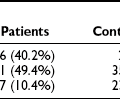Hypoglycemia and Hypoglycemic Syndromes
Neurohumoral Regulation During Hypoglycemia
Syndromes of Disordered Counterregulatory Responses During Hypoglycemia
Hypoglycemia-Associated Neuroendocrine and Autonomic Failure
Strategies to Improve Counterregulatory Responses During Hypoglycemia
Treatment and Strategies to Reduce Hypoglycemia
Mitochondrial Fatty Acid Oxidation Disorders
Renal and Hepatic Disease: General Considerations
Cortisol and Growth Hormone Deficiency
Hypoglycemia
Hypoglycemia, a condition of low plasma glucose, has many varied causes, which are discussed below. The definition of hypoglycemia is often debated, but in this chapter, the glucose values adopted by the hypoglycemia working panel of the American Diabetes Association1 will be used. Hypoglycemia will be defined as any plasma glucose value below 70 mg/dL (3.9 mmol/L). Severe hypoglycemia is reserved for occasions when the plasma glucose is very low (usually below 50 mg/dL, or 2.9 mmol/L) and is accompanied by significant neurologic deficits.
Hypoglycemia is associated with significant morbidity and can be fatal.2,3,9 Hypoglycemia occurring in a nondiabetic individual always warrants attention and should be investigated (Table 21-1). Before one embarks on a comprehensive workup for possible hypoglycemia, certain pitfalls need to be considered. Whole blood glucose values are 10% to 15% lower than plasma glucose levels. Thus, the lower limit of normal for a whole blood glucose value would be about 60 mg/dL (3.3 mmol/L). Keeping this in mind, it is important to determine whether the glucose value is blood or plasma. This will also apply to glucose meters that can provide blood or plasma glucose values. Another important consideration is that mixed venous blood samples can be dramatically lower than arterial glucose values. This will vary depending upon the insulin sensitivity of the individual and the prevailing insulinemia. Thus, mixed venous glucose levels could be 25 to 30 mg/dL (≈1.5 mmol/L) lower than arterial levels in lean healthy individuals during conditions of physiologic hyperinsulinemia. Therefore, hypoglycemic values obtained from mixed venous blood during a 2- to 3-hour oral glucose tolerance test should be interpreted with caution. Similarly, mixed venous glucose measurements obtained during periods of non–steady state (e.g., after a meal, after exercise) can significantly underestimate arterial glucose values.4 In fact, very low glucose levels of between 30 and 50 mg/dL (1.7 to 2.8 mmol/L) have been measured in healthy adults after prolonged exercise.5
Table 21-1
Artifactual hypoglycemia can also occur if a blood glucose sample is not collected in a tube containing fluoride and/or oxalate to inhibit glycolysis. Without appropriate sample collection, glucose values can decrease by 10 to 20 mg/dL (≈0.5 to 1.0 mmol/L) per hour at room temperature.6 In addition, even if glucose samples are collected in tubes containing glycolytic inhibitors, artifactually low readings can be obtained if the sample contains large quantities of blood cells,7 if the sample remains unmeasured for many hours, or if the sample is heavily lipemic with triglycerides.8
Physiology of Hypoglycemia
During typical physiologic conditions, the brain requires a constant and adequate supply of glucose. Under normal postabsorptive conditions (e.g., an overnight fast), the brain accounts for ≈65% of whole body glucose uptake. Following feeding, the amount of glucose taken up by the brain can increase, but insulin does not influence brain glucose kinetics in a similar fashion to other organs such as liver or muscle. Although the brain was classically considered an insulin-insensitive organ, recent work has challenged this concept.10 Several studies have elegantly demonstrated that insulin can regulate appetite and feeding mechanisms in rodent models.10 Additionally, insulin administration into areas of the hypothalamus has been demonstrated to regulate hepatic glucose output.11 Furthermore, studies in dogs12 and mice13 have demonstrated direct CNS effects of insulin to amplify autonomic nervous system (ANS) counterregulatory responses to hypoglycemia. Thus, accumulating data indicate that insulin can have direct metabolic effects on certain areas of the brain.
Although typically dependent upon glucose as a fuel, the brain can adapt and utilize other substrates. Thus during periods of fasting, ketone bodies, lactate, and alanine can be used as alternative brain fuels.14 Several studies have demonstrated, experimentally, that high levels of alternative substrates can be infused during acute hypoglycemia with a concomitant reduction in neuroendocrine and ANS responses. This indicates that the brain has the capacity to switch from glucose to alternative substrates in a matter of hours. However, it should be noted that the concentrations of substrate infused experimentally are far higher than levels observed during most physiologic conditions (the exception is levels of ketone bodies that occur during prolonged fasting).
During hypoglycemia, brain glucose uptake falls. The exact glycemic value for the start of decreased blood-to-brain glucose transport is debated but is thought to be around 3.6 to 3.8 mmol/L in humans. As hypoglycemia deepens (−3 mmol/L), blood-to-brain glucose transport becomes rate limiting for brain glucose metabolism. Glycolytic derived lactate and a small amount of stored astrocytic glycogen can provide a short duration of fuel supply. Recent work has estimated that stored glycogen could provide the brain oxidative fuel for about 20 minutes15; based on an estimate that blood-to-brain glucose transport could provide up to 90% of the brain’s oxidative requirements during moderate hypoglycemia,16 it can be seen that the remaining 10% of energy requirements obtained from lactate and glycogen would last only ≈200 minutes, thus emphasizing the need for a continuous and adequate supply of glucose to the brain from the circulation.
Physiologic Responses to Hypoglycemia
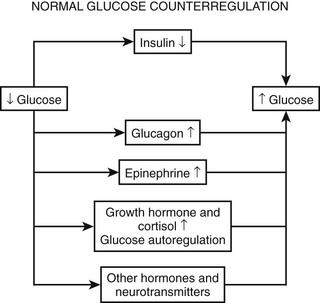
As plasma glucose falls, a well-orchestrated response of multiple physiologic mechanisms is activated. The initial defense is a reduction in endogenous insulin secretion. This occurs as plasma glucose levels fall to below 80 mg/dL (4.4 mmol/L). The reduction in endogenous insulin in response to falling plasma glucose is often overlooked in the hierarchy of defenses against hypoglycemia. Data from the Diabetes Control and Complications Trial (DCCT) demonstrate that the presence of even small amounts of C-peptide (i.e., endogenous insulin) are protective against severe hypoglycemia.17 Similarly, experience from recent islet cell transplantations clearly demonstrates that the ability to modulate endogenous insulin levels is also protective against episodes of hypoglycemia.18 As glucose levels continue to fall at or around 70 mg/dL (3.9 mmol/L), a coordinated release of anti-insulin (counterregulatory hormones) occurs. Epinephrine (from the adrenal medulla), glucagon (pancreatic α cells), norepinephrine (sympathetic nerve endings and adrenal medulla), growth hormone (anterior pituitary), and cortisol (adrenal cortex) all have been demonstrated to have protective metabolic effects during acute or prolonged hypoglycemia. It should be noted that the release of neuroendocrine counterregulatory hormones and the inhibition of endogenous insulin secretion occur before a healthy adult can feel any symptoms of hypoglycemia. If plasma glucose continues to fall, at ≈60 mg/dL (3.3 mmol), a series of autonomic (sometimes called neurogenic) signs and symptoms is activated. Autonomic warning responses to hypoglycemia include adrenergic and cholinergic symptoms. Adrenergic symptoms include palpitations, tremor, dry mouth, warmth, and anxiety.19 Cholinergic symptoms include sweating, hunger, and paresthesias.20–22 Signs of adrenergic activation include sweating and pallor. If the glucose level continues to fall, neuroglycopenic symptoms are activated at ≈50 to 55 mg/dL (2.8 to 3.1 mmol/L). These include blurred vision, drowsiness, slurred speech, confusion, and difficulty in concentrating. Defects in cognitive function are also apparent at this level of glycemia. If plasma glucose continues to fall, individuals can become drowsy, enter into a coma, and suffer seizures. Alternatively, individuals can become aggressive, which can be difficult to control and can be distinct from their usual personality. If severe hypoglycemia is prolonged, life-threatening events such as arrhythmias, myocardial infarction, and stroke, can occur.23,24 Long-term cognitive damage and even death can occur if very severe hypoglycemia continues for longer than a few hours.25 Although the above represents typical responses to falling glucose levels, it should be appreciated that many patients have idiosyncratic neurologic and symptomatic responses to hypoglycemia that fall outside the classical description. Thus, it is worthwhile to measure the glucose level in anyone who presents with neurologic deficits and/or strange and uncharacteristic behavior. It also should be noted that the typical physiologic responses to hypoglycemia can be modified by a number of factors, including antecedent hypoglycemia, long duration diabetes, age, gender, pregnancy, autonomic neuropathy, and use of certain drugs (Table 21-2). These altered physiologic and pathophysiologic responses are discussed in detail in the following sections.
Neurohumoral Regulation During Hypoglycemia
Insulin is the principal physiologic factor that lowers plasma glucose. Insulin is secreted primarily in response to glucose, but amino acids, nonesterified fatty acids, β2-adrenergic stimulation, and acetylcholine can also activate secretion of the hormone. Insulin secretion can be inhibited by hypoglycemia, insulin itself, somatostatin, and α2-adrenergic activity.75
Glucagon
Glucagon is released from the α cells in the islet of Langerhans. Similar to insulin, a number of physiologic factors can regulate secretion. These include hypoglycemia, amino acids, catecholamines (epinephrine and norepinephrine via β2-adrenergic mechanisms), and free fatty acids.26 Inhibitors of glucagon release include insulin and somatostatin. The regulation of glucagon release during hypoglycemia in humans is still undecided. Hypoglycemia, per se, can stimulate glucagon release in humans with cervical transection, individuals with a transplanted pancreas, and in vitro pancreas preparations.27 These data would point to the fact that direct α cell sensing of hypoglycemia would be the mechanism responsible for glucagon release. However, convincing data demonstrate that autonomic input (both sympathetic and parasympathetic) into the pancreas also can result in glucagon secretion.27 More recently, a third hypothesis has been proposed, which argues that a reduction in islet cell insulin levels is the mechanistic trigger for glucagon release during hypoglycemia.28
Epinephrine and Norepinephrine
Epinephrine (adrenaline) is released from the adrenal medulla. Similar to glucagon, the hormone can act rapidly to increase hepatic glucose output via elevations in hepatic glycogenolysis. If hypoglycemia continues and three-carbon precursors are present, epinephrine will stimulate gluconeogenesis in the liver and kidneys.29 Unlike glucagon, epinephrine has important effects on peripheral tissues. Epinephrine can restrict insulin-stimulated glucose uptake in skeletal muscle, which, when quantified in terms of maintaining glucose in the circulation, is greater than the contribution made by any increase in endogenous glucose production. This latter property of epinephrine is especially important in the defense against hypoglycemia that typically is encountered in clinical practice. Unlike the model of rapid hypoglycemia produced during an insulin tolerance test, the usual clinical course involves a slower decline into hypoglycemia and a more protracted duration of low glycemia, which, during the night, can last up to several hours. In the acute induction of hypoglycemia caused by a large bolus of rapid acting insulin, it is the activation of endogenous glucose production that is the primary physiologic defense against hypoglycemia, but in a model of more prolonged hypoglycemia, the restriction to insulin-mediated glucose uptake is paramount.30 Epinephrine’s important metabolic effects, which are mediated via β2-adrenoreceptors, also include stimulation of lipolysis to provide substrate (glycerol) and energy (NEFA) for gluconeogenesis. Additional effects on muscle provide lactate, pyruvate, and amino acids for gluconeogenic precursors.
Norepinephrine has similar metabolic actions to those of epinephrine. However, because 90% of norepinephrine is taken up at the level of sympathetic clefts, and 90% of catecholamine is taken up by the gut and then the liver, the increase in circulating norepinephrine during hypoglycemia is relatively modest (about 50% as compared with the 30-fold elevations that can occur with epinephrine). Therefore, quantifying the effects of norepinephrine at a tissue level in humans is problematic. However, recent work in conscious dogs has demonstrated that a 2.5-fold greater infusion of norepinephrine as compared with epinephrine is needed to produce similar increases in hepatic glucose production.31
Cortisol and Growth Hormone
The acute metabolic effects of these hormones are similar. Both can increase glucose production through increases in gluconeogenesis. Both hormones also can inhibit insulin-stimulated peripheral glucose uptake and can increase lipolysis and proteolysis. However, prolonged hypoglycemia (3 to 5 hours) is needed before the metabolic effects of growth hormone or cortisol are measurable, and even at that time represents only (≈20% to 25%) the action of epinephrine.32 Thus although chronic deficiencies of cortisol and/or growth hormone can cause hypoglycemia, these hormones serve no demonstrable function in the defense against acute hypoglycemia.
Syndromes of Disordered Counterregulatory Responses During Hypoglycemia
Almost immediately after the discovery of the hormone in the early 1920s, hypoglycemia was recognized as an unpleasant and dangerous side effect of insulin therapy. More than 90% of all patients with type 1 diabetes mellitus (DM) have suffered an episode of hypoglycemia. Typically, a patient with type 1 DM will experience 2 to 10 episodes of glucose below 70 mg/dL (3.9 mmol/L) per week and may have glycemic values below 50 mg/dL (2.9 mmol/L) at least 10 times each week. Episodes of major hypoglycemia requiring resuscitative measures from an additional person or hypoglycemic events resulting in seizures or coma have been reported to occur about once every other year to up to three times a year.17 The duration of type 1 DM plays an important role in increasing the frequency of severe hypoglycemia. Recent data from the UK hypoglycemia study group33 demonstrate that the frequency of severe hypoglycemia increases from 110 per 100 patient-years after less than 5 years of insulin therapy to 320 episodes per hundred patient-years after insulin therapy for longer than 15 years.
The risk and frequency of hypoglycemia are considered to be much lower in type 2 DM as compared with type 1 DM. This is undoubtedly true in patients with type 2 DM treated with monotherapy or combination therapy of metformin, a thiazolidineodione, a DPP-4 inhibitor, and/or a GLP-1 agonist. However, the incidence of hypoglycemia increases sharply in longer duration (insulin-deficient) type 2 DM. Recent clinical trials investigating insulin replacement strategies in type 2 DM report hypoglycemia in more than 70% of participants.34 Furthermore, the UK hypoglycemia study group reported that patients with type 2 DM receiving insulin for longer than 5 years had rates of severe hypoglycemia of about 70 episodes per 100 patient-years.33 This rate is slightly higher than the rate of 60 episodes per 100 patient-years reported for patients with type 1 DM in the intensive group of the DCCT.17 Other studies examining the frequency of severe hypoglycemia in type 2 DM have reported rates of one-third or similar to those occurring in type 1 DM. Furthermore, large recent multicenter studies investigating the effects of glucose control on complications of diabetes in type 2 DM have reported a significant incidence and prevalence of hypoglycemia.35,36 Thus, severe hypoglycemia continues to be a significant clinical problem for patients with type 1 DM and longer duration type 2 DM (Fig. 21-1).
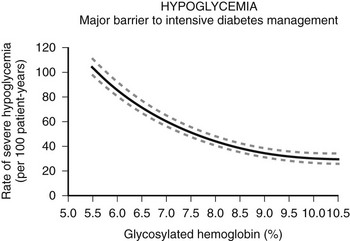
FIGURE 21-1 Hypoglycemia—Major barrier to intensive diabetes management. (Data from Diabetes Control and Complications Trial Research Group. N Engl J Med 1993;329:977–986.)
Death from hypoglycemia can be considered on the one hand to be rare when one considers the very large number of episodes of hypoglycemia that occur in clinical practice. However, mortality does occur during severe hypoglycemia in both type 1 and type 2 DM. Studies investigating the cause of death in patients with type 1 DM have reported that 2% to 10% may have died because of hypoglycemia.2,3 Similar death rates from hypoglycemia have been reported in patients with type 2 DM. The specific mechanism responsible for hypoglycemia-induced death is not currently understood. Possible suggested causes include cardiac arrhythmias, thrombotic events, and brain death. However, it should be noted that in primates, several hours of profoundly low glucose <1 mmol/L is required to induce irreversible brain damage and death.37
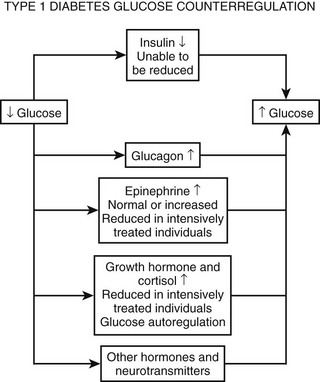
Hypoglycemia-Associated Neuroendocrine and Autonomic Failure
As discussed earlier, the four primary physiologic defenses against falling plasma glucose include inhibition of endogenous insulin secretion, release of glucagon and epinephrine, and symptomatic cues to ingest carbohydrate (Fig. 21-2). Unfortunately in patients with diabetes, all of these physiologic defenses can become defective and/or deficient. In type 1 DM and in long duration type 2 DM, the individual becomes critically insulin deficient, and thus the first physiologic line of defense (modulation of endogenous insulin) becomes lost. After varying periods of duration of disease (≈5 years), the ability of individuals with type 1 DM to release glucagon in response to hypoglycemia is lost (Fig. 21-3). This defect also occurs to a similar extent in long duration type 2 DM (Fig. 21-4).38 The mechanism for this finding is currently under intense investigation in humans. Hypotheses include lack of β cell turnoff, a possible specific autonomic nervous system dysfunction, and another as yet unidentified local signaling defect at the level of the α cell. The defect involved in releasing glucagon in type 1 DM is specific for hypoglycemia as the α cells in these individuals are present in normal number and size. In fact underscoring this point, glucagon responses to other metabolic stressors in type 1 DM such as exercise or amino acid infusions are preserved.39 The fact that glucagon responses are preserved during exercise in type 1 DM is interesting in that exercise is also typically associated with a physiologic “β cell switch off.” Whatever the mechanism, unfortunately after a few years’ duration, individuals with type 1 DM lose two of the four primary defenses against falling blood glucose. This leaves a functioning ANS (sympathoadrenal and sympathetic nervous systems) to serve as the primary defense against hypoglycemia in type 1 DM. In some individuals with type 1 DM, the ability to secrete epinephrine in response to hypoglycemia is preserved and can compensate for the lack of glucagon release.40 However, it is now clear that epinephrine responses to hypoglycemia are significantly reduced in patients with type 1 DM with intensive metabolic control.41 This epinephrine deficiency has been determined to be due to previous episodes of hypoglycemia42 and is separate from the syndrome of classic diabetic autonomic neuropathy that can occur after many years of suboptimal glycemic control. Models of repeated antecedent hypoglycemia have been demonstrated to produce acute reductions (30% to 50%) in epinephrine, pancreatic polypeptide (a marker of parasympathetic nervous system activity), and muscle sympathetic nerve activity (a direct marker of sympathetic nerve system activation) in individuals with type 1 DM, in those with type 2 DM, and in nondiabetic individuals.43 Additionally, recent (within 24 hours) antecedent hypoglycemia has been found to blunt a wide spectrum of neuroendocrine responses such as glucagon, growth hormone, ACTH, and cortisol during subsequent hypoglycemia44 in healthy and diabetic men. Confirming the role of antecedent hypoglycemia in causing blunted counterregulatory responses are a number of studies that prospectively investigated the effects of avoiding hypoglycemia in type 1 DM and following successful removal of an insulinoma.45–48 In all cases, there were initial blunted, neuroendocrine, ANS, and symptomatic responses to hypoglycemia. However, when patients with type 1 DM were restudied several months later, all showed improved counterregulatory responses to hypoglycemia, and patients with previous insulinomas had counterregulatory defenses restored to normal. Symptomatic responses, the fourth critical physiologic counterregulatory response, were significantly improved in all studies following a period of hypoglycemia avoidance.45–48 The blunting effects of antecedent hypoglycemia on subsequent counterregulatory responses have been termed by Cryer as “hypoglycemia-associated autonomic failure” (HAAF).
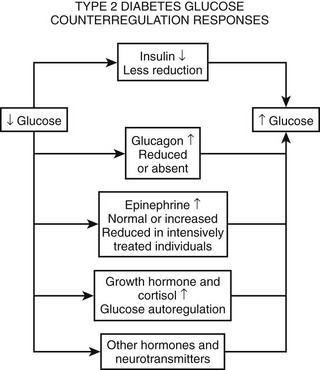
Following identification of this syndrome, a great deal of work has been performed in both animal and human models to further elucidate the mechanisms responsible for and the characteristics of hypoglycemia-associated counterregulatory failure. However, it should be appreciated that HAAF does not occur only in type 1 DM. Work from two independent laboratories has determined that this syndrome also occurs in type 2 DM.49,50 Segel and colleagues clearly demonstrated that moderate antecedent hypoglycemia of 50 mg/dL (2.8 mmol/L) can blunt ANS responses to subsequent hypoglycemia in moderately controlled (HbA1c 8.4%) individuals with type 2 DM.49 More recently, Davis and coworkers have demonstrated that even milder antecedent hypoglycemia of only 60 mg/dL (3.3 mmol/L) can blunt ANS responses to subsequent hypoglycemia in patients with type 2 DM with suboptimal (HbA1c ≈10.0%) or intensive glycemic control (HbA1c ≈6.7%).50
The great challenge in determining the mechanisms responsible for HAAF is explaining the simultaneous reduction in ANS-neuroendocrine responses and the change in glycemic thresholds that activate the physiologic defenses against falling plasma glucose. As described earlier, the usual physiologic thresholds for activation of ANS-neuroendocrine and symptom responses during hypoglycemia occur in the plasma glucose range between 50 and 80 mg/dL. In individuals with chronic hyperglycemia, symptoms of hypoglycemia can occur at plasma glucose levels between 90 and 140 mg/dL, depending upon the severity of the prevailing hyperglycemia. Simultaneous measurements of ANS-neuroendocrine hormones are in fact elevated during these symptoms, thus the individuals are experiencing the condition of “relative hypoglycemia.” This syndrome occurs because the thresholds for activation of ANS-neuroendocrine responses have been pushed to a higher plasma glucose level by the chronic hyperglycemia. On the other hand, individuals with intensive glucose control and multiple episodes of hypoglycemia often find that the activation of physiologic responses to hypoglycemia is pushed to a lower plasma glucose level. This dangerous condition, called hypoglycemic unawareness, results in inability of patients to recognize a falling plasma glucose until the value is below 50 mg/dL (2.9 mmol/L). In some individuals, a falling plasma glucose level is not recognized at plasma glucose levels of 30 mg/dL. This reduces the interval between first recognition of hypoglycemia and the onset of serious sequelae (such as coma or seizure). Thus, thresholds for the activation of physiologic defenses against hypoglycemia are labile and can change rapidly. The duration and depth of antecedent hypoglycemia required to induce HAAF have been characterized. Repeated episodes or relatively mild (3.9 mmol/L, or 70 mg/dL) and only brief durations (15 to 20 minutes) of hypoglycemia can independently blunt counterregulatory responses to subsequent hypoglycemia.51 However, one prolonged episode (2 hours) of moderate hypoglycemia (50 mg/dL, or 2.9 mmol/L) is sufficient to induce HAAF within a few hours on the same day.52
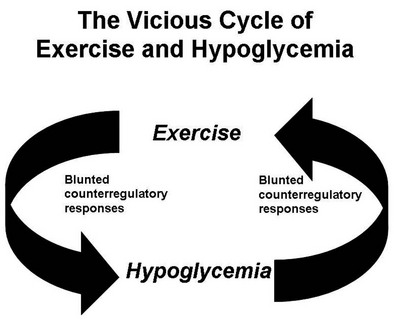
Numerous mechanisms responsible for the syndrome of HAAF have been proposed over recent years, with data both supporting and at times contrary to any given hypothesis. Boyle et al. proposed that repeated hypoglycemia increased cerebral glucose uptake in both healthy individuals and patients with type 1 DM, thereby reducing the stimulus for neuroendocrine counterregulatory responses during subsequent hypoglycemia.53 This finding was later challenged by work reporting no increase in blood-to-brain glucose transport following antecedent hypoglycemia.54 Other mechanisms that have been proposed include activation of the hypothalamic-pituitary-adrenal axis,44,55 increases in neurotransmitters such as GABA, and changes in hypothalamic fuel sensors such as glucokinase or AMP kinase (increases and decreases, respectively).56 Additionally, experimental evidence demonstrates that alcohol and opioids can downregulate subsequent ANS and neuroendocrine responses to hypoglycemia.57 Other physiologic mechanisms also have been found to cause forms of HAAF. These include sleep, exercise, and gender (Fig. 21-5).43,58 The association between exercise and hypoglycemia in type 1 DM has been both problematic and perplexing. Hypoglycemia can occur during, 1 to 2 hours after, or up to 21 hours after exercise. Traditionally, the explanation for this phenomenon was either a relative or absolute excess of subcutaneously injected insulin (due to an increase in insulin sensitivity following exercise) and/or incomplete glycogen repletion following exercise. Although these factors are important contributors to exercise-associated hypoglycemia, they cannot explain some of the profound episodes of hypoglycemia that occur during or after exercise. Studies from our own laboratory and others have demonstrated that exercise and hypoglycemia could reciprocally blunt subsequent ANS responses to either stress (Fig. 21-6).59,60 Thus, exercise blunts ANS responses (by 30% to 50%) to subsequent hypoglycemia, and vice versa. This feed-forward vicious cycle of blunted ANS responses between exercise and hypoglycemia can occur after only a few hours and persists for at least 24 hours following either stress (Fig. 21-7).39 Appreciation that deficient counterregulatory responses are also involved in the pathogenesis of exercise-related hypoglycemia explains why this phenomenon can occur many hours after exercise and provides a platform for therapeutic intervention. In general, in an individual with type 1 DM who is experiencing exercise-related hypoglycemia, it is appropriate to consider reducing both basal and mealtime insulin doses. Additionally, slightly raising glycemic targets and ensuring adequate carbohydrate repletion of glycogen stores are useful recommendations.
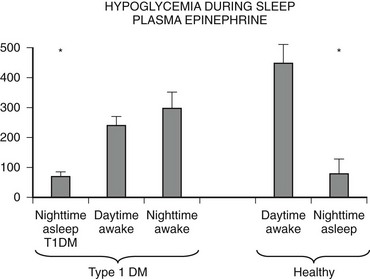
FIGURE 21-5 Hypoglycemia and plasma epinephrine responses during sleep/plasma epinephrine. (Data from Jones TW, et al. N Engl J Med 1998;338:1657–1662.)
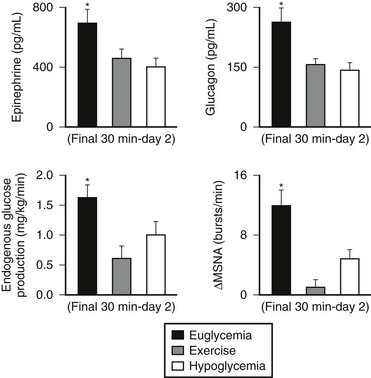
FIGURE 21-6 Reciprocal effects of antecedent hypoglycemia and exercise on subsequent neuroendocrine, autonomic nervous system, and metabolic (endogenous glucose production) responses to either stress. (Data from Galassetti P, et al. Am J Physiol Endocrinol Metab 2001;280:E908–E917; Davis SN, et al. Diabetes 1997;46:1328–1335.)
Gender can also play a large role in modulating ANS and neuroendocrine responses during hypoglycemia.58 In premenopausal, nondiabetic, and type 1 DM women, moderate hypoglycemia of ≈50 mg/dL (2.9 mmol/L) produces 30% to 50% reduced ANS responses compared with that seen in age- and body mass index (BMI)-matched men.58 However, when postmenopausal women, not on estrogen replacement, were compared with postmenopausal women receiving estrogen and age- and BMI-matched men, it was found that the large sexual dimorphism in ANS (epinephrine, muscle sympathetic nerve activity) and neuroendocrine (glucagon, growth hormone) responses was no longer present in estrogen-deficient women. It therefore would appear that in healthy humans, estrogen is a major mechanism responsible for sexual dimorphic counterregulatory responses during hypoglycemia.61 It should also be mentioned that women appear to be more resistant than men to the blunting effects of prior hypoglycemia. Thus, antecedent hypoglycemia can have up to a twofold greater suppressive effect on subsequent counterregulatory responses in men than in women.62 The mechanism for this intriguing finding is as yet unknown. Many studies have focused on the blunting effects of antecedent hypoglycemia on neuroendocrine and symptom responses during subsequent hypoglycemia. However, an additional component in the spectrum of deficient counterregulatory responses deserves mention. Several laboratories have identified that adrenergic receptors and particularly epinephrine action are downregulated by intensive glucose control and prior hypoglycemia.63,64 These reduced metabolic (lipolytic, endogenous glucose production, glycogenolysis) and cardiovascular responses contribute to defective counterregulatory defenses against a falling plasma glucose in patients with diabetes. Thus, it should be noted that strategies aimed at increasing epinephrine levels during hypoglycemia will be only partially successful if tissue resistance or adrenoreceptor downregulation to the action of the hormone is present. What is clear from the above wealth of data is that multiple mechanisms can downregulate ANS responses to hypoglycemia, and that this complex model presents numerous targets for therapeutic interventions to stimulate and restore counterregulatory responses during hypoglycemia in patients with diabetes.
Strategies to Improve Counterregulatory Responses During Hypoglycemia
Parallel with studies investigating the mechanisms responsible for HAAF, a number of laboratories have been exploring strategies for improving ANS and neuroendocrine responses during hypoglycemia. These have included preclinical approaches in rodents through to interventions in humans with type 1 DM. As mentioned above, hypothalamic kinases can act as important fuel sensors. Recent work in rats has shown that methods to reduce AMP-activated protein kinase (AMPK) in the ventral medial nucleus of the hypothalamus reduce epinephrine and glucagon responses during hypoglycemia, whereas increases in AMP kinase action can increase responses of these key neuroendocrine hormones during hypoglycemia.65 Other approaches to increase neuroendocrine responses during hypoglycemia in man have included amino acid infusions (glucagon) and use of caffeine (epinephrine and symptom responses). Terbutaline before bed has been demonstrated recently to increase plasma glucose levels during the night and to prevent nocturnal hypoglycemia.66 Recently, peroxisome-proliferator–activated receptor-γ (PPAR-γ) agonists, which are known to activate AMP kinase and fructose, which under certain conditions can inhibit glucokinase,67 have been demonstrated to increase counterregulatory responses in both healthy men and those with type 1 DM. Although at first glance these latter studies may seem unrelated, both may have mechanisms working through hypothalamic fuel sensing. Most recently, studies in conscious rats and healthy and type 1 DM men have highlighted the possible role of serotonergic transmission in modulating counterregulatory responses during hypoglycemia. Prolonged (i.e., weeks) use of two different selective serotonin reuptake inhibitors (sertraline and fluoxetine) has led to dramatic (30% to 60%) increases in ANS (epinephrine) responses during hypoglycemia.68
Drug-Induced Hypoglycemia
By far the most common cause of drug-induced hypoglycemia is insulin followed by sulfonylurea, meglitinides, and benzoic acid derivatives (i.e., oral insulin-producing agents). Hypoglycemia induced by oral insulin secretagogues is much less frequent than that caused by insulin but in certain instances can still be common. For example, hypoglycemia can occur in up to ≈35% of patients receiving glyburide or repaglinide. Hypoglycemia rates are less in agents with glucose-dependent insulin secretion (i.e., stimulation of insulin release is reduced during periods of hypoglycemia). Thus, the percentage of patients who experience hypoglycemia when receiving glimepiride, glipizide XL, or nateglinide is lower and is in the range of 5% to 10%. However, combination of even glucose-dependent insulin secretagogues with agents such as insulin or glucagon-like peptide-1 agonists can result in a much higher frequency of hypoglycemia (≥35%). Generally, newer sulfonylureas produce less hypoglycemia than do older first-generation sulfonylureas such as chlorpropamide. Severe hypoglycemia remains relatively uncommon with oral insulin secretagogues at a rate of ≈1.5 per 100 patient-years.33 Alcohol might be a more common cause of severe hypoglycemia in the United States than sulfonylureas.69 Alcohol can cause hypoglycemia in overnight fasted normal volunteers,70,71 with plasma glucose values as low as 5 mg/dL (0.3 mmol/L)72 and mortality rates ranging from 10% in adults to 25% in children.119 In a series of deaths caused by hypoglycemia, alcohol was the most common causative agent.74 The most common situation is a glycogen-depleted state, such as occurs in an individual who drinks after a considerable fast, or who drinks and then fasts. In the latter situation, blood alcohol levels can be low or undetectable.
Alcohol induces hypoglycemia by inhibiting gluconeogenesis72; as little as 50 g might be sufficient.73,76 Its mechanism of action is complex, with evidence of impaired counterregulatory hormone responses71 and impaired uptake of gluconeogenic precursors,77 but the predominantly accepted mechanism is its inhibition of the gluconeogenic process stemming from an increased reduced nicotinamide adenine dinucleotide (NADH)/NAD ratio as a result of the oxidation of alcohol to acetaldehyde and acetate, thus reducing the ability of the liver and kidney to oxidize lactate and glutamate to pyruvate and α-ketoglutarate, respectively.78–80 Although plasma insulin levels are appropriately suppressed in this condition, because of this inhibition of gluconeogenesis, glucagon and catecholamines are ineffective in increasing glucose release and raising plasma glucose levels.81 Thus, in a patient with suspected alcohol-induced hypoglycemia, oral or intravenous glucose is the treatment of choice.
Only about 10% of reported cases of drug-induced hypoglycemia have occurred without concomitant insulin, sulfonylurea, or alcohol.82 Of these, propranolol,83 sulfonamides,84 and salicylates169 have been reported most frequently. Propranolol and other nonselective β-blockers decrease the ability of the liver and kidney to increase their release of glucose,85,86 enhance peripheral insulin sensitivity,87 and mask symptoms of impending hypoglycemia. The adverse effects of β-adrenergic β-blockers are mediated through β2-receptors. Recent studies indicate that β1-selective blockers do not present an increased risk for severe hypoglycemia and therefore should not be considered as being contraindicated in diabetic patients.87,88
Salicylates can act by inhibiting hepatic glucose release and increasing insulin secretion, although their exact mechanism remains to be determined. Sulfonamides probably act by stimulating insulin release in a manner similar to that of sulfonylureas. Angiotensin-converting enzyme inhibitors89and pentamidine90 are associated more frequently with hypoglycemia, as their use increases in diabetic patients and those with AIDS, respectively. Angiotensin-converting enzyme inhibitors can increase tissue insulin sensitivity91 and can decrease the degradation of bradykinin, which has certain insulin-mimetic actions.92 Pentamidine is cytotoxic to pancreatic β cells, and hypoglycemia occurs with the release of insulin from degenerating cells, often with subsequent permanent diabetes mellitus.93 Many of the drugs listed in Table 21-2 have been reported to cause hypoglycemia only in association with the use of antidiabetic medications or have been the subject of isolated case reports, and their etiologic significance remains to be established. However, their use in a patient with otherwise unexplained hypoglycemia should be discontinued whenever possible.
Treatment and Strategies to Reduce Hypoglycemia
Clinical Strategies to Reduce Hypoglycemia
Over the last generation, interest has increased in replacing insulin in the most physiologic manner for patients with diabetes. In the 1980s, the introduction of recombinant human insulin reduced the formation of antibodies and provided more predictable pharmacokinetic profiles. The next decade produced analogue insulins that initially were designed to provide a quicker onset and a shorter duration of action. These insulins (lispro, aspart, glulisine) were designed to reproduce more closely the typical physiologic prandial spikes of insulin observed following meals. The second wave produced long-acting basal types of insulin (glargine, detemir) designed to mimic the background constitutive release of the hormone that regulates nocturnal and interprandial glycemia. Studies in type 1 DM have demonstrated that hypoglycemia (particularly nocturnal) can be reduced when short-acting analogues versus regular (soluble) insulin are used.94,96 Similarly, long-acting analogues have been demonstrated to reduce hypoglycemia by 20% to 33% in patients with type 2 DM when compared with NPH-based regimens.34 Thus, current recommendations are to use analogue-based insulin replacement regimens whenever possible.
Insulin pump development began in the 1970s and over the past 20 years has become a major method of insulin replacement in the United States. Although widely acknowledged for its usefulness, the cost of treatment (pump and supplies) has restricted a more widespread acceptance of this technology. Nevertheless, studies in children94 and pregnant women have demonstrated a reduction in hypoglycemia when compared with multiple daily insulin injection regimens.
Most recently, continuous, real-time glucose monitoring has been introduced into clinical practice. Currently, a variety of devices can be worn in combination with an insulin pump or independently. In a multicenter, randomized, controlled trial, the use of a continuous glucose monitor has been shown to reduce both glycosylated hemoglobin (HbA1c) and, as a secondary end point, the incidence of severe hypoglycemia in type 1 DM adults and children but not adolescents95 when compared with conventional self-blood glucose monitoring. Currently, another large randomized study is under way that will test the hypothesis that a continuous glucose sensor–driven insulin pump replacement approach will provide better HbA1c and less hypoglycemia than multiple daily injections of insulin and traditional self-blood glucose monitoring in both children and adults with type 1 DM.
Pancreas Transplantation and Hypoglycemia
Pancreas transplantation has been performed in patients with type 1 DM for over 25 years. Typically, patients have experienced episodes of severe hypoglycemia, and this is one of the major criteria for consideration of pancreas transplantation. Generally, rates of hypoglycemia improve dramatically in the first year after transplantation. However, ≈30% of patients report episodes of minor hypoglycemia. Most (but not all) studies also demonstrate that counterregulatory defenses are improved after pancreatic transplantation.97–99 Most notably, glucagon responses to hypoglycemia increase, accompanied at an early stage by some improvement in epinephrine and symptomatic responses. Improvements in glucagon responses appear to be persistent and have been documented in long-standing pancreas transplantation recipients of up to 19 years post transplant.100 Although the improvement in glucagon responses to hypoglycemia appears to be durable after long-term pancreatic transplantation, it is clear that improvements in epinephrine and symptomatic responses to hypoglycemia are not sustained.100 Along similar lines, it has been reported that physiologic insulin and glucagon responses during exercise are maintained for the most part following pancreas transplantation.101 Robertson and colleagues have investigated whether living donors of pancreas segments have normal counterregulatory responses to hypoglycemia.102 This has particular relevance, as ≈25% of donors can develop diabetes within 1 year after the procedure. It is interesting to note that despite deficient insulin and glucagon responses to glucose or arginine infusion, it was found that glucagon responses during hypoglycemia were preserved among donors.
A history of recurrent severe hypoglycemic events is also a major indication for islet cell transplantation. Although the number of transplanted individuals who are insulin independent is low (≈10%) up to 5 years after transplantation,103 it generally is reported that major episodes of hypoglycemia are significantly improved following islet cell transplantation.104 This finding appears to be due to the fact that there is some restoration of the ability to modulate insulin levels during hypoglycemia, as no reports have described significant increases in glucagons during hypoglycemia following islet cell transplantation.105 These findings underscore the importance of modulating insulin in the defense against hypoglycemia, but they also pose the intriguing question as to why there is an absent glucagon response. One plausible explanation is that the intrahepatic site for the islet transplant is the cause of the deficient glucagon response to hypoglycemia.106 In a recent series of elegant experiments in rats, Zhou et al.106 demonstrated that in the normal postprandial state, glucagon responses to hypoglycemia were absent in intrahepatially transplanted islets. However, when the animals were fasted for 48 hours and thus intrahepatic glucose flux was reduced, normal glucagon responses to hypoglycemia were obtained in the islet-transplanted animals.106 Supporting the theory that placing islets in the liver may be cause for the absent glucagon response during hypoglycemia are data indicating that alternate sites for islet cell transplantation away from the liver may produce improved glucagon responses to hypoglycemia.107
Hypoglycemia and Gastric Bypass Surgery
Accompanying the rapid increase in obesity is an increase in the number of bariatric surgical procedures performed. Recently, accumulating reports are demonstrating an increase in severe postprandial hypoglycemia following gastric bypass surgery.108–110 Although the disorder was initially thought to represent a version of the adult nesidioblastosis syndrome, subsequent analysis of the pancreata have revealed potentially alternative pathologic causes.111 It is interesting to that that there is a strong female preponderance, and this condition can develop months or even years after bypass surgery is performed. Treatment for hypoglycemia has proved to be problematic. Neither acarbose given to try to alleviate dumping syndrome symptoms by slowing glucose absorption nor somatostatin given to inhibit endogenous insulin secretion has been successful.112 Consequently, pancreatic resection was needed to reduce the occurrence of hypoglycemia in the six patients originally reported with this syndrome.108 Subsequent reports have highlighted that patients with this syndrome have exaggerated glucagon-like peptide-1 (GLP-1) and insulin responses to a mixed meal.113 In late 2007, Vella and Service provided an update on the Mayo Clinic’s experience regarding hypoglycemia following gastric bypass surgery.112 They reported that 43 patients required an ≈60% gradient-guided pancreatic surgical resection to alleviate hypoglycemia. Pathologic inspection revealed that most of the pancreata had islet hypertrophy with nesidioblastosis, although some also had one or more insulinomas. It appears that this syndrome of hypoglycemia is specifically associated with bypass surgery, as a recent report of gastric banding to induce weight loss did not report any significant hypoglycemia over a 2-year postprocedure follow-up period.114
Neonatal Hypoglycemia
Plasma glucose levels below 70 mg/dL (3.9 mmol/L) commonly occur in newborn infants. It has been estimated that up to 50% of neonates can have plasma glucose levels below 50 mg/dL (2.9 mmol/L) following short-term fasting (up to 8 hours) after birth.115 Over the first 72 hours of life, gluconeogenic pathways mature, and the risk of hypoglycemia in normal infants is removed. However, in addition to the above physiologic causes of neonatal hypoglycemia, numerous pathologic conditions can cause hypoglycemia (Table 21-3).
Table 21-3
Causes of Neonatal Hypoglycemia
• IV glucose during labor and delivery
• Maternal oral hypoglycemic agents, propranolol, labetalol
The most common causes of persistent neonatal hypoglycemia are the congenital hyperinsulinism syndromes. These syndromes have been estimated to occur at a rate of about 1:30,000 births worldwide. However, in certain areas (e.g., Finland, the Arabian Peninsula), these conditions have been reported to occur at much higher rates (≈1:2,100).116,117 Over the past 50 years, congenital hyperinsulinism syndromes have been given a number of titles (e.g., nesidioblastosis, islet dysregulation syndrome, persistent hyperinsulinemic hypoglycemia of infancy). Recently, it has been discovered that nesiodioblastosis can be a normal feature of the pancreas in the neonatal period and is histologically different from the pathologic processes that cause congenital hyperinsulinism. The pathophysiology and the molecular and genetic bases for congenital hyperinsulinism were reviewed recently by De Leon and Stanley.115 Of these, the most frequently occurring involve loss-of-function mutations in the pancreatic β-cell K-ATP channel, Kir6.2 and Sur-1 receptors.
The clinical presentation of infants with congenital hyperinsulinism may be varied but typically involves prolonged and severe hypoglycemia with lethargy, seizures, and apnea, and babies are often large for gestational dates. Typically, hypoglycemia occurs in the fasting or postprandial state, and these infants require large rates of glucose infusion >10 mg/kg/min to prevent severe hypoglycemia. In the glutamate dehydrogenase version of congenital hyperinsulinism, hypoglycemia can occur during both the fasting and the postprandial state. Hypoglycemia in this condition can be precipitated by a protein meal and is characteristically accompanied by elevated ammonia levels. Diagnosis of glutamate dehydrogenase hyperinsulinism typically occurs when the child is several months of age.118
Diagnostic Investigation of an Infant With Persistent Hypoglycemia
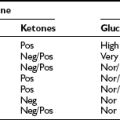
Because of the wide range of conditions that can cause hypoglycemia in an infant, a variety of blood and urine tests can be helpful in identifying metabolic and endocrine causes of the disorder (Table 21-4).
Table 21-4
Blood and Urine Tests for Metabolic and Endocrine Causes of Neonatal Hypoglycemia
It is surprising that plasma insulin levels often are not strikingly elevated in the congenital hyperinsulinism syndromes. Similar to insulinomas, plasma insulin levels often are modestly increased but are inappropriately raised in relation to the prevailing hypoglycemia. However, measurement of free fatty acids and β-hydroxybutyrate levels reveals dramatic suppression and evidence of significant insulin action. An increase in plasma glucose greater than 30 mg/dL following glucagon is also supportive of the diagnosis. Infants with the GDH-M1 form of congenital hyperinsulinism exhibit increased responses to leucine118 and thus can be distinguished from those with the KATP channel forms of hyperinsulinism. Additionally, genetic testing is now widely available for identification of four of the five genes associated with congenital hyperinsulinism. Initially, treatment consists of diazoxide, which is a KATP channel agonist and has a suppressive affect on insulin secretion. If diazoxide does not produce a clinical response, octreotide has been used to suppress insulin release. However, in children who are unresponsive to medical therapy, the next treatment is surgery. This can comprise a local resection or a near total (98%) pancreatectomy, depending upon the presence of focal or diffuse disease.115,118 The application of 18F-DOPA positron emission tomography (PET) scans to differentiate focal from diffuse pancreatic involvement in the congenital hyperinsulinism syndromes has provided a clinical breakthrough in the management of these children.119
Mitochondrial Fatty Acid Oxidation Disorders
Eleven different mitochondrial fatty acid oxidation disorders120 can present with fasting (hypoketotic) or postprandial hypoglycemia. Hepatic oxidation of fatty acids produces energy and acetyl-CoA, both of which are essential for gluconeogenesis. Additionally, fatty acid oxidation is required for hepatic ketone production. However, because of specific defects in mitochondrial fatty acid oxidation, gluconeogenesis is reduced, and this results in hypoglycemia and a lack of ketone bodies. The combination creates a scenario whereby the brain becomes starved of glucose and its primary alternative substrate (ketones), which creates conditions for severe neurologic consequences. Plasma membrane carnitine transporter deficiency is an autosomal recessive disorder that occurs in ≈1:40,000 infants. Low plasma carnitine and acyl carnitine are indicative of this disorder, and treatment with dietary carnitine prevents hypoglycemia. Carnitine palmitoyl transferase I and II (CPT I and CPT II) deficiencies can present as severe, life-threatening events that require frequent feeding and supplementation with dietary medium-chain triglycerides and l-carnitine (CPT II deficiency). Unfortunately, the most severe form of CPT II deficiency can present soon after birth and can be fatal. Medium-chain acetyl-CoA dehydrogenase (MCAD) deficiency is the most common fatty acid oxidation defect and is inherited as an autosomal recessive trait. Hypoglycemia usually occurs during fasting and conditions of stress such as viral illness.121,122 The presentation can be serious with vomiting, apnea, coma, encephalopathy, and death. It has been estimated that if left undiagnosed, up to 25% of children may die during the presenting event. Blood tests demonstrate elevated medium-chain acylcarnitine. As with other mitochondrial fatty acid oxidation disorders, the enzyme deficiency can be demonstrated diagnostically in fibroblasts. Treatment consists of avoidance of fasting and precipitation of attacks that could lead to neurologic defects and even death. Thus, frequent meals, bedtime snacks, uncooked cornstarch (to avoid nocturnal hypoglycemia), and carnitine supplementation have been used successfully in these children.
Long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD) and mitochondrial trifunctional protein (MTP) deficiency are also autosomal recessive disorders. Both conditions can present with severe hypoglycemia, encephalopathy, neurologic complications, and death. Both conditions can result in peripheral neuropathy, and LCHAD deficiency can result in pigmentary retinopathy and blindness. Treatment is similar to that provided for the above fatty acid oxidation defects acyl-CoA dehydrogenase and carnitine/acylcarnitine translocase (CACT). Long-chain ketothilase (LCKAT) and 2-4-dienoyl-CoA reductase deficiency are very rare conditions that have been described recently.120
Glycogen Storage Diseases
Glycogen synthase and glucose-6-phosphatase deficiencies often present in the neonatal period with hypoglycemia. Additionally, glycogen-phosphorylase deficiency can present later in childhood. Of the three disorders, glucose-6-phosphate deficiency presents with the most severe hypoglycemic phenotype. Treatment to prevent hypoglycemia is similar in all of the glycogen storage disorders.123 Avoidance of hypoglycemia during stress, exercise, and the night involves frequent carbohydrate-containing meals and bedtime snacks. In times of severe stress, intravenous glucose may be required to maintain plasma glucose levels above 70 mg/dL (3.9 mmol/L).
Sepsis, Trauma, and Burns
Initially, the response to the stress of infection is an increase in glucose turnover, with glucose production often exceeding glucose utilization and resulting in mild hyperglycemia. This response involves increases in both glycogenolysis and gluconeogenesis and is largely mediated by glucagon124 because adrenergic blockade has no effect on glucose turnover.125 As the infection worsens, increased release of endotoxin and its derivatives, complement activation, endoperoxide activation, and release of endogenous inflammatory mediators (tumor necrosis factor-α, interleukins, and other monokines) compromise cardiovascular integrity and cause central venous pooling, inadequate tissue perfusion, and microvascular protein transudation.126 At this stage, a decrease in splanchnic and renal blood flow occurs. Despite concomitantly reduced peripheral tissue perfusion, glucose utilization is increased.127–129 Decreased tissue oxygenation causes increased anaerobic glycolysis, which perpetuates the increased glucose utilization.
The inability of glucose release to keep pace with increased tissue demands results in hypoglycemia. Hepatic glycogen stores are rapidly exhausted; consequently, glucose release becomes solely dependent on gluconeogenesis. However, gluconeogenesis fails to stimulate this process because of a reduction in ANS and neuroendocrine effects.130–132 Factors such as acidosis (which inhibits hepatic gluconeogenesis), increased intracellular calcium (which impairs mitochondrial function and inhibits gluconeogenic enzymes), and siphoning of available energy from gluconeogenesis to support ion transport might be the mechanisms responsible for diminished ANS and neuroendocrine responsiveness.
Cardiac Failure
Spontaneous hypoglycemia can occur with severe heart failure133–135; it is rare in adults but not uncommon in infants and children,136 in whom reduced hepatic glycogen levels (but normal phosphorylase and glucose-6-phosphatase activity) have been found in liver biopsy specimens. The condition has been attributed to a variety of mechanisms, including reduced gluconeogenesis, poor dietary intake, and gastrointestinal malabsorption, which are present in cardiac failure.
Mellinkoff and Tumulty first described the hypoglycemia of cardiac failure and attributed it to associated hepatic disease.137 However, chronic lung disease with right and left heart failure is seen in most patients.135 Thus, hypoxemia and low cardiac output may produce hepatic ischemia. Marks and Rose postulated that decreased availability of oxygen would suppress gluconeogenesis by increasing hepatic anaerobic glycolysis (Pasteur effect) and lactate production, thereby resulting in an increased NADH/NAD ratio.138 This decreased ratio could compromise gluconeogenesis because NAD is an essential cofactor for several of the enzymatic steps of gluconeogenesis. This attractive hypothesis could explain the association between hypoglycemia and the lactic acidosis of cardiac139 and liver140 disease, as well as the hypoglycemia accompanying other conditions associated with tissue anoxia, such as sepsis and shock.141 Low cardiac output would be expected to limit substrate delivery to the kidneys. In addition to a reduced capacity to produce glucose, increased glucose utilization from increased anaerobic glycolysis and increased energy demands from labored breathing and malnutrition (anorexia) are probably additional important factors. At the present time, no evidence indicates that abnormal counterregulatory hormone responses play a role in the pathogenesis of the hypoglycemia associated with these conditions.
Renal and Hepatic Disease: General Considerations
The liver and kidneys are the only organs that are capable of releasing glucose into the circulation, inasmuch as other tissues generally lack or have minimal amounts of the enzyme glucose-6-phosphatase. Consequently, it would not be surprising that patients with hepatic or renal disease should be prone to hypoglycemia. Nevertheless, it is uncommon for hypoglycemia to occur simply as a result of loss of mass or function of these organs, and when it does occur, the cause is usually multifactorial.72,142 The large capacities of these organs to release glucose into the circulation and their ability to compensate for each other’s shortcomings appear to provide an explanation for this phenomenon.
Normally, the liver accounts for 80% to 85% of all glucose released into the circulation; it can increase its output (initially mainly by glycogenolysis, later by gluconeogenesis) over a sustained period by twofold to threefold (at least for several days, as exemplified by burn patients130). Thus, hypoglycemia with an appropriate compensatory increase in hepatic glucose release would be unlikely to develop in anephric individuals because the kidney normally contributes only 15% to 20% of all glucose that is released into the circulation. On the other hand, the kidney can increase its output over a prolonged period by twofold to threefold, as exemplified in humans who have fasted for several weeks.143 Animal and human studies indicate that the kidney can acutely increase its output to compensate for decreased hepatic glucose release and vice versa,144–146 a phenomenon that is referred to as hepatorenal reciprocity.147 For example, during the anhepatic phase of human liver transplantation, the kidney can maintain normoglycemia without the need for exogenous glucose.148 Thus, hypoglycemia would be unlikely to develop in patients with hepatic disease until the liver’s capacity to release glucose was reduced beyond the ability of the kidney to compensate. In fact, animal studies indicate that more than 80% of the liver must be removed for hypoglycemia to occur.149
Liver Disease
Although hypoglycemia has been associated with a wide range of liver diseases (hepatocellular carcinoma, cirrhosis, fatty metamorphosis, toxic and infectious hepatitis, cholangitis, and biliary obstruction), its occurrence is actually uncommon in the absence of other complicating factors (i.e., infection).72,138 For example, Zimmerman and coworkers found fasting hypoglycemia levels of 60 mg/dL (3.3 mmol/L) or less in only 6 of 269 patients with a variety of liver diseases.150 In humans, the insult to hepatic function must be acute, or the loss of parenchyma must be widespread. Felig and coworkers found that about 25% (4 of 15) of patients with acute viral hepatitis had a fasting blood glucose level less than 50 mg/dL (2.8 mmol/L).151 In chronic liver disease associated with hypoglycemia, additional factors, such as malnutrition and infection, usually are involved. A 50% incidence of hypoglycemia was found in patients with liver disease associated with sepsis and circulatory collapse.152,153 In such situations, liver function tests might not parallel the severity of hypoglycemia. Infiltrative diseases such as metastatic disease, amyloidosis, sarcoidosis, and hemachromatosis rarely replace sufficient parenchyma to cause hypoglycemia.154
Hyperinsulinemia often accompanies hepatic disease as a consequence of decreased insulin degradation by the liver,155 but the hypoglycemia of hepatic disease is almost always accompanied by appropriate suppression of plasma insulin concentrations.151 Likewise, little evidence has been found to implicate overproduction of insulin-like growth factors (IGFs) by the liver. Gorden and colleagues reported one patient with hemangiopericytoma of the liver in their series of 52 patients with extrapancreatic tumors, hypoglycemia, and IGFs, but no excessive IGFs were associated with primary hepatocellular carcinoma, the hepatic neoplasm most frequently associated with hypoglycemia.156
Renal Disease
Except in infants, hypoglycemia rarely occurs with acute renal failure. However, with chronic renal failure, hypoglycemia is not uncommon in adult patients. It can occur as an isolated event or can be repetitive. In general, neuroglycopenia rather than autonomic symptoms predominates.142
Although it has long been recognized that uremia reduced the insulin requirement in diabetic humans,157 it was not until 1970 that Block and Rubinstein reported three diabetic patients with renal failure who had suffered severe hypoglycemia after insulin and sulfonylurea therapy had been stopped.158 Shortly afterward, spontaneous hypoglycemia associated with renal failure was described in nondiabetic patients,159–162 with an incidence of 1% to 3% in two large studies.162,163
The origin of renal hypoglycemia is complex.142,164 Many factors, including altered drug metabolism, delayed gastric emptying, malnutrition, infection, dialysis, altered insulin sensitivity, associated hepatic and cardiac disease, and impaired renal and hepatic glucose release, predispose uremic patients to hypoglycemia. Drugs are probably the most common immediate cause. Any drug that can cause hypoglycemia is more likely to do so in a uremic patient because of a prolonged half-life (e.g., insulin, certain sulfonylureas, especially chlorpropamide) or decreased protein binding secondary to hypoalbuminuria.165 Although hypoglycemia occurs in nondiabetic as well as diabetic patients with renal failure, it is more likely to occur in the latter because of the use of hypoglycemic agents, and because patients with long-standing diabetes have autonomic neuropathy and defects in glucose counterregulation. Most patients have been malnourished, although the condition has been reported in well-nourished patients.160 Malnutrition secondary to anorexia or vomiting, which can reduce hepatic glycogen stores and the availability of gluconeogenic precursors, is a common feature that increases the risk for hypoglycemia.166
Fatal hypoglycemia can occur with peritoneal dialysis or hemodialysis when high glucose–containing dialysate is used, because of exaggerated insulin release in conjunction with impaired renal insulin degradation.167 Other factors such as the use of glucose-deficient solutions in diabetic patients and loss of alanine during hemodialysis may contribute to the development of hypoglycemia.168,169
Several reports suggest that renal failure per se predisposes to the development of hypoglycemia.158,161,162,170–176 Most evidence points to diminished glucose release rather than increased glucose utilization as a cause of the hypoglycemia.160,162,177–179 Accompanying malnutrition and acidosis would also contribute to diminished hepatic glycogenolytic and gluconeogenic potential.134,171,173,180–182 The expected compensatory increase in renal gluconeogenesis in response to acidosis would be compromised by loss of renal mass and exacerbated by inappropriate plasma insulin levels caused by reduced renal insulin degradation.
Glucagon Deficiency
Pancreatectomized individuals, patients with long-standing type 1 diabetes, and those in whom insulin-requiring diabetes develops as a result of chronic pancreatitis or cystic fibrosis are glucagon deficient182 and prone to severe hypoglycemia during treatment for their diabetes.183 An imbalance between insulin and glucagon secretion (relative glucagon deficiency) has been reported in reactive postprandial hypoglycemia.184 Because of glucagon’s importance, it might have been expected to be a common counterregulatory hormone deficiency. However, the condition is extremely rare, with only two poorly substantiated cases of neonatal hypoglycemia185,186 and two reported cases in adults.187,188
Catecholamine Deficiency
Patients with long-standing type 1 diabetes mellitus, adrenalectomized individuals, and those with autonomic neuropathy have impaired catecholamine responses during insulin-induced hypoglycemia,189–191 but their increased risk for hypoglycemia can be compensated for by increases in the secretion of other counterregulatory hormones, in particular, glucagon.192 Subtle defects in recovery from hypoglycemia have been demonstrated when such compensatory increases have been prevented experimentally.394 However, if glucagon responses to hypoglycemia are impaired simultaneously (e.g., in the patient with type 1 DM), the risk for hypoglycemia markedly increases.183
Several cases of neonatal ketotic hypoglycemia and one case in a 5-year-old boy have been attributed to epinephrine deficiency on the basis of low urinary epinephrine excretion.193,194 No cases of hypoglycemia secondary to isolated catecholamine deficiency have been reported in an adult. The hypoglycemia that occurs with propranolol may relate to the drug’s inhibition of lipolysis, which would reduce gluconeogenesis, an important counterregulatory process, and its promotion of increased glucose clearance by peripheral tissues.85 It is important to note that acute hypoglycemia can occur during surgical removal of a pheochromocytoma, presumably because of disinhibition of insulin release and abrupt withdrawal of the anti-insulin actions of catecholamines.195
Cortisol and Growth Hormone Deficiency
Although cortisol and growth hormone have been demonstrated to contribute independently to glucose counterregulation via their actions to promote glucose release and limit glucose uptake,196–199 hypoglycemia does not develop in most adults who lack these hormones. However, serious hypoglycemia often develops in infants and children, who lack these hormones, especially after a period of fasting or during an intercurrent illness.199–203 In a review of 76 adults with isolated adrenocorticotropic hormone (ACTH) deficiency,204 24 (≈33%) had hypoglycemia. During prolonged fasting (6 days), adult growth hormone–deficient dwarfs become hypoglycemic,205 as can hypopituitary pregnant women.206 On the other hand, overnight fasting plasma glucose levels have been reported to be normal in glucocorticoid-withdrawn patients with primary adrenal insufficiency207 or panhypopituitarism.208 Acute adrenal insufficiency such as in Sheehan’s syndrome can be manifested as severe hypoglycemia,209 and autoimmune Addison’s disease may be the cause of severe recurrent hypoglycemia in a patient with type 1 diabetes mellitus.210
It is important to be aware that malabsorption of glucocorticoids can occur in patients with bowel disease and in patients treated with drugs such as bile acid sequestrants.211
Autoimmune Hypoglycemia
Of the two types of autoimmune hypoglycemia, one is due to autoantibodies against the insulin receptor, and the other is due to autoantibodies against insulin itself in individuals who have never received exogenous insulin. Both are rare and can produce fasting, as well as postprandial reactive hypoglycemia—primarily the former.212–220
Anti-Insulin Receptor Antibodies
Fewer than 30 patients in whom hypoglycemia developed as a result of antibodies directed against the insulin receptor have been reported. These antibodies act as agonists and produce hypoglycemia similar to insulin. Most patients have had evidence of other conditions associated with altered autoimmunity (systemic lupus erythematosus, scleroderma, primary biliary cirrhosis, immune thrombocytopenic purpura, celiac disease, Hashimoto’s thyroiditis, and Hodgkin’s lymphoma).216 Some patients have developed severe insulin resistance as a result of the antibodies blocking the insulin receptor before the antibodies became agonists. Patients with this condition have low circulating insulin and C-peptide levels, normal IGF-1, and appropriate counterregulatory hormone responses.
Although experience is limited, antibody titers generally decrease over time, and remission eventually occurs in most patients. However, because of the severity of the hypoglycemia, aggressive treatment is indicated. High-dose glucocorticoids,221,222 plasmapheresis,223 and alkylating agents224 all have been tried with variable success.
Anti-Insulin Antibody Hypoglycemia
Since 1970, approximately 200 cases of anti-insulin antibody hypoglycemia have been reported, nearly 90% occurring in Japanese patients.213 Associated autoimmune disorders and plasma cell dyscrasias (Graves’ disease, rheumatoid arthritis, polymyositis, and systemic lupus erythematosus) are common. The use of certain drugs (hydralazine, procainamide, penicillamine, interferon-α, and methimazole) has been implicated in initiating the syndrome.216
Pregnancy
Despite a decrease in insulin sensitivity and an increase in glucose turnover to accommodate the needs of the fetus, fasting plasma glucose levels are normally 10% to 15% lower during the third trimester of pregnancy.225,226 Nevertheless, a great number of metabolic changes occur during pregnancy to make a woman more vulnerable to hypoglycemia.227 Any condition that can cause hypoglycemia in a nonpregnant woman can do so in a pregnant woman. In addition to insulinoma,228,229 non–islet cell tumors,230 severe infection,231 poor nutrition,232 drug-induced sources (e.g., insulin in diabetic patients), and a condition called the HELLP syndrome (characterized by hemolysis, elevated liver enzymes, and a low platelet count) can cause hypoglycemia as a result of fulminating hepatic dysfunction.233–235
Exercise
Hypoglycemia can develop after prolonged strenuous exercise and has been reported in marathon runners,236 in normal volunteers after exercise on a bicycle ergometer for 3 hours at 56% of maximal capacity,237 and in a healthy male subject taking a β-blocker after skiing 15 km in 2 hours.238 Although coma developed in the latter instance, most instances of exercise-associated hypoglycemia are asymptomatic, self-limited, and readily reversed by carbohydrates.239
The increased fuel demands of the working muscle necessitate compensatory metabolic processes in the liver and kidney.240–243 Changes in hepatic glycogenolysis and gluconeogenesis have been found to be closely coupled to the increase in glucose uptake produced by the working muscle because of the actions of the pancreatic hormones.240 The exercise-induced increase in glucagon secretion and the concomitant decrease in insulin secretion interact to stimulate hepatic glycogenolysis, whereas the increase in hepatic gluconeogenesis is determined primarily by glucagon’s action to increase hepatic gluconeogenic precursor fractional extraction and the efficiency of intrahepatic conversion to glucose. On the other hand, no evidence has shown that hepatic innervation is essential for the rise in hepatic glucose production. Epinephrine and norepinephrine become important in increasing glucose production during prolonged or heavy exercise, when levels are particularly high. Catecholamines can produce this effect by directly stimulating both hepatic and renal glucose release, by increasing the availability of gluconeogenic precursors, and by increasing lipolysis.
Factitious Hypoglycemia
Factitious hypoglycemia refers to a situation in which an individual intentionally attempts to create the impression of the presence of a hypoglycemic disorder.244–246 This includes diabetic individuals who falsify their self-glucose monitoring, or who overdose or underdose themselves to intentionally create the impression of better than actual glycemic control or brittle diabetes,247,248 and in cases of child abuse.12 Generally excluded are inadvertent sulfonylurea ingestion (see drug-induced hypoglycemia), homicides,249,250 suicide attempts,251,252 and substance abuse in which the additional use of insulin or sulfonylureas is intended to “obtain a high,” rather than to create the impression of a hypoglycemic disorder.253–255 These situations nevertheless can present diagnostic challenges.
Since 1947, more than 80 cases of insulin-induced or sulfonylurea-induced factitious hypoglycemia have been described in the literature,245,256–259 but the condition is probably more common than this figure would indicate because most cases go unreported. Indeed, in a survey from the United Kingdom, it was estimated that 12% of cases of spontaneous hypoglycemia referred for investigation were in fact probably factitious.260 In nearly all instances, the individuals had had diabetes; had been a relative, spouse, or friend of a diabetic patient; or were in the medical or paramedical profession. Thus, factitious hypoglycemia should be suspected in individuals or their relatives who have unexplained hypoglycemia and knowledge of and/or access to insulin, sulfonylureas, or meglitinides261 and unexplained hypoglycemia. Such patients have generally been healthy women younger than 50 years of age who have an underlying psychological disorder. Because the main differential diagnosis is insulinoma, it is important to consider this condition as a possible cause of unexplained hypoglycemia to avoid unnecessary laboratory workup, which such patients have undergone.
Previously, autoimmune hypoglycemia was a possibility because of the presence of insulin antibodies in patients who are taking animal or impure insulin. Currently, the use of human insulin does not generally result in antibody production, so autoimmune hypoglycemia is no longer a major consideration. Because the main differential is insulinoma, most patients with this condition should undergo a 72-hour fast. Those in whom hypoglycemia develops as a result of surreptitious insulin injection will have inappropriate plasma insulin levels and a suppressed plasma C-peptide level (because of inhibition of endogenous insulin secretion). This finding is diagnostic. Patients taking sulfonylureas or a meglitinide will have inappropriate plasma insulin and C-peptide levels because of stimulation of endogenous insulin secretion, and this pattern will mimic that seen in patients with insulinoma. However, these conditions can be distinguished by a positive urine or plasma assay for sulfonylureas and meglitinides. It is important that samples be collected during hypoglycemia or as close as possible to the event, and that they be analyzed by a laboratory with a sufficiently sensitive assay.250
Many patients who are documented biochemically to have self-induced hypoglycemia will adamantly deny doing so when confronted with the evidence. Nevertheless, psychological counseling with other physicians is warranted to prevent subsequent episodes or substitution of other potentially self-destructive behaviors.246
Non–Islet Cell Tumors
The development of hypoglycemia in a patient with a non–islet cell tumor was first reported in 1930—a mediastinal fibrosarcoma.262 Since that time, it has become apparent that a large variety of non–islet cell tumors are associated with hypoglycemia.156,230,263–285
Tumors of mesenchymal origin are the most commonly reported in Western countries.264 Such tumors generally are large and slow growing, but often malignant. About one third are retroperitoneal, one third are intra-abdominal, and one third are intrathoracic. In South Africa and Asia, hepatomas are the most common non–islet cell tumors associated with hypoglycemia.272
With one possible exception (a small cell carcinoma of the cervix),286 ectopic production of insulin has never been demonstrated convincingly in patients with this condition.280,287 Characteristically, circulating plasma insulin and C-peptide levels are suppressed. In some cases, hypoglycemia results mainly from increased glucose utilization by the tumor, and debulking by surgery or by radiation treatment can alleviate or ameliorate the hypoglycemia.264,268,279,283 In patients with rapidly growing hepatomas, hypoglycemia can occur as a terminal or near-terminal event, mainly because of inanition. However, in the great majority of cases, hypoglycemia is explained by tumor production of IGF-like molecules,270 in particular, IGF-2 and its isoforms.285 Excessive release of IGF-like molecules by the tumor can increase glucose utilization in tissues such as muscle,271,274,284 can suppress endogenous glucose production,278,284 and can reduce or overcome the secretion of counterregulatory hormones.230
Reactive Hypoglycemia
Reactive hypoglycemia refers to hypoglycemia that occurs after meals. Any condition that causes fasting hypoglycemia, for example, insulinoma,288 hypopituitarism,289 alcoholism,71,290 sulfonylurea ingestion, hypothyroidism,291 growth hormone deficiency,292 and cortisol deficiency, can also cause postprandial hypoglycemia.184,291 Nevertheless, some conditions are associated with hypoglycemia only after meal ingestion. These conditions fall into four categories: alimentary, prediabetes, idiopathic, and functional hypoglycemia.184
Alimentary
Hypoglycemia can occur 2 to 4 hours after meal ingestion in patients who have undergone gastrectomy,293,294 vagotomy and pyloroplasty,295,296 or esophageal resection,297 and in patients with altered gastric motility,298 peptic ulcer disease,299,300 or renal glycosuria.301 Repeated episodes can lead to hypoglycemia unawareness, such as occurs in insulinoma and diabetic patients, thus making interpretation of counterregulatory hormone responses difficult.302
The pathogenesis in most of these conditions involves rapid gastric emptying and absorption of glucose, both of which cause hyperglycemia and stimulation of the release of gut insulin secretagogues, resulting in excessive secretion of insulin; the biological actions of insulin to suppress endogenous glucose release and to stimulate tissue glucose uptake persist after the carbohydrate in the meal has been absorbed. This disequilibrium leads to postprandial hypoglycemia.304 Studies have implicated GLP-1 as the gut insulin secretagogue that is most likely responsible for the excessive insulin secretion observed in most of these conditions.184,297,304,305
Treatment involves prevention of rapid absorption of large amounts of carbohydrate, frequent small feedings, avoidance of large amounts of simple sugars, addition of fiber to the diet, and use of β-adrenergic antagonists, anticholinergics, and intestinal α-glucosidase inhibitors.184,301
Prediabetes
Individuals with impaired glucose tolerance306,307 characteristically have a delay in early insulin release that impairs suppression of endogenous glucose release and reduces the early efficiency of glucose uptake, which leads to hyperglycemia and late hyperinsulinemia. Because absorption of glucose is not affected,306 a disequilibrium such as that observed in patients with alimentary hypoglycemia can occur and may lead to late (3 to 5 hours) and often asymptomatic hypoglycemia during oral glucose tolerance tests (OGTTs).303,308–310 How often this disequilibrium leads to symptomatic hypoglycemia after meals is not known, but the hypoglycemia is mild, and treatment is directed at improvement in glucose tolerance, that is, weight loss in an obese patient and/or pharmacologic intervention with sulfonylureas, metformin, and α-glucosidase inhibitors, as in patients with alimentary hypoglycemia. Use of high-fiber diets, anticholinergics, doxepin, cornstarch, and chromium has been proposed but with little scientific support.311
Idiopathic
Idiopathic/functional reactive hypoglycemia is an extremely rare condition,184,298,312,313 in contrast to the number of patients evaluated who think that they have the condition. For this diagnosis to be made, patients must demonstrate arterial/capillary (not venous) hypoglycemia (<50 mg/dL, 2.8 mmol/L) after everyday meals (not OGTTs!), which is associated with symptoms of hypoglycemia that are relieved by carbohydrate ingestion and do not have any other known cause (e.g., prior gastrointestinal surgery, peptic ulcer disease, glucose intolerance, endocrine deficiency).314 Various causes such as impaired glucagon counterregulatory responses,184,315–317 excessive GLP-1 secretion,318 abnormal neuroendocrine regulation of insulin release,319 increased insulin sensitivity,320 and increased β-adrenergic sensitivity have been proposed.184 The disorder is not life threatening and is treated in the same manner as alimentary hypoglycemia.
However, it has recently become evident that some of these patients might have an adult form of nesidioblastosis321,322 (not related to mutation of the Kir6.2 and SUR1 genes323); this rare disorder, which has also been referred to as noninsulinoma pancreatogenous hypoglycemia, has a 4 : 1 male predominance and is characterized by symptomatic hypoglycemia occurring only postprandially in association with hyperinsulinemia, increased plasma C-peptide levels, and negative sulfonylurea screens.324 In this variant of adult nesidioblastosis, the 72-hour fast is usually negative, as are conventional localization tests. The condition is a diagnostic problem in that plasma insulin and C-peptide levels often are elevated only marginally during postprandial hypoglycemia. In contrast, the calcium stimulation test is positive, and there is a gradient among different sites of venous drainage from the pancreas, indicating diffuse oversecretion of insulin.325 Treatment entails partial pancreatectomy.311
Functional (Nonhypoglycemic)
It is common practice for physicians to refer patients who have symptoms suggesting hypoglycemia that occur 1 to 4 hours after meal ingestion. These symptoms generally include chronic fatigue, light-headedness, shakiness, sweating, weakness, blurred vision, blackouts, headaches, depression, anxiety, confusion, and poor concentration and memory.326 When such patients are administered a meal similar to meals that elicited the symptoms, the symptoms can be reproduced, but hypoglycemia is rarely if ever observed.327,328 Thus, in virtually all these patients, the symptoms are not due to hypoglycemia367 but rather are better explained by psychological profiles, which have indicated a wide variety of personality and psychiatric disorders, prominent among which are somatization, obsessive-compulsive behavior, depression, anxiety, and hysteria.326,330,331
Normally, blood glucose levels do not decrease below 50 mg/dL (2.8 mmol/L) during everyday life314,329; however, during OGTTs, normal volunteers can have decreases in their plasma glucose level below 30 mg/dL (1.6 mmol/L).332 In fact, blood glucose levels below 50 mg/dL (2.8 mmol/L) will develop in about 10%, and as many as 25% will have values below 60 mg/dL (3.3 mmol/L).184,332,333
Therefore, it is not surprising that “hypoglycemia” can develop during OGTTs in individuals complaining of hypoglycemic symptoms in everyday life, and this test for diabetes mellitus should never be used to assess the presence of reactive hypoglycemia. The first step in the workup of such patients is to establish Whipple’s triad during everyday life; this can be done with self-glucose monitoring that is readily available for diabetic patients. If established, the next step would be to reproduce hypoglycemia with a standardized meal. If the diagnosis is confirmed, one should exclude impaired glucose tolerance or mild diabetes, renal glycosuria, peptic ulcer disease, and other gastrointestinal disorders. In the absence of these conditions, and if the diagnosis is not confirmed, treatment might include psychological consultation and the use of β-adrenergic antagonists, as well as restriction of large carbohydrate-containing meals.301
Insulin-Producing Islet Cell Tumors and Nesidioblastosis
Nesidioblastosis, or diffuse hyperplasia of the islets, as a cause of hypoglycemia was first described by Laidlaw in 1937.325,334 In contrast to multiple adenomas that accompany the multiple endocrine neoplasia type I (MEN-I) syndrome, the condition is characterized by diffuse, although not necessarily uniform, hypertrophy and hyperplasia of islet cells (insulin- as well as glucagon- and somatostatin-producing cells), usually associated with differentiation of ductal cells into insulin-producing cells.335 This condition can cause hypoglycemia in infants as a result of mutations in the sulfonylurea receptor323 or in the anatomically linked potassium channel.323 However, it has become apparent that the condition can occur in adults independent of these genetic mutations.322,324,325,336–340
Treatment generally consists of 60% to 90% distal pancreatectomy. Fifty percent of patients undergoing this procedure are cured of hypoglycemia and are normoglycemic. Ten percent develop insulin-requiring diabetes, and the remaining 40% require medication for treatment of hyperglycemia (insulin secretagogue) or persistent hypoglycemia (calcium channel blockers).322,325
Incidence
Insulinoma is a very rare disorder, and nesidioblastosis is even rarer.322,339,340 On the basis of a 60-year experience at the Mayo Clinic, it was estimated that the incidence in Olmsted County, Minnesota, was about eight cases per million patient-years; however, half these cases were found incidentally at autopsy.341 Other estimates from Seattle, Washington342; and Auckland, New Zealand,343 which emphasize clinically apparent cases, yield an incidence of about one to two cases per million patient-years. The incidence of adult-onset nesidioblastosis would be expected to be only about 0.3 case per million patient-years.322,344,345
Demographics
Insulinoma occurs somewhat more frequently in women than in men, with an average/median onset at about 45 years of age; most cases occur between the ages of 30 and 70 years. Younger patients generally have had a greater occurrence in association with MEN-I,346 whereas carcinoma has been more frequent in older patients. However, insulinoma is uncommon in those older than 80 years. Although extraordinarily rare in patients with type 1 diabetes mellitus, insulinomas have occurred in several patients with type 2 diabetes.347–349
Pathophysiology
Hypoglycemia in patients with insulinoma is the result of dysregulated insulin release. Normally, increased plasma insulin levels and hypoglycemia per se350 suppress insulin release. In patients with insulinoma, suppression of insulin release by insulin and hypoglycemia is abnormal, and insulin release can be described as chaotic, often not appropriately increased by hyperglycemia; some patients occasionally have impaired glucose tolerance. Further evidence of abnormal function of the tumorous islets consists of the increased proinsulin-to-insulin ratios and the decreased insulin concentration relative to normal islet tissue.351–353
In most patients with insulinoma, hypoglycemia occurs as the result of suppression of glucose release rather than increased glucose utilization, inasmuch as plasma insulin concentrations are usually only 50% to threefold above normal levels (but of course inappropriate for the plasma glucose concentration).354 The frequently observed finding that large infusions of glucose are necessary to maintain normoglycemia in patients with insulinoma is probably a result of the creation of a slight hyperglycemia that can stimulate insulin release by some insulinomas. Suppression of normal β cells in islets by insulin released by the tumor, or by repetitive hypoglycemia, can result in glucose intolerance or transient diabetes after removal of the tumor.
Symptoms and Signs
Except in late-diagnosed malignant insulinoma cases in which an abdominal mass and signs of metastasis may be present, the physical examination is usually normal. Symptoms are the result of hypoglycemia. Patients with MEN-I also can exhibit symptoms as a result of hypercalcemia and accompanying hyperparathyroidism or other excessive hormone secretions (islet production of ACTH, gastrin, and vasoactive intestinal peptide).355 Common initial symptoms of patients with insulinoma are confusion, weight gain, weakness, fatigue, headaches, faintness, altered mental state, and altered behavior. Because of their nonspecific and insidious nature, the time between onset of symptoms and diagnosis is about 3 to 5 years, with the world record being 26 years.356 Initially, autonomic symptoms predominate; these are followed later by neuroglycopenic symptoms. Frequency and/or severity increases over time. Patients with insulinoma, similar to those with diabetes mellitus, can acquire the syndrome of hypoglycemia unawareness,357 which is characterized by diminished symptoms, counterregulatory hormone responses, and β-adrenergic sensitivity,358 which are reversed after successful surgical cure.357,359 Thus, it is not uncommon for a patient with insulinoma with a plasma glucose concentration less than 36 mg/dL (2 mmol/L) to be completely asymptomatic. The development of this condition can make it difficult to exclude hypopituitarism as an initial cause of the hypoglycemia.
Although insulinoma has long been classified as one of the so-called fasting hypoglycemias, it is important to remember that such patients can experience hypoglycemia at any time of day, even 2 to 4 hours after a meal (Table 21-5). Only about a quarter of patients have hypoglycemia episodes solely after an overnight fast during real-life activity, and postprandial hypoglycemia has been reported to be the sole initial feature.288 In contrast, an appreciable proportion of patients with nesidioblastosis may have symptoms only after meal ingestion.324 Seizures tend to be more common in children, but permanent neurologic sequelae have been observed in about 7% of adults.360 Patients often learn that eating frequently reduces episodes; therefore, weight gain has been observed in about 50% of patients.360
Table 21-6
Admit before the evening meal. Discontinue all nonessential medications. Insert an intravenous line for blood sampling.
Begin blood sampling (plasma glucose, insulin, C-peptide) just before the meal, and continue every 30 minutes for 6 hours and thereafter every 2 to 3 hours.
Baseline samples should also include growth hormone, cortisol, glucagon, catecholamines, IGF-1, and sulfonylureas/meglitinides.
Patient may consume calorie- and caffeine-lacking liquids and should ambulate.
The fast is ended at 72 hours or earlier if the patient has a plasma glucose level below 40 mg/dL (2.5 mmol/L) associated with symptoms. Do not end the fast for symptoms if hypoglycemia is not documented.
At the end of the fast, draw samples for all the above measurements plus an oral hypoglycemic agent screen and α-hydroxybutyrate level and an extra tube for insulin antibodies, anti-insulin receptor antibodies, and IGF-1.
Pathology
About 80% of insulinomas are due to benign single adenomas, which average about 2 cm in diameter; 10% are due to multiple benign adenomas. Diffuse hyperplasia and/or nesidioblastosis is uncommon in adults (i.e., one to two cases per 100 million patient-years) and usually accounts for only 1% to 2% of cases.361,362 Carcinomas account for about 8%. Five percent of insulinomas are associated with MEN-I, in which case the tumor is more likely to be multiple and malignant and to secrete additional hormones ectopically, such as gastrin or ACTH.
Diagnosis
The diagnosis of insulinoma is readily established by the demonstration of fasting hypoglycemia (<50 mg/dL, 2.8 mmol/L), inappropriate plasma insulin (>5 µU/mL, 30 pmol/L) and C-peptide (>0.25 nmol/L, 0.75 pg/mL), and a negative sulfonylurea/meglitinide blood/urine screen. A plasma proinsulin concentration greater than 5 pmol/L can be useful if plasma insulin and C-peptide values are borderline; additionally, measurement of a high proinsulin-to-insulin ratio can be diagnostic.363,364 Various tests have been proposed over the years to rule in or rule out the presence of an insulinoma: the intravenous tolbutamide tolerance test, the glucagon test, the leucine test, and the C-peptide suppression test.344,365–368 All suffer from frequent false positives and false negatives and are not recommended. Additionally, a 3- to 5-hour prolonged OGTT can often produce misleading results. In an insulin-sensitive individual, an oral glucose load will evoke an insulin response that will stimulate glucose uptake into peripheral muscles. This will create an arteriovenous difference, with lower glucose levels in mixed venous blood. Thus, sampling venous blood from a peripheral vein after an OGGT can produce glucose values in the hypoglycemic range that are in fact 10 to 30 mg/dL lower than simultaneous arterial glucose values. The “gold standard” remains the classic 72-hour fast. Hypoglycemia will develop in essentially all patients with insulinoma during this test; in fact, 75% will become hypoglycemic within 24 hours and up to 90% over 72 hours.369 The test should be conducted in a hospital under standardized and supervised conditions. It is worth emphasizing that many patients with nesidioblastosis will have a normal 72-hour fast, and their only biochemical abnormality will be postprandial hypoglycemia associated with abnormal plasma insulin and C-peptide levels, which is why it is preferable to begin the 72-hour fast with a standard meal.
The 72-hour Fast
Successful completion of a 72-hour fast without the development of hypoglycemia effectively excludes a serious hypoglycemic disorder except for nesidioblastosis (Table 21-6).370 It is recommended that the patient be hospitalized and supervised to prevent inadvertent caloric consumption or surreptitious drug administration. Admission is preferred before a standard evening meal so that the response to a meal can be assessed, as well as the response to a fast. The patient should be encouraged to be active to simulate real-life situations and prevent sedentary decreases in glucose utilization. Baseline samples for counterregulatory hormones are drawn to assess the adequacy of a response if hypoglycemia should occur subsequently. Blood for a β-hydroxybutyrate level is drawn at the end of the fast to exclude the consumption of calories. Should hypoglycemia occur, anti-insulin antibodies, anti-insulin receptor antibodies, and IGF-2 levels may be useful in assessing the presence of an autoimmune cause or an occult IGF-2–secreting non–islet cell tumor. Interpretation of the results of these determinations is given in Table 21-7.
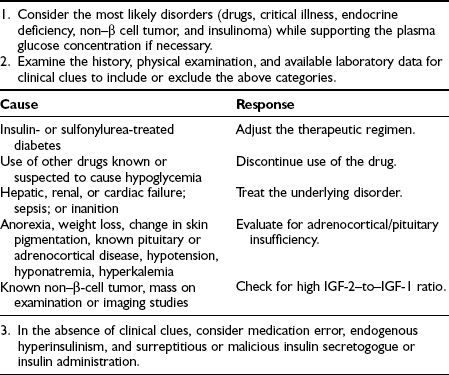
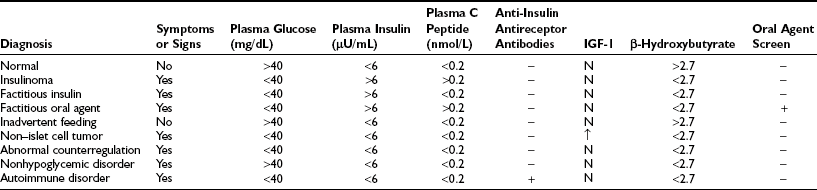
Localization
Only after the diagnosis of hypoglycemia associated with inappropriate hyperinsulinemia has been established biochemically should localization studies be performed. Ninety-nine percent of insulinomas occur within the pancreas. Of the rare insulinomas that are not found in the pancreas, most are found in the wall of the duodenum or gastrosplenic omentum.371 They average about 2 cm in diameter and appear with equal frequency in the head, body, and tail.360,372 Although about 75% to 90% can be identified correctly by palpation at surgery,373,374 preoperative localization generally is recommended to minimize manipulation of the pancreas, shorten operative time, avoid blind partial pancreatectomy when the tumor is not palpable, and assist in reoperations when scarring and fibrosis from prior surgery make palpation difficult.344 Computed tomography and NMR imaging can detect large tumors and stage malignant ones but yield false positives and false negatives; these techniques correctly localize tumors only 50% to 70% of the time.344,373–375 Celiac arteriography and transsplenic portal venous sampling have had variable results and are invasive and probably not needed.344,374,375 Currently, preoperative transabdominal ultrasonography followed by intraoperative ultrasonography is considered the most sensitive and specific approach and has been recommended for routine use; this approach along with palpation can be used to detect over 95% of tumors.373,375–377 Recently, dual-phase spiral computed tomography has been reported to detect six of seven tumors ranging in size from 6 to 18 mm.378 In patients who are suspected of having nesidioblastosis, the tests of choice are 18F-DOPA PET scans and the selective calcium infusion procedure,339,375 because this test will confirm the diagnosis of pancreatic hyperinsulinism as the probable cause of the hypoglycemia when reliable localization procedures are negative.322
Treatment
Surgery is the treatment of choice for insulinoma and nesidioblastosis.322,360,373 For patients with solitary adenomas, enucleation is curative; in cases that are thought to be due to a single adenoma, recurrence or lack of cure could be due to the presence of multiple adenomas. In 5% to 10% of patients, the adenoma is not found; nevertheless, in such cases, partial pancreatectomy can result in cure. Reoperation still might result in cure, but recurrence has been noted up to 18 years after the initial surgery.341 Recurrences are more common in patients with MEN-I (up to 20%).341 Multiple adenomas, hyperplasia, and malignancy require more extensive surgery. Debulking of a malignant tumor is worthwhile in helping to control hypoglycemia. The major complications of surgery, which include acute pancreatitis (≈10% to 15%), wound infection (5% to 10%), fistulas (10% to 15%), and pseudocysts (5%), are related to the extent of surgery. Reoperations have greater complication and mortality rates (≈15% vs. 5%). Commonly, transient hyperglycemia occurs and lasts up to 2 to 3 weeks because of suppression of normal islet function. Permanent diabetes mellitus can occur after partial pancreatectomy and reoperation. In patients with nesidioblastosis, gradient-directed resection can result in cure of hypoglycemia.
Medical therapy is reserved for operated patients with recurrence who refuse another exploration and for those with inoperable malignant tumors. Diazoxide (100 to 200 mg, three times daily), which inhibits insulin secretion, has been used most widely.379 Approximately 60% (mainly those with benign disease) can be maintained nearly free of symptoms with only occasional hypoglycemia. The main side effects are fluid retention (≈15%) and hirsutism (≈5%); thiazide diuretics can be used to combat fluid retention and enhance hyperglycemic effects. The somatostatin analogue octreotide has been found to be effective in some patients but must be injected.380,381 Continuous subcutaneous glucagon infusion has also been used.382 Glucocorticoids (to induce insulin resistance) and verapamil, the calcium channel blocker, and phenytoin, the anticonvulsant, both of which inhibit insulin release at high doses, have been used when other measures were ineffective.344
Malignant insulinomas respond poorly to chemotherapy.383 Streptozocin has been reported to reduce tumor size in about 50% of patients, with fewer than 20% achieving complete remission; although its use prolongs life, streptozocin has considerable renal, hepatic, and hematopoietic toxicity. The addition of fluorouracil has been reported to have advantages over streptozocin alone.384 Mithramycin,385 doxorubicin (Adriamycin),386 and hepatic embolization387 have been tried with some success in refractory cases.
General Approach to the Patient
The causes, clinical features, consequences, and need for immediate treatment of hypoglycemia will vary, depending on whether the patient to be evaluated is seen in the clinic, emergency room, or hospital ward. In two series,388,389 diabetes, alcohol, sepsis, and combinations thereof accounted for about 90% of emergency room cases. Virtually all patients had stupor, coma, confusion, or bizarre behavior or were postictal. About 10% were hypothermic. Death occurred in 10%, and 3% had permanent neurologic sequelae. Among diabetic patients, about 80% were taking insulin; strenuous exercise (7%), accidental (6%) or deliberate insulin overdose (13%), skipped meals (28%), and alcohol ingestion (19%) were identified as precipitating factors. In 27% of cases, no immediate cause was identified. Some patients were also taking other agents with hypoglycemic potential or had chronic renal failure, psychiatric disorders, and previous emergency room visits for hypoglycemia. In the emergency room, prompt treatment (usually with intravenous glucose) is more of a concern than an etiologic diagnosis.
In hospitalized patients, the incidence of hypoglycemia ranges from 0.5% in elderly nondiabetic patients8 to 28% in diabetic patients390; about 1.5% were found in studies that included diabetic and nondiabetic patients.6,7 Although hypoglycemia is rarely the direct cause of death, mortality in hospitalized patients who are experiencing severe hypoglycemia (excluding those with obvious insulin reactions) has ranged from 22% to 48%.6–9 Renal insufficiency, diabetes mellitus, malnutrition, liver disease, infection, and malignancy were the most common risk factors or contributing conditions. Most patients had multiple risk factors. Episodes were frequently repetitive, and a great majority were asymptomatic because they occurred in patients with reduced sensorium. Among diabetic patients, decreased caloric intake (missed meals, vomiting, withheld enteral feedings, meals withheld for procedures) and inappropriate insulin doses accounted for nearly 90% of occurrences.
The physical examination can be useful in excluding carotid disease, orthostatic hypotension, severe cardiac and hepatic disease, hypothyroidism, cachexia, and cancer, which should be obvious at the stage at which they would cause hypoglycemia. Routine laboratory testing can exclude renal insufficiency or can raise the possibility of adrenal insufficiency (hyponatremia). If all of the above are negative and prior hypoglycemia has not been documented unequivocally, this must be done before one proceeds with other diagnostic tests. Table 21-7 gives a suggested approach.193
Treatment and Strategies to Reduce Hypoglycemia
Treatment is aimed at restoring euglycemia, preventing recurrences, and, if possible, alleviating the underlying cause. In an insulin-taking diabetic patient with mild hypoglycemia because of a skipped meal, treatment can simply entail 15 g of oral carbohydrate every 15 minutes until the blood glucose level is above 80.391,392 With more severe hypoglycemia resulting in obtundation, and when oral administration of carbohydrate might result in aspiration, 1 mg of glucagon given subcutaneously or intramuscularly might be sufficient to raise the blood glucose concentration and revive the patient so that oral carbohydrate can be given. Comatose patients should receive intravenous glucose (a 25 g bolus, followed by an infusion to maintain glucose at around 4.3 to 5.5 mmol/L [80 to 100 mg/dL]). A sulfonylurea or meglitinide overdose can result in prolonged hypoglycemia requiring sustained intravenous glucose infusion, which also is aimed at keeping the blood glucose level at approximately 4.5 to 5.5 mmol/L (≈80 mg/dL) to avoid hyperglycemia, causing further stimulation of insulin secretion and setting in motion a vicious cycle. Blood glucose levels should be monitored initially every 30 minutes and subsequently at 1- to 2-hour intervals. Occasionally, diazoxide or a somatostatin analogue might be needed to inhibit insulin secretion.393 When other drugs are involved, their use should be discontinued if possible (e.g., sulfonamides in a patient with renal insufficiency). In other conditions, the underlying disorder (e.g., sepsis, heart failure, endocrine deficiency) should be treated, and the blood glucose level should be supported.
References
1. Cryer, PE, Shamoon, H, Davis, SN, et al. Defining and reporting hypoglycemia in diabetes. Diabetes Care. 2005;28:1245–1249.
2. Deckert, T, Poulsen, JE, Larsen, M. Prognosis of diabetes with diabetes before the age of 31. Survival, casue of deaths and complications. Diabetologia. 1978;14:363–370.
3. Laing, SP, Swerdlow, A, Slater, S, et al. The British Diabetic Association Cohort Study 1: all cause mortality in patients with insulin treated diabetes mellitus. Diabetic Medicine. 1999;16:459–465.
4. Klatt, E, Beatie, C, Noguchi, T. Evaluation of death from hypoglycemia. Am J Forensic Med Pathol. 1988;9:122–125.
5. Huic, M, Mucolic, V, Vrhovac, B, et al. Adverse drug reactions resulting in hospital admission. Int J Clin Pharmacol Ther. 1994;32:675–682.
6. Fischer, K, Lees, J, Newman, J. Hypoglycemia in hospitalized patients: causes and outcomes. N Engl J Med. 1986;315:1245–1250.
7. Stagnaro-Green, A, Barton, M, Linekin, P, et al. Mortality in hospitalized patients with hypoglycemia and severe hyperglycemia. Mt Sinai J Med. 1995;62:422–426.
8. Shilo, S, Berezovsky, S, Friedlander, Y, et al. Hypoglycemia in hospitalized nondiabetic older patients. J Am Geriatr Soc. 1998;46:978–982.
9. Cruz Jentoft, A, Villar, I, Carreras, P, et al. Unexpected hypoglycemia in hospitalized patients [in Spanish]. Rev Clin Esp. 1992;191:295–298.
10. Porte, D, Jr., Baskin, D, Schwartz, M. Insulin signalling in the central nervous system: a critical role in metabolic homeostasis and disease from C elegans to humans. Diabetes. 2005;54:1264–1276.
11. Obici, S, Zhang, B, Karkanias, G, et al. Hypothalamic insulin signaling is required for inhibition of glucose production. Nat Med. 2002;8:1376–1382.
12. Davis, SN, Colburn, C, Dobbins, R, et al. Evidence that the brain of the conscious dog is insulin sensitive. J Clin Invest. 1995;95:593–602.
13. Fisher, SJ, Bruning, JC, Lannon, S, et al. Insulin signaling in the central nervous sytem is critical for the normal sympathoadrenal response to hypoglcyemia. Diabetes. 2005;54:1441–1451.
14. Veneman, T, Mitrakou, A, Mokan, M, et al. Effect of hyperketonemia and hyperlacticacidemia on symptoms, cognitive function, and counterregulatory hormone responses during hypoglycemia in normal humans. Diabetes. 1994;43:1311–1317.
15. Oz, G, Seaquist, E, Kumar, A, et al. Human brain glycogen content and metabolism: implications on its role in brain energy metabolism. Am J Physiol Endo. 2007;292:E946–E951.
16. Wahren, J, Ekberg, K, Fernquist-Forbes, E, et al. Brain substrate utilisation during acute hypoglycaemia. Diabetologia. 1999;42:812–818.
17. The Diabetes Control and Complications Trial Research Group. Hypoglycemia in the diabetes control and complications trial. Diabetes. 1997;46:271–286.
18. Ryan, E, Lakey, J, Rajotte, R, et al. Clinical outcomes and insulin secretion after islet transplantation with the Edmonton protocol. Diabetes. 2001;50:710–719.
19. Veneman, T, Mitrakou, A, Mokan, M, et al. Induction of hypoglycemia unawareness by asymptomatic nocturnal hypoglycemia. Diabetes. 1993;42:1233–1237.
20. Mitrakou, A, Ryan, C, Veneman, T, et al. Hierarchy of glycemic thresholds for counterregulatory hormone secretion, symptoms, and cerebral dysfunction. Am J Physiol. 1991;260:E67–E74.
21. Schwartz, N, Clutter, W, Shah, S, et al. The glycemic thresholds for activation of glucose counterregulatory systems are higher than the threshold for symptoms. J Clin Invest. 1987;79:777–781.
22. Hepburn, D, Deary, I, Frier, B, et al. Symptoms of acute insulin-induced hypoglycemia in humans with and without IDDM: factor-analysis approach. Diabetes Care. 1991;14:949–957.
23. Pladziewicz, D, Nesto, R. Hypoglycemia-induced silent myocardial ischemia. Am J Cardiol. 1989;63:1531–1532.
24. Duh, E, Feinglos, M. Hypoglycemia-induced angina pectoris in a patient with diabetes mellitus. Ann Intern Med. 1994;121:945–946.
25. Patrick, A, Campbell, I. Fatal hypoglycaemia in insulin-treated diabetes mellitus: clinical features and neuropathological changes. Diabetic Med. 1990;7:349–354.
26. Cryer, PE, Glucose homeostasis and hypoglycemia in Williams Textbook of Endocrinology. Kronenberg, HM, Melmed, S, Polonsky, K, Larsen, P, eds., ed 11. Philadephia: Saunders, an imprint of Elsevier, Inc., 2008:1503–1533.
27. Cherrington, AD. Control of glucose production by in-vivo insulin and glucagon. In: Jefferson LS, Cherrington AD, eds. Handbook of Physiology, Section 7. The Endocrine System, Volume II. The Endocrine Pancreas and Regulation of Metabolism. New York: Oxford University Press; 2001:759–785.
28. Raju, B, Cryer, PE. Loss of the decrement in intraislet insulin plasibly explains loss of the glucagon response to hypoglycemia in insulin-deficient diabetes. Diabetes. 2005;54:757–764.
29. Cersosimo, E, Garlick, P, Ferretti, J. Renal glucose production during insulin-induced hypoglycemia in humans. Diabetes. 1999;48:261–266.
30. Defeo, P, Perriello, A, DeCosmo, S, et al. Comparison of glucose counterregulation during short term and prolonged hypoglycemia in normal humans. Diabetes. 1986;35:563–569.
31. Chu, CA, Sindelar, D, Igawa, K, et al. The direct effects of catecholamines on hepatic glucose production occur via alpha (1) and beta (2) receptors in the dog. Am J Physiol Endocrinol Metab. 2000;279:E463–E473.
32. Defeo, P, Perriello, G, Torlone, E, et al. Demonstration of a role for growth hormone in glucose counterregulation. Am J Physiol Endocrinol Metab. 1989;256:E835–E843.
33. UK Hypoglycaemia Study Group. Risk of hypoglycaemia in types 1 and 2 diabetes: effects of treatment modalities and their duration. Diabetologia. 2007;50:1140–1147.
34. Riddle, MC, Rosenstock, J, Gerich, J. The Insulin Glargine 4002 Study Investigators. The Treat to Target Trial. Diabetes Care. 2003;26:3080–3086.
35. Duckworth, W, Abraira, C, Moritz, T, et al. Glucose control and vascular complications in veterans with type 2 diabetes. N Engl J Med. 2009;360:129–139.
36. The Action to Control Cardiovascular Risk in Diabetes Study Group. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med. 2008;258:2545–2559.
37. Cryer, PE. Hypoglycemia, functional brain failure and brain death. J Clin Invest. 2007;117:868–870.
38. Cryer, PE. Hypoglycemia: the limiting factor in the glycaemic management of type I and type II diabetes. Diabetologia. 2002;45:937–948.
39. Ertl, A, Davis, SN. Evidence for a vicious cycle of exercise and hypoglcyemia in type 1 diabetes mellitus. Diabetes Metab Res Rev. 2004;20:124–130.
40. Defeo P, Perriello G, Torlone E, et al: Contribution of adrenergic mechanisms to glucose counterregulation in humans, Am J Physiol 261:E725–E736, 1991.
41. Amiel, SA, Tamborlane, W, Simonson, D, et al. Defective glucose counterregulation after strict glycemic control of insulin dependent diabetes mellitus. N Engl J Med. 1987;316:1376–1383.
42. Dagogo-Jack, S, Craft, S, Cryer, PE. Hypoglycemia associated autonomic failure in insulin-dependent diabetes mellitus. J Clin Invest. 1993;91:819–828.
43. Cryer, PE. Diverse causes of hypoglycemia-associated autonomic failure in diabetes. N Engl J Med. 2004;350:2272–2279.
44. Davis, SN, Shaver, C, Costa, F, et al. Role of cortisol in the pathogenesis of deficient counterregulation after antecedent hypoglycemia in normal humans. J Clin Invest. 1996;96:680–691.
45. Mitrakou, A, Fanelli, C, Veneman, T, et al. Reversibility of unawareness of hypoglycemia in patients with insulinomas. N Engl J Med. 1993;329:834–839.
46. Cranston, I, Lomas, J, Maran, A. Restoration of hypoglycemia awareness in patients with long duration insulin dependent diabetes. Lancet. 1994;344:283–287.
47. Fanelli, C, Pampanelli, S, Epitano, L, et al. Long term recovery from unawareness, deficient counterregulation and lack of cognitive dysfunction during hypoglycemia following institution of rational, intensive therapy in IDDM. Diabetologia. 1994;37:1265–1276.
48. Dagogo-Jack, S, Rattarasarn, C, Cryer, PE. Reversal of hypoglycemia unawareness but not defective glucose counterregulation in IDDM. Diabetes. 1994;43:1426–1434.
49. Segel, SA, Paramore, D, Cryer, PE. Hypoglycemia associated autonomic failure in advanced type 2 diabetes. Diabetes. 2002;51:724–733.
50. Davis, SN, Mann, S, Briscoe, V, et al. The effects of intensive therapy and antecedent hypoglcyemia on counterregulatory responses to hypoglcyemia in type 2 DM. Diabetes. 2009;58:701–709.
51. Davis, SN, Shavers, C, Mosqueda-Garcia, R, et al. Effects of differing antecedent hypoglcyemia on subsequent counterregulation in normal man. Diabetes. 1997;46:1328–1335.
52. Davis, SN, Tate, D. Effects of morning hypoglcyemia on neuroendocrine and metabolic responses to subsequent afternoon hypoglycemia. J Clin Endocrinol Metab. 2001;86:2043–2050.
53. Boyle, P, Kempers, S, O’Connor, A, et al. Brain glucose uptake and unawareness of hypoglcyemia in patients with insulin dependent diabetes mellitus. N Engl J Med. 1995;333:1726–1731.
54. Fanelli, C, Dence, C, Markham, J, et al. Blood to brain glucose transport and cerebral glucose metabolism are not reduced in poorly controlled type 1 diabetes. Diabetes. 1998;47:1444–1450.
55. McCrimmon, R, Song, Z, Cheng, M, et al. Corticotrophin releasing factor receptors within the ventromedial hypothalamus regulate hypoglcyemia induced hormonal counterregualtion. J Clin Invest. 2006;118:1723–1730.
56. McCrimmon, R. The mechanisms that underlie glucose sensing during hypoglycaemia in humans. Diabet Med. 2008;25:513–522.
57. Sandoval, D, Gong, B, Davis, SN. Forebrain and hindbrain effects of alcohol on counterregulatory responses to hypoglycemia in conscious rats. Metabolism. 2007;12:1623–1628.
58. Davis, SN, Fowler, S, Costa, F. Hypoglycemic counterregulatory responses differ between men and women with type 1 diabetes. Diabetes. 2000;49:65–72.
59. Cryer, PE. Mechanisms of sympathoadrenal failure and hypoglycemia in diabetes. J Clin Invest. 2006;116:1470–1473.
60. Galassetti, P, Mann, S, Tate, D, et al. Effects of antecedent prolonged exercise on subsequent counterregulatory responses to hypoglycemia. Am J Physiol Endocrinol Metab. 2001;280:E908–E917.
61. Sandoval, D, Ertl, A, Richardson, A, et al. Estrogen blunts neuroendocrine and metabolic responses to hypoglycemia. Diabetes. 2003;52:1749–1755.
62. Davis, SN, Shavers, C, Costa, F. Gender-related differences in counterregulatory responses to antecedent hypoglycemia in normal humans. J Clin Endocrinol Metab. 2000;85:2148–2157.
63. Aftab-Guy, D, Sandoval, D, Richardson, A, et al. Differing physiologic effects of epinephrine in type 1 DM and healthy subjects. American Journal of Physiology Endo-Metab. 2005;288:E178–E186.
64. Fritsche, A, Stefan, N, Haring, M, et al. Avoidance of hypoglycemia restores hypoglycemia awareness by increasing β-adrenergic sensitivity in type 1 diabetes. Ann Intern Med. 2001;134:729–736.
65. McCrimmon, R, Shaw, M, Fan, X, et al. Key role for AMP-activated protein kinase in regulating counterregulatory hormone responses to acute hypoglycemia. Diabetes. 2008;57:444.
66. Wiethrop, B, Cryer, P. Glycemic actions of alanine and terbutaline in IDDM. Diabetes Care. 1993;16:1124–1130.
67. Gabriely, I, Wosniak, R, Hawkins, M, et al. Troglitazone amplifies counterregulatory responses to hypoglycemia in nondiabetic subjects. J Clin Endocrinol Metab. 2001;86:521–528.
68. Briscoe, V, Ertl, A, Tate, D, et al. Effects of selective serotonin reuptake inhibitors on counterregulatory responses to hypoglycemia in healthy man. Diabetes. 2008;57:2453–2460.
69. Seltzer, H. Drug-induced hypoglycemia: a review of 1418 cases. Endocrinol Metab Clin North Am. 1989;18:163–183.
70. Arky, R, Freinkel, N. Alcohol infusion to test gluconeogenesis in starvation with special reference to obesity. N Engl J Med. 1966;274:426–433.
71. Flanagan, D, Wood, P, Sherwin, R, et al. Gin and tonic and reactive hypoglycemia: what is important—the gin, the tonic, or both? J Clin Endocrinol Metab. 1998;83:796–800.
72. Arky, R. Hypoglycemia associated with liver disease and ethanol. Endocrinol Metab Clin North Am. 1989;18:75–90.
73. Madison, L, Ethanol-induced hypoglycemia. Advances in Metabolic Disorders. Levine, R, Luft, R, eds. Advances in Metabolic Disorders, vol 3. New York: Academic Press, 1968:85–109.
74. Klatt, E, Beatie, C, Noguchi, T. Evaluation of death from hypoglycemia. Am J Forensic Med Pathol. 1988;9:122–125.
75. Cryer, PE. Catecholamines, pheochromocytoma and diabetes. Diabetes Reviews. 1993;1:309–317.
76. Marks, V, Teale, J. Drug-induced hypoglycemia. Endocrinol Metab Clin North Am. 1999;28:555–577.
77. Siler, S, Neese, R, Christiansen, M, et al. The inhibition of gluconeogenesis following alcohol in humans. Am J Physiol. 1998;275:E897–E907.
78. Krebs, H, Freedland, R, Stubbs, M. Inhibition of hepatic gluconeogenesis by ethanol. Biochem J. 1969;112:117–124.
79. Kreisberg, R, Owen, W, Siegal, A. Ethanol-induced hyperlacticacidemia: inhibition of lactate utilization. J Clin Invest. 1971;50:166–174.
80. Kreisberg, R, Siegal, A, Owen, W. Alanine and gluconeogenesis in man: effect of ethanol. J Clin Endocrinol Metab. 1972;34:876–883.
81. Lecavalier, L, Bolli, G, Cryer, P, et al. Contributions of gluconeogenesis and glycogenolysis during glucose counterregulation in normal humans. Am J Physiol. 1989;256:E844–E851.
82. Chan, J, Cockram, C, Critchley, J. Drug-induced disorders of glucose metabolism: mechanisms and management. Drug Saf. 1996;15:135–157.
83. Reith, D, Dawson, A, Epid, D, et al. Relative toxicity of beta blockers in overdose. Clin Toxicol. 1996;34:273–278.
84. Poretsky, L, Moses, A. Hypoglycemia associated with trimethoprim/sulfamethoxazole therapy. Diabetes Care. 1984;7:508–509.
85. Fanelli, C, Calderone, S, Epifano, L, et al. Demonstration of critical role for FFA in mediating counterregulatory stimulation of gluconeogenesis and suppression of glucose utilization in man. J Clin Invest. 1993;92:1617–1622.
86. Fanelli, C, DeFeo, P, Perriello, G, et al. Adrenergic mechanisms contribute to the late phase of glucose counterregulation in humans by stimulating lipolysis. J Clin Invest. 1992;89:2005–2013.
87. Rizza, R, Cryer, P, Haymond, M, et al. Adrenergic mechanisms for the effect of epinephrine on glucose production and clearance in man. J Clin Invest. 1980;65:682–689.
88. Sawicki, PT, Siebenhofer, A. Betablocker treatment in diabetes mellitus. J Intern Med. 2001;250:11–17.
89. Washio, M, Onoyama, K, Makita, Y, et al. Hypoglycemia associated with the administration of angiotensin-converting enzyme inhibitor in a diabetic hemodialysis patient. Nephron. 1991;59:341–342.
90. Perronne, C, Bricaire, F, Leport, C, et al. Hypoglycaemia and diabetes mellitus following parenteral pentamidine mesylate treatment in AIDS patients. Diabet Med. 1990;7:585–589.
91. Pollare, T, Lithell, H, Berne, C. A comparison of the effects of hydrochlorothiazide and captopril on glucose and lipid metabolism in patients with hypertension. N Engl J Med. 1989;321:868–873.
92. Jauch, K, Hartl, W, Georgieff, M, et al. Low dose bradykinin infusion reduces endogenous glucose production in surgical patients. Metabolism. 1988;37:185–190.
93. Bouchard, P, Sai, P, Reach, G, et al. Diabetes mellitus following pentamidine-induced hypoglycemia in humans. Diabetes. 1982;31:40–45.
94. Shalitin, S, Phillip, M. Hypoglycemia in type 1 diabetes. Diabetes Care. 2008;31(Suppl 2):S121–S124.
95. Juvenile Diabetes Research Foundation Glucose Monitoring Study Group. Continuous glucose monitoring and intensive treatment of type 1 diabetes. N Engl J Med. 2008;359:1464–1476.
96. Rolla, A. Pharmacokinetic and pharmacodynamic advantages of insulin analogues and premixed insulin analogues over human insulins: impact on efficacy and safety. Am J Med. 2008;121(6 Suppl):S9–S19.
97. Redmon, JB, Teuscher, A, Robertson, RP. Hypoglycemia after pancreas transplantation. Diabetes Care. 1998;21:1944–1950.
98. Robertson, RP. Pancreas and islet transplants for patients with diabetes: taking positions and making decisions. Endocrine Practice. 1999;5:24–28.
99. Diem, P, Redmon, JB, Abid, M, et al. Glucagon, catecholamine and pancreatic polypeptide secretion in type 1 diabetic recipients of pancreas allografts. J Clin Invest. 1990;86:2008–2013.
100. Paty, B, Lanz, K, Kendall, D, et al. Restored hyopglycemic counterregulation is stable in successful pancreas transplant recipients for up to 19 years after transplantation. Transplantation. 2001;72:1103–1107.
101. Redmon, J, Kubo, S, Robertson, RP. Glucose, insulin and glucagon levels during exercise in pancreas transplant recipients. Diabetes Care. 1995;4:457–462.
102. Robertson, RP, Sutherland, D, Seaquist, E, et al. Glucagon, catecholamine and symptom responses to hypoglycemia in living donors of pancreas segments. Diabetes. 2003;52:1689–1694.
103. Keymeulen, B, Gillard, P, Mathieu, C, et al. Correlation between β-cell mass and glycemic control in type 1 diabetic recipients of islet cell graft. PNAs. 2006;103:17444–17449.
104. O’Connell, P, Hawthorne, W, Holmes-Walker, DJ, et al. Clinical islet transplantation in type 1 diabetes mellitus: results of Australia’s first trial. MJA. 2006;184:221–225.
105. Rickels, M, Schutts, M, Mueller, R, et al. Islet cell hormonal responses to hypoglycemia after human islet transplantation. Diabetes. 2005;54:3205–3211.
106. Zhou, H, Zhang, T, Bogdani, M, et al. Intrahepatic glucose flux as a mechanism for defective intrahepatic islet α-cell response to hypoglycemia. Diabetes. 2008;57:1567–1574.
107. Gupta, V, Wahoff, D, Rooney, D, et al. The defective glucagon response from transplanted intrahepatic pancreatic islets during hypoglycemia is transplantation site-determined. Diabetes. 1997;46:28–33.
108. Service, G, Thompson, G, Service, J, et al. Hyperinsulinemic hypoglycemia with nesidioblastosis after gastric-bypass surgery. N Engl J Med. 2005;353:249–254.
109. Patti, M, McMahon, G, Mun, E, et al. Severe hypoglycemia post gastric bypass requiring partial pancreatectomy: evidence for inapprorpriate insulin secretion and pancreatic iselt hyperplasia. Diabetologia. 2005;48:2236–2240.
110. Z’graggen, K, Guweidhi, A, Steffen, R, et al. Severe recurent hyopglycemia after gastric bypass surgery. Obes Surg. 2008;18:981–988.
111. Meier, J, Butler, A, Galasso, B, et al. Hyperinsulinemic hypoglycemia after gastric bypass surgery is not accompanied by islet hyperplasia or increased β-cell turnover. Diabetes Care. 2006;29:1554–1559.
112. Vella, A, Service, FJ. Incretin hypersecretion in post gastric bypass hypoglycemia: primary problem or red herring? J Clin Endocrinol Metab. 2007;92:4563–4565.
113. Goldfine, A, Mun, E, Devine, E, et al. Patients with neuroglycopenia after gastric bypass surgery have exaggerated incretin and insulin secretory responses to a mixed meal. J Clin Endocrinol Metab. 2007;92:4678–4685.
114. Dixon, J, O’Brien, P, Playfair, J, et al. Adjustable gastric banding and conventional therapy for type 2 diabetes: a randomized controlled trial. JAMA. 2008;299:316–323.
115. De Leon, DD, Stanley, C. Mechanisms of disease: advances in diagnosis and treatment of hyperinsulinism in neonates. Nature Clinical Practice Endocrinology and. Metabolism. 2007;9:57–68.
116. Otonkoski, T, Ammälä, C, Huopio, H, et al. A point mutation inactivating the sulfonylurea receptor causes the severe form of persistent hyperinsulinemic hypoglycemia of infancy in Finland. Diabetes. 1999;48:408–415.
117. Thomas, PM, Cote, GJ, Wohllk, N, et al. Mutations in the sulfonylurea receptor gene in familial persistent hyperinsulinemic hypoglycemia of infancy. Science. 1995;268:426–429.
118. Palladino, A, Bennett, M, Stanley, C. Hyperinsulinism in infancy and childhood: when insulin level is not always enough. Clin Chemistry. 2008;54:256–263.
119. Hardy, O, Hernandez-Pampaloni, M, Saffer, J, et al. Diagnosis and localization of focal congenital hyperinsulinism by 18F-fluorodopa PET Scan. J Pediatr. 2007;150:140–145.
120. Kompare, M, Rizzo, W. Mitrochondrial fatty-acid oxidation disorders. Semin Pediatr Neurol. 2008;15:140–149.
121. Angelini, C, Trevisan, G, Isaya, G, et al. Clinical varieties of carnitine and carnitine palmitoyl transferase deficiency. Clin Biochem. 1987;20:1–7.
122. Bonnefont, J-P, Djoundi, F, Prip-Buuo, C, et al. Carnitine palmitoyltranserases 1 and 2: biochemical, molecular and medical aspects. Molecular Aspects of Medicine. 2004;25:495–520.
123. Heller, S, Worona, L, Consuelo, A. Nutritional therapy for glycogen storage diseases. Journal of Pediatric Gastroenterology and Nutrition. 2008;47:S15–S21.
124. Lang, C, Bagby, G, Blakesley, H, et al. Importance of hyperglucagonemia in eliciting the sepsis-induced increase in glucose production. Circ Shock. 1989;29:181–191.
125. Hargrove, D, Bagby, G, Lang, C, et al. Adrenergic blockade does not abolish elevated glucose turnover during bacterial infection. Am J Physiol. 1988;254:E16–E22.
126. Fettman, M. Endotoxemia in Yucatan miniature pigs: Metabolic derangements and experimental therapies. Lab Anim Sci. 1986;36:370–374.
127. Wichterman, K, Chaudry, I, Baue, A. Studies of peripheral glucose uptake during sepsis. Arch Surg. 1979;114:740–745.
128. Meszaros, K, Lang, C, Bagby, G, et al. In vivo glucose utilization by individual tissues during nonlethal hypermetabolic sepsis. FASEB J. 1988;2:3083–3086.
129. Romanosky, A, Bagby, G, Bockman, E, et al. Increased muscle glucose uptake and lactate release after endotoxin administration. Am J Physiol. 1980;239:E311–E316.
130. Durkot, M, Wolfe, R. Effects of adrenergic blockade on glucose kinetics in septic and burned guinea pigs. Am J Physiol. 1981;241:R222–R227.
131. Wannemacher, R, Pace, J, Beall, F, et al. Role of the liver in regulation of ketone body production during sepsis. J Clin Invest. 1979;64:1565–1572.
132. Wannemacher, R, Beall, F, Canonico, P, et al. Glucose and alanine metabolism during bacterial infections in rats and rhesus monkeys. Metabolism. 1980;29:201–212.
133. Alderfer, H, Richardson, J. Hepatic hypoglycemia and infarction of the bowel. Arch Intern Med. 1963;112:50–55.
134. Medalle, R, Webb, R, Waterhouse, C. Lactic acidosis and associated hypoglycemia. Arch Intern Med. 1971;128:273–278.
135. Fuchs, S, Bogomolski-Yahalom, V, Paltiel, O, et al. Ischemic hepatitis. J Clin Gastroenterol. 1998;26:183–186.
136. Hedayati, H, Beheshti, M. Profound spontaneous hypoglycaemia in congestive heart failure. Curr Med Res Opin. 1977;4:501–504.
137. Mellinkoff, S, Tumulty, P. Hepatic hypoglycemia: its occurrence in congestive heart failure. N Engl J Med. 1952;247:745–750.
138. Marks, V, Rose, F. Hypoglycemia, vol 2. Oxford, UK: Blackwell Scientific, 1981.
139. Medalle, R, Webb, R, Waterhouse, C. Lactic acidosis and associated hypoglycemia. Arch Intern Med. 1952;128:273–278.
140. Seltzer, H. Severe drug-induced hypoglycemia: a review. Compr Ther. 1979;5:21–29.
141. Miller, S, Wallace, R, Musher, D, et al. Hypoglycemia as a manifestation of sepsis. Am J Med. 1980;68:649–654.
142. Arem, R. Hypoglycemia associated with renal failure. Endocrinol Metab Clin North Am. 1989;18:103–121.
143. Owen, O, Felig, P, Morgan, A, et al. Liver and kidney metabolism during prolonged starvation. J Clin Invest. 1969;48:574–583.
144. Woerle, HJ, Meyer, C, Popa, E, et al. Renal compensation for impaired hepatic glucose release during hypoglycemia in type 2 diabetes: further evidence for hepatorenal reciprocity. Diabetes. 2003;52:1386–1392.
145. Reinecke, R. The kidney as a source of glucose in the eviscerated rat. Am J Physiol. 1943;140:276–285.
146. Lupianez, J, Faus, M, Munoz-Clares, R, et al. Stimulation of rat kidney gluconeogenic ability by inhibition of liver gluconeogenesis. FEBS Lett. 1976;61:277–281.
147. Gerich, J. Hepatorenal glucose reciprocity in physiologic and pathologic conditions. Diab Nutr Metab. 2002;15:298–302.
148. Battezzati, A, Fattorini, A, Caumo, A, et al. Non-hepatic glucose production in humans. Diabetes. 1999;48(Suppl 1):A49.
149. Mann, F. Effects of complete and partial removal of the liver. Medicine (Baltimore). 1927;6:419–467.
150. Zimmerman, H, Thomas, L, Scherr, E. Fasting blood sugar in hepatic disease with reference to infrequency of hypoglycemia. Arch Intern Med. 1953;91:577–584.
151. Felig, P, Brown, W, Levine, R, et al. Glucose homeostasis in viral hepatitis. N Engl J Med. 1970;283:1436–1440.
152. Heinig, R, Clarke, E, Waterhouse, C. Lactic acidosis and liver disease. Arch Intern Med. 1979;139:1229–1232.
153. Nouel, O, Bernuau, J, Rueff, B, et al. Hypoglycemia: a common complication of septicemia in cirrhosis. Arch Intern Med. 1981;141:1477–1478.
154. Younus, S, Soterakis, J, Sossi, A, et al. Hypoglycemia secondary to metastases to the liver. Gastroenterology. 1977;72:334–337.
155. Johnson, D, Alberti, K, Faber, O, et al. Hyperinsulinism of hepatic cirrhosis: diminished degradation or hypersecretion? Lancet. 1977;1:10–13.
156. Gorden, P, Hendricks, C, Kahn, C. Hypoglycemia associated with non-islet cell tumor and insulin-like growth factors. N Engl J Med. 1981;305:1452–1455.
157. Zubrod, C, Eversole, S, Dane, G. Amelioration of diabetes and striking rarity of acidosis in patients with Kimmelstiel-Wilson lesions. N Engl J Med. 1951;245:518–525.
158. Block, M, Rubinstein, A. Spontaneous hypoglycemia in diabetic patients with renal insufficiency. JAMA. 1970;213:1863–1866.
159. Hultman, E, Nilsson, L. Liver glycogen in man: effect of different diets and muscular exercise. Adv Exp Med Biol. 1971;11:143–151.
160. Frizzel, M, Larsen, R, Field, J. Spontaneous hypoglycemia associated with chronic renal failure. Diabetes. 1973;22:493–498.
161. Peitzman, S, Agarwal, B. Spontaneous hypoglycemia in end-stage renal failure. Nephron. 1977;19:131–139.
162. Avram, M, Wolf, R, Gan, A, et al. Uremic hypoglycemia: a preventable life-threatening complication. N Y St J Med. 1984;84:593–596.
163. Rutsky, E, McDaniel, H, Tharpe, D, et al. Spontaneous hypoglycemia in chronic renal failure. Arch Intern Med. 1978;138:1364–1368.
164. Toth, E, Lee, D. “Spontaneous”/uremic hypoglycemia is not a distinct entity: substantiation from a literature review. Nephron. 1991;58:325–329.
165. Mühlhauser, I, Toth, G, Sawicki, P, et al. Severe hypoglycemia in type I diabetic patients with impaired kidney function. Diabetes Care. 1991;14:344–346.
166. Fürst, P. Amino acid metabolism in uremia. J Am Coll Nutr. 1989;8:310–323.
167. Greenblatt, D. Fatal hypoglycaemia occurring after peritoneal dialysis. Br Med J. 1972;2:270–271.
168. Tzamaloukas, A, Murata, G, Eisenberg, B, et al. Hypoglycemia in diabetics on dialysis with poor glycemic control: hemodialysis versus continuous ambulatory peritoneal dialysis. Int J Artif Organs. 1992;15:390–392.
169. Mak, RH. Impact of end-stage renal disease and dialysis on glycemic control. Semin Dial. 2000;13:4–8.
170. Garber, A, Bier, D, Cryer, P, et al. Hypoglycemia in compensated chronic renal insufficiency. Substrate limitation of gluconeogenesis. Diabetes. 1976;23:982–986.
171. Langlois, M, Robert, G, Nawar, T, et al. Spontaneous hypoglycemia and chronic kidney insufficiency [in French]. Can Med Assoc J. 1978;118:1083–1086.
172. Rabau, M, Dor (Dershovitz), J, Adar, R, et al. Spontaneous hypoglycemia in a diabetic patient with renal failure. Isr J Med Sci. 1973;9:1036–1039.
173. Rutsky, E, McDaniel, H, Tharpe, D, et al. Spontaneous hypoglycemia in chronic renal failure. Arch Intern Med. 1978;138:1364–1368.
174. Bonapart, I, Diderich, P, Elte, J, et al. Spontaneous hypoglycaemia in chronic renal failure. Neth J Med. 1996;48:180–184.
175. Bansal, V, Brooks, M, York, J, et al. Intractable hypoglycemia in a patient with renal failure. Arch Intern Med. 1979;139:101–102.
176. Grimaldi, A, Massin, P, Champigneulle, A, et al. Spontaneous hypoglycemia in a non-insulin-dependent diabetic with advanced renal failure [in French]. Presse Med. 1987;16:36.
177. Baylor, P, Shilo, S, Zonszein, J, et al. β-Adrenergic contribution to glucagon-induced glucose production and insulin secretion in uremia. Am J Physiol. 1986;251:E322–E327.
178. Ramirez, G, Brueggemeyer, C, Ganguly, A. Counterregulatory hormonal response to insulin-induced hypoglycemia in patients on chronic hemodialysis. Nephron. 1988;49:231–236.
179. Greenblatt, D. Insulin sensitivity in renal failure. N Y St J Med. 1974;74:1040–1041.
180. Metcoff, J, Furst, P, Scharer, K, et al. Energy production, intracellular amino acid pools, and protein synthesis in chronic renal disease. J Am Coll Nutr. 1989;8:271–284.
181. Riegel, W, Stepinski, J, Hörl, W, et al. Effect of hormones on hepatocyte gluconeogenesis in different models of acute uraemia. Nephron. 1982;32:67–72.
182. Gerich, J, Langlois, M, Noacco, C, et al. Lack of glucagon response to hypoglycemia in diabetes: evidence for an intrinsic pancreatic alpha-cell defect. Science. 1973;182:171–173.
183. White, N, Skor, D, Cryer, P, et al. Identification of type I diabetic patients at increased risk for hypoglycemia during intensive therapy. N Engl J Med. 1983;308:485–491.
184. Brun, JF, Fedou, C, Mercier, J. Postprandial reactive hypoglycemia. Diabetes Metab. 2000;26:337–351.
185. Vidnes, J, Oyasaeter, S. Glucagon deficiency causing severe neonatal hypoglycemia in a patient with normal insulin secretion. Pediatr Res. 1977;11:943–949.
186. Kollee, L, Monnens, L, Cecjka, V, et al. Persistent neonatal hypoglycaemia due to glucagon deficiency. Arch Dis Child. 1978;53:422–424.
187. Abs, R, Verbist, L, Moeremans, M, et al. Hypoglycemia owing to inappropriate glucagon secretion treated with a continuous subcutaneous glucagon infusion system. Acta Endocrinol (Copenh). 1990;122:319–322.
188. Starke, A, Valverde, I, Bottazzo, G, et al. Glucagon deficiency associated with hypoglycaemia and the absence of islet cell antibodies in the polyglandular failure syndrome before the onset of insulin-dependent diabetes mellitus: a case report. Diabetologia. 1983;25:336–339.
189. Gerich, J. Glucose counterregulation and its impact on diabetes mellitus. Diabetes. 1988;37:1608–1617.
190. Polinsky, R, Kopin, I, Ebert, M, et al. The adrenal medullary response to hypoglycemia in patients with orthostatic hypotension. J Clin Endocrinol Metab. 1980;51:1401–1406.
191. Gerich, J, Davis, J, Lorenzi, M, et al. Hormonal mechanisms of recovery from insulin-induced hypoglycemia in man. Am J Physiol. 1979;236:E380–E385.
192. Rizza, R, Cryer, P, Gerich, J. Role of glucagon, catecholamines, and growth hormone in human glucose counterregulation: effects of somatostatin and combined α- and β-adrenergic blockade on plasma glucose recovery and glucose flux rates after insulin-induced hypoglycemia. J Clin Invest. 1979;64:62–71.
193. Cryer, P. Hypoglycemia. Pathophysiology, Diagnosis, and Treatment. New York: Oxford University Press; 1997.
194. Seagall, M. Spontaneous hypoglycemia with failure to increase adrenal output. Proc R Soc Med. 1967;60:50.
195. Levin, H, Heifetz, M. Phaeochromocytoma and severe protracted postoperative hypoglycaemia. Can J Anaesth. 1990;37:477–478.
196. Gerich, J, Campbell, P. Overview of counterregulation and its abnormalities in diabetes mellitus and other conditions. Diabetes Metab Rev. 1988;4:93–111.
197. McMahon, M, Gerich, J, Rizza, R. Effects of glucocorticoids on carbohydrate metabolism. Diabetes Metab Rev. 1988;4:17–30.
198. DeFeo, P, Perriello, G, Torlone, E, et al. Contribution of cortisol to glucose counterregulation in man. Am J Physiol. 1989;257:E35–E42.
199. DeFeo, P, Perriello, G, Torlone, E, et al. Demonstration of a role of growth hormone in glucose counterregulation. Am J Physiol. 1989;256:E835–E843.
200. Goodman, H, Grumbach, M, Kaplan, S. Growth and growth hormone: II. A comparison of isolated growth-hormone deficiency and multiple pituitary-hormone deficiencies in 35 patients with idiopathic hypopituitary dwarfism. N Engl J Med. 1968;278:57–68.
201. Artavia-Loria, E, Chaussain, J, Bougneres, P, et al. Frequency of hypoglycemia in children with adrenal insufficiency. Acta Endocrinol Suppl (Copenh). 1986;279:275–278.
202. Wolfsdorf, J, Sadeghi-Nejad, A, Senior, B. Hypoketonemia and age-related fasting hypoglycemia in growth hormone deficiency. Metabolism. 1983;32:457–462.
203. Haymond, M, Karl, I, Weldon, V, et al. The role of growth hormone and cortisone on glucose and gluconeogenic substrate regulation in fasted hypopituitary children. J Clin Endocrinol Metab. 1976;42:846–856.
204. Yamamoto, T, Fukuyama, J, Hasegawa, K, et al. Isolated corticotropin deficiency in adults. Report of 10 cases and review of literature. Arch Intern Med. 1992;152:1705–1712.
205. Merimee, T, Felig, P, Marliss, E, et al. Glucose and lipid homeostasis in the absence of human growth hormone. J Clin Invest. 1971;50:574–582.
206. Smallridge, R, Corrigan, D, Thomason, A, et al. Hypoglycemia in pregnancy: occurrence due to adrenocorticotropic hormone and growth hormone deficiency. Arch Intern Med. 1980;140:564–565.
207. Malerbi, D, Liberman, B, Giurno-Filho, A, et al. Glucocorticoids and glucose metabolism: hepatic glucose production in untreated Addisonian patients and on two different levels of glucocorticoid administration. Clin Endocrinol. 1988;28:415–422.
208. Boyle, P, Cryer, P. Growth hormone, cortisol, or both are involved in defense against, but are not critical to recovery from, hypoglycemia. Am J Physiol. 1991;260:E395–E402.
209. Zuker, N, Bissessor, M, Korber, M, et al. Acute hypoglycaemic coma: a rare, potentially lethal form of early onset Sheehan syndrome. Aust N Z J Obstet Gynaecol. 1995;35:318–320.
210. Hardy, K, Burge, M, Boyle, P, et al. A treatable cause of recurrent severe hypoglycemia. Diabetes Care. 1994;17:722–724.
211. Johansson, C, Adamsson, U, Stierner, U, et al. Interaction by cholestyramine on the uptake of hydrocortisone in the gastrointestinal tract. Acta Med Scand. 1978;204:509–512.
212. Taylor, S, Barbetti, F, Accili, D, et al. Syndromes of autoimmunity and hypoglycemia. Autoantibodies directed against insulin and its receptor. Endocrinol Metab Clin North Am. 1989;18:123–143.
213. Hirata, Y, Uchigata, Y. Insulin autoimmune syndrome in Japan. Diabetes Res Clin Pract. 1994;24(Suppl):S153–S157.
214. Burch, H, Clement, S, Sokol, M, et al. Reactive hypoglycemic coma due to insulin autoimmune syndrome: case report and literature review. Am J Med. 1992;92:681–685.
215. Archambeaud-Mouveroux, F, Huc, M, Nadalon, S, et al. Autoimmune insulin syndrome. Biomed Pharmacother. 1989;43:581–586.
216. Redmon, J, Nuttall, F. Autoimmune hypoglycemia. Endocrinol Metab Clin North Am. 1999;28:603–618.
217. Kim, CH, Park, JH, Park, TS, et al. Autoimmune hypoglycemia in a type 2 diabetic patient with anti-insulin and insulin receptor antibodies. Diabetes Care. 2004;27:288–289.
218. Arioglu, E, Andewelt, A, Diabo, C, et al. Clinical course of the syndrome of autoantibodies to the insulin receptor (type B insulin resistance): a 28-year perspective. Medicine (Baltimore). 2002;81:87–100.
219. Okabe, R, Inaba, M, Hosoi, M, et al. Remission of insulin autoimmune syndrome in a patient with Grave’s disease by treatment with methimazole. Intern Med. 1999;38:482–485.
220. Buysschaert, M. Coeliac disease in patients with type 1 diabetes mellitus and auto-immune thyroid disorders. Acta Gastroenterol Belg. 2003;66:237–240.
221. Flier, J, Bar, R, Muggeo, M, et al. The evolving clinical course of patients with insulin receptor autoantibodies: spontaneous remission of receptor proliferation with hypoglycemia. J Clin Endocrinol Metab. 1978;47:985–995.
222. Taylor, S, Grunberger, G, Marcus-Samuels, B, et al. Hypoglycemia associated with antibodies to the insulin receptor. N Engl J Med. 1982;307:1422–1426.
223. Muggeo, M, Flier, J, Abrams, R, et al. Treatment by plasma exchange of a patient with autoantibodies to the insulin receptor. N Engl J Med. 1979;300:477–480.
224. Kawanishi, K, Kawamura, K, Nishina, Y, et al. Successful immunosuppressive therapy in insulin resistant diabetes caused by anti-insulin receptor autoantibodies. J Clin Endocrinol Metab. 1977;44:15–21.
225. Victor, A. Normal blood sugar variation during pregnancy. Acta Obstet Gynecol Scand. 1974;53:37–40.
226. Kalhan, S, D’Angelo, L, Savin, S, et al. Glucose production in pregnant women at term gestation: sources of glucose for human fetus. J Clin Invest. 1979;63:388–394.
227. Reece, E, Homko, C, Wiznitzer, A. Metabolic changes in diabetic and nondiabetic subjects during pregnancy. Obstet Gynecol Surv. 1994;49:64–71.
228. Garner, P, Tsang, R. Insulinoma complicating pregnancy presenting with hypoglycemic coma after delivery: a case report and review of the literature. Obstet Gynecol. 1989;73:847–849.
229. Takacs, CA, Krivak, TC, Napolitano, PG. Insulinoma in pregnancy: a case report and review of the literature. Obstet Gynecol Surv. 2002;57:229–235.
230. Schweichler, M, Hennessey, J, Cole, P, et al. Hypoglycemia in pregnancy secondary to a non-islet cell tumor of the pleura and ectopic insulin-like growth factor II hormone production. Obstet Gynecol. 1995;85:810–813.
231. White, N, Warrell, D, Chanthavanich, P, et al. Severe hypoglycemia and hyperinsulinemia in falciparum malaria. N Engl J Med. 1983;309:61–66.
232. Long, P, Abell, D, Beischer, N. Importance of abnormal glucose tolerance (hypoglycaemia and hyperglycaemia) in the aetiology of pre-eclampsia. Lancet. 1977;1:923–925.
233. Neuman, M, Ron-El, R, Langer, R, et al. Maternal death caused by HELLP syndrome (with hypoglycemia) complicating mild pregnancy-induced hypertension in a twin gestation. Am J Obstet Gynecol. 1989;162:372–373.
234. Egley, C. Severe hypoglycemia associated with HELLP syndrome. Am J Obstet Gynecol. 1985;152:576–577.
235. Aarnoudse, J, Houthoff, H, Weits, J, et al. A syndrome of liver damage and intravascular coagulation in the last trimester of normotensive pregnancy: a clinical and histopathological study. Br J Obstet Gynaecol. 1986;93:145–155.
236. Levine, A, Burgess, G, Derick, C. Some changes in the chemical constituents of the blood following a marathon race. JAMA. 1924;82:1778–1782.
237. Ahlborg, G, Felig, P. Lactate and glucose exchange across the forearm, legs, and splanchnic bed during and after prolonged leg exercise. J Clin Invest. 1982;69:45–54.
238. Uusitupa, M, Aro, A, Pietikainen, M. Severe hypoglycaemia caused by physical strain and pindolol therapy: a case report. Ann Clin Res. 1980;12:25–27.
239. Field, J. Exercise and deficient carbohydrate storage and intake as causes of hypoglycemia. Endocrinol Metab Clin North Am. 1989;18:155–161.
240. Wasserman, D, Cherrington, A. Hepatic fuel metabolism during muscular work: role and regulation. Am J Physiol. 1991;260:E811–E824.
241. Wahren, J, Felig, P, Hagenfeldt, L. Physical exercise and fuel homeostasis in diabetes mellitus. Diabetologia. 1978;14:213–222.
242. Marker, J, Hirsch, I, Smith, L, et al. Catecholamines in prevention of hypoglycemia during exercise in humans. Am J Physiol. 1991;260:E705–E712.
243. Hirsch, I, Marker, J, Smith, L, et al. Insulin and glucagon in prevention of hypoglycemia during exercise in humans. Am J Physiol. 1991;260:E695–E704.
244. Horwitz, D. Factitious and artifactual hypoglycemia. Endocrinol Metab Clin North Am. 1989;18:203–210.
245. Service, F, Moore, G. Factitial Hypoglycemia. In: Service F, ed. Hypoglycemic Disorders: Pathogenesis, Diagnosis and Treatment. Boston: GK Hall; 1983:129–141.
246. Marks, V, Teale, J. Hypoglycemia: factitious and felonious. Endocrinol Metab Clin North Am. 1999;28:579–601.
247. Sheehy, T. Case report: factitious hypoglycemia in diabetic patients. Am J Med Sci. 1992;304:298–302.
248. Schade, D, Drumm, D, Eaton, R, et al. Factitious brittle diabetes mellitus. Am J Med. 1985;78:777–784.
249. Marks, V. Hypoglycaemia—real and unreal, lawful and unlawful: The 1994 Banting Lecture. Diabet Med. 1995;12:850–864.
250. Manning, PJ, Espiner, EA, Yoon, K, et al. An unusual cause of hyperinsulinaemic hypoglycaemia syndrome. Diabet Med. 2003;20:772–776.
251. Kaminer, Y, Robbins, D. Insulin misuse: a review of an overlooked psychiatric problem. Psychosomatics. 1989;30:19–24.
252. Marchetti, P, Faloppa, C, Zappella, A, et al. A case of factitious hypoglycemia with unusual presentation [in Italian]. Minerva Med. 1988;79:1101–1103.
253. Scarlett, J, Mako, M, Rubenstein, A, et al. Factitious hypoglycemia: diagnosis by measurement of serum C-peptide immunoreactivity and insulin-binding antibodies. N Engl J Med. 1977;297:1029–1032.
254. Retsas, S. Insulin abuse by a drug addict. Br Med J. 1972;4:792–793.
255. Jordan, R, Kammer, H, Riddle, M. Sulfonylurea-induced factitious hypoglycemia: a growing problem. Arch Intern Med. 1977;137:390–393.
256. Grunberger, G, Weiner, J, Silverman, R, et al. Factitious hypoglycemia due to surreptitious administration of insulin. Diagnosis, treatment, and long-term follow-up. Ann Intern Med. 1988;108:252–257.
257. Rynearson, E. Hyperinsulinism among malingerers. Med Clin North Am. 1947;31:477–480.
258. Siegel, E, Mayer, G, Nauck, M, et al. Factitious hypoglycemia caused by taking a sulfonylurea drug [in German]. Dtsch Med Wochenschr. 1987;112:1575–1579.
259. Charlton, R, Smith, G, Day, A. Munchausen’s syndrome manifesting as factitious hypoglycaemia. Diabetologia. 2001;44:784–785.
260. Teale, J, Starkey, B, Marks, V, et al. The prevalence of factitious hypoglycaemia due to sulphonylurea abuse in the UK: a preliminary report. Practical Diabetes. 1989;6:177–178.
261. Hirshberg, B, Skarulis, MC, Pucino, F, et al. Repaglinide-induced factitious hypoglycemia. J Clin Endocrinol Metab. 2001;86:475–477.
262. Doege, K. Fibro-sarcoma of the mediastinum. Ann Surg. 1930;92:955–960.
263. Daughaday, W. Hypoglycemia in patients with non-islet cell tumors. Endocrinol Metab Clin North Am. 1989;18:91–101.
264. Anderson, N, Lokich, J. Mesenchymal tumors associated with hypoglycemia: case report and review of the literature. Cancer. 1979;44:785–790.
265. Baxter, R. The role of insulin-like growth factors and their binding proteins in tumor hypoglycemia. Horm Res. 1996;46:195–201.
266. Benn, J, Firth, R, Sönksen, P. Metabolic effects of an insulin-like factor causing hypoglycaemia in a patient with a haemangiopericytoma. Clin Endocrinol. 1990;32:769–780.
267. Chandalia, H, Boshell, B. Hypoglycemia associated with extrapancreatic tumors. Arch Intern Med. 1972;129:447–456.
268. Chowdhury, F, Bleicher, S. Studies of tumor hypoglycemia. Metabolism. 1973;22:663–674.
269. Daughaday, W, Emanuele, M, Brooks, M, et al. Synthesis and secretion of insulin-like growth factor II by a leiomyosarcoma with associated hypoglycemia. N Engl J Med. 1988;319:1434–1440.
270. Daughaday, W. The pathophysiology of IGF-II hypersecretion in non-islet cell tumor hypoglycemia. Diab Rev. 1995;3:62–72.
271. Eastman, R, Carson, R, Orloff, D, et al. Glucose utilization in a patient with hepatoma and hypoglycemia. J Clin Invest. 1992;89:1958–1963.
272. McFadzean, A, Yeung, R. Further observations on hypoglycaemia in hepatocellular carcinoma. Am J Med. 1969;47:220–235.
273. Millard, P, Jerrome, D, Millward-Sadler, G. Spindle-cell tumours and hypoglycaemia. J Clin Pathol. 1976;29:520–529.
274. Moller, N, Blum, W, Mengel, A, et al. Basal and insulin stimulated substrate metabolism in tumour induced hypoglycaemia: evidence for increased muscle glucose uptake. Diabetologia. 1991;34:17–20.
275. Nissan, S, Bar-Maor, A, Shafrir, E. Hypoglycemia associated with extrapancreatic tumors. N Engl J Med. 1968;278:177–183.
276. Reeve, A, Eccles, M, Wilkins, R, et al. Expression of insulin-like growth factor-II transcripts in Wilms’ tumour. Nature. 1985;317:258–260.
277. Scott, J, Cowell, J, Robertson, M, et al. Insulin-like growth factor-II gene expression in Wilms’ tumour and embryonic tissues. Nature. 1985;317:260–262.
278. Silbert, C, Rossini, A, Ghazvinian, S, et al. Tumor hypoglycemia: deficient splanchnic glucose output and deficient glucagon secretion. Diabetes. 1976;25:202–206.
279. Silverstein, M. Tumor hypoglycemia. Cancer. 1969;23:142–144.
280. Skrabanek, P, Powell, D. Ectopic insulin and Occam’s razor: reappraisal of the riddle of tumour hypoglycaemia. Clin Endocrinol. 1978;9:141–154.
281. Widmer, U, Zapf, J, Froesch, E. Is extrapancreatic tumor hypoglycemia associated with elevated levels of insulin-like growth factor II? J Clin Endocrinol Metab. 1982;55:833–839.
282. Horecker, B, Hiatt, H. Pathways of carbohydrate metabolism in normal and neoplastic cells. N Engl J Med. 1958;258:177–184.
283. Phuphanich, S, Jacobs, L, Poulos, J, et al. Case report: hypoglycemia secondary to a meningioma. Am J Med Sci. 1995;309:317–321.
284. Chung, J, Henry, R. Mechanisms of tumor-induced hypoglycemia with intraabdominal hemangiopericytoma. J Clin Endocrinol Metab. 1996;81:919–925.
285. Ko, AH, Bergsland, EK, Lee, GA. Tumor-associated hypoglycemia from metastatic colorectal adenocarcinoma: case report and review of the literature. Dig Dis Sci. 2003;48:192–196.
286. Seckl, M, Mulholland, P, Bishop, A, et al. Hypoglycemia due to an insulin-secreting small-cell carcinoma of the cervix. N Engl J Med. 1999;341:733–736.
287. Marks, V, Teale, J. Tumours producing hypoglycaemia. Endocr Relat Cancer. 1998;5:111–129.
288. Del Sindaco, P, Casucci, G, Pampanelli, S, et al. Late post-prandial hypoglycaemia as the sole presenting feature of secreting pancreatic beta-cell adenoma in a subtotally gastrectomized patient. Eur J Endocrinol. 1997;136:96–99.
289. Brasel, J, Wright, J, Wilkins, L, et al. An evaluation of seventy-five patients with hypopituitarism beginning in childhood. Am J Med. 1965;38:484–498.
290. O’Keefe, S, Marks, V. Lunchtime gin and tonic a cause of reactive hypoglycaemia. Lancet. 1977;1:1286–1288.
291. Hofeldt, F, Lufkin, E, Hagler, L, et al. Are abnormalities in insulin secretion responsible for reactive hypoglycemia? Diabetes. 1974;23:589–596.
292. Hopwood, N, Forsman, P, Kenny, F, et al. Hypoglycemia in hypopituitary children. Am J Dis Child. 1975;129:918–926.
293. Leichter, S, Permutt, M. Effect of adrenergic agents on postgastrectomy hypoglycemia. Diabetes. 1975;24:1005–1010.
294. Shultz, K, Neelon, F, Nilsen, L, et al. Mechanism of postgastrectomy hypoglycemia. Arch Intern Med. 1971;128:240–246.
295. Wiznitzer, T, Shapira, N, Stadler, J, et al. Late hypoglycemia in patients following vagotomy and pyloroplasty. Int Surg. 1974;59:229–232.
296. Hall, W, Snaders, L. Hypoglycemic convulsions after vagotomy and pyloroplasty. South Med J. 1973;66:502–504.
297. Miholic, J, Orskov, C, Holst, J, et al. Postprandial release of glucagon-like peptide-1, pancreatic glucagon, and insulin after esophageal resection. Digestion. 1993;54:73–78.
298. Veverbrants, E, Olsen, W, Arky, R. Role of gastro-intestinal factors in reactive hypoglycemia. Metabolism. 1969;18:6–12.
299. Zieve, L, Jones, D, Aziz, M. Functional hypoglycemia and peptic ulcer. Postgrad Med. 1966;40:159–170.
300. O’Brien, T, Tijtgat, G, Ensinck, J. Alimentary hypoglycemia associated with the Zollinger-Ellison syndrome. Am J Med. 1973;54:637–644.
301. Luyckx, A, Lefebvre, P. Plasma insulin in reactive hypoglycemia. Diabetes. 1971;20:435–442.
302. Bellini, F, Sammicheli, L, Ianni, L, et al. Hypoglycemia unawareness in a patient with dumping syndrome: report of a case. J Endocrinol Invest. 1998;21:463–467.
303. Freinkel, N, Metzger, B. Oral glucose tolerance curve and hypoglycemias in the fed state. N Engl J Med. 1969;280:820–828.
304. Andreasen, J, Orskov, C, Holst, J. Secretion of glucagon-like peptide-1 and reactive hypoglycemia after partial gastrectomy. Digestion. 1994;55:221–228.
305. Miholic, J, Orskov, C, Holst, J, et al. Emptying of the gastric substitute, glucagon-like peptide-1 (GLP-1), and reactive hypoglycemia after total gastrectomy. Dig Dis Sci. 1991;36:1361–1370.
306. Mitrakou, A, Kelley, D, Mokan, M, et al. Role of reduced suppression of glucose production and diminished early insulin release in impaired glucose tolerance. N Engl J Med. 1992;326:22–29.
307. Seltzer, H, Allen, E, Herror, A, et al. Insulin response to glycemic stimulus: relation of delayed initial release to carbohydrate intolerance in mild diabetes. J Clin Invest. 1967;46:323–335.
308. Faludi, G, Bendersky, G, Gerber, P. Functional hypoglycemia in early latent diabetes. Ann N Y Acad Sci. 1968;148:868–874.
309. Permutt, M. Postprandial hypoglycemia. Diabetes. 1976;25:719–733.
310. Hofeldt, F. Reactive hypoglycemia. Endocrinol Metab Clin North Am. 1989;18:185–201.
311. Service, F. Classification of hypoglycemic disorders. Endocrinol Metab Clin North Am. 1999;28:501–517.
312. Owada, K, Wasada, T, Miyazono, Y, et al. Highly increased insulin secretion in a patient with postprandial hypoglycemia: role of glucagon-like peptide-1 (7-36) amide. Endocr J. 1995;42:147–151.
313. Permutt, M, Kelly, J, Berstein, R, et al. Alimentary hypoglycemia in the absence of gastrointestinal surgery. N Engl J Med. 1973;288:1206–1210.
314. Palardy, J, Havrankova, J, Lepage, R, et al. Blood glucose measurements during symptomatic episodes in patients with suspected postprandial hypoglycemia. N Engl J Med. 1989;321:1421–1425.
315. Foa, P, Dunbar, J, Jr., Klein, S, et al. Reactive hypoglycemia and A-cell (“pancreatic”) glucagon deficiency in the adult. JAMA. 1980;244:2281–2285.
316. Ahmadpour, S, Kabadi, U. Pancreatic alpha-cell function in idiopathic reactive hypoglycemia. Metabolism. 1997;46:639–643.
317. Leonetti, F, Morviducci, L, Giaccari, A, et al. Idiopathic reactive hypoglycemia: a role for glucagon? J Endocrinol Invest. 1992;15:273–278.
318. Shima, K, Tabata, M, Tanaka, A, et al. Exaggerated response of plasma glucagon-like immunoreactivity (GLI) to oral glucose in patients with reactive hypoglycemia. Endocrinol Jpn. 1981;28:249–256.
319. Wasada, T, Katsumori, K, Saeki, A, et al. Lack of C-peptide suppression by exogenous hyperinsulinemia in subjects with symptoms suggesting reactive hypoglycemia. Endocr J. 1996;43:639–644.
320. Leonetti, F, Foniciello, M, Iozzo, P, et al. Increased nonoxidative glucose metabolism in idiopathic reactive hypoglycemia. Metabolism. 1996;45:606–610.
321. Lteif, A, Schwenk, W. Hypoglycemia in infants and children. Endocrinol Metab Clin North Am. 1999;28:619–646.
322. Witteles, RM, Straus, FH, II., Sugg, SL, et al. Adult-onset nesidioblastosis causing hypoglycemia: an important clinical entity and continuing treatment dilemma. Arch Surg. 2001;136:656–663.
323. Thomas, P. Genetic mutations as a cause of hyperinsulinemic hypoglycemia in children. Endocrinol Metab Clin North Am. 1999;28:647–656.
324. Service, F, Natt, N, Thompson, G, et al. Noninsulinoma pancreatogenous hypoglycemia: a novel syndrome of hyperinsulinemic hypoglycemia in adults independent of mutations in Kir6.2 and SUR1 genes. J Clin Endocrinol Metab. 1999;84:1582–1589.
325. Kaczirek, K, Soleiman, A, Schindl, M, et al. Nesidioblastosis in adults: a challenging cause of organic hyperinsulinism. Eur J Clin Invest. 2003;33:488–492.
326. Johnson, D, Dorr, K, Swenson, W, et al. Reactive hypoglycemia. JAMA. 1980;243:1151–1155.
327. Hogan, M, Service, F, Sharbrough, F, et al. Oral glucose tolerance test compared with a mixed meal in the diagnosis of reactive hypoglycemia: a caveat on stimulation. Mayo Clin Proc. 1983;58:491–496.
328. Charles, M, Hofeldt, F, Shackelford, A, et al. Comparison of oral glucose tolerance tests and mixed meals in patients with apparent idiopathic postabsorptive hypoglycemia: absence of hypoglycemia after meals. Diabetes. 1981;30:465–470.
329. Snorgaard, O, Binder, C. Monitoring of blood glucose concentration in subjects with hypoglycaemic symptoms during everyday life. Br Med J. 1990;300:16–18.
330. Ford, C, Bray, G, Swerdloff, R. A psychiatric study of patients referred with a diagnosis of hypoglycemia. Am J Psychiatry. 1976;133:290–294.
331. Berlin, I, Grimaldi, A, Landault, C, et al. Suspected postprandial hypoglycemia is associated with beta-adrenergic hypersensitivity and emotional distress. J Clin Endocrinol Metab. 1994;79:1428–1433.
332. Fariss, B. Prevalence of post-glucose load glycosuria and hypoglycemia in a group of healthy young men. Diabetes. 1974;23:189–191.
333. Lev-Ran, A, Anderson, R. the diagnosis of postprandial hypoglycemia. Diabetes. 1981;30:996–999.
334. Laidlaw, G. Nesidioblastoma: the islet cell tumor of the pancreas. Am J Pathol. 1937;14:125–139.
335. Yakovac, W, Baker, L, Hummeler, K. Beta cell nesidioblastosis in idiopathic hypoglycemia of infancy. J Pediatr. 1971;79:226–231.
336. Harness, J, Geelhoed, G, Thompson, N, et al. Nesidioblastosis in adults: a surgical dilemma. Arch Surg. 1981;116:575–580.
337. Gould, V, Chejfec, G, Shah, K, et al. Adult nesidiodysplasia. Semin Diagn Pathol. 1984;1:43–53.
338. Walmsley, D, Matheson, N, Ewen, S, et al. Nesidioblastosis in an elderly patient. Diabet Med. 1995;12:542–545.
339. Harrison, T, Fajans, S, Floyd, J, Jr., et al. Prevalence of diffuse pancreatic beta islet cell disease with hyperinsulinism: problems in recognition and management. World J Surg. 1984;8:583–589.
340. Stefanini, P, Carboni, M, Patrassi, N, et al. Hypoglycemia and insular hyperplasia: review of 148 cases. Ann Surg. 1974;180:130–135.
341. Service, F, McMahon, M, O’Brien, P, et al. Functioning insulinoma: incidence, recurrence, and long-term survival of patients: a 60-year study. Mayo Clin Proc. 1991;66:711–719.
342. Kavlie, H, White, T. Pancreatic islet beta cell tumors and hyperplasia: experience in 14 Seattle hospitals. Ann Surg. 1972;175:326–335.
343. Cullen, R, Ong, C. Insulinoma in Auckland 1970–1985. N Z Med J. 1987;100:560–562.
344. Fajans, S, Vinik, A. Insulin-producing islet cell tumors. Endocrinol Metab Clin North Am. 1989;18:45–74.
345. Dizon, A, Kowalyk, S, Hoogwerf, B. Neuroglycopenic and other symptoms in patients with insulinomas. Am J Med. 1999;106:307–310.
346. Perry, R, Vinik, A. Clinical review 72: diagnosis and management of functioning islet cell tumors. J Clin Endocrinol Metab. 1995;80:2273–2278.
347. Kane, L, Grant, C, Nippoldt, T, et al. Insulinoma in a patient with NIDDM. Diabetes Care. 1993;16:1298–1300.
348. Sakurai, A, Aizawa, T, Katakura, M, et al. Insulinoma in a patient with non-insulin-dependent diabetes mellitus. Endocr J. 1997;44:473–477.
349. Wildbrett, J, Nagel, M, Theissig, F, et al. An unusual picture of insulinoma in type-2 diabetes mellitus and morbid obesity [in German]. Dtsch Med Wochenschr. 1999;124:248–252.
350. Gerich, J, Charles, M, Grodsky, G. Regulation of pancreatic insulin and glucagon secretion. Annu Rev Physiol. 1976;38:353–388.
351. Creutzfeldt, W, Arnold, R, Creutzfeldt, C, et al. Biochemical and morphological investigations of 30 human insulinomas: correlation between the tumour content of insulin and proinsulin-like components and the histological and ultrastructural appearance. Diabetologia. 1973;9:217–231.
352. Lindall, A, Steffes, M, Wong, E. Comparison of insulin and proinsulin storage in an islet adenoma and adjacent pancreas. Metabolism. 1974;23:249–256.
353. Alsever, R, Stjernholm, M, Sussman, K, et al. Clinical correlations of serum proinsulin-like material in islet cell tumours. Diabetologia. 1976;12:527–530.
354. Rizza, R, Haymond, M, Verdonk, C, et al. Pathogenesis of hypoglycemia in insulinoma patients: suppression of hepatic glucose production by insulin. Diabetes. 1981;30:377–381.
355. Wynick, D, Williams, S, Bloom, S. Symptomatic secondary hormone syndromes in patients with established malignant pancreatic endocrine tumors. N Engl J Med. 1988;319:605–607.
356. Fonseca, V, Ames, D, Ginsburg, J. Hypoglycaemia for 26 years due to an insulinoma. J R Soc Med. 1989;82:437–438.
357. Mitrakou, A, Fanelli, C, Veneman, T, et al. Reversibility of unawareness of hypoglycemia in patients with insulinomas. N Engl J Med. 1993;329:834–839.
358. Vea, H, Trovik, T, Sager, G, et al. Return of beta-adrenergic sensitivity in a patient with insulinoma after removal of the tumour. Diabet Med. 1997;14:979–984.
359. Vea, H, Jorde, R, Sager, G, et al. Pre- and postoperative glucose levels for eliciting hypoglycemic responses in a patient with insulinoma. Diabetic Med. 1992;9(10):950–953.
360. Stefanini, P, Carboni, M, Patrassi, N, et al. Beta-islet cell tumors of the pancreas: results of a study on 1,067 cases. Surgery. 1974;75:597–609.
361. Lloyd, R, Caceres, V, Warner, T, et al. Islet cell adenomatosis: a report of two cases and review of the literature. Arch Pathol Lab Med. 1981;105:198–202.
362. Fong, T, Warner, N, Kumar, D. Pancreatic nesidioblastosis in adults. Diabetes Care. 1989;12:108–114.
363. Service, F. Hypoglycemic disorders. N Engl J Med. 1995;332:1144–1152.
364. Marks, V, Teale, J. Investigation of hypoglycaemia. Clin Endocrinol (Oxf). 1996;44:133–136.
365. Service, F, O’Brien, P, Kao, P, et al. C-peptide suppression test: effects of gender, age, and body mass index; implications for the diagnosis of insulinoma. J Clin Endocrinol Metab. 1992;74:204–210.
366. Marks, V, Somols, E. Diagnostic tests for evaluating hypoglycemia. In: Rodriguez R, Vallance-Owen J, eds. Diabetes. Amsterdam: Excerpta Medica; 1971:864–872.
367. Kumar, D, Mehtalia, S, Miller, L. Diagnostic use of glucagon-induced insulin response. Studies in patients with insulinoma or other hypoglycemic conditions. Ann Intern Med. 1974;80:697–701.
368. Floyd, J, Jr., Fajans, S, Knopf, R, et al. Plasma insulin in organic hyperinsulinism: comparative effects of tolbutamide, leucine and glucose. J Clin Endocrinol. 1964;24:747–760.
369. Scholz, D, ReMine, W, Priestley, J. Hyperinsulinism: review of 95 cases of functioning pancreatic islet cell tumors. Mayo Clin Proc. 1960;35:545–550.
370. Service, F. Diagnostic approach to adults with hypoglycemic disorders. Endocrinol Metab Clin North Am. 1999;28:519–532.
371. Filipi, C, Higgins, G. Diagnosis and management of insulinoma. Am J Surg. 1973;125:231–239.
372. Galbut, D, Markowitz, A. Insulinoma: diagnosis, surgical management and long-term follow-up: review of 41 cases. Am J Surg. 1980;139:682–690.
373. Boukhman, M, Karam, J, Shaver, J, et al. Insulinoma: experience from 1950 to 1995. West J Med. 1998;169:98–104.
374. Daggett, P, Goodburn, E, Kurtz, A, et al. Is preoperative localisation of insulinomas necessary? Lancet. 1981;1:483–486.
375. Grant, C. Surgical aspects of hyperinsulinemic hypoglycemia. Endocrinol Metab Clin North Am. 1999;28:533–554.
376. Norton, J, Whitman, E. Insulinoma. Endocrinologist. 1995;3:258–267.
377. Huai, J-C, Zhang, W, Niu, H-O, et al. Localization and surgical treatment of pancreatic insulinomas guided by intraoperative ultrasound. Am J Surg. 1998;175:18–21.
378. King, A, Ko, G, Yeung, V, et al. Dual phase spiral CT in the detection of small insulinomas of the pancreas. Br J Radiol. 1998;71:20–23.
379. Gill, G, Rauf, O, MacFarlane, I. Diazoxide treatment for insulinoma: a national UK survey. Postgrad Med J. 1997;73:640–641.
380. Osei, K, O’Dorisio, T. Malignant insulinoma: effects of a somatostatin analog (compound 201-995) on serum glucose, growth, and gastro-entero-pancreatic hormones. Ann Intern Med. 1985;103:223–225.
381. Hearn, P, Ahmed, M, Woodhouse, N. The use of SMS 201-995 (somatostatin analogue) in insulinomas: additional case report and literature review. Horm Res. 1988;29:211–213.
382. Richter, W, Otto, C. Continuous subcutaneous glucagon infusion as a symptomatic therapy in two patients with organic hyperinsulinemia. Endocrinol Metab. 1996;3:63–65.
383. Broder, L, Carter, S. Pancreatic islet cell carcinoma: I. Clinical features of 52 patients. Ann Intern Med. 1973;79:101–107.
384. Moertel, C, Hanley, J, Johnson, L. Streptozocin alone compared with streptozocin plus fluorouracil in the treatment of advanced islet-cell carcinoma. N Engl J Med. 1980;303:1189–1194.
385. Kiang, D, Frenning, D, Bauer, G. Mithramycin for hypoglycemia in malignant insulinoma. N Engl J Med. 1978;299:134–135.
386. Eastman, R, Come, S, Strewler, G, et al. Adriamycin therapy for advanced insulinoma. J Clin Endocrinol Metab. 1977;44:142–148.
387. Ajani, J, Carrasco, C, Charnsangavej, C, et al. Islet cell tumors metastatic to the liver: effective palliation by sequential hepatic artery embolization. Ann Intern Med. 1988;108:340–344.
388. Hart, S, Frier, B. Causes, management and morbidity of acute hypoglycaemia in adults requiring hospital admission. Q J Med. 1998;91:505–510.
389. Malouf, R, Brust, J. Hypoglycemia: causes, neurological manifestations, and outcome. Ann Neurol. 1985;17:421–430.
390. Kresevic, D, Slavin, S. Incidence of hypoglycemia and nutritional intake in patients on a general medical unit. Nurs Connections. 1989;2:33–40.
391. Gaston, S. Outcomes of hypoglycemia treated by standardized protocol in a community hospital. Diabetes Educ. 1992;18:491–494.
392. Slama, G, Traynard, P, Desplanque, N, et al. The search for an optimized treatment of hypoglycemia: carbohydrates in tablets, solution, or gel for the correction of insulin reactions. Arch Intern Med. 1990;150:589–593.
393. Palatnick, W, Meatherall, R, Tenenbein, M. Clinical spectrum of sulfonylurea overdose and experience with diazoxide therapy. Arch Intern Med. 1991;151:1859–1862.
394. DeFeo, P, Perriello, G, Torlone, E, et al. Contribution of adrenergic mechanisms to glucose counterregulation in humans. Am J Physiol. 1991;261:E725–E736.