatment on adult height in peripubertal children with idiopathic short stature: a randomized, double-blind, placebo-controlled trial. J Clin Endorinol Metab. 2004;89:3140-3148.
The Medical Letter. Insulin-like growth factor-1 for severe growth failure. Med Lett. 2007;49:43-44.
Rappold G, Blum WF, Shavrikova EP, et al. Genotypes and phenotypes in children with short stature: clinical indicators of SHOX haploinsufficiency. J Med Genet. 2007;44:306-313.
Rosenfeld RG, Rosenbloom AL, Guevara-Aguirre J. Growth hormone (GH) insensitivity due to primary GH receptor deficiency. Endocr Rev. 1994;15:369-390.
Ross JL, Quigley CA, Cao D, et al. Growth hormone plus childhood low-dose estrogen in Turner’s syndrome. N Engl J Med. 2011;364(13):1230-1242.
Schmiegelow M, Lassen S, Weber L, et al. Dosimetry and growth hormone deficiency following cranial irradiation of childhood brain tumors. Med Pediatr Oncol. 1999;33:564-571.
Schneider HJ, Aimaretti G, Kreitschmann-Andermahr I, et al. Hypopituitarism. Lancet. 2007;369:1461-1470.
Simon D, Hadjiathanasiou C, Garel C, et al. Phenotypic variability in children with growth hormone deficiency associated with posterior pituitary ectopia. Clin Endocrinol. 2006;64:416-422.
Traggiai C, Stanhope R. Endocrinopathies associated with midline cerebral and cranial malformations. J Pediatr. 2002;140:252-255.
Tajima T, Ohtake A, Hoshio M, et al. OTX2 loss of function mutation causes anophthalmia and combined pituitary hormone deficiency with a small anterior and ectopic posterior pituitary. J Clin Endocr Metab. 2009;94:314-319.
Woods KA, Camacho-Hubner C, Savage MO, et al. Intrauterine growth retardation and postnatal growth failure associated with deletion of the insulin-like growth factor gene. N Engl J Med. 1996;35:1363-1367.