Hypopituitarism and Growth Hormone Deficiency
Epidemiology
Limited information is available on the epidemiology of hypopituitarism. A Swedish survey estimates the prevalence of hypopituitarism to be 175 cases per million.1 A Spanish study has reported a prevalence of hypopituitarism of 290 and 450 cases per million from two cross-sectional surveys in 1992 and 1999, respectively, and a corresponding incidence of 60 per million per year.2 Sixty percent of the patients were GH deficient, giving a prevalence of GH deficiency of 114 to 270 cases per million and an incidence of 24 per million per year. A recent nationwide study in Denmark of GH deficiency identified an average incidence of approximately 20 million per year.3
Mortality
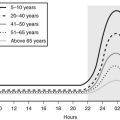
Mortality is increased in hypopituitarism. Data from six epidemiologic studies,4–9 comprising patients aged between 46 and 52 years who were followed for 10 to 13 years, report increased mortality with standardized mortality rates (SMRs) from 1.2 to 2.2. The higher mortality arises from cardiovascular and cerebrovascular disease and appears to be greater in women10 (Fig. 6-1).5–8,11,12 Risk ratios for malignancies and respiratory disease varied. Comparison of these studies is difficult because of different definitions, causes, and degrees of hypopituitarism. For example, patients with craniopharyngioma and/or patients treated with radiotherapy were included in some4,6,8,9 but not all studies.5,7 Craniopharyngiomas carry a worse prognosis than pituitary adenomas,13 and radiotherapy has been identified as a factor that increases mortality.8 However, it is unclear whether it is radiotherapy or the result of more aggressive disease requiring radiotherapy that reduces survival.8 A recent study, which included only postoperative hypopituitarism from pituitary adenoma followed for a mean duration of 12.4 years, demonstrated increased mortality only in women, with an SMR of 1.97.14
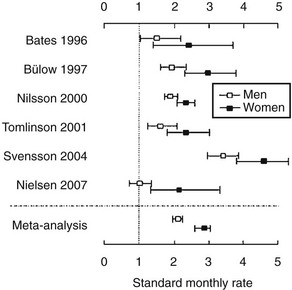
FIGURE 6-1 Standard mortality rates (SMR) and 95% confidence intervals (CI) in individual studies on patients with nonmalignant pituitary diseases not associated with excess adrenocorticotropic hormone (ACTH) or growth hormone (GH) secretion, and in the weighted meta-analysis (bottom line). Results are shown for men (open boxes) and women (black boxes) separately. (From Nielsen EH, Lindholm J, Laurberg P. Excess mortality in women with pituitary disease: a meta-analysis. Clin Endocrinol 67:693–697, 2007.)
Causes
Major causes of hypopituitarism are shown in Table 6-1. The most common cause is a pituitary adenoma or treatment with pituitary surgery or radiotherapy.
Pituitary and Hypothalamic Mass Lesions
Pituitary adenomas account for the vast majority of pituitary mass lesions, although secondary tumors do occur, from metastases to the pituitary gland from carcinomas of the breast, lung, colon, and prostate. Pituitary microadenomas are surprisingly common and are found in between 1.5% and 27% of patients at autopsy15; these tumors are very rarely, if at all, associated with hypopituitarism and tend to run a benign course. Macroadenomas are less common but are more frequently associated with pituitary hormone deficiencies; some 30% of patients with pituitary macroadenomas have one or more anterior pituitary hormone deficiencies. The causative mechanism of hypopituitarism is compression of the portal vessels in the pituitary stalk, secondary to the expanding tumor mass directly or to increased intrasellar pressure,16 which explains the potential reversibility of pituitary dysfunction after surgery in some patients.
Pituitary Surgery
The incidence and degree of hypopituitarism after surgery depend on the size of the original tumor, the degree of infiltration, and the experience of the surgeon. About 50% of patients already had evidence of GH, gonadotropin, or cortisol deficiency17 before surgery. The patient should also be warned of a possible deterioration of postoperative pituitary function, and assessment of pituitary function should be performed promptly after surgery. However, a postoperative decline in pituitary function is not universal. After surgery, about 80% had evidence of GH or gonadotropin deficiency. In patients who received postoperative radiotherapy, evaluation after 5 years revealed that all patients were GH deficient.17 On the other hand, surgery for pituitary adenomas may be associated with significant recovery of pituitary function. About half of the patients recover at least one pituitary insufficiency after transsphenoidal surgery. Postoperative improvement is more likely if no tumor is found on postoperative imaging, or if the tumor is not invasive.18 The pituitary hormone most likely to recover is thyroid-stimulating hormone (TSH), followed in order by adrenocorticotropic hormone (ACTH), gonadotropins, and GH.19 Recovery of pituitary function occurs early, within 8 weeks after surgery.20
Radiotherapy
Deficiency of one or more anterior pituitary hormones is almost invariable when the hypothalamic-pituitary axis lies within the fields of radiation. Hypopituitarism also develops in patients who received radiation therapy for nasopharyngeal carcinomas, parasellar tumors, and primary brain tumors, as well as in children who underwent prophylactic cranial irradiation for acute lymphoblastic leukemia or total body irradiation (TBI) for a variety of tumors and other diseases.21
The radiobiological impact of an irradiation schedule is dependent on the total dose, the number of fractions, and the duration and length of follow-up. Somatotrophs are the most sensitive to radiation damage, and thus, after lower radiation doses (<30 Gy), isolated GH deficiency ensues, whereas higher doses (30 to 50 Gy) increase the frequency of GH deficiency to 50% to 100% and may produce panhypopituitarism (Fig. 6-2). Radiation dose also determines the speed of onset of hormonal deficiency. The greater the dose, the earlier is the occurrence of GH deficiency, so that between 2 and 5 years after irradiation, 100% of children receiving more than 30 Gy (over a 3-week period) to the hypothalamic-pituitary axis developed subnormal GH responses to an insulin tolerance test (ITT), whereas 35% of those receiving less than 30 Gy (over a 3-week period) still showed a normal GH response22 (Fig. 6-3). However, interpretation of the impact of radiation-induced damage to the hypothalamic-pituitary axis on GH status is complicated in the early years after irradiation. Discordant results to different GH-provocative agents may be seen, such that up to 50% of patients classified as severely GH deficient with use of the ITT showed normal or mildly insufficient GH response during the combined GH-releasing hormone (GHRH) plus arginine stimulation test.23,24 The discordant response to dynamic testing in patients with GH deficiency is discussed in greater detail later in the chapter, under Diagnosis in Growth Hormone Deficiency.
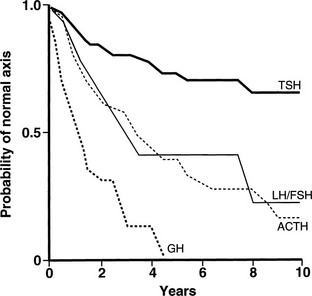
FIGURE 6-2 Life-table analysis indicating probabilities of initially normal hypothalamic-pituitary-target gland axes remaining normal after radiotherapy (3750 to 4250 cGy). Growth hormone (GH) secretion is the most sensitive of the anterior pituitary hormones to the effects of external radiotherapy, and thyroid-stimulating hormone (TSH) secretion is the most resistant. In two thirds of patients, gonadotropin deficiency develops before adrenocorticotropic hormone (ACTH) deficiency. FSH, Follicle-stimulating hormone; LH, luteinizing hormone. (From Littley MD, Shalet SM, Beardwell CG, et al: Hypopituitarism following external radiotherapy for pituitary tumors in adults. Q J Med 70:145–160, 1989.)
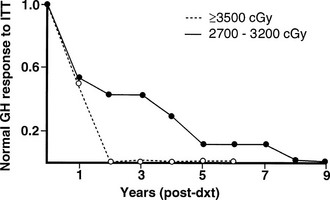
FIGURE 6-3 The incidence of growth hormone (GH) deficiency in children receiving 27 to 32 Gy or ≥35 Gy of cranial irradiation for a brain tumor in relation to time from irradiation (dxt). This illustrates that the speed at which individual pituitary hormone deficits develop is dose dependent; the higher the radiation dose, the earlier GH deficiency occurs. (Courtesy the Department of Medical Illustrations, Withington Hospital, Manchester, England.)
Fractionated stereotactic conformal radiotherapy (SCRT) is a more precise technique of localized irradiation that may reduce radiation damage to normal structures in the brain. However, despite this theoretical advantage of normal-tissue sparing, hypopituitarism remains a common complication. At a median follow-up of 32 months, 22% of patients with previously normal pituitary function or partial hypopituitarism developed new hormonal deficit, and 18% developed panhypopituitarism.25 The incidence of hypopituitarism was not different between conventional radiotherapy and SCRT,25 and it may occur as late as 10 years after therapy, reaching as high as 66% after a median follow-up of 17 years.26,27 Therefore, with increased survival, follow-up evaluation of patients irradiated for tumors of the brain and surrounding structures must focus equally on the possibility of tumor recurrence and on the delayed effects of therapy, including the endocrine effects. Endocrine testing should be performed on a yearly basis for at least 10 years and again at 15 years.
Genetic Causes
Combined Pituitary Hormone Deficit
A cascade of pituitary transcription factors regulates the differentiation of cells of Rathke’s pouch into somatotrophs, lactotrophs, thyrotrophs, gonadotrophs, and corticotrophs. Mutations in early appearing transcription factors tend to cause more extensive hormone deficiencies (Table 6-2).
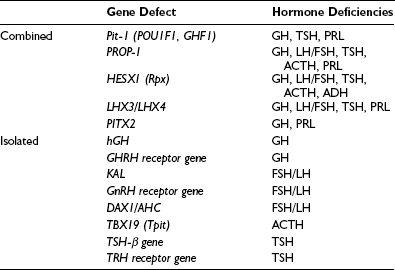
PROP1 (Prophet of Pit-1) is a novel pituitary paired-like homeodomain factor, which regulates the expression of Pit-1. Several multicenter studies of patients have reported that PROP1 mutations are the most common genetic cause of multiple pituitary hormone deficiencies, accounting for up to 40% to 50% of cases. PROP1 abnormalities result in similar phenotypic abnormalities to Pit-1 mutations but with associated gonadotropin deficiency. The degree of gonadotropin deficiency is variable, even among individuals within the same family with identical mutations. In individuals with a mutation of the PROP1 gene, progressive hypopituitarism develops, with GH, TSH, and gonadotropin deficiency typically present by the end of the second decade.28 The pituitary gland may pass through a hyperplastic phase before undergoing a phase of degeneration and late appearance of partial ACTH deficiency.
Other genetic causes of combined pituitary hormone deficiencies include mutations of HESX1, LHX3/LHX4, and Pitx1/Pitx2, which are transcription factors engaged in early pituitary cell development before the appearance of Pit-1 and PROP1.29–31 For example, HESX-1 is a homeobox gene expressed early in the ectoderm, which is the precursor of Rathke’s pouch. It plays an important role in optic nerve and anterior pituitary development. HESX-1 mutations in humans are associated with septo-optic dysplasia and GH deficiency. Other genes (e.g., LHX3) bind to Pit-1 and can enhance Pit-1 activity, or synergistically activate prolactin and TSH genes (e.g., Pitx1).
Isolated Pituitary Hormone Deficiency
Isolated Growth Hormone Deficiency: Isolated GH deficiency (IGHD) can arise from mutations of the GH gene and of the GH-releasing hormone (GHRH) receptor gene.32 The human GH (hGH) gene is located on chromosome 17 in a cluster of five genes: hGH-N encodes the gene for pituitary GH, hGH-V encodes the gene for placental GH, and three genes encode for human chorionic somatotropin (hCS). There are four types of Mendelian disorders of the GH gene that are due to deletion of these genes: IGHD IA and IB are both inherited in an autosomal recessive manner, resulting in absent or low GH levels. In patients with absent GH (IGHD IA), anti-GH antibodies often develop when they are treated with GH. IGHD II has an autosomal dominant mode of inheritance with variable clinical severity. IGHD III is an X-linked disorder that often is associated with hypogammaglobulinemia. Mutations of the gene encoding the GHRH receptor have been identified in a number of kindreds with severe GH deficiency in Pakistan and in Brazil.33,34
Isolated Gonadotropin Deficiency: Several gene mutations have been identified as causes of idiopathic hypogonadotropic hypogonadism (IHH) in humans.35 Gonadotropin-releasing hormone (GnRH) neurons originate in the olfactory placode and migrate during embryogenesis with the olfactory nerves to the hypothalamus. The KAL protein is necessary for this process to occur. Kallmann’s syndrome is characterized by the combination of IHH and anosmia or hyposmia, which usually is caused by defective GHRH secretion. It is a heterogeneous condition that manifests in an X chromosome–linked or autosomal dominant form. The X-linked form of the disorder is due to mutations in the KAL gene. Recently, several growth factors, including fibroblast growth factors (FGFs) and adhesion molecules, have been identified to play important regulatory roles in the development and migration of GHRH neurons. Mutation of the FGF receptor 1 (FGFR1) gene causes the autosomal dominant form of Kallmann’s syndrome.36 Additional clinical features associated with KAL mutations include bimanual synkinesia and renal agenesis; FGFR1 mutations typically lead to cleft lip palate and dental agenesis.
Isolated ACTH and TSH Deficiencies: Isolated deficiencies of TSH or ACTH are very rare; however, in a number of cases, a genetic abnormality has been described or proposed. Mutations of the coding region of the TSH-β subunit gene37 and the thyrotropin-releasing hormone (TRH)-receptor gene38 have been found in a number of families to be the cause of hereditary isolated TSH deficiency.
Recently, a pituitary transcription factor causing isolated ACTH deficiency was identified. TBX19 (the human T-box pituitary transcription factor, analogous to Tpit in the mouse) plays an essential role in differentiation of pro-opiomelanocortin (POMC) cells in the pituitary. At least two TBX19 gene mutations causing isolated ACTH deficiency have been described,39,40 and this may underlie the cause of a neonatal-onset form of congenital isolated ACTH deficiency,41 which is fatal unless diagnosed early and replaced with glucocorticoid.
Traumatic Brain Injury
Traumatic brain injury (TBI) is an under-appreciated cause of hypopituitarism. It was first reported in 1918. Meta-analysis of 19 studies, which included more than 1000 patients, demonstrated a pooled prevalence of hypopituitarism following TBI of 27.5% (1 pituitary hormone deficiency), and 7.7% of patients had multiple deficiencies. GH deficiency was the most common with a prevalence of 12.4%, followed by secondary hypogonadism (12.5%), hypoadrenalism (8.2%), and hypothyroidism (4.1%). Prevalence of diabetes insipidus is 26% in the acute phase and is decreased to 6.9% among long-term survivors.42
TBI is common, with an overall incidence of 235 per 100,000 persons per year.43 Traumatic hypopituitarism therefore is a major public health issue with significant clinical implications. The incidence of hypopituitarism following TBI is more than 30 patients per 100,000 population per year.44 Patients therefore should be screened for hypopituitarism following TBI. Early (<3 months) hormonal dysfunction, including central hypothyroidism and hypogonadism, was not predictive of long-term development of hypopituitarism.45 Assessment for GH deficiency, hypogonadism, and hypothyroidism is not necessary in the acute phase, but adrenal insufficiency should not be missed, as it can be fatal if untreated. All patients should undergo screening for hypopituitarism between 3 and 6 months after injury.
Lymphocytic Hypophysitis
Lymphocytic hypophysitis, an immune-mediated diffuse infiltration of the anterior pituitary with lymphocytes and plasma cells, occurs predominantly in women and often is first evident in pregnancy or after delivery. The classic presentation is peripartum hypopituitarism, often with a pituitary mass and visual failure. ACTH deficiency is an almost universal feature that, when undiagnosed, has proved fatal. At an early stage, the pituitary gland is enlarged and cannot be distinguished from a pituitary tumor by computed tomography (CT) or magnetic resonance imaging (MRI), whereas in later stages, the gland may atrophy, leaving an empty sella. Lymphocytic hypophysitis is more common in patients with other autoimmune endocrine diseases. Cytosolic autoantigens against the pituitary can be demonstrated in some cases but also are present in normal patients; thus the definitive diagnosis of this condition remains difficult without pituitary biopsy. Recently, identification of more specific autoantigens, such as chorionic somatomammotropin, may allow noninvasive diagnosis of hypophysitis in the future, with a sensitivity of 64% and a specificity of 86% by immunoblotting.46 Spontaneous resolution of both the mass and the hypopituitarism has been reported, and in some cases, neurosurgical intervention has led to irreversible pituitary failure. Therefore, conservative management is appropriate in most patients. Spontaneous recovery with physiologic hydrocortisone replacement can happen,47 and high-dose methylprednisolone pulse therapy may improve adrenopituitary function and shrinkage of the sellar mass.48
Hypopituitarism
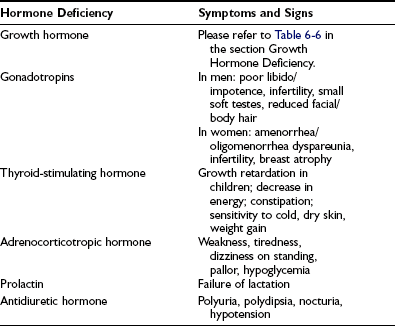
In cases arising from loss of function caused by an expanding silent mass lesion, the onset of symptoms is insidious, typically occurring with mild headaches, lethargy, fatigue, disinterest, weight gain, low mood, and declining libido—symptoms mimicking depression. Rarely, anorexia and weight loss may arise from ACTH deficiency and may be mistaken for and lead to extensive investigations for occult malignancy. Progressive mass expansion causing increasingly severe headaches or visual symptoms from chiasmal compression usually leads to radiologic investigations that clinch the diagnosis. A high index of suspicion is required to diagnose hypopituitarism. The symptoms and signs of individual hormone deficiency are listed in Table 6-3. The features of isolated deficiencies of each axis are described below. GH deficiency is addressed separately in the section dedicated to GH deficiency in adults.
Adrenocorticotropic Hormone Deficiency
ACTH deficiency is the most life-threatening component of hypopituitarism. If the onset is abrupt, as in pituitary apoplexy, the clinical picture may be dominated by profound shock in the most serious form. Patients with chronic ACTH deficiency usually present with chronic progressive symptoms of chronic fatigue, anorexia, and weight loss, sometimes mimicking anorexia nervosa or an occult malignancy. Patients on long-term glucocorticoid therapy can develop adrenal atrophy secondary to ACTH suppression.49,50 Examination may reveal pallor of the skin, in contrast to the hyperpigmentation of Addison’s disease, and in female patients particularly, loss of secondary sexual hair occurs. In severe ACTH deficiency, particularly in childhood, hypoglycemia can occur: Cortisol deficiency results in increased insulin sensitivity and a decrease in hepatic glycogen reserves. Hyponatremia, although less commonly seen than in Addison’s disease because of preservation of aldosterone secretion, may be the presenting feature of ACTH deficiency, particularly in the elderly.
Diagnosis and Endocrine Evaluation
Radiologic Phenotype of GH Deficiency
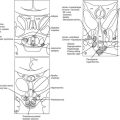
GH deficiency can be divided broadly into either genetic GH deficiency, when proven genetic mutations, such as GH-1, GHRH-R, and PROP-1, are identified, or idiopathic GH deficiency, when a genetic abnormality cannot be identified. On MRI, cases of genetic GH deficiency typically reveal a pituitary gland that is small or of normal size. The pituitary stalk is intact, and the location of the posterior lobe is normal. In contrast, idiopathic GH deficiency frequently is associated with a small pituitary gland, with evidence of stalk hypoplasia or interruption and an ectopic posterior lobe (Fig. 6-4). It has been suggested that perinatal trauma may be responsible for these abnormalities on MRI, resulting in idiopathic GH deficiency.51,52 This is supported by the observed higher frequency of breech delivery and birth hypoxemia with idiopathic GH deficiency than in genetic cases.53
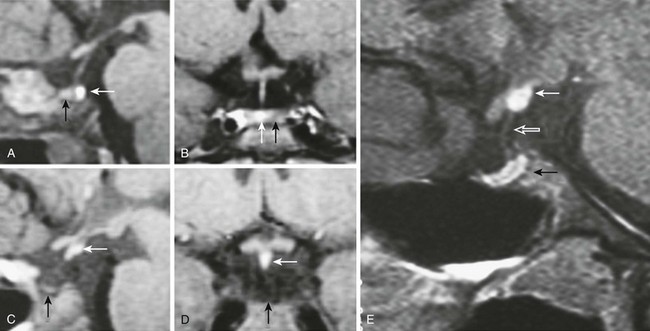
FIGURE 6-4 Magnetic resonance imaging (MRI) of congenital causes of growth hormone deficiency: Sagittal (A) and coronal (B) pituitary imaging studies of a patient with isolated growth hormone (GH) deficiency caused by GH-1 gene deletion of 6.7 kb showing intact stalk, normal pituitary gland (black arrow), and posterior lobe (white arrow); sagittal (C) and coronal (D) views of a patient with idiopathic isolated GH deficiency showing stalk interruption, an ectopic posterior lobe (white arrow), and pituitary gland hypoplasia (black arrow). Sagittal (E) view of pituitary MRI of a 17-year-old patient with idiopathic GH deficiency, demonstrating a hypoplastic gland (black arrow), stalk hypoplasia (hollow white arrow), and an ectopic posterior pituitary gland (white arrow). (From Osorio MG, Marui S, Jorge AA, Latronico AC, Lo LS, Leite CC, Estefan V, Mendonca BB, Arnhold IJ: Pituitary magnetic resonance imaging and function in patients with growth hormone deficiency with and without mutations in GHRH-R, GH-1, or PROP-1 genes. J Clin Endocrinol Metab 87:5076–5084, 2002.)
The stalk provides vascular communication, and its presence is significant in relation to the evaluation of diagnostic testing for GH deficiency. Maghnie and colleagues have reported that integrity of the hypothalamic-pituitary connection is necessary for GHRH-arginine to stimulate GH release, as the GH response is markedly impaired in patients with stalk agenesis.54
Endocrine Evaluation
Adult Gonadotropin Deficiency.: In women of postmenopausal age, gonadotropin levels are clearly low or undetectable, whereas in premenopausal women, amenorrhea (or less commonly, oligomenorrhea), in addition to low estradiol levels and low or normal gonadotropin levels, provides sufficient evidence of the diagnosis. In adult men, a similar picture of low testosterone levels and low or inappropriately normal gonadotropin levels is seen.
Adrenocorticotropic Hormone Deficiency.: A high index of clinical suspicion is most important in establishing the diagnosis. In normal people, the highest plasma cortisol levels are found between 6:00 am and 8:00 am, and the lowest before midnight. Plasma cortisol and ACTH concentrations are elevated during physical and emotional stress, including acute illness, trauma, surgery, infection, and starvation.
If a 9:00 am cortisol level is less than 100 nmol/L, particularly in an unwell patient, cortisol deficiency is highly likely, whereas a baseline level greater than 500 nmol/L indicates normality; many authors suggest that dynamic assessment of the hypothalamic-pituitary-adrenal (HPA) axis is not necessary under these circumstances.55 Unless the patient is known to have pituitary disease, a paired plasma ACTH level will help distinguish between primary and secondary glucocorticoid deficiency: In primary cortisol deficiency (Addison’s disease), the ACTH level will be high, whereas in secondary glucocorticoid deficiency, the ACTH level will be low or inappropriately normal.
The insulin tolerance test (ITT) is the test of choice in those suspected of secondary adrenal failure. The ITT evaluates the response of the HPA axis to the potent stressor of hypoglycemia, and it is generally the “gold standard” in the confirmation of secondary adrenal failure. It has the advantage of also being a test of growth hormone reserve in patients with pituitary disease.56 Following injection of a standard dose of intravenous insulin (0.1 unit/kg),57 cortisol concentrations are measured serially. Upon achievement of adequate hypoglycemia (<2.2 mmol/L), a peak cortisol response of between 500 and 600 nmol/L generally is accepted as adequate.58
Thyroid-Stimulating Hormone Deficiency.: In secondary hypothyroidism, one might expect to find reduced concentrations of free or total T4 in association with a serum TSH concentration below the normal range, analogous to the biochemical findings in secondary hypogonadism. Most have normal or occasionally elevated TSH levels. The mechanism behind this apparent contradiction is poorly understood, but it may be due to reduced bioactivity of TSH,59 which suggests that TRH regulates not only the secretion of TSH but also its specific molecular and conformational features. Dynamic testing with thyrotropin-releasing hormone has little diagnostic value other than distinguishing a hypothalamic cause, which is indicated by a delayed TSH peak.
Antidiuretic Hormone Deficiency.: The diagnosis of ADH deficiency first requires confirmation of polyuria, which is defined as the excretion of more than 3 L of urine per 24 hours (40 mL/kg/24 hours). Any patient with normal serum sodium and plasma osmolality who has a fluid output of <2 L/24 hours is likely to be normal and does not warrant further investigation.
Management
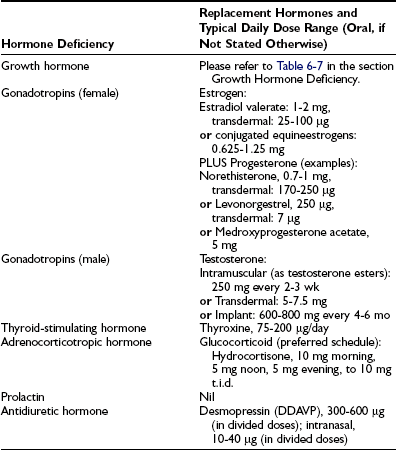
Treatment for hypopituitarism can be separated into those therapies directed at the underlying disease process and endocrine replacement therapy (Table 6-4). Management of the underlying condition is particularly challenging for craniopharyngiomas because randomized studies are lacking and their growth pattern is often unpredictable.60 Surgical excision in combination with external beam irradiation forms the mainstay of treatment. A recent trial demonstrated no recurrence of tumors during an average follow-up of 6.5 years in patients who received combined surgery with radiotherapy at diagnosis. Sixty percent of patients who received surgery alone had tumor recurrence.61
Hormone Replacement in Hypopituitarism
Gonadotropin Deficiency: In both sexes, sex steroid replacement therapy is important for the maintenance of normal body composition, skeletal health, and sexual function, and it is the most appropriate form of replacement therapy in patients not desirous of fertility.
Estrogen Replacement.: In women, this can be provided by many standard hormone replacement therapy preparations. Progesterone must be given (cyclically or continuously) in all women with an intact uterus to prevent the possible effect of unopposed estrogen on the endometrium, that is, dysfunctional bleeding or endometrial cancer. The dose of estrogen should not be supraphysiologic (as in the oral contraceptive pill) unless a clear indication, such as strong patient preference, exists, or a patient with partial gonadotropin deficiency still has occasional menstrual cycles, along with a desire for contraception. Although estrogen can be delivered as a tablet, patch, gel, or implant, a nonoral route is recommended because of reduction of insulin-like growth factor (IGF)-1 and fat oxidation by oral estrogen. The pathophysiology of the interaction of oral estrogen with the GH axis is discussed separately under the section on adult GH deficiency. However, an international surveillance study on 315 hypopituitary women taking estrogen replacement demonstrated significant predominance of oral versus transdermal estrogen use (86% vs. 14%). Women on oral estrogen had a significantly greater waist/hip ratio after GH treatment, with lower IGF-1 levels at the end of the study period on twice the GH dose received by women on transdermal estrogen.62 Therefore a nonoral route is highly recommended.
Androgen Replacement.: The choice of preparation of androgen replacement depends on local availability and patient preference. IM injection of testosterone can be associated with disturbing fluctuations in sexual function, energy level, and mood, mirroring the changes in testosterone concentrations. Transdermal testosterone systems, which are an alternative, are available as patch systems (nonscrotal or scrotal) or as the recently introduced testosterone gel. Both formulations are able to maintain physiologic testosterone profiles in most patients, but skin irritation, the need for scrotal shaving, and drying time after gel application are some of the potential drawbacks of both transdermal systems. Testosterone undecanoate has become available as an intramuscular injection, which achieves stable serum testosterone levels over a 10- to 14-week period. This new preparation has essentially replaced testosterone implants as replacement therapy.63
An area of investigative concern now is the therapeutic use of testosterone in women. In postmenopausal women, particularly those who have undergone bilateral oophorectomy, evidence exists that combined estrogen and testosterone replacement improves libido and sexual function. The rationale behind such therapy is that after bilateral oophorectomy, circulating testosterone levels decrease by 50%. This reduction tends to be even greater in women with hypopituitarism, who are likely to be more severely androgen deficient from loss of adrenal androgen production. Thus symptomatic patients may benefit from androgen supplementation. Such patients demonstrate improvement in sense of well-being, libido, lean body mass, and bone mineral density with androgen supplementation with dehydroepiandrosterone64 or low-dose testosterone.65
Gonadotropin and Gonadotropin-Releasing Hormone Therapy.: In the hypogonadotropic hypogonadal patient, fertility can be achieved with gonadotropin therapy in both men and women. The choice of therapy lies between gonadotropin replacement and GnRH. The former is the traditional therapeutic approach; initially, LH “activity” is provided by human chorionic gonadotropin (hCG) administered subcutaneously (SC) or IM at a dose of between 1000 and 2000 IU, two to three times weekly. Spermatogenesis is unlikely within the first 3 months of therapy. Treatment with hCG alone is continued for 6 months, with regular sperm counts to monitor progress. If adequate spermatogenesis is not achieved, then FSH in the form of human menopausal gonadotropin (hMG) or a recombinant FSH is added. The dose of FSH is increased if adequate spermatogenesis is not achieved after 6 months of combination therapy. The alternative regimen in patients with idiopathic hypogonadotropic hypogonadism and Kallmann’s syndrome is pulsatile GnRH therapy. GnRH is administered SC via a catheter attached to a minipump. This regimen appears to offer few advantages over gonadotropin therapy in men but may cause less gynecomastia. Both regimens may take up to 2 years to achieve adequate spermatogenesis; thus once effective, consideration should be given to storing several samples of frozen sperm for any future attempts at pregnancy.
Adrenocorticotropic Hormone Replacement.: Different forms of glucocorticoids are available for replacement, and each has its merits and disadvantages. The modern approach to glucocorticoid replacement is to mimic physiologic levels, ensuring sufficiency during times of acute illness, and to prevent over-replacement, which is associated with adverse metabolic outcomes.
Hydrocortisone directly replaces the missing hormone. Cortisone acetate is metabolized to cortisol and has a slower onset of action with longer biological activity. Both prednisolone and dexamethasone have longer half-lives, thus allowing daily administration. Recent estimation of a cortisol production rate of 9 mg/m2/day demonstrated by the isotope dilution method is lower than previously shown, and the traditional cortisol replacement dose of 30 mg/day is supraphysiologic and may lead to adverse metabolic outcomes.66,67 Generally, the lowest replacement dose tolerated by the patient is preferred (10 to 20 mg/day). Indeed higher serum concentrations of total cholesterol, low-density lipoprotein, triglycerides, and waist circumference were observed with increasing doses of glucocorticoid levels in a study comparing the metabolic phenotypes of patients with growth hormone deficiency treated with different formulations and doses of glucocorticoids. Metabolic end points were not worsened compared with ACTH-sufficient patients only in those treated with hydrocortisone-equivalent doses <20 mg/day.68 Doses should be divided to suit individual needs.
Thyroid-Hormone Stimulating Hormone Deficiency.: Secondary hypothyroidism is treated with thyroxine (T4) replacement therapy, as is primary hypothyroidism. The normal starting dose in a young patient without evidence of cardiac disease is 1.5 mcg/kg/day. Lower starting doses are used in the elderly and in patients with evidence of ischemic heart disease. In patients with suspected hypopituitarism, thyroxine therapy should be delayed until ACTH deficiency has been excluded or treated, because the risk for worsening the features of cortisol deficiency is present. Goal of replacement is to be to restore the serum-free T4 concentration to the normal range. Measurement of TSH is unhelpful in the monitoring of T4 replacement therapy in secondary hypothyroidism.
Antidiuretic Hormone Deficiency.: Desmopressin is the drug of choice for the treatment of ADH deficiency. It is available in a number of preparations, including oral, intranasal, parenteral, and the recently available oral form. Dosages vary as much as 10-fold between individuals, with no apparent relation to age, sex, weight, or degree of polyuria. The drug should be started at low dose and increased gradually until urine output is controlled. Overdosage carries a risk for hyponatremia, and sodium levels should be checked after therapy is commenced or changed.
Growth Hormone Deficiency in Adults
Causes
The causes of adult GH deficiency from four series totaling 1798 patients are shown in Table 6-5.69–72 Approximately 50% arise from pituitary tumors, 18% from extrapituitary tumors, and 5% from inflammatory or infiltrative lesions; up to 15% are idiopathic. Treatment for pituitary and extrapituitary tumors is the most common cause of deficiency, accounting for nearly two thirds of cases. The frequency of causes differs between patients with childhood-onset and adult-onset GH deficiency.70 Idiopathic causes, representing the largest group in childhood disease, are likely to represent a heterogeneous collection of congenital developmental abnormalities, including mutations of pit-1 and PROP genes, which cause multiple pituitary hormone deficiencies.
Table 6-5
Causes of Growth Hormone Deficiency in 1798 Patients
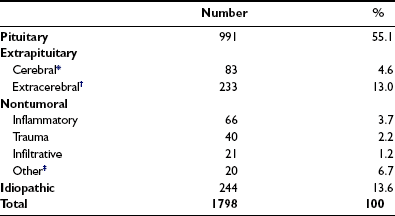
*Includes gliomas, pinealomas, dysgerminomas.
†Includes craniopharyngiomas, meningiomas, epidermoid cysts, Rathke’s cysts.
‡Includes developmental malformations, irradiation other than for pituitary treatment, empty sella.
Data are derived from four studies (References 5-8).
Clinical Features
Adults with GH deficiency, whether dating from childhood or acquired in later adult life, have a range of metabolic, body compositional, and functional abnormalities. These patients have a recognizable clinical syndrome, associated with a characteristic history, symptoms, signs, and investigative findings (Table 6-6).
Table 6-6
Syndrome of Adult Growth Hormone Deficiency
Symptoms
Signs
Investigations
Metabolism
Hypopituitary patients in whom GH has not been replaced display biochemical abnormalities that are linked strongly to the development of vascular disease. These patients have higher concentrations of total and low-density lipoprotein (LDL) cholesterol, as well as apolipoprotein B.73–75 Evidence is available from ultrasonographic studies of intima and media thickening and premature atherosclerosis of large vessels.76,77 These patients also have a higher level of plasminogen inhibitory activity and a higher concentration of fibrinogen,78 both of which are markers of increased atherothrombotic propensity. Fibrinogen also is a risk factor for stroke and myocardial infarction. Circulating levels of proinflammatory factors linked to the development of vascular disease, such as C-reactive protein (CRP), also are increased.79
Body Composition
Marked abnormality of body composition is characterized by increased proportion of body fat and reduced lean mass.80,81 These changes are a consequence of the loss of lipolytic and anabolic actions of GH. The effects of GH on body fat and muscle can be seen in Fig. 6-5, which shows striking changes in body physique in a man before and 5 years after acquisition of GH deficiency after surgery for a pituitary tumor.
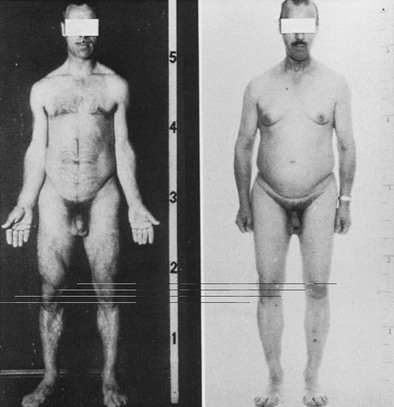
FIGURE 6-5 Body physique in a normal man before and 5 years after acquiring growth hormone deficiency as a result of pituitary surgery for a macroadenoma. Note the striking change in body composition with an accumulation of body fat, particularly in the abdomen, and marked loss of musculature. (Courtesy Professor Peter Sonksen.)
These patients are more obese and display a disproportionate increase in central abdominal fat.81 The tendency toward central fat deposition is important because visceral obesity is linked to the development of insulin resistance, diabetes, and cardiovascular disease.82 Adults with GH deficiency have evidence of insulin resistance83 and a higher prevalence of impaired glucose tolerance.84
The reduction of lean body mass in adult GH deficiency arises from the combined reduction of bone, muscle and visceral mass, and extracellular fluid volume. Bone mass at different skeletal sites is reduced in patients with childhood-onset and adult-onset GH deficiency.85,86 The risk for fracture is increased between twofold and threefold.87,88
Physical Performance
Patients show significant impairment of physical performance and muscle strength.89,90 Physical fitness, as determined by cycle ergometry, has been shown to be reduced consistently in adult GH deficiency, with rates of maximal oxygen uptake reduced on average by about 30%.91 Exercise performance is a complex parameter that is dependent on numerous factors, including cardiorespiratory and neuromuscular muscle function. These patients have impaired cardiac function with reduced ventricular muscle mass, reduced ejection fraction, impaired ventricular filling,92–94 and reduced lung size, all of which contribute to reduced exercise capacity. As the skin is a target tissue of GH action, another likely contributing factor to reduced exercise endurance is impairment of sweating, which arises from hypoplasia of the eccrine sweat glands. The skin of GH-deficient subjects is atrophic and dry.73 Reduced sweating increases susceptibility to hyperthermia during exercise and may limit exercise performance.95
Quality of Life
Metabolic, body compositional, and functional abnormities in adult GH deficiency are accompanied by significant impairment of psychological well-being and reduced quality of life. Fatigue, easy exhaustion, and lack of vitality are common symptoms. Early studies using generic questionnaires revealed lower self-perception of quality of life, with patients regarding themselves as having reduced health, self-control, and vitality, and experiencing increased anxiety.96,97 A Dutch survey of social integration reported that GH-deficient adults had impaired social status.98 These patients were on a lower professional scale, had lower income, and generally were without partners and were living at home with their parents. Recent studies based on disease-specific questionnaires evaluating life satisfaction revealed marked impairment in quality of life, regardless of country and cultural background.99 On average, scores from GH-deficient patients were approximately half those of the normal population.100 A recent study demonstrated significantly increased morbidity in patients with GHD, with hazard ratios approximately three times higher than in the normal population.101
Diagnosis
Although the features of GH deficiency are recognizable, they are not particularly distinct and mimic body compositional and biochemical changes of the aging process.102 GH secretion itself decreases progressively with aging, associated with a progressive increase in adiposity, which itself reduces GH secretion.103,104 Thus, clinical suspicion must be confirmed by accurate biochemical diagnosis to ensure that GH-deficient patients are accurately identified and treated.
Who to Treat
GH deficiency should be defined biochemically within an appropriate clinical context. Biochemical testing for GH deficiency should be considered in patients with a high probability of hypothalamic-pituitary disease who manifest clinical features of the syndrome.105 This includes patients with a history of organic hypothalamic-pituitary dysfunction, cranial irradiation, known childhood-onset GH deficiency, and TBI. Patients with childhood-onset GH deficiency should be retested as adults before they are committed to long-term GH replacement.105
Biochemical Diagnosis
Provocative Test
The diagnosis of adult GH deficiency is established by provocative testing of GH secretion. Patients should be receiving adequate and stable hormone replacement for other hormonal deficits before they undergo testing.105 Provocative tests include the insulin tolerance test (ITT); arginine, glucagon, clonidine, and growth hormone-releasing hormone (GHRH) alone or in combination with arginine or pyridostigmine are available for testing.
In 1997, the Growth Hormone Research Society recommended the ITT as the diagnostic test of choice.106 It is superior to measuring integrated 24 hour GH concentration or IGF-157 (Fig. 6-6). Provided adequate hypoglycemia (<2.2 mmol/L or 40 mg/dL) is achieved, the ITT distinguishes GH deficiency from the reduced GH secretion that accompanies normal aging and obesity. The ITT should be performed in experienced endocrine units under supervision. The test is contraindicated in patients with electrocardiographic evidence or history of ischemic heart disease, and in those with seizure disorder. Given these precautions, the ITT is safe, with a risk for adverse events of less than 1 in 450.107 Normal subjects respond to insulin-induced hypoglycemia with a peak GH concentration greater than 5 mg/L.107,108 Severe GH deficiency is defined by a peak GH response to hypoglycemia of less than 3 mg/L. These cutoff values were defined in GH assays in which polyclonal competitive radioimmunoassays were use.56 However, GH immunoassay results vary between different methods; therefore the cutoff value may require appropriate adjustment.
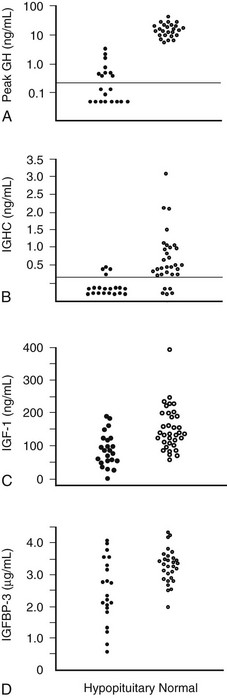
FIGURE 6-6 Comparison of peak growth hormone (GH) concentration obtained during an insulin tolerance test (A), integrated GH concentration (IGHC) obtained from blood withdrawal every 20 minutes for 24 hours (B), insulin-like growth factor (IGF)-1 (C), and IGF-binding protein (IGFBP)-3 concentrations (D) in patients with organic hypopituitarism and age- and sex-matched normal subjects. Dotted line, Limit of reading. (From Hoffman DM, O’Sullivan AJ, Baxter RC, Ho KKY: Diagnosis of growth hormone deficiency in adults. Lancet 343:1064–1068, 1994.)
One stimulation test is sufficient for the diagnosis of adult GH deficiency. A 2007 workshop of the Growth Hormone Society has endorsed the GHRH plus arginine test, the GHRH plus GH-releasing peptide (GHRP) test, and the glucagon stimulation test as validated alternatives to the ITT.105 The ITT evaluates the integrity of the hypothalamic-pituitary axis and has the added advantage of stimulating ACTH. Diagnostic tests employing GHRH and/or GHRP, both of which directly stimulate GH release from the pituitary gland, may miss GH deficiency due to hypothalamic disease.109 This is exemplified by studies in those treated with cranial irradiation, in which the ITT shows the greatest sensitivity and specificity within the first 5 years after irradiation. If the peak GH level during a GHRH plus arginine test is normal in those who have received irradiation, then an ITT should also be performed. In irradiated patients and in those with inflammatory and infiltrative lesions, GH deficiency may develop many years after the initial insult. Therefore, this group should be followed over the longer term with repeat testing as clinically indicated.24
Biochemical Markers of GH Action
These markers include IGF-1, IGFBP-3, and the acid-labile subunit of the IGF-1-BP complex. Of the three biochemical markers, the merit of IGF-1 has been the most intensively studied. Serum IGF-1 concentrations are useful only when age-adjusted normal ranges are used. Although IGF-1 levels are reduced in adult GH deficiency, a normal concentration does not exclude the diagnosis (see Fig. 6-6). A subnormal IGF-1 level in an adult patient with coexisting pituitary hormone deficits is strongly suggestive of GH deficiency, particularly in the absence of conditions known to reduce IGF-1 levels, such as malnutrition, liver disease, poorly controlled diabetes mellitus, and hypothyroidism. The separation of IGF-1 values between GH-deficient and normal subjects is greatest in the young. As IGF-1 levels decline in normal subjects with aging, IGF-1 becomes less reliable as a biochemical marker of GH deficiency in patients older than 50 years, when the values are merged with those of normal subjects.110 Measurement of IGFBP-3 or the acid-labile subunit does not offer any advantage over IGF-1.111
In patients with organic hypothalamic-pituitary disease, the prevalence of GH deficiency is strongly linked to the number of pituitary hormone deficits, ranging from approximately 25% to 40% with no deficit to virtually 95% to 100% when more than three pituitary hormone deficiencies are present112 (Fig. 6-7). Patients with three or more pituitary hormone deficiencies and an IGF-1 level below the reference range have a >97% chance of being GH deficient and therefore do not need a GH stimulation test.105
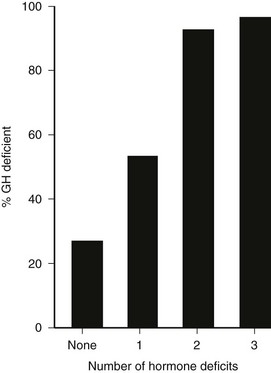
FIGURE 6-7 Relation between the number of anterior pituitary hormone deficits and the prevalence of growth hormone deficiency in 190 patients with known pituitary disease. (From Toogood AA, Beardwell C, Shalet SM: The severity of growth hormone deficiency in adults with pituitary disease is related to the degree of hypopituitarism. Clin Endocrinol 41:511–516, 1994.)
Growth Hormone Replacement
The benefit effects of GH replacement in adults with hypopituitarism were first reported in 1989.81,113 Since then, the impact of GH replacement has been studied extensively, and long-term experience of up to 10 years indicates sustained benefits.114
Metabolism
GH treatment induces profound effects on protein and fat metabolism, which result in significant changes in body composition. The anabolic effects arise from direct stimulation of protein synthesis and reduction of protein oxidation. GH stimulates lipolysis and fat oxidation, enhancing the utilization of fat for energy metabolism. The marked effects of GH on substrate metabolism are accompanied by significant stimulation of resting energy expenditure.81
In addition to exerting effects on the oxidative metabolism of fat, GH reduces a significant shift in lipoprotein metabolism to a less atherogenic profile. Most studies report a decrease in total cholesterol.75 Less consistently reported are effects on increasing high-density lipoprotein (HDL) cholesterol and reducing levels of LDL cholesterol and apolipoprotein.115–117 The favorable effects of improving the lipoprotein profile are more evident after treatment is provided for longer than a year.117,118 Most studies report little effect on triglyceride levels.75 Mechanisms that account for a less atherogenic profile include GH induction of hepatic LDL receptors and reduction in central adiposity, accompanied by an improvement in insulin sensitivity.
GH treatment reduces intima-media thickness of the carotid arteries and improves flow-mediated endothelium-dependent dilatation.15,119 The mean intima-media thickness of the carotid vessels was significantly less after 10 years of GH treatment when compared with that of an untreated GH-deficient group.114 The changes are not correlated to the reductions in plasma lipids. Proinflammatory factors such as CRP and interleukin (IL)-6, strongly implicated in the pathogenesis of vascular disease, decrease significantly with GH treatment.120 It is yet to be established that improvement in these risk markers translates to a reduction in cardiovascular mortality.
Body Composition
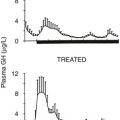
GH replacement induces striking changes in body composition.75,81,113,116 One of the first studies of adult replacement reported a significant reduction in body fat of 18% and a corresponding increase in lean body mass of 10% over a 6-month treatment period (Fig. 6-8). These changes in body composition occurred without a significant change in body weight. The greatest reduction in body fat occurs in abdominal and visceral fat.121 Restoration of body composition is largely completed within the first 12 months of treatment. Significant increases in extracellular water also occur as a consequence of the antinatriuretic properties of GH, which are dose dependent and involve activation of the renin-angiotensin system, as well as a direct renal tubular effect.122 Renal plasma flow and glomerular filtration rate are increased.
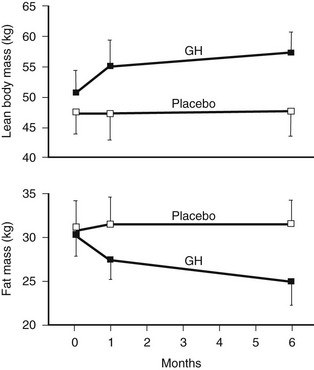
FIGURE 6-8 Body composition in growth hormone (GH)-deficient adult during treatment with GH or placebo for 6 months. A, Body fat. B, Lean body mass. (From Salomon F, Cuneo RC, Hesp R, Sonksen PH: The effects of treatment with recombinant human growth hormone on body composition and metabolism in adults with growth hormone deficiency. N Engl J Med 321:1797–1803, 1989.)
Bone remodeling is activated by GH. Markers of bone formation such as osteocalcin, alkaline phosphatase, and bone Gla protein, along with markers of resorption such as urinary hydroxyproline, are increased by GH treatment.75 Initial studies reporting changes in BMD over 6 to 12 months of treatment yielded conflicting results. However, more recent studies reporting long-term data show a progressive increase in BMD beyond 12 to 18 months of treatment123–125 (Fig. 6-9), with a plateau reached after 3 years.118,126 Markers of bone turnover increase over the first 12 months but return to baseline after 3 to 4 years.127
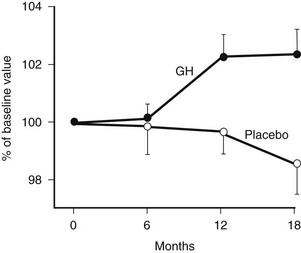
FIGURE 6-9 Bone mineral density of the lumbar spine in growth hormone (GH)-deficient adults during treatment with GH or placebo for 18 months. (From Baum HB, Biller BM, Finkelstein JS, Klibanski A: Effect of physiologic growth hormone therapy on bone density and body composition in patients with adult onset growth hormone deficiency: a randomised, placebo-controlled trial. Ann Intern Med 125:883–890, 1996.)
Physical Performance
Several studies reported that the increase in lean body and muscle mass during GH treatment is accompanied by an improvement in muscle strength. Quadriceps or hip muscle strength improves significantly after 6 months of treatment.90,113 Muscle strength is normalized after 2 years, without further significant change at 5 years.128,129
Many studies reported improvement in exercise capacity and performance in parallel with an increase in maximal oxygen uptake90,130 (Fig. 6-10). Exercise training alone significantly improves the aerobic capacity of GH-deficient adults, and this is not additive to the effects of GH treatment alone.131
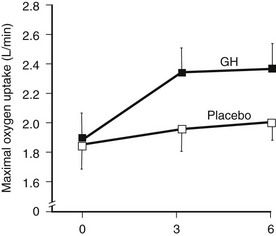
FIGURE 6-10 Maximal exercise capacity in growth hormone (GH)-deficient adults during treatment with GH or placebo for 6 months. Exercise capacity was measured as maximal oxygen uptake during incremental cycle ergometry. (From Cuneo RC, Salomon F, Wiles CM, et al: Growth hormone treatment of growth hormone deficient adults, II. Effects on exercise performance. J Appl Physiol 70:695–700, 1991.)
In patients with GH deficiency, submaximal exercise performance, estimated as anaerobic threshold, increases significantly during GH treatment, suggesting that physical activities of daily living may be accomplished with less metabolic stress and subjective perception of effort.132 Maximal workload and oxygen consumption increased progressively over a 5 year period of GH treatment.125 In addition to increases in muscle strength, many factors may contribute to an improvement in exercise performance. These include enhanced cardiac function and improved heat dissipation through increased sweating. The data supporting a positive effect of GH on cardiac function are strong. Stroke volume, cardiac output, and diastolic function improve during GH treatment.90,133,134
Quality of Life
Several double-blind, placebo-controlled studies have reported improvement in mood, energy, sleep, and vitality scores with GH treatment,96,116,121 with continued improvement in these domains noted during the open phase. In general, GH replacement improved perceived health status and subjective well-being in the domains of health-related quality of life within 6 months. These findings were confirmed in a large randomized placebo-controlled blinded trial based on partner evaluation by questionnaire. According to the partners, patients were more alert, active, and industrious and had greater vitality and endurance during GH treatment.135 Disease-specific tools have reported unequivocal improvement in measures of life satisfaction after GH treatment.100 A large survey of 304 patients showed not only an improvement in quality of life, but also significant reduction in the numbers of sick leave and doctor visits during 12 months of GH therapy136 (Fig. 6-11).
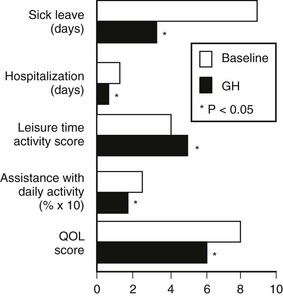
FIGURE 6-11 Changes in health care utilization and quality of life (QOL) measured at baseline and after 12 months of growth hormone replacement. The numbers of days of sick leave and hospitalization in the previous 6 months were taken as the baseline measures. QOL was assessed by a disease-specific questionnaire. (From Hernberg-Stahl E, Luger A, Abs R, et al: Healthcare consumption decreases in parallel with improvements in quality of life during GH replacement in hypopituitary adults with GH deficiency. J Clin Endocrinol Metab 86:5277–5281, 2001.)
Significant differences are noted in clinical and biochemical presentation and responses to GH therapy between patients with childhood-onset and those with adult-onset GH deficiency.70,129 Height, body weight, and lean body mass are lower in those with childhood-onset GH deficiency. The quality of life appears to be less disrupted in childhood-onset disease. During GH treatment, this group displays greater changes in body composition and greater increases in BMD and muscle strength, but lesser improvement in lipid profiles and quality-of-life measures, when compared with their adult-onset counterparts. The interesting differences at baseline and in responses to GH are likely to reflect the biological roles of GH at difference phases of life, as well as the psychological impact of GH injections given to the developing child. A patient with adult-onset disease is likely to recognize the restoration of quality of life to a level experienced before GH deficiency was acquired. In contrast, adults who received GH as a developing child have grown up with and adapted to the condition and are likely to harbor negative recollections of enforced daily injections. As GH therapy is terminated on epiphyseal closure, which occurs before somatic maturation, conventionally GH-treated children may not reach their physical and developmental potential on termination of GH treatment for dwarfism.137 The data suggest the existence of two clinical entities, developmental and metabolic, which reflect the function of GH at different stages of life.70
Transition Age Patients
GH treatment of the GH-deficient child normally is terminated when final height and epiphyseal closure are reached. Strong evidence indicates that biological maturity is attained after the postpubertal period during the early years of adulthood. Muscle mass and strength continue to increase in normal subjects after puberty; this does not occur in GH-deficient subjects.137,138 The bone mineral content of GH-treated subjects doubled that of unreplaced GH-deficient subjects after 2 years in the postpubertal period.139
A significant proportion of patients with childhood-onset GH deficiency exhibit normal GH response when retested at the end of completion of GH treatment. The requirement for continued GH treatment should be evaluated carefully, as proposed by the European Society for Paediatric Endocrinology140 (Fig. 6-12). GH testing is not required for those with a transcription factor mutation (e.g., Pit-1, PROP-1), those with more than three pituitary hormone deficits, and those with isolated GH deficiency associated with an identified mutation (e.g., GH-1, GHRH-R). All other patients should undergo GH testing after at least 1 month off GH treatment. Compelling reasons exist for GH-deficient children to continue GH treatment after puberty, to optimize not only physical maturation but also cardiovascular health and quality of life.
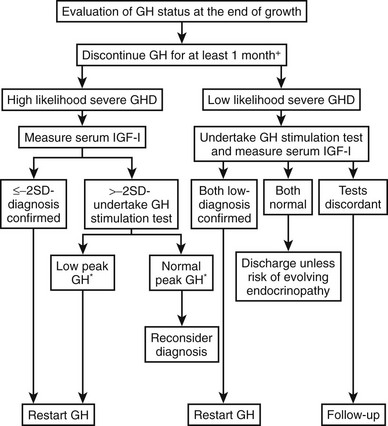
FIGURE 6-12 An approach to the reevaluation of growth hormone (GH) status for consideration of resuming GH treatment in GH deficiency diagnosed in childhood at the end of growth. *Peak GH < 5 µg/L. (Adapted from Clayton PE, Cuneo RC, Juul A, Monson JP, Shalet SM, Tauber M: European Society of Paediatric Endocrinology. Consensus statement on the management of the GH-treated adolescent in the transition to adult care. Eur J Endocrinol. 152:165–170, 2005.)
Treatment
GH secretion is greater in younger individuals than in older ones, and in women than in men. It therefore is recommended that the starting dose of GH in young men and women be 0.2 and 0.3 mg/day, respectively, and in older individuals 0.1 mg/day.105 Dose determination based on body weight is not recommended because of large interindividual variation in absorption and in sensitivity to GH, as well as the lack of evidence that a larger replacement is required for heavier individuals in adults. It is recommended that GH be administered in the evening to mimic the greater secretion of GH at night. Dose escalation should be gradual, individualized, and guided by clinical and biochemical response. Long-acting preparations of human GH are under evaluation for long-term safety and efficacy.
Interaction
GH may influence the metabolism of many substances, including hormones and medications. GH stimulates the activity of the hepatic cytochrome P-450 system, a major pathway of the oxidative metabolism of several drugs, including anticonvulsants. It is likely that dosage adjustments may be necessary in patients commencing GH treatment. Cortisol also is metabolized by the hepatic cytochrome P-450 system. Biochemical evidence indicates that GH increases the metabolic disposition of cortisol and may increase the risk for adrenal insufficiency,141 which has been reported in some studies.142 Although a causal relation remains unproved, GH-deficient patients receiving GH therapy should be strongly advised to increase the dosage of glucocorticoids when unwell, as is generally recommended.
Recently, the importance of 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) in determining tissue exposure to glucocorticoids has been recognized increasingly.66,143 Hepatic 11β-HSD1 converts cortisone to cortisol. Its activity is inhibited by GH and therefore is increased in GHD individuals. Some studies demonstrated supraphysiologic tissue cortisol exposure in hydrocortisone-treated GHD subjects. This may explain the observation in one study of higher waist circumference and glycated hemoglobin (HbA1c) in hydrocortisone-treated GHD patients, compared with GHD patients on cortisone.66
GH stimulates the peripheral conversion of T4 to triiodothyronine (T3). This effect may be seen frequently as a decrease in circulating T4 levels, particularly in patients taking thyroid hormone replacement for hypopituitarism.144 If T3 is not monitored during GH replacement, a decrease in T4 may be misinterpreted as inadequate substitution, and this may lead to an unnecessary increase in the dosage of thyroid hormone replacement.
Sex steroids exert significant modulatory effects on GH action. Estrogen exerts significant effects on hepatic function that are dependent on the route of administration. When compared with estrogen given by the transdermal route, oral estrogen reduces IGF-1 and fat oxidation—effects that are opposite to those of GH.145 GH-deficient women who are also hypogonadal should receive estrogen by a nonoral route during GH replacement, because oral but not transdermal estrogen attenuates the biological effects of GH.146 Fifty percent more GH was required during oral estrogen treatment than during transdermal administration to maintain an equivalent IGF-1 level (Fig. 6-13). In contrast, androgens enhance the metabolic effects of GH. The divergent effects of estrogens and androgens on GH action provide a likely explanation for the observation that women are less responsive than men to GH118,147 (Fig. 6-14). Over a 5-year treatment period, the average weight-adjusted dose for women was 30% higher than for men, whose IGF-1 was one standard deviation higher than that in women118 (Fig. 6-14). Thus, women require a larger dose of GH than is required by men.
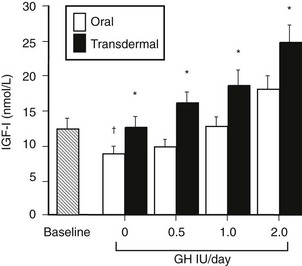
FIGURE 6-13 Mean insulin-like growth factor (IGF)-1 levels before and during incremental doses of growth hormone (GH; 0.5, 1.0, and 2.0 Units/day) during oral and transdermal estrogen therapy in eight GH-deficient adult women. *p < 0.05 oral versus transdermal. †p < 0.05 versus baseline. (From Wolthers T, Hoffman DM, Nugent AG, et al: Oral estrogen therapy impairs the metabolic effects of growth hormone [GH] in GH deficient women. Am J Physiol 281:E1191–E1196, 2001.)
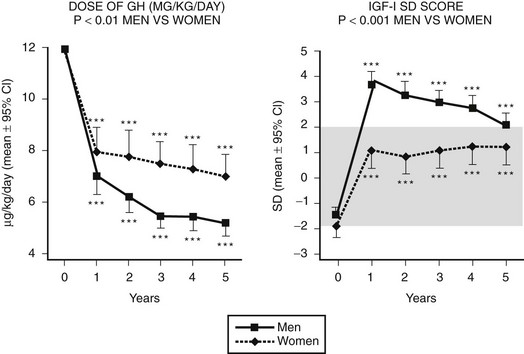
FIGURE 6-14 Growth hormone (GH) dose (µg/kg/day) and insulin-like growth factor (IGF)-1 standard deviation score in a group of 70 men and 48 women with GH deficiency treated for 5 years. The mean GH dose was lower, yet men attained higher IGF-1 responses than did women. (From Gotherstrom G, Svensson J, Koranyi J, et al: A prospective study of 5 years of GH replacement therapy in GH-deficient adults: sustained effects on body composition, bone mass and metabolic indices. J Clin Endocrinol Metab 86:4657–4665, 2001.)
Safety
The experience from several large multicenter clinical trials indicates that GH treatment is safe and well tolerated.116,148,149 The most common side effects arise from the antinatriuretic action of GH, which causes fluid retention (Table 6-7). These manifest as dependent edema, paresthesia, and carpal tunnel syndrome and occur with greater frequency in older patients. However, the symptoms are mild and dose related and resolve in most patients either spontaneously or with dosage reduction.150
Table 6-7
Treatment Guidelines for Growth Hormone (GH) Replacement in Adult GH Deficiency
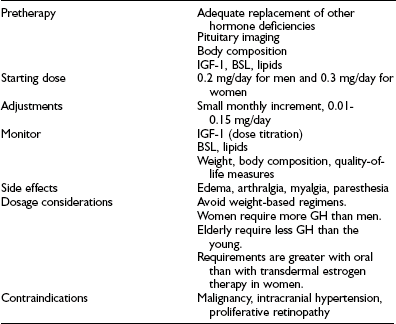
BSL, Blood sugar level; IGF-1, insulin-like growth factor-1.
Although GH antagonizes insulin action, the risk for developing hyperglycemia is very low. Only two case of reversible diabetes were reported from two European multicenter trials with a combined total of 400 patients,148,149 whereas diabetes developed in none of 166 patients in an Australian study.88 Insulin sensitivity did not change after 7 years of GH treatment.151
Because GH promotes the growth of tissues, concern has been expressed that GH therapy may increase the risk for pituitary tumor recurrence or the development of neoplasia. Analysis of the extensive pediatric experience shows no convincing evidence for a causal link between GH treatment and tumor recurrence or the development of neoplasia,152 including leukemia.153 In a retrospective comparison between hypopituitary adults with or without GH replacement and the normal population, an increased rate of malignancy, with colorectal cancer being the most common, was observed in hypopituitary adults without GH replacement. The rate of malignancy in patients on GH replacement was not different from that seen in the normal population.13 In addition, overall cancer incidence rates were lower in acromegalic patients than in the general population.154 These data on acromegaly provide the strongest evidence against a causal association between IGF-1 and malignancy.
References
1. Rosen T, Bengtsson BA: Epidemiology of adult onset hypopituitarism in Goteborg, Sweden during 1956–1987. Presented at the International Symposium on Growth Hormone and Growth Factors, Gothenburg, 1994, pp A3–A60 [Abstract]
2. Regal, M, Paramo, C, Sierra, SM, et al. Prevalence and incidence of hypopituitarism in an adult Caucasian population in northwestern Spain. Clin Endocrinol (Oxf). 2001;55:735–740.
3. Stochholm, K, Gravholt, CH, Laursen, T, et al. Incidence of GH deficiency—a nationwide study. Eur J Endocrinol. 2006;155(July 1):61–71.
4. Rosén, T, Bengtsson, BA. Premature mortality due to cardiovascular disease in hypopituitarism. Lancet. 1990;336:285–288.
5. Bates, AS, Van’t Hoff, W, Jones, PJ, et al. The effect of hypopituitarism on life expectancy. J Clin Endocrinol Metab. 1996;81:1169–1172.
6. Bülow, B, Hagmar, L, Mikoczy, Z, et al. Increased cerebrovascular mortality in patients with hypopituitarism. Clin Endocrinol (Oxf). 1997;46:75–81.
7. Nilsson, B, Gustavasson-Kadaka, E, Bengtsson, BA, et al. Pituitary adenomas in Sweden between 1958 and 1991: incidence, survival, and mortality. J Clin Endocrinol Metab. 2000;85:1420–1425.
8. Tomlinson, JW, Holden, N, Hills, RK, et al. Association between premature mortality and hypopituitarism: West Midlands Prospective Hypopituitary Study Group. Lancet. 2001;357:425–431.
9. Bates, AS, Bullivant, B, Sheppard, MC, et al. Life expectancy following surgery for pituitary tumours. Clin Endocrinol (Oxf). 1999;50:315–319.
10. Nielsen, EH, Lindholm, J, Laurberg, P. Excess mortality in women with pituitary disease: a meta-analysis. Clin Endocrinol (Oxf). 2007 Nov;67(5):693–697.
11. Svensson, J, Bengtsson, BA, Rosén, T, et al. Malignant disease and cardiovascular morbidity in hypopituitary adults with or without growth hormone replacement therapy. J Clin Endocrinol Metab. 2004 Jul;89(7):3306–3312.
12. Nielsen, EH, Lindholm, J, Laurberg, P, et al. Nonfunctioning pituitary adenoma: incidence, causes of death and quality of life in relation to pituitary function. Pituitary. 2007;10(1):67–73.
13. Karavitaki, N, Wass, JA:. Craniopharyngiomas. Endocrinol Metab Clin North Am. 2008 Mar;37(1):173–193.
14. Lindholm, J, Nielsen, EH, Bjerre, P, et al. Hypopituitarism and mortality in pituitary adenoma. Clin Endocrinol (Oxf). 2006 Jul;65(1):51–58.
15. Molitch, ME, Russell, EJ. The pituitary “incidentaloma,”. Ann Intern Med. 1990;112:925–931.
16. Arafah, BM, Prunty, D, Ybarra, J, et al. The dominant role of increased intrasellar pressure in the pathogenesis of hypopituitarism, hyperprolactinemia, and headaches in patients with pituitary adenomas. J Clin Endocrinol Metab. 2000;85:1789–1793.
17. Littley, MD, Shalet, SM, Beardwell, CG, et al. Hypopituitarism following external radiotherapy for pituitary tumours in adults. Q J Med. 1989;70:145–160.
18. Webb, SM, Rigla, M, Wägner, A, et al. Recovery of hypopituitarism after neurosurgical treatment of pituitary adenomas. J Clin Endocrinol Metab. 1999;84:3696–3700.
19. Arafah, BM. Reversible hypopituitarism in patients with large nonfunctioning pituitary adenomas. J Clin Endocrinol Metab. 1986;62:1173–1179.
20. Arafah, BM, Kailani, SH, Nekl, KE, et al. Immediate recovery of pituitary function after transsphenoidal resection of pituitary macroadenomas. J Clin Endocrinol Metab. 1994;79:348–354.
21. Littley, MD, Shalet, SM, Beardwell, CG. Radiation and hypothalamic-pituitary function. Baillieres Clin Endocrinol Metab. 1990;4:147–175.
22. Shalet, SM, Beardwell, CG, Pearson, D, et al. The effect of varying doses of cerebral irradiation on growth hormone production in childhood. Clin Endocrinol (Oxf). 1976;5:287–290.
23. Darzy, KH, Shalet, SM. Radiation-induced growth hormone deficiency. Horm Res. 2003;59(Suppl 1):1–11.
24. Darzy, KH, Aimaretti, G, Wieringa, G, et al. The usefulness of the combined growth hormone (GH)-releasing hormone and arginine stimulation test in the diagnosis of radiation-induced GH deficiency is dependent on the post-irradiation time interval. J Clin Endocrinol Metab. 2003 Jan;88(1):95–102.
25. Minniti, G, Traish, D, Ashley, S, et al. Fractionated stereotactic conformal radiotherapy for secreting and nonsecreting pituitary adenomas. Clin Endocrinol (Oxf). 2006 May;64(5):542–548.
26. Hoybye, C, Grenback, E, Rahn, T, et al. Adrenocorticotropic hormone-producing pituitary tumours: 12- to 22-year follow-up after treatment with stereotactic radiosurgery. Neurosurgery. 2001;49:284–291.
27. Besson, A, Salemi, S, Gallati, S, et al. Reduced longevity in untreated patients with isolated growth hormone deficiency. J Clin Endocrinol Metab. 2003;88:3664–3667.
28. Deladoëy, J, Flück, C, Büyükgebiz, A, et al. “Hot spot” in the PROP1 gene responsible for combined pituitary hormone deficiency. J Clin Endocrinol Metab. 1999;84:1645–1650.
29. Thomas, PQ, Dattani, MT, Brickman, JM, et al. Heterozygous HESX1 mutations associated with isolated congenital pituitary hypoplasia and septo-optic dysplasia. Hum Mol Genet. 2001;10:39–45.
30. Netchine, I, Sobrier, ML, Krude, H, et al. Mutations in LHX3 result in a new syndrome revealed by combined pituitary hormone deficiency. Nat Genet. 2000;25:182–186.
31. Machinis, K, Pantel, J, Netchine, I, et al. Syndromic short stature in patients with a germline mutation in the LIM homeobox LHX4. Am J Hum Genet. 2001;69:961–968.
32. Binder, G. Isolated growth hormone deficiency and the GH-1 gene: update 2002. Horm Res. 2002;58(Suppl 3):2–6.
33. Netchine, I, Talon, P, Dastot, F, et al. Extensive phenotypic analysis of a family with growth hormone (GH) deficiency caused by a mutation in the GH-releasing hormone receptor gene. J Clin Endocrinol Metab. 1998;83:432–436.
34. Salvatori, R, Hayashida, CY, Aguiar-Oliveira, MH, et al. Familial dwarfism due to a novel mutation of the growth hormone-releasing hormone receptor gene. J Clin Endocrinol Metab. 1999 Mar;84(3):917–923.
35. Quinton, R, Duke, VM, Robertson, A, et al. Idiopathic gonadotrophin deficiency: genetic questions addressed through phenotypic characterization. Clin Endocrinol (Oxf). 2001;55:163–174.
36. Falardeau, J, Chung, WC, Beenken, A, et al. Decreased FGF8 signaling causes deficiency of gonadotropin-releasing hormone in humans and mice. J Clin Invest. August 1 2008;118(8):2822–2831.
37. Doeker, BM, Pfäffle, RW, Pohlenz, J, et al. Congenital central hypothyroidism due to a homozygous mutation in the thyrotropin beta-subunit gene follows an autosomal recessive inheritance. J Clin Endocrinol Metab. 1998;83:1762–1765.
38. Collu, R, Tang, J, Castagné, J, et al. A novel mechanism for isolated central hypothyroidism: inactivating mutations in the thyrotropin-releasing hormone receptor gene. J Clin Endocrinol Metab. 1997;82:1561–1565.
39. Asteria, C. T-box and isolated ACTH deficiency. Eur J Endocrinol. 2002;146:463–465.
40. Tateno, T, Izumiyama, H, Doi, M, et al. Differential gene expression in ACTH-secreting and non-functioning pituitary tumors. Eur J Endocrinol. 2007;157:717–724.
41. Vallette-Kasic, S, Pulichino, AM, Gueydan, M, et al. A neonatal form of isolated ACTH deficiency frequently associated with Tpit gene mutations. Endocr Res. 2004 Nov;30(4):943–944.
42. Schneider, HJ, Kreitschmann-Andermahr, I, Ghigo, E, et al. Hypothalamopituitary dysfunction following traumatic brain injury and aneurysmal subarachnoid hemorrhage: a systematic review. JAMA. 2007 Sep 26;298(12):1429–1438.
43. Tagliaferri, F, Compagnone, C, Korsic, M, et al. A systematic review of brain injury epidemiology in Europe. Acta Neurochir (Wien). 2006;148(3):255–268.
44. Schneider, HJ, Aimaretti, G, Kreitschmann-Andermahr, I, et al. Hypopituitarism. Lancet. 2007;369(9571):1461–1470.
45. Klose, M, Juul, A, Struck, J, et al. Acute and long-term pituitary insufficiency in traumatic brain injury: a prospective single-centre study. Clin Endocrinol (Oxf). 2007 Oct;67(4):598–606.
46. Lupi, I, Broman, KW, Tzou, SC, et al. Novel autoantigens in autoimmune hypophysitis. Clin Endocrinol (Oxf). 2008;69(2):269–278.
47. Cosman, F, Post, KD, Holub, DA, et al. Lymphocytic hypophysitis. Report of 3 new cases and review of the literature. Medicine. 1989;68:240–256.
48. O’Dwyer, DT, Smith, AI, Matthew, ML, et al. Identification of the 49-kDa autoantigen associated with lymphocytic hypophysitis as alpha-enolase. J Clin Endocrinol Metab. 2002;87:752.
49. Simpson, ER, Waterman, MR:. Regulation of the synthesis of steroidogenic enzymes in adrenal cortical cells by ACTH. Annu Rev Physiol. 1988;50:427–440.
50. Cronin, CC, Callaghan, N, Kearney, PJ, et al. Addison disease in patients treated with glucocorticoid therapy. Arch Intern Med. 1997;157:456–458.
51. Fujisawa I, Kikuchi K, Nishimura K, et al: Transection of the pituitary stalk: development of an ectopic posterior lobe assessed with MR imaging, Radiology 165:487–489, 1987
52. Kikuchi, K, Fujisawa, I, Momoi, T, et al. Hypothalamic-pituitary function in growth hormone-deficient patients with pituitary stalk transection. J Clin Endocrinol Metab. 1988;67:817–823.
53. Osorio, MG, Marui, S, Jorge, AA, et al. Pituitary magnetic resonance imaging and function in patients with growth hormone deficiency with and without mutations in GHRH-R, GH-1, or PROP-1 genes. J Clin Endocrinol Metab. 2002 Nov;87(11):5076–5084.
54. Maghnie, M, Salati, B, Bianchi, S, et al. Relationship between the morphological evaluation of the pituitary and the growth hormone (GH) response to GH-releasing hormone plus arginine in children and adults with congenital hypopituitarism. J Clin Endocrinol Metab. 2001 Apr;86(4):1574–1579.
55. Le Roux, CW, Meeran, K, Alaghband-Zadeh, J. Is a 0900-h serum cortisol useful prior to a short Synacthen test in outpatient assessment? Ann Clin Biochem. 2002;39:148–150.
56. Hoffman, DM, O’Sullivan, AJ, Baxter, RC, et al. Diagnosis of growth-hormone deficiency in adults. Lancet. April 30 1994;343(8905):1064–1068.
57. Trainer, PJ, Besser, M. The Bart’s Endocrine Protocols. Livingstone: Churchchill; 1995.
58. Nieman, LK. Dynamic evaluation of adrenal hypofunction. J Endocrinol Invest. 2003;26:74–82.
59. Beck-Peccoz, P, Amr, S, Menezes-Ferreira, MM, et al. Decreased receptor binding of biologically inactive thyrotropin in central hypothyroidism: Effect of treatment with thyrotropin-releasing hormone. N Engl J Med. 1985;312:1085–1090.
60. Karavitaki, N, Wass, JA. Craniopharyngiomas. Endocrinol Metab Clin North Am. 2008 Mar;37(1):173–193.
61. Lin, LL, El Naqa, I, Leonard, JR, et al. Long-term outcome in children treated for craniopharyngioma with and without radiotherapy. J Neurosurg Pediatrics. 2008 Feb;1(2):126–130.
62. Mah, PM, Webster, J, Jönsson, P, et al. Estrogen replacement in women of fertile years with hypopituitarism. J Clin Endocrinol Metab. 2005 Nov;90(11):5964–5969.
63. Schubert, M, Minnemann, T, Hübler, D, et al. Intramuscular testosterone undecanoate: pharmacokinetic aspects of a novel testosterone formulation during long-term treatment of men with hypogonadism. J Clin Endocrinol Metab. 2004 Nov;89(11):5429–5434.
64. Arlt, W, Callies, F, van Vlijmen, JC, et al. Dehydroepiandrosterone replacement in women with adrenal insufficiency. N Engl J Med. 1999 Sep 30;341(14):1013–1020.
65. Miller, KK, Biller, BM, Beauregard, C, et al. Effects of testosterone replacement in androgen-deficient women with hypopituitarism: a randomized, double-blind, placebo-controlled study. Clin Endocrinol Metab. 2006 May;91(5):1683–1690.
66. Filipsson, H, Monson, JP, Koltowska-Haggstrom, M, et al. The impact of glucocorticoid replacement regimens on metabolic outcome and comorbidity in hypopituitary patients. J Clin Endocrinol Metab. 2006;91:3954–3961.
67. Lukert, BP. Editorial: glucocorticoid replacement—how much is enough? J Clin Endocrinol Metab. 2006;91:793–794.
68. Filipsson, H, Monson, JP, Koltowska-Häggström, M, et al. The impact of glucocorticoid replacement regimens on metabolic outcome and comorbidity in hypopituitary patients. J Clin Endocrinol Metab. 2006 Oct;91(10):3954–3961.
69. Rosen, T, Bengtsson, B-A. Premature mortality due to cardiovascular disease in hypopituitarism. Lancet. 1990;336:285–288.
70. Attanasio, AF, Lamberts, SWJ, Matranga, AMC. Adult growth hormone deficient patients demonstrate heterogeneity between childhood-onset and adult-onset before and during human GH treatment. J Clin Endocrinol Metab. 1997;82:82–88.
71. Abs, R, Verhelst, J, Maiter, D, et al. Cabergoline in the treatment of acromegaly: a study in 64 patients. J Clin Endocrinol Metab. 1998 Feb;83(2):374–378.
72. Christ, ER, Carroll, PV, Sonksen, PH. The etiology of growth hormone deficiency in human adult. In: Bengtsson B-A, ed. Growth Hormone. Boston: Kluwer Academic Publishers; 1999:97–108.
73. Besson, A, Salemi, S, Gallati, S, et al. Reduced longevity in untreated patients with isolated growth hormone deficiency. J Clin Endocrinol Metab. 2003;88:3664–3667.
74. De Boer, H, Blok, GJ, Voerman, HJ, et al. Serum lipid levels in growth hormone deficient men. Metabolism. 1994;43:199–203.
75. Carroll, PV, Christ, ER, Bengtsson, BA, et al. Growth hormone deficiency in adulthood and the effects of growth hormone replacement: a review. J Clin Endocrinol Metab. 1998;83:382–395.
76. Markussis, V, Beyshah, SA, Fisher, C, et al. Detection of premature atherosclerosis by high resolution ultrasonography in symptom-free hypopituitary adults. Lancet. 1992;340:1188–1192.
77. Pfeiffer, M, Verhovec, R, Zizek, B, et al. Growth hormone (GH) treatment reverses early atherosclerotic changes in GH-deficient adults. J Clin Endocrinol Metab. 1991;84:453–457.
78. Johansson, JO, Landin, K, Tengborn, L, et al. High fibrinogen and plasminogen activator inhibitory activity in growth hormone-deficiency adults. Arterioscler Thromb. 1994;14:434–437.
79. Sesmilo, G, Miller, KK, Hayden, D, et al. Inflammatory cardiovascular risk markers in women with hypopituitarism. J Clin Endocrinol Metab. 2001;86:5774–5781.
80. Hoffman, DM, O’Sullivan, AJ, Freund, J, et al. Adults with growth hormone deficiency have abnormal body composition but normal energy metabolism. J Clin Endocrinol Metab. 1995;80:72–77.
81. Salomon, F, Cuneo, RC, Hesp, R, et al. The effects of treatment with recombinant human growth hormone on body composition and metabolism in adults with growth hormone deficiency. N Engl J Med. 1989;321:1797–1803.
82. Reaven, G. Banting Lecture 1988: role of insulin resistance in human disease. Diabetes. 1988;37:1595–1607.
83. Hew, FL, Koschmann, M, Christopher, M, et al. Insulin resistance in growth hormone deficient adults: defects in glucose utilisation and glycogen synthetase activity. J Clin Endocrinol Metab. 1996;81:555–564.
84. Beshyah, SA, Henderseon, A, Nithayanathan, R, et al. Metabolic abnormalities in growth hormone deficient adults: carbohydrate tolerance and lipid metabolism. Endocrinol Metab. 1994;1:173–180.
85. Kaufman, J, Taelman, P, Vermelen, A, et al. Bone mineral status in growth hormone deficient males with isolated and multiple pituitary deficiencies. J Clin Endocrinol Metab. 1932;74:118–123.
86. Holmes, SJ, Economou, G, Whitehouse, RW, et al. Reduced bone mineral densities in patients with adult onset growth hormone deficiency. J Clin Endocrinol Metab. 1994;78:669–674.
87. Wuster, C, Abs, R, Bengtsson, BA, et al. The influence of growth hormone deficiency, growth hormone replacement therapy, and other aspects of hypopituitarism on fracture rate and bone mineral density. J Bone Miner Res. 2001;16:398–405.
88. Rosen, T, Wilhelmsen, L, Landin-Wilhelmsen, K, et al. Increased fracture frequency in adults with hypopituitarism and growth hormone deficiency. Eur J Endocrinol. 1998;137:240–245.
89. Rutherford, OM, Beshyah, SA, Johnston, DG. Quadriceps strength before and after growth hormone replacement in growth hormone deficient adults. Endocrinol Metab. 1994;1:44–47.
90. Cuneo, RC, Salomon, F, Wiles, CM, et al. Growth hormone treatment of growth hormone deficient adults, I: effects on muscle mass and strength. J Appl Physiol. 1991;70:688–694.
91. Cuneo, RC, Salomon, F, Wiles, CM, et al. Growth hormone treatment of growth hormone deficient adults, II: effects on exercise performance. J Appl Physiol. 1991;70:695–700.
92. Amato, G, Carella, C, Fazio, S, et al. Body composition, bone metabolism, and heart structure and function in growth hormone (GH) deficient adults before and after GH replacement therapy. J Clin Endocrinol Metab. 1993;77:1671–1676.
93. Merola, B, Cittadini, A, Coloa, A, et al. Cardiac structural and functional abnormalities in adult patients with growth hormone deficiency. J Clin Endocrinol Metab. 1993;77:1658–1661.
94. Shahi, M, Beshyah, SA, Hackett, D, et al. Myocardial dysfunction in treated adult hypopituitarism: a possible explanation for increased cardiovascular mortality. Br Heart J. 1992;67:92–96.
95. Juul, A, Behrenscheer, A, Tims, T, et al. Impaired thermoregulation in adults with growth hormone deficiency during heat exposure and exercise. Clin Endocrinol. 1993;38:237–244.
96. McGauley, GA, Cuneo, RC, Salomon, FC, et al. Psychological well-being before and after growth hormone treatment in adults with growth hormone deficiency. Horm Res. 1990;33(Suppl):52–54.
97. Rosen, T, Wiren, L, Wilhemsen, L, et al. Decreased psychological well-being in adult patients with growth hormone deficiency. Clin Endocrinol. 1994;40:111–116.
98. Rikken, B, Van Busschbach, J, Le Cessie, S, et al. Impaired social status of growth hormone deficient adults as compared to controls with short or normal stature. Clin Endocrinol. 1995;43:205–211.
99. Blum, WF, Shavrikova, EP, Edwards, DJ, et al. Decreased quality of life in adult patients with growth hormone deficiency compared with general populations using the new, validated, self-weighted questionnaire, questions on life satisfaction hypopituitarism module. J Clin Endocrinol Metab. 2003;88:4158–4167.
100. Rosilio, M, Blum, WF, Edwards, DJ, et al. Long-term improvement of quality of life during growth hormone (GH) replacement therapy in adults with GH deficiency, as measured by questions on life satisfaction-hypopituitarism (QLS-H). J Clin Endocrinol Metab. 2004;89:1684–1693.
101. Stochholm, K, Laursen, T, Green, A, et al. Morbidity and GH deficiency: a nationwide study. Eur J Endocrinol. 2008 Apr;158(4):447–457.
102. Rudman, D. Growth hormone, body composition and aging. J Am Geriatr Soc. 1985;33:800–807.
103. Ho, KY, Evans, WS, Blizzard, RM, et al. Effects of sex and age on the 24 hour secretory profile of GH secretion in man: importance of endogenous estradiol concentrations. J Clin Endocrinol Metab. 1987;64:51–58.
104. Iranmanesh, A, Lizarralde, G, Veldhuis, JD. Age and relative adiposity are specific negative determinants of the frequency and amplitude of growth hormone (GH) secretory bursts and the half-life of endogenous GH in healthy men. J Clin Endocrinol Metab. 1991;73:1081–1088.
105. Ho, KK. GH Deficiency Consensus Workshop participants. Consensus guidelines for the diagnosis and treatment of adults with GH deficiency II: a statement of the GH Research Society in association with the European Society for Pediatric Endocrinology, Lawson Wilkins Society, European Society of Endocrinology, Japan Endocrine Society, and Endocrine Society of Australia. Eur J Endocrinol. 2007 Dec;157(6):695–700.
106. Growth Hormone Research Society. Consensus guidelines for the diagnosis and treatment of adults with growth hormone deficiency: summary statement of the Growth Hormone Research Society Workshop on Adult Growth Hormone Deficiency. J Clin Endocrinol Metab. 1998;83:379–381.
107. Hoffman, DM, Ho, KKY. Diagnosis of GH deficiency in adults. In: Juul A, Jorgensen JOL, eds. Growth hormone in adults. Cambridge: Cambridge University Press; 1996:168–185.
108. Ho, KKY. Diagnosis of GH deficiency in adults [Editorial]. Lancet. 2000;356:1125–1126.
109. Leal-Cerro, A, Garcia, E, Astorga, R, et al. Growth hormone (GH) responses to the combined administration of GH-releasing hormone plus GH-releasing peptide 6 in adults with GH deficiency. Eur J Endocrinol. 1995 Jun;132(6):712–715.
110. Ghigo, E, Aimaretti, G, Gianotti, L, et al. New approach to the diagnosis of growth hormone deficiency in adults. Eur J Endocrinol. 1996;134:352–356.
111. Arosio, M, Garrone, S, Bruzzi, P, et al. Diagnostic value of the acid-labile subunit in acromegaly: evaluation in comparison with insulin-like growth factor (IGF) I, and IGF binding protein-1, 2, 3. J Clin Endocrinol Metab. 2001;86:1091–1098.
112. Toogood, AA, Beardwell, C, Shalet, SM. The severity of growth hormone deficiency in adults with pituitary disease is related to the degree of hypopituitarism. Clin Endocrinol. 1994;41:511–516.
113. Jorgensen, JOL, Theusen, L, Ingemann-Hansen, T, et al. Beneficial effects of growth hormone treatment in GH-deficient adults. Lancet. 1989;1:1221–1225.
114. Gibney, J, Wallace, JD, Spinks, T, et al. The effect of 10 years of recombinant human growth hormone (GH) in adult GH-deficient patients. J Clin Endocrinol Metab. 1999;184:2596–2602.
115. Weaver, JU, Monson, JP, Noonan, K, et al. The effect of low dose recombinant human growth hormone replacement on regional fat distribution, insulin sensitivity and cardiovascular risk factors in hypopituitary patients. J Clin Endocrinol Metab. 1995;80:153–159.
116. Cuneo, RC, Judd, S, Wallace, JD, et al. The Australian multicentre trial of growth hormone treatment in GH-deficient adults. J Clin Endocrinol Metab. 1998;83:107–116.
117. Beshyah, SA, Henderson, A, Niththyananthan, R, et al. The effects of short and long term growth hormone replacement in hypopituitary adults on lipid metabolism and carbohydrate tolerance. J Clin Endocrinol Metab. 1995;80:356–363.
118. Gotherstrom, G, Svensson, J, Koranyi, J, et al. A prospective study of 5 years of GH replacement therapy in GH-deficient adults: sustained effects on body composition, bone mass and metabolic indices. J Clin Endocrinol Metab. 2001;86:4657–4665.
119. Borson-Chazot, F, Serusclat, A, Kalfallah, Y, et al. Decrease in carotid intima-media thickness after one year growth hormone (GH) treatment in adults with GH deficiency. J Clin Endocrinol Metab. 1999;84:1329–1333.
120. Sesmilo, G, Biller, BM, Llevadot, J, et al. Effects of growth hormone administration on inflammatory and other cardiovascular risk markers in men with growth hormone deficiency. A randomized, controlled clinical trial. Ann Intern Med. 2000;133:111–122.
121. Bengtsson, B-A, Eden, S, Lonn, L, et al. Treatment of adults with growth hormone (GH) deficiency with recombinant human GH. J Clin Endocrinol Metab. 1993;76:309–317.
122. Hoffman, DM, Crampton, L, Sernia, C, et al. Short term growth hormone (GH) treatment of GH deficient adults increases body sodium and extracellular water but not blood pressure. J Clin Endocrinol Metab. 1996;81:1123–1128.
123. Johannsson, G, Rosen, T, Bosaues, I, et al. Two years of growth hormone treatment increases bone mineral content and density in hypopituitary patients with adult-onset growth hormone deficiency. J Clin Endocrinol Metab. 1996;81:2865–2873.
124. Baum, HB, Biller, BM, Finkelstein, JS, et al. Effect of physiologic growth hormone therapy on bone density and body composition in patients with adult onset growth hormone deficiency: a randomised, placebo-controlled trial. Ann Intern Med. 1996;125:883–890.
125. Vandeweghe, M, Taelman, P, Kaufman, J-M. Short and long term effects of growth hormone treatment on bone turnover and bone mineral content in adult growth hormone-deficient males. Clin Endocrinol. 1993;39:409–415.
126. Ter Maaten, JC, Be Boer, H, Kamp, O, et al. Long-term effects of growth hormone (GH) replacement in men with childhood-onset GH deficiency. J Clin Endocrinol Metab. 1999;84:2373–2380.
127. Valimaki, MJ, Salmela, PI, Salmi, J, et al. Effects of 42 months of GH treatment on bone mineral density and bone turnover in GH-deficient adults. Eur J Endocrinol. 1999;140:545–554.
128. Johannsson, G, Grimby, G, Sunnerhagen, KS, et al. Two years of growth hormone (GH) treatment increase isometric and isokinetic muscle strength in GH-deficient adults. J Clin Endocrinol Metab. 1997;82:2877–2884.
129. Koranyi, J, Svensson, J, Gotherstrom, G, et al. Baseline characteristics and the effects of five years of GH replacement therapy in adults with GH deficiency of childhood or adulthood onset: a comparative, prospective study. J Clin Endocrinol Metab. 2001;86:4693–4699.
130. Nass, R, Huber, RM, Klauss, V, et al. Effect of growth hormone (hGH) replacement therapy on physical work capacity and cardiac and pulmonary function in patients with hGH deficiency acquired in adulthood. J Clin Endocrinol Metab. 1995;80:552–557.
131. Thomas, SG, Esposito, JG, Ezzat, S. Exercise training benefits growth hormone (GH)-deficient adults in the absence or presence of GH treatment. J Clin Endocrinol Metab. 2003;88:5734–5738.
132. Woodhouse, LJ, Asa, SL, Thomas, SG, et al. Measures of submaximal aerobic performance evaluate and predict functional response to growth hormone (GH) treatment in GH-deficient adults. J Clin Endocrinol Metab. 1999;84:4570–4577.
133. Caidahl, K, Eden, S, Bengtsson, BA. Cardiovascular and renal effects of growth hormone. Clin Endocrinol. 1994;40:393–400.
134. Valcavi, R, Gaddi, O, Zini, M, et al. Cardiac performance and mass in adults with hypopituitarism: effect of one year of growth hormone treatment. J Clin Endocrinol Metab. 1995;80:659–666.
135. Burman, P, Broman, JE, Hetta, J, et al. Quality of life in adults with growth hormone (GH) deficiency: response to treatment with recombinant GH in a placebo-controlled 21 month trial. J Clin Endocrinol Metab. 1995;80:3585–3590.
136. Hernberg-Stahl, E, Luger, A, Abs, R, et al. Healthcare consumption decreases in parallel with improvements in quality of life during GH replacement in hypopituitary adults with GH deficiency. J Clin Endocrinol Metab. 2001;86:5277–5281.
137. Rutherford, OM, Jones, DA, Round, JM, et al. Changes in skeletal muscle and body composition after discontinuation of growth hormone treatment in growth hormone deficient young adults. Clin Endocrinol. 1991;34:469–475.
138. Hulthen, L, Bengtsson, BA, Sunnerhagen, KS, et al. GH is needed for the maturation of muscle mass and strength in adolescents. J Clin Endocrinol Metab. 2001;86:4765–4770.
139. Shalet, SM, Shavrikova, E, Cromer, M, et al. Effect of growth hormone (GH) treatment on bone in postpubertal GH-deficient patients: a 2-year randomized, controlled, dose-ranging study. J Clin Endocrinol Metab. 2003;88:4124–4129.
140. Clayton, PE, Cuneo, RC, Juul, A, et al. European Society of Paediatric Endocrinology. Consensus statement on the management of the GH-treated adolescent in the transition to adult care. Eur J Endocrinol. 2005 Feb;152(2):165–170.
141. Orme, SM, McNally, RI, Cartwright, RA, et al. Mortality and cancer incidence in acromegaly: a retrospective cohort study. J Clin Endocrinol Metab. 1998;83:2730–2734.
142. Chan, JM, Stampfer, MJ, Giovannucci, E, et al. Plasma insulin-like growth factor-I and prostate cancer risk: a prospective study. Science. January 23 1998;279(5350):563–566.
143. Sigurjónsdóttir, HA, Koranyi, J, Axelson, M, et al. GH effect on enzyme activity of 11betaHSD in abdominal obesity is dependent on treatment duration. Eur J Endocrinol. 2006 Jan;154(1):69–74.
144. Jorgensen, JOL, Pedersen, SA, Lauberg, P, et al. Effects of growth hormone therapy on thyroid function of growth hormone-deficient adults with and without concomitant thyroxine-substituted central hypothyroidism. J Clin Endocrinol Metab. 1989;69:1127–1132.
145. O’Sullivan, AJ, Crampton, L, Freund, J, et al. Route of estrogen replacement confers divergent effects on energy metabolism and body composition in postmenopausal women. J Clin Invest. 1998;102:1035–1040.
146. Wolthers, T, Hoffman, DM, Nugent, AG, et al. Oral estrogen therapy impairs the metabolic effects of growth hormone (GH) in GH deficient women. Am J Physiol. 2001;281:E1191–E1196.
147. Burman, P, Johansson, AG, Siegbahn, A, et al. Growth hormone (GH)-deficient men are more responsive to GH replacement therapy than women. J Clin Endocrinol Metab. 1997;82:550–555.
148. Mardh, G, Lundin, K, Borg, G, et al. Growth hormone replacement therapy in adult hypopituitary patients with growth hormone deficiency: combined data from 12 European placebo-controlled trials. Endocrinol Metab. 1994;1(Suppl A):43–49.
149. Chipman, JJ, Attansio, AF, Birkett, MA, et al. The safety profile of growth hormone replacement therapy in adult based on measurement of serum markers. J Clin Endocrinol Metab. 1996;80:2069–2076.
150. De Boer, H, Blok, GJ, Popp-Snijders, C, et al. Monitoring of growth hormone replacement therapy in adult based on measurement of serum markers. J Clin Endocrinol Metab. 1996;80:2069–2076.
151. Svensson, J, Fowelin, J, Landin, K, et al. Effects of seven years of GH-replacement therapy on insulin sensitivity in GH-deficient adults. J Clin Endocrinol Metab. 2002;87:2121–2127.
152. Allen, D. National Cooperative Growth Study Safety Symposium: safety of human growth hormone therapy. J Pediatr. 1996;128:S8–S13.
153. Nishi, Y, Tanaka, T, Takano, K, et al. Recent status in the occurrence of leukemia in growth hormone-treated patients in Japan. J Clin Endocrinol Metab. 1999;84:1961–1965.
154. Orme, SM, McNally, RI, Cartwright, RA, et al. Mortality and cancer incidence in acromegaly: a retrospective cohort study. J Clin Endocrinol Metab. 1998;83:2730–2734.