CHAPTER 39 Histocompatibility
HLA and other systems
The human major histocompatibility complex
History
What has become known as the major histocompatibility complex (MHC) was initially identified in the early 1900s, but it was not until the late 1930s that studies began to focus on graft acceptance (histocompatibility) and antigen response phenotypes (H-2) in different strains of mice.1,2 In the 1950s, Dausset detected the first histocompatibility antigens in humans, the MHC class I antigens, with antibodies from multiply transfused patients.3,4 These antibodies revealed in the human population differing patterns of binding to white blood cells (leukocytes) and each pattern of binding came to define a human leukocyte antigen (HLA) specificity.5,6 These HLA specificities were later determined to be encoded by three distinct polymorphic loci, HLA-A, HLA-B and HLA-C. The human MHC class II antigens were initially described via their ability to stimulate the proliferation of T-cells from one individual when mixed with lymphocytes from a second individual.7 Each pattern of T-cell reactivity (allorecognition) to a panel of homozygous typing cells (HTC) was assigned an HLA-D phenotype.8 It is now known that the HLA-D phenotypes are due to T-cell allorecognition of the products of the MHC class II loci, primarily HLA-DR. HLA-DQ and HLA-DP make minor contributions to these phenotypes. The genes specifying both class I and class II antigens are tightly clustered in a single chromosomal region, the MHC.
Genomic organization
The human MHC is a genetic region located on the short arm of chromosome 6 (6p21.3) extending approximately 4 megabases (Mb) (Fig. 39.1). The MHC encodes over 250 genes and pseudogenes of which at least 150 are expressed as proteins.9,10 This genetic complex is divided into three regions: (centromeric) class II, class III and class I (telomeric). Although the proteins encoded within the MHC participate in a variety of functions, approximately 40% are devoted to immune system functions.
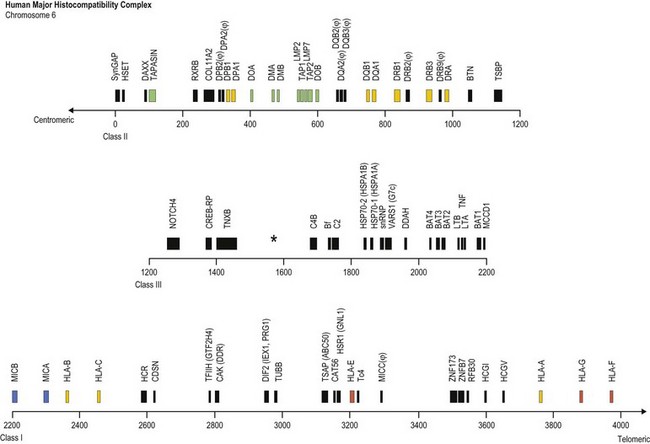
Fig. 39.1 Map of the human major histocompatibility complex (MHC) located on the short arm of chromosome 6. The map (drawn closely to scale) is divided into three regions (class II, class III and class I) and shows the relative positions of some of the MHC encoded genes and pseudogenes (ψ). The class II region encodes the expressed classical HLA class II molecules (yellow). The number of HLA-DR B (DRB) genes and pseudogenes differs for different haplotypes (DR52 haplotype shown; Fig. 39.2). Genes encoding protein products intimately involved in MHC class I and class II molecule assembly and in peptide processing and binding (green) also are shown. The class III region contains several immune system relevant genes and varies in length among individual chromosomes in the area encoding C4B (asterisk). The class I region encodes the classical HLA class I heavy chain proteins (yellow). This region also encodes the expressed nonclassical HLA class I genes (red) and MHC class I chain-related genes (blue).
(The figure was generated from data contained in references.9,13)
The class II region (~1.2 Mb) contains at least 34 expressed genes and 16 pseudogenes and spans from SynGAP (Ras-GTPase-activating protein, centromeric) to TSBP (testis-specific basic protein, telomeric).11 This region includes the genes that encode for the classical class II molecules (HLA-DR, -DQ and -DP). In addition, gene products involved in MHC class I antigen processing (LMP2 and LMP7, the large multifunctional proteosome genes), peptide transport (TAP1 and TAP2, the transporter associated with antigen processing genes) and complex assembly (tapasin) and gene products involved in MHC class II complex assembly (HLA-DM and HLA-DO) are encoded in this region.
The class III region (~1 Mb) extends from NOTCH4 (transmembrane receptor involved in cell differentiation and development, centromeric) to MCCD1 (mitochondrial coiled-coil domain protein 1, telomeric).12,13 On average, this region contains 1 gene every 10 kilobases (kb) and is the most gene dense region in the human genome with approximately 72% of it being transcribed. The class III region encodes at least 62 expressed genes including complement components (e.g., C2 and C4B), heat shock proteins (e.g., Hsp70-1 and Hsp70-2) and cytokines of the tumor necrosis factor family (e.g., TNF and LTA). Some individuals have duplications in the area encoding complement component C4B; thus, this area can vary in length.
The class I region (~1.85 Mb) from MICB (centromeric) to HLA-F (telomeric) encodes at least 118 genes; 57 expressed genes and 61 pseudogenes.14 In some instances, an additional ~0.95 Mb to TRIM27 (tripartite motif 27, a transcription factor, telomeric) is included and called the extended class I region.9 This region includes the classical class I genes (HLA-A, -B and -C) and the nonclassical class I genes (HLA-E, -F, and -G). This region also encodes the MHC class I chain-related (MIC) genes (MICA and MICB). The focus of this chapter is on the MHC encoded gene products involved in histocompatibility, primarily the classical human leukocyte antigens. Other MHC and nonMHC encoded genes that participate in histocompatibility are also covered.
The human leukocyte antigens
Genomic organization
The heavy chains of the classical MHC class I (class Ia) molecules are encoded in the class I region of the MHC and associate with beta 2 microglobulin (encoded on chromosome 15) to form the mature class I molecule. The gene order within the MHC is shown in Fig. 39.1. Each of the classical MHC class I heavy chains is encoded by a single gene that is divided into 8 exons.15 Exon 1 encodes the 5′ untranslated region (UTR) and the hydrophobic signal sequence. The signal sequence directs insertion of the protein into the membrane at the cell surface and is cleaved from the mature protein. The extracellular portion of the class I heavy chain is encoded by exons 2–4. Exon 5 encodes the transmembrane region and exons 6 and 7 encode the intracellular cytoplasmic tail. The 3′ UTR and the polyadenylation (poly(A)) site are encoded by exon 8. The mRNA, which is translated into protein, includes all eight exons after removal by splicing of the intervening sequences (introns).
The class II region of the MHC contains three subregions (centromeric) HLA-DP, -DQ and -DR (telomeric). Each encodes at least one cell surface class II molecule (Fig. 39.1). The class II molecules are noncovalently associated heterodimers that consist of an α chain and a β chain.15,16 Each chain is encoded by a separate gene, an A gene for the α chain and a B gene for the β chain. The expressed HLA-DP heterodimer is encoded by the DPA1 and DPB1 genes. The HLA-DP subregion contains two DP pseudogenes, DPA2 and DPB2. The HLA-DQ subregion contains two A (DQA1 and DQA2) and three B (DQB1, DQB2 and DQB3) genes. The expressed HLA-DQ heterodimer is encoded by the DQA1 and DQB1 genes, while the remaining DQ genes are pseudogenes. Each individual has two copies of chromosome 6 and, thus, two copies of each of the expressed HLA-DP and HLA-DQ genes. These genes are polymorphic and, consequently, an individual can have two different expressed A genes and two different expressed B genes for HLA-DP and for HLA-DQ. While not all combinations form,17 the products of some of these genes can associate in several αβ combinations, regardless of chromosomal origin. Therefore, an individual could express up to four different HLA-DP and up to four different HLA-DQ molecules.
The HLA-DR subregion is more complex.18,19 A HLA-DR molecule composed of a conserved α chain encoded by the DRA gene and a polymorphic β chain encoded by the DRB1 gene is almost always present. This is the major class II molecule expressed on the cell surface. An additional eight DRB genes and pseudogenes have been identified in the HLA-DR subregion. The number of DRB genes present and the number of expressed DRB gene products is characteristic of each chromosome (haplotype) a person inherits (Fig. 39.2). For example, the DR1 haplotype carries two DRB genes, the expressed DRB1 gene and a DRB6 pseudogene. The DR8 haplotype carries only one DRB gene, the expressed DRB1 gene. Other DR subregion haplotypes can encode a second expressed HLA-DR molecule composed of the DRA gene product associated with a DRB3 gene product (DR52 molecule), a DRB4 gene product (DR53 molecule) or a DRB5 gene product (DR51 molecule) and can contain one to three DRB pseudogenes. The designations DR51, DR52 and DR53 are antibody (serologically) defined.
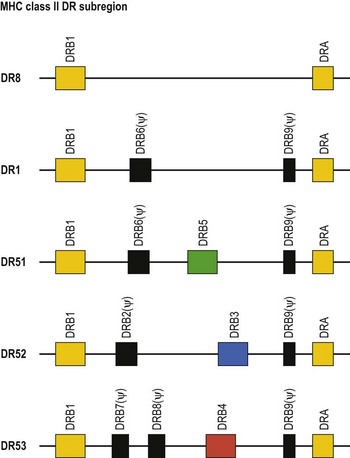
Fig. 39.2 Relative gene organization of the DR subregion of different DR haplotype groups. The protein products of the DRA and DRB1 genes (yellow) combine to form the primary expressed HLA-DR molecule. The protein products of the DRB5 (green), DRB3 (blue) and DRB4 (red) genes encoded by the DR51, DR52 and DR53 haplotype groups, respectively, also form expressed functional DR molecules in combination with the DRA polypeptide. The DRB pseudogenes (ψ) are shown in black. Table 39.4 lists the alleles associated with each haplotype.
The A genes, which encode the α chains of class II molecules, contain five exons.15,16 The 5′ UTR and hydrophobic signal sequence are encoded by exon 1, like the class I genes. Exons 2 and 3 encode the extracellular domains. Exon 4 encodes the connecting peptide, the transmembrane region, the intracellular cytoplasmic tail and a portion of the 3′ UTR. The remainder of the 3′ UTR and the poly(A) signal are encoded by exon 5. Each class II β chain is encoded by a B gene divided into six exons. Exons 1–3 are similar to that of the A genes. Exon 4 encodes the connecting peptide and transmembrane region, while exon 5 encodes the cytoplasmic tail. The 3′ UTR and poly(A) signal are encoded by exon 6. All exons and introns are transcribed into RNA for the class II A and B genes. Again, introns are removed by splicing to form the mRNA that is translated into protein.
Diversity
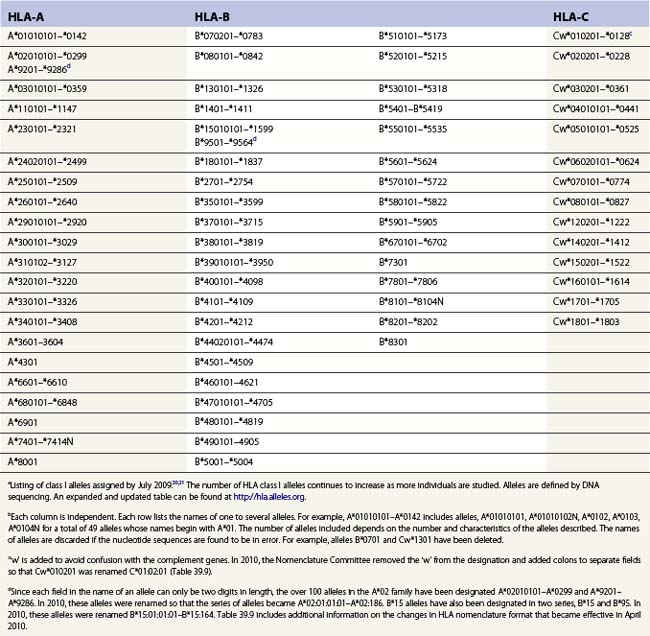
The nucleotide sequences of many of the HLA genes differ among individuals. These sequence variants are termed alleles; genes with many alleles are termed polymorphic. Alleles of a locus may differ by a single nucleotide to many nucleotides potentially resulting in changes in the amino acid sequence of the protein specified by that gene. The classical HLA class I and class II loci are the most polymorphic loci in humans. The HLA-B and HLA-DRB1 loci have over 1100 and 650 known alleles, respectively (Tables 39.1, 39.2).20,21 In contrast to non-HLA genes, the nucleotide differences found among HLA alleles are usually nonsynonymous (alter the amino acid sequence) and are focused in the exon(s) encoding the most functionally important region of the HLA molecule, the antigen binding site.18 It is thought that this diversity has been maintained to provide the human population with the capacity to recognize a diverse repertoire of pathogenic peptides.22–24 Unfortunately, the allelic differences in HLA molecules expressed on the cells of different individuals can be recognized as foreign when tissue is grafted from one individual to another.
| DR | DQ | DP |
|---|---|---|
| DRA*0101–*010202c | DQA1*010101–0107h | DPA1*010301–*0110j |
| DRB1*010101–*0122d | DQA1*0201 | DPA1*020101–*0204 |
| DRB1*03010101–*0348 | DQA1*030101–*0303 | DPA1*0301–*0303 |
| DRB1*040101–*0478 | DQA1*040101–*0404 | DPA1*0401 |
| DRB1*07010101–*0717 | DQA1*050101–*0509 | DPB1*010101–*9901k |
| DRB1*080101–*0836 | DQA1*060101–*0602 | |
| DRB1*090102–*0908 | DQB1*020101–*0205i | |
| DRB1*100101–*1003 | DQB1*030101–*0325 | |
| DRB1*110101–*1181 | DQB1*0401–*0403 | |
| DRB1*120101–*1219 | DQB1*050101–*0505 | |
| DRB1*130101–*1392 | DQB1*060101–*0635 | |
| DRB1*140101–*1490 | ||
| DRB1*15010101–*1533 | ||
| DRB1*160101–*1613N | ||
| DRB3*01010201–*0113e | ||
| DRB3*0201–*0224 | ||
| DRB3*030101–*0303 | ||
| DRB4*01010101–*0107f | ||
| DRB4*0201N | ||
| DRB4*0301N | ||
| DRB5*010101–*0113g | ||
| DRB5*0202–*0205 |
a Listing of class II alleles assigned by July 2009.20,21 The number of HLA class II alleles continues to increase as more individuals are studied. Alleles are defined by DNA sequencing. An expanded and updated table can be found at http://hla.alleles.org. A description of the nomenclature in this table can be found in Table 39.9.
c Alleles of the DRA locus. The differences among these DR alpha chain alleles are not considered important for transplantation matching.
d Alleles of the DRB1 locus. Most haplotypes contain a DRB1 locus.
e Alleles of the DRB3 locus, the second expressed DR molecule in haplotypes carrying DRB1*03, *11, *12, *13, *14 alleles.
f Alleles of the DRB4 locus, the second expressed DR molecule in haplotypes carrying DRB1*04, *07, *09 alleles.
g Alleles of the DRB5 locus, the second expressed DR molecule in haplotypes carrying DRB1*15, *16 alleles.
h Alleles of the DQA1 locus. DQA1 allelic products pair with DQB1 allelic products to form the DQ molecule.
j Alleles of the DPA1 locus. DPA1 allelic products pair with DPB1 allelic products to form the DP molecule.
k Alleles of the DPB1 locus. The approach to naming DPB1 alleles was slightly different than that used for other loci because of the lack of serologic information.20
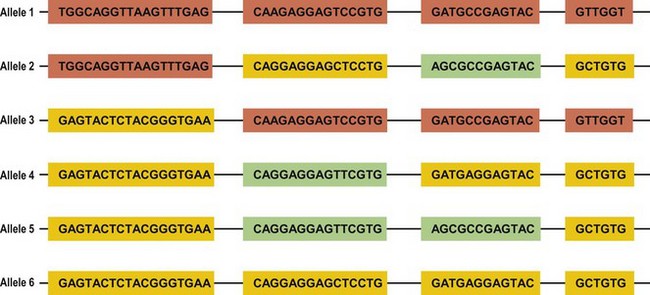
Several mechanisms are hypothesized to have generated this HLA diversity over evolutionary time. The majority of the polymorphism is hypothesized to have arisen by the non-reciprocal exchange of short polymorphic regions or cassettes among alleles, a process referred to as gene conversion. As a result, the HLA alleles are patchworks of polymorphic cassettes, each cassette shared by some of the other alleles at the locus, embedded in a conserved framework (Fig. 39.3). Exchange of cassettes among alleles at different loci, reciprocal recombination involving the exchange of entire exons, and mutation also have contributed to HLA diversity.18,25
Each individual inherits two copies of the chromosome carrying the MHC, one from each parent, and thus has two copies of each gene in the MHC. Individuals who carry two identical alleles at a locus are homozygous; individuals with two different alleles at a locus are heterozygous. Because the HLA genes are located within a small genetic distance, they are usually inherited as a block unless separated by recombination. The block, a specific set of alleles at the multiple HLA loci inherited together from a parent, is termed a HLA haplotype. Fig. 39.4 illustrates the inheritance of HLA-A, -B, and -DRB1 alleles within a family. By convention, the paternal haplotypes are generally designated a and b and the maternal haplotypes, c and d. Thus, there are four possible MHC genotypes in the offspring: ac, ad, bc, and bd. Because the chances of inheriting a given genotype are random, the probability of occurrence of any one of the four genotypes is one in four in a mating. In a family with five children, at least two of the children will be MHC identical unless recombination has occurred.
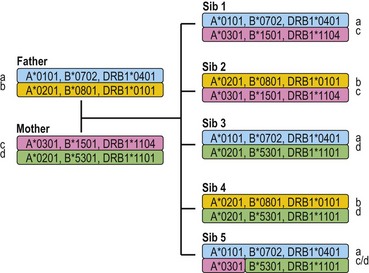
Fig. 39.4 The inheritance of HLA alleles and haplotypes within a family. Paternal haplotypes are labeled a,b; maternal haplotypes, c,d. Sibling 5 inherits a recombinant chromosome from the mother. Not all HLA genes are shown. The HLA nomenclature used in the figure is described in Table 39.9.
The alleles in the MHC complex can be reshuffled by crossing over between homologous chromosomes during the generation of sperm or eggs. The frequency of recombination across the MHC from HLA-A to HLA-DPB1 can range from 0.7 to 4.3%.26 Studies in humans suggest that there are several sites at which recombination preferentially occurs within the MHC, particularly between HLA-B and HLA-DRB1 and between HLA-DQB1 and HLA-DPB1.26,27 Studies comparing MHC-identical sib pairs versus haplotype mismatched sib pairs and unrelated individuals show that recombination rates can vary up to sixfold between individuals with different MHC haplotypes and suggest a genetic influence within the MHC on recombination rates.26 On average, the frequency of recombination between HLA-A and HLA-B is 0.7%, between HLA-B and HLA-DRB1 is 1.0%, and between DQB1 and DPB1 is 0.8%. Recombinations between DQA1 and DRB1 loci and between B and C loci are very rare.
The HLA alleles and haplotypes found in individuals depend on their racial and ethnic backgrounds.28,29 For example, the allele DRB1*0302 is found in African Americans, but is only rarely observed in individuals of northern European or Asian descent. Likewise, the frequency of a combination of alleles on a single copy of chromosome 6 can differ among population groups. Table 39.3 lists the ten most common haplotypes identified in the African American population compared to the ranking of these haplotypes in several other US populations.29 For example, the most common haplotype in African Americans is A*3001, Cw*1701, B*4201, DRB1*0302 found at a frequency of 1.5%. This haplotype was ranked 37th in Hispanic Americans but was not observed in this sampling of European Americans and Asian Americans.
Table 39.3 The ten most common African American haplotypes and their ranking in individuals of other ethnicities from a US registrya,b
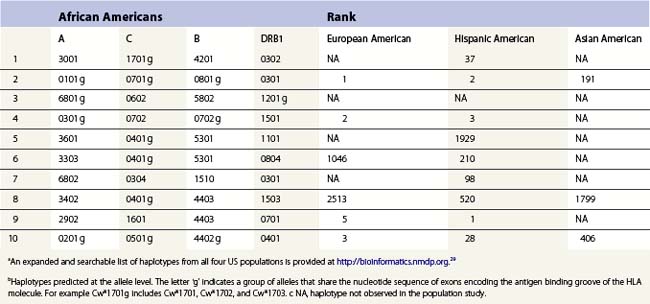
There are, however, some alleles and haplotypes that are found in many populations. For example, the allele A*0201 is a common allele in many US populations, being found in 29.6% of European Americans, 12.5% of African Americans, 9.5% of Asian Americans and 19.4% of Hispanic Americans.29 The most common haplotype in European Americans, A*0101, Cw*0701, B*0801, DRB1*0301, is the second most frequent haplotype in African Americans and Hispanic Americans but is the 191st most frequent haplotype in Asian Americans.
When large databases of HLA typed individuals are analyzed, only a small percent of potential HLA phenotypes are found. Using serologic assignments from an unrelated donor registry, of the predicted 19 536 660 HLA-A,-B,-DR phenotypes, only 1.6% were observed.30 This suggests that not all HLA allele combinations will be found. Indeed, some HLA haplotypes appear more frequently than expected. Linkage disequilibrium measures the degree of non-random association between alleles of separate loci. Apparently high disequilibrium across the DR-DQ subregion coupled with a lack of recombination have resulted in specific associations between DQA1 and DQB1 alleles and between DRB1 and DQ alleles although a single allele such as DQB1 may be associated with one of several partner alleles. For example, DQB1*0602 is found on the same chromosome as the DQA1 alleles DQA1*0102, or *0103 or *0104, but has not been observed with DQA1*0201, *0301, *0401, or *0501.31,32 These associations may differ among individuals of different racial/ethnic backgrounds. For example, DRB1*0901 is associated with DQB1*0201 in African Americans but with DQB1*0303 in individuals of northern European descent. Within the DR subregion, specific allele combinations at the several DRB loci are associated with families of DR haplotypes (Fig. 39.2). For example, the DRB3 locus is found in haplotypes carrying specific DRB1 alleles including DRB1*0301, *1101, *1201, *1301, and *1401 alleles (Table 39.4). In the class I region, associations between HLA-B and -C alleles have also been noted.29
| DR molecules encoded | Expressed DR loci included in haplotype | DRB1 alleles associated with haplotype |
|---|---|---|
| DR, DR51 | DRA, DRB1, DRB5 | DRB1*15, *16 |
| DR, DR52 | DRA, DRB1, DRB3 | DRB1*03, *11, *12, *13, *14 |
| DR, DR53 | DRA, DRB1, DRB4 | DRB1*04, *07, *09 |
| DR | DRA, DRB1 | DRB1*01, *08, *10 |
a It should be noted that there are exceptions to these associations. For examples, DRB1*15 haplotypes have been observed that lack a DRB5 locus and some DRB5 positive haplotypes lack a DRB1 locus. In addition, some DRB1*01 haplotypes carry a DRB5 locus.
Extension of linkage disequilibrium across longer regions of the MHC has resulted in associations between specific class I and class II alleles. The associations of multiple alleles result in extended haplotypes.33,34 The most well known extended haplotype is A*0101, Cw*0701, B*0801, DRB1*0301 which is common in northern Europe, appearing at a frequency of approximately 5–15%.28 It has been hypothesized that these associations may have been maintained in the population by selection, that is, associations between DR and DQ as well as associations within an extended haplotype might represent optimal combinations of immune response molecules. It is also likely that features of the genome structure limiting recombination or changes in the structure of the population, such as through admixture of different ethnic groups, have caused the linkage disequilibrium. Because alleles at various HLA loci are non-randomly associated, these associations enhance the frequency with which individuals share alleles across multiple HLA loci facilitating the selection of HLA identical individuals as tissue donors.
Expression
Classical MHC class I proteins are expressed by most nucleated cells, but the level of expression on the cell surface varies for different cell types. Cis-acting sequence blocks (enhancer A, interferon-stimulated response element (ISRE), W/S box, X box (previously known as site α) and Y box (previously known as enhancer B)) in the regulatory (promoter) region upstream of each class I gene control gene expression (Fig. 39.5A).35 Each promoter sequence block binds numerous proteins (transcription factors) that regulate the level of transcription of the gene and ultimately the amount of protein at the cell surface. For example, enhancer A binds stimulating protein 1 (Sp1) and various members of the NF-κB/rel family of transcription factors. Collectively, the complex is termed NF-κB. Normal class I gene expression requires the coordinated action of each of these regulatory elements; however, disruption of any one sequence block reduces, but does not appear to ablate, MHC class I expression.
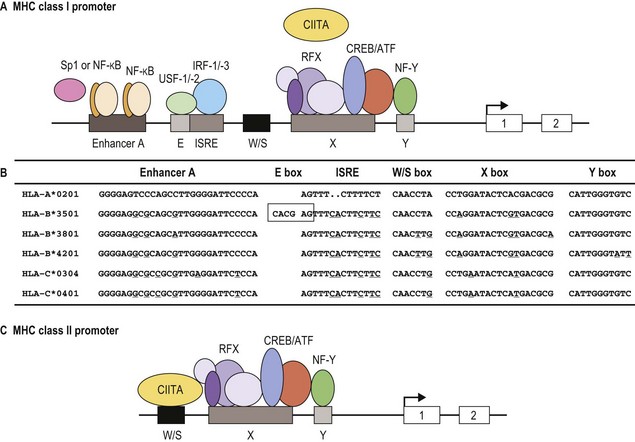
The amounts of HLA-A, -B and -C molecules expressed at the surface of a cell are not equal.36 HLA-A and -B are abundant with HLA-A expressed at somewhat higher levels than HLA-B, in many instances. HLA-C is expressed at very low levels in comparison accounting for about 10% of cell surface class I molecules. This is due to sequence variations in the regulatory blocks of each class I locus (Fig. 39.5B) which alter the type and affinity of transcription factor binding. For example, only NF-κB/rel family members bind to enhancer A in the HLA-A promoter, while enhancer A in the HLA-B promoter binds SP1 in addition to NF-κB/rel family members. The inclusion of SP1 binding may lead to less efficient expression of the HLA-B gene. There are also allele specific differences in the nucleotide sequence of these regulatory elements such that, for example, different HLA-B alleles are expressed at different levels. Additionally, some HLA-B allele promoter regions encode an E box regulatory element that binds the transcription factors upstream stimulatory factor 1 (USF-1) and USF-2. The presence of the E box correlates with reduced basal expression levels of these HLA-B alleles.35
MHC class I gene expression can be up-regulated by various cytokines.35,36 Interferon-γ (IFN-γ) performs a fundamental role in enhancing MHC class I expression by inducing increased gene transcription via transactivation of the ISRE. Again, locus and allele specific sequence differences in the ISRE result in different levels of transcriptional enhancement for each of the class I genes. Additionally, IFN-γ up-regulates expression of the class II transactivator (CIITA) further enhancing MHC class I gene expression. Other cytokines, such as tumor necrosis factor (TNF), can enhance the stimulatory effect of INF-γ on MHC class I gene expression via up-regulation of NF-κB that acts through enhancer A.
MHC class II protein expression is more limited than that of MHC class I. Cell surface HLA-DR, -DQ and -DP molecules are found primarily on professional antigen presenting cells (APC) and on other immune system cells such as T-lymphocytes.36,37 Professional APC are bone marrow derived cells dedicated to the task of peptide presentation by MHC molecules and include B-lymphocytes, macrophages, dendritic cells, thymic epithelial cells and Kupffer cells. IFN-γ can induce class II expression in other cell types. Like the class I genes, the promoter regions of the class II genes contain the cis-acting sequence blocks W/S box, X box and Y box (termed the SXY module) that regulate gene expression (Fig. 39.5C). This is a conserved regulatory module present in the promoters of a wide range of genes involved in antigen presentation which functions as a single regulatory block. The X and Y boxes bind several ubiquitous transcription factors, such as regulatory factor X (RFX) and nuclear factor Y (NF-Y), respectively, in a complex termed the enhanceosome.38,39
Occupancy of the class II gene regulatory elements by the enhanceosome is absolutely required, but not adequate for expression and IFN-γ induction of the A and B genes of HLA-DR, -DQ and -DP. CIITA also is required for class II gene expression.35,38 Cell type specific and IFN-γ induction of class II expression is the direct result of CIITA expression patterns. Constitutive expression of CIITA is confined to professional APC and other immune system cells and can be induced by IFN-γ in other cell types, paralleling expression of MHC class II. CIITA is recruited to the enhanceosome of the class II promoter by the S box. This is not the case for class I gene expression for which the S box apparently plays no role. All of the other transcription factors that regulate class II gene transcription are ubiquitously expressed and constitutively occupy the regulatory sequence blocks in the MHC class II gene promoters. Of interest, patients with bare lymphocyte syndrome (MHC class II deficiency) can have a defect in any one of a number of the transcription factors that bind to the SXY module or in CIITA. These patients still express MHC class I albeit at reduced levels.
HLA-DR, -DQ and -DP are not expressed at the same levels on cell surfaces, similar to expression of the different MHC class I molecules. HLA-DR is the most abundant MHC class II molecule expressed by cells. DRB1 is expressed at a higher level compared to DRB3 and DRB4.40,41 HLA-DQ is expressed at reduced levels and HLA-DP is the least abundant cell surface class II molecule. Like the regulatory elements in the promoters of class I genes, there are both locus and allele specific sequence differences in the regulatory elements of the class II genes.36 These sequence differences account for the dissimilar levels of class II molecule expression in two ways: 1) alter the binding affinity of the transcription factors and 2) allow binding of proteins that repress gene transcription of specific class II loci. For example, the X box in the HLA-DPA1 gene promoter region specifically binds the X box repressor protein which diminishes transcription of the HLA-DPA1 gene and reduces the overall level of HLA-DP on the cell surface.
Some pathogenic micoorganisms and many malignant cells down-regulate HLA gene expression to avoid recognition by the immune system.42–44 For example, human cytomegalovirus (CMV) interferes with IFN-γ induction of MHC class I and class II gene expression.45 To avoid detection by T-cells, many carcinomas and lymphomas lack cell surface HLA-A and HLA-B molecules due to defects in the expression or the binding of specific transcription factors to the promoter regulatory blocks of these genes. In many instances, expression of HLA-C in these malignant cells is unaffected, allowing the cell to avoid recognition by natural killer (NK) cells. In fact, alterations in MHC class I expression is very common (reported to be 70% or greater) in a variety of tumor types such as cervical, breast and colorectal cancers.42
Structure of class I and class II molecules
The classical class I molecules (HLA-A, -B and -C) are expressed on cell surfaces as a trimolecular complex. This complex is composed of the HLA class I polypeptide (heavy chain), beta 2 microglobulin (β2m) and a peptide. The MHC encoded class I heavy chains are glycosylated transmembrane proteins of approximately 340 amino acids (~44 kilodaltons (kD)) that belong to the immunoglobulin (Ig) superfamily of proteins.15,46 The extracellular portion of the class I heavy chain is composed of the amino-terminal 275 amino acids. The following 40 amino acids make up the hydrophobic transmembrane region and the carboxy-terminal 25 amino acids comprise the intracellular cytoplasmic tail. As an aside, soluble isoforms of the classical HLA molecules are produced and may have immunoregulatory roles.47
The extracellular portion of the class I heavy chain is divided into three domains, termed α1, α2 and α3 (Fig. 39.6A). Each domain is encoded by a separate exon (exons 2–4, respectively) and is approximately 90 amino acids long. The 3D structures of the extracellular portion of several class I molecules were resolved by X-ray crystallography.48,49 The α1 and α2 domains fold together to form a groove (termed the antigen binding groove) distal to the cell membrane that consists of a floor of eight antiparallel beta strands topped by two alpha helices fashioned into the walls. The membrane proximal α3 domain folds into a structure which is similar to that of the constant region domains of immunoglobulins (antibodies). This domain is composed of two antiparallel beta sheets, one with four strands and one with three strands. The sheets are linked by a disulfide bond. β2m (~12 kD) is non-covalently associated with the α3 domain of the class I heavy chain and is required for cell surface expression. Its 3D structure is identical to that of the α3 domain of the heavy chain.
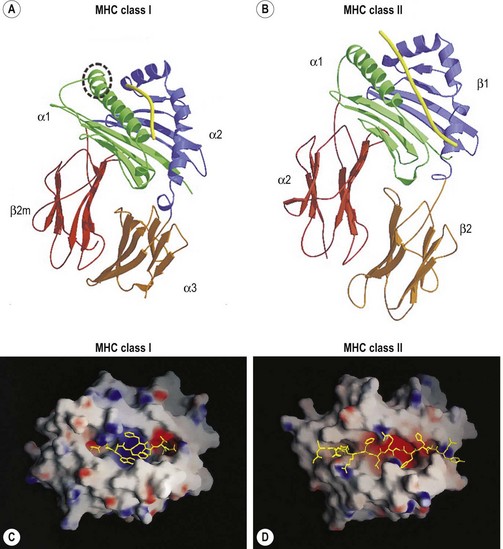
The peptide, the third component of the trimolecular complex, is generally 8–10 amino acids in length and non-covalently bound to the class I heavy chain.48,49 It lies in the groove formed by the α1 and α2 domains (Fig. 39.6C). The peptide is anchored at its amino- and carboxy-terminal ends by non-covalent bonds to amino acid residues in the class I heavy chain. There are pockets along the groove which accommodate amino acid side chains at various positions along the peptide. The pockets are unique for each class I molecule because polymorphic residues from the α1 and α2 domains participate in their formation. Each pocket has specific physical and chemical characteristics that are determined by the conserved and polymorphic class I residues that form the pocket. These characteristics, in turn, dictate which amino acid side chains are accommodated at the corresponding peptide position. This defines the peptide binding motif of each class I molecule and defines the overall character of the set of peptides bound (Table 39.5).50,51 For example, the protein encoded by A*1101 will accommodate a variety of ‘small’ amino acids at peptide postions 2, 3 and 6 and prefers basic amino acids at peptide position 9. However, each pocket does not make an equal contribution to peptide binding. Certain pockets, specific to each class I molecule, play a more predominant role and the corresponding peptide position is termed an anchor position. The preferred amino acids at an anchor position are termed anchor residues. Using the protein encoded by A*1101 again as an example, although peptide positions 2, 3 and 6 contribute to peptide binding, it is peptide position 9 that is the anchor position. Lysine and arginine are the anchor residues at this position with lysine preferred over arginine. It is of note that not all peptides that bind to an HLA molecule fully adhere to the defined peptide binding motif and that amino acids other than anchor residues can be found at anchor positions in these peptides. In the end, each class I molecule does bind a unique, large set of peptides and the peptide set shares particular sequence characteristics which are dictated by the amino acid residues that make up the groove of the HLA class I heavy chain.
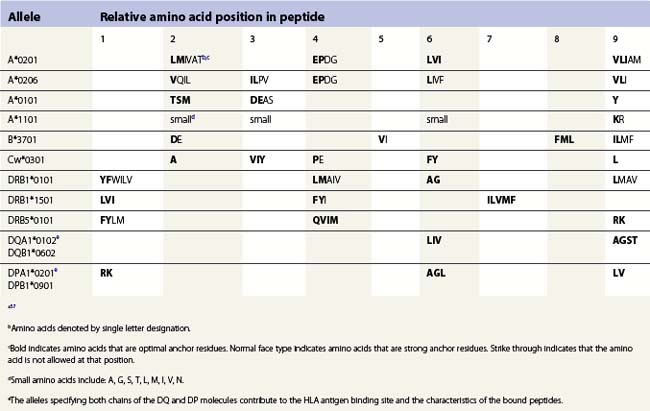
There are benefits to determining the peptide binding motif for HLA molecules. For example, these motifs can be used to identify antigenic peptides from pathogen proteins as candidates for use in peptide based vaccines. As another example, expression of specific HLA allelic products is associated with an increased risk of developing many autoimmune diseases.52,53 In most cases, this is thought to be the result of the differential binding capacity of HLA molecules for particular peptides. Thus, knowing the binding motif for a HLA molecule aids in the identification of the culprit peptide and allows for the design of synthetic peptides that mimic disease associated peptides for use in blocking autoimmune responses. Because the number of HLA allelic products is so large, algorithms have been developed for prediction of peptide binding to HLA molecules.54–57
The class II molecules are expressed on cell surfaces as a trimolecular complex, structurally analogous to the class I molecules, and consist of the class II α chain, the class II β chain and a peptide. Both the class II α (34 kD) and β (28 kD) chains are transmembrane glycoproteins and are Ig superfamily members, comparable to the class I heavy chain.15,16 The extracellular portion of the α and β chains are divided into two domains, the membrane distal α1 and β1 domains and the membrane proximal α2 and β2 domains. Similar to the class I heavy chains, each domain is encoded by a separate exon (exons 2 and 3, respectively) and is about 90 amino acids in length. Three additional regions complete each chain of the class II molecule; a connecting peptide of 12 amino acids which is highly hydrophilic and links the membrane proximal domain to the transmembrane region, a 23 amino acid hydrophobic transmembrane region and an intracellular cytoplasmic tail that consists of the carboxy terminal 8–15 amino acids.
The 3D structures of the extracellular portion of several class II molecules have been determined by X-ray crystallography.48,49,58 These structures are strikingly similar to that of class I molecules. The α1 and β1 domains fold together to form a peptide binding groove like the groove formed by the class I heavy chain α1 and α2 domains (Fig. 39.6B). The α2 and β2 domains of the class II chains fold to form Ig constant region domain like structures similar to that of the class I α3 domain and β2m.
The peptides that bind to class II molecules are anchored to the class II antigen binding groove by non-covalent bonds to the peptide backbone and by binding of peptide amino acid side chains into pockets along the groove (Fig. 39.6D), similar to the class I molecules.49,58,59 Because of the polymorphic nature of the MHC class II proteins, each class II molecule, like each class I molecule, also binds a large set of peptides which share a peptide binding motif specific to that class II molecule (Table 39.5). The peptides that bind to class II molecules are heterogenous in length and generally 13–25 amino acids long. The low and open ends of the class II groove allow peptides of varying lengths to bind in an extended conformation with the ends of the peptide overhanging the ends of the groove. This is in contrast to the class I molecules which bind peptides of 8–10 amino acids. The ends of the class I groove are high and closed; thus, MHC class I molecules optimally accommodate shorter peptides whose ends are tucked into the groove (compare Fig. 39.6C and 39.6D).
Function
Peptide processing and binding
Mature cell surface class I molecules are formed in the endoplasmic reticulum (ER) with the aid of several resident ER proteins including tapasin, calnexin and calreticulin.60,61 Initially, the class I heavy chain and β2m fold and associate facilitated by calnexin and calreticulin (Fig. 39.7). This complex then transiently associates with the transporter associated with antigen processing (TAP) where a peptide is loaded into the groove of the class I heavy chain. Finally, the trimolecular complex is dispatched to the cell surface.
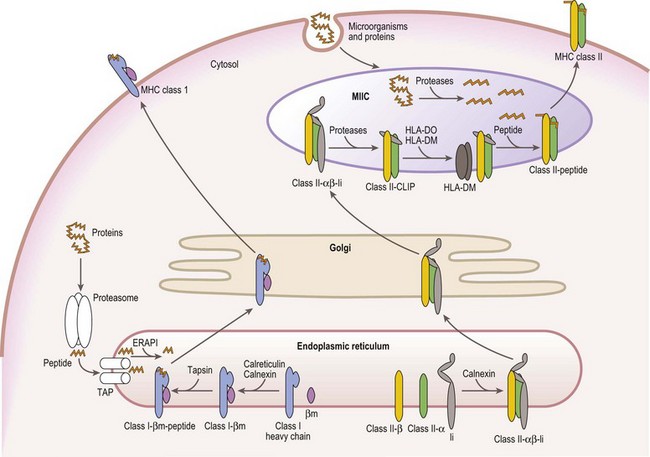
Peptides, derived from both self (normal cellular) proteins (potential autoantigens) or foreign proteins (antigens), are generated in the cytosol by the proteosome.60,61 The proteosome is a macromolecular structure that proteolytically cleaves proteins into peptides (a process termed antigen processing) and consists of members of the large multifunctional proteosome (LMP) family and other protein subunits. Two LMP family members (LMP2 and LMP7) are encoded in the class II region of the MHC. The proteosome is tightly associated with the TAP molecule which shuttles the peptides into the lumen of the ER.60,61 TAP is formed by the association of the products of the TAP1 and TAP2 genes also encoded in the MHC class II region. Another MHC encoded gene product, tapasin, stabilizes the TAP heterodimers, links the class I heavy chain to TAP for peptide loading and facilitates loading of peptides onto the class I molecule. An endoplasmic reticulum aminopeptidase plays a dual function in class I antigen presentation. It trims longer peptides that are more readily shuttled by TAP to the optimal length (8–9 amino acids) enhancing binding to class I molecules, but also destroys many peptides limiting the pool available for binding to class I.62,63
The class II α and β chains fold and associate in the ER with the assistance of resident ER chaperones such as calnexin (Fig. 39.7),64 similar to class I molecules. Unlike class I, full maturation of the class II molecule does not take place in the ER. Instead, class II heterodimers are directed primarily to specialized endosomal compartments (MHC class II compartments, MIIC), or, in some instance, first traffic to the cell surface and then to the MIIC.65 Once in the MIIC, peptides are loaded into the antigen binding groove and mature class II molecules are dispatched to the cell surface.
MHC class II molecules bind peptides derived from endocytosed microorganisms and from self and foreign proteins degraded by proteases in the endocytic pathway.66 This is in contrast, yet complementary, to the peptides bound by MHC class I molecules. In general, class II molecules bind peptides from cell surface and extracellular sources, while class I molecules bind peptides from intracellular sources. Thus, proteins from the whole environment of a cell can be surveyed by the immune system. Invariant chain (Ii), a nonMHC encoded glycoprotein, plays a key role in facilitating this division of function.
Ii performs several functions in assuring proper antigen presentation by class II molecules. Ii chain serves as a chaperone in the folding and assembly of class II αβ heterodimers and protects the class II peptide binding groove from binding peptide in the ER via a 25 residue internal peptide segment termed CLIP (class II associated invariant chain peptide).65,66 Ii also provides the intracellular targeting signals that direct the complex to the MIIC. Under the acidic conditions of the MIIC, Ii is proteolytically cleaved and dissociates from the class II molecule, while CLIP remains bound to the class II antigen binding groove. CLIP is exchanged for antigenic peptides in a reaction catalyzed by HLA-DM, a resident MIIC protein that tightly associates with the class II heterodimer.67–69 HLA-DM is a critical component in shaping the repertoire of peptides bound to class II molecules as it retains the class II molecules in the MIIC until a stable, high affinity complex between the class II molecule and a peptide is formed. In certain APC types, B-cells and thymic epithelial cells, another resident MIIC protein, HLA-DO, negatively regulates the actions of HLA-DM.67,68
Analogous to the MHC class II molecules, both HLA-DM and HLA-DO are class II related Ig superfamily members expressed as heterodimers that consist of an α chain and a β chain.67–69 The HLA-DM and HLA-DO α and β chains are encoded by A and B genes, respectively, in the MHC class II region and regulation of these genes is similar to that of the MHC class II genes.69 The 3D structure of HLA-DM resembles that of the MHC class II molecules except that its peptide binding groove is almost entirely obscured.48
Components of the peptide processing and binding pathways of MHC class I and class II molecules are a favorite target of disruption by many pathogens and malignant cells to avoid detection by the immune system.42–44 For example, two proteins (US3 and US6) encoded by human CMV block cell surface expression of MHC class I molecules and thus, detection of the infected cell by cytotoxic T-cells. US3 binds to MHC class I molecules and retains them in the ER and US6 inhibits peptide transport into the ER by TAP. Lack of cell surface MHC class I, however, renders the CMV infected cell susceptible to lysis by NK cells. To circumvent NK cell recognition, CMV encodes a class I like decoy termed UL18 which is recognized by NK cells and inhibits their function.45 Other pathogens and malignant cells employ a variety of unique strategies to block MHC expression.
T lymphocyte recognition of HLA molecules
The predominant function of classical MHC class I and class II molecules is to bind peptides from the cell’s environment and present these peptides on the surface of the cell for inspection by the immune system. The archetypical receptor involved in the inspection process is the T-cell receptor (TCR) on T lymphocytes (Fig. 39.8). There are two types of TCR (αβ and γδ) both of which are multipolypeptide complexes that consist of a TCR α chain covalently paired with a TCR β chain or of a TCR γ chain covalently paired with a TCR δ chain tightly, but noncovalently, associated with several chains of the CD3 family (α, δ, ε, ζ, η).70,71 The TCR chains are involved in the direct recognition of the HLA molecule and of the peptide bound to the HLA molecule.71,72 CD3 is involved in the signaling process which activates the T-cell after recognition of the ligand by the TCR chains.70
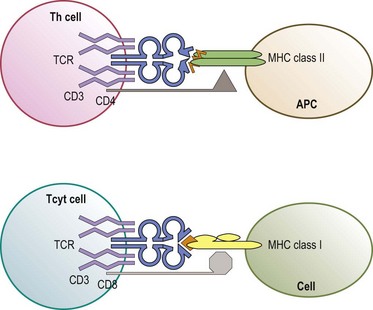
T lymphocytes are classically divided into two groups based on expression of co-receptors (CD4 and CD8) which are intimately involved in recognition of the HLA molecule.71,73 CD4 expressing (CD4+) T-cells are usually MHC class II restricted; that is, their TCR recognizes either an HLA-DR, -DQ or -DP molecule which all have a CD4 binding site (Fig. 39.8). CD8 expressing (CD8+) T-cells are generally MHC class I restricted recognizing either an HLA-A, -B or -C molecule which all have a CD8 binding site. CD4 and CD8 enhance the interaction between the TCR and HLA molecule and provide signals, like the CD3 chains, to activate the T-cell.
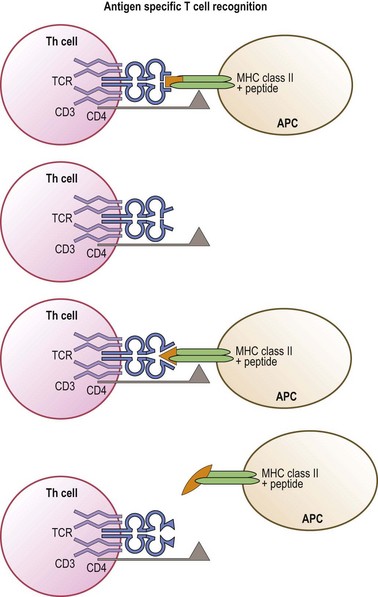
Like immunoglobulins, the TCR chains are encoded by novel genes formed by the combining of gene segments termed variable, diversity, joining and constant.74,75 Multiple gene segment alternatives and variable joining lead to an enormous potential repertoire of TCR structures. The outcome of this diversity is the generation of a large pool of T-cells expressing a wide range of TCR that can recognize a wide variety of antigenic peptides. Simplistically, each T-cell, through its TCR, recognizes a specific MHC molecule (MHC restriction) in combination with a distinct peptide (antigen specific recognition) (Fig. 39.9). In actuality, a single TCR can recognize with different affinities many (estimated to be approximately 106) different MHC/peptide complexes that share common structural features.72
MHC class I and class II molecules are essential to the creation of the T-cell pool available for immune surveillance. T-cell maturation and selection occurs in the thymus in a process called thymic education.76,77 In the thymus, T-cells are in contact with MHC class I and class II molecules that are complexed with a variety of self peptides. The TCR must be able to interact with a self MHC molecule complexed with a self peptide. Many T-cells have a TCR that is unable to bind to self MHC plus self peptides complexes and these die in the thymus. T-cells whose TCR has a low affinity interaction with and are partially activated by self MHC plus self peptide complexes (positive selection) are self tolerant and leave the thymus for peripheral tissues. These T-cells maintain some degree of partial autoreactivity and constitute the pool available for immune responses to foreign peptides. T-cells whose TCR has a high affinity for and are reactive to (autoreactive) these complexes are deleted (negative selection) from the T-cell pool via the generation of a variety of intracellular signals. It has become apparent in recent years that a subset of these autoreactive cells are not deleted from the thymus, but instead develop into regulatory T-cells (Treg). Treg-cells are not themselves reactive to self antigens, but instead suppress the activity of other potentially autoreactive T-cells.76,78
The diversity of MHC molecule + self peptide complexes is central not only to the selection of a diverse repertoire of T-cells in the thymus, but also to the maintenance of the multiplicity of available T-cells in peripheral tissues.79 Disruptions in this process can result in autoreactivity and autoimmune diseases. Other histocompatibility antigens (nonclassical MHC class I molecules, MHC class I related chains and CD1 molecules) are expressed in the thymus and by other tissues and likely help to select and maintain the available T-cell pool. In a human tissue transplant, amino acid sequence differences (mismatches) between any of the HLA molecules expressed by the cells of the donor and of the recipient, theoretically, can cause T-cell reactivity to the foreign HLA molecule. This reactivity can manifest itself as either graft rejection or GVHD and results because the T-cells were not educated for tolerance to the disparate HLA molecule.
Allorecognition and transplantation
Allorecognition results when T-cells of one individual react to foreign (allogeneic) MHC molecules of another individual. There are three distinct pathways of allorecognition termed direct, indirect, and semi-direct80,81 (Fig. 39.10). Direct allorecognition occurs when recipient T-cells recognize as foreign an intact allogeneic MHC molecule expressed by a donor cell.81 Indirect allorecognition occurs when recipient T-cells recognize a self MHC molecule that has bound a peptide derived from the foreign MHC molecule which contains the amino acid difference(s). This type of allorecognition involves the uptake of grafted cellular material, from which the foreign MHC molecules are processed into peptides and bound to self MHC molecules via the normal MHC class I and class II peptide processing and binding pathways. The semi-direct pathway involves the uptake and surface expression of intact foreign MHC+peptide complexes by recipient APC. Since these APC can also process and present foreign MHC molecules as peptides, this enables both direct and indirect recognition of foreign MHC on the surface of the same APC by recipient T-cells. When immune cells are present in the graft, donor T-cells may recognize recipient MHC by these same pathways. These pathways may contribute differentially to the alloresponse over time.
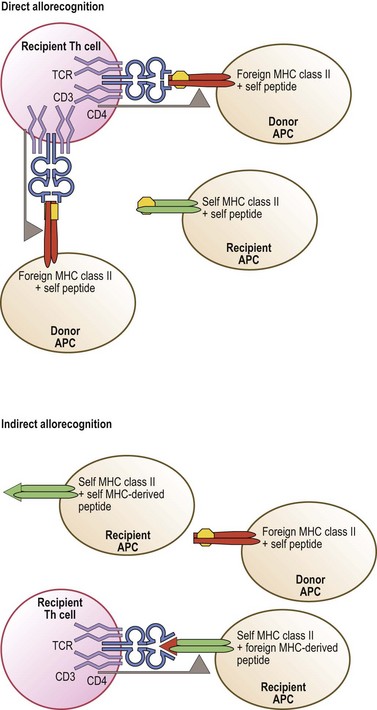
Allorecognition is purely a transplantation phenomenon. Successful transplantation of tissues, especially HSC grafts, relies heavily on the HLA identity of the donor and the recipient.82 Unfortunately, HLA mismatches between a donor and recipient are often unavoidable. Any HLA disparity has the potential to cause allorecognition which increases the risk of graft rejection or GVHD. However, not all HLA disparity leads to destructive alloreactive T-cell responses.
The frequency of T-cells of any individual that respond to allogeneic MHC is substantially greater than the frequency of T-cells that respond to a specific foreign antigen.83 The high frequency of potentially alloreactive T-cells is most likely due to the fact that the available T-cell pool consists of T-cells that are inherently reactive to MHC molecules complexed with self peptides.81,84 Furthermore, because the TCR itself is flexible, a single TCR is able to recognize multiple MHC+peptide complexes and to accommodate differences in structure.
Non-classical MHC class I molecules
The non-classical MHC class I (class Ib) molecules, HLA-E, HLA-F and HLA-G, are encoded in the class I region of the MHC (Fig. 39.1) and the proteins expressed by these genes are structurally similar to the classical class I molecules (class Ia). However, the class Ib molecules, in general, are noticeably less polymorphic and expressed at lower levels on cell surfaces than the class Ia molecules. These glycoproteins also display unique tissue distribution patterns and participate in specialized immune system functions. The impact of the class Ib molecules on immune responses directed toward foreign tissue is not yet known.
HLA-E
HLA-E shares many features with the classical class I molecules. Its gene is organized and regulated in a manner nearly identical to that of the class Ia genes and is highly transcribed in most nucleated cells.85–87 HLA-E is expressed on cell surfaces as a trimolecular complex that includes the HLA-E heavy chain, β2m and peptide and has a similar overall 3D conformation.88–90 Assembly of the HLA-E molecule and binding of peptides requires all of the same ER components. Like the class Ia molecules, the peptides that bind to HLA-E are optimal at 9 amino acids in length.
In contrast to classical MHC class I genes, HLA-E is the least polymorphic of the MHC class I genes; nine alleles encoding three unique proteins.21,87 Although high levels of HLA-E gene transcripts and intracellular protein are found in most cells, very little HLA-E protein is detected at cell surfaces. These observations are likely the consequence of the unique function of HLA-E, which has been well conserved across species. The peptide binding groove of HLA-E is highly hydrophobic and specializes in the binding of hydrophobic peptides primarily derived from the signal sequences of many of the classical class I molecules and HLA-G. These HLA-E + signal sequence complexes serve as the ligand recognized by some members of the CD94/NKG2 family of NK cell receptors. Recognition of HLA-E by CD94/NKG2 family members regulates NK cell mediated lysis of a potential target cell. The coordinate expression of HLA-E with classical MHC class I molecules and the binding of peptides derived from their signal sequences provides a means for NK cells to monitor the efficacy of expression and translation of the classical class I genes which is often disrupted in malignant and virally infected cells.
Recently, it has been documented that HLA-E also plays a role in antigen presentation to T-cells expressing αβ TCR. HLA-E restricted CD8+ cytotoxic T-cells have been isolated that recognize peptides from pathogens such as CMV, Salmonella and Mycobacterium.91 Further, the crystal structure of the interaction of a TCR with HLA-E/CMV peptide complex was recently solved.89 Thus, HLA-E is important not only in NK cell (innate), but also in T-cell (adaptive) immune responses. Whether HLA-E affects transplantation outcome is yet to be determined.
HLA-F
HLA-F remains the most mysterious of the class Ib molecules. Like HLA-E, HLA-F also shares properties associated with the classical MHC class I molecules.85 Its gene is highly transcribed in a variety of cell types such as B-cells, tonsil, spleen and thymus, but not as ubiquitously as expression of the classical MHC class I genes.92 The HLA-F heavy chain associates with β2m and possibly to several of the ER components involved in class Ia molecule assembly, but these data are conflicting.92,93
Contrary to the class Ia genes, polymorphisms reported in the HLA-F gene are limited; 21 alleles have been described.21 The HLA-F protein has only been detected intracellularly in normal tissues in which its gene is transcribed, but it has been detected on the surface of transformed B-cell and monocyte cell lines93 and may have a unique pathway for its transport to the cell surface.94 It has not been determined whether HLA-F binds peptides, although 3D models suggest that the HLA-F antigen binding groove could accommodate peptides. One report has shown an interaction of HLA-F with leukocyte immunoglobulin-like receptors on the surface of B-cells and monocytes suggesting that HLA-F may have a role in the regulation of these cell types.92 However, more work needs to be done before the function of HLA-F is fully discerned.
HLA-G
HLA-G is less analogous to the classical class I molecules than HLA-E and HLA-F. Regulation of the HLA-G gene is unique.85 First, the sequence of the SXY module is divergent from the other class Ia and class Ib genes, is not responsive to CIITA and may not bind components of the enhansosome. Second, part of the ISRE is deleted and is replaced with a SP1 binding site. Thus, HLA-G is not IFN-γ inducible. Although less polymorphic than the classical class I genes, HLA-G is the most polymorphic of the class Ib genes with 44 alleles that encode 14 unique proteins.21,91 Assembly and expression of the HLA-G trimolecular complex (HLA-G heavy chain, β2m and peptide) is identical to classical MHC class I molecules.95 The 3D structure of HLA-G is analogous to that of the class Ia molecules and HLA-E, but the number of contacts between HLA-G and the bound peptide is greater than class Ia molecules and is more deeply buried.96 The peptides bound to HLA-G appear to be less diverse than class Ia molecules and their presentation to T-cells is not known as HLA-G restricted T-cells have not been identified.97
HLA-G has other unique features. Six different HLA-G proteins (isoforms) are expressed as a result of alternative splicing of its mRNA. HLA-G1 is the full length isoform that is membrane bound and associated with β2m. There are three other membrane bound isoforms of HLA-G (HLA-G2, -G3 and -G4) that lack various extracellular domains. Alternative mRNA that includes intron 4 produces the two soluble isoforms of HLA-G, HLA-G1sol and HLA-G2sol. All isoforms appear to bind peptides and, thus, may be functional, but the G1 isoform is the most predominant in vivo.97
HLA-G is the primary histocompatibility molecule expressed during pregnancy and its expression appears to be restricted to trophoblasts of the fetal placenta.97 HLA-E and HLA-F are also expressed, but classical class I molecules are not expressed in the trophoblasts.85 HLA-G has a dual role in maternal tolerance of the fetus.97 First, HLA-G provides a signal sequence peptide for expression of HLA-E which, in turn, regulates maternal NK cell recognition of fetal tissue. Second, HLA-G, particularly the homodimeric form, regulates the functional interaction of a variety of immune system cells with fetal tissue by serving directly as the ligand for members of the leukocyte immunoglobulin-like receptor family, LILRB1 and LILRB2, which are expressed by professional APC of the myeloid lineage and by some lymphocytes.98,99 HLA-G also may serve as the ligand for the NK cell receptor KIR2DL4.100
Other potential histocompatibility molecules
MHC class I chain-related molecules (MIC)
The genes encoding the MHC class I chain-related molecules (MIC) are located in the class I region of the MHC centromeric to HLA-B (Fig. 39.1).101 MICA and MICB are relatively polymorphic.21,102 To date, 67 alleles of MICA that encode 56 unique proteins and 30 alleles of MICB that encode 19 unique proteins have been described.
Several features are unique to MICA and MICB. Expression of MICA and MICB is up-regulated in response to stress because of a heat shock response element in the promoter region of their genes.101 In fact, the promoter regions of the MICA and MICB genes do not contain any of the classical MHC class I regulatory elements (Fig. 39.5). Instead, their promoters share homologies with the heat shock protein 70 promoter. MICA and MICB protein expression is limited to endothelial cells, fibroblasts and epithelial cells primarily in the gastrointestinal tract and thymus and does not require association with β2m, peptide or TAP.103 The 3D structure of the extracellular domains of MICA revealed that the folding resembles that of the class Ia molecules, but that the overall orientation of these domains is dissimilar.104 The MICA α1 and α2 domains are tilted to expose a portion of the underside of the groove, as well as the groove itself to interactions with receptors. The peptide binding groove is mostly obscured and unlikely to bind peptide antigens.
MICA and MICB serve as ligands for NKG2D, an activating NK cell receptor.101,103 The interaction of NKG2D with the MIC molecules leads to NK cell mediated lysis of the epithelial cell. In fact, MIC expression is up-regulated in a wide variety of epithelial tumors such as melanoma, colon and kidney and it has been shown that the response to these cells is mediated through the interaction of NKG2D with MICA and MICB.105 MICA and MICB also may play a role in solid organ transplantation. Both molecules have been found to be expressed on epithelial and endothelial cells of transplanted kidney, heart and pancreas that show evidence of rejection and/or cellular injury.106 Further, the presence of MIC-specific antibodies in solid organ transplant recipients shows an association with graft failure.107 Thus, MIC molecules appear to be transplantation antigens that can mediate an alloresponse by NK cells and B-cells that leads to graft rejection.
CD1 molecules
CD1 is a family of proteins whose nonpolymorphic genes are encoded on an MHC paralogous region on chromosome 1.108 CD1 molecules are structurally close to MHC class I molecules at the gene and protein levels, while they are functionally close to MHC class II molecules as CD1 molecules associate with invariant chain and utilize the class II antigen presentation pathway.65,109 Like both MHC class I and class II molecules, CD1 molecules present antigens to T-lymphocytes for immune recognition.
Five CD1 molecules have been described which are classified into two groups based on sequence homology and tissue distribution.108 Members of group I (CD1a, -b, -c, -e) are expressed primarily on professional APC, such as dendritic cells and B-cells, and on cortical thymocytes; while members of group II (CD1d) are expressed on most cells of hematopoietic origin and on hepatocytes, keratinocytes and intestinal epithelium. The crystal structures of CD1a, CD1b and CD1d reveal a general conformation similar to MHC class Ia molecules except that the antigen-binding grooves are narrower, deeper and very hydrophobic.108,110 There are two deep pockets at either end of the groove that accommodate the lipid tails of lipid antigens. The depth of these pockets varies (CD1d has the deepest and CD1a has the shallowest), conferring different lipid antigen binding characteristics on each of the CD1 molecules. Several lipid antigens have been identified from bacterial, synthetic and self sources. For example, CD1b binds mycolates derived from mycolic acid of Mycobacterium species, while CD1a binds lipopeptides also from mycobacteria. Lipid antigens also have been identified from Borrelia burgdorferi (Lyme disease) and Sphingomonas.
Group I CD1 molecules present lipid antigens to T-cells that express either γδ TCR or αβ TCR.108,110 CD1d (group II) is the ligand primarily for invariant NK T-cells (iNKT). iNKT cells are usually double negative (CD4−/CD8−) or CD4+ T-cells that express a specific αβ TCR (Vα24-Jα18 with Vβ11) together with receptors found on NK cells, CD161 (NK-RP1A) in particular. iNKT cells develop in the thymus from double positive (CD4+/CD8+) T-cells like other T-cells, but require interaction with CD1d in the thymus for their differentiation and maturation. iNKT cells play a large role in enhancing the immune response to certain microbial pathogens by stimulating APCs, T-cells and B-cells. These cells can also be directly cytotoxic. Because the CD1 molecules are not polymorphic, they are unlikely to have an impact on donor selection for tissue transplantation.
Minor histocompatibility antigens
Clinically significant GVHD is observed in HLA identical sibling transplants of HSC implicating histocompatibility antigens specified by genes other than the classical HLA genes in the allorecognition of foreign tissue.111 These so-called minor histocompatibility antigens (mHag) were identified coincident with the major histocompatibility antigens. mHag are peptides derived from self proteins, other than HLA, whose sequence differs among individuals in the population. Theoretically, any polymorphic protein that differs between the tissue donor and recipient has the potential to provide a peptide which functions as a mHag. Many mHag loci have been described in humans utilizing mHag specific T-cell clones in cytotoxicity assays. However, only a handful of peptides derived from these loci have been identified (Table 39.6).112,113 The frequency of the mHags alleles varies in different population groups and some can be quite common.114 The peptides containing the variant amino acid(s) are processed through the normal MHC class I and class II antigen presentation pathways, are bound to an HLA molecule and are recognized as foreign by T-cells.111,115
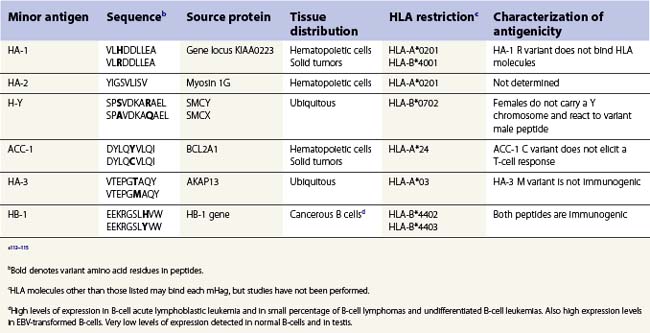
The HLA alleles expressed by a donor and recipient determine whether a mHag might contribute to allorecognition in transplantation. Furthermore, the contribution of a mHag on transplant outcome might be contingent on the sex of the donor and recipient and on the type of tissue transplanted (Table 39.6). HLA alleles are important because each HLA allelic product binds peptides that fit a specific binding motif and, thus, each mHag is only bound by certain HLA molecules. Therefore, the mHag(s) of significance differ widely depending on which HLA alleles are carried by the tissue donor and recipient (transplant pair). For example, mHag HA-1 could have an impact if the donor or recipient expresses HLA-A*0201 and there is HA-1 disparity between the transplant pair.113,115 Recognition of the H allelic form of the HA-1 peptide bound to the A*0201 encoded molecule stimulates allorecognition. HA-1 would be of no consequence to the graft when, for instance, both donor and recipient are homozygous for HLA-A*0101, as HA-1 is not able to bind to this HLA-A molecule and consequently is never presented to T-cells. The HA-1 R peptide does not elicit an immune response. HA-1 also can be used to demonstrate the importance of the type of tissue involved in the transplant. HA-1 is expressed only on cells of hematopoietic origin. Therefore, HA-1 most likely would not be pertinent in the case of a kidney transplant, even if the transplant pair expresses HLA-A*0201, since HA-1 would not be expressed by the transplanted kidney.
It is now apparent that mHags not only have an impact on the development of GVHD, but also can be beneficial in generating a graft versus tumor (GVT) response111–113115 as cancer cells of both hematopoietic and epithelial origin have been found to aberrantly express mHag. Once characterized, mHags can be included in matching protocols when they are pertinent to transplantation of tissue or to the elimination of residual malignant disease.112
Other immune system receptors that recognize histocompatibility molecules as their ligands
Killer cell immunoglobulin-like receptors
The killer cell immunoglobulin-like receptors (KIR) are, at least, a 14 member family of proteins expressed by NK cells and a subset of T-cells.116 Like the HLA molecules and TCR, KIR belong to the immunoglobulin (Ig) protein superfamily and possess either 2 (KIR2D) or 3 (KIR3D) glycosylated extracellular Ig like domains (Fig. 39.11). The type of signal (activating or inhibiting) generated by a KIR is dictated by its cytoplasmic tail. Members that generate inhibitory signals, preventing cytotoxicity and cytokine release, have a long cytoplasmic tail (e.g., KIR3DL1 and KIR2DL2) which contains immunoreceptor tyrosine based inhibitory motifs (ITIM). Binding of the inhibitory KIR to its ligand leads to phosphorylation of the tyrosine residue in each ITIM and ultimately to inhibition of NK and T-cell functions. Activating KIR family members have a short cytoplasmic tail without ITIM (e.g., KIR2DS1 and KIR3DS1). Instead, these members associate via a conserved positively charged residue in their transmembrane regions with an adaptor protein, often DAP12, that contains immunoreceptor tyrosine-based activation motifs (ITAM) in its cytoplasmic tail. Interaction of an activating KIR with its ligand causes phosphorylation of the ITAM in the adaptor which may lead to activation of NK and T-cell functions. Since NK and T-cells express multiple activating and inhibiting receptors including KIR, cellular function is finely regulated and represents an integration of signals.117
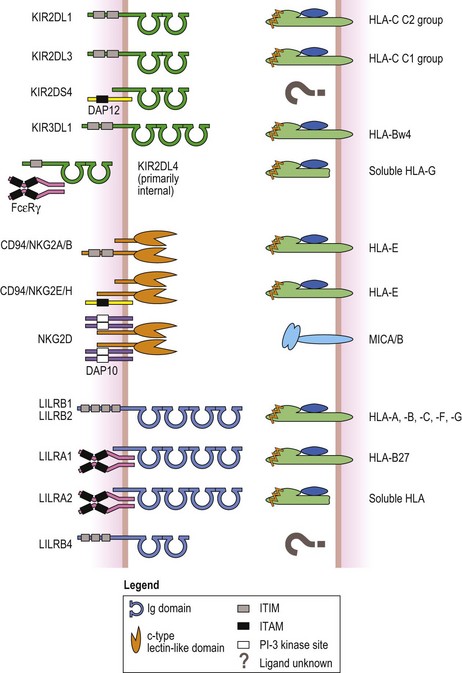
Fig. 39.11 Examples of the three receptor families that recognize HLA molecules as their ligands. The killer cell immunoglobulin-like receptors (KIR, green, top) have either two or three extracellular Ig like domains. Inhibitory KIR have long cytoplasmic tails and signal via immunoreceptor tyrosine based inhibitory motifs (ITIM, gray box). Activating KIR with short cytoplasmic tails associate with a signaling protein called DAP12 (yellow) that has an immunoreceptor tyrosine based activation motif (ITAM) (black box) in its cytoplasmic tail. KIR2DL4 has an ITIM but is activating by associating with Fc epsilon receptor gamma chains (FcεRγ, magenta) that contain ITAM in their cytoplasmic tails. The ligands for many of the KIR are groups of class I HLA molecules (heavy chain, light green; β2m, blue; peptide, orange). For example, KIR2DL1 recognizes group 2 HLA-C class I molecules; KIR2DL3 recognizes group 1 HLA-C class I molecules; KIR3DL3 recognizes HLA-B class I molecules with a HLA-Bw4 public epitope (Fig. 39.6, Table 39.8); and KIR2DL4 recognizes soluble HLA-G. The ligands for some KIR are unknown (denoted by question mark). NKG2A, -B, -C, -E and -H (orange, center) are expressed as heterodimers with CD94. NKG2D is expressed as a homodimer. Like the KIR, inhibitory members contain ITIM in their cytoplasmic tails, while activating members associate with DAP12. The exception is NKG2D, an activating receptor, which associates with another signaling protein called DAP10 (lavender) that possesses a phosphatidylinositol-3 (PI-3) kinase binding site (white box) instead of an ITAM. The ligand for all CD94/NKG2 heterodimers is the non-classical MHC class I molecule, HLA-E. NKG2D recognizes the MHC class I chain-related A and B (MICA/B) molecules (light blue). The leukocyte immunoglobulin-like receptors (LILR) have either two or four extracellular Ig like domains (cyan, bottom). Again, inhibitory members possess ITIM in their cytoplasmic tails. Activating members associate with FcεRγ (magenta) like KIR2DL4. LILRB1 and LILRB2 recognize a variety of HLA molecules. HLA-B27 allelic products are the ligand for LILRA1 and LILRA2 recognizes soluble HLA molecules. The ligand(s) for other family members is not known.
Classical MHC class I molecules serve as the ligand for several of the KIR family members (Fig. 39.11).116,118 Some KIR with two extracellular Ig like domains interact with groups of HLA-C molecules.116,119 Almost all HLA-C molecules can be divided into one of two groups (C1 and C2) based on the presence of a shared (public) epitope determined by amino acid residue 80 in the α1 domain of the HLA-C molecule.120 The C1 group (e.g. Cw1 encoded by Cw*010201) has an asparagine at residue 80 and the C2 group (e.g. Cw2 encoded by Cw*020201) has a lysine at residue 80. C1 group molecules are ligands for KIR2DL2 and KIR2DL3, while C2 group molecules are ligands for KIR2DL1. A KIR with three extracellular Ig like domains, KIR3DL1, interacts with a group of HLA-B molecules carrying the Bw4 public epitope. HLA-G is recognized by KIR2DL4.100 Ligands for some KIR have not been identified.
The genes encoding KIR are located in the leukocyte receptor complex on chromosome 19.121 Haplotypes vary in the number of KIR genes they encode.122 One common haplotype includes only seven of the 14 KIR genes: KIR3DL3, KIR2DL3, KIR2DL1, KIR2DL4, KIR3DL1, KIR2DS4, and KIR3DL2. Other haplotypes include more of the stimulatory KIR genes. In addition to gene content variability, the KIR genes are polymorphic.123 For example, there are over 50 KIR3DL1 alleles. KIR3DL1 allelic variation alters its level on the cell surface and impacts the affinity of interaction with its HLA ligand.124
At the level of the individual NK cell, only a subset of the KIR genes carried by an individual are expressed.125 During development, individual NK cells acquire specific KIR, apparently at random so that an individual who carries KIR2DL1 and KIR2DL3, for example, will have NK cells that express only KIR2DL1, only KIR2DL3, and KIR2DL1+KIR2DL3. This variegated expression increases the sensitivity of the NK cells to the loss of individual HLA molecules during tumorgenesis or viral infection. Because KIR and HLA genes are found on different chromosomes, individuals may carry a KIR gene yet lack its cognate HLA ligand. For example, an individual may express KIR3DL1 but not express an HLA-B molecule carrying a Bw4 epitope. To avoid autoreactivity by absence of a ligand, it is thought that individual NK cells are educated by interaction with MHC class I molecules and that this process controls the functional responsiveness of the NK cells.126 In KIR3DL1 positive, HLA-Bw4 negative individuals, NK cells expressing KIR3DL1 as their only inhibitory receptor will be hyporesponsive, unable to react to the absence of the HLA-Bw4 ligand.127
Specific KIR genes have been associated with susceptibility or resistance to a variety of infectious and autoimmune diseases.118 The impact of NK cells, and inhibitory KIR in specific, in eradicating residual malignant disease was first noted in T-cell depleted haploidentical HSC transplants.128 Subsequent studies have underscored the difficulty in predicting NK alloreactivity in the context of differing transplantation protocols and recent studies have continued to investigate the utility of considering KIR when selecting a HSC donor.129 In general, algorithms to predict NK alloreactivity have focused on KIR and HLA genotypes of the donor and the recipient.130 The goal is to select a donor expressing an inhibitory KIR ligand for a patient who does not express the ligand for that receptor, for example, to select a KIR2DL1 positive donor for a patient who is negative for the HLA-C2 group. It is then expected that donor NK cells, released from their inhibition, will attack residual malignant cells expressing NK cell activating ligands. Observations of NK reactivity in HSC transplantation have lead to clinical trials using adoptive immunotherapy with NK cells for patients with myeloid leukemia and with solid tumors.131
CD94 and NKG2
CD94 and the NKG2 family (NKG2A-NKG2F and NKG2H) are primarily transmembrane glycoproteins and their genes are encoded in the NK complex on chromosome 12.132 NKG2A, NKG2C, NKG2E and NKG2F share greater than 94% protein sequence homology in their extracellular domains. NKG2A, 2C and 2E are expressed as covalently linked heterodimers with CD94. NKG2B and NKG2H are alternative mRNA splice products of the NKG2A and NKG2E genes, respectively, and also are expressed as heterodimers with CD94. NKG2F does not form heterodimers with CD94 and is expressed in intracellular compartments. NKG2D is discussed separately below.
Like the KIR, CD94/NKG2 heterodimers and NKG2F are expressed by NK cells and subsets of CD8+ T-cells and can either inhibit or activate cellular functions.132 The action of the CD94/NKG2 heterodimeric receptors is determined by the NKG2 family member. CD94/NKG2A and CD94/NKG2B are inhibitory with two ITIM in their cytoplasmic tails. NKG2C, NKG2E and NKG2H lack ITIM in their cytoplasmic tails and, like activating KIR, are associated noncovalently with DAP12 and are activating receptors. NKG2F, although intracellular, also associates with DAP12 and is an activating receptor.
All of the CD94/NKG2 heterodimers recognize the non-classical class I molecule HLA-E as their ligand; NKG2A/B and NKG2E/H with equal affinity and NKG2C with tenfold less affinity.132 Polymorphism has not been demonstrated in these receptors, nor is it likely since their ligand, HLA-E, is highly conserved in the population. On the other hand, it has been demonstrated that the sequence of the bound peptide can affect CD94/NKG2 recognition of HLA-E.132,133 This could have consequences on the regulation of NK and T-cell recognition of tissues in transplantation, particularly stimulating a GVT response, especially since there is polymorphism in the HLA class I signal sequences bound by HLA-E.132
NKG2D shares only ~20% protein identity with other NKG2 family members and is expressed as a homodimer on the cell surface of NK, γδ T-cells and subsets of CD4+ and CD8+ T-cells.134,135 NKG2D is an activating receptor and signaling is achieved by association with two dimers of DAP10 carrying phosphatidylinositol-3 kinase binding sites in their cytoplasmic tails. The ligands for NKG2D are MICA and MICB. There is now evidence that the interaction of NKG2D with its ligands may play a role in the transplantation of tissues, particularly with respect to graft rejection of solid organ transplants.134
Leukocyte immunoglobulin-like receptors
The leukocyte immunoglobulin-like receptors (LILR) or immunoglobulin-like transcripts (ILT) are a multigene family located on the long arm of chromosome 19 closely linked to the KIR genes.136 Eleven expressed LILR members have been identified, ten of which are glycosylated transmembrane proteins and one being a glycosylated soluble molecule.136,137 The LILR, like the KIR, belong to the Ig superfamily and have either two or four Ig-like extracellular domains (see Fig. 39.11). There are inhibitory (LILRB1-5) and activating (LILRA1-6) receptors, similar to the KIR and CD94/NKG2 receptor families. Inhibitory LILR have long cytoplasmic tails that contain two to four ITIM and activating LILR have short cytoplasmic tails devoid of known signaling motifs. Activating LILR associate with the Fc epsilon receptor γ chain which contains an ITAM in its cytoplasmic tail.
LILR expression varies widely on immune system cells, but they are found primarily on APC of the myeloid lineage such as macrophages and dendritic cells136–138 as well as on B-cells and on subsets of NK and T-cells. Two of the inhibitory LILR, LILRB1 and LILRB2, interact with HLA-A, -B, -C, -F, and -G molecules, but their specificities for HLA allelic products and affinities differ. Two of the activating LILR also bind class I molecules. LILRA1 binds some allelic products of HLA-B*27 and LILRA2 binds soluble class I molecules. There is a fair degree of polymorphism in the LILR genes which clusters in several discrete areas of the extracellular domains136 which could be the reason for the differences in allelic specificity and affinity for class I molecules.
Evidence is beginning to mount for a possible role of LILRs in transplantation. The inhibitory LILR mediate potent tolerogenic effects on immune responses.137,138 Thus, engagement of these receptors on APCs presenting alloantigens can result in down-regulation of the alloresponse. For instance, LILR recognition of HLA-G in kidney transplant patients was shown to be associated with a reduction in T-cell alloproliferation and induction of a regulatory/suppressive T-cell population. The full extent of LILR involvement in transplantation requires further study.
Techniques for the identification of HLA polymorphism
Selection of individuals expressing the same HLA molecules (an HLA identical or matched donor) has been used to decrease the detrimental immune response during transplantation of human tissues. HLA alleles and consequent differences in the HLA proteins can be identified through a variety of testing methods including serology, cellular assays, and DNA based detection methods. This process is termed tissue or HLA typing. The methods and quality control for typing have been described.139 HLA typing and other histocompatibility testing is categorized as high complexity testing in the US. Clinical laboratory improvement amendments (CLIA) and histocompatibility testing laboratories are accredited through the American Society for Histocompatibility and Immunogenetics (ASHI), the European Federation for Immunogenetics (EFI) and other organizations.
Serologic detection of class I and class II allelic products
Lymphocyte microcytotoxicity testing has been used for HLA typing since the 1960s; however, typing for HSC transplantation is performed mainly by DNA-based testing. The HLA serologic phenotype is determined by testing unseparated lymphocyte preparations or T-lymphocytes (for HLA-A, -B, -C) or enriched B-lymphocytes (for HLA-DR, -DQ) for the presence of specific HLA molecules as detected by a panel of well-characterized HLA antibody preparations (alloantisera or monoclonal antibodies).139 Lymphocytes are incubated with a panel of antibody reagents. If the lymphocytes carry a cell surface HLA molecule recognized by a complement-fixing antibody, the antibody binds to the cells and the cells are subsequently lysed by the addition of complement in excess. Following termination of the reaction and staining to distinguish live from dead cells, the percent lysis of the cells is determined for each antibody reagent using a microscope and a numerical grade is assigned (e.g., 1 = 0–10% lysis, 6 = 51–80%, 8 = 81–100%). Scores of 6–8 indicate that the specific HLA molecule detected by the antibody reagent is present.
Cellular detection of HLA disparity
Cellular assays were historically used to detect HLA differences between individuals, but are not commonly used today. The response of one cell (responder) in tissue culture to the foreign histocompatibility molecules on the surface of a second cell (stimulator) is called the mixed leukocyte culture (MLC) or mixed lymphocyte reaction (MLR).140 The response is made unidirectional by preventing cells from one of the two individuals from replicating by treating those cells with radiation or an alkylating agent prior to addition to the culture. The MLC represents a summation response of a responder cell to differences in the HLA class II molecules (HLA-DR, -DQ, and -DP) encoded by the irradiated stimulator cell haplotypes. The response to DR molecules predominates. The use of cellular assays to determine the similarity between two individuals has declined significantly because DNA based testing procedures can now be used to identify class II disparity between individuals and because the MLC can be influenced by a variety of factors including the health of the individuals contributing the cells, the patient’s disease, and the history of prior transfusion.141
Other cellular assays measure the frequency of cytotoxic or helper T-lymphocytes in the potential HSC donor. Limiting dilution is used to isolate and measure the frequency of T-lymphocyte precursors which respond to recipient cells. The differences stimulating these responses are thought to be HLA class I differences or minor histocompatibility antigens for cytotoxic precursors and HLA class II differences for helper precursors. The correlation between the precursor frequency and transplantation outcome is controversial;142,143 thus, DNA-based testing is more commonly used to identify HLA disparity between potential donor and recipient.
DNA-based identification of class I and class II alleles
DNA-based methods target differences in the nucleotide sequences of alleles for HLA typing. Commercial kits employing all of the methodologies described below are available although some laboratories continue to design their own reagents. Any cell with a nucleus can be used as a source of DNA; however, DNA is usually prepared from a small quantity (0.2–1 ml) of whole blood. Alternative sample types such as epithelial cells from a buccal swab might provide a more reliable source of genomic DNA in patients with hematologic malignancies,144 in patients treated with immune suppressive agents or in patients who have been transfused.145 The sensitivity of detection of HLA alleles is enhanced greatly by the amplification of DNA encoding HLA genes using the polymerase chain reaction (PCR) (Fig. 39.12). Many typing reactions utilize PCR primer sequences that are broadly specific and are shared by all alleles at an HLA locus. Other typing procedures utilize primer sets that are narrowly specific and are shared by only a subset of alleles at a locus. In this way, the laboratory can isolate large quantities of specific HLA alleles for identification as described below.
Sequence specific priming (SSP)
One method of typing uses a large panel of pairs of amplification primers.146,147 Each pair of primers anneals to a limited number of potential alleles. In the subsequent PCR, only certain primer pairs will amplify the DNA from a given individual. Amplification is detected by electrophoresis in an agarose gel or, if the DNA is labeled with a dye during amplification, by fluorescence. The HLA alleles carried by an individual are then determined by the pattern of positive and negative amplifications with the primer panel using a knowledge of which alleles amplify with which primer pairs.
Sequence-specific oligonucleotide probe hybridization (SSOPH)
It is possible to use the binding of synthetic single stranded DNA molecules (i.e., probes) to identify HLA alleles.148,149 A panel of probes, each capable of identifying a short polymorphic region found in a group of HLA alleles, is hybridized to denatured PCR amplified DNA. Specific alleles are identified through the use of several probes and/or the use of SSP followed by hybridization with several probes. Fig. 39.12 illustrates the technique using a format where the oligonucleotide probes are bound to a solid support. The pattern of hybridization, including which probes bound and which did not, can be read to determine the nucleotide polymorphisms present or absent. Comparison of this result with the known sequences of alleles at the locus will predict the HLA alleles present or absent.
Sequence-based typing (SBT) of genomic DNA
A final method of identification of HLA alleles involves the direct determination of the DNA sequences of the HLA alleles carried by an individual.150,151 Alleles are identified by sequencing following PCR amplication. The choice of PCR primers determines whether the alleles will be isolated separately based on sequence polymorphisms (using SSP) or whether the two alleles of a locus will be characterized as a mixture. The choice of primers also determines which segments of the gene are sequenced; most testing focuses on the exons encoding the antigen binding site of the HLA molecules (exons 2 and 3 for class I genes and exon 2 for class II genes). The presence of two alleles in the sequencing reaction is indicated by the presence of two half-height peaks at each polymorphic nucleotide position. Sequencing is the most powerful approach to determine if two individuals share the same HLA alleles.
Measurement of sensitization to histocompatibility differences
Individuals may become sensitized to foreign histocompatibility molecules through transfusion, pregnancy or prior transplantation. Sensitization may result in the presence of HLA specific antibodies and/or significant numbers of cytotoxic T-cells which may cause lysis of cells expressing the sensitizing foreign HLA molecules. If a pre-sensitized patient, such as a patient with severe aplastic anemia who has received prior blood transfusions, undergoes transplantation with tissue expressing the foreign HLA molecules targeted by the immune system, the pre-existing antibodies or T-cells may cause destruction of the graft. Antibodies are detected prospectively by incubation of serum from the putatively sensitized individual with a panel of HLA antigens.152 There are several approaches used to measure alloreactive antibodies which vary in sensitivity and in the level of information provided on the specificity of the antibodies. Assays today are likely to utilize isolated HLA molecules bound to a solid support, often a multi-wall plate or microspheres, in ELISA, flow cytometry, or Luminex153 formats. The screening assay identifies the presence of IgG antibodies specific for particular HLA antigens and the extent of sensitization to foreign HLA molecules in general (i.e., a measurement of panel (or percent) reactive antibody (PRA)).
Once a potential tissue donor has been selected, patient antibodies specific for cells of the donor are detected in a donor-specific cross-match in a microcytotoxicity or flow cytometry-based assay.152 In some centers, a patient with an HLA mismatched donor with a positive donor directed cross-match (i.e., patient sensitized to donor) would be considered at increased risk for graft failure after HSC transplantation.154,155 As an aside, while sensitization to paternal HLA antigens inherited by the child can occur in the mother during pregnancy, exposure of the child to the non-inherited maternal antigens (NIMA) may lead to tolerance.156
HLA assignments
HLA terminology is assigned by a World Health Organization (WHO) Committee for HLA Nomenclature. Several different naming systems are in use, each the result of the technology used to identify HLA diversity: serologic, cellular, or DNA based. The nomenclature is described on the website http://hla.alleles.org.21 Since not all tissue typing techniques identify specific HLA alleles, the assignments are called HLA ‘types’. Individuals who differ at the level of resolution of each test are assigned different HLA ‘types’. For example, an individual tested by serology might express HLA types HLA-A1 and -A2 (heterozygous) while another individual might express HLA-A2 and -A74. Within the limits of resolution of the serologic technique, the two individuals in this example share one HLA-A type (A2) but differ for the second (A1 vs. A74). Within the limits of DNA-based testing, however, these individuals may differ in the A2 allele carried by each individual, for example A*0204 and A*0207. Likewise, two individuals typed by DNA-based methods as DRB1*04, *11 may differ for the alleles they carry.
Serologic specificities
Historically, serological and cellular reagents were used to define HLA types. These reagents defined specificities (or determinants) localized to one or several HLA molecules; these specificities were used as surrogates of alleles. The specificities identified through these methods were defined and standardized during international workshops in which typing reagents and cells were exchanged among participating laboratories and a consensus reached on the definition of each specificity6 (Table 39.7). These specificities were named by the HLA molecule and by a numerical designation based on the order of discovery. As additional reagents were identified, serologically defined specificities were often subdivided (i.e., ‘split’). For example, the serologic specificity HLA-B5 defined in the 1967 international workshop was subdivided into HLA-B51 and -B52 as a result of the reagents tested in the 1975 workshop. Table 39.7 lists the broader, shared (supertypic) specificities for the current ‘split’ (subtypic) specificities.
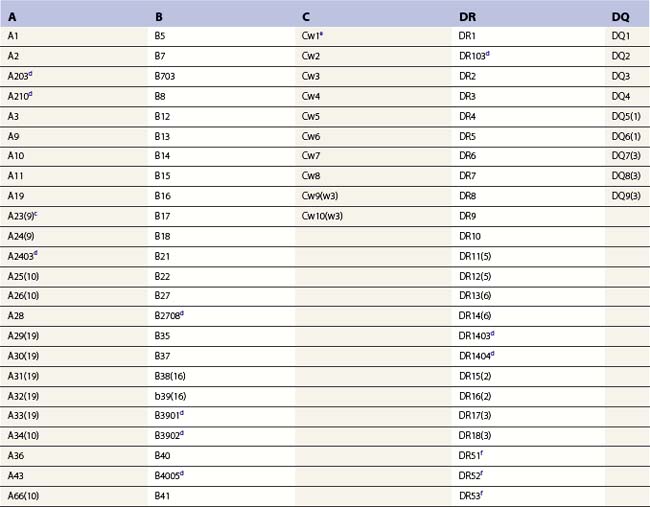
The numbering system used to define HLA ‘types’ is based on the date of assignment so that the relationships among various types is not immediately clear. Thus, for example, subdivisions of B5 were assigned as B51 and B52 since they were the 51st and 52nd serologic specificities to be assigned. Their relationship to B5 is not immediately clear unless their more complete nomenclature, B51(5) and B52(5), is provided. The assignments associated with the HLA-A and HLA-B serologic specificities are further complicated because it was not initially appreciated that these were specified by two different loci. Thus, the naming system is based on a single numerical series5,6 such that numerical designations of HLA-A include HLA-A1, -A2, -A3, -A9, -A10, -A11, etc. while the HLA-B designations include the remaining numbers in the series, HLA-Bw4, -B5, -Bw6, -B7, -B8, -B12, etc.
The HLA-Bw4 and -Bw6 serologic specificities are broad, supertypic determinants which reside on the same molecule as the B locus subtypic specificities. HLA-B locus molecules (and some HLA-A locus molecules) characteristically carry either Bw4 or Bw6 specificities. In the case of Bw4 and Bw6, the use of the ‘w’ in the designation indicates the shared nature of the determinant. The region of the HLA-B molecule carrying the Bw4/Bw6 specificities has been identified to lie in the α1 alpha helix between amino acid residues 77 and 83 (Fig. 39.6A).157,158 (A similarly located epitope identified by NK cell reactivity divides the HLA-C molecules in to group 1 and group 2116,119,120.)
Antibody reactivity patterns have been used to group HLA-A and HLA-B molecules into cross-reactive epitope groups (CREG).159–161 Examples of CREGs are listed in Table 39.8. An antigenic determinant (epitope) shared among members of a CREG is called a public specificity. Class I molecules within a CREG share one or more determinants that are not shared by molecules in another CREG. These groupings have been used as the basis for matching schemes for selection of solid organ donors162,163 and to predict the reactivity of HLA-specific antibodies in sensitized patients.160 In contrast, in HSC transplantation, mismatching schemes based on CREGs or other shared epitopes have been shown not to impact survival or the occurrence of GVHD.164,165
| CREG | Antigen specificities included |
|---|---|
| 1C | A1, 3, 11, 23, 24, 29, 30, 31, 36, 80 |
| 2C | A2, 23, 24, 68, 69, B57, 58 |
| 5C | B51, 52, 18, 35, 46,49, 50, 53,57, 58, 62, 63, 75, 76, 77, 71, 72, 73, 78 |
| 7C | B7, 8, 13, 27, 54, 55, 56, 60, 61, 41, 42, 47, 48, 59, 67, 81, 82 |
| 8C | B8, 18, 38, 39, 64, 65, 59, 67 |
| 10C | A25, 26, 34, 66, 11, 68, 69, 32, 33, 43, 74 |
| 12C | B44, 45, 13, 49, 50, 37, 60, 61, 41, 47 |
| Bw4b | B13, 27, 37, 38, 44, 47, 49, 51, 52, 53, 57, 58, 59, 63, 77, A23, A24, 25, 32 |
| Bw6b | B7, 8, 18, 35, 39, 41, 42, 45, 46, 48, 50, 54, 55, 56, 60, 61, 62, 64, 65, 67, 71, 72, 73, 75, 76, 78, 81, 82 |
a 159,160 The inclusion of HLA specificities within a CREG is not standard (i.e., CREG nomenclature has not been assigned by consensus of the HLA community) although there is considerable overlap between the various groupings reported in the literature.
b A listing of antigens carrying Bw4 and Bw6 specificities can be found at http://hla.alleles.org. Not all alleles encoding a specific antigen may carry the usual Bw4 or Bw6 epitope. For example, B*0736, an allele whose product appears to carry the B7 serologic specificity,168 carries a Bw4 epitope rather than the Bw6 epitope carried by most of the B*07 allelic products. See also B*2715 in Table 39.11.
Limitations in serologic testing reagents have resulted in inconsistent assignments of some HLA types.166 This can complicate the selection of HLA matched donors from HSC donor registries and umbilical cord blood banks. With the increased use of DNA based HLA typing and its increased resolution and consistency, these difficulties are expected to disappear as the number of DNA typed donors and cord blood units increases.
DNA-based allele designations
Nucleotide sequencing has been used to identify the many alleles encoding the HLA molecules. Each HLA allele is designated by the name of the gene followed by an asterisk and a multiple digit number indicating the allele.20 Additional and optional letters added to the end of the numerical designation relate to the expression of the allele at the protein level. The present nomenclature system is summarized in Tables 39.1, 39.2, and 39.9 and is described below.
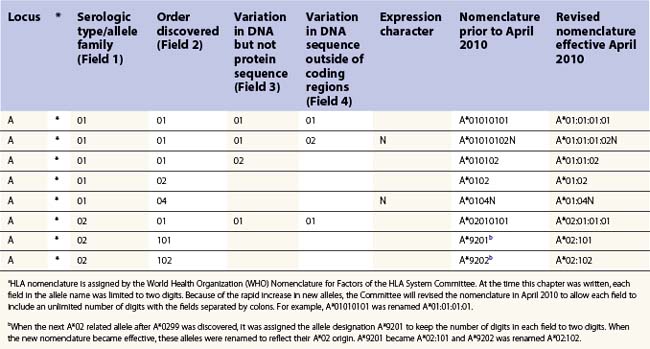
Alleles that are not expressed as proteins at the cell surface may have a single letter, N, appended to their names (e.g., A*01010102N) to indicate ‘null’. Null alleles are important to consider in transplantation since individuals who carry a null allele are functional homozygotes. For example, an individual who carries A*01010102N, A*0207 alleles would be serologically typed as A2 only compared to an individual who carries A*01010101, A*0207 who would be serologically typed as A1,A2. Other designations (e.g., L, S, Q) signify alterations in HLA expression and are described on the HLA nomenclature website (http://hla.alleles.org).21
While this chapter was being written, the HLA nomenclature underwent a revision to allow an extension of the number of digits designating each field in an allele name and to use colons to separate the fields making up the designation. Examples of the new nomenclature are shown in Table 39.9.
National Marrow Donor Program® (NMDP) DNA nomenclature
The ability to distinguish among alleles (the level of resolution) by DNA-based typing methods is controlled by the choice and number of reagents used and the typing technique. Large scale volunteer donor typing for a HSC registry or umbilical cord blood bank is usually carried out at low to intermediate resolution in which the alleles at each HLA locus are narrowed down to a few possibilities. For example, the HLA-DRB1 test results of a volunteer might narrow the possible alleles at this locus to DRB1*1301 or 1320 and to DRB1*0403 or *0406 or *0407 or *0411 or *0417. In order to include these possibilities within their database, the NMDP has developed a letter coding system for these multiple allele combinations. Thus, the combination DRB1*1301 or 1320 is designated DRB1*13VS where VS means 01 or 20 and DRB1*0403 or *0406 or *0407 or *0411 or *0417 is designated DRB1*04DZ (DZ means 03 or 06 or 07 or 11 or 17). The letter codes are purely descriptive, with codes being created only for allele combinations which have been observed during HLA testing. A listing of codes can be found on the NMDP website (http://bioinformatics.nmdp.org); the NMDP codes are used internationally in the exchange of HLA information among registries and cord blood banks.167
Limitations in the assignment of HLA alleles
Since identification of HLA alleles may often be based on partial nucleotide sequence information obtained through the use of a small set of primers and probes, it is possible that the interpretation of the assay results will miss alleles which were unknown at the time of the typing.168 For example, the identification of the DNA sequence GGTAAGTATAAG encompassing codons 11–14 in the DRB1 gene was once interpreted to mean that the individual carried the allele DRB1*0701 since only DRB1*0701 was known to carry that polymorphic sequence cassette. In 1998, two new alleles were described which also carry this sequence, DRB1*0703 and DRB1*0704. Thus, individuals tested prior to 1998 and characterized as carrying DRB1*0701 might alternatively carry DRB1*0703 or DRB1*0704. The continued discovery of new HLA alleles has significant implications for the HLA assignments since HLA types of individuals are stored and used for many years in HSC registries or cord blood banks. Since a patient may be HLA typed during the initial evaluation of treatment options but donor selection and transplant may not be the first line of therapy, the clinical laboratory should update the HLA assignments for that patient when the decision to seek an unrelated donor is made. Patients with newly described or rare alleles may require additional search strategy assistance.
Correlation between serologic specificities and DNA-based allele assignments
Today, patients requiring HSC transplants are often typed by DNA-based technologies while registries and banks of potential donors include many serologically typed volunteers or cord blood units. Thus, a HSC registry’s matching algorithm must be designed to compare a variety of HLA assignments and to determine the potential for an allele match. Table 39.10 provides some examples of the association between HLA types. In many cases, digits in the first field of the allele name are based on the serologic assignment of the HLA molecule so, for example, alleles such as A*0101 and A*6801 correspond to A1 and A68, respectively. In addition, several alleles may encode the same serologic type, such as B*2701, *2702 and *2703, which all encode a HLA-B molecule carrying a B27 serologic specificity (Table 39.11).
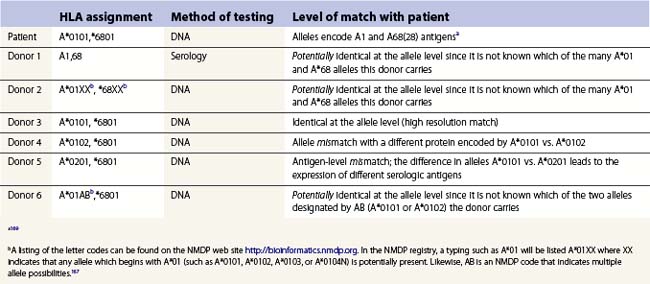
Table 39.11 Association between some of the HLA-B*27 alleles and serologic specificitiesa
| Allele | Serologic specificityb |
|---|---|
| B*2701 | B27 |
| B*2702 | B27 |
| B*2703 | B27 |
| B*2708 | B2708 [Sera detecting B7 are positive; most B27 sera are negative; neural network predicts B27.] |
| B*2711 | B27 [B27 sera are positive; some B40 sera are positive; neural network predicts an undefinedc antigen.] |
| B*2712 | Undefined [Most B27 sera are negative, sera detecting both B7 and B27 are positive; some B40 sera are positive; neural network predicts an undefined antigen.] |
| B*2715 | Undefined [B27 sera are negative, Bw4 instead of usual Bw6; neural network predicts B27.] |
b Based on serologic typing of specific B*27 allelic products. Data in [ ] describes unusual serologic patterns or the prediction of serologic typing based on an artificial neural network.
c Undefined indicates that the allelic product does not have a known serologic specificity.
It is important to note, however, the many examples where the associations between an allele and the serologic type of the HLA molecule encoded by that allele are not known or are apparently different. For example, even though B*2708 encoded a molecule which bore a B7 serologic determinant, it was found to have a closer structural relationship to alleles in the B*27 group which took precedence in the assignment of the allele name. In this case, the name of the allele cannot be used to predict the serologic type of its allelic product. Information on the serologic assignments associated with each HLA allele is routinely updated and reported.169 This ‘dictionary’ can be found in a searchable format at http://www.ebi.ac.uk/imgt/hla/dictionary.html.
Identification of HLA in clinical situations
Autoimmunity
Autoimmune diseases are characterized by an abnormal immune response to self antigens. Many of these diseases are associated with the presence of specific HLA types.52,53 The association of HLA-DQ2 and DQ8 with celiac disease is one example. This disease is a chronic inflammatory disease developing in response to ingested wheat gluten or related proteins.170 The majority of patients carry DQ2 and a minority DQ8. Yet, since only a fraction of individuals with DQ2 or DQ8 develop celiac disease, HLA-DQ is most likely just one out of several genetic and/or environment factors that combine to cause susceptibility to the disease.
Drug hypersensitivity
Hypersensitivity to several drugs has been associated with specific HLA alleles: 1) the HIV reverse transcriptase inhibitor abacavir and B*5701; 2) the gout prophylactic allopurinol and B*5801; and 3) the antiepileptic carbamazepine and B*1502. Hypersensitivity to abacavir has been linked to HLA antigen presentation to T-lymphocytes although it is not yet known how abacavir or its by-products create the novel antigen triggering the response.171
Cell, tissue and organ transplantation
Allogeneic grafts of HSCs, organs and other tissues have been everyday life-saving procedures since the mid 20th century.4 The necessity for HLA matching of donor and recipient differs for each type of graft. For HSC transplantation, close matching for HLA is required for successful outcome. For kidney transplantation, close matching increases the likelihood of long-term organ survival,172 but survival of grafts in mismatched donor–recipient pairs is also good. For logistical reasons, HLA matching plays a smaller role in transplantation of organs other than HSC and kidneys and outcome relies on management of graft rejection using immune suppression. Grafts of non-viable tissues rarely take HLA matching into consideration, as these non-viable tissues are generally used either temporarily (skin) or as ‘scaffolding’ for replacement with autologous tissue.
Solid organ transplantation
According to the US Department of Health’s Organ Procurement and Transplantation Network (OPTN) data, as of 22 May 2009 (http://optn.transplant.hrsa.gov), there were 27 961 transplants of organs in 2008 in the United States. Of these, 16 517 were kidney transplants. Included in this number are 6217 transplants from living donors. All of the other reported transplants were from cadaveric donors.
The OPTN is a nationwide system for organ sharing authorized by the US Congress through the National Organ Transplant Act of 1984. One of the primary purposes for nationwide organ sharing is to provide an opportunity for avoiding HLA mismatches that might result in graft loss in a sensitized patient.173,174 Organs are allocated based on the best opportunity for successful outcome both for patients local to the organization that obtained the cadaveric organ and for patients located nationwide.
Platelets
Platelet transfusions are required for clinical conditions where bleeding occurs or is threatened and there is a low platelet count. The clinical decision to transfuse platelets is made more difficult because patients can become refractory to platelet transfusion due to allosensitization to HLA antigens and platelet-specific antigens.175 Platelets express significant amounts of HLA class I antigens (but not class II antigens) and can be readily destroyed by platelet-directed antibodies. Immune sensitization can be reduced by removing leukocytes from the transfused product; however, the patients may be allosensitized prior to platelet transfusions because of pregnancies or prior transfusion of blood products.
HSC transplantation
Successful HSC grafts could not be achieved until donors adequately matched for HLA could be identified. The first successful grafts in the United States were performed in 1968 using HLA-matched sibling donors to treat patients with immune deficiency diseases.4,176 By the mid 1970s, it was established that marrow transplants from sibling donors could save the lives of people with a number of diseases. Unfortunately, only 30% of patients needing a transplant had HLA-matched siblings. Transplant physicians and families began to seek ways to identify alternate donors. The first successful transplant from an unrelated donor was in 1975, for a patient with severe combined immunodeficiency.177
Unrelated donor registries and umbilical cord blood banks
Today, over 60 registries of HLA typed potential adult HSC donors in 44 countries and 42 umbilical cord blood banks in 26 countries include over 13 million volunteer donors and cord blood units (http://www.bmdw.org). In the United States, the C. W. Bill Young Cell Transplantation Program, authorized by Congress through the Stem Cell Therapeutic and Research Act of 2005, is a nationwide system supporting those patients seeking an unrelated donor. The ‘Be The Match’ national registry, operated by the NMDP, lists 7 million donors in its database and has facilitated over 30 000 transplants (http://www.marrow.org). In any given month, there is an average of 3000 patients searching the ‘Be The Match’ registry. While a donor search usually begins within the country of origin of the patient, searches failing to identify a donor can extend to foreign registries and banks. Many registries have established cooperative relationships with one another to facilitate searches of these international resources. In 2007, almost 40% of transplants facilitated through the NMDP were performed with bone marrow or peripheral blood stem cells (PBSC) from a donor in another country. US donors of marrow or PBSC facilitated 623 transplants in other countries; this was 30% of the donations through the NMDP. An international voluntary organization of registries and banks worldwide, the World Marrow Donor Association (WMDA), has published policies and procedures for these international exchanges178,179 (http://www.worldmarrow.org).
Search strategies for donor identification
Ideally, HLA testing and a donor search are initiated for a patient with the diagnosis of one of over 20 diseases routinely treatable through HSC transplantation.180 Testing of the patient, siblings and parents should be used to identify a HLA matched sibling donor since the observed probability of a sibling match is 30%. The family typing will also confirm the patient’s HLA types and define the linkage of alleles at multiple loci, that is, define haplotypes. If the patient does not have a matched sibling, testing of the extended family, such as cousins, may identify an HLA matched related donor.181 Selection of an HLA haploidentical relative may also be an alternative.182
An additional tool in the search process is a database listing HLA phenotypes available on most registries worldwide provided through Bone Marrow Donors Worldwide (BMDW)183 (http://www.bmdw.org). This listing is frequently useful for rapidly estimating the likelihood of finding an HLA matched donor for a specific patient. BMDW should be used to provide supplementary information since not all registry updates are incorporated into the BMDW database and not all donors listed in BMDW will be suitable or available. The BMDW search report, coupled with a knowledge of allele and haplotype frequencies in world populations, can assist the physician in determining the usefulness of and strategy for a search of worldwide registries.
A list of HLA types from potentially matched volunteers is provided by a registry or bank to the patient’s physician. From this list of potential matches, the physician selects donors for additional HLA testing. Guidelines for unrelated adult donor selection have been published by the NMDP.184 Guidelines for HLA matching for the transplantation of umbilical cord blood are not yet clear; a literature review and summary of the current status has been published by the NMDP.185 Adult volunteer donors are contacted to determine whether they are interested in continuing participation. More than one potential donor should be evaluated since some may be unavailable for any of a number of reasons such as inability to locate the donor or donor’s poor health. Extended HLA typing may include a higher level of resolution for the four primary HLA loci or the testing of additional loci such as HLA-DQB1 and/or -DPB1. These additional tests are used to identify the best matched donor if several HLA-A, -B, -C, -DRB1 matched donors are available. To ensure that the selected donor or umbilical cord blood unit carries the HLA type shown on the search report (i.e., to confirm identity), a low resolution HLA typing of two or more loci should be performed on a fresh blood sample from the adult donor or on an attached segment from the umbilical cord blood unit. Searches for the best HLA match for a patient must be balanced with the timing of the transplant in relationship to disease stage since survival is reduced as the disease advances.
Histocompatible matches for HSC transplantation
At a minimum, HSC transplant centers attempt to match HLA-A, -B, -C and -DRB1 assignments of patient and potential donor since these loci have been shown to be clinically important in outcome.82,184 This level of match is sometimes referred to as an 8/8 allele match – two assignments at HLA-A, two at HLA-B, two at HLA-C, and two at HLA-DRB1. (A mismatch for a single allele is termed a 7 of 8 match, and so on.) The impact of mismatching likely differs depending on a variety of recipient factors including disease, treatment and age, as well as the source of the HSC; however, comprehensive guidelines are lacking at present.184 One source of stem cells which may offer a reduced risk of severe GVHD is umbilical cord blood. The relative immaturity of these cells may permit more HLA mismatching.185 In addition, an umbilical cord blood unit is more readily available than an adult donor for a patient with an urgent need for transplantation. Selection factors that HSC transplant centers use to identify the best donor for a particular patient are based on criteria that improve the likelihood of successful transplant outcome. In addition to HLA, these may include donor age, cytomegalovirus infection status, large donor size for large patients, and matching for ABO red blood cell type, although the relative importance of these criteria is still unclear.184
Today, most patients can find closely matched volunteer donors, although the likelihood of finding a matched donor varies depending on the frequency that various HLA haplotypes are found. Some individuals have thousands of identical volunteers listed on donor registries, while other individuals have no matches. Lack of a well-matched donor is not a contraindication to transplantation, however. Although HLA matching for alleles of HLA-A,-B,-C, and -DRB1 is preferred, an unrelated donor with a limited number of mismatches or a suitable cord blood unit may be acceptable alternatives compared to alternative therapies.184,185
1 Snell GD. Studies in histocompatibility. Science. 1981;213:172-178.
2 Benacerraf B. Role of MHC gene products in immune regulation. Science. 1981;212:1229-1238.
3 Dausset J. The Nobel Lectures in Immunology. Lecture for the Nobel Prize for Physiology or Medicine, 1980. The major histocompatibility complex in man. Past, present, and future concepts. Science. 1981;213:1469-1474.
4 Groth CG, Brent LB, Calne RY, et al. Historic landmarks in clinical transplantation: Conclusions from the consensus conference at the University of California, Los Angeles. World J Surg. 2000;24:834-843.
5 Payne R, Tripp M, Weigle J, et al. A new leukocyte isoantigen system in man. Cold Spring. Harbor Symp Quant Biol. 1964;29:285-295.
6 van Rood JJ. HLA and I. Annu Rev Immunol. 1993;11:1-28.
7 Bach FH, Hirschhorn K. Lymphocyte interaction: a potential histocompatibility test in vitro. Science. 1964;143:813-814.
8 Thorsby E, Piazza A. Joint report from the sixth international histocompatibility workshop conference. II. Typing for HLA-D (LD-1 or MLC) determinants. In: Kissmeyer-Nielsen F, editor. Histocompatibility Testing. Copenhagen: Munksgaard; 1975:414-458.
9 Horton R, Gibson R, Coggill P, et al. Variation analysis and gene annotation of eight MHC haplotypes: the MHC Haplotype Project. Immunogenetics. 2008;60(1):1-18.
10 MHC Sequencing Consortium. Complete sequence and gene map of a human major histocompatibility complex. Nature. 1999;401:921-923.
11 Beck S, Trowsdale J. Sequence organization of the class II region of the human MHC. Immunol Rev. 1999;167:201-210.
12 Xie T, Rowen L, Aguado B, et al. Analysis of the gene-dense major histocompatibility complex class III region and its comparison to mouse. Genome Res. 2003;13(12):2621-2636.
13 Milner CM, Campbell RD. Genetic organization of the human MHC class III region. Front Biosci. 2001;6:D914-D926.
14 Shiina T, Tamiya G, Oka A, et al. Genome sequence analysis of the 1.8Mb entire human MHC class I region. Immunol Rev. 1999;167:193-199.
15 Auffray C, Strominger JL. Molecular genetics of the human major histocompatibility complex. Adv Hum Genet. 1986;15:197-247.
16 Kappes D, Strominger JL. Human class II major histocompatibility complex genes and proteins. Annu Rev Biochem. 1988;57:991-1028.
17 Kwok WW, Kovats S, Thurtle P, Nepom GT. HLA-DQ allelic polymorphisms constrain patterns of class II heterodimer formation. J Immunol. 1993;150:2263-2272.
18 Little A-M, Parham P. Polymorphism and evolution of HLA class I and class II genes and molecules. Rev Immunogenetics. 1999;1:105-123.
19 Bontrop RE. Comparative genetics of MHC polymorphisms in different primate species: duplications and deletions. Hum Immunol. 2006;67(6):388-397.
20 Marsh SG, Albert ED, Bodmer WF, et al. Nomenclature for factors of the HLA system, 2004. Tissue Antigens. 2005;65(4):301-369.
21 Robinson J, Waller MJ, Parham P, et al. IMGT/HLA and IMGT/MHC: sequence databases for the study of the major histocompatibility complex. Nucleic Acids Res. 2003;31(1):311-314.
22 Martin MP, Carrington M. Immunogenetics of viral infections. Curr Opin Immunol. 2005;17(5):510-516.
23 Meyer D, Single RM, Mack SJ, et al. Signatures of demographic history and natural selection in the human major histocompatibility complex loci. Genetics. 2006;173(4):2121-2142.
24 Prugnolle F, Manica A, Charpentier M, et al. Pathogen-driven selection and worldwide HLA class I diversity. Curr Biol. 2005;15(11):1022-1027.
25 Yeager M, Hughes AL. Evolution of the mammalian MHC: natural selection, recombination, and convergent evolution. Immunol Rev. 1999;167:45-58.
26 Cullen M, Perfetto SP, Klitz W, et al. High-resolution patterns of meiotic recombination across the human major histocompatibility complex. Am J Hum Genet. 2002;71(4):759-776.
27 Jeffreys AJ, Holloway JK, Kauppi L, et al. Meiotic recombination hot spots and human DNA diversity. Philos Trans R Soc Lond B Biol Sci. 2004;359(1441):141-152.
28 Solberg OD, Mack SJ, Lancaster AK, et al. Balancing selection and heterogeneity across the classical human leukocyte antigen loci: a meta-analytic review of 497 population studies. Hum Immunol. 2008;69(7):443-464.
29 Maiers M, Gragert L, Klitz W. High-resolution HLA alleles and haplotypes in the United States population. Hum Immunol. 2007;68(9):779-788.
30 Mori M, Beatty PG, Graves M, et al. HLA gene and haplotype frequencies in the North American population – the National Marrow Donor Program Donor Registry. Transplantation. 1997;64(7):1017-1027.
31 Begovich AB, Klitz W, Steiner LL, et al. HLA-DQ haplotypes in 15 different populations. In: Kasahara M, editor. The major histocompatibility complex: evolution, structure and function. Tokyo: Springer-Verlag; 2000:412-426.
32 Klitz W, Maiers M, Spellman S, et al. New HLA haplotype frequency reference standards: high-resolution and large sample typing of HLA DR-DQ haplotypes in a sample of European Americans. Tissue Antigens. 2003;62(4):296-307.
33 Yunis EJ, Larsen CE, Fernandez-Vina M, et al. Inheritable variable sizes of DNA stretches in the human MHC: conserved extended haplotypes and their fragments or blocks. Tissue Antigens. 2003;62(1):1-20.
34 Traherne JA, Horton R, Roberts AN, et al. Genetic analysis of completely sequenced disease-associated MHC haplotypes identifies shuffling of segments in recent human history. PLoS Genet. 2006;2(1):e9.
35 Van den Elsen PJ, Holling TM, Kuipers HF, van der Stoep N. Transcriptional regulation of antigen presentation. Curr Opin Immunol. 2004;16(1):67-75.
36 Van den Elsen PJ, Gobin SJP, Van Eggermond MC, Peijnenburg A. Regulation of MHC class I and II gene transcription: differences and similarities. Immunogenetics. 1998;48(3):208-221.
37 Mach B, Steimle V, Martinez-Soria E, Reith W. Regulation of MHC class II genes: lessons from a disease. Annu Rev Immunol. 1996;14:301-331.
38 Muhlethaler-Mottet A, Krawczyk M, Masternak K, et al. The S box of major histocompatibility complex class II promoters is a key determinant for recruitment of the transcriptional co-activator CIITA. J Biol Chem. 2004;279(39):40529-40535.
39 Gobin SJ, van Zutphen M, Westerheide SD, et al. The MHC-specific enhanceosome and its role in MHC class I and beta(2)-microglobulin gene transactivation. J Immunol. 2001;167(9):5175-5184.
40 Emery P, Mach B, Reith W. The different level of expression of HLA-DRB1 and -DRB3 genes is controlled by conserved isotypic differences in promoter sequence. Hum Immunol. 1993;38:137-147.
41 Leën MPJM, Gorski J. DRB4 promoter polymorphism in DR7 individuals: correlation with DRB4 pre-mRNA and mRNA levels. Immunogenetics. 1997;45(6):371-378.
42 Garcia-Lora A, Algarra I, Garrido F. MHC class I antigens, immune surveillance, and tumor immune escape. J Cell Physiol. 2003;195(3):346-355.
43 Lilley BN, Ploegh HL. Viral modulation of antigen presentation: manipulation of cellular targets in the ER and beyond. Immunol Rev. 2005;207:126-144.
44 Kaufmann SH, Schaible UE. Antigen presentation and recognition in bacterial infections. Curr Opin Immunol. 2005;17(1):79-87.
45 Lin A, Xu H, Yan W. Modulation of HLA expression in human cytomegalovirus immune evasion. Cell Mol Immunol. 2007;4(2):91-98.
46 Kimball ES, Coligan JE. Structure of class I major histocompatibility antigens. Contemp Top Mol Immunol. 1983;9:1-63.
47 Adamashvili I, Kelley RE, Pressly T, McDonald JC. Soluble HLA: patterns of expression in normal subjects, autoimmune diseases, and transplant recipients. Rheumatol Int. 2005;25(7):491-500.
48 Maenaka K, Jones EY. MHC superfamily structure and the immune system. Curr Opin Struct Biol. 1999;9:745-753.
49 Madden DR. The three-dimensional structure of peptide-MHC complexes. Annu Rev Immunol. 1995;13:587-622.
50 Stevanovic S. Structural basis of immunogenicity. Transpl Immunol. 2002;10(2–3):133-136.
51 Hickman HD, Luis AD, Buchli R, et al. Toward a definition of self: proteomic evaluation of the class I peptide repertoire. J Immunol. 2004;172(5):2944-2952.
52 Jones EY, Fugger L, Strominger JL, Siebold C. MHC class II proteins and disease: a structural perspective. Nat Rev Immunol. 2006;6(4):271-282.
53 Thorsby E, Lie BA. HLA associated genetic predisposition to autoimmune diseases: genes involved and possible mechanisms. Transpl Immunol. 2005;14(3–4):175-182.
54 Hattotuwagama CK, Doytchinova IA, Flower DR. Toward the prediction of class I and II mouse major histocompatibility complex-peptide-binding affinity: in silico bioinformatic step-by-step guide using quantitative structure-activity relationships. Methods Mol Biol. 2007;409:227-245.
55 Sturniolo T, Bono E, Ding J, et al. Generation of tissue-specific and promiscuous HLA ligand databases using DNA microarrays and virtual HLA class II matrices. Nat Biotechnol. 1999;17(6):555-561.
56 Nielsen M, Lundegaard C, Blicher T, et al. Quantitative predictions of peptide binding to any HLA-DR molecule of known sequence: NetMHCIIpan. PLoS Comput Biol. 2008;4(7):e1000107.
57 Stevanovic S, Lemmel C, Hantschel M, Eberle U. Generating data for databases – the peptide repertoire of HLA molecules. Novartis Found Symp. 2003;254:143-155.
58 Lee KH, Wucherpfennig KW, Wiley DC. Structure of a human insulin peptide-HLA-DQ8 complex and susceptibility to type 1 diabetes. Nat Immunol. 2001;2(6):501-507.
59 Zavala-Ruiz Z, Strug I, Anderson MW, et al. A polymorphic pocket at the P10 position contributes to peptide binding specificity in class II MHC proteins. Chem Biol. 2004;11(10):1395-1402.
60 Garbi N, Tanaka S, van den BM, et al. Accessory molecules in the assembly of major histocompatibility complex class I/peptide complexes: how essential are they for CD8(+) T-cell immune responses? Immunol Rev. 2005;207:77-88.
61 Van Kaer L. Major histocompatibility complex class I-restricted antigen processing and presentation. Tissue Antigens. 2002;60(1):1-9.
62 York IA, Chang SC, Saric T, et al. The ER aminopeptidase ERAP1 enhances or limits antigen presentation by trimming epitopes to 8–9 residues. Nat Immunol. 2002;3(12):1177-1184.
63 Serwold T, Gonzalez F, Kim J, et al. ERAAP customizes peptides for MHC class I molecules in the endoplasmic reticulum. Nature. 2002;419(6906):480-483.
64 Pieters J. MHC class II restricted antigen presentation. Curr Opin Immunol. 1997;9:89-96.
65 Gelin C, Sloma I, Charron D, Mooney N. Regulation of MHC II and CD1 antigen presentation: from ubiquity to security. J Leukoc Biol. 2009;85(2):215-224.
66 Watts C. The exogenous pathway for antigen presentation on major histocompatibility complex class II and CD1 molecules. Nat Immunol. 2004;5(7):685-692.
67 Brocke P, Garbi N, Momburg F, Hammerling GJ. HLA-DM, HLA-DO and tapasin: functional similarities and differences. Curr Opin Immunol. 2002;14(1):22-29.
68 Karlsson L. DM and DO shape the repertoire of peptide-MHC-class-II complexes. Curr Opin Immunol. 2005;17(1):65-70.
69 Alfonso C, Karlsson L. Nonclassical MHC class II molecules. Annu Rev Immunol. 2000;18:113-142.
70 Kuhns MS, Davis MM, Garcia KC. Deconstructing the form and function of the TCR/CD3 complex. Immunity. 2006;24(2):133-139.
71 Rudolph MG, Stanfield RL, Wilson IA. How TCRs bind MHCs, peptides, and coreceptors. Annu Rev Immunol. 2006;24:419-466.
72 Armstrong KM, Piepenbrink KH, Baker BM. Conformational changes and flexibility in T-cell receptor recognition of peptide-MHC complexes. Biochem J. 2008;415(2):183-196.
73 Gao GF, Rao Z, Bell JI. Molecular coordination of alphabeta T-cell receptors and coreceptors CD8 and CD4 in their recognition of peptide-MHC ligands. Trends Immunol. 2002;23(8):408-413.
74 Rowen L, Koop BF, Hood L. The complete 685-kilobase DNA sequence of the human β T cell receptor locus. Science. 1996;272:1755-1762.
75 Xiong N, Raulet DH. Development and selection of gammadelta T cells. Immunol Rev. 2007;215:15-31.
76 Von Boehmer H. Selection of the T-cell repertoire: receptor-controlled checkpoints in T-cell development. Adv Immunol. 2004;84:201-238.
77 Starr TK, Jameson SC, Hogquist KA. Positive and negative selection of T cells. Annu Rev Immunol. 2003;21:139-176.
78 Maggi E, Cosmi L, Liotta F, et al. Thymic regulatory T cells. Autoimmun Rev. 2005;4(8):579-586.
79 Goldrath AW, Bevan MJ. Selecting and maintaining a diverse T-cell repertoire. Nature. 1999;402:255-262.
80 Afzali B, Lombardi G, Lechler RI. Pathways of major histocompatibility complex allorecognition. Curr Opin Organ Transplant. 2008;13(4):438-444.
81 Felix NJ, Allen PM. Specificity of T-cell alloreactivity. Nat Rev Immunol. 2007;7(12):942-953.
82 Lee SJ, Klein J, Haagenson M, Baxter-Lowe LA, et al. High-resolution donor-recipient HLA matching contributes to the success of unrelated donor marrow transplantation. Blood. 2007;110(13):4576-4583.
83 Lindahl KF, Wilson DB. Histocompatibility antigen-activated cytotoxic T lymphocytes. II. Estimates of the frequency and specificity of precursors. J Exp Med. 1977;145(3):508-522.
84 Archbold JK, Macdonald WA, Burrows SR, et al. T-cell allorecognition: a case of mistaken identity or deja vu? Trends Immunol. 2008;29(5):220-226.
85 Gobin SJ, Van den Elsen PJ. Transcriptional regulation of the MHC class Ib genes HLA-E, HLA-F, and HLA-G. Hum Immunol. 2000;61(11):1102-1107.
86 Posch PE, Borrego F, Brooks AG, Coligan JE. HLA-E is the ligand for the natural killer cell CD94/NKG2 receptors. J Biomed Sci. 1998;5:321-331.
87 Sullivan LC, Clements CS, Rossjohn J, Brooks AG. The major histocompatibility complex class Ib molecule HLA-E at the interface between innate and adaptive immunity. Tissue Antigens. 2008;72(5):415-424.
88 Petrie EJ, Clements CS, Lin J, et al. CD94-NKG2A recognition of human leukocyte antigen (HLA)-E bound to an HLA class I leader sequence. J Exp Med. 2008;205(3):725-735.
89 Hoare HL, Sullivan LC, Pietra G, et al. Structural basis for a major histocompatibility complex class Ib-restricted T cell response. Nat Immunol. 2006;7(3):256-264.
90 O’Callaghan CA, Bell JI. Structure and function of the human class Ib molecules HLA-E, HLA-F and HLA-G. Immunol Rev. 1998;163:129-138.
91 Sullivan LC, Hoare HL, McCluskey J, et al. A structural perspective on MHC class Ib molecules in adaptive immunity. Trends Immunol. 2006;27(9):413-420.
92 Lepin EJ, Bastin JM, Allan DS, et al. Functional characterization of HLA-F and binding of HLA-F tetramers to ILT2 and ILT4 receptors. Eur J Immunol. 2000;30(12):3552-3561.
93 Lee N, Geraghty DE. HLA-F surface expression on B cell and monocyte cell lines is partially independent from tapasin and completely independent from TAP. J Immunol. 2003;171(10):5264-5271.
94 Boyle LH, Gillingham AK, Munro S, Trowsdale J. Selective export of HLA-F by its cytoplasmic tail. J Immunol. 2006;176(11):6464-6472.
95 Munz C, Nickolaus P, Lammert E, et al. The role of peptide presentation in the physiological function of HLA-G. Cancer Biol. 1999;9:47-54.
96 Clements CS, Kjer-Nielsen L, Kostenko L, et al. Crystal structure of HLA-G: a nonclassical MHC class I molecule expressed at the fetal-maternal interface. Proc Natl Acad Sci USA. 2005;102(9):3360-3365.
97 Apps R, Gardner L, Moffett A. A critical look at HLA-G. Trends Immunol. 2008;29(7):313-321.
98 Shiroishi M, Kuroki K, Rasubala L, et al. Structural basis for recognition of the nonclassical MHC molecule HLA-G by the leukocyte Ig-like receptor B2 (LILRB2/LIR2/ILT4/CD85d). Proc Natl Acad Sci U S A. 2006;103(44):16412-16417.
99 Apps R, Gardner L, Sharkey AM, et al. A homodimeric complex of HLA-G on normal trophoblast cells modulates antigen-presenting cells via LILRB1. Eur J Immunol. 2007;37(7):1924-1937.
100 Rajagopalan S, Bryceson YT, Kuppusamy SP, et al. Activation of NK cells by an endocytosed receptor for soluble HLA-G. PLoS Biol. 2006;4(1):e9.
101 Bahram S. MIC genes: from genetics to biology. Adv Immunol. 2000;76:1-60.
102 Bahram S, Inoko H, Shiina T, Radosavljevic M. MIC and other NKG2D ligands: from none to too many. Curr Opin Immunol. 2005;17(5):505-509.
103 Collins RW. Human MHC class I chain related (MIC) genes: their biological function and relevance to disease and transplantation. Eur J Immunogenet. 2004;31(3):105-114.
104 Li P, Willie ST, Bauer S, et al. Crystal structure of the MHC class I homolog MIC-A, a gamma/delta T cell ligand. Immunity. 1999;10:577-584.
105 Seliger B, Abken H, Ferrone S. HLA-G and MIC expression in tumors and their role in anti-tumor immunity. Trends Immunol. 2003;24(2):82-87.
106 Suarez-Alvarez B, Lopez-Vazquez A, Baltar JM, et al. Potential role of NKG2D and its ligands in organ transplantation: new target for immunointervention. Am J Transplant. 2009;9(2):251-257.
107 Zou Y, Stastny P. The role of major histocompatibility complex class I chain-related gene A antibodies in organ transplantation. Curr Opin Organ Transplant. 2009;14(4):414-418.
108 Silk JD, Salio M, Brown J, et al. Structural and functional aspects of lipid binding by CD1 molecules. Annu Rev Cell Dev Biol. 2008;24:369-395.
109 Lawton AP, Kronenberg M. The third way: progress on pathways of antigen processing and presentation by CD1. Immunol Cell Biol. 2004;82(3):295-306.
110 Cohen NR, Garg S, Brenner MB. Antigen presentation by CD1 lipids, T cells, and NKT cells in microbial immunity. Adv Immunol. 2009;102:1-94.
111 Simpson E, Scott D, James E, et al. Minor H antigens: genes and peptides. Transpl Immunol. 2002;10(2–3):115-123.
112 Hambach L, Spierings E, Goulmy E. Risk assessment in haematopoietic stem cell transplantation: minor histocompatibility antigens. Best Pract Res Clin Haematol. 2007;20(2):171-187.
113 Goulmy E. Minor histocompatibility antigens: from transplantation problems to therapy of cancer. Hum Immunol. 2006;67(6):433-438.
114 Spierings E, Hendriks M, Absi L, et al. Phenotype frequencies of autosomal minor histocompatibility antigens display significant differences among populations. PLoS Genetics. 2007;3(6):e103.
115 Falkenburg JH, van de Corput L, Marijt EW, Willemze R. Minor histocompatibility antigens in human stem cell transplantation. Exp Hematol. 2003;31(9):743-751.
116 Lanier LL. NK cell recognition. Annu Rev Immunol. 2005;23:225-274.
117 Bryceson YT, March ME, Ljunggren HG, Long EO. Activation, coactivation, and costimulation of resting human natural killer cells. Immunol Rev. 2006;214:73-91.
118 Bashirova AA, Martin MP, McVicar DW, Carrington M. The killer immunoglobulin-like receptor gene cluster: tuning the genome for defense. Annu Rev Genomics Hum Genet. 2006;7:277-300.
119 Colonna M, Spies T, Strominger JL, et al. Alloantigen recognition by two human natural killer cell clones is associated with HLA-C or a closely linked gene. Proc Natl Acad Sci USA. 1992;89:7983-7985.
120 Mandelboim O, Reyburn HT, Vales-Gomez M, et al. Protection from lysis by natural killer cells of group 1 and 2 specificity is mediated by residue 80 in human histocompatibility leukocyte antigen C alleles and also occurs with empty major histocompatibility complex molecules. J Exp Med. 1996;184(3):913-922.
121 Trowsdale J. Genetic and functional relationships between MHC and NK receptor genes. Immunity. 2001;15(3):363-374.
122 Hsu KC, Chida S, Dupont B, Geraghty DE. The killer cell immunoglobulin-like receptor (KIR) genomic region: gene-order, haplotypes and allelic polymorphism. Immunological Reviews. 2002;190(1):40-52.
123 Robinson J, Waller MJ, Stoehr P, Marsh SG. IPD – the Immuno Polymorphism Database. Nucleic Acids Res. 2005;33(Database Issue):D523-D526.
124 Yawata M, Yawata N, Draghi M, et al. Roles for HLA and KIR polymorphisms in natural killer cell repertoire selection and modulation of effector function. J Exp Med. 2006;203(3):633-645.
125 Andersson S, Fauriat C, Malmberg JA, et al. KIR acquisition probabilities are independent of self-HLA class I ligands and increase with cellular KIR expression. Blood. 2009;114(1):95-104.
126 Jonsson AH, Yokoyama WM. Natural killer cell tolerance licensing and other mechanisms. Adv Immunol. 2009;101:27-79.
127 Brodin P, Karre K, Hoglund P. NK cell education: not an on-off switch but a tunable rheostat. Trends Immunol. 2009;30(4):143-149.
128 Ruggeri L, Capanni M, Urgani E, et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science. 2002;295:2097-2100.
129 Witt CS. The influence of NK alloreactivity on matched unrelated donor and HLA identical sibling haematopoietic stem cell transplantation. Curr Opin Immunol. 2009;21(5):531-537.
130 Grzywacz B, Miller JS, Verneris MR. Use of natural killer cells as immunotherapy for leukaemia. Best Pract Res Clin Haematol. 2008;21(3):467-483.
131 Passweg JR, Huard B, Tiercy JM, Roosnek E. HLA and KIR polymorphisms affect NK-cell anti-tumor activity. Trends Immunol. 2007;28(10):437-441.
132 Borrego F, Masilamani M, Marusina AI, et al. The CD94/NKG2 family of receptors: from molecules and cells to clinical relevance. Immunol Res. 2006;35(3):263-278.
133 Sullivan LC, Clements CS, Beddoe T, et al. The heterodimeric assembly of the CD94-NKG2 receptor family and implications for human leukocyte antigen-E recognition. Immunity. 2007;27(6):900-911.
134 Burgess SJ, Maasho K, Masilamani M, et al. The NKG2D receptor: immunobiology and clinical implications. Immunol Res. 2008;40(1):18-34.
135 Lopez-Larrea C, Suarez-Alvarez B, Lopez-Soto A, et al. The NKG2D receptor: sensing stressed cells. Trends Mol Med. 2008;14(4):179-189.
136 Brown D, Trowsdale J, Allen R. The LILR family: modulators of innate and adaptive immune pathways in health and disease. Tissue Antigens. 2004;64(3):215-225.
137 Anderson KJ, Allen RL. Regulation of T-cell immunity by leucocyte immunoglobulin-like receptors: innate immune receptors for self on antigen-presenting cells. Immunology. 2009;127(1):8-17.
138 Katz HR. Inhibition of inflammatory responses by leukocyte Ig-like receptors. Adv Immunol. 2006;91:251-272.
139 American Society of Histocompatibility and Immunogenetics Laboratory Manual. 4.2 ed. 2008.
140 Reinsmoen NL, Zeevi A. Evaluation of the cellular immune response in transplantation. In: Detrick B, Hamilton RG, Folds JD, editors. Manual of Molecular and Clinical Laboratory Immunology. Washington DC: ASM Press; 2006:1228-1243.
141 Mickelson EM, Longton G, Anasetti C, et al. Evaluation of the mixed lymphocyte culture (MLC) assay as a method for selecting unrelated donors for marrow transplantation. Tissue Antigens. 1996;47:27-36.
142 Spencer A, Szydlo RM, Brookes PA, et al. Bone marrow transplantation for chronic myeloid leukemia with volunteer unrelated donors using ex vivo or in vivo T-cell depletion: major prognostic impact of HLA class I identity between donor and recipient. Blood. 1995;86:3590-3597.
143 Wang XN, Taylor PR, Skinner R, et al. T-cell frequency analysis does not predict the incidence of graft-versus-host disease in HLA-matched sibling bone marrow transplantation. Transplantation. 2000;70(3):488-493.
144 Sayer DC, Smith LK, Krueger R, Chrisitansen FT. DNA sequencing-based HLA typing detects a B-cell ALL blast-specific mutation in HLA-A(*)2402 resulting in loss of HLA allele expression. Leukemia. 2004;18(1):174-176.
145 Jacobbi LM, Blackwell P. Guidelines for specimen collection, storage and transportation. In: Hahn AB, Land GA, Strothman RM, editors. ASHI Laboratory Manual. American Society for Histocompatibility and Immunogenetics, 2000.
146 Schaffer M, Olerup O. HLA-AB typing by polymerase-chain reaction with sequence-specific primers: more accurate, less errors, and increased resolution compared to serological typing. Tissue Antigens. 2001;58(5):299-307.
147 Welsh K, Bunce M. Molecular typing for the MHC with PCR-SSP. Rev Immunogenetics. 1999;1:157-176.
148 Kimura A, Dong RP, Harada H, Sasazuki T. DNA typing of HLA class II genes in B-lymphoblastoid cell lines homozygous for HLA. Tissue Antigens. 1992;40:5-12.
149 Cao K, Chopek M, Fernandez-Vina MA. High and intermediate resolution DNA typing systems for class I HLA-A, -B, -C genes by hybridization with sequence specific oligonucleotide probes (SSOP). Rev. Immunogenetics. 1999;1:177-208.
150 Scheltinga SA, Johnston-Dow LA, White CB, et al. A generic sequencing based typing approach for the identification of HLA-A diversity. Hum Immunol. 1997;57(2):120-128.
151 McGinnis MD, Conrad MP, Bouwens AGM, et al. Automated, solid-phase sequencing of DRB region genes using T7 sequencing chemistry and dye-labeled primers. Tissue Antigens. 1995;46:173-179.
152 Zachary AA, Houp JA, Vega R, et al. Evaluation of the humoral response in transplantation. In: Detrick B, Hamilton RG, Folds JD, editors. Manual of Molecular and Clinical Laboratory Immunology. Washington, DC: ASM Press; 2006:1215-1227.
153 Fulton RJ, McDade RL, Smith PL, et al. Advanced multiplexed analysis with the FlowMetrix system. Clinical Chemistry. 1997;43:1749-1756.
154 Mickelson EM, Petersdorf E, Anasetti C, et al. HLA matching in hematopoietic cell transplantation. Hum Immunol. 2000;61:92-100.
155 Ottinger HD, Rebmann V, Pfeiffer KA, et al. Positive serum crossmatch as predictor for graft failure in HLA-mismatched allogeneic blood stem cell transplantation. Transplantation. 2002;73:1280-1285.
156 van den Boogaardt DE, van Rood JJ, Roelen DL, Claas FH. The influence of inherited and noninherited parental antigens on outcome after transplantation. Transpl Int. 2006;19(5):360-371.
157 Wan AM, Ennis PD, Parham P, Holmes N. The primary structure of HLA-A32 suggests a region involved in formation of the Bw4/Bw6 epitopes. J Immunol. 1986;137:3671-3674.
158 Toubert A, Raffoux C, Boretto J, et al. Epitope mapping of HLA-B27 and HLA-B7 antigens by using intradomain recombinants. J Immunol. 1988;141(7):2503-2509.
159 Rodey GE, Fuller TC. Public epitopes and the antigenic structure of the HLA molecules. Crit Rev Immunol. 1987;7:229-267.
160 Rodey GE, Neylan JF, Whelchel JD, et al. Epitope specificity of HLA class I alloantibodies. I. Frequency analysis of antibodies to private versus public specificities in potential transplant recipients. Hum Immunol. 1994;39:272-280.
161 Rodey GE. HLA Beyond Tears, 2nd ed. Durango CO: De Novo; 2000.
162 Thompson JS, Thacker LR. CREG matching for first cadaveric kidney transplants (TNX) performed by SEOPF centers between October 1987 and September 1995. Southeastern Organ Procurement Foundation. Clin Transplant. 1996;10:586-593.
163 Takemoto SK, Cecka JM, Terasaki PI. Benefits of HLA-CREG matching for sensitized recipients as illustrated in kidney regrafts. Transplant Proc. 1997;29(1–2 PT 5):1417.
164 Wade JA, Hurley CK, Takemoto SK, et al. HLA mismatching within or outside of cross-reactive groups (CREGs) is associated with similar outcomes after unrelated hematopoietic stem cell transplantation. Blood. 2007;109(9):4064-4070.
165 Duquesnoy R, Spellman S, Haagenson M, et al. HLAMatchmaker-defined triplet matching is not associated with better survival rates of patients with class I HLA allele mismatched hematopoietic cell transplants from unrelated donors. Biol Blood Marrow Transplant. 2008;14(9):1064-1071.
166 Noreen HJ, Yu N, Setterholm M, et al. Validation of DNA-based HLA-A and HLA-B testing of volunteers for a bone marrow registry through parallel testing with serology. Tissue Antigens. 2001;57(3):221-229.
167 Bochtler W, Maiers M, Oudshoorn M, et al. World Marrow Donor Association guidelines for use of HLA nomenclature and its validation in the data exchange among hematopoietic stem cell donor registries and cord blood banks. Bone Marrow Transplant. 2007;39(12):737-741.
168 Hurley CK. Acquisition and use of DNA-based HLA typing data in bone marrow registries. Tissue Antigens. 1997;49:323-328.
169 Holdsworth R, Hurley CK, Marsh SG, et al. The HLA dictionary 2008: a summary of HLA-A, -B, -C, -DRB1/3/4/5, and -DQB1 alleles and their association with serologically defined HLA-A, -B, -C, -DR, and -DQ antigens. Tissue Antigens. 2009;73(2):95-170.
170 Sollid LM. Coeliac disease: dissecting a complex inflammatory disorder. Nat Rev Immunol. 2002;2(9):647-655.
171 Chessman D, Kostenko L, Lethborg T, et al. Human leukocyte antigen class I-restricted activation of CD8+ T cells provides the immunogenetic basis of a systemic drug hypersensitivity. Immunity. 2008;28(6):822-832.
172 Opelz G, Dohler B. Effect of human leukocyte antigen compatibility on kidney graft survival: comparative analysis of two decades. Transplantation. 2007;84(2):137-143.
173 Takemoto SK, Zeevi A, Feng S, et al. National conference to assess antibody-mediated rejection in solid organ transplantation. Am J Transplant. 2004;4(7):1033-1041.
174 Zachary AA, Montgomery RA, Leffell MS. Defining unacceptable HLA antigens. Curr Opin Organ Transplant. 2008;13(4):405-410.
175 Hod E, Schwartz J. Platelet transfusion refractoriness. Br J Haematol. 2008;142(3):348-360.
176 Thomas ED. A history of haematopoietic cell transplantation. Br J Haematol. 1999;105(2):330-339.
177 O’Reilly RJ, Dupont B, Pahwa S, et al. Reconstitution in severe combined immundeficiency by transplantation of marrow from an unrelated donor. N Engl J Med. 1977;297:1311-1318.
178 Hurley CK, Raffoux C. World Marrow Donor Association: international standards for unrelated hematopoietic stem cell donor registries. Bone Marrow Transplant. 2004;34(2):103-110.
179 Gahrton G, van Rood JJ, Oudshoorn M. The World Marrow Donor Association (WMDA): its goals and activities. Bone Marrow Transplant. 2003;32(2):121-124.
180 Ljungman P, Urbano-Ispizua A, Cavazzana-Calvo M, et al. Allogeneic and autologous transplantation for haematological diseases, solid tumours and immune disorders: definitions and current practice in Europe. Bone Marrow Transplant. 2006;37(5):439-449.
181 Schipper RF, D’Amaro J, Oudshoorn M. The probability of finding a suitable related donor for bone marrow transplantation in extended families (see comment by Kollman). Blood. 1996;87:800-804.
182 Koh LP, Chao N. Haploidentical hematopoietic cell transplantation. Bone Marrow Transplant. 2008;42(Suppl 1):S60-S63.
183 Oudshoorn M, van Leeuwen A, v.d.Zanden HG, van Rood JJ. Bone Marrow Donors Worldwide: a successful exercise in international cooperation. Bone Marrow Transplant. 1994;14:3-8.
184 Bray RA, Hurley CK, Kamani NR, et al. National marrow donor program HLA matching guidelines for unrelated adult donor hematopoietic cell transplants. Biol Blood Marrow Transplant. 2008;14(9 Suppl):45-53.
185 Kamani N, Spellman S, Hurley CK, et al. State of the art review: HLA matching and outcome of unrelated donor umbilical cord blood transplants. Biol Blood Marrow Transplant. 2008;14(1):1-6.