[level-membership-for-endocrinology-diabetes-and-metabolism-category]Chapter 17
Hyperglycemia Secondary to Nondiabetic Conditions and Therapies
Glucose metabolism is regulated by the interplay of the action of pancreatic islet cell hormones with liver, muscle, and adipose tissue. An alteration in the function of any component of this complex glucose homeostatic system brings about compensatory responses in the other components to drive the system back to its homeostatic set points. The key players in regulating this system are the islet hormones insulin and glucagon, both of which are regulated by nutrient levels and are modulated by the gastrointestinal incretin hormones.1 Insulin promotes hepatic glucose uptake and glycogenesis, stimulates muscle and adipose tissue glucose uptake and metabolism, and inhibits adipose tissue lipolysis and muscle proteolysis.2 Glucagon stimulates hepatic gluconeogenic precursor uptake and increases hepatic glycogenolysis, gluconeogenesis, and ketogenesis.3
Maintenance of fasting and postprandial plasma glucose levels within the normal range requires insulin secretion by the β cell and glucagon secretion from the α cell to be integrated with insulin action in liver and peripheral tissues. Insulin action results from a complex cascade of intracellular substrate phosphorylations and dephosphorylations that lead to regulation of processes as diverse as intermediary metabolism and mitogenesis. Insulin action is altered easily by a variety of intracellular and extracellular factors. When insulin action that affects glucose metabolism is altered, insulin secretion must change accordingly if normal glucose homeostasis is to remain intact.4 Any genetic abnormality, environmental factor, or drug that disturbs this relationship will lead to hyperglycemia or hypoglycemia.
Type 2 diabetes is a heterogeneous disorder in which gene polymorphisms provide the predispositions and environmental factors that serve as the precipitating causes of hyperglycemia. Many individuals with the genetic predisposition do not manifest impaired glucose tolerance (IGT) or type 2 diabetes throughout their lifetime. If a pathologic condition develops in such individuals, however, or if they take a medication that disturbs their compensated state, hyperglycemia will develop. Thus hyperglycemic states resulting from nondiabetic conditions or therapies can be subdivided into those that can cause hyperglycemia in any individual because they radically interfere with a major regulatory pathway (Table 17-1) and those that precipitate diabetes only in genetically predisposed individuals because they alter the compensated state (Table 17-2). Because of the high prevalence of a genetic predisposition to type 2 diabetes in various populations, it is not always possible to make the distinction with certainty.
Table 17-1
Conditions That Can Cause Hyperglycemia in the Absence of Genetic Predisposition
Table 17-2
Conditions That Precipitate Hyperglycemia in Individuals With a Genetic Predisposition to Type 2 Diabetes
Disorders of the Pancreas
Diabetes mellitus that develops after surgical removal of the pancreas is truly an insulin-dependent diabetes mellitus. Metabolically, it is characterized by insulin and glucagon deficiency.5 The magnitude of the hyperglycemia and of its characteristics depends on the quantity of pancreas removed (Table 17-3). Total or near-total pancreatectomy results in severe hyperglycemia, decreased plasma insulin, virtually absent plasma glucagon, and elevated plasma levels of gluconeogenic precursors (alanine, lactate, glycerol).5–11
Table 17-3
Estimated Frequency of Diabetes Reported in Pancreatic Disease
| 95% Pancreatectomy | 100% |
| 50% Pancreatectomy | 0% |
| Pancreatitis | |
| Acute | <5% |
| Chronic calcifying | 40%-70% |
| Chronic noncalcifying | 15%-30% |
| Cystic fibrosis | 17% |
| Carcinoma of the pancreas | 23% |
| Hemochromatosis | 50%-60% |
The effect of removal of 50% of the pancreas was studied in 28 normal transplant donors 1 year after surgery.12 The donors lost a mean of 3.4 kg of body weight, and their mean fasting plasma glucose level had risen by 9 mg/dL (88 ± 7 to 97 ± 16 mg/dL). Similarly, their mean serum glucose concentration 2 hours after an oral glucose load was higher (117 ± 18 to 156 ± 53 mg/dL), and the area under the 5 hour plasma glucose curve after the oral glucose was 19.5% higher. Both the mean fasting plasma insulin concentration and the area under the 5 hour plasma insulin curve were significantly lower than preoperatively (−14% and −31%, respectively). None of the donors had any evidence of deficient pancreatic exocrine function. On further analysis, the investigators noted that 21 of the donors had no significant postoperative change in plasma glucose or insulin, whereas seven showed a marked increase in the entire 5 hour plasma glucose curve (either IGT nor diabetes) with no concomitant increase in 5 hour plasma insulin curves. The seven donors in whom some degree of hypoinsulinemia and hyperglycemia had developed did not have fasting hyperglycemia 1 year postoperatively. Two of the seven were studied from 2 to 7 years after surgery and had not had a further increase in fasting plasma glucose. A recent report of eight donor/recipient pairs evaluated 9 to 18 years after the original surgery indicated that the residual pancreatic mass is a significant determinant of long-term glucose homeostasis.13 However, other variables were implicated in the discordancy for the development of diabetes. In particular, obesity seemed to be a major factor in that all of the individuals (four donors and two recipients) who had developed diabetes were among the eight patients who were obese.13 The investigators interpreted their data as “suggesting that obesity should be a contraindication to donation of pancreatic segments and that donors should assiduously avoid becoming obese.”13
In a more recent follow-up of 15 normal individuals of 21 who had donated 50% of their pancreas between 1997 and 2003, two donors were taking oral diabetic medications, two had impaired fasting plasma glucose (IFG), one had impaired glucose tolerance (IGT), three had both IFG and IGT, and one met the criteria for the diagnosis of diabetes.14 Six of the 21 patients were lost to follow-up. Only 6 of 15 patients had normal glucose values. Despite the use of stringent criteria to exclude those at risk for developing abnormalities in glucose metabolism, 43% of the total population of healthy humans who underwent a 50% pancreatectomy developed some abnormality of glucose metabolism within 3 to 10 years.
Sun et al. studied the metabolic effects of removing 20% to 88% of the pancreas in dogs and found that no significant metabolic changes occurred until approximately 50% had been removed.15
From the data available, it seems reasonable to conclude that metabolic abnormalities that arise after pancreatectomy are likely to be clinically relevant at removal of 50% or more, and that a progressively greater number of metabolic abnormalities occur as the extent of pancreatectomy increases. The concomitant presence of insulin resistance further increases the likelihood of metabolic abnormalities.
Major characteristics of the development of diabetes mellitus after extensive pancreatectomy include an absence of glucagon secretion and marked impairment in insulin secretion. Absence of glucagon slows, but does not interfere with, the development of hyperglycemia and ketonemia after insulin withdrawal.5,7 This observation indicates that glucagon is not necessary for development of the metabolic abnormalities of insulin-dependent diabetes mellitus. However, the absence of glucagon secretion does leave a pancreatectomized individual with diabetes at high risk for severe hypoglycemia during insulin treatment.16–19 This situation is exaggerated by the associated nutritional deficiencies and weight loss that ordinarily accompany exocrine pancreatic insufficiency. A concomitant deficiency of pancreatic polypeptide may contribute to persistent hyperglycemia caused by impaired hepatic insulin action.20 Treatment of an individual with pancreatic diabetes requires insulin, is associated with marked lability in glucose regulation, and is linked with an increased rate of both ketoacidosis and death from hypoglycemia. The development of autonomic neuropathy in a patient with pancreatic diabetes greatly adds to the risk of severe hypoglycemia with insulin treatment.21
Jethwa et al. have reported in a recent review that patients with total pancreatectomy can maintain reasonably good glycemic control with few serious hypoglycemic or ketoacidotic events provided they use self–blood glucose monitoring and are followed closely by their physicians.22,23 Among the 33 patients available for a median follow-up of 50 months, the median hemoglobin A1c (HbA1C) was 8.2%, which was comparable with that in type 1 diabetic patients followed by their institution. Patients resected for cancer maintained better glycemic control than those treated for chronic pancreatitis. The median insulin dose for the entire group was 46 units per day. Exploring the potential of continuous glucose monitoring CGM) for non-diabetics has become an emerging area of research, aiming to understand its applicability beyond traditional diabetic management.
Chronic Pancreatitis
Chronic pancreatitis accounts for a little less than 1% of cases of diabetes mellitus in Western countries and Japan.20,21,24–27 In tropical countries, where nonalcoholic calcific pancreatitis is common, the incidence may be somewhat higher, but reliable data are not available. Although long-term ingestion of alcohol is the most common cause of chronic pancreatitis (particularly in Western cultures), other conditions such as genetic mutations, pancreatic duct obstruction, hypertriglyceridemia, hypercalcemia, autoimmunity, calcific tropical pancreatitis, and idiopathic pancreatitis are not uncommon.28 The development of diabetes mellitus in patients with chronic pancreatitis is most frequent in those with calcific disease (55% to 70%) and occurs less often in those with noncalcific disease (30%).29 The prevalence of diabetes in patients with chronic pancreatitis increases with increasing duration of pancreatitis and with increasing exocrine deficiency.26,30–34
The inflammatory response causes loss of exocrine tissue, extensive fibrosis, and distorted and blocked ducts. The islets of Langerhans are relatively resistant and undergo pathologic changes only late in the disease. Chronic pancreatitis is associated with loss of functioning β cells and a somewhat lesser loss of α cells.17,35–41 Hormonal alterations seen include a decrease in insulin secretion in response to nutrients, followed later by a decrease in fasting C peptide levels. Plasma C peptide levels rather than insulin levels may be a better means of assessment of insulin secretion because associated liver disease may change hepatic extraction rates of insulin. With progressive chronic pancreatitis, insulin secretion falls even lower. Glucagon secretion is impaired in moderate to severe chronic pancreatitis. Insulin resistance frequently develops in such individuals.
Diabetes mellitus is seen after several years of chronic pancreatitis. In an unselected series of patients with chronic pancreatitis, 35% had type 1 diabetes, 31% had type 2 diabetes or IGT, and 34% had normal glucose tolerance.42 The nature of the diabetes reflects the severity of the chronic pancreatitis. Mild pancreatitis may be associated only with IGT, whereas severe pancreatitis is associated primarily with insulin-dependent (type 1) diabetes. Patients with chronic pancreatitis and diabetes mellitus fail to secrete glucagon in response to hypoglycemia. If patients have concomitant autonomic neuropathy, they are extremely susceptible to severe and prolonged hypoglycemia.
Treatment of diabetes mellitus in patients with chronic pancreatitis should entail the use of small doses of short-acting or rapid-acting insulins to manage the hyperglycemia, replacement enzymes for the malnutrition and malabsorption, and elimination of the use of alcoholic beverages.29,39,43,44 Surgery44 with subtotal resection or near-total resection may be necessary to relieve severe pain.45,46 Following pancreatic surgery for pain or other complications, a significant incidence of diabetes mellitus occurs; however, whether diabetes develops is dependent on the type of surgical intervention used. Whipple procedures usually are associated with an incidence of 25% to 40%, a Frey or Berne procedure 8% to 22%, and a distal pancreatectomy approximately 60%.47 In such patients, successful islet allotransplants and autotransplants in the liver have been able to maintain near normoglycemia.48 The hepatic islet cell transplants were able to secrete insulin in response to nutrients but were unable to secrete glucagon in response to hypoglycemia.18,19 A recent report of the results of pancreas allotransplants in patients who had undergone total pancreatectomy in the past showed a 70% survival rate and successful correction of both exocrine and endocrine deficiencies.49
Pancreatic exocrine function has been reported to be reduced in some type 1 and type 2 diabetic patients. This has been explained as a complication of the diabetes. A retrospective analysis of pancreatograms of patients with known diabetes has suggested that chronic pancreatitis may be much more common as a cause of diabetes than was previously thought.48 A total of 38 type 1 and 118 type 2 diabetic patients underwent endoscopic retrograde cholangiopancreatography (ERCP) studies for varying reasons. Pancreatic ducts were classified as normal in 23.3% and as exhibiting chronic pancreatitis degree I, II, and III in 22.7%, 32.7%, and 21.3%, respectively.50 The investigators suggested that a substantial number of patients with primary diabetes mellitus may have a concomitant chronic pancreatitis, or perhaps that many cases of primary diabetes mellitus may be related to chronic pancreatitis.
Pancreatic Cancer
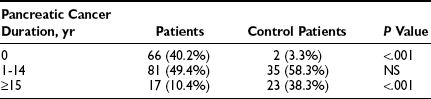
Diabetes mellitus is known to occur more frequently in patients with pancreatic cancer than in the general population.51–53 Published data indicate that as many as 70% of patients with pancreatic carcinoma have impaired glucose tolerance or frank diabetes mellitus, and that 60% demonstrate improved glucose metabolism after surgery.54 Wakasugi et al. reported that 53.1% of patients with invasive ductal pancreatic carcinoma had diabetes mellitus, and in 45.9% it was thought to be secondary to the carcinoma.55 The reasons for this association have been the subject of much speculation. Some studies show that diabetes mellitus is associated with increased risk for susceptibility to pancreatic cancer (2.15 to 4.9 in men).56,57 Other studies have indicated that in most patients with pancreatic carcinoma, the diabetes is secondary to some effect of the cancer, which causes insulin resistance and impairs the function of normal β cells.58–60 A multicenter case-control study of 720 patients with pancreatic cancer addressed this issue.51 The prevalence of diabetes in the patients with pancreatic cancer was 22.8%, whereas that in the matched control population was 8.3%. The patients with pancreatic cancer were characterized as having type 2 diabetes. A recent diagnosis of diabetes had been made in 40.2% of the patients with pancreatic cancer with diabetes, as contrasted with only 3.3% of the control population with diabetes (Table 17-4). A higher percentage of the control population with diabetes than of the pancreatic cancer population with diabetes had had their diabetes for longer than 15 years (see Table 17-4). These data support the idea that in a small number of patients, diabetes predisposes to the development of diabetes mellitus.
Table 17-4
Interval Between Diagnosis of Diabetes and Diagnosis of Pancreatic Cancer or Date of Examination of Control Population
Possible mechanisms by which pancreatic cancer could contribute to the development of type 2 diabetes include the following: (1) destruction of islets, (2) impairment of the insulin secretory mechanism, (3) development of insulin resistance, and (4) tumor-related pancreatitis. Insulin and C-peptide measurements during oral glucose tolerance testing in patients with pancreatic cancer have shown abnormal β cell function with reduced plasma C-peptide responses in 50% of patients and increased plasma proinsulin-to-C-peptide ratios61,62 Insulin resistance has been demonstrated in most patients with pancreatic carcinoma.63–65 Morphometric studies of tumor-free regions of the pancreas have shown reduced β cell populations.66 An inverse correlation was noted between the number of β cells and the fasting plasma glucose concentration. These data can be interpreted as indicating that pancreatic cancers produce substances or responses that destroy normal β cells.64–67 The presence of diabetes in patients with pancreatic cancer predicts that the tumor is less likely to be respectable and the patient has a poorer prognosis than if diabetes is not present.53 Patients with pancreatic cancer typically have type 2 diabetes; most have been treated with oral antihyperglycemic agents.52,53
Several very intriguing observations that were reported recently make this relationship between pancreatic cancer and diabetes mellitus even more complex and confusing. Li et al. reported that metformin treatment of diabetic patients was associated with a decreased risk of developing pancreatic carcinoma, while insulin or insulin secretogogue treatment was associated with an increased risk.68 In a large, multicenter clinical trial that compared the treatment of type 2 diabetic patients with rosiglitazone-based therapy versus metformin plus sulfonylurea therapy, the development of pancreatic cancer during the study was statistically less frequent in the rosiglitazone arm than in the active comparator arm (2 cases versus 13 cases; P < .007).69
Hemochromatosis
Hemochromatosis is an autosomal recessive genetic disorder that results in excessive deposition of iron in parenchymal cells of the liver, pancreas, muscle, heart, anterior pituitary, and other organs.70,71 The clinical diagnosis in the past was made by the findings of diabetes mellitus, hepatomegaly, and skin pigmentation. More recently, it has been recognized through biochemical and genetic testing.72 The first phenotypic expression of the disease is an elevation in serum transferrin saturation. This abnormality is followed by iron accumulation in the tissues and an elevation in the serum ferritin concentration. Early clinical findings are related to hepatic dysfunction and joint symptoms. Clinical diabetes mellitus and skin pigmentation occur relatively late in the course of the disease.71 A candidate gene for human leukocyte antigen (HLA)-linked hemochromatosis has been cloned and a mutation (C282Y) of the HFE gene identified that may account for 60% or more of cases of hereditary hemochromatosis. Another mutation (H63D) has been identified more recently.73
Diabetes mellitus has been reported in 50% to 60% of patients with hemochromatosis.71,74 Another 20% to 30% had glucose intolerance. These figures represent data from older series in which the diagnosis was made late in the course of the disease. Diabetes mellitus is more frequent in patients who have a family history of diabetes mellitus.
Follow-up of 237 patients who were given a diagnosis of hemochromatosis and treated at a single center from 1983 through 2005 has diabetes mellitus associated with the disease.73 Before 1996, hemochromatosis was diagnosed by the classic clinical and laboratory features of the disease. Since 1996, genetic testing, which is now available commercially, made it possible for them to diagnose hemochromatosis in asymptomatic patients. Of 45 patients diagnosed before 1996, 30.2% had cirrhosis of the liver and 35.6% had diabetes mellitus. After 1996, 192 patients were given the diagnosis of hemochromatosis (34% by family screening and 40% by clinical suspicion) and only 7.5% had cirrhosis of the liver and 17.7% had diabetes mellitus at the time of diagnosis. It is obvious that early through genetic testing and effective treatment can significantly reduce development of the complications of hemochromatosis. Of note was the observation that performing phlebotomy and reducing tissue iron in well-established clinical disease did not improve diabetes or lessen its management requirements. Thus early diagnosis and treatment is primarily preventative.73
Before 1996, hemochromatosis was diagnosed by the classic clinical and laboratory features of the disease. Since 1996, genetic testing, which is now available commercially, has made it possible to diagnose hemochromatosis in asymptomatic patients. Of the 45 patients diagnosed before 1996, 30.2% had cirrhosis of the liver and 35.6% had diabetes mellitus. After 1996, 192 patients were given the diagnosis of hemochromatosis (34% by family screening and 40% by clinical suspicion); only 7.5% had cirrhosis of the liver and 17.7% had diabetes at the time of diagnosis. It is obvious that early diagnosis through genetic testing and effective treatment have significantly reduced development of the complications. Of note is the observation that performing phlebotomy and reducing tissue iron did not improve diabetes or lessen its management requirements. Thus early diagnosis and treatment is primarily preventative.73
The metabolic studies that have been done show that patients with hemochromatosis have marked insulin resistance. Histologic study of the pancreas shows iron deposits that are greatest in the acinar cells but do involve islet cells. Insulin secretion in response to glucose or arginine is decreased; however, glucagon secretory responses to arginine are increased and unaffected by glucose.75,76 The data are compatible with a marked reduction in β cell function and no disturbance in α cell function. The hyperglycemia is a result of insulin resistance and decreased β cell function. The prevalence of diabetes mellitus probably could be reduced by early diagnosis of hemochromatosis and initiation of phlebotomy therapy.
Therapy for patients with hemochromatosis and clinical diabetes frequently requires insulin (40% to 50% of patients), although no systematic studies of therapy have been done.72 Reduction of tissue iron stores, although most beneficial in the early stages of disease, nonetheless can help improve glycemic control in 35% to 45% of patients.70,71
Hemosiderosis
Excessive iron deposition occurs in a variety of conditions other than primary hemochromatosis. In thalassemia major, frequent blood transfusions are necessary and may lead to massive iron overload. The reported prevalence of diabetes mellitus in treated thalassemia major is about 16%. This figure is highly correlated with the number of blood transfusions given and the duration of the disease. The incidence of IGT is reported to be 60%.77
Further evidence that deposits of excess tissue iron themselves are responsible for many of the metabolic abnormalities seen in hemochromatosis and thalassemia major comes from studies in rural male Bantus.78 Many Bantus drink alcoholic beverages that are brewed in iron containers and ingest in excess of 100 mg of iron per day. In those individuals, the prevalence of diabetes mellitus is 10-fold higher than in non–alcoholic beverage–consuming males.
Mechanistic studies in patients with thalassemia major with normal, impaired, and diabetic glucose tolerance tests show that increased iron stores are associated with the development of insulin resistance and a delay in early insulin secretion.79 A correlation between increased iron stores in normal women and the development of type 2 diabetes was demonstrated recently in the Nurses Health Study.80
Cystic Fibrosis
Cystic fibrosis (CF) is a monogenetic disorder with abnormal cyclic adenosine monophosphate–regulated chloride channel activity. It is an autosomal recessive genetic disease with an incidence of 1 in 2500 live births in Caucasian populations. More than 1000 gene mutations have been identified in the CF gene.81 Organs as diverse as the lung, exocrine pancreas, large and small intestine, hepatobiliary system, and sweat glands are involved. Failure to secrete Na+, HCO3−, and water leads to retention of enzymes in the pancreas and ultimately to destruction of pancreatic tissue.82–84 Histologic examination of the pancreas in patients with CF shows fatty infiltration, necrosis, and fibrosis of the exocrine pancreas. Islet cell architecture is disrupted and the absolute number of pancreatic islets diminished. Islets that are present show significant decreases in β cells, α cells, and pancreatic polypeptide–producing cells and increases in δ (somatostatin-producing) cells. Islet amyloid deposits have been found in 69% of diabetic CF cases examined.
Diabetes mellitus requiring medical therapy (usually insulin) has been reported in 4.9% of patients of all ages with CF in a large European study of 1348 patients85 and in 5.1% of 18,627 patients of all ages monitored at CF centers in the United States and Canada.82 Diabetes occurs more often in individuals who are homozygous for the most common CF mutation, ΔF508.82,86,87 Diabetes also occurs with greater frequency with increasing age; it has been reported in 32% of Danish patients who were older than 25 years. Routine oral glucose tolerance testing suggests that of the total CF population aged 5 years or older, 35% have normal glucose tolerance, 37% have IGT, 17% have CF-related diabetes without fasting hyperglycemia, and 11% have CF-related diabetes with fasting hyperglycemia.
Several features of CF-related diabetes are noteworthy. Autoantibodies to pancreatic heat shock protein 60 have been found to precede the development of glucose intolerance (IGT and diabetes) and to decline subsequently with the onset of glucose intolerance.88 The development of diabetes in patients with CF is characterized initially by abnormal oral glucose tolerance and a delay in oral glucose–stimulated insulin secretion, followed later by a decrease in total insulin, glucagon, and pancreatic polypeptide secretion.89 First-phase insulin secretion after intravenous glucose is reduced markedly in patients with CF compared with matched controls. Patients with CF with IGT have normal plasma free fatty acid levels as compared with matched controls.90 Their plasma tumor necrosis factor-α levels are elevated, and they have insulin resistance as measured by the hyperinsulinemic euglycemic clamp.90 Decreased translocation of Glut-4 glucose transporters in muscle was observed during the peak insulin effect. Patients with CF have an increase in hepatic glucose production and are resistant to suppression of hepatic glucose production by insulin even in the nondiabetic state.91 Peripheral insulin sensitivity is increased in healthy nondiabetic individuals with CF, but insulin resistance occurs later as IGT and diabetes develop, and patients experience additional complications related to their CF.92 The development of diabetes worsens pulmonary function and other clinical manifestations of CF and may increase mortality by up to sixfold.93 Insulin treatment appears to reduce the extent of this deterioration. Hyperglycemia in patients with CF can be intermittent or permanent. Intermittent hyperglycemia occurs with glucocorticoid therapy, infection, or stress and must be treated with insulin until it resolves. Permanent hyperglycemia is always treated with insulin.82,84 It is likely that patients with CF progress from intermittent hyperglycemia to permanent hyperglycemia as pancreatic destruction continues to occur.
A small short-term study (12 weeks) suggests that treatment of CF-related diabetes with glargine insulin may provide some advantages over treatment with NPH insulin.94
Microvascular complications have been observed in patients with CF-related diabetes with fasting hyperglycemia for longer than 10 years. Fourteen percent had microalbuminuria and 16% had retinopathy.95 Macrovascular disease appears to be uncommon.
Hyperglycemia Associated With Endocrinopathies
In the complex regulation of fuel homeostasis, many hormones other than insulin play a complementary role. Growth hormone (GH) by itself and through its synthesis of insulin-like growth factor-1 (IGF-1) controls many aspects of amino acid transport, protein synthesis, and lipid metabolism. Glucagon and catecholamines are counterregulatory hormones that protect against hypoglycemia and provide extra glucose when needed during stress states. Glucocorticoids exert both a permissive role in the normal physiologic regulation of gluconeogenesis and a pharmacologic role in providing increased glucose availability during stress. Somatostatin is a paracrine hormone that appears to act locally to help regulate the normal secretory patterns of GH, insulin, glucagon, and several gastrointestinal hormones.
This section will address the unique characteristics of hyperglycemia as it relates to each endocrinopathy and its treatment.
Acromegaly
Acromegaly is characterized by excessive and autonomous secretion of growth hormone and IGF-1.96,97 The prevalence of overt diabetes mellitus reported in different series of acromegalic patients ranges from 30% to 56%.96,98 IGT may be present in as many as 36% of acromegalic patients.96 In a specific population, the percentage of acromegalic patients in whom diabetes mellitus will develop depends on the prevalence of predisposition to type 2 diabetes in the population and the magnitude of elevation of serum IGF-1 levels.
Elevated GH and IGF-1 levels cause excessive hepatic glucose production and impaired insulin-mediated muscle glucose uptake.99–101 This insulin resistance is correlated with circulating IGF-1 levels and has been demonstrated by the euglycemic hyperinsulinemic clamp and the minimal model techniques.
Reduction in circulating GH and IGF-1 levels by successful surgical removal of the tumor producing growth hormone or GH-releasing factor results in significant improvement in glycemic control in acromegalic patients with diabetes mellitus.100,102 Recent data suggest that circulating GH must be lowered to 2 ng/L and IGF-1 lowered to the normal range to be considered curative.97,103,104 Transsphenoidal surgery achieves growth hormone levels less than 5 ng/L in approximately 60% of patients. Curative levels are attained in about 70% of patients with microadenomas (<10 mm in diameter) but are attained considerably less often in those with macroadenomas of the pituitary.97,103
Use of the somatostatin analogue octreotide to treat acromegaly as the primary medical therapy or to supplement prior inadequate surgical treatment or radiotherapy has allowed attainment of greater and more consistent reductions in circulating GH and IGF-1 levels (GH levels ≤5 ng/L in 65% and ≤2 ng/L in 40%, and IGF-1 levels in the normal range in 64% of patients).103–105
Treatment of acromegalic patients with octreotide presents several issues with respect to glucose metabolism.103–106 Reductions in circulating GH and IGF-1 levels will decrease insulin resistance and should lead to improvement in glycemic control in subjects with diabetes mellitus or IGT. However, pharmacologic doses of a somatostatin analogue also reduce insulin secretion (decreased insulinogenic index), and such a reduction should cause a deterioration in glucose tolerance. Thus in any particular patient, octreotide therapy will modify glucose metabolism in accordance with these competing effects. Approximately two thirds of acromegalic patients with diabetes mellitus are treated with insulin and one third with oral hypoglycemic agents. Octreotide treatment in patients with diabetes mellitus and acromegaly frequently leads to improvement in glycemic control as measured by a reduction in the insulin dose, conversion from insulin therapy to oral hypoglycemic agent therapy, or conversion from oral hypoglycemic agent therapy to dietary management.103,106 Some patients (those with more severe insulin deficiency), however, will have significant deterioration in glycemic control.104,106 When higher doses of octreotide are given, IGT and even frank diabetes mellitus (as high as 20% and 29%, respectively) may develop in acromegalic patients with normal glucose tolerance before octreotide treatment.106 An alternative to treatment with somatostatin analogues is treatment with the GH receptor blocker Pegvisomant. This agent will decrease IGF-1 levels and can improve glycemic control.107,108 However, it does cause an increase in visceral fat and occasionally is associated with increased liver enzymes.109
Appropriate treatment for acromegaly is necessary to reduce the increased mortality that has been seen in the past.110 This increased mortality is due to cardiovascular, cerebrovascular, and neoplastic diseases. The best determinants of outcome in acromegalic patients are age at diagnosis, interval between symptoms and diagnosis, and mean long-term circulating GH and IGF-1 levels. Because insulin resistance and diabetes mellitus contribute significantly to cardiovascular risk, aggressive diagnosis and management of the diabetes mellitus associated with acromegaly are essential.
Growth Hormone Treatment
The availability of recombinant DNA technology to make human GH has provided an opportunity to treat many GH-deficient individuals using this hormone. One consideration in treatment with recombinant human GH is the question of whether long-term treatment can lead to the development of diabetes mellitus.111 A recent study of glucose metabolism in 23,333 children and adolescents treated with human GH found a sixfold greater frequency of type 2 diabetes mellitus than predicted (34.4 cases per 100,000 years of GH treatment).112 In contrast, the frequency of type 1 diabetes was the same as expected. The data suggest that GH treatment accelerates the development of type 2 diabetes in individuals who have a genetic predisposition.
Cushing’s Syndrome
Glucocorticoids are insulin antagonistic hormones.113 When administered in pharmacologic doses, they increase basal hepatic glucose production and decrease the insulin-mediated effects caused by suppressing hepatic glucose production and increasing muscle glucose uptake.114–117 Insulin secretion is increased as a consequence of hepatic and peripheral insulin resistance.
Pharmacologic concentrations of glucocorticoids occur in disease states associated with autonomous secretion of adrenal cortical hormones (Cushing’s syndrome) or as the result of administration of such agents for the treatment of nonendocrine diseases. When sustained pharmacologic concentrations of glucocorticoids occur in normal individuals, increased insulin secretion maintains fasting plasma glucose within the normal range, but the postprandial plasma glucose concentration is elevated above normal in 25% to 90% of such individuals, depending on the magnitude of plasma glucocorticoid elevation.118 Individuals with limited β cell insulin secretory reserve are more subject to fasting hyperglycemia and type 2 diabetes mellitus. Ten percent to 20% of patients with Cushing’s syndrome have overt type 2 diabetes mellitus.119–123 Among renal transplant recipients receiving long-term corticosteroid therapy, steroid-induced diabetes mellitus has been reported to develop in as few as 5.5% and as many as 46% of patients.124,125 Factors that influence the development of diabetes mellitus during corticosteroid therapy include a family history of diabetes mellitus, increasing age, obesity, and both average daily and total cumulative corticosteroid dose.126,127 In individuals with a previous onset of diabetes mellitus, administration of glucocorticoids significantly worsens glycemic control and requires modification of diabetes management.
Recent studies have documented that steroid-induced diabetes mellitus occurs in adrenal disorders other than those associated with frank Cushing’s syndrome. Measurement of oral glucose tolerance in 64 consecutive patients with “nonfunctioning” adrenal adenomas identified normal glucose tolerance in 25, glucose intolerance in 17, and diabetes mellitus in 22, including 6 patients with previously diagnosed diabetes mellitus.128 Autonomous cortisol secretion without the clinical stigmata of Cushing’s syndrome has been recognized recently as preclinical Cushing’s syndrome. A retrospective study of 63 such individuals found that 17.5% had diabetes mellitus.129 A cross-sectional study of 90 obese, poorly controlled type 2 diabetic patients found that 3 had the preclinical Cushing’s syndrome abnormality.110 Excess IGT and diabetes mellitus have been reported in hypopituitary adults receiving conventional replacement therapy and may be related to the intermittently higher plasma levels that occur after dosing than would occur under normal hypothalamic-pituitary-adrenal axis function.130
Some insight into the mechanisms by which glucocorticoids cause diabetes mellitus was obtained by investigating the effects of administration of dexamethasone on oral glucose tolerance, on glucose turnover under basal conditions and during glucose infusion, and on the insulin response during hyperglycemic clamp studies in normal individuals who had been characterized previously as low insulin responders or high insulin responders.131 Dexamethasone caused a higher fasting plasma glucose concentration, a greater rise in plasma glucose, and a lesser rise in plasma insulin during the oral glucose tolerance test in the low insulin responders than in the higher insulin responders. A diabetic oral glucose tolerance test result was obtained in three of the six low and none of the six high insulin responders. Dexamethasone increased hepatic glucose production only in the low insulin responders and increased insulin secretion during the hyperglycemic clamp study only in the high insulin responders. The conclusion drawn from these studies is that type 2 diabetes develops when plasma glucocorticoids are elevated in individuals with limited β cell secretory function.
Steroid-induced diabetes may be permanent or transient. In general, insulin is required for treatment if the fasting plasma glucose concentration exceeds 180 mg/dL.132 At lesser levels of fasting hyperglycemia, many physicians treat the hyperglycemia with oral antihyperglycemic agents. Very few studies have evaluated the efficacy of pharmacologic treatment of steroid-induced diabetes mellitus. Because insulin resistance is a major abnormality, perhaps the combination of insulin sensitizers and insulin would be most effective. A reduction in corticosteroid dose or secretion improves glycemic control and in some individuals may even reverse the diabetes. The more severely elevated the fasting plasma glucose concentration, the less likely it is that reducing corticosteroid levels will reverse the diabetes. Ketoacidosis is very uncommon with steroid-induced diabetes or Cushing’s syndrome. Hyperosmolar nonketotic coma, however, is not uncommon.133
Glucagonoma Syndrome
Glucagon plays a primary role in facilitating the uptake of amino acids by the liver and their conversion into glucose by gluconeogenesis. Excess and unregulated glucagon secretion alone or in conjunction with other islet hormones occurs in some islet cell tumors. A classic syndrome has been described in individuals who have tumors secreting high quantities of glucagon. This syndrome was initially recognized in 1974 and is referred to as the glucagonoma syndrome.134 Features of this syndrome include necrolytic migratory erythema, mild non–insulin-requiring type 2 diabetes mellitus, glossitis, angular cheilitis, weight loss, and anemia.135,136 Laboratory studies show markedly elevated plasma glucagon levels and severe hypoaminoacidemia (<25% normal).
Glucagonomas are rare, with a reported incidence of 1 case per 20 to 200 million population. Several reviews of the literature indicate that most tumors occur in the tail of the pancreas (reported in 54% to 68%); they have an average tumor diameter of 3.6 cm (but as many as one third are less than 2 cm), are malignant in about two thirds of cases, and have metastases in other organs in 51% to 54% of patients at the time of diagnosis.137,138 The diabetes mellitus is characterized by mild hyperglycemia and is nonketotic. The tumors are relatively slow growing, and the 10-year survival rate is 52% in those with metastases and 64% in those without metastases.
The hyperglycemia is due to excess glucose production by the liver. The hypoaminoacidemia results from an increase in amino acid clearance.139 Necrolytic migratory erythema, glossitis, weight loss, and anemia are in large part a consequence of the protein malnutrition.136 Deep venous thrombosis that is not associated with coagulation disorders is common.
All components of the syndrome are improved if the hyperglucagonemia can be reduced. Treatment consists of surgical removal of the tumor, followed by hepatic artery embolization if necessary for liver metastases.136,140 Octreotide treatment has been very effective in reducing residual plasma glucagon levels.136,140 Cytotoxic agents such as streptozotocin and fluorouracil may be valuable as additional modes of therapy. Zinc and amino acid supplementation has been used to treat the rash but is relatively ineffective if plasma glucagon levels remain very high. Antiplatelet therapy should be used to prevent venous thrombosis. The hyperglycemia, although mild, generally requires treatment with an antihyperglycemic agent. Insulin would appear to be the most appropriate agent, although few or no clinical outcome data are available to support this hypothesis. Treatment of the hyperglucagonemia will ameliorate the hyperglycemia in most cases.140
Somatostatinoma
Case reports of patients with hyperglycemia and a pancreatic tumor containing large quantities of somatostatin first appeared in 1977.141,142 One of those patients became euglycemic after complete resection of the tumor was performed. Since that time, it has been recognized that large somatostatin-producing pancreatic tumors may be associated with a clinical syndrome consisting of hyperglycemia, cholelithiasis, steatorrhea, and hypochlorhydria.143
Somatostatin-producing tumors arising from the gastrointestinal tract and the pancreatic islets have been reported.144 Duodenal somatostatinomas with and without von Recklinghausen’s disease are seldom associated with recognizable somatostatinoma syndrome, often contain psammoma bodies, and may be associated with demonstrable metastases at the time of surgery.144 The clinical features of pancreatic somatostatin-producing tumors are variable, and this variation is related to differences in the quantity and qualitative features of somatostatin variants that are synthesized and secreted by these tumors.145–147 Marked differences in the degree to which insulin, glucagon, and growth hormone secretion are affected in various patients with pancreatic somatostatin-producing tumors probably account for some of the variation in the clinical syndrome. Hyperglycemia in patients with pancreatic somatostatinomas can vary from mild to modest hyperglycemia to severe diabetic ketoacidosis.144,148,149
Somatostatin infusions in humans are associated with a pronounced decrease in bile flow and bile acid secretion and an increase in bile cholesterol saturation.150 In vitro, somatostatin has a direct inhibitory effect on cholecystokinin stimulation of gallbladder contraction.151 These observations provide a basis for understanding the cholelithiasis and steatorrhea commonly seen with pancreatic somatostatinomas. Additionally, they explain the development of gallstones in 23.5% of acromegalic patients treated with octreotide during the first year of treatment.103
The hyperglycemia seen as part of the somatostatinoma syndrome most likely is related to suppression of insulin secretion. In some patients, a relative insulin deficiency leads to reduced peripheral glucose utilization without impairing suppression of hepatic glucose production.152 In more severe suppression of insulin secretion, both features of insulin action are reduced.
Somatostatin-producing tumors are rare, usually asymptomatic, or only mildly symptomatic and frequently remain undiagnosed for many years. The diagnosis frequently is made late and the prognosis is poor because of extensive metastases. The use of somatostatin receptor scintigraphy with indium 111–labeled pentetreotide promises to improve the ability to detect somatostatinomas earlier.153 Early diagnosis and surgical removal can lead to cure, but medical treatment has produced questionable results. A recent study suggests that somatostatinomas possess functioning somatostatin receptors and that octreotide therapy (0.5 mg/day subcutaneously) can effectively decrease somatostatin production by the tumor and improve diabetes and diarrhea.153
Pheochromocytoma
Glucose intolerance occurs in about 30% of patients with pheochromocytoma, but overt diabetes mellitus is uncommon.154 Mechanisms responsible for glucose intolerance include suppression of insulin by α-adrenergic receptor stimulation of β cells; an increase in insulin resistance, probably related to elevated plasma free fatty acid levels; and increased hepatic glucose output as a result of β-adrenergic stimulation of hepatocytes. α-Adrenergic receptor blockade improves glucose tolerance and insulin secretion.155,156 Removal of the pheochromocytoma restores glucose tolerance to normal in most cases. Treatment of glucose intolerance with antihyperglycemic agents is rarely required.153
Drugs That Can Cause Hyperglycemia
Blood glucose is regulated by the balance between insulin secretion and insulin action. A drug that destroys β cells or blocks their insulin secretory function will cause hyperglycemia in any individual (Table 17-5). A drug that directly or indirectly increases insulin resistance can cause hyperglycemia only in individuals with β cells that have limited insulin secretory reserve (individuals with a predisposition to type 2 diabetes). Several recent reviews on drug-induced disorders of glucose metabolism are available.157–159
Table 17-5
Drugs That Affect β Cell Function
Pentamidine and pyriminil (Vacor) are substances that resemble streptozotocin and alloxan chemically. Pentamidine is an antiprotozoal agent that is used extensively to treat Pneumocystis carinii infection. Pyriminyl is a nitrosourea-derived rodenticide that has been accidentally or intentionally ingested by humans. These agents cause necrosis of β cells, leading initially to hyperinsulinemia and hypoglycemia, followed by permanent hyperglycemia and an insulin-dependent diabetes mellitus.160–164 In a series of 128 patients with AIDS who were treated with pentamidine for P carinii pneumonia, severe glucose homeostasis disorders developed in 48 patients (37.5%), hypoglycemia in 7, hypoglycemia and then diabetes in 18, and diabetes alone in 23.162 Of the 41 patients in whom diabetes developed, 26 required insulin therapy. Risk factors for the development of dysglycemia include higher pentamidine doses, higher plasma creatinine, and more severe anoxia. Whereas most of the dysglycemic patients received parenteral pentamidine, six were treated exclusively with pentamidine aerosols. Pyriminyl ingestion has been followed by severe insulinopenic diabetes mellitus in numerous instances.164 Several fluoroquinolones have been reported to cause hypoglycemia or hyperglycemia. This effect occurs most dramatically with gatifloxacin. In a review of patients who were being admitted to hospitals in Ontario, Canada, for dysglycemia, gatifloxacin had an adjusted odds ratio for causing hypoglycemia of 4.3 and for causing hyperglycemia of 16.7 compared with macrolides or other non-fluoroquinolone antibiotics.165 This effect was not shared by all fluoroquinolones. Gatifloxacin stimulates insulin release by blocking the Kir 6.2 subunit of the β cell adenosine triphosphate (ATP)-dependent potassium channel.166 The mechanism for the cause of hyperglycemia is not known.
Drugs That Inhibit Increases in β Cell Cytosolic Ca2+
An increase in the β cell cytosolic Ca2+ concentration is the major mechanism responsible for insulin secretion. Calcium ion entry into the β cell is controlled by several calcium ion channels. A voltage-dependent L-type calcium channel has its activity linked to an ATP-dependent potassium channel (KATP channel). The KATP channel is closed by increases in plasma glucose. Drugs that keep the KATP channel open such as diazoxide (a KATP channel opener) block glucose-mediated insulin secretion and lead to hyperglycemia.167,168
Phenytoin (Dilantin) and other phenylhydantoins can interfere with Na+, K+, and Ca2+ ion transport. Dilantin administration has been reported to increase plasma glucose and decrease plasma insulin.168–171 Several studies indicate that it interferes with Ca2+ ion entry into the β cell, but by a process different from the KATP channel–related mechanism. Dilantin has been reported to cause hyperosmolar nonketotic hyperglycemia or diabetes mellitus. This complication occurs infrequently and probably in individuals with some preceding genetic susceptibility.
Drugs That Cause K+ Depletion
The effect of diuretics on glucose tolerance has been debated for close to two decades. Numerous older studies have shown that diuretics can cause a deterioration in glucose tolerance in nondiabetic individuals and worsening of glycemic control in patients with type 2 diabetes mellitus.172–175 The effect appears to be dose related and is either absent or markedly reduced with low-dose diuretic therapy.176–178 The Atherosclerosis Risk in Communities (ARIC) study compared 458 hypertensive patients treated with a thiazide diuretic for 6 years versus a control hypertensive cohort on no medications and found that the rates of development of new diabetes were no different.179 In contrast, the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT), which randomized 15,255 hypertensive patients to 12.5 to 25 mg/day of a diuretic, found that 11.6% of patients developed new-onset diabetes over 4 years of treatment.180 In contrast, the rate for patients randomized to a calcium channel blocker was 9.8% and to an angiotensin-converting enzyme (ACE) inhibitor 8.1%. In humans, diuretics appear to worsen glycemia primarily by inhibiting insulin secretion, although they also have a modest effect in increasing insulin resistance. Several studies have shown that the decreased insulin secretory response is due to intracellular K+ depletion and can be restored toward normal with potassium repletion.181–183 In the ALLHAT study, 8.5% of the cohort randomized to the diuretic had a serum potassium less than 3.5 mEq/L at 4 years of treatment, but only 1.9% of the cohort randomized to the calcium channel blocker had this degree of hypokalemia.180 Diabetogenic effects are seen with most diuretics. Therefore, if diuretics are used in diabetic or prediabetic patients to control blood pressure, doses should be restricted to those equivalent to 12.5 to 25.0 mg hydrochlorothiazide, and potassium supplements should be prescribed as needed.
Mechanisms Unknown
β-Adrenergic antagonists have been shown to worsen glycemic control in type 2 diabetic patients and to impair glucose tolerance or even precipitate type 2 diabetes in nondiabetic patients.172,184 A 6-year follow-up of hypertensive patients treated with β-adrenergic receptor antagonists in the ARIC study showed a 28% higher risk for development of diabetes than that in hypertensive patients not taking drug therapy.180 The deleterious effect of β-adrenergic blockade on glucose tolerance is worse with nonspecific β-adrenergic receptor blockade than with specific β2-adrenergic antagonists.184,185 Combination therapy with β-adrenergic antagonists and diuretics has an additive effect of worsening glucose tolerance. β-Adrenergic antagonists impair glucose tolerance and worsen hyperglycemia by blocking nutrient-mediated insulin secretion. β2-Adrenergic receptor activation stimulates and β2-adrenergic receptor blockade inhibits insulin secretion.186,187 Drugs with both α- and β-adrenergic receptor antagonist activity have no effect or may slightly improve glucose metabolism.
Cyclosporine and tacrolimus treatment for immunosuppression in renal transplant recipients is associated with an increased incidence of diabetes mellitus.188,189 Direct inhibitory effects of cyclosporine on β cells in vitro have been associated with decreased insulin secretion. Although it is likely that cyclosporine has direct effects in causing deterioration in glucose tolerance in renal transplant patients, it is not possible to exclude a contributory role of the associated corticosteroid therapy. The onset of diabetes mellitus in cyclosporine-treated patients occurs within the first several months of treatment and often requires insulin treatment. The reported incidence of diabetes mellitus and glucose intolerance in cyclosporine-treated transplant patients ranges from 13% to 47%.159
Other agents that have been reported to cause hyperglycemia by inhibiting insulin secretion are asparaginase and some opiates.157–159 Many of the newer cancer chemotherapeutic agents such as temsirolimus cause hyperglycemia as a significant but manageable side effect.190
Drugs That Cause Insulin Resistance
Drugs that increase insulin resistance will not affect glucose metabolism in individuals with normal β cell function. However, when administered to individuals with limited β cell reserve, they will cause glucose intolerance or overt diabetes mellitus. Agents that cause an increase in body weight and particularly those associated with an increase in central obesity will cause insulin resistance. Drugs or hormones that elevate plasma free fatty acid levels will lead to insulin resistance. Counterregulatory hormones or drugs that raise circulating levels of counterregulatory hormones will cause impairment of insulin action. The most commonly used agents that can increase insulin resistance are glucocorticoids, estrogens, progestogens, and nicotinic acid. Hyperglycemia associated with glucocorticoid therapy has been discussed in the section on Cushing’s syndrome.
Oral Contraceptives and Sex Hormones
Older studies investigating the effects of oral contraceptives, estrogens, and progestogens showed that deterioration of glucose tolerance and development of type 2 diabetes were occasional complications of long-term therapy.191 Most studies attributed the diabetogenic effects of these steroids to an increase in insulin resistance. More recent studies with natural estrogens and lower-dose administration indicate that these regimens produce little or no increase in glucose intolerance.192–194 Progesterone derivatives have been shown consistently to cause insulin resistance and impair glucose tolerance.195
Nicotinic Acid
Nicotinic acid is used extensively to treat mixed hyperlipidemias and has been shown to decrease morbidity and mortality from cardiovascular disease. Short-term administration of nicotinic acid reduced plasma free fatty acid levels by 30% to 40%, but as the drug effect wears off, the plasma free fatty acids rebound to 50% to 100% above baseline concentrations. Long-term administration of nicotinic acid, 1 to 4.5 g/day, causes severe insulin resistance, presumably as the result of elevated plasma free fatty acid levels. Normal individuals have a compensatory rise in insulin secretion, and glucose tolerance remains normal. In individuals with diminished insulin secretory reserve, nicotinic acid causes glucose intolerance and type 2 diabetes.196 In type 2 diabetic patients, nicotinic acid administration results in marked deterioration in glycemic control.197,198 Newer extended-release niacin preparations at doses of 1000 and 1500 mg/day have been shown to be very effective in raising plasma high-density lipoprotein (HDL) cholesterol and lowering plasma triglyceride levels with markedly reduced flushing and hepatotoxicity. The Assessment of Diabetes Control and Evaluation of the Efficacy of Niaspan Trial (ADVENT), which evaluated the effect of extended-release niacin in type 2 diabetic patients, showed no effect on glycemic control at 1000 mg/day and only a minor increase in HbA1c (+0.29%) over 16 weeks of therapy.199 These data indicate that the advantages of extended-release niacin in improving diabetic dyslipidemia far outweigh any detrimental effects on glycemic control.
Protease Inhibitors
The protease inhibitors indinavir, nelfinavir, ritonavir, and saquinavir have been used to treat patients with AIDS and have been reported to cause an unusual lipodystrophy. The main clinical features of this lipodystrophy are peripheral lipoatrophy of the face, limbs, and buttocks and central fat accumulation, including over the dorsocervical spine.200 Patients with HIV who were newly exposed to highly active antiretroviral therapy (HAART) were found to have prevalences of HIV lipodystrophy between 20% and 35% after 12 to 18 months of therapy.200 In addition to lipodystrophy, protease inhibitor therapy has been associated with the development of new hyperglycemia, hypercholesterolemia, and hypertriglyceridemia.200–203 The prevalence of new-onset hyperglycemia with protease inhibitor therapy has been reported to vary from 1% to approximately 6%. Tsiodras et al. presented data from a 5-year cohort study of 221 HIV-infected patients treated with protease inhibitors.204 The cumulative incidences of new-onset hyperglycemia, hypercholesterolemia, hypertriglyceridemia, and lipodystrophy were 5%, 24%, 19%, and 13%, respectively. The prevalence and incidence of hyperglycemia in HAART patients with HIV are influenced by concomitant hepatitis C virus infection. Retrospective analysis of a cohort of 1230 persons on their first HAART regimen revealed a prevalence of hyperglycemia in hepatitis C–coinfected patients of 5.9% versus 3.3% in uninfected patients.205 Hyperglycemia occurred in only one patient, who was neither infected with hepatitis C nor treated with protease inhibitors. The mechanism responsible for the protease-induced hyperglycemia appears to be the development of peripheral insulin resistance in skeletal muscle and adipose tissue.206 Pancreatic β cell function is also impaired, so that it cannot compensate for the insulin resistance.206,207 At the present time, it is unclear how the protease inhibitors cause lipodystrophy and other related metabolic side effects. The data indicate that protease inhibitor–mediated metabolic side effects are not related to virologic suppression, CD4 cell count, or changes in weight. Dube et al. reported seven new cases of diabetes occurring in a population of 1050 patients treated with protease inhibitors over a 9-month period, for a rate of less than 1%.202 Two patients were treated successfully with insulin and two with sulfonylureas. Two patients had resolution of their hyperglycemia by stopping indinavir therapy. Kilby and Tabereaux determined the frequency of severe hyperglycemia in a university clinic for HIV-1–infected patients and found a prevalence of less than 2%.202a Preexisting diabetes was present in 12 of 1392 adults, and new cases of hyperglycemia occurred in 13 of these 1392 adults.203 Most of the incident cases could be attributed to megestrol or corticosteroid treatment. The multiple factors that influence which HAART patients will develop new-onset hyperglycemia require further study because all series show a much lower prevalence of hyperglycemia than dyslipidemia or lipodystrophy.
Atypical Antipsychotic Agents
The atypical antipsychotic drugs represent several chemical classes of drugs that have been shown to have beneficial effects in patients with schizophrenia and manic-depressive disorder. They differ from older agents in that they are associated with significantly fewer extrapyramidal side effects and they provide superior beneficial effects.208 They bind to a variety of serotonin, dopamine, and histamine receptors.209 Atypical antipsychotic drugs include clozapine, olanzapine, risperidone, quetiapine, ziprasidone, and aripiprazole.
Shortly after their introduction into the market for the treatment of schizophrenia, case reports appeared in the literature documenting the development of new cases of diabetes mellitus during therapy with clozapine and olanzopine.210 Because clozapine and olanzapine cause considerable weight gain,211 the question was posed as to whether the weight gain might be responsible for the development of diabetes. It also was speculated that schizophrenia itself is associated with an increased prevalence of type 2 diabetes. Subsequently, numerous reports, large health care databases, and reviews have appeared in the literature citing new cases of diabetes and hypertriglyceridemia as complications of therapy with clozapine and olanzapine.209,212,213
As of 2004, the following data were available. Clozapine and olanzapine treatment was associated with a mean weight gain of approximately 10 kg over the first year of treatment. Risperidone and quetiapine cause weight gain of about 2 to 3 kg. In contrast, ziprasidone and aripiprazole treatment caused minor amounts of weight gain (0.5 to 1.0 kg over 1 year). Clozapine and olanzapine treatment have been shown in several studies to cause insulin resistance.214 Ziprasidone and aripiprazole did not appear to cause insulin resistance. The data on risperidone and quetiapine were not sufficient to confirm, but these agents appeared to have little effect. Patients taking clozapine showed a 5-year cumulative prevalence of diabetes of 35%.215 Published reports and MedWatch data from the FDA have been reviewed by Koller,216–218 who found new-onset cases of diabetes in 188 patients on olanzapine, 132 on risperidone, and 242 on clozapine. Approximately 40% of those cases presented as diabetic ketoacidosis. Seventy percent of patients developed diabetes within 6 months of starting olanzapine. When olanzapine was discontinued, 76% of patients exhibited improvement or remission of their diabetes. Among 10 patients who had had a remission and were rechallenged with olanzapine, a recurrence of diabetes occurred in 80%. Although the development of new-onset diabetes was frequently associated with significant weight gain, this was not always the case. Ziprasidone and aripiprazole have been on the market for a shorter time than the other atypical antipsychotic drugs, but patient exposure was sufficient to reveal that they are unlikely to cause the metabolic complications associated with the other atypical antipsychotic drugs.
In addition to diabetes, clozapine and olanzapine appeared to cause hypertriglyceridemia in a modest number of patients.
Because no randomized, controlled comparative studies have investigated the effects of the different atypical antipsychotic agents on the development of diabetes, current conclusions must be viewed with some reservations. The FDA has taken the position that all atypical antipsychotic agents should be viewed as creating a risk for new-onset diabetes. A recent joint consensus conference sponsored by the American Diabetes Association and the American Psychiatric Society came to a different conclusion. Their conclusions can be summarized as follows219:
The prevalence of the three adverse conditions (obesity, diabetes, dyslipidemia) differs among the second-generation antipsychotics:
1. Clozapine and olanzapine are associated with the greatest weight gain and with diabetes and dyslipidemia.
2. Risperidone and quetiapine appear to have intermediate effects.
3. Aripiprazole and ziprasidone are associated with little or no significant weight gain, diabetes, or dyslipidemia but have less usage exposure.
The consensus panel made the following recommendations:
1. If a patient gains 5% of his or her initial weight during therapy, one should consider switching the specific antipsychotic agent.
2. For patients who develop worsening glycemia or dyslipidemia while on antipsychotic therapy, the panel recommends considering switching to an agent that has not been associated with weight gain or diabetes.
Unfortunately, little progress has been made in further defining the risks or the mechanisms of the metabolic abnormalities associated with the different atypical antipsychotics. A large randomized, initially double-blind, controlled clinical trial comparing the effectiveness and safety of olanzapine, risperidone, quetiapine (a first-generation antipsychotic), perphenazine, and ziprasidone was carried out in 1493 patients from January 2001 through December 2004 under the auspices of the National Institute of Mental Health; it was called the CATIE (Clinical Antipsychotic Trial of Intervention Effectiveness).220 The first phase was projected to last 18 months; however, 74.5% of patients discontinued their medications before the end of the first phase. The results suggested that olanzapine was somewhat more effective in controlling the symptoms of schizophrenia; however, its use was associated with a mean weight gain of 9.4 kg, an increase in plasma glucose of 15 mg/dL, an increase in HbA1c of 0.41%, an increase in plasma cholesterol of 9.7 mg/dL, and an increase in plasma triglyceride of 42.9 mg/dL. Quetiapine and risperidone showed minimal increases compared with the first-generation agent, and ziprasidone caused no weight, glycemic, or lipid effects. Because of the high rate of discontinuation of initial medications, the validity of the data must be interpreted with caution. Scheen and De Hert performed a prospective open nonrandomized study comparing the metabolic effects of six atypical antipsychotics in 238 schizophrenic patients who underwent a fasting analysis and an oral glucose tolerance test at baseline and after 3 months of treatment.221 Weight gain, glucose alterations, incidence of new diabetes, and development of metabolic syndrome were greatest with clozapine, followed by olanzapine then quetiapine, risperidone, and amisulpride. Aripiprazole was not associated with weight gain and actually improved glucose tolerance and reduced the occurrence of metabolic syndrome. Thus the newer data, although not ideal, confirm the conclusions derived from previous reports.
Another 6-month multicenter randomized, double-blind trial in 59 patients attempted to determine the mechanisms by which olanzapine and risperidone caused their metabolic effects.222 Patients had baseline anthropomorphic measurements and measurement of total body fat by dual-energy x-ray absorptiometry (DEXA) analysis, visceral fat by abdominal computed tomography (CT), and insulin sensitivity and disposition index by frequently sampled intravenous glucose tolerance using the Bergman technique. Patients were randomized to olanzapine or risperidone and the studies repeated at 6 months. Both drugs induced weight gain and increased total body and visceral fat. Insulin sensitivity did not deteriorate, nor was a significant change noted in disposition index (a composite measure of changes in insulin sensitivity and insulin secretion). A subset analysis of African American and Hispanic subjects suggested that they had greater susceptibility to the detrimental effects of olanzapine in increasing insulin resistance and decreasing pancreatic compensation to insulin resistance.
Thus it seems fair to conclude that weight gain plays some role in the development of the metabolic effects of atypical antipsychotic agents. However, other unknown factors also contribute and the factors contributing to β cell failure are still unknown. Clozapine and olanzapine are the worst offenders in causing metabolic side effects, quetiapine and risperidone cause significantly fewer metabolic alterations, and ziprasidone and aripiprazone seem to produce no significant metabolic side effects.
References
1. Baggio, LL, Drucker, DJ. Biology of incretins: GLP-1 and GIP. Gastroenterology. 2007;132:2131–2157.
2. Yki-Jarvinen, H. Action of insulin on glucose metabolism in vivo. Baillieres Clin Endocrinol Metab. 1993;7:903–927.
3. Lefebvre, PJ. Biosynthesis and action of glucagon. In: Alberti KGMM, Zimmet P, DeFronzo RA, et al, eds. International textbook of diabetes mellitus. ed 2. New York: John Wiley & Sons; 1997:383–389.
4. Gerich, JE. The genetic basis of type 2 diabetes mellitus: impaired insulin secretion versus impaired insulin sensitivity. Endocr Rev. 1998;19:491–503.
5. Barnes, AJ, Bloom, SR, George, K, et al. Ketoacidosis in pancreatectomized man. N Engl J Med. 1977;296:1250–1253.
6. Barnes, AJ, Bloom, SR, Mashiter, K, et al. Persistent metabolic abnormalities in diabetes in the absence of glucagon. Diabetologia. 1977;13:71–75.
7. Barnes, AJ, Bloom, SR. Pancreatectomized man: a model for diabetes without glucagon. Lancet. 1976;1:219–221.
8. Morrow, CE, Cohen, JI, Sutherland, DER, et al. Chronic pancreatitis: long-term surgical results of pancreatic duct drainage, pancreatic resection, and near-total pancreatectomy and islet transplantation. Surgery. 1984;90:608–915.
9. Yasugi, H, Mizumoto, R, Sakurai, H, et al. Changes in carbohydrate metabolism and endocrine function of remnant pancreas after major pancreatic resection. Am J Surg. 1976;132:577–580.
10. Tiengo, A, Bessioud, M, Valverde, I, et al. Absence of islet alpha cell function in pancreatectomized patients. Diabetologia. 1982;22:25–32.
11. Nakamura, T, Takebe, K, Kudoh, K, et al. Increased plasma gluconeogenic and system A amino acids in patients with pancreatic diabetes due to chronic pancreatitis in comparison with primary diabetes. Tohoku J Exp Med. 1994;173:413–420.
12. Kendall, DM, Sutherland, DER, Najarian, JS, et al. Effects of hemipancreatectomy on insulin secretion and glucose tolerance in healthy humans. N Engl J Med. 1990;322:898–903.
13. Robertson, RP, Lanz, KJ, Sutherland, DE, et al. Relationship between diabetes and obesity 9 to 18 years after hemipancreatectomy and transplantation in donors and recipients. Transplantation. 2002;73:736–741.
14. Kumar, AF, Gruessner, RWG, Seaquist, ER. Risk of glucose intolerance and diabetes in hemipancreatectomized donors selected for normal preoperative glucose metabolism. Diabetes Care. 2008;31:1639–1643.
15. Sun, AM, Coddling, JA, Haist, RE. A study of glucose tolerance and insulin response in partially depancreatized dogs. Diabetes. 1974;23:424–432.
16. Polonsky, KS, Herold, KC, Gilden, JL, et al. Glucose counterregulation in patients after pancreatectomy: comparison with other clinical forms of diabetes. Diabetes. 1984;33:1112–1119.
17. Del Prato, S, Tiengo, A, Baccaglini, U, et al. Effect of insulin replacement on intermediary metabolism in diabetes secondary to pancreatectomy. Diabetologia. 1983;25:252–259.
18. Kendall, DM, Teuscher, AU, Robertson, RP. Defective glucagon secretion during sustained hypoglycemia following successful islet allo- and autotransplantation in humans. Diabetes. 1997;46:23–27.
19. Redmon, JB, Teuscher, AU, Robertson, RP. Hypoglycemia after pancreas transplantation. Diabetes Care. 1998;12:1944–1950.
20. Slezak, LA, Andersen, DK. Pancreatic resection: effects on glucose metabolism. World J Surg. 2001;25:452–460.
21. Nakamura, T, Takebe, K, Kudoh, K, et al. Decreased counter-regulatory hormone responses to insulin-induced hypoglycemia in patients with pancreatic diabetes having autonomic neuropathy. Tohoku J Exp Med. 1994;174:305–315.
22. Jethwa, P, Sodergren, M, Lala, A, et al. Diabetic control after total pancreatectomy. Dig Liver Dis. 2006;38:415–419.
23. Pezzilli, R. Commentary—diabetic control after pancreatectomy. Dig Liver Dis. 2006;38:420–422.
24. Ganda, OP. Secondary forms of diabetes. In: Kahn CR, Weir GC, eds. Joselin’s diabetes mellitus. ed 13. Philadelphia: Lea & Febiger; 1994:300–316.
25. Sarles, H. Chronic pancreatitis and diabetes. Baillieres Clin Endocrinol Metab. 1992;64:745–775.
26. Larsen, S. Diabetes mellitus secondary to chronic pancreatitis. Dan Med Bull. 1993;40:153–162.
27. Koizumi, M, Yoshida, Y, Abe, N, et al. Pancreatic diabetes in Japan. Pancreas. 1998;16:385–391.
28. Witt, H, Apte, MV, Keim, V, et al. Chronic pancreatitis: challenges and advances in pathogenesis, genetics, diagnosis and therapy. Gastroenterology. 2007;132:1557–1573.
29. DelPrato, S, Tiengo, A. Diabetes secondary to acquired diseases of the pancreas. In: Alberti KGMM, Zimmet P, DeFronzo RA, et al, eds. International textbook of diabetes mellitus. ed 2. New York: John Wiley & Sons; 1997:189–212.
30. Sjoberg, RJ, Kidd, GS. Pancreatic diabetes. Diabetes Care. 1989;12:715–724.
31. Banks, S, Marks, IN, Vinik, AL. Clinical and hormonal aspects of pancreatic diabetes. Am J Gastroenterol. 1975;64:13–22.
32. Nakamura, T, Imamura, K, Takebe, K, et al. Correlation between pancreatic endocrine and exocrine function and characteristics of pancreatic endocrine function in patients with diabetes mellitus owing to chronic pancreatitis. Int J Pancreatol. 1996;20:169–175.
33. Anagnostides, AA, Cos, TM, Adrian, TE, et al. Pancreatic exocrine and endocrine response in chronic pancreatitis. Am J Gastroenterol. 1984;79:206–212.
34. Kalk, WJ, Vinik, AI, Jackson, WPU, et al. Insulin secretion and pancreatic exocrine function in patients with chronic pancreatitis. Diabetologia. 1979;16:355–358.
35. Kalk, WJ, Vinik, AI, Bank, S, et al. Selective loss of beta cell response to glucose in chronic pancreatitis. Horm Metab Res. 1974;6:95–98.
36. Joffe, BI, Bank, S, Jackson, WP, et al. Insulin reserve in patients with chronic pancreatitis. Lancet. 1968;2:890–892.
37. McKiddie, MT, Buchanan, KD, McBain, GC, et al. The insulin response to glucose in patients with pancreatic disease. Postgrad Med J. 1969;45:726–730.
38. Nyboe Andersen, B, Krarup, T, Thorsgaard Pedersen, N, et al. β cell function in patients with chronic pancreatitis and its relation to exocrine pancreatic function. Diabetologia. 1982;23:86–89.
39. Larsen, S, Hilsted, J, Tronier, B, et al. Metabolic control and β cell function in patients with insulin-dependent diabetes mellitus secondary to chronic pancreatitis. Metabolism. 1987;36:964–967.
40. Duckworth, WC, Solomon, SS, Jallepalli, P, et al. Hormonal response to intravenous glucose and arginine in patients with pancreatitis. Horm Res. 1983;17:65–73.
41. Nealon, WH, Townsend, CM, Thompson, JC. The time course of beta cell dysfunction in chronic ethanol-induced pancreatitis: a prospective analysis. Surgery. 1988;104:1074–1079.
42. Larsen, S, Hilsted, J, Tronier, B, et al. Metabolic control and β cell function in patients with insulin-dependent diabetes mellitus secondary to chronic pancreatitis. Metabolism. 1987;36:964–967.
43. Yasida, H, Harand, Y, Ohgaku, S, et al. Insulin sensitivity in pancreatitis, liver disease, steroid treatment and hyperthyroidism assessed by glucose, insulin and somatostatin infusions. Horm Metab Res. 1984;16:3–6.
44. Marks, V. Alcohol and carbohydrate metabolism. Clin Endocrinol Metab. 1978;7:333–349.
45. Schoenberg, MH, Schlosser, W, Ruck, W, et al. Distal pancreatectomy in chronic pancreatitis. Dig Surg. 1999;16:130–136.
46. Buhler, L, Schimdlin, F, de Perrot, M, et al. Long-term results after surgical management of chronic pancreatitis. Hepatogastroenterology. 1999;46:1986–1989.
47. Gaia, E, Salacone, P. Medical complications of pancreatic resections: conference report. J Pancreas. 2007;8(suppl 1):114–117.
48. Teuscher, AU, Kendell, DM, Smets, FC, et al. Successful islet autotransplantation in humans. Diabetes. 1998;47:324–330.
49. Gruessner, RWG, Sutherland, DER, Drangstveit, MB, et al. Pancreas allotransplants in patients with a previous total pancreatectomy for chronic pancreatitis. J Am Coll Surg. 2008;206:458–465.
50. Hardt, PD, Killinger, A, Nalop, J, et al. Chronic pancreatitis and diabetes mellitus: a retrospective analysis of 156 ERCP investigations in patients with insulin-dependent and non-insulin-dependent diabetes mellitus. Pancreatology. 2002;2:30–33.
51. Karmody, AJ, Kyle, J. The association between carcinoma of the pancreas and diabetes mellitus. Br J Surg. 1969;56:362–364.
52. Gullo, L, Pezzilli, R, Morselli-Labate, AM, et al. Diabetes and the risk of pancreatic cancer. N Engl J Med. 1994;331:81–84.
53. Rosewicz, S, Wiedenmann, B. Pancreatic carcinoma. Lancet. 1997;349:485–489.
54. Suruc, M, Pour, PM. Diabetes and its relationship to pancreatic carcinoma. Pancreas. 2003;26:381–387.
55. Wakasugi, H, Funakoshi, A, Iguchi, H. Clinical observations of pancreatic diabetes caused by pancreatic carcinoma, and survival period. Int J Clin Oncol. 2001;6:50–54.
56. Gapstur, SM, Gann, PH, Lowe, W, et al. Abnormal glucose metabolism and pancreatic cancer mortality. JAMA. 2000;283:2552–2558.
57. Balkau, B, Barrett-Connor, E, Eschwege, E, et al. Diabetes and pancreatic carcinoma. Diabetes Metab. 2003;19:458–462.
58. Gullo, L, Ancona, D, Pezzilli, R, et al. Glucose tolerance and insulin secretion in pancreatic cancer. Ital J Gastroenterol. 1993;25:487–489.
59. Permert, J, Ihse, I, Jorfeldt, L, et al. Pancreatic cancer is associated with impaired glucose metabolism. Eur J Surg. 1993;159:101–107.
60. Fogar, P, Basso, D, Panozzo, MP, et al. C-peptide pattern in patients with pancreatic cancer. Anticancer Res. 1993;13:2577–2580.
61. Basso, D, Plebani, M, Fogar, P, et al. Beta-cell function in pancreatic adenocarcinoma. Pancreas. 1994;9:332–335.
62. Nakamori, S, Ishikawa, O, Ohigashi, H, et al. Increased blood proinsulin and decreased C-peptide levels in patients with pancreatic cancer. Hepatogastroenterology. 1999;46:16–24.
63. Schwartz, SS, Zeidler, A, Moossa, AR, et al. A prospective study of glucose intolerance, insulin, C-peptide and glucagon response in patients with pancreatic carcinoma. Am J Dig Dis. 1978;23:1107–1114.
64. Ahren, B, Andren-Sandberg, A. Capacity to secrete islet hormones after subtotal pancreatectomy for pancreatic cancer. Eur J Surg. 1993;159:223–227.
65. Perhert, J, Ihse, I, Jorfeldt, L, et al. Improved glucose metabolism after subtotal pancreatectomy for pancreatic cancer. Br J Surg. 1993;80:1047–1050.
66. Hayashida, CY, Suzuki, K, Fujiya, H, et al. Morphometrical quantitation of pancreatic endocrine cells in patients with carcinoma of the pancreas. Tohoku J Exp Med. 1983;141:311–322.
67. Noy, A, Bilezikian, JP. Clinical review 63: diabetes and pancreatic cancer: clues to early diagnosis of pancreatic malignancy. J Clin Endocrinol Metab. 1994;79:1223–1231.
68. Li, D, Yeung, S-CJ, Hassan, MM, et al. Antidiabetic therapies affect risk of pancreatic cancer. Gastroenterology. 2009;137:482–488.
69. Home, PD, Pocock, SJ, Beck-Nielsen, H, et al. Rosiglitazone evaluated for cardiovascular outcomes in oral agent combination therapy for type 2 diabetes (RECORD): a multicentre, randomized, open-label trial. Lancet. 2009;373:2125–2135.
70. Powell, LW, George, KD, McDonnell, SM, et al. Diagnosis of hemochromatosis. Ann Intern Med. 1998;129(suppl):925–931.
71. Milman, N. Hereditary haemochromatosis in Denmark 1950–1985: clinical, biochemical and histologic features in 179 patients and 13 preclinical cases. Dan Med Bull. 1991;38:385–393.
72. O’Brien, T, Barrett, B, Murray, DM, et al. Usefulness of biochemical screening of diabetic patients for hemochromatosis. Diabetes Care. 1990;13:532–534.
73. O’Sullivan, EP, McDermott, JH, Murphy, MS, et al. Declining prevalence of diabetes mellitus in hereditary haemochromatosis—the results of earlier diagnosis. Diabetes Res Clin Pract. 2008;81:316–320.
74. Saddi, R, Feingold, J. Idiopathic hemochromatosis and diabetes mellitus. Clin Genet. 1974;5:242–247.
75. Passa, P, Luyckx, AS, Carpentier, JL, et al. Glucagon secretion in diabetic patients with hemochromatosis. Diabetologia. 1977;13:509–513.
76. Nelson, RL, Baldus, WP, Rubenstein, AH, et al. Pancreatic alpha-cell function in diabetic hemochromatotic subjects. J Clin Endocrinol Metab. 1979;49:412–416.
77. Saudek, CD, Hemm, RM, Peterson, CM. Abnormal glucose tolerance in β-thalassemia major. Metabolism. 1977;26:43–52.
78. Isaacson, C, Seftel, SC, Keeley, KJ, et al. Siderosis in the Bantu: the relationship between iron overload and cirrhosis. J Lab Clin Med. 1961;58:845–853.
79. Cario, H, Holl, RW, Debatin, KM, et al. Insulin sensitivity and beta-cell secretion in thalassaemia major with secondary haemochromatosis: assessment by oral glucose tolerance test. Eur J Pediatr. 2003;162:139–146.
80. Jiang, R, Manson, JE, Meigs, JB, et al. Body iron stores in relation to risk of type 2 diabetes in apparently healthy women. JAMA. 2004;291:711–717.
81. Alves, CDAD, Aguiar, RA, Alves, AC, et al. Diabetes mellitus in patients with cystic fibrosis. J Bras Pneumol. 2007;33:213–221.
82. Moran, A, Doherty, L, Wang, X, et al. Abnormal glucose metabolism in cystic fibrosis. J Pediatr. 1998;133:10–17.
83. Cucinotta, D, DeLuca, F, Scoglio, R, et al. Factors affecting diabetes mellitus onset in cystic fibrosis: evidence from a 10-year follow-up study. Acta Paediatr. 1999;88:339–343.
84. Lanng, S. Glucose intolerance in cystic fibrosis. Dan Med Bull. 1997;44:23–29.
85. Rosenecker, J, Eichler, I, Kuhn, L, et al. Genetic determination of diabetes mellitus in patients with cystic fibrosis. J Pediatr. 1995;127:441–443.
86. Allen, HF, Gay, EC, Klingensmith, GJ, et al. Identification and treatment of cystic fibrosis related diabetes. Diabetes Care. 1998;21:943–948.
87. Yung, B, Hodson, ME. Diabetes in cystic fibrosis. J R Soc Med. 1999;92(suppl 37):35–40.
88. Jensen, P, Johansen, HK, Carmi, P, et al. Autoantibodies to pancreatic hsp60 precede the development of glucose intolerance in patients with cystic fibrosis. J Autoimmun. 2001;17:165–172.
89. Lanng, S. Glucose intolerance in cystic fibrosis patients. Paediatr Respir Rev. 2001;2:253–259.
90. Holl, RW, Heinze, E, Wolf, A, et al. Reduced pancreatic insulin release and reduced peripheral insulin sensitivity contribute to hyperglycaemia in cystic fibrosis. Eur J Pediatr. 1995;154:356–361.
91. Hardin, DS, LeBlanc, A, Para, L, et al. Hepatic insulin resistance and defects in substrate utilization in cystic fibrosis. Diabetes. 1999;48:1082–1087.
92. Hardin, DS, Leblanc, A, Marshall, G, et al. Mechanisms of insulin resistance in cystic fibrosis. Am J Physiol Endocrinol Metab. 2001;281:E1022–E1028.
93. Mackie, AD, Thornton, SJ, Edenborough, FP. Cystic fibrosis-related diabetes. Diabet Med. 2003;20:425–436.
94. Grover, P, Thomas, W, Moran, A. Glargine versus NPH insulin in cystic fibrosis related diabetes. J Cyst Fibros. 2008;7:134–136.
95. Schwarzenberg, SJ, Thomas, W, Olsen, TW, et al. Microvascular complications in cystic fibrosis–related diabetes. Diabetes Care. 2007;30:1056–1061.
96. Ezzat, S, Forster, MJ, Berchtold, P, et al. Acromegaly, clinical and biochemical features in 500 patients. Medicine (Baltimore). 1994;73:233–240.
97. Melmed, S, Ho, K, Klibanski, A, et al. Recent advances in pathogenesis, diagnosis and management of acromegaly (Clinical Review 75). J Clin Endocrinol Metab. 1995;80:3395–3402.
98. Arya, KR, Pathare, AV, Chadda, M, et al. Diabetes in acromegaly—a study of 34 cases. J Indian Med Assoc. 1997;95:546–547.
99. Moller, N, Jorgensen, JO, Abildgard, N, et al. Effects of growth hormone on glucose metabolism. Horm Res. 1991;36(suppl 1):32–35.
100. Wasada, T, Aoki, K, Sato, A, et al. Assessment of insulin resistance in acromegaly associated with diabetes mellitus before and after transsphenoidal adenomectomy. Endocr J. 1997;44:617–620.
101. Garcia-Estevez, DA, Araujo-Vilar, D, Cabelas-Cerato, J. Non–insulin-mediated glucose uptake in several insulin-resistant states in the postabsorptive period. Diabetes Res Clin Pract. 1998;39:107–113.
102. Szeto, CC, Li, KY, Ko, GT, et al. Acromegaly in a woman presenting with diabetic ketoacidosis and insulin resistance. Int J Clin Pract. 1997;51:476–477.
103. Colao, A, Ferone, D, Cappabianca, P, et al. Effect of octreotide pretreatment on surgical outcome in acromegaly. J Clin Endocrinol Metab. 1997;82:3308–3314.
104. Newman, CB, Melmed, S, Snyder, PJ, et al. Safety and efficacy of long-term octreotide therapy of acromegaly: results of a multicenter trial in 103 patients—a clinical research center study. J Clin Endocrinol Metab. 1995;80:2768–2775.
105. Arosio, M, Macchelli, S, Ross, CM, et al. Effects of treatment with octreotide in acromegalic patients—a multicenter Italian study, Italian Multicenter Octreotide Study Group. Eur J Endocrinol. 1995;133:430–439.
106. Koop, BL, Harris, AG, Ezzat, S. Effect of octreotide on glucose tolerance in acromegaly. Eur J Endocrinol. 1994;130:581–586.
107. Hodish, I, Barkan, A. Long-term effects of pegvisomant in patients with acromegaly. Nat Clin Pract Endocrinol Metab. 2008;4:324–332.
108. Barkin, AL, Burman, P, Clemmons, DR, et al. Glucose homeostasis and safety in patients with acromegaly converted from long acting octreotide to pegvisomant. J Clin Endocrinol Metab. 2005;90:5684–5691.
109. Plockinger, U, Reuter, T. Pegvisomant increases intra-abdominal fat in patients with acromegaly: a pilot study. Eur J Endocrinol. 2008;158:467–471.
110. Bates, AS, Van’t Hoff, W, Jones, JM, et al. An audit of outcome of treatment in acromegaly. Q J Med. 1993;86:293–299.
111. Sonksen, PH, Russell-Jones, D, Jones, RH. Growth hormone and diabetes mellitus: a review of sixty-three years of medical research and a glimpse into the future? Horm Res. 1993;40:68–79.
112. Cutfield, WS, Wilton, P, Bennmarker, H, et al. Incidence of diabetes mellitus and impaired glucose tolerance in children and adolescents receiving growth-hormone treatment. Lancet. 2000;355:610–613.
113. McMahon, M, Gerich, J, Rizza, R. Effect of glucocorticoids on carbohydrate metabolism. Diabetes Metab Rev. 1988;4:17–30.
114. Rizza, RA, Mandarino, LJ, Gerich, JE. Cortisol-induced insulin resistance in man: impaired suppression of glucose production and stimulation of glucose utilization due to postreceptor defect of insulin action. J Clin Endocrinol Metab. 1982;54:131–138.
115. Shamoon, H, Soman, V, Sherwin, RS. The influence of acute physiological increments of cortisol on fuel metabolism and insulin binding to monocytes in normal humans. J Clin Endocrinol Metab. 1980;50:495–501.
116. Olefsky, JM, Kimmerling, G. Effects of glucocorticoids on carbohydrate metabolism. Am J Med Sci. 1976;271:201–210.
117. Nosadini, R, Del Prato, S, Tiengo, A, et al. Insulin resistance in Cushing’s syndrome. J Clin Endocrinol Metab. 1983;57:529–536.
118. Conn, JM, Fajans, SS. Influence of adrenal cortical steroids on carbohydrate metabolism in man. Metabolism. 1956;5:114–127.
119. Boyle, PJ. Cushing’s disease, glucocorticoid excess, glucocorticoid deficiency and diabetes. Diabetes Rev. 1993;1:301–308.
120. Plotz, CM, Knowlton, AI, Ragan, C. The natural history of Cushing’s syndrome. Am J Med. 1952;13:597–614.
121. Urbanic, RC, George, JM. Cushing’s disease—18 years experience. Medicine (Baltimore). 1961;60:14–24.
122. Ross, EJ, Linch, DC. Cushing’s syndrome—killing disease: discriminatory value of signs and symptoms aiding early diagnosis. Lancet. 1982;2:646–649.
123. Soffer, L, Iannaccone, A, Gabrilove, J. Cushing’s syndrome: A study of fifty patients. Arch Intern Med. 1961;300:215–219.
124. Roth, D, Milgram, M, Esquenazi, V, et al. Posttransplant hyperglycemia: increased incidence in cyclosporin-treated renal allograft recipients. Transplantation. 1989;47:278–281.
125. Arner, P, Gunnarsson, R, Blomdahl, S, et al. Some characteristics of steroid diabetes: a study in renal-transplant recipients receiving high dose corticosteroid therapy. Diabetes Care. 1983;6:23–25.
126. Gurwitz, JH, Bohn, RL, Glynn, RJ, et al. Glucocorticoids and the risk for initiation of hypoglycemic therapy. Arch Intern Med. 1994;154:97–101.
127. Weissman, DE, Dufer, D, Vogel, V, et al. Corticosteroid toxicity in neuro-oncology patients. J Neurooncol. 1987;5:125–128.
128. Fernandez-Real, JM, Engel, WR, Simo, R, et al. Study of glucose tolerance in consecutive patients harbouring incidental adrenal tumors. Clin Endocrinol. 1998;49:53–61.
129. Leibowitz, G, Tsur, A, Chayen, SD, et al. Pre-clinical Cushing’s syndrome: an unexpected frequent cause of poor glycaemic control in obese diabetic patients. Clin Endocrinol. 1996;44:717–722.
130. Al-Shoumer, KA, Beshyah, SA, Niththyananthan, R, et al. Effect of glucocorticoid replacement therapy on glucose tolerance and intermediary metabolites in hypopituitary adults. Clin Endocrinol. 1995;42:85–90.
131. Wajngot, A, Giacca, A, Grill, V, et al. The diabetogenic effects of glucocorticoids are more pronounced in low than in high insulin responders. Proc Natl Acad Sci U S A. 1992;89:6035–6039.
132. Hirsh, IB, Paauw, DS. Diabetes management in special situations. Endocrinol Metab Clin North Am. 1997;26:631–646.
133. Umpierrez, GE, Khajavi, M, Kitabchi, AE. Review: Diabetic ketoacidosis and hyperglycemic hyperosmolar nonketotic syndrome. Am J Med Sci. 1996;311:225–233.
134. Mallison, CN, Bloom, SR, Warin, AP, et al. A glucagonoma syndrome. Lancet. 1974;2:1–5.
135. Stacpoole, PW. The glucagonoma syndrome: clinical features, diagnosis and treatment. Endocr Rev. 1981;2:347–361.
136. Bloom, SR, Polak, JM. Glucagonoma syndrome. Am J Med. 1987;82(suppl 5B):25–36.
137. Soga, J, Yakawa, Y. Glucagonomas/diabetico-dermatogenic syndrome (DDS): a statistical evaluation of 407 reported cases. J Hepatobiliary Pancreat Surg. 1998;5:312–319.
138. Shyr, YM, Su, CH, Lee, CH, et al. Glucagonoma syndrome: a case report. Chin Med J. 1999;62:639–643.
139. Barazzoni, R, Zanetti, M, Tiengo, A, et al. Protein metabolism in glucagonoma. Diabetologia. 1999;42:326–329.
140. Frankton, S, Bloom, SR. Gastrointestinal endocrine tumours. Glucagonomas. Baillieres Clin Gastroenterol. 1996;10:697–705.
141. Larsen, LI, Hirsch, MA, Holst, JJ, et al. Pancreatic somatostatinoma: clinical features and physiological implications. Lancet. 1977;1:666–668.
142. Ganda, OP, Weir, GC, Soeldner, JS, et al. “Somatostatinoma”: a somatostatin-containing tumor of the endocrine pancreas. N Engl J Med. 1977;296:963–967.
143. Krejs, GJ, Orci, L, Conlon, JM, et al. Somatostatin syndrome, biochemical, morphologic and clinical features. N Engl J Med. 1979;301:285–292.
144. Mao, C, Shah, A, Hanson, DJ, et al. Von Recklinghausen’s disease associated with duodenal somatostatinoma: contrast of duodenal versus pancreatic somatostatinomas. J Surg Oncol. 1995;59:67–73.
145. Penman, E, Lowry, PJ, Wass, JA. Molecular forms of somatostatin in normal subjects and in patients with somatostatinoma. Clin Endocrinol. 1980;12:611–620.
146. Conlon, JM, McCarthy, D, Krejs, G, et al. Characterization of somatostatin-like components in the tumors and plasma of a patient with a somatostatinoma. J Clin Endocrinol Metab. 1981;52:66–73.
147. Patel, YC, Ganda, OP, Benoit, R. Pancreatic somatostatinoma: abundance of somatostatin-28(1–12)-like immunoreactivity in tumor and plasma. J Clin Endocrinol Metab. 1983;57:1048–1053.
148. Willcox, PA, Immelman, EJ, Barron, JL, et al. Pancreatic somatostatinoma: presentation with recurrent episodes of severe hyperglycemia and ketoacidosis. Q J Med. 1988;68:559–571.
149. Jackson, JA, Raju, BU, Fachnie, JD, et al. Malignant somatostatinoma presenting with diabetic ketoacidosis. Clin Endocrinol. 1987;26:609–621.
150. Yamasaki, T, Chijiiwa, K, Chijiiwa, Y. Somatostatin inhibits cholecystokinin-induced contraction of isolated gallbladder smooth muscle cells. J Surg Res. 1995;59:743–746.
151. Marteau, P, Chretien, Y, Calmus, Y, et al. Pharmacological effect of somatostatin on bile secretion in man. Digestion. 1989;42:16–21.
152. Lowry, SF, Burt, ME, Brennan, MF. Glucose turnover and gluconeogenesis in a patient with somatostatinoma. Surgery. 1981;89:309–313.
153. Angeletti, S, Corleto, VD, Schillaci, O, et al. Use of the somatostatin analogue octreotide to localize and manage somatostatin-producing tumors. Gut. 1998;42:792–794.
154. Stenstrom, G, Sjostrom, I, Smith, U. Diabetes mellitus in pheochromocytoma: fasting blood glucose level before and after surgery in 60 patients with pheochromocytoma. Acta Endocrinol. 1984;106:511–515.
155. Hamaji, M. Pancreatic α and β cell function in pheochromocytoma. J Clin Endocrinol Metab. 1979;49:322–325.
156. Vance, JE, Buchanan, KD, O’Hara, D, et al. Insulin and glucagon responses in subjects with pheochromocytoma: effect of alpha adrenergic blockade. J Clin Endocrinol Metab. 1969;29:911–916.
157. Pandit, MK, Burke, J, Gustafson, AB, et al. Drug-induced disorders of glucose metabolism. Ann Intern Med. 1993;118:529–539.
158. Chan, JC, Cockran, CS, Critchley, JAHJ. Drug-induced disorders of glucose metabolism: mechanisms and management. Drug Saf. 1996;15:136–157.
159. Bressler, P, DeFronzo, RA. Drug effects on glucose homeostasis. In: Alberti KGMM, Zimmet P, DeFronzo RA, et al, eds. International textbook of diabetes mellitus. ed 2. New York: John Wiley & Sons; 1997:214–254.
160. Bouchard, P, Sai, P, Reach, G, et al. Diabetes mellitus following pentamidine-induced hypoglycemia in humans. Diabetes. 1982;31:40–45.
161. Osei, K, Falko, JM, Nelson, KP, et al. Diabetogenic effect of pentamidine: in vitro and in vivo studies in a patient with malignant insulinoma. Am J Med. 1984;77:41–46.
162. Assan, R, Mayaud, C, Perronne, C, et al. Pentamidine-induced derangements of glucose homeostasis. Diabetes Care. 1995;18:47–55.
163. LeWitt, PA. The neurotoxicity of the rat poison Vacor: a clinical study of 12 cases. N Engl J Med. 1980;302:73–77.
164. Karam, JH, Lewitt, PA, Young, CW, et al. Insulinopenic diabetes after rodenticide (Vacor) ingestion: a unique model of acquired diabetes in man. Diabetes. 1980;29:971–978.
165. Park-Wyllie, LY, Juurlink, DN, Kopp, A, et al. Outpatient gatifloxacin therapy and dysglycemia in older adults. N Engl J Med. 2006;354:1352–1361.
166. Saraya, A, Yokokura, M, Gonoi, T, et al. Effects of fluoroquinolones on insulin secretion and beta cell ATP-sensitive potassium channels. Eur J Pharmacol. 2004;497:111–117.
167. Danforth, E, Jr. Hyperglycemia after diazoxide. N Engl J Med. 1971;285:1487.
168. Milsap, RL, Auld, PA. Neonatal hyperglycemia following maternal diazoxide administration. JAMA. 1980;243:144–145.
169. Goldberg, EM, Sanbar, SS. Hyperglycemic non-ketotic coma following administration of Dilantin. Diabetes. 1969;18:101–106.
170. Fariss, BL, Lutcher, CL. Diphenylhydantoin-induced hyperglycemia and impaired insulin release: effect of dosage. Diabetes. 1971;20:177–181.
171. Malherbe, C, Burrill, KC, Levin, SR, et al. Effect of diphenylhydantoin on insulin secretion in man. N Engl J Med. 1972;286:339–342.
172. Bengtsson, C, Blohme, G, Lapidus, L, et al. Do antihypertensive drugs precipitate diabetes. BMJ. 1984;289:1495–1497.
173. Gurwitz, JH, Bohn, RL, Glynn, RJ, et al. Antihypertensive drug therapy and the initiation of treatment for diabetes mellitus. Ann Intern Med. 1993;118:273–278.
174. Murphy, MB, Kohner, E, Lewis, PJ, et al. Glucose intolerance in hypertensive patients treated with diuretics: a fourteen year follow-up. Lancet. 1982;2:1293–1295.
175. Donahue, R, Abbott, R, Wilson, P. Effect of diuretics on the development of diabetes mellitus: the Framingham study. Horm Metab Res. 1990;22(suppl 1):46–48.
176. Kaplan, NM. The case for low-dose diuretic therapy. Am J Hypertens. 1991;4:970–971.
177. Berglund, G, Andersson, O, Widgren, B. Low dose antihypertensive treatment with a thiazide diuretic is not diabetogenic: a 10-year controlled trial with bendroflumethiazide. Acta Med Scand. 1986;220:419–424.
178. Luna, B, Feinglos, MN. Drug-induced hyperglycemia. JAMA. 2001;286:1945–1948.
179. Gress, TW, Nieto, FJ, Shahar, E, et al. Hypertension and antihypertensive therapy as risk factors for type 2 diabetes mellitus. N Engl J Med. 2000;342:905–912.
180. ALLHAT Officers. Major outcomes in high-risk hypertensive patients randomized to angiotensin-converting enzyme inhibitor or calcium channel blocker vs diuretic. JAMA. 2002;288:2981–2997.
181. Rowe, JW, Tobin, JD, Rosa, RM, et al. Effects of experimental potassium deficiency on glucose and insulin metabolism. Metabolism. 1980;29:493–502.
182. Helderman, JH, Elahi, D, Anderson, DK, et al. Prevention of the glucose intolerance of thiazide diuretics by maintenance of body potassium. Diabetes. 1983;32:106–111.
183. Shafi, T, Appel, LJ, Miller, ER, et al. Changes in serum potassium mediate thiazide-induced diabetes. Hypertension. 2008;52:1022–1029.
184. Micossi, P, Pollavini, G, Piaggi, U, et al. Effects of metoprolol and propranolol on glucose metabolism and insulin secretion in diabetes mellitus. Horm Metab Res. 1984;16:59–63.
185. Whitcroft, I, Wilkinson, N, Ranthorne, A, et al. Beta-adrenoreceptor antagonist impairs long-term glucose control in hypertensive diabetes: role of beta-adrenoreceptor selectivity and lipid solubility. Br J Clin Pharm. 1986;22:236–237.
186. Cerasi, E, Luft, R, Effendic, S. Effect of adrenergic blocking agents on insulin response to glucose infusion in man. Acta Endocrinol. 1972;69:335–346.
187. Totterman, K, Groop, L, Groop, PH, et al. Effect of beta blocking drugs on beta cell function and insulin sensitivity in hypertensive non-diabetic patients. Eur J Clin Pharmacol. 1984;26:13–17.
188. Bending, JJ, Ogg, CS, Viberti, GC. Diabetogenic effect of cyclosporin. BMJ. 1987;294:401–402.
189. Ost, L, Tyden, G, Fehrman, I. Impaired glucose tolerance in cylcosporin-prednisolone–treated renal graft recipients. Transplantation. 1988;46:370–372.
190. Hudes, G, Carducci, M, Tomczak, P, et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med. 2007;356:2271–2281.
191. Perlman, JA, Russell-Briefel, R, Ezzati, T, et al. Oral glucose tolerance and the potency of contraceptive progestins. J Chronic Dis. 1985;38:857–864.
192. Spellacy, WN, Tsibris, JC, Ellingson, AB. Carbohydrate metabolic studies in women using a levonorgestrel/ethinyl estradiol containing triphasic oral contraceptive for 18 months. Int J Gynaecol Obstet. 1991;35:69–71.
193. Scheen, AJ, Jandrain, BJ, Humblet, DM, et al. Effects of a 1-year treatment with low-dose combined oral contraceptive containing ethinyl estradiol and cyproterone acetate on glucose and insulin metabolism. Fertil Steril. 1993;59:797–802.
194. Spellacy, WN. Carbohydrate metabolism during treatment with oestrogen, progestogen and low-dose oral contraceptives. Am J Obstet Gynecol. 1982;142:732–734.
195. Bettino, JC, Tashima, CK. Medroxyprogesterone acetate and diabetes mellitus. Ann Intern Med. 1976;84:341–342.
196. Lithell, H, Vessby, B, Hellsing, K. Changes in glucose tolerance and plasma insulin during lipid-lowering treatment with diet, clofibrate and niceritrol. Atherosclerosis. 1982;43:177–184.
197. Garg, A, Grundy, SM. Nicotinic acid as therapy for dyslipidemia in non–insulin-dependent diabetes. JAMA. 1990;264:723–726.
198. Molnar, GD, Berge, KG, Rosevear, JW, et al. The effect of nicotinic acid in diabetes mellitus. Metabolism. 1964;13:181–189.
199. Grundy, SM, Vega, GL, McGovern, ME, et al. Efficacy, safety, and tolerability of once-daily niacin for the treatment of dyslipidemia associated with type 2 diabetes. Arch Intern Med. 2002;162:1568–1576.
200. Carr, A. HIV lipodystrophy: risk factors, pathogenesis, diagnosis and management. AIDS. 2003;17(suppl 1):S141–S148.
201. Tershakovec, AM, Frank, I, Rader, D. HIV-related lipodystrophy and related factors. Atherosclerosis. 2004;174:1–10.
202. Dube, MP, Johnson, DL, Currier, JS, et al. Protease inhibitor–associated hyperglycaemia. Lancet. 1997;350:713–714.
202a. Kilby, JM, Tabereaux, PB. Severe hyperglycemia in an HIV clinic: preexisting versus drug associated diabetes mellitus. J Acquir Immune Syndr Human Retrovirol. 1998;17:46–50.
203. Kaufman, MB, Simionatta, C. A review of protease inhibitor–induced hyperglycemia. Pharmacotherapy. 1999;19:114–117.
204. Tsiodras, S, Mantzoros, C, Hammer, S, et al. Effects of protease inhibitors on hyperglycemia, hyperlipidemia, and lipodystrophy. Arch Intern Med. 2000;160:2050–2056.
205. Mehta, SH, Moore, RD, Thomas, DL, et al. The effect of HAART and HCV infection on the development of hyperglycemia among HIV-infected persons. J Acquir Immune Defic Syndr. 2003;33:577–584.
206. Woerle, HJ, Mariuz, PR, Meyer, C, et al. Mechanisms for the deterioration in glucose tolerance associated with HIV protease inhibitor regimens. Diabetes. 2003;52:918–925.
207. Yarasheski, KE, Tebas, P, Sigmund, C, et al. Insulin resistance in HIV protease inhibitor–associated diabetes. J Acquir Immun Defic Syndr Hum Retrovirol. 1999;21:209–216.
208. Casey, DE, Haupt, DW, Newcomer, JW, et al. Antipsychotic-induced weight gain and metabolic abnormalities: implications for increased mortality in patients with schizophrenia. J Clin Psychiatry. 2004;65(suppl 7):4–18.
209. Lebovitz, HE. Metabolic consequences of atypical antipsychotic drugs. Psychiatr Q. 2003;74:277–290.
210. Jin, H, Meyer, JM, Jeste, DV. Phenomenology of and risk factors for new-onset diabetes mellitus and diabetic ketoacidosis associated with atypical antipsychotics: an analysis of 45 published cases. Ann Clin Psychiatry. 2002;14:59–64.
211. Allison, DB, Mentore, JL, Heo, M, et al. Antipsychotic-induced weight gain: a comprehensive research synthesis. Am J Psychiatry. 1999;156:1686–1696.
212. Sernyak, MJ, Leslie, DL, Alarcon, RD, et al. Association of diabetes mellitus with use of atypical neuroleptics in the treatment of schizophrenia. Am J Psychiatry. 2002;159:561–566.
213. Koro, CE, Fedder, DO, L’Italien, GJ, et al. An assessment of the independent effects of olanzapine and risperidone exposure on the risk of hyperlipidemia in schizophrenic patients. Arch Gen Psychiatry. 2002;59:1021–1026.
214. Newcomer, JW, Haupt, DW, Fucetola, R, et al. Abnormalities in glucose regulation during antipsychotic treatment of schizophrenia. Arch Gen Psychiatry. 2002;59:337–345.
215. Henderson, DC, Cagliero, E, Gray, C, et al. Clozapine, diabetes mellitus, weight gain, and lipid abnormalities: a five-year naturalistic study. Am J Psychiatry. 2000;157:975–981.
216. Koller, E, Schneider, B, Bennett, K, et al. Clozapine-associated diabetes. Am J Med. 2001;111:716–723.
217. Koller, EA, Doraiswamy, PM. Olanzapine-associated diabetes mellitus. Pharmacotherapy. 2002;22:841–852.
218. Koller, EA, Cross, JT, Doraiswamy, PM, et al. Risperidone-associated diabetes mellitus: a pharmacovigilance study. Pharmacotherapy. 2003;23:735–744.
219. American Diabetes AssociationAmerican Psychiatric AssociationAmerican Association of Clinical EndocrinologistsNorth American Association for the Study of Obesity. Consensus development conference on antipsychotic drugs and obesity and diabetes. Diabetes Care. 2004;27:596–601.
220. Lieberman, JA, Stroup, TS, McEvoy, JP, et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. 2005;353:1209–1223.
221. Scheen, AJ, De Hert, MA. Abnormal glucose metabolism in patients treated with antipsychotics. Diabetes Metab. 2007;33:169–175.
222. Ader, M, Garvey, T, Phillips, LS, et al. Ethnic heterogeneity in glucoregulatory function during treatment with atypical antipsychotics in patients with schizophrenia. J Psychiatr Res. 2008;42:1076–1085.
[/level-membership-for-endocrinology-diabetes-and-metabolism-category][not-level-membership-for-endocrinology-diabetes-and-metabolism-category]Chapter 17
Hyperglycemia Secondary to Nondiabetic Conditions and Therapies
Glucose metabolism is regulated by the interplay of the action of pancreatic islet cell hormones with liver, muscle, and adipose tissue. An alteration in the function of any component of this complex glucose homeostatic system brings about compensatory responses in the other components to drive the system back to its homeostatic set points. The key players in regulating this system are the islet hormones insulin and glucagon, both of which are regulated by nutrient levels and are modulated by the gastrointestinal incretin hormones.1 Insulin promotes hepatic glucose uptake and glycogenesis, stimulates muscle and adipose tissue glucose uptake and metabolism, and inhibits adipose tissue lipolysis and muscle proteolysis.2 Glucagon stimulates hepatic gluconeogenic precursor uptake and increases hepatic glycogenolysis, gluconeogenesis, and ketogenesis.3
Maintenance of fasting and postprandial plasma glucose levels within the normal range requires insulin secretion by the β cell and glucagon secretion from the α cell to be integrated with insulin action in liver and peripheral tissues. Insulin action results from a complex cascade of intracellular substrate phosphorylations and dephosphorylations that lead to regulation of processes as diverse as intermediary metabolism and mitogenesis. Insulin action is altered easily by a variety of intracellular and extracellular factors. When insulin action that affects glucose metabolism is altered, insulin secretion must change accordingly if normal glucose homeostasis is to remain intact.4 Any genetic abnormality, environmental factor, or drug that disturbs this relationship will lead to hyperglycemia or hypoglycemia.
Type 2 diabetes is a heterogeneous disorder in which gene polymorphisms provide the predispositions and environmental factors that serve as the precipitating causes of hyperglycemia. Many individuals with the genetic predisposition do not manifest impaired glucose tolerance (IGT) or type 2 diabetes throughout their lifetime. If a pathologic condition develops in such individuals, however, or if they take a medication that disturbs their compensated state, hyperglycemia will develop. Thus hyperglycemic states resulting from nondiabetic conditions or therapies can be subdivided into those that can cause hyperglycemia in any individual because they radically interfere with a major regulatory pathway (Table 17-1) and those that precipitate diabetes only in genetically predisposed individuals because they alter the compensated state (Table 17-2). Because of the high prevalence of a genetic predisposition to type 2 diabetes in various populations, it is not always possible to make the distinction with certainty.
Table 17-1
Conditions That Can Cause Hyperglycemia in the Absence of Genetic Predisposition
Table 17-2
Conditions That Precipitate Hyperglycemia in Individuals With a Genetic Predisposition to Type 2 Diabetes
Disorders of the Pancreas
Diabetes mellitus that develops after surgical removal of the pancreas is truly an insulin-dependent diabetes mellitus. Metabolically, it is characterized by insulin and glucagon deficiency.5 The magnitude of the hyperglycemia and of its characteristics depends on the quantity of pancreas removed (Table 17-3). Total or near-total pancreatectomy results in severe hyperglycemia, decreased plasma insulin, virtually absent plasma glucagon, and elevated plasma levels of gluconeogenic precursors (alanine, lactate, glycerol).5–11
Table 17-3
Estimated Frequency of Diabetes Reported in Pancreatic Disease
| 95% Pancreatectomy | 100% |
| 50% Pancreatectomy | 0% |
| Pancreatitis | |
| Acute | <5% |
| Chronic calcifying | 40%-70% |
| Chronic noncalcifying | 15%-30% |
| Cystic fibrosis | 17% |
| Carcinoma of the pancreas | 23% |
| Hemochromatosis | 50%-60% |
The effect of removal of 50% of the pancreas was studied in 28 normal transplant donors 1 year after surgery.12 The donors lost a mean of 3.4 kg of body weight, and their mean fasting plasma glucose level had risen by 9 mg/dL (88 ± 7 to 97 ± 16 mg/dL). Similarly, their mean serum glucose concentration 2 hours after an oral glucose load was higher (117 ± 18 to 156 ± 53 mg/dL), and the area under the 5 hour plasma glucose curve after the oral glucose was 19.5% higher. Both the mean fasting plasma insulin concentration and the area under the 5 hour plasma insulin curve were significantly lower than preoperatively (−14% and −31%, respectively). None of the donors had any evidence of deficient pancreatic exocrine function. On further analysis, the investigators noted that 21 of the donors had no significant postoperative change in plasma glucose or insulin, whereas seven showed a marked increase in the entire 5 hour plasma glucose curve (either IGT nor diabetes) with no concomitant increase in 5 hour plasma insulin curves. The seven donors in whom some degree of hypoinsulinemia and hyperglycemia had developed did not have fasting hyperglycemia 1 year postoperatively. Two of the seven were studied from 2 to 7 years after surgery and had not had a further increase in fasting plasma glucose. A recent report of eight donor/recipient pairs evaluated 9 to 18 years after the original surgery indicated that the residual pancreatic mass is a significant determinant of long-term glucose homeostasis.13 However, other variables were implicated in the discordancy for the development of diabetes. In particular, obesity seemed to be a major factor in that all of the individuals (four donors and two recipients) who had developed diabetes were among the eight patients who were obese.13 The investigators interpreted their data as “suggesting that obesity should be a contraindication to donation of pancreatic segments and that donors should assiduously avoid becoming obese.”13
In a more recent follow-up of 15 normal individuals of 21 who had donated 50% of their pancreas between 1997 and 2003, two donors were taking oral diabetic medications, two had impaired fasting plasma glucose (IFG), one had impaired glucose tolerance (IGT), three had both IFG and IGT, and one met the criteria for the diagnosis of diabetes.14 Six of the 21 patients were lost to follow-up. Only 6 of 15 patients had normal glucose values. Despite the use of stringent criteria to exclude those at risk for developing abnormalities in glucose metabolism, 43% of the total population of healthy humans who underwent a 50% pancreatectomy developed some abnormality of glucose metabolism within 3 to 10 years.
Sun et al. studied the metabolic effects of removing 20% to 88% of the pancreas in dogs and found that no significant metabolic changes occurred until approximately 50% had been removed.15
From the data available, it seems reasonable to conclude that metabolic abnormalities that arise after pancreatectomy are likely to be clinically relevant at removal of 50% or more, and that a progressively greater number of metabolic abnormalities occur as the extent of pancreatectomy increases. The concomitant presence of insulin resistance further increases the likelihood of metabolic abnormalities.
Major characteristics of the development of diabetes mellitus after extensive pancreatectomy include an absence of glucagon secretion and marked impairment in insulin secretion. Absence of glucagon slows, but does not interfere with, the development of hyperglycemia and ketonemia after insulin withdrawal.5,7 This observation indicates that glucagon is not necessary for development of the metabolic abnormalities of insulin-dependent diabetes mellitus. However, the absence of glucagon secretion does leave a pancreatectomized individual with diabetes at high risk for severe hypoglycemia during insulin treatment.16–19 This situation is exaggerated by the associated nutritional deficiencies and weight loss that ordinarily accompany exocrine pancreatic insufficiency. A concomitant deficiency of pancreatic polypeptide may contribute to persistent hyperglycemia caused by impaired hepatic insulin action.20 Treatment of an individual with pancreatic diabetes requires insulin, is associated with marked lability in glucose regulation, and is linked with an increased rate of both ketoacidosis and death from hypoglycemia. The development of autonomic neuropathy in a patient with pancreatic diabetes greatly adds to the risk of severe hypoglycemia with insulin treatment.21
Jethwa et al. have reported in a recent review that patients with total pancreatectomy can maintain reasonably good glycemic control with few serious hypoglycemic or ketoacidotic events provided they use self–blood glucose monitoring and are followed closely by their physicians.22,23 Among the 33 patients available for a median follow-up of 50 months, the median hemoglobin A1c (HbA1C) was 8.2%, which was comparable with that in type 1 diabetic patients followed by their institution. Patients resected for cancer maintained better glycemic control than those treated for chronic pancreatitis. The median insulin dose for the entire group was 46 units per day.
Chronic Pancreatitis
Chronic pancreatitis accounts for a little less than 1% of cases of diabetes mellitus in Western countries and Japan.20,21,24–27 In tropical countries, where nonalcoholic calcific pancreatitis is common, the incidence may be somewhat higher, but reliable data are not available. Although long-term ingestion of alcohol is the most common cause of chronic pancreatitis (particularly in Western cultures), other conditions such as genetic mutations, pancreatic duct obstruction, hypertriglyceridemia, hypercalcemia, autoimmunity, calcific tropical pancreatitis, and idiopathic pancreatitis are not uncommon.28 The development of diabetes mellitus in patients with chronic pancreatitis is most frequent in those with calcific disease (55% to 70%) and occurs less often in those with noncalcific disease (30%).29 The prevalence of diabetes in patients with chronic pancreatitis increases with increasing duration of pancreatitis and with increasing exocrine deficiency.26,30–34
The inflammatory response causes loss of exocrine tissue, extensive fibrosis, and distorted and blocked ducts. The islets of Langerhans are relatively resistant and undergo pathologic changes only late in the disease. Chronic pancreatitis is associated with loss of functioning β cells and a somewhat lesser loss of α cells.17,35–41 Hormonal alterations seen include a decrease in insulin secretion in response to nutrients, followed later by a decrease in fasting C peptide levels. Plasma C peptide levels rather than insulin levels may be a better means of assessment of insulin secretion because associated liver disease may change hepatic extraction rates of insulin. With progressive chronic pancreatitis, insulin secretion falls even lower. Glucagon secretion is impaired in moderate to severe chronic pancreatitis. Insulin resistance frequently develops in such individuals.
Diabetes mellitus is seen after several years of chronic pancreatitis. In an unselected series of patients with chronic pancreatitis, 35% had type 1 diabetes, 31% had type 2 diabetes or IGT, and 34% had normal glucose tolerance.42 The nature of the diabetes reflects the severity of the chronic pancreatitis. Mild pancreatitis may be associated only with IGT, whereas severe pancreatitis is associated primarily with insulin-dependent (type 1) diabetes. Patients with chronic pancreatitis and diabetes mellitus fail to secrete glucagon in response to hypoglycemia. If patients have concomitant autonomic neuropathy, they are extremely susceptible to severe and prolonged hypoglycemia.
Treatment of diabetes mellitus in patients with chronic pancreatitis should entail the use of small doses of short-acting or rapid-acting insulins to manage the hyperglycemia, replacement enzymes for the malnutrition and malabsorption, and elimination of the use of alcoholic beverages.29,39,43,44 Surgery44 with subtotal resection or near-total resection may be necessary to relieve severe pain.45,46 Following pancreatic surgery for pain or other complications, a significant incidence of diabetes mellitus occurs; however, whether diabetes develops is dependent on the type of surgical intervention used. Whipple procedures usually are associated with an incidence of 25% to 40%, a Frey or Berne procedure 8% to 22%, and a distal pancreatectomy approximately 60%.47 In such patients, successful islet allotransplants and autotransplants in the liver have been able to maintain near normoglycemia.48 The hepatic islet cell transplants were able to secrete insulin in response to nutrients but were unable to secrete glucagon in response to hypoglycemia.18,19 A recent report of the results of pancreas allotransplants in patients who had undergone total pancreatectomy in the past showed a 70% survival rate and successful correction of both exocrine and endocrine deficiencies.49
Pancreatic exocrine function has been reported to be reduced in some type 1 and type 2 diabetic patients. This has been explained as a complication of the diabetes. A retrospective analysis of pancreatograms of patients with known diabetes has suggested that chronic pancreatitis may be much more common as a cause of diabetes than was previously thought.48 A total of 38 type 1 and 118 type 2 diabetic patients underwent endoscopic retrograde cholangiopancreatography (ERCP) studies for varying reasons. Pancreatic ducts were classified as normal in 23.3% and as exhibiting chronic pancreatitis degree I, II, and III in 22.7%, 32.7%, and 21.3%, respectively.50 The investigators suggested that a substantial number of patients with primary diabetes mellitus may have a concomitant chronic pancreatitis, or perhaps that many cases of primary diabetes mellitus may be related to chronic pancreatitis.
Pancreatic Cancer
Diabetes mellitus is known to occur more frequently in patients with pancreatic cancer than in the general population.51–53 Published data indicate that as many as 70% of patients with pancreatic carcinoma have impaired glucose tolerance or frank diabetes mellitus, and that 60% demonstrate improved glucose metabolism after surgery.54 Wakasugi et al. reported that 53.1% of patients with invasive ductal pancreatic carcinoma had diabetes mellitus, and in 45.9% it was thought to be secondary to the carcinoma.55 The reasons for this association have been the subject of much speculation. Some studies show that diabetes mellitus is associated with increased risk for susceptibility to pancreatic cancer (2.15 to 4.9 in men).56,57 Other studies have indicated that in most patients with pancreatic carcinoma, the diabetes is secondary to some effect of the cancer, which causes insulin resistance and impairs the function of normal β cells.58–60 A multicenter case-control study of 720 patients with pancreatic cancer addressed this issue.51 The prevalence of diabetes in the patients with pancreatic cancer was 22.8%, whereas that in the matched control population was 8.3%. The patients with pancreatic cancer were characterized as having type 2 diabetes. A recent diagnosis of diabetes had been made in 40.2% of the patients with pancreatic cancer with diabetes, as contrasted with only 3.3% of the control population with diabetes (Table 17-4). A higher percentage of the control population with diabetes than of the pancreatic cancer population with diabetes had had their diabetes for longer than 15 years (see Table 17-4). These data support the idea that in a small number of patients, diabetes predisposes to the development of diabetes mellitus.
Table 17-4
Interval Between Diagnosis of Diabetes and Diagnosis of Pancreatic Cancer or Date of Examination of Control Population
Possible mechanisms by which pancreatic cancer could contribute to the development of type 2 diabetes include the following: (1) destruction of islets, (2) impairment of the insulin secretory mechanism, (3) development of insulin resistance, and (4) tumor-related pancreatitis. Insulin and C-peptide measurements during oral glucose tolerance testing in patients with pancreatic cancer have shown abnormal β cell function with reduced plasma C-peptide responses in 50% of patients and increased plasma proinsulin-to-C-peptide ratios61,62 Insulin resistance has been demonstrated in most patients with pancreatic carcinoma.63–65 Morphometric studies of tumor-free regions of the pancreas have shown reduced β cell populations.66 An inverse correlation was noted between the number of β cells and the fasting plasma glucose concentration. These data can be interpreted as indicating that pancreatic cancers produce substances or responses that destroy normal β cells.64–67 The presence of diabetes in patients with pancreatic cancer predicts that the tumor is less likely to be respectable and the patient has a poorer prognosis than if diabetes is not present.53 Patients with pancreatic cancer typically have type 2 diabetes; most have been treated with oral antihyperglycemic agents.52,53
Several very intriguing observations that were reported recently make this relationship between pancreatic cancer and diabetes mellitus even more complex and confusing. Li et al. reported that metformin treatment of diabetic patients was associated with a decreased risk of developing pancreatic carcinoma, while insulin or insulin secretogogue treatment was associated with an increased risk.68 In a large, multicenter clinical trial that compared the treatment of type 2 diabetic patients with rosiglitazone-based therapy versus metformin plus sulfonylurea therapy, the development of pancreatic cancer during the study was statistically less frequent in the rosiglitazone arm than in the active comparator arm (2 cases versus 13 cases; P < .007).69
Hemochromatosis
Hemochromatosis is an autosomal recessive genetic disorder that results in excessive deposition of iron in parenchymal cells of the liver, pancreas, muscle, heart, anterior pituitary, and other organs.70,71 The clinical diagnosis in the past was made by the findings of diabetes mellitus, hepatomegaly, and skin pigmentation. More recently, it has been recognized through biochemical and genetic testing.72 The first phenotypic expression of the disease is an elevation in serum transferrin saturation. This abnormality is followed by iron accumulation in the tissues and an elevation in the serum ferritin concentration. Early clinical findings are related to hepatic dysfunction and joint symptoms. Clinical diabetes mellitus and skin pigmentation occur relatively late in the course of the disease.71 A candidate gene for human leukocyte antigen (HLA)-linked hemochromatosis has been cloned and a mutation (C282Y) of the HFE gene identified that may account for 60% or more of cases of hereditary hemochromatosis. Another mutation (H63D) has been identified more recently.73
Diabetes mellitus has been reported in 50% to 60% of patients with hemochromatosis.71,74 Another 20% to 30% had glucose intolerance. These figures represent data from older series in which the diagnosis was made late in the course of the disease. Diabetes mellitus is more frequent in patients who have a family history of diabetes mellitus.
Follow-up of 237 patients who were given a diagnosis of hemochromatosis and treated at a single center from 1983 through 2005 has diabetes mellitus associated with the disease.73 Before 1996, hemochromatosis was diagnosed by the classic clinical and laboratory features of the disease. Since 1996, genetic testing, which is now available commercially, made it possible for them to diagnose hemochromatosis in asymptomatic patients. Of 45 patients diagnosed before 1996, 30.2% had cirrhosis of the liver and 35.6% had diabetes mellitus. After 1996, 192 patients were given the diagnosis of hemochromatosis (34% by family screening and 40% by clinical suspicion) and only 7.5% had cirrhosis of the liver and 17.7% had diabetes mellitus at the time of diagnosis. It is obvious that early through genetic testing and effective treatment can significantly reduce development of the complications of hemochromatosis. Of note was the observation that performing phlebotomy and reducing tissue iron in well-established clinical disease did not improve diabetes or lessen its management requirements. Thus early diagnosis and treatment is primarily preventative.73
Before 1996, hemochromatosis was diagnosed by the classic clinical and laboratory features of the disease. Since 1996, genetic testing, which is now available commercially, has made it possible to diagnose hemochromatosis in asymptomatic patients. Of the 45 patients diagnosed before 1996, 30.2% had cirrhosis of the liver and 35.6% had diabetes mellitus. After 1996, 192 patients were given the diagnosis of hemochromatosis (34% by family screening and 40% by clinical suspicion); only 7.5% had cirrhosis of the liver and 17.7% had diabetes at the time of diagnosis. It is obvious that early diagnosis through genetic testing and effective treatment have significantly reduced development of the complications. Of note is the observation that performing phlebotomy and reducing tissue iron did not improve diabetes or lessen its management requirements. Thus early diagnosis and treatment is primarily preventative.73
The metabolic studies that have been done show that patients with hemochromatosis have marked insulin resistance. Histologic study of the pancreas shows iron deposits that are greatest in the acinar cells but do involve islet cells. Insulin secretion in response to glucose or arginine is decreased; however, glucagon secretory responses to arginine are increased and unaffected by glucose.75,76 The data are compatible with a marked reduction in β cell function and no disturbance in α cell function. The hyperglycemia is a result of insulin resistance and decreased β cell function. The prevalence of diabetes mellitus probably could be reduced by early diagnosis of hemochromatosis and initiation of phlebotomy therapy.
Therapy for patients with hemochromatosis and clinical diabetes frequently requires insulin (40% to 50% of patients), although no systematic studies of therapy have been done.72 Reduction of tissue iron stores, although most beneficial in the early stages of disease, nonetheless can help improve glycemic control in 35% to 45% of patients.70,71
Hemosiderosis
Excessive iron deposition occurs in a variety of conditions other than primary hemochromatosis. In thalassemia major, frequent blood transfusions are necessary and may lead to massive iron overload. The reported prevalence of diabetes mellitus in treated thalassemia major is about 16%. This figure is highly correlated with the number of blood transfusions given and the duration of the disease. The incidence of IGT is reported to be 60%.77
Further evidence that deposits of excess tissue iron themselves are responsible for many of the metabolic abnormalities seen in hemochromatosis and thalassemia major comes from studies in rural male Bantus.78 Many Bantus drink alcoholic beverages that are brewed in iron containers and ingest in excess of 100 mg of iron per day. In those individuals, the prevalence of diabetes mellitus is 10-fold higher than in non–alcoholic beverage–consuming males.
Mechanistic studies in patients with thalassemia major with normal, impaired, and diabetic glucose tolerance tests show that increased iron stores are associated with the development of insulin resistance and a delay in early insulin secretion.79 A correlation between increased iron stores in normal women and the development of type 2 diabetes was demonstrated recently in the Nurses Health Study.80
Cystic Fibrosis
Cystic fibrosis (CF) is a monogenetic disorder with abnormal cyclic adenosine monophosphate–regulated chloride channel activity. It is an autosomal recessive genetic disease with an incidence of 1 in 2500 live births in Caucasian populations. More than 1000 gene mutations have been identified in the CF gene.81 Organs as diverse as the lung, exocrine pancreas, large and small intestine, hepatobiliary system, and sweat glands are involved. Failure to secrete Na+, HCO3−, and water leads to retention of enzymes in the pancreas and ultimately to destruction of pancreatic tissue.82–84 Histologic examination of the pancreas in patients with CF shows fatty infiltration, necrosis, and fibrosis of the exocrine pancreas. Islet cell architecture is disrupted and the absolute number of pancreatic islets diminished. Islets that are present show significant decreases in β cells, α cells, and pancreatic polypeptide–producing cells and increases in δ (somatostatin-producing) cells. Islet amyloid deposits have been found in 69% of diabetic CF cases examined.
Diabetes mellitus requiring medical therapy (usually insulin) has been reported in 4.9% of patients of all ages with CF in a large European study of 1348 patients85 and in 5.1% of 18,627 patients of all ages monitored at CF centers in the United States and Canada.82 Diabetes occurs more often in individuals who are homozygous for the most common CF mutation, ΔF508.82,86,87 Diabetes also occurs with greater frequency with increasing age; it has been reported in 32% of Danish patients who were older than 25 years. Routine oral glucose tolerance testing suggests that of the total CF population aged 5 years or older, 35% have normal glucose tolerance, 37% have IGT, 17% have CF-related diabetes without fasting hyperglycemia, and 11% have CF-related diabetes with fasting hyperglycemia.
Several features of CF-related diabetes are noteworthy. Autoantibodies to pancreatic heat shock protein 60 have been found to precede the development of glucose intolerance (IGT and diabetes) and to decline subsequently with the onset of glucose intolerance.88 The development of diabetes in patients with CF is characterized initially by abnormal oral glucose tolerance and a delay in oral glucose–stimulated insulin secretion, followed later by a decrease in total insulin, glucagon, and pancreatic polypeptide secretion.89 First-phase insulin secretion after intravenous glucose is reduced markedly in patients with CF compared with matched controls. Patients with CF with IGT have normal plasma free fatty acid levels as compared with matched controls.90 Their plasma tumor necrosis factor-α levels are elevated, and they have insulin resistance as measured by the hyperinsulinemic euglycemic clamp.90 Decreased translocation of Glut-4 glucose transporters in muscle was observed during the peak insulin effect. Patients with CF have an increase in hepatic glucose production and are resistant to suppression of hepatic glucose production by insulin even in the nondiabetic state.91 Peripheral insulin sensitivity is increased in healthy nondiabetic individuals with CF, but insulin resistance occurs later as IGT and diabetes develop, and patients experience additional complications related to their CF.92 The development of diabetes worsens pulmonary function and other clinical manifestations of CF and may increase mortality by up to sixfold.93 Insulin treatment appears to reduce the extent of this deterioration. Hyperglycemia in patients with CF can be intermittent or permanent. Intermittent hyperglycemia occurs with glucocorticoid therapy, infection, or stress and must be treated with insulin until it resolves. Permanent hyperglycemia is always treated with insulin.82,84 It is likely that patients with CF progress from intermittent hyperglycemia to permanent hyperglycemia as pancreatic destruction continues to occur.
A small short-term study (12 weeks) suggests that treatment of CF-related diabetes with glargine insulin may provide some advantages over treatment with NPH insulin.94
Microvascular complications have been observed in patients with CF-related diabetes with fasting hyperglycemia for longer than 10 years. Fourteen percent had microalbuminuria and 16% had retinopathy.95 Macrovascular disease appears to be uncommon.
Hyperglycemia Associated With Endocrinopathies
In the complex regulation of fuel homeostasis, many hormones other than insulin play a complementary role. Growth hormone (GH) by itself and through its synthesis of insulin-like growth factor-1 (IGF-1) controls many aspects of amino acid transport, protein synthesis, and lipid metabolism. Glucagon and catecholamines are counterregulatory hormones that protect against hypoglycemia and provide extra glucose when needed during stress states. Glucocorticoids exert both a permissive role in the normal physiologic regulation of gluconeogenesis and a pharmacologic role in providing increased glucose availability during stress. Somatostatin is a paracrine hormone that appears to act locally to help regulate the normal secretory patterns of GH, insulin, glucagon, and several gastrointestinal hormones.
This section will address the unique characteristics of hyperglycemia as it relates to each endocrinopathy and its treatment.
Acromegaly
Acromegaly is characterized by excessive and autonomous secretion of growth hormone and IGF-1.96,97 The prevalence of overt diabetes mellitus reported in different series of acromegalic patients ranges from 30% to 56%.96,98 IGT may be present in as many as 36% of acromegalic patients.96 In a specific population, the percentage of acromegalic patients in whom diabetes mellitus will develop depends on the prevalence of predisposition to type 2 diabetes in the population and the magnitude of elevation of serum IGF-1 levels.
Elevated GH and IGF-1 levels cause excessive hepatic glucose production and impaired insulin-mediated muscle glucose uptake.99–101 This insulin resistance is correlated with circulating IGF-1 levels and has been demonstrated by the euglycemic hyperinsulinemic clamp and the minimal model techniques.
Reduction in circulating GH and IGF-1 levels by successful surgical removal of the tumor producing growth hormone or GH-releasing factor results in significant improvement in glycemic control in acromegalic patients with diabetes mellitus.100,102 Recent data suggest that circulating GH must be lowered to 2 ng/L and IGF-1 lowered to the normal range to be considered curative.97,103,104 Transsphenoidal surgery achieves growth hormone levels less than 5 ng/L in approximately 60% of patients. Curative levels are attained in about 70% of patients with microadenomas (<10 mm in diameter) but are attained considerably less often in those with macroadenomas of the pituitary.97,103
Use of the somatostatin analogue octreotide to treat acromegaly as the primary medical therapy or to supplement prior inadequate surgical treatment or radiotherapy has allowed attainment of greater and more consistent reductions in circulating GH and IGF-1 levels (GH levels ≤5 ng/L in 65% and ≤2 ng/L in 40%, and IGF-1 levels in the normal range in 64% of patients).103–105
Treatment of acromegalic patients with octreotide presents several issues with respect to glucose metabolism.103–106 Reductions in circulating GH and IGF-1 levels will decrease insulin resistance and should lead to improvement in glycemic control in subjects with diabetes mellitus or IGT. However, pharmacologic doses of a somatostatin analogue also reduce insulin secretion (decreased insulinogenic index), and such a reduction should cause a deterioration in glucose tolerance. Thus in any particular patient, octreotide therapy will modify glucose metabolism in accordance with these competing effects. Approximately two thirds of acromegalic patients with diabetes mellitus are treated with insulin and one third with oral hypoglycemic agents. Octreotide treatment in patients with diabetes mellitus and acromegaly frequently leads to improvement in glycemic control as measured by a reduction in the insulin dose, conversion from insulin therapy to oral hypoglycemic agent therapy, or conversion from oral hypoglycemic agent therapy to dietary management.103,106 Some patients (those with more severe insulin deficiency), however, will have significant deterioration in glycemic control.104,106 When higher doses of octreotide are given, IGT and even frank diabetes mellitus (as high as 20% and 29%, respectively) may develop in acromegalic patients with normal glucose tolerance before octreotide treatment.106 An alternative to treatment with somatostatin analogues is treatment with the GH receptor blocker Pegvisomant. This agent will decrease IGF-1 levels and can improve glycemic control.107,108 However, it does cause an increase in visceral fat and occasionally is associated with increased liver enzymes.109
Appropriate treatment for acromegaly is necessary to reduce the increased mortality that has been seen in the past.110
[/not-level-membership-for-endocrinology-diabetes-and-metabolism-category]
