Chapter 43 Hereditary Choroidal Diseases
 For additional online content visit http://www.expertconsult.com
For additional online content visit http://www.expertconsult.com
Introduction
Nonetheless, the hereditary choroidal dystrophies can be classified in the following manner: (1) choroidal atrophy phenotypes, which can be further subdivided into: (a) central areolar choroidal dystrophy (CACD); (b) peripapillary choroidal dystrophy; and (c) diffuse choroidal dystrophy; (2) gyrate atrophy of the choroid and retina; and (3) choroideremia (CHM). While each of the choroidal dystrophies has characteristic fundus features, in certain instances at advanced stages of disease an overlap in fundus appearance may be observed (Table 43.1).
Choroidal atrophy phenotypes
According to Sorsby,1 this group of disorders can be subdivided into three clinical phenotypes based on their geographical distribution. They include: central areolar, peripapillary, and more diffuse or generalized choroidal dystrophy. All can be inherited as either autosomal dominant or autosomal recessive traits.
Central areolar choroidal dystrophy
CACD was first described by Nettleship in 1884. It is inherited primarily as an autosomal dominant trait,2,3 although autosomal recessive cases have been occasionally reported.4,5 Yanagihashi and colleagues6 identified a novel mutation in the peripherin/RDS (retinal degeneration slow) gene in a Japanese family with an autosomal dominant form of CACD.
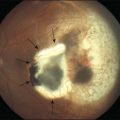
The initial symptoms of diminished central vision generally begin in the latter part of the second to the early part of the fourth decade. Characteristic bilateral macular lesions are solitary with well-defined margins, and circular or ovoid in shape (Fig. 43.1). Although they may increase in size and become irregular in shape, they do not involve the peripapillary region or extend beyond the vascular arcades.
Peripapillary choroidal dystrophy
The peripapillary form of choroidal dystrophy is usually inherited as an autosomal recessive trait,7 although in some instances autosomal dominant transmission may be encountered.
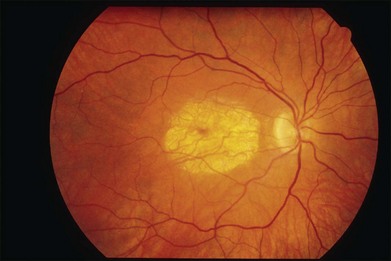
The fundus findings in this form of choroidal dystrophy initially include changes seen in the RPE and later an ophthalmoscopically apparent loss of RPE and choroidal tissue. The important distinction between CACD and the peripapillary phenotype is their location. The peripapillary form begins in the region surrounding the optic disc and slowly enlarges, in finger-like projections, nasally, temporally, and into the macula, eventually occupying the entire posterior pole (Fig. 43.2). In some instances the peripapillary form can progress to a phenotype similar to the diffuse form.
Visual field and dark-adapted final thresholds testing indicate that peripheral to the fundoscopically involved area, retinal function is either normal or mildly impaired. The full-field ERG is either normal or only slightly reduced, reflecting the extent of the disease.7,8
The differential diagnosis includes peripapillary pigment epithelial dystrophy in which there are well-defined areas of RPE loss, with direct visualization of the underlying choroidal vasculature. Fluorescein angiography shows an intact choriocapillaris that differentiates it from those with choriocapillaris loss.7 Another disorder that mimics peripapillary choroidal dystrophy is serpiginous choroiditis, which usually begins in the peripapillary region and then extends into the retina in pseudopod-like extensions, sometimes involving the macula.
Diffuse choroidal dystrophy
This diffuse disorder of the RPE and choriocapillaris is most often inherited as an autosomal dominant trait;9 however, autosomal recessive transmission may occur. The onset of symptoms occurs most often in the fourth and fifth decade and is usually manifested by poor central vision, impairment of night vision, or both.
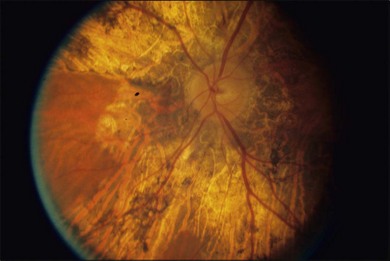
The early fundus changes include retinal pigment mottling and hypopigmentation. The disease may initially show a predilection for the posterior pole of the retina before progressing to a more diffuse phenotype. Later there is diffuse atrophy of both the RPE and choriocapillaris while the larger choroid vessels appear sclerotic as yellowish-white bands. Both the posterior pole and the periphery are involved to varying degrees. Even with diffuse involvement in the more advanced stages, the retinal vessels usually remain normal10 (Fig. 43.3). In the end stages, diffuse choroidal dystrophy cannot be easily differentiated from other diffuse chorioretinal diseases such as thioridazine (Mellaril) retinal toxicity, advanced stages of both pattern dystrophy and Stargardt disease, in addition to the advanced retinopathy seen in the Kearns–Sayre syndrome.
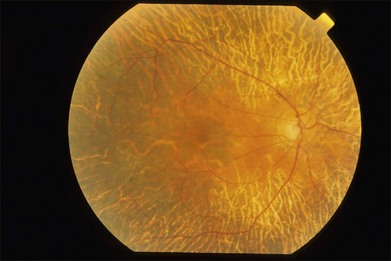
Psychophysical and electrophysiological studies reflect the diffuse involvement. Visual fields show a concentric peripheral constriction, while ERG recordings are either subnormal10 or undetectable.7 Fluorescein angiography shows a loss of the choriocapillaris and visualization of the larger choroidal vessels beneath atrophic-appearing RPE.7,11 A few scattered areas show a patchy choroidal flush pattern indicative of some remnants of the choriocapillaris.
Gyrate atrophy of the choroid and retina
The first case of this disease was described in 1888 by Jacobsohn12 as an example of “atypical retinitis pigmentosa”; however, Cutler13 in 1895 and Fuchs14 in 1896 were the first to recognize the disease as a distinct clinical entity. Gyrate atrophy is a rare choroidal disease with a prevalence of about 1 in 50 000 in Finland.15 It is inherited as an autosomal recessive trait, although dominant pedigrees have also been reported.10 The biochemical abnormalities observed in this disorder were initially described by Simell and Takki16 in 1973; these included a deficiency of the enzyme ornithine-delta-aminotransferase (OAT), which results in an increase in the plasma ornithine concentration (10–15 times the normal levels). The enzyme OAT is a mitochondrial-encoded enzyme with pyridoxal phosphate (vitamin B6) enzyme as a cofactor that catalyzes the interconversion of ornithine, glutamate, and proline. This results in systemic biochemical abnormalities, including hyperornithinemia, and reductions in plasma lysine, glutamine, glutamate, and creatine.17–19 Either an absence or a marked reduction of OAT in cultured skin fibroblasts and in lymphocytes has been observed.20 A number of different mutations have been identified within the OAT gene on chromosome 10.21–23 Kellner and colleagues24 previously described a gyrate atrophy-like phenotype in 6 male patients, 3 of whom were patients of the same family, with normal serum ornithine levels.
The onset of visual symptoms, including poor night vision and constricted peripheral vision, usually begins in the second and third decades. Since both structural and visual functional changes spread from more peripheral to a central location, loss of visual acuity is a later complaint in the disease. Myopia and posterior subcapsular cataracts are frequently observed and vitreous opacities may also be present.15
The fundus changes begin in the midperipheral and peripheral retina with a thinning and atrophic appearance of the RPE in which the underlying choroidal vessels may appear either normal or sclerotic. These areas are typically scalloped in shape and are initially separate but tend to become confluent as they slowly progress both centrally and peripherally (Fig. 43.4). Progression of the disease leads to pigment clumping, RPE and choriocapillaris atrophy, and eventual total atrophy of the choroid exposing the white sclera. In the late stages, an annular ring of choroidal atrophy may be seen from the periphery to the posterior pole, usually sparing the macula. The retinal vessels may appear normal initially or attenuated in later stages of the disease when the optic nerve may appear pale.7,10
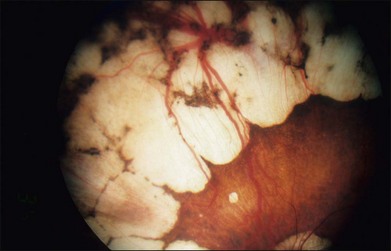
There are reports on the presence of cystoid macular edema in patients with gyrate atrophy.25–27 A study by Vasconcelos-Santos et al.27 showed a short-term therapeutic effect with the use of a 4-mg intravitreal triamcinolone acetonide injection for gyrate atrophy-related macular edema. After drug clearance, the edema recurred, with return of visual acuity to the pretreatment level.
Visual function varies considerably from case to case and seems to be related to the extent of fundus involvement. Visual field testing shows a concentric peripheral constriction of the visual field as the most often observed abnormality. However, an annular ring and paracentral scotomas may develop as the disease progresses. Eventually, if the fovea becomes involved, a central scotoma will be seen.7,28
Early in the disease, dark adaptation testing shows only mild threshold elevations while significant elevation of the final rod thresholds is eventually noted in most patients. In the early stage, full-field ERG recordings may show only a mild abnormality in rod and cone amplitudes, while, as the disease progress, the ERG responses deteriorate and may eventually become undetectable. The rod responses are affected more severely in the early stages, but later both cone as well as rod function is severely impaired.29,30 The electro-oculogram (EOG) is normal or only mildly reduced at very early stages. The EOG light peak to dark trough ratio becomes markedly reduced in the later stages. Electromyograms are often abnormal, although only a few patients complain of mild muscle weakness. Muscle biopsy shows atrophic type 2 muscle fibers with tubular aggregates visible on electron microscopy.31,32 Both electrocardiographic and electroencephalographic abnormalities may be observed in some patients.33,34 Histopathologic studies show early changes in RPE cells, with subsequent loss of photoreceptors and choriocapillaris, suggesting that these latter changes may be secondary to the loss of RPE cell integrity.35 A report of a histologic study in the ornithine-deficient mouse model of gyrate atrophy36 is available online.
A histopathologic study in the ornithine-deficient mouse model of gyrate atrophy showed earliest changes in the RPE cells in the form of sporadic degeneration of scattered cells. By 6 months, there were more diffuse abnormalities of the RPE with accumulation of large phagosomes and crystalloid inclusions. Although morphologically normal at 2 months, the photoreceptor outer segments became highly disorganized and shortened to 60% of mouse control lengths by 10 months. Additionally, there was a cumulative loss of the photoreceptor cells, which reached 33% by 10 months.36
An arginine-restricted diet has been pursued as a form of therapy in patients with gyrate atrophy of the choroid and retina.17,37–39 Since ornithine is produced from other amino acids, mainly arginine, some investigators advocate that patients be restricted to a rigid low-protein diet, including near-total elimination of arginine with supplementation of essential amino acids. Orally administered pyridoxal phosphate (vitamin B6), can result in a reduction in plasma ornithine levels in some patients, while others are nonresponsive to B6.29 Overall, ERG responses are better maintained by B6-responsive patients compared to those who are nonresponders.29,37 While Kaiser-Kupfer et al.38 concluded that a dietary approach to reducing plasma ornithine was effective, this was not similarly observed to occur in patients with gyrate atrophy in studies by Vannas-Sulonen et al.39 or Berson et al.37
Choroideremia
Mauthner in 187240 was the first to describe the clinical features of CHM, which is a generalized degeneration of the retina, inherited as an X-linked recessive trait,41 with an estimated prevalence of 1 in 50 000.42 The onset of symptoms occurs in the first and second decades with impairment of night vision and peripheral visual field loss. Central vision is most often preserved until later in life. Males in their 40s generally have useful visual acuity, but typically only a small residual visual field. Later (ages 50–70 years), central vision is more substantially reduced. In a study of 115 males with CHM (mean age 39 years), a slow rate of visual acuity loss with the retention of central visual acuity until the seventh decade was found.43
Fundus changes are readily apparent by the second decade of life or earlier. A 22-month-old infant with fundus changes has been described.41 The clinical diagnosis of affected males is characterized by the following:43
• A history of defective dark adaptation, manifesting as poor visual function in dim illumination is commonly the first symptom. Males may not show this impairment until their early teens.
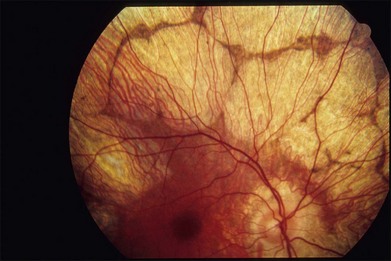
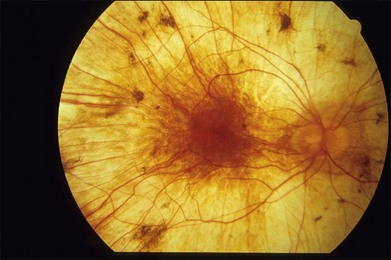
• The fundus changes in affected males undergo a characteristic progression. The initial appearance is a fine, peppery-like retinal pigment mottling at the midperipheral retina and posterior pole. At this stage, the ERG is abnormal, showing reduced or absent scotopic responses.10 Focal disturbances in the RPE consisting of pigmentary loss or a metallic sheen follow the “salt and pepper” mottling while the underlying choroid may appear normal or show choriocapillaris atrophy. Occasionally, these areas of focal disturbances assume a shape similar to gyrate atrophy, and the distinction between the two diseases may be difficult on the basis of the fundus appearance (Fig. 43.5). Atrophy of the choroid follows with eventual loss of the entire layer and exposure of bare sclera. The rate of progression will vary from individual to individual and from family to family. These changes are initially most apparent in the midperipheral retina and progress centrally, the macula being the last affected, with central vision preserved until later in the disease. In the final stage, the fundus shows an extensive yellowish-white reflex from the sclera (Fig. 43.6). Cystoid macular edema has recently been described by Genead and Fishman44 to be present in 63% of patients in a small (n = 16) cohort.
1. Peripheral visual field loss manifests as a ring scotoma that corresponds to areas of chorioretinal degeneration.
2. Even in the early stages of the disease, the ERG is most often abnormal under both light- and dark-adapted conditions. The ERG may be normal early in the course of the disease when only a few focal lesions are present,45 but eventually becomes undetectable in most.45 Nonetheless, a wide intrafamilial and interfamilial variability in ERG amplitudes with age has been observed.46 EOG recordings show an abnormally low light peak to dark trough ratio.
Histopathologic examination of a 30-year-old man with CHM showed diffuse abnormalities of the retina, RPE, and choriocapillaris that varied from different areas and appeared to occur independently of each other. In addition, mild T-lymphocyte infiltration was found within the choroid.47 Prior pathological specimens41,48–50 from patients afflicted with CHM showed the following:
1. Extensive atrophy of the choroidal vasculature and Bruch’s membrane was found in all subjects. The choroid was most recognizable at the macula.
2. Extensive atrophy of the RPE and photoreceptors was common even in the macula. However, in the eye from the youngest patient, distinct photoreceptor nuclei were seen in the macula.
3. In general, the retinal bipolar, ganglion, and nerve fiber layers were normal.
4. The optic nerve showed an increase in glial tissue within the septa and mild cystoid degeneration among the axons in the neural channels.
The CHM gene was localized to the long arm of the X chromosome (Xq21).51,52 The gene encodes the Rab escort protein-1 (REP-1) that is involved in the prenylation of Rabs. The REP-1 protein facilitates posttranslational modification of Rab proteins, which regulate intracellular trafficking in the RPE and photoreceptors and is likely involved in the removal of outer-segment disc membranes by the RPE.53 All currently described mutations in REP-1/CHM, including deletions, translocations, and mutations, result in protein truncation due to replacement of an arginine residue with a stop codon.54 In 1998, MacDonald et al.55 observed the absence of REP-1 in peripheral lymphocytes of affected individuals that led to the development of an immunoblot assay using anti-REP-1 antibodies to diagnose patients with CHM.
The carrier females of X-linked CHM show the following:
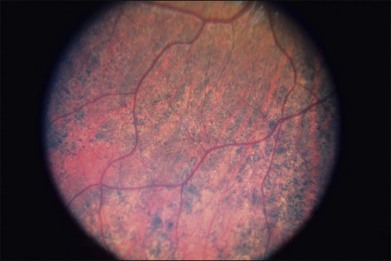
1. With few exceptions, fundus changes are considerably milder than those observed in affected males. Typically there is pigment mottling, best seen in the midperipheral retina, which may also be apparent in the macula. This finding becomes more readily evident after the second decade and has been described as showing a “moth-eaten appearance.” Some of the pigmentary changes in carriers consist of radial bands that course from the midperipheral retina toward the ora serrata (Fig. 43.7). There is no apparent relation between the degree of fundus pigmentary changes and the age of a carrier. The pigmentary changes observed in carriers are due to a skewed X-chromosome inactivation or the presence of an X-chromosome translocation involving Xq21.56
2. In most instances, carrier females do not experience significant visual impairment and in general are asymptomatic. However, carrier females may show changes on ERG, dark adaptation, visual field, and macular microperimetry testing.57 The ERG may be normal, even in carriers with pigmentary fundus changes. EOG recordings characteristically show no abnormality in the light peak to dark trough ratio.58 Measurement of fundus autofluorescence may demonstrate patchy areas of autofluorescence loss.59
3. There are occasional case reports60,61 in which the carrier female may have retinal and functional changes similar to affected male patients, but such findings are a rarity.
A histopathologic study62 of an eye from an 88-year-old carrier of CHM showed a patchy degeneration of photoreceptors and RPE cells that were not strictly concordant. The choriocapillaris was described as normal, except in regions of severe retinal degeneration. Immunofluorescence analysis, with a mouse monoclonal antibody, localized the CHM gene product, REP-1, to the rod cytoplasm and amacrine cells but not in cone cells.62 This observation suggests the possibility that the primary site of this disease may reside in the rods rather than in the RPE or choroid. Labeling observed within small vesicles in the rod cytoplasm is consistent with the association of REP-1 with intracellular vesicular transport.
Clinical phenotypes resembling hereditary choroidal diseases
X-linked retinitis pigmentosa (XLRP)
RP is a group of inherited disorders in which abnormalities of the RPE and photoreceptors lead to progressive visual loss. The initial symptoms of RP include night vision impairment and restriction of the peripheral visual field. A diagnosis of RP includes abnormalities on ERG testing. RP can be inherited in an autosomal dominant, autosomal recessive, or X-linked manner. Mutations in RPGR (also called RP3) and RP2 genes are the most common causes of XLRP. Linkage studies suggest that they account for 70–90% and 10–20%, respectively, of XLRP. In the later stages of particularly CHM, when the loss of choroid and retina is significant, the fundus appearance may be confused with end-stage XLRP; however, the degree and appearance of pigment migration into the retina that typifies RP are not characteriscally seen in individuals with CHM (Figs 43.8 and 43.9).
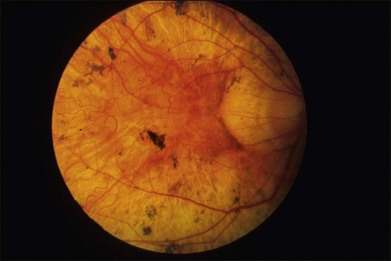
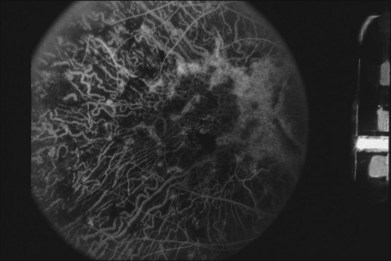
Kearns–Sayre syndrome (KSS)
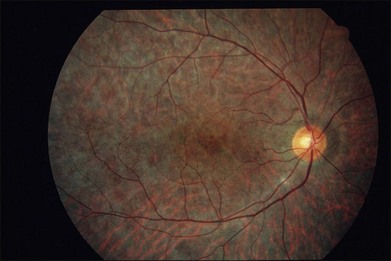
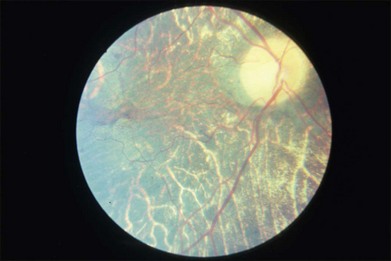
This disease represents a multisystem mitochondrial DNA deletion syndrome that is observed before 20 years of age and consists of a pigmentary retinopathy (Figs 43.10 and 43.11), progressive external ophthalmoplegia, as well as ptosis. In addition, patients may have at least one of the following: cardiac conduction block, cerebellar ataxia, or cerebrospinal fluid protein concentration greater than 100 mg/dL. Muscle biopsy in such patients shows ragged red fibers by light microscopy and mitochondrial abnormalities by high-resolution microscopy. Three phenotypes were described by Bastiaensen et al.,63 including infantile, juvenile, and adult-onset forms. In some patients with advanced disease, a generalized atrophy of the RPE and choriocapillaris can be encountered (Fig. 43.11). Subnormal cone and rod a- and b-wave amplitudes are most frequently observed in patients with Kearns–Sayre syndrome.
Bietti’s crystalline dystrophy
Patients affected with Bietti’s crystalline dystrophy most often present within the third decade of life with visual impairment and glistening crystalline-like changes in the posterior pole of the retina. Approximately one-third will also show crystals in the superficial stromal layer of the paralimbal region of the cornea. These cholesterol or cholesterol esters can also be found in fibroblasts as well as circulating lymphocytes, suggesting that this disorder may be a systemic abnormality of lipid metabolism. Both diffuse and more localized forms of retinal degenerative changes may be encountered. These changes involve both RPE and choriocapillaris atrophy and in this sense represent an overlap with the changes observed in choroidal dystrophies. ERG amplitude reduction often parallels the degree of fundus pigmentary changes. The disease is most often transmitted as an autosomal recessive trait. Gekka and colleagues64 identified a mutation in the CYP4V2 gene in two Japanese patients with Bietti’s crystalline dystrophy.
Thioridazine (Mellaril) retinal toxicity
Thioridazine was initially introduced in 1959 for the treatment of psychosis. Patients who receive relatively high doses of the drug can experience a decrease in their visual acuity as well as night blindness. Both central and ring scotomas have been observed. In earlier stages, a pigmentary granularity or mottling occurs in the macular or paramacular regions. Subsequently, extensive degenerative changes of the RPE, choriocapillaris, and photoreceptors are seen (Fig. 43.12). A phenotype with geographic, scalloped regions of hypopigmentation and loss of choriocapillaris vessels may become evident. Both this intermediate and a more advanced and more diffuse disease stage can mimic fundus changes seen in CHM, gyrate atrophy and diffuse choroidal dystrophy phenotypes. ERG recordings show various degrees of diminished photopic and scotopic a- and b-wave responses that parallel in severity the clinically evident fundus changes.
Stargardt disease
This disease is typically characterized by impairment of central vision within the first 10–20 years of life that often progresses to the level of legal blindness. The peripheral vision is most often preserved. There is a characteristic “beaten bronze” appearance of the macula, with small pisciform yellow-white flecks scattered within the posterior pole and, to a lesser extent, in the midperipheral retina. In the majority of affected patients, fluorescein angiography shows a masking of the fluorescence from the choroidal circulation (dark choroid).65–68 Although the inheritance is usually autosomal recessive,69 rare families have been reported with autosomal dominant inheritance.70–72 The autosomal dominant form has been attributed to mutations in the gene ELOVL4 (chromosome 6q),73 whereas the recessive form has been linked to mutations in ABCA4 (chromosome 1p).69,74 More information is available online.
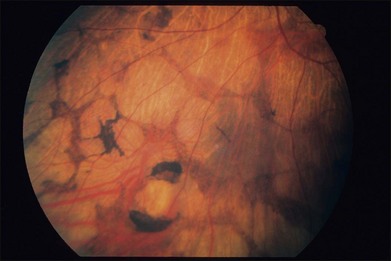
The advanced stages of Stargardt disease75 may appear as extensive atrophy of the RPE and choroid in the posterior pole and anterior to the vascular arcades (Fig. 43.13). Although the ERG is most often normal to modestly subnormal in the early to middle course of typical autosomal recessive Stargardt disease, it is often more notably abnormal by the advanced stage of disease.
Pattern macular dystrophy
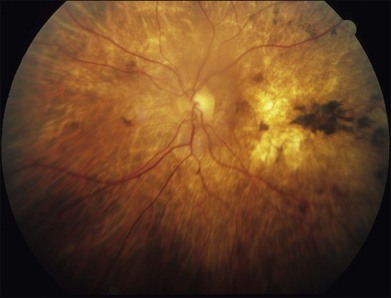
This disease is an autosomal dominant, fundoscopically variable disorder that involves the accumulation of lipofuscin within RPE cells with subsequent cell degeneration and secondary choriocapillaris loss.76 The lesions often begin in midlife and can be associated with mild to moderate visual acuity loss.77,78 Pattern dystrophy can, in late stages, occasionally produce diffusely atrophic changes of the RPE and choroid that resembles CHM or localized RPE and choroidal atrophy resembling CACD.79
Conclusion
Gene therapy for individuals with hereditary choroidal dystrophies is likely to be a treatment strategy that is available in the future. Introduction of recombinant adenovirus containing the full-length REP-1 CHM coding region can restore protein levels and REP-1 activity in enzyme-deficient lymphocytes and fibroblasts in vitro.80 A recent report by Genead et al.81 showed that treatment of cystoid macular edema in CHM patients with a topical dorzolamide 2% ophthalmic formulation can reduce central macular thickness associated with cystoid macular edema on spectral-domain optical coherence tomography testing with a potential improvement for visual acuity, while the study by Vasconcelos-Santos et al.27 showed a short-term beneficial effect on cystoid macular edema in patients with gyrate atrophy and the use of intravitreal triamcinolone acetonide.
 Bonus material for this chapter is available online at http://www.expertconsult.com
Bonus material for this chapter is available online at http://www.expertconsult.com
1 Sorsby A. Choroidal angiosclerosis with special reference to its hereditary character. Br J Ophthalmol. 1939;23:433–444.
2 Carr RE. Central areolar choroidal dystrophy. Arch Ophthalmol. 1965;73:32–35.
3 Sandvig K. Familial, central, areolar, choroidal atrophy of autosomal dominant inheritance. Acta Ophthalmol (Copenh). 1955;33:71–78.
4 Waardenburg PJ. Familial angiosclerosis of the choroid. J Genet Hum. 1952;1:83–90.
5 Sorsby A, Crick RP. Central areolar choroidal sclerosis. Br J Ophthalmol. 1953;37:129–139.
6 Yanagihashi S, Nakazawa M, Kurotaki J, et al. Autosomal dominant central areolar choroidal dystrophy and a novel Arg195Leu mutation in the peripherin/RDS gene. Arch Ophthalmol. 2003;121:1458–1461.
7 Krill AE, Archer D. Classification of the choroidal atrophies. Am J Ophthalmol. 1971;72:562–585.
8 Carr RE, Mittl RN, Noble KG. Choroidal abiotrophies. Trans Sect Ophthalmol Am Acad Ophthalmol Otolaryngol. 1975;79:OP796–OP816.
9 Sorsby A, Davey JB. Generalized choroidal sclerosis; course and mode of inheritance. Br J Ophthalmol. 1955;39:257–276.
10 Franceschetti A, Francois J, Babel J. Chorioretinal heredodegenerations. Springfield, Ill: Charles C Thomas; 1974.
11 Curry HF, Jr., Schonberg SS. Fluorescein photography in choroidal sclerosis. Arch Ophthalmol. 1969;81:177–183.
12 Jacobsohn E. Ein fall von Retinitis pigmentosa atypica. Klin Monatsbl Augenheilkd. 1888;26:202–206.
13 Cutler C. Dri ungewohnliche Falle von retino-choroideak Degeneration. Arch Augenheilkd. 1895;30:117.
14 Fuchs E. Ueber awei der Retinitis pigmentosa verwandte Krankheiten (retinitis punctate albescens und atrophia gyrate chorioideae et retinae). Arch Augenheilkd. 1896;32:111.
15 Takki KK, Milton RC. The natural history of gyrate atrophy of the choroid and retina. Ophthalmology. 1981;88:292–301.
16 Simell O, Takki K. Raised plasma-ornithine and gyrate atrophy of the choroid and retina. Lancet. 1973;1:1031–1033.
17 Valle D, Walser M, Brusilow SW, et al. Gyrate atrophy of the choroid and retina: amino acid metabolism and correction of hyperornithinemia with an arginine-deficient diet. J Clin Invest. 1980;65:371–378.
18 Valle D, Walser M, Brusilow S, et al. Gyrate atrophy of the choroid and retina. Biochemical considerations and experience with an arginine-restricted diet. Ophthalmology. 1981;88:325–330.
19 Kaiser-Kupfer MI, de Monasterio FM, Valle D, et al. Gyrate atrophy of the choroid and retina: improved visual function following reduction of plasma ornithine by diet. Science. 1980;210:1128–1131.
20 Heinänen K, Näntö-Salonen K, Leino L, et al. Gyrate atrophy of the choroid and retina: lymphocyte ornithine-delta-aminotransferase activity in different mutations and carriers. Pediatr Res. 1998;44:381–385.
21 Inana G, Hotta Y, Zintz C, et al. Expression defect of ornithine aminotransferase gene in gyrate atrophy. Invest Ophthalmol Vis Sci. 1988;7:1001–1005.
22 Mitchell GA, Brody LC, Siplia I, et al. At least two mutant alleles of ornithine delta-aminotransferase cause gyrate atrophy of the choroid and retina in Finns. Proc Natl Acad Sci USA. 1989;86:197–201.
23 McClatchey AI, Kaufman DL, Berson EL, et al. Splicing defect at the ornithine amino-transferase (OAT) locus in gyrate atrophy. Am J Hum Genet. 1990;47:790–794.
24 Kellner U, Weleber RG, Kennaway NG, et al. Gyrate atrophy-like phenotype with normal plasma ornithine. Retina. 1997;17:403–413.
25 Feldman RB, Mayo SS, Robertson DM, et al. Epiretinal membranes and cystoid macular edema in gyrate atrophy of the choroid and retina. Retina. 1989;9:139–142.
26 Oliveira TL, Andrade RE, Muccioli C, et al. Cystoid macular edema in gyrate atrophy of the choroid and retina: a fluorescein angiography and optical coherence tomography evaluation. Am J Ophthalmol. 2005;140:147–149.
27 Vasconcelos-Santos DV, Magalhães EP, Nehemy MB. Macular edema associated with gyrate atrophy managed with intravitreal triamcinolone: a case report. Arq Bras Oftalmol. 2007;70:858–861.
28 Kurstjens JH. Choroideremia and gyrate atrophy of the choroid and retina. Docum. Ophtal. 1965;19:1.
29 Weleber RG, Kennaway NG. Clinical trial of vitamin B6 for gyrate atrophy of the choroid and retina. Ophthalmology. 1981;88:316–324.
30 Raitta C, Carlson S, Vannas-Sulonen K. Gyrate atrophy of the choroid and retina: ERG of the neural retina and the pigment epithelium. Br J Ophthalmol. 1990;74:363–367.
31 Sipilä I, Simell O, Rapola J, et al. Gyrate atrophy of the choroid and retina with hyperornithinemia: tubular aggregates and type 2 fiber atrophy in muscle. Neurology. 1979;29:996–1005.
32 Kaiser-Kupfer MI, Kuwabara T, Askanas V, et al. Systemic manifestations of gyrate atrophy of the choroid and retina. Ophthalmology. 1981;88:302–306.
33 McCulloch JC, Arshinoff SA, Marliss EB, et al. Hyperornithinemia and gyrate atrophy of the choroid and retina. Ophthalmology. 1978;85:918–928.
34 Takki K. Gyrate atrophy of the choroid and retina associated with hyperornithinaemia. Br J Ophthalmol. 1974;58:3–23.
35 Wilson DJ, Weleber RG, Green WR. Ocular clinicopathologic study of gyrate atrophy. Am J Ophthalmol. 1991;111:24–33.
36 Wang T, Milam AH, Steel G, et al. A mouse model of gyrate atrophy of the choroid and retina. Early retinal pigment epithelium damage and progressive retinal degeneration. J Clin Invest. 1996;97:2753–2762.
37 Berson EL, Hanson AH, 3rd., Rosner B, et al. A two year trial of low protein, low arginine diets or vitamin B6 for patients with gyrate atrophy. Birth Defects Orig Artic Ser. 1982;18:209–218.
38 Kaiser-Kupfer MI, Caruso RC, Valle D. Gyrate atrophy of the choroid and retina. Long-term reduction of ornithine slows retinal degeneration. Arch Ophthalmol. 1991;109:1539–1548.
39 Vannas-Sulonen K, Simell O, Sipilä I. Gyrate atrophy of the choroid and retina. The ocular disease progresses in juvenile patients despite normal or near normal plasma ornithine concentration. Ophthalmology. 1987;94:1428–1433.
40 Mauthner L. Ein Fall von Chorioideremie. Berl Natur-med. Ver Innsbruck. 1872;2:191.
41 McCulloch C, McCulloch RJP. A hereditary and clinical study of choroideremia. Trans. Am. Acad. Ophthal. Otolaryngol. 1948;52:160.
42 MacDonald IM, Sereda C, McTaggart K, et al. Choroideremia gene testing. Expert Rev Mol Diagn. 2004;4:478–484.
43 Roberts MF, Fishman GA, Roberts DK, et al. Retrospective, longitudinal, and cross sectional study of visual acuity impairment in choroideraemia. Br J Ophthalmol. 2002;86:658–662.
44 Genead MA, Fishman GA. Cystic macular oedema on spectral-domain optical coherence tomography in choroideremia patients without cystic changes on fundus examination. Eye (Lond). 2011;25:84–90.
45 Francis PJ, Fishman GA, Trzupek KM, et al. Stop mutations in exon 6 of the choroideremia gene, CHM, associated with preservation of the electroretinogram. Arch Ophthalmol. 2005;123:1146–1149.
46 Ponjavic V, Abrahamson M, Andréasson S, et al. Phenotype variations within a choroideremia family lacking the entire CHM gene. Ophthalmic Genet. 1995;16:143–150.
47 MacDonald IM, Russell L, Chan CC. Choroideremia: new findings from ocular pathology and review of recent literature. Surv Ophthalmol. 2009;54:401–407.
48 Rafuse EV, McCulloch C. Choroideremia. A pathological report. Can J Ophthalmol. 1968;3:347–352.
49 McCulloch C. Choroideremia: a clinical and pathological review. Trans Am Ophthalmol Soc. 1969;67:142–195.
50 McCulloch JC. The pathologic findings in two cases of choroideremia. Trans Am Acad Ophthalmol Otolaryngol. 1950;54:565–572.
51 Lewis RA, Nussbaum RL, Ferrell R. Mapping X-linked ophthalmic diseases. Provisional assignment of the locus for choroideremia to Xq13-q24. Ophthalmology. 1985;92:800–806.
52 Nussbaum RL, Lewis RA, Lesko JG, et al. Choroideremia is linked to the restriction fragment length polymorphism DXYS1 at XQ13–21. Am J Hum Genet. 1985;37:473–481.
53 Seabra MC, Brown MS, Slaughter CA, et al. Purification of component A of Rab geranylgeranyl transferase: possible identity with the choroideremia gene product. Cell. 1992;70:1049–1057.
54 van den Hurk JA, Schwartz M, van Bokhoven H, et al. Molecular basis of choroideremia (CHM): mutations involving the Rab escort protein-1 (REP-1) gene. Hum Mutat. 1997;9:110–117.
55 MacDonald IM, Mah DY, Ho YK, et al. A practical diagnostic test for choroideremia. Ophthalmology. 1998;105:1637–1640.
56 Lorda-Sanchez IJ, Ibañez AJ, Sanz RJ, et al. Choroideremia, sensorineural deafness, and primary ovarian failure in a woman with a balanced X-4 translocation. Ophthalm Genet. 2000;21:185–189.
57 Thobani A, Anastasakis A, Fishman GA. Microperimetry and OCT findings in female carriers of choroideremia. Ophthalmic Genet. 2010;31:235–239.
58 Yau RJ, Sereda CA, McTaggart KE, et al. Choroideremia carriers maintain a normal electro-oculogram (EOG). Doc Ophthalmol. 2007;114:147–151.
59 Preising MN, Wegscheider E, Friedburg C, et al. Fundus autofluorescence in carriers of choroideremia and correlation with electrophysiologic and psychophysical data. Ophthalmology. 2009;116:1201–1209.
60 Fraser GR, Friedmann AI. Choroideremia in a female. Br Med J. 1968;2:732–734.
61 Harris GS, Miller JR. Choroideremia. Visual defects in a heterozygote. Arch Ophthalmol. 1968;80:423–429.
62 Syed N, Smith JE, John SK, et al. Evaluation of retinal photoreceptors and pigment epithelium in a female carrier of choroideremia. Ophthalmology. 2001;108:711–720.
63 Bastiaensen LA, Notermans SL, Ramaekers CH, et al. Kearns syndrome or Kearns disease: further evidence of a genuine entity in a case with uncommon features. Ophthalmologica. 1982;184:40–50.
64 Gekka T, Hayashi T, Takeuchi T, et al. CYP4V2 mutations in two Japanese patients with Bietti’s crystalline dystrophy. Ophthalm Res. 2005;37:262–269.
65 Fishman GA, Farber M, Patel BS, et al. Visual acuity loss in patients with Stargardt’s macular dystrophy. Ophthalmology. 1987;94:809–814.
66 Rotenstreich Y, Fishman GA, Anderson RJ. Visual acuity loss and clinical observations in a large series of patients with Stargardt disease. Ophthalmology. 2003;110:1151–1158.
67 Armstrong JD, Meyer D, Xu S, et al. Long-term follow-up of Stargardt’s disease and fundus flavimaculatus. Ophthalmology. 1998;105:448–457.
68 Aaberg TM. Stargardt’s disease and fundus flavimaculatus: evaluation of morphologic progression and intrafamilial co-existence. Trans Am Ophthalmol Soc. 1986;84:453–487.
69 Allikmets R, Singh N, Sun H, et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet. 1997;15:236–246.
70 Cibis GW, Morey M, Harris DJ. Dominantly inherited macular dystrophy with flecks (Stargardt). Arch Ophthalmol. 1980;98:1785–1789.
71 Zhang K, Bither PP, Park R, et al. A dominant Stargardt’s macular dystrophy locus maps to chromosome 13q34. Arch Ophthalmol. 1994;112:759–764.
72 Stone EM, Nichols BE, Kimura AE, et al. Clinical features of a Stargardt-like dominant progressive macular dystrophy with genetic linkage to chromosome 6q. Arch Ophthalmol. 1994;112:765–772.
73 Zhang K, Kniazeva M, Han M, et al. A 5-bp deletion in ELOVL4 is associated with two related forms of autosomal dominant macular dystrophy. Nat Genet. 2001;27:89–93.
74 Kaplan J, Gerber S, Larget-Piet D, et al. A gene for Stargardt’s disease (fundus flavimaculatus) maps to the short arm of chromosome 1. Nat Genet. 1993;5:308–311.
75 Fishman GA. Fundus flavimaculatus: a clinical classification. Arch Ophthalmol. 1976;94:2061–2067.
76 Zhang K, Garibaldi DC, Li Y, et al. Butterfly-shaped pattern dystrophy: a genetic, clinical, and histopathological report. Arch Ophthalmol. 2002;120:485–490.
77 Marmor MF, Byers B. Pattern dystrophy of the pigment epithelium. Am J Ophthalmol. 1977;84:32–44.
78 de PTVM, Delleman JW. Pigment epithelial pattern dystrophy: four different manifestations in a family. Arch Ophthalmol. 1982;100:1416–1421.
79 Watzke RC, Folk JC, Lang RM. Pattern dystrophy of the retinal pigment epithelium. Ophthalmology. 1982;89:1400–1406.
80 Anand V, Barral DC, Zeng Y, et al. Gene therapy for choroideremia: in vitro rescue mediated by recombinant adenovirus. Vision Res. 2003;43:919–926.
81 Genead MA, McAnany JJ, Fishman GA. Topical dorzolamide for treatment of cystoid macular edema in patients with choroideremia. Retina. 2012;32:826–833.