[level-membership-for-hematology-oncology-and-palliative-medicine-category]
Hemorrhagic Coagulation Disorders
After completion of this chapter, the reader will be able to:
1. Distinguish among the causes of localized versus generalized, soft tissue versus mucocutaneous, and acquired versus congenital bleeding.
2. List laboratory tests to differentiate among acquired hemorrhagic disorders of trauma, liver disease, vitamin K deficiency, and kidney failure, and interpret results when given.
3. Interpret laboratory assay results to diagnose and subtype von Willebrand disease.
4. Use the results of laboratory tests to identify and monitor the treatment of congenital single coagulation factor deficiencies such as deficiencies of factors VIII, IX, and XI.
5. Explain the principle and rationale for the use of each laboratory test for the detection and monitoring of hemorrhagic disorders.
6. Discuss the laboratory monitoring of therapy for hemorrhagic disorders.
Symptoms of Coagulopathies
Hemorrhage is a severe form of bleeding that requires intervention. Hemorrhage may be localized or generalized, acquired or congenital. To establish the cause of either a bleeding event or a patient’s tendency to bleed, the physician must obtain a complete patient and family history and do a thorough physical examination before ordering diagnostic laboratory tests.1
Mucocutaneous versus Anatomic Hemorrhage
Generalized bleeding may exhibit a mucocutaneous or a soft tissue (anatomic) pattern.
Mucocutaneous hemorrhage may be manifested as bruises or purpura, purple lesions of the skin caused by extravasated (seeping) red blood cells (RBCs).2 Petechiae and ecchymoses are purpura of less than or greater than 3 mm, respectively, and the presence of more than one such lesion may indicate a disorder of primary hemostasis. Other symptoms of defects of primary hemostasis are menorrhagia (profuse menstrual flow), bleeding from the gums, hematemesis (vomiting of blood), and epistaxis (uncontrolled nosebleed). Although nosebleeds are common and mostly innocent, especially in children, they suggest a primary hemostatic defect when they occur repeatedly, last longer than 10 minutes, involve both nostrils, or require physical intervention or transfusions.
Mucocutaneous hemorrhage tends to be associated with thrombocytopenia (platelet count lower than 150,000/mcL), qualitative platelet disorders, von Willebrand disease (VWD), or vascular disorders such as scurvy or telangiectasia (see Chapters 43 and 44). A careful history and physical examination distinguishes between mucocutaneous and anatomic bleeding, and the distinction helps direct investigative laboratory testing and treatment.
Anatomic (soft tissue) hemorrhage is seen in acquired or congenital defects in secondary hemostasis, or plasma coagulation factor deficiencies. Examples of anatomic bleeding include recurrent or excessive bleeding after minor trauma, dental extraction, or a surgical procedure. In such cases, hemorrhage may immediately follow a traumatic event, but is often delayed or recurs minutes or hours after the initial blood flow is stopped. In other patients, anatomic bleeding episodes are spontaneous. Most anatomic bleeds are internal, such as bleeds into joints, body cavities, muscles, or the central nervous system, and may initially have few visible signs. Because joint bleeds cause swelling and acute pain, they may not be immediately recognized as hemorrhages, although hemophilia patients usually know what is happening from experience. Repeated joint bleeds (hemarthroses) cause painful inflammation, with swelling and permanent cartilage damage that immobilize the joint. Repeated joint bleeds cause painful inflammation and permanent cartilage damage that swells and immobilizes the joint. Bleeds into soft tissues such as muscle or fat may cause nerve compression and subsequent temporary or permanent loss of function.3 When the bleeding involves body cavities, it causes symptoms related to the organ that is affected. Bleeding into the central nervous system, for instance, may cause headaches, confusion, seizures, and coma and must be managed as a medical emergency. Bleeds into the kidney may present as hematuria and may potentially be associated with acute kidney failure.
Whenever a soft tissue or mucocutaneous generalized hemostatic disorder is suspected, hemostasis laboratory testing is essential. Emergency department physicians often must begin to intervene in cases of acute hemorrhage, even before taking a history or waiting for the results of laboratory assays. In these instances, group-matched or O-negative RBCs, frozen plasma (FP), platelet concentrate, cryoprecipitate, recombinant activated coagulation factor VII (rFVIIa; NovoSeven, Novo Nordisk, Inc., Princeton, N.J.) or the antifibrinolytic tranexamic acid (Cyklokapron, Pfizer, New York, N.Y.) may be used to correct coagulation factor and platelet deficiencies to stop hemorrhage.4–6 Box 41-1 lists symptoms that suggest generalized hemorrhagic disorders.
Acquired versus Congenital Bleeding Disorders
Liver disease, kidney failure, chronic infections, autoimmune disorders, obstetric complications, dietary deficiencies such as vitamin C or vitamin K deficiency, blunt or penetrating trauma, and inflammatory disorders may all be associated with coagulopathy. If a patient’s bleeding episodes began after infancy, are associated with some disease or physical trauma, and are not duplicated in relatives, they probably are caused by an acquired, not a congenital, condition. When an adult patient comes for treatment of generalized hemorrhage, the physician first looks for an underlying disease or event and takes a personal and family history. The important elements of patient history are age, sex, current or past pregnancy, any systemic disorders such as diabetes or cancer, trauma, and exposure to drugs, including over the counter drugs or drugs of abuse. The physician determines the trigger, location, and volume of bleeding, then orders laboratory assays (Table 41-1). The initial coagulopathy test profile should consist of a complete blood count (CBC) that includes a platelet count, prothrombin time (PT), partial thromboplastin time (PTT), and fibrinogen assay.7 These tests take on clinical significance when the history and physical examination already have established the existence of abnormal bleeding. They are not effective as screens for healthy individuals.
TABLE 41-1
Screening Tests for a Generalized Hemostatic Disorder
| Test | Assesses for |
| Hemoglobin, hematocrit, reticulocyte count | Anemia associated with chronic bleeding; bone marrow response |
| Platelet count | Thrombocytopenia |
| PT | Deficiencies of factors II (prothrombin), V, VII, or X (clotting time prolonged) |
| PTT | Deficiencies of all factors except VII and XIII (clotting time prolonged) |
| Thrombin time | Hypofibrinogenemia and dysfibrinogenemia |
Congenital hemorrhagic disorders are uncommon, occurring in 1 per 100 people, and are usually diagnosed in infants or young children, who often have relatives with similar symptoms. Congenital bleeding disorders lead to repeated hemorrhages that may be spontaneous or occur following minor injury or may occur in unexpected locations, such as joints, body cavities, retinal veins and arteries, or the central nervous system. Patients with mild congenital hemorrhagic disorders may have no symptoms until they reach adulthood or experience some physical challenge, such as trauma, dental extraction, or a surgical procedure. The most common congenital deficiencies are VWD, factor VIII and IX deficiencies (hemophilia A and B), and platelet function disorders. Inherited deficiencies of fibrinogen, prothrombin, and factors V, VII, X, XI, and XIII also exist, but are rare (Box 41-2).
Acquired Coagulopathies
Far more patients have acquired bleeding disorders secondary to trauma, drug exposure, or chronic disease than have congenital or inherited coagulopathies. Chronic diseases or disorders commonly associated with bleeding are liver disease, vitamin K deficiency, and renal failure. In all cases, laboratory test results are necessary to confirm the diagnosis and guide the management of acquired hemorrhagic disorders.8
Acute Coagulopathy of Trauma-Shock
In North America, unintentional injury is the leading cause of death among those aged 1 to 45 years. The total rises when felonious, self-inflicted, and combat injuries are included: in the United States alone, trauma causes 93,000 deaths per year.9 Severe neurologic displacement accounts for 50% of deaths, with most occurring before the patient arrives at the hospital; however, of initial survivors, 20,000 die of hemorrhage within 48 hours.10 Acute coagulopathy of trauma-shock (ACOTS) accounts for fatal hemorrhage; 3000 to 4000 of these deaths are preventable. ACOTS is triggered by the combination of injury-related acute inflammation, platelet activation, tissue factor release, hypothermia, acidosis, and hypoperfusion (poor distribution of blood to tissues caused by low blood pressure), all of which are elements of systemic shock. For in-transit fluid resuscitation, colloid plasma expanders such as 5% dextrose are used to counter hypoperfusion.11 The administration of plasma expanders has a dilutional effect that is compounded by emergency department transfusion of RBCs, and although these measures are life saving, they intensify the coagulopathy, as does subsequent surgical intervention.
Massive transfusion, defined as administration of 4 to 5 RBC units within 1 hour or 8 to 10 units within 24 hours, is essential in otherwise healthy trauma victims when the systolic blood pressure is less than 110 mm Hg, pulse is greater than 105 beats/min, pH is less than 7.25, hematocrit is less than 32%, hemoglobin is less than 10 g/dL, and PT is prolonged to greater than 1.5 times the mean of the reference interval or generates an international normalized ratio (INR) of 1.5.12
Management of Acute Coagulopathy of Trauma-Shock
According to the American Society of Anesthesiologists surgical practice guidelines, RBC transfusions are required when the hemoglobin is less than 6.0 g/dL and are contraindicated when the hemoglobin is greater than 10.0 g/dL.13 For hemoglobin concentrations between 6.0 and 10.0 g/dL, the decision to transfuse is based upon the acuity of the patient’s condition as determined by physical evidence of blood loss; current blood loss rate; blood pressure; arterial blood gas values, especially pH and oxygen saturation; urine output; and laboratory evidence for coagulopathy, provided there is time for specimens to be collected and laboratory assays completed.
During surgery, coagulopathy is assessed based on microvascular bleeding, which is evaluated by measuring the volume of blood filling suction canisters, surgical sponges, and surgical drains. Platelet concentrate is ordered when the platelet count is less than 50,000/mcL unless the surgeon anticipates limited blood loss. Platelet concentrate therapy is generally ineffective when the patient is known to have immune thrombocytopenic purpura, thrombotic thrombocytopenic purpura, or heparin-induced thrombocytopenia. In patients with these conditions, therapeutic platelets are rapidly consumed, and their administration may therefore be contraindicated, although they may provide temporary rescue. Platelet concentrate is never ordered when the platelet count is greater than 100,000/mcL. Platelet administration may be necessary when the platelet count is between 50,000 and 100,000/mcL provided there is bleeding into a confined space such as the brain or eye; if the patient is taking aspirin or clopidogrel; if there is a known platelet disorder such as a release defect, Glanzmann thrombasthenia, or Bernard-Soulier syndrome (see Chapter 43); or if the surgery involves cardiopulmonary bypass, which suppresses platelet activity.
Use of Frozen Plasma in Acute Coagulopathy of Trauma-Shock
Although many refer to frozen plasma as “fresh frozen plasma,“ this term no longer applies, because most FP is prepared up to 24 hours after the time of collection rather than within 8 hours, as was the traditional practice.14 High-volume trauma centers such as combat facilities may maintain a small inventory of thawed FP ready for administration. Thawed FP may be stored at 1° C to 6° C for up to 5 days subsequent to thawing. The activity of von Willebrand factor (VWF) and coagulation factors V and VIII decline to approximately 60% after 5 days of refrigerator storage; however, the product may still be transfused.15
Most trauma center transfusion service directors require surgeons and emergency department physicians to transfuse 1 unit of FP for every 4 units of RBCs, although there is battlefield evidence that a ratio of 1 unit of FP per single unit of RBCs may lead to more favorable mortality statistics.16 The latter approach places a strain on the FP supply, however.
Use of Activated Prothrombin Complex Concentrate, Recombinant Activated Factor VII, and Tranexamic Acid in Acute Coagulopathy of Trauma-Shock
When administration of RBCs, platelets, cryoprecipitate, and FP fails to achieve hemostasis, activated prothrombin complex concentrate (FEIBA [factor eight inhibitor bypassing activity], Baxter Healthcare Corp., Deerfield, Ill.; Autoplex T, Nabi Biopharmaceuticals, Inc., Boca Raton, Fla.), recombinant activated coagulation factor VII (NovoSeven), or the antifibrinolytic drug tranexamic acid (Cyklokapron) may be employed. Autoplex T or FEIBA may be used at a dosage of 50 units/kg every 12 hours not to exceed 200 units/kg in 24 hours. The dose-response relationship of FEIBA is variable, and because FEIBA contains activated coagulation factors it raises the risk of disseminated intravascular coagulation (DIC). NovoSeven has been cleared by the Food and Drug Administration (FDA) for use in treating hemophilia A or B when factor VIII or factor IX inhibitors are present, respectively; its application in the treatment of ACOTS is off label. A NovoSeven dosage of 30 mcg/kg is instantly effective in stopping microvascular hemorrhage in nonhemophilic trauma victims, and NovoSeven does not cause DIC.17,18 However, one study found a possible link between off-label NovoSeven use and arterial and venous thrombosis in patients with existing thrombotic risk factors.19 Cyklokapron may be used at a loading dose of 1 g administered over 10 minutes followed by infusion of 1 g over 8 hours. First approved in 1986 to prevent bleeding in hemophilic patients who are about to undergo invasive procedures, Cyklokapron is effective for ACOTS, but this is an off-label use.
Liver Disease
Procoagulant Deficiency in Liver Disease
Liver disease predominantly alters the production of the vitamin K–dependent factors II (prothrombin), VII, IX, and X and control proteins C, S, and Z. In liver disease, these seven factors are produced in their des-γ-carboxyl forms, which cannot participate in coagulation (see Chapter 40). At the onset of liver disease, factor VII, which has the shortest plasma half-life at 6 hours, is the first coagulation factor to exhibit decreased activity. Because the PT is particularly sensitive to factor VII activity, it is characteristically prolonged in mild liver disease, serving as a sensitive early marker.20 Vitamin K deficiency caused by dietary insufficiency independent of liver disease produces a similar effect on the PT (see the case study in this chapter).
Declining coagulation factor V activity is a more specific marker of liver disease than deficient factor II, VII, IX or X because factor V is non–vitamin K dependent and is not affected by dietary vitamin K deficiency. The factor V activity assay, performed in conjunction with the factor VII assay, may be used to distinguish liver disease from vitamin K deficiency.21
Fibrinogen is an acute phase reactant that is frequently elevated in early or mild liver disease. Moderately and severely diseased liver produces fibrinogen that is coated with excessive sialic acid, a condition called dysfibrinogenemia in which the fibrinogen functions poorly. Dysfibrinogenemia causes generalized soft tissue bleeding associated with a prolonged thrombin time and an exceptionally prolonged reptilase clotting time.22 In end-stage liver disease, the fibrinogen level may fall to less than 100 mg/dL, which is a mark of liver failure.23
VWF and factors VIII and XIII are acute phase reactants that may be unaffected or elevated in mild to moderate liver disease.24,25 In contrast to the other coagulation factors, VWF is produced from endothelial cells and megakaryocytes and is stored in endothelial cells and platelets.
Platelet Abnormalities in Liver Disease
Moderate thrombocytopenia occurs in a third of patients with liver disease. Platelet counts of less than 150,000/mcL may result from sequestration and shortened platelet survival associated with portal hypertension and resultant hepatosplenomegaly. In alcoholism-related hepatic cirrhosis, acute alcohol toxicity also suppresses platelet production. Platelet aggregation and secretion properties are often suppressed; this is reflected in reduced platelet aggregometry and lumiaggregometry results. Occasionally platelets are hyperreactive. Aggregometry may be used to predict bleeding and thrombosis risk.26
Disseminated Intravascular Coagulation in Liver Disease
Chronic or compensated DIC (see Chapter 42) is a significant complication of liver disease that is caused by decreased liver production of regulatory antithrombin, protein C, or protein S and by the release of activated procoagulants from degenerating liver cells. In addition, the failing liver cannot clear activated coagulation factors that are regularly produced by abdominal organs and transported through the portal circulation. In primary or metastatic liver cancer, hepatocytes also may produce procoagulant substances that trigger chronic DIC, leading to ischemic complications.
In acute, uncompensated DIC, the PT, PTT, and thrombin time are prolonged; the fibrinogen level is reduced to less than 100 mg/dL; and fibrin degradation products, including D-dimers, are significantly increased.27 If the DIC is chronic and compensated, the only elevated test result may be the D-dimer assay value, a hallmark of unregulated coagulation and fibrinolysis.28 Although DIC can be resolved only by removing its underlying cause, its hemostatic deficiencies may be temporarily corrected by administering RBCs, FP, platelet concentrates, activated protein C, or antithrombin concentrates.29,30
Hemostasis Laboratory Tests in Liver Disease
The PT, PTT, thrombin time, fibrinogen concentration, platelet count, and D-dimer concentration are useful in characterizing the hemostatic abnormalities in liver disease (Table 41-2). Factor V and VII assays may be used in combination to differentiate liver disease from vitamin K deficiency. Both factors are decreased in liver disease, but only factor VII is decreased in vitamin K deficiency.
TABLE 41-2
Hemostasis Laboratory Tests in Liver Disease
| Assay | Interpretation |
| Fibrinogen | >400 mg/dL in early, mild liver disease; <200 mg/dL in moderate to severe liver disease causing hypofibrinogenemia or dysfibrinogenemia |
| Thrombin time | Prolonged in dysfibrinogenemia, hypofibrinogenemia, elevation of fibrin degradation products, and therapy with unfractionated heparin |
| Reptilase time | Prolonged in hypofibrinogenemia, significantly prolonged in dysfibrinogenemia; assay rarely used |
| PT | Prolonged even in mild liver disease due to presence of des-γ-carboxyl factors in place of normal factors II (prothrombin), VII, IX, and X. Report PT in seconds, not INR, when testing for liver disease. |
| PTT | Mildly prolonged in severe liver disease due to DIC or des-γ-carboxyl factors II (prothrombin), IX, and X |
| Platelet count | Mild thrombocytopenia, platelet count <150,000/mcL |
| Platelet aggregometry | Mild suppression of platelet aggregation and secretion to most agonists |
| D-dimer | >240 ng/mL by quantitative assay |
| Euglobulin clot lysis time | Lysis in <2 hr in primary or secondary systemic fibrinolysis; assay rarely used |
Renal Failure and Hemorrhage
Acute renal failure may be associated with acute gastrointestinal bleeding caused by specific anatomic lesions, but this does not necessarily indicate that a coagulopathy or platelet dysfunction is present.31 Chronic renal failure of any cause is commonly associated with platelet dysfunction and mild to moderate mucocutaneous bleeding.32 Platelet adhesion to blood vessels and platelet aggregation are suppressed, perhaps because the platelets are coated by guanidinosuccinic acid or dialyzable phenolic compounds.33 Decreased RBC mass (anemia) and thrombocytopenia contribute to the bleeding and may be corrected with dialysis, erythropoietin or RBC transfusions, and interleukin-11 therapy.34,35
Hemostasis activation syndromes that deposit fibrin in the renal microvasculature reduce the function of the glomeruli. Examples of such disorders are DIC, hemolytic uremic syndrome, and thrombotic thrombocytopenic purpura. Although these are by definition thrombotic disorders, they invariably cause thrombocytopenia, which may lead to mucocutaneous bleeding. Fibrin also may be deposited in renal transplant rejection and in the glomerulonephritis syndrome of systemic lupus erythematosus. This may be associated with a rise in quantitative plasma D-dimer, thrombin-antithrombin complexes, or prothrombin fragment 1 + 2, which are markers of coagulation activation (see Chapter 45).36
Laboratory tests for bleeding in renal disease provide only modest information with little predictive or management value. The bleeding time may be prolonged, but is too unreliable to provide an accurate diagnosis or to assist in monitoring treatment.37 Platelet aggregometry test results may predict bleeding. The PT and PTT are expected to be normal.
Management of renal failure–related bleeding typically focuses on the severity of the hemorrhage without reliance on laboratory test results. Renal dialysis improves platelet function, particularly when anemia is well controlled. Desmopressin acetate may be administered intravenously (DDAVP) or intranasally (Stimate) to increase the plasma concentration of high-molecular-weight multimers of VWF, which also aids platelet adhesion and aggregation.38 Patients with renal failure should not take aspirin, clopidogrel, or other platelet inhibitors, because these drugs increase the risk of hemorrhage.
Nephrotic Syndrome and Hemorrhage
Nephrotic syndrome is a state of increased glomerular permeability associated with a variety of conditions, such as chronic glomerulonephritis, diabetic glomerulosclerosis, systemic lupus erythematosus, amyloidosis, and renal vein thrombosis.39 In nephrotic syndrome, low-molecular-weight proteins are lost through the glomerulus into the glomerular filtrate and the urine. Coagulation factors II (prothrombin), VII, IX, X and XII have been detected in the urine, as have the coagulation regulatory proteins antithrombin and protein C. In 25% of cases, loss of regulatory proteins takes precedence over loss of procoagulants and leads to a tendency toward venous thrombosis.40
Vitamin K Deficiency
Hemorrhagic Disease of the Newborn Caused by Vitamin K Deficiency
Because of their sterile intestines and the minimal concentration of vitamin K in human milk, newborns are constitutionally vitamin K deficient.41 Hemorrhagic disease of the newborn was common in the United States before routine administration of vitamin K to infants was legislated in the 1960s, and it still occurs in developing countries. The activity levels of factors II (prothrombin), VII, IX, and X are lower in normal newborns than in adults, and premature infants have even lower concentrations of these factors.42 Breastfeeding prolongs the deficiency, because passively acquired maternal antibodies delay the establishment of gut flora.
Vitamin K Antagonists
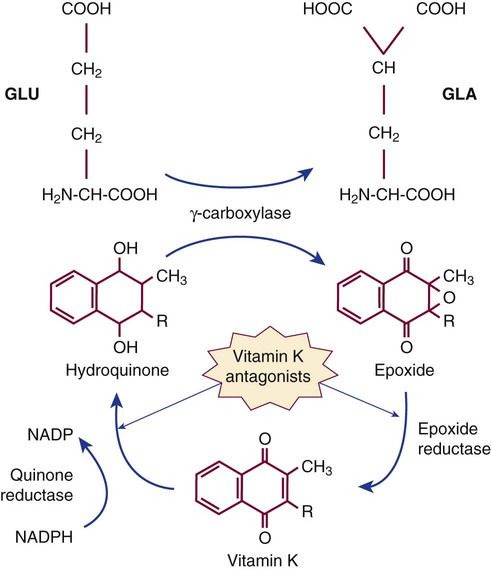
The γ-carboxylation cycle of coagulation factors is interrupted by coumarin-type oral anticoagulants such as warfarin that disrupt the vitamin K epoxide reductase and vitamin K quinone reductase reactions (Figure 41-1). In deficient γ-carboxylation, the liver releases dysfunctional des-γ-carboxyl factors II (prothrombin), VII, IX, and X, and proteins C, S, and Z; these inactive forms are called proteins in vitamin K antagonism (PIVKA). Therapeutic overdose or the accidental or felonious administration of warfarin-containing rat poisons may result in moderate to severe hemorrhage because of the lack of functional factors. The effect of brodifacoum or “superwarfarin,“ often used as a rodenticide, lasts for weeks to months, and treatment of poisoning with this substance requires repeated administration of vitamin K with follow-up PT monitoring.43 Warfarin overdose is the single most common reason for hemorrhage-associated emergency department visits.
Autoanti-VIII Inhibitor and Acquired Hemophilia
Acquired autoantibodies that specifically inhibit factors II (prothrombin), V, VIII, IX, and XIII and VWF have been described in nonhemophilic patients.44 Anti-VIII is the most common. Patients who develop an autoantibody to factor VIII, which is diagnostic of acquired hemophilia, are frequently older than age 60 and have no apparent underlying disease. Acquired hemophilia is occasionally associated with rheumatoid arthritis, inflammatory bowel disease, systemic lupus erythematosus, or lymphoproliferative disease. It also may occur in women of childbearing age 2 to 5 months after delivery. Patients with inhibitor autoantibodies are given immunosuppressive therapy, although autoantibodies that develop after pregnancy typically disappear spontaneously. Acquired hemophilia has an overall incidence of 1 per 1 million people per year. Patients experience sudden and severe bleeding in soft tissues or bleeding in the gastrointestinal or genitourinary tract. Acquired hemophilia, even when treated, remains fatal in at least 20% of cases. Autoantibodies to other procoagulants are less frequent, but create similar symptoms.45
Clot-Based Studies in Acquired Hemophilia
The in vitro kinetics of factor VIII neutralization by inhibitor are nonlinear. Although there is early rapid loss of factor VIII activity, residual activity remains, which indicates that the reaction has reached equilibrium. This is called type II kinetics (Figure 41-2). In contrast, alloantibodies to factor VIII, which develop in 20% to 25% of patients with severe hemophilia in response to factor VIII therapy, exhibit type I kinetics. In the latter, there is linear in vitro neutralization of factor VIII activity over 1 to 2 hours, which results in complete inactivation. In type I kinetics in vitro measurement is relatively accurate, whereas in type II kinetics the titration of inhibitor activity is semiquantitative.46
Quantitation of autoanti-VIII inhibitor is accomplished using the Bethesda assay, which is ordinarily employed to measure inhibitor in hemophilic patients with alloantibodies to factor VIII (see Hemophilia A and Factor VIII Inhibitors). Titer results help the clinician choose the proper therapy to control bleeding. Repeat titers are used to follow the response to immunosuppressive drugs, but are not needed for management of the bleeding symptoms.
Other Factor Inhibitors
Anti–coagulation factor II (antiprothrombin) antibodies, detectable by immunoassay, develop in approximately 30% of patients with lupus anticoagulant.47 Although lupus anticoagulant is associated with thrombosis, some patients with antiprothrombin antibodies experience bleeding and have a prolonged PT. A finding of reduced activity on prothrombin assay and a positive test result for lupus anticoagulant confirm the diagnosis. Antiprothrombin antibodies not associated with lupus anticoagulant are rare. Factor V and XIII inhibitors have been documented in patients receiving isoniazid treatment for tuberculosis.48,49 Antibodies to factor IIa (thrombin) and factor V may arise after exposure to topical bovine thrombin or fibrin glue.50 Autoanti-X antibodies are rare; however, factor X deficiency in amyloidosis may be caused by what seems to be an absorptive mechanism.51 In many cases of acquired inhibitors, mixing studies show uncorrected prolongation without incubation (immediate mixing study), and inhibitor titers may be determined by the Bethesda procedure.
Management of Acquired Hemophilia
Autoplex T, FEIBA, or NovoSeven may bypass the acquired coagulation factor VIII or factor IX inhibitor in acquired hemophilia and control acute bleeding. Patients who have low titers of inhibitor (less than 5 Bethesda units) may respond to administration of desmopressin acetate (DDAVP) or factor VIII concentrates, but close monitoring of their response to therapy with serial coagulation factor VIII activity assays is warranted. Once bleeding is controlled, immunosuppressive therapy may reduce the inhibitor titer.52 Plasma exchange may also be used in severe cases, but the response is less reliable than the response to immunosuppression.
Acquired von Willebrand Disease
Acquired VWF deficiency, with symptoms similar to those of congenital VWD, has been described in association with hypothyroidism; autoimmune, lymphoproliferative, and myeloproliferative disorders; benign monoclonal gammopathies; Wilms tumor; intestinal angiodysplasia; congenital heart disease; pesticide exposure; and hemolytic uremic syndrome.53 The pathogenesis of acquired VWD varies and may involve decreased production of VWF, presence of an autoantibody, or adsorption of VWF to abnormal cell surfaces, as seen in association with lymphoproliferative disorders and Wilms tumor.54
Congenital Coagulopathies
von Willebrand Disease
VWD is a common mucocutaneous bleeding disorder first described by Finnish Professor Erik von Willebrand in 1926. VWD is caused by any one of dozens of germline mutations that result in quantitative or structural abnormalities of VWF. Both quantitative and structural abnormalities lead to decreased adhesion by platelets to injured vessel walls, causing impaired primary hemostasis. VWD is the most prevalent of the congenital bleeding disorders and is found in approximately 1% of the population. It affects both sexes because of its autosomal dominant inheritance pattern. The parameters of and laboratory testing guidelines for VWD are established and defined in the National Heart, Lung, and Blood Institute (NHLBI) publication The Diagnosis, Evaluation, and Management of von Willebrand Disease.55
Molecular Biology and Functions of von Willebrand Factor
VWF is a glycoprotein whose molecular mass ranges from 800,000 to 20,000,000 D, the largest molecule in human plasma. Its plasma concentration is 0.5 to 1.0 mg/dL, but a great deal more is readily available on demand from storage organelles. VWF is synthesized in the endoplasmic reticulum of endothelial cells and stored in cytoplasmic Weibel-Palade bodies of endothelial cells. It is also synthesized in megakaryocytes and stored in the α-granules of platelets (see Chapter 13). Weibel-Palade bodies and α-granules release VWF in response to a variety of hemostatic stimuli.56
The VWF gene consists of 52 exons spanning 178 kilobase pairs (kb) on chromosome 12.57 The translated protein is a monomer of 2813 amino acids that, after glycosylation, forms dimers that are transferred to the aforementioned storage organelles, where the dimers polymerize to form enormous multimers. At the time of storage, a signal sequence and a propeptide, known as VWF antigen II, are cleaved so that the mature monomers, already polymerized, consist of 2050 amino acids.58
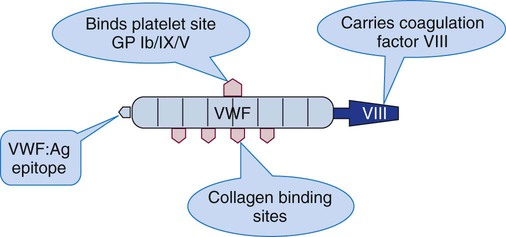
Each VWF monomer has four functional domains that bind factor VIII, platelet glycoprotein Ib/V/IX, platelet glycoprotein IIb/IIIa, and collagen, respectively (see Chapter 13).59 Upon release from intracellular stores, VWF forms a complex with coagulation factor VIII named VIII/VWF (Figure 41-3). VWF protects factor VIII from proteolysis, prolonging its plasma half-life from a few minutes without VWF to 8 to 12 hours with VWF. Table 41-3 lists the nomenclature for the structural and functional components of the factor VIII/VWF molecule.
TABLE 41-3
Nomenclature Relating to the VIII/VWF Complex
| Term | Meaning |
| VIII/VWF | Customary term for the plasma combination of factor VIII and VWF. |
| VIII | Procoagulant factor VIII, the protein transported on VWF. Factor VIII binds activated factor IX to form the complex of VIIIa-IXa, which digests and activates factor X. Factor VIII deficiency is called hemophilia A. |
| VWF : Ag | Epitope that is the antigenic basis for the VWF immunoassay. |
| VWF : RCo | Ristocetin cofactor activity, also called VWF activity. VWF activity is measured by the ability of ristocetin to cause agglutination of reagent platelets by the patient’s VWF. |
| VIII : C | Factor VIII coagulant activity as measured in factor-specific clot-based assays. |
von Willebrand Disease Types and Subtypes
Type 1 von Willebrand Disease
Type 1 VWD is a quantitative VWF deficiency caused by one of several autosomal dominant frameshifts, nonsense mutations, or deletions.60,61 Type 1 is seen in approximately 75% of VWD patients. The plasma concentrations of VWF and factor VIII are variably, although proportionally, reduced.62 There is mild to moderate bleeding, usually following a hemostatic challenge such as dental extraction or surgery. In women, menorrhagia is a common complaint that leads to the diagnosis of VWD.
Subtype 2A von Willebrand Disease
Ten percent to 20% of VWD patients have subtype 2A, which arises from well-characterized autosomal dominant point mutations in the A2 structural domain of the VWF molecule. These mutations render VWF more susceptible to proteolysis, which leads to a predominance of small-molecular-weight multimers in the plasma. The smaller multimers support less platelet adhesion activity than the normal high- or intermediate-molecular-weight multimers. Patients with subtype 2A VWD have normal or slightly reduced VWF : Ag levels with markedly reduced VWF activity as a result of the loss of the high-molecular-weight and intermediate-molecular-weight multimers essential for platelet adhesion.63
Subtype 2B von Willebrand Disease
A platelet mutation that increases glycoprotein Ib/V/IX affinity for normal VWF multimers creates a clinically similar disorder called platelet-type VWD or pseudo-VWD. In this instance, the large multimers also are lost from the plasma, and platelets become adhesive (Figure 41-4). Clinically and in the laboratory, the two entities are indistinguishable.
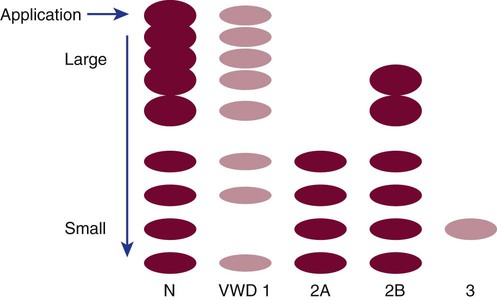
Laboratory Detection and Classification of von Willebrand Disease
VWF multimer analysis by sodium dodecyl sulfate–polyacrylamide gel electrophoresis further differentiates between VWD subtypes 2A and 2B. Although both 2A and 2B lack high-molecular-weight multimers, intermediate multimers are present in the electrophoretic pattern of subtype 2B VWD samples but are absent from the pattern of a patient with subtype 2A VWD. Multimer analysis is technically demanding and is performed mainly in reference laboratories for differentiation of VWD subtypes. Table 41-4 lists the expected test results for all VWD types and subtypes.
TABLE 41-4
Laboratory Detection and Classification of von Willebrand Disease: Typical Results*
| Laboratory Test | Type 1 | Type 2A | Type 2B | Type 3 |
| VWF activity | Low | Low | Low | Very low |
| VWF antigen | Low | Normal to slightly decreased | Normal to slightly decreased | Very low |
| VWF activity to VWF antigen ratio | >0.5 | <0.5 | <0.5 | N/A |
| Platelet count | Normal | Normal | Decreased | Normal |
| PTT | Normal to slightly prolonged | Normal | Normal | Prolonged |
| RIPA | Decreased | Decreased | Increased | Absent |
| Factor VIII activity | Mildly low | Normal | Normal | <5% |
| VWF multimers | Normal pattern | Large and intermediate forms absent | Large forms absent | All forms absent |
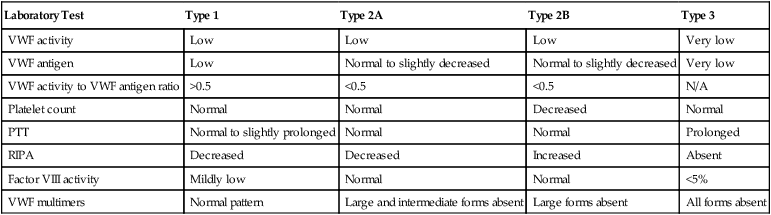
*Expected results are given, but results vary over time and within affected kindreds.
Pitfalls in the Diagnosis of von Willebrand Disease
VWF activity is influenced by varying genetic penetrance, ABO blood group, inflammation, hormones, age, and physical stress.64 Increased estrogen levels during the second and third trimesters of pregnancy nearly normalize plasma VWF activity even in women with moderate VWF deficiency. However, VWF function and concentration decrease rapidly after delivery, which may lead to acute postpartum hemorrhage, for which the obstetrician is watchful. Oral contraceptive use and hormone replacement therapy also raise VWF activity, and activity waxes and wanes with the menstrual cycle.
VWF activity rises substantially in acute inflammation such as occurs postoperatively, subsequent to trauma, or during an infection. Physical stress such as cold, exertion, or children’s crying or struggling during venipuncture causes VWF activity to rise. VWF activity rises when the tourniquet remains tied for more than 1 minute prior to venipuncture and drops if the specimen is stored in the refrigerator prior to testing. VWD patients experience fluctuation in disease severity over time, and the clinical manifestations of the disease vary from person to person within kindreds, despite the assumption that everyone in the family possesses the same mutation. When the clinical presentation suggests VWD, the VWD laboratory assay panel should be repeated until the results are conclusive.65
Adding to VWD diagnostic confusion is concern over the variability in the results of the VWF : RCo assay, which is based on platelet aggregometry but which is performed using a variety of instruments and methods, including at least one automated device, the BCS XP system with von Willebrand reagent (Siemens Healthcare Diagnostics, Inc., Deerfield, Ill.). Ristocetin avidity varies from lot to lot. In the United States, proficiency surveys consistently reveal VWF : RCo assays to have an unimpressive interlaboratory coefficient of variation of 30% and a least detectable activity range of 6% to 12%.66
Concern over the poor reproducibility of results of the VWF activity assay has led to attempts at developing alternative assays, the most promising of which is the VWF collagen binding (VWF : CB) assay. VWF : CB employs type III collagen as its solid-phase target antigen. Developed in 1990, the VWF : CB assay produces results that more closely match those of the VWF : RCo assay than of VWF : Ag assays, because collagen type III binds predominantly large VWF multimers; however, the target collagen composition needs to be standardized before this assay achieves standard screening status.67 The VWF : CB assay has better precision than the VWF : RCo assay.68
VWF activity varies by ABO blood group, and expert panels have in the past recommended that laboratory directors maintain separate reference intervals for each group, as provided in Table 41-5. The 2009 NHLBI VWD guidelines have recommended against this practice, noting that “despite the ABO blood grouping and associated reference ranges, the major determinant of bleeding risk is low VWF.“ Therefore, VWF test result reference intervals are now population based rather than ABO stratified. The internationally generalized cutoff of 50% for low VWF and 30% for VWD is clinically reasonable, although laboratory directors may choose to validate and adjust this cutoff in internal reference interval studies.
TABLE 41-5
Von Willebrand Factor (VWF) Reference Intervals by Blood Group
| Blood Group | Reference Interval |
| O | 36-157% |
| A | 48-234% |
| B | 57-241% |
| AB | 64-238% |
| Population based: “Low VWF” | <50% |
| Population based: von Willebrand disease | <30% |
Treatment of von Willebrand Disease
Mild bleeding may resolve with the use of local measures, such as limb elevation, pressure, and application of ice packs (the athlete’s acronym is RICE for rest, ice, compression, and elevation).69 Moderate bleeding may respond to estrogen and desmopressin acetate, which trigger the release of VWF from storage organelles. Therapeutic dosages are monitored when necessary using serial VWF : Ag concentration assays. Desmopressin acetate (1-desamino-8-D arginine vasopressin) is an antidiuretic hormone analogue used to control incontinence in diabetes mellitus and bedwetting; release of VWF from storage organelles is a side-effect. Desmopressin acetate in its oral form, DDAVP, or nasal spray form, Stimate (both from CSL Behring, King of Prussia, Pa.), is consistently effective for type 1 VWD and generally useful for subtype 2A. It is contraindicated for subtype 2B, however, because it causes the release of abnormal VWF with increased affinity for platelet receptors, which may intensify thrombocytopenia and lead to increased platelet activation and thrombosis. Because of its antidiuretic property, repeated doses may lead to hyponatremia (low serum sodium). For this reason, it is necessary to monitor and regulate electrolytes during desmopressin acetate therapy.
For treatment of severe VWD (type 3) and subtype 2B, three commercially prepared human plasma–derived high-purity preparations are available that provide a mixture of VWF and coagulation factor VIII. These are Humate-P, Alphanate, and Wilate (FDA-cleared December 2009).70 The calculation of the proper dosage follows principles identical to those used for treatment of hemophilia A provided in the next section. Laboratory monitoring by the VWF : Ag assay is essential to determine if the given amount produced the target level of VWF and to follow its degradation between doses.
Recombinant and affinity-purified factor VIII preparations contain no VWF and cannot be used to treat VWD. Cryoprecipitate and FP are less desirable alternatives because of the risk of virus transmission, and the necessary FP volume per dose may cause TACO. Therapy for bleeding secondary to acquired VWD follows the same principles as delineated previously plus treatment of the primary disease, if applicable. Therapeutic recommendations for VWD are summarized in Table 41-6.
TABLE 41-6
Therapeutic Strategies in von Willebrand Disease
| Type | Primary Approach | Other Options |
| 1 | Estrogen, DDAVP, EACA | Factor VIII/VWF concentrate |
| 2A | Estrogen, DDAVP, EACA | Factor VIII/VWF concentrate |
| 2B | Factor VIII/VWF concentrate | EACA |
| 2N | Factor VIII/VWF concentrate | EACA |
| 3 | Factor VIII/VWF concentrate | Platelet transfusions |
DDAVP, Desmopressin acetate; EACA, ε-aminocaproic acid; VWF, von Willebrand factor.
Hemophilia A
The hemophilias are congenital single-factor deficiencies marked by anatomic soft tissue bleeding. Second to VWD in prevalence among congenital bleeding disorders, hemophilias occur in 1 in 10,000 individuals. Of those affected, 85% are deficient in factor VIII, 14% are deficient in factor IX, and 1% are deficient in factor XI or one of the other coagulation factors, such as factor II (prothrombin), V, VII, X, or XIII. Congenital deficiency of factor VIII is called classic hemophilia or hemophilia A.71
Hemophilia A Genetics
The gene for factor VIII spans 186 kb of the X chromosome and is the site of various deletions, stop codons, and nonsense and missense mutations.72 Most of these mutations result in quantitative disorders in which the factor VIII coagulant activity and antigen concentration are in agreement, but in rare cases low activity is seen despite normal antigen concentration.73 The latter cases are due to qualitative or structural factor VIII abnormalities traditionally known as cross-reacting material positive disorders.74
Male hemizygotes, whose sole X chromosome contains a factor VIII gene mutation, experience anatomic bleeding, but female heterozygotes, who are carriers, do not. When a female carrier has children with an unaffected man the chances of hemophilia A inheritance can be described as follows: 25% chance of a normal daughter, 25% chance of a carrier daughter, 25% chance of a normal son, and 25% chance of a hemophilic son. All sons of men with hemophilia A and noncarrier women are normal, whereas all daughters are obligate carriers of the disease.75 In addition, approximately 30% of newly diagnosed cases arise as a result of spontaneous germline mutations in individuals who have no family history of hemophilia A.76 Rarely, the symptoms of hemophilia A may be seen in females. This phenomenon could be due to true homozygosity or double heterozygosity, such as in the female offspring of a hemophilic father and a carrier mother. Other possibilities include a spontaneous germline mutation in the otherwise normal allele of a heterozygous female or a disproportional inactivation of the X chromosome with the normal gene, termed extreme lyonization. Finally, VWD of the Normandy subtype may present as mild hemophilia A in males and females.
Hemophilia A Complications
As a result of frequent bleeds, hemophilic patients often have debilitating and progressive musculoskeletal lesions and deformities, and neurologic deficiencies after intracranial hemorrhage. In addition, other effects of chronic diseases, such as limited productivity, low self-esteem, poverty, drug dependency, and depression, are common problems. Before the advent of sterilized and recombinant factor concentrates, chronic hepatitis often resulted from repeated exposure to blood products. Tragically, 70% of hemophilics treated before 1984 are human immunodeficiency virus (HIV) positive or have died from acquired immunodeficiency syndrome.77
Laboratory Diagnosis of Hemophilia A
The laboratory workup for a suspected congenital single coagulation factor deficiency starts with the PT, PTT, and thrombin time and continues with factor assays based on the results of the screening tests. Before the physician initiates laboratory testing, however, a clear history of the patient’s hemorrhagic symptoms must be recorded. In hemophilia A, the PT and thrombin time are normal, and the PTT is prolonged, provided that the PTT reagent is sensitive to factor VIII deficiencies at or less than the 30% plasma level. Table 41-7 lists the expected results for each clot-based screening assay for any single-factor deficiency associated with bleeding, including deficiencies of fibrinogen and factors II (prothrombin), V, VII, VIII, IX, X, and XI. Deficiencies of contact factors (factor XII, high-molecular-weight kininogen, and prekallikrein) have no relationship to bleeding, and these factors do not appear in Table 41-7.
TABLE 41-7
Results of Clot-Based Screening Assays in Congenital Single-Factor Deficiencies*
| Deficient Factor | PT | PTT | TCT | Reflex Test |
| Fibrinogen | Prolonged | Prolonged | Prolonged | Fibrinogen assay |
| Prothrombin | Prolonged | Prolonged | Normal | Prothrombin, V, VII, and X assays |
| V | Prolonged | Prolonged | Normal | Prothrombin, V, VII, and X assays |
| VII | Prolonged | Normal | Normal | VII assay |
| VIII | Normal | Prolonged | Normal | VIII, IX, and XI assays |
| IX | Normal | Prolonged | Normal | VIII, IX, and XI assays |
| X | Prolonged | Prolonged | Normal | Prothrombin, V, VII, and X assays |
| XI | Normal | Prolonged | Normal | VIII, IX, and XI assays |
| XIII | Normal | Normal | Normal | Factor XIII quantitative assay |
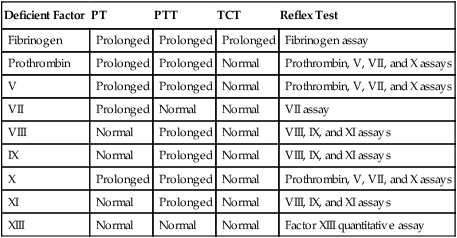
PT, Prothrombin time; PTT, partial thromboplastin time; TCT, thrombin clotting time.
Hemophilia A Treatment
With the availability of abundant recombinant factor VIII concentrate, many hemophilic patients maintain themselves on a steady prophylactic dosage designed to constantly keep their factor VIII activity at hemostatic levels.78 Although it is initially more expensive, the prophylactic approach saves downstream resources by ameliorating the adverse effects of repeated hemorrhages and their long-term consequences.79
Many hemophilic patients’ factor VIII activity rises upon administration of desmopressin acetate in the form of DDAVP or Stimate (nasal formulation), alone or in combination with an antifibrinolytic such as Amicar or Cyklokapron. When desmopressin acetate treatment proves ineffective, intravenous factor VIII concentrates are the next option. High-purity synthetic factor VIII concentrates are produced from mammalian cells using recombinant deoxyribonucleic acid (DNA) technology or from human plasma using factor VIII–specific monoclonal antibodies and column separation.80,81 The human plasma–derived concentrates Alphanate, Humate-P, and Wilate are prepared by chemical fractionation of human plasma and contain VWF, fibrinogen, and noncoagulant proteins, in addition to factor VIII. All plasma-derived concentrates undergo viral inactivation steps, and since 1985, none has transmitted lipid-envelope viruses such as HIV, hepatitis B virus, or hepatitis C virus. Plasma-derived factor VIII concentrates may transmit nonlipid viruses such as parvovirus B19 and hepatitis A virus, however.82 Recombinant products may use human albumin in the manufacturing process, which introduces the theoretical risk of Creutzfeldt-Jakob disease transmission; however, manufacturers more recently have developed products free of all human protein, such as Bioclate (Pfizer, New York, N.Y.).83
When hematologists treat a hemophilic patient, they base their factor VIII concentrate dosage calculations on the definition of a unit of factor VIII activity, which is taken as the amount present in 1 mL of normal plasma and is synonymous with 100% activity. They further calculate the desired increase after factor VIII concentrate infusion by subtracting the patient’s preinfusion factor activity from the target activity level. The desired increase is multiplied by the patient’s plasma volume to compute the dosage. The patient’s plasma volume may be estimated from his or her blood volume and hematocrit.84 The blood volume is approximately 65 mL/kg of body weight, and the plasma volume is the plasmacrit (100% − hematocrit %) as in the following formulas:
< ?xml:namespace prefix = "mml" />


Hemophilia A and Factor VIII Inhibitors
Alloantibody inhibitors of factor VIII arise in response to treatment in 30% of patients with severe hemophilia and 3% of those with moderate hemophilia. The presence of an inhibitor is suspected when bleeding persists or when the plasma factor VIII activity fails to rise to the target level after appropriate concentrate administration. Most factor VIII inhibitors are immunoglobulin G4, non–complement-fixing, warm-reacting antibodies. It is impossible to predict which patients are likely to develop inhibitors based on genetics, demographics, or the type of concentrate used.85
Treatment of Hemophilia A in Patients with Inhibitors
Every hemophilic patient with an inhibitor needs an individualized treatment plan to control bleeding episodes. Low responders often experience cessation of bleeding upon administration of large doses of factor VIII concentrate and may be so maintained. High responders may gain no benefit from factor VIII concentrates and instead are treated with Autoplex T or FEIBA, which generates thrombin in the presence of factor VIII inhibitors. FEIBA dosage should not exceed 200 units/kg per day, distributed in two to four injections, because the activated factors may trigger DIC. NovoSeven also bypasses the physiologic factor VIII requirement, because it promotes thrombin formation through the tissue factor pathway.86 The discussions in this chapter of ACOTS and acquired hemophilia provide additional detail on FEIBA and NovoSeven dosages.
Hemophilia B (Factor IX Deficiency)
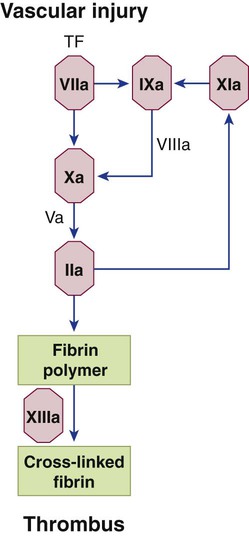
Hemophilia B, also called Christmas disease, represents approximately 14% of hemophilia cases in the United States, although its incidence in India nearly equals that of hemophilia A.87 Hemophilia B is caused by deficiency of factor IX, one of the vitamin K–dependent serine proteases. Factor IX is a substrate for both factors XIa and VIIa because it is cleaved by either to form dimeric factor IXa (Figure 41-5). Subsequently, factor IXa complexes with factor VIIIa to cleave and activate its substrate, factor X. Factor IX deficiency reduces thrombin production and causes soft tissue bleeding that is indistinguishable from that in hemophilia A. It also is a sex-linked, markedly heterogeneous disorder involving more than 220 separate mutations resulting in a range of mild to severe bleeding manifestations. Determination of carrier status is less successful in hemophilia B than in hemophilia A because of the large number of factor IX mutations and the lack of a linked molecule such as VWF that can be used as a normalization index. DNA analysis occasionally may be used to establish carrier status when hemophilia B has been diagnosed and its specific mutation identified in a relative.
Hemophilia C (Rosenthal Syndrome, Factor XI Deficiency)
Factor XI deficiency is an autosomal dominant hemophilia with mild to moderate bleeding symptoms. More than half of the cases have been described in Ashkenazi Jews, but individuals of any ethnic group may be affected. The frequency and severity of bleeding episodes do not correlate with factor XI levels, and laboratory monitoring of treatment serves little purpose after the diagnosis is established. The physician treats hemophilia C with frequent infusions of FP during times of hemostatic challenge.88 In the laboratory, the PTT is prolonged and the PT is normal.
Other Congenital Single-Factor Deficiencies
The remaining congenital single-factor deficiencies listed in Table 41-8 are rare, are caused by autosomal recessive mutations, and are often associated with consanguinity. The PT, PTT, and thrombin time may be employed to distinguish among these disorders, as shown in Table 41-7. In addition, immunoassays may be performed to distinguish among the more prevalent quantitative and the less prevalent qualitative abnormalities. In qualitative disorders, often called dysproteinemias, the ratio of factor activity to antigen is less than 0.7. The bleeding symptoms in the dysproteinemias may be more severe than in quantitative deficiencies, but the risk of inhibitor formation is theoretically lower. The clot-based measurement of factors II (prothrombin) and X may be supplemented or replaced by more reproducible chromogenic substrate assays. Fibrinogen is usually measured using the Clauss assay, a modification of the thrombin time, but may be measured by turbidimetry or immunoassay.
TABLE 41-8
Rare Congenital Single-Factor Deficiencies
| Deficiency | Factor Levels | Symptoms | Therapy |
| Afibrinogenemia | No measurable fibrinogen | Severe anatomic bleeding | CRYO to raise to >100 mg/dL |
| Hypofibrinogenemia | Fibrinogen activity assay <100 mg/dL | Moderate systemic bleeding | CRYO to raise to >100 mg/dL |
| Dysfibrinogenemia | Fibrinogen activity assay <100 mg/dL | Mild systemic bleeding | CRYO to raise to >100 mg/dL |
| Prothrombin deficiency | Factor II <30% | Mild systemic bleeding | PCC or plasma to raise to 75% |
| Factor V deficiency | Factor V <30% | Mild systemic bleeding | Plasma to raise to 75% |
| Factor VII deficiency | Factor VII <30% | Moderate to severe anatomic bleeding | Activated factor VII, plasma |
| Factor X deficiency | Factor X <30% | Severe anatomic bleeding | PCC or plasma to raise to 75% |
| Factor XI deficiency | Factor XI <50% | Anatomic bleeding | Plasma to raise to 75% |
| Factor XIII deficiency | Factor XIII <1% | Moderate to severe systemic bleeding, poor wound healing | Plasma or CRYO every 3 wk |
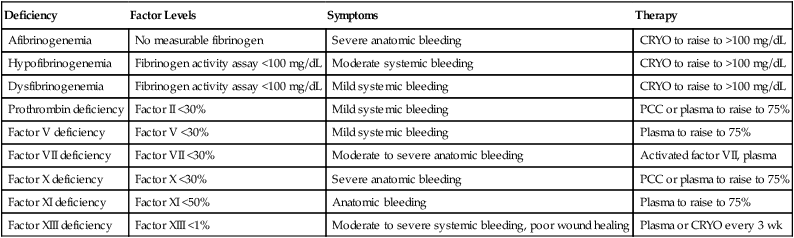
CRYO, Cryoprecipitate; PCC, prothrombin complex concentrate.
Because platelets transport about 20% of circulating factor V, the platelet function in factor V deficiency may be diminished, which is reflected in a prolonged bleeding time but normal platelet aggregation.89 The PT and PTT are prolonged. Because of the concentration of factor V in platelet α-granules, normal platelet concentrate is an effective form of therapy for factor V deficiency. A combined factor V and VIII deficiency may be caused by a genetic defect traced to chromosome 18 that affects transport of both factors by a common protein in the Golgi apparatus.90
Factor VII deficiency causes moderate to severe anatomic hemorrhage. The bleeding does not necessarily reflect the factor VII activity level. The half-life of factor VII is approximately 6 hours, which affects the frequency of therapy. NovoSeven at 30 mcg/mL and prothrombin complex concentrate such as Autoplex T preparations are effective and may provide a target level of 10% to 30%. Many factor VII deficiencies are dysproteinemias.91 The PT, but not the PTT, is prolonged in factor VII deficiency.
Factor X deficiency causes moderate to severe anatomic hemorrhage that may be treated with FP or prothrombin complex concentrate to produce therapeutic levels of 10% to 40%.92 The half-life of factor X is 24 to 40 hours. Acquired factor X deficiency has been described in amyloidosis, in paraproteinemia, and in association with antifungal drug therapy. The hemorrhagic symptoms are severe and life-threatening. The PT and PTT are both prolonged in factor X deficiency. In the time-honored Russell viper venom time test, which activates the coagulation mechanism at the level of factor X, clotting time is prolonged in deficiencies of factors X and V, prothrombin, and fibrinogen. The venom used is harvested from the Russell viper, the most dangerous snake in Asia. This test may be useful in distinguishing a factor VII deficiency, which does not affect the results, from deficiencies in the common pathway, although specific factor assays are the standard approach.
Plasma factor XIII is a tetramer of paired α and β monomers. The intracellular form is a homodimer (two α chains) and is stored in platelets, monocytes, placenta, prostate, and uterus. The α chain contains the active enzyme site, and the β chain is a binding and stabilizing portion. Factor XIII deficiency occurs in three forms related to the affected chain, as shown in Table 41-9. Patients with factor XIII deficiency have a normal PT, PTT, and thrombin time despite anatomic bleeds and poor wound healing. They form weak (non–cross-linked) clots that dissolve within 2 hours when suspended in a 5-M urea solution, a traditional factor XIII screening assay.93 To confirm factor XIII deficiency, factor activity may be measured accurately using a chromogenic substrate assay such as the Behring Behrichrom FXIII assay (Behring Diagnostics, King of Prussia, Pa.).
TABLE 41-9
| Type of Deficiency | Incidence | Factor XIII Activity | β-Protein | α-Protein |
| Type I | Rare | Absent | Absent | Absent |
| Type II | Frequent | Absent | Normal | Low |
| Type III | Rare | Low | Absent | Low |
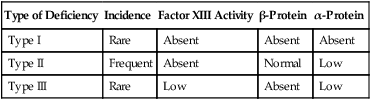
Summary
• Hemorrhage can be classified as localized versus generalized, acquired versus congenital, and anatomic soft tissue versus mucocutaneous.
• Acquired hemorrhagic disorders that are diagnosed in the hemostasis laboratory include thrombocytopenia of various etiologies, ACOTS, liver and renal disease, vitamin K deficiency, scurvy, acquired hemophilia or VWD, and DIC.
• VWD is the most common congenital bleeding disorder, and the diagnosis and classification of the type and subtypes require a series of clinical laboratory assays.
• The hemophilias are congenital single-factor deficiencies that cause moderate to severe anatomic hemorrhage. The clinical laboratory plays a key role in diagnosis, classification, and treatment monitoring in hemophilia.
Review Questions
1. What is the most common acquired bleeding disorder?
2. Which is a typical form of anatomic bleeding?
3. To a deficiency of which factor is the PT most sensitive?
4. Which of the following conditions causes a prolonged thrombin time?
5. In what subtype of VWD is the RIPA test result positive when ristocetin is used at a concentration of less than 0.5 mg/mL?
6. What is the typical treatment for vitamin K deficiency when the patient is bleeding?
7. If a patient has anatomic soft tissue bleeding and poor wound healing, but the PT, PTT, thrombin time, platelet count, and platelet functional assay results are normal, a deficiency of what factor could exist?
8. What therapy may be used for a hemophilic boy who is bleeding and who has a high titer of factor VIII inhibitor?
9. What is the most prevalent form of VWD?
10. Which of the following assays can be used to distinguish vitamin K deficiency from liver disease?
*The author acknowledges the substantial contributions of Marisa B. Marques, MD, to chapter 41 of the third edition of this textbook, many of which continue in this edition.
[/level-membership-for-hematology-oncology-and-palliative-medicine-category][not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
Hemorrhagic Coagulation Disorders
After completion of this chapter, the reader will be able to:
1. Distinguish among the causes of localized versus generalized, soft tissue versus mucocutaneous, and acquired versus congenital bleeding.
2. List laboratory tests to differentiate among acquired hemorrhagic disorders of trauma, liver disease, vitamin K deficiency, and kidney failure, and interpret results when given.
3. Interpret laboratory assay results to diagnose and subtype von Willebrand disease.
4. Use the results of laboratory tests to identify and monitor the treatment of congenital single coagulation factor deficiencies such as deficiencies of factors VIII, IX, and XI.
5. Explain the principle and rationale for the use of each laboratory test for the detection and monitoring of hemorrhagic disorders.
6. Discuss the laboratory monitoring of therapy for hemorrhagic disorders.
Symptoms of Coagulopathies
Hemorrhage is a severe form of bleeding that requires intervention. Hemorrhage may be localized or generalized, acquired or congenital. To establish the cause of either a bleeding event or a patient’s tendency to bleed, the physician must obtain a complete patient and family history and do a thorough physical examination before ordering diagnostic laboratory tests.1
Mucocutaneous versus Anatomic Hemorrhage
Generalized bleeding may exhibit a mucocutaneous or a soft tissue (anatomic) pattern.
Mucocutaneous hemorrhage may be manifested as bruises or purpura, purple lesions of the skin caused by extravasated (seeping) red blood cells (RBCs).2 Petechiae and ecchymoses are purpura of less than or greater than 3 mm, respectively, and the presence of more than one such lesion may indicate a disorder of primary hemostasis. Other symptoms of defects of primary hemostasis are menorrhagia (profuse menstrual flow), bleeding from the gums, hematemesis (vomiting of blood), and epistaxis (uncontrolled nosebleed). Although nosebleeds are common and mostly innocent, especially in children, they suggest a primary hemostatic defect when they occur repeatedly, last longer than 10 minutes, involve both nostrils, or require physical intervention or transfusions.
Mucocutaneous hemorrhage tends to be associated with thrombocytopenia (platelet count lower than 150,000/mcL), qualitative platelet disorders, von Willebrand disease (VWD), or vascular disorders such as scurvy or telangiectasia (see Chapters 43 and 44). A careful history and physical examination distinguishes between mucocutaneous and anatomic bleeding, and the distinction helps direct investigative laboratory testing and treatment.
Anatomic (soft tissue) hemorrhage is seen in acquired or congenital defects in secondary hemostasis, or plasma coagulation factor deficiencies. Examples of anatomic bleeding include recurrent or excessive bleeding after minor trauma, dental extraction, or a surgical procedure. In such cases, hemorrhage may immediately follow a traumatic event, but is often delayed or recurs minutes or hours after the initial blood flow is stopped. In other patients, anatomic bleeding episodes are spontaneous. Most anatomic bleeds are internal, such as bleeds into joints, body cavities, muscles, or the central nervous system, and may initially have few visible signs. Because joint bleeds cause swelling and acute pain, they may not be immediately recognized as hemorrhages, although hemophilia patients usually know what is happening from experience. Repeated joint bleeds (hemarthroses) cause painful inflammation, with swelling and permanent cartilage damage that immobilize the joint. Repeated joint bleeds cause painful inflammation and permanent cartilage damage that swells and immobilizes the joint. Bleeds into soft tissues such as muscle or fat may cause nerve compression and subsequent temporary or permanent loss of function.3 When the bleeding involves body cavities, it causes symptoms related to the organ that is affected. Bleeding into the central nervous system, for instance, may cause headaches, confusion, seizures, and coma and must be managed as a medical emergency. Bleeds into the kidney may present as hematuria and may potentially be associated with acute kidney failure.
Whenever a soft tissue or mucocutaneous generalized hemostatic disorder is suspected, hemostasis laboratory testing is essential. Emergency department physicians often must begin to intervene in cases of acute hemorrhage, even before taking a history or waiting for the results of laboratory assays. In these instances, group-matched or O-negative RBCs, frozen plasma (FP), platelet concentrate, cryoprecipitate, recombinant activated coagulation factor VII (rFVIIa; NovoSeven, Novo Nordisk, Inc., Princeton, N.J.) or the antifibrinolytic tranexamic acid (Cyklokapron, Pfizer, New York, N.Y.) may be used to correct coagulation factor and platelet deficiencies to stop hemorrhage.4–6 Box 41-1 lists symptoms that suggest generalized hemorrhagic disorders.
Acquired versus Congenital Bleeding Disorders
Liver disease, kidney failure, chronic infections, autoimmune disorders, obstetric complications, dietary deficiencies such as vitamin C or vitamin K deficiency, blunt or penetrating trauma, and inflammatory disorders may all be associated with coagulopathy. If a patient’s bleeding episodes began after infancy, are associated with some disease or physical trauma, and are not duplicated in relatives, they probably are caused by an acquired, not a congenital, condition. When an adult patient comes for treatment of generalized hemorrhage, the physician first looks for an underlying disease or event and takes a personal and family history. The important elements of patient history are age, sex, current or past pregnancy, any systemic disorders such as diabetes or cancer, trauma, and exposure to drugs, including over the counter drugs or drugs of abuse. The physician determines the trigger, location, and volume of bleeding, then orders laboratory assays (Table 41-1). The initial coagulopathy test profile should consist of a complete blood count (CBC) that includes a platelet count, prothrombin time (PT), partial thromboplastin time (PTT), and fibrinogen assay.7 These tests take on clinical significance when the history and physical examination already have established the existence of abnormal bleeding. They are not effective as screens for healthy individuals.
TABLE 41-1
Screening Tests for a Generalized Hemostatic Disorder
| Test | Assesses for |
| Hemoglobin, hematocrit, reticulocyte count | Anemia associated with chronic bleeding; bone marrow response |
| Platelet count | Thrombocytopenia |
| PT | Deficiencies of factors II (prothrombin), V, VII, or X (clotting time prolonged) |
| PTT | Deficiencies of all factors except VII and XIII (clotting time prolonged) |
| Thrombin time | Hypofibrinogenemia and dysfibrinogenemia |
Congenital hemorrhagic disorders are uncommon, occurring in 1 per 100 people, and are usually diagnosed in infants or young children, who often have relatives with similar symptoms. Congenital bleeding disorders lead to repeated hemorrhages that may be spontaneous or occur following minor injury or may occur in unexpected locations, such as joints, body cavities, retinal veins and arteries, or the central nervous system. Patients with mild congenital hemorrhagic disorders may have no symptoms until they reach adulthood or experience some physical challenge, such as trauma, dental extraction, or a surgical procedure. The most common congenital deficiencies are VWD, factor VIII and IX deficiencies (hemophilia A and B), and platelet function disorders. Inherited deficiencies of fibrinogen, prothrombin, and factors V, VII, X, XI, and XIII also exist, but are rare (Box 41-2).
Acquired Coagulopathies
Far more patients have acquired bleeding disorders secondary to trauma, drug exposure, or chronic disease than have congenital or inherited coagulopathies. Chronic diseases or disorders commonly associated with bleeding are liver disease, vitamin K deficiency, and renal failure. In all cases, laboratory test results are necessary to confirm the diagnosis and guide the management of acquired hemorrhagic disorders.8
Acute Coagulopathy of Trauma-Shock
In North America, unintentional injury is the leading cause of death among those aged 1 to 45 years. The total rises when felonious, self-inflicted, and combat injuries are included: in the United States alone, trauma causes 93,000 deaths per year.9 Severe neurologic displacement accounts for 50% of deaths, with most occurring before the patient arrives at the hospital; however, of initial survivors, 20,000 die of hemorrhage within 48 hours.10 Acute coagulopathy of trauma-shock (ACOTS) accounts for fatal hemorrhage; 3000 to 4000 of these deaths are preventable. ACOTS is triggered by the combination of injury-related acute inflammation, platelet activation, tissue factor release, hypothermia, acidosis, and hypoperfusion (poor distribution of blood to tissues caused by low blood pressure), all of which are elements of systemic shock. For in-transit fluid resuscitation, colloid plasma expanders such as 5% dextrose are used to counter hypoperfusion.11 The administration of plasma expanders has a dilutional effect that is compounded by emergency department transfusion of RBCs, and although these measures are life saving, they intensify the coagulopathy, as does subsequent surgical intervention.
Massive transfusion, defined as administration of 4 to 5 RBC units within 1 hour or 8 to 10 units within 24 hours, is essential in otherwise healthy trauma victims when the systolic blood pressure is less than 110 mm Hg, pulse is greater than 105 beats/min, pH is less than 7.25, hematocrit is less than 32%, hemoglobin is less than 10 g/dL, and PT is prolonged to greater than 1.5 times the mean of the reference interval or generates an international normalized ratio (INR) of 1.5.12
Management of Acute Coagulopathy of Trauma-Shock
According to the American Society of Anesthesiologists surgical practice guidelines, RBC transfusions are required when the hemoglobin is less than 6.0 g/dL and are contraindicated when the hemoglobin is greater than 10.0 g/dL.13 For hemoglobin concentrations between 6.0 and 10.0 g/dL, the decision to transfuse is based upon the acuity of the patient’s condition as determined by physical evidence of blood loss; current blood loss rate; blood pressure; arterial blood gas values, especially pH and oxygen saturation; urine output; and laboratory evidence for coagulopathy, provided there is time for specimens to be collected and laboratory assays completed.
During surgery, coagulopathy is assessed based on microvascular bleeding, which is evaluated by measuring the volume of blood filling suction canisters, surgical sponges, and surgical drains. Platelet concentrate is ordered when the platelet count is less than 50,000/mcL unless the surgeon anticipates limited blood loss. Platelet concentrate therapy is generally ineffective when the patient is known to have immune thrombocytopenic purpura, thrombotic thrombocytopenic purpura, or heparin-induced thrombocytopenia. In patients with these conditions, therapeutic platelets are rapidly consumed, and their administration may therefore be contraindicated, although they may provide temporary rescue. Platelet concentrate is never ordered when the platelet count is greater than 100,000/mcL. Platelet administration may be necessary when the platelet count is between 50,000 and 100,000/mcL provided there is bleeding into a confined space such as the brain or eye; if the patient is taking aspirin or clopidogrel; if there is a known platelet disorder such as a release defect, Glanzmann thrombasthenia, or Bernard-Soulier syndrome (see Chapter 43); or if the surgery involves cardiopulmonary bypass, which suppresses platelet activity.
Use of Frozen Plasma in Acute Coagulopathy of Trauma-Shock
Although many refer to frozen plasma as “fresh frozen plasma,“ this term no longer applies, because most FP is prepared up to 24 hours after the time of collection rather than within 8 hours, as was the traditional practice.14 High-volume trauma centers such as combat facilities may maintain a small inventory of thawed FP ready for administration. Thawed FP may be stored at 1° C to 6° C for up to 5 days subsequent to thawing. The activity of von Willebrand factor (VWF) and coagulation factors V and VIII decline to approximately 60% after 5 days of refrigerator storage; however, the product may still be transfused.15
Most trauma center transfusion service directors require surgeons and emergency department physicians to transfuse 1 unit of FP for every 4 units of RBCs, although there is battlefield evidence that a ratio of 1 unit of FP per single unit of RBCs may lead to more favorable mortality statistics.16 The latter approach places a strain on the FP supply, however.
Use of Activated Prothrombin Complex Concentrate, Recombinant Activated Factor VII, and Tranexamic Acid in Acute Coagulopathy of Trauma-Shock
When administration of RBCs, platelets, cryoprecipitate, and FP fails to achieve hemostasis, activated prothrombin complex concentrate (FEIBA [factor eight inhibitor bypassing activity], Baxter Healthcare Corp., Deerfield, Ill.; Autoplex T, Nabi Biopharmaceuticals, Inc., Boca Raton, Fla.), recombinant activated coagulation factor VII (NovoSeven), or the antifibrinolytic drug tranexamic acid (Cyklokapron) may be employed. Autoplex T or FEIBA may be used at a dosage of 50 units/kg every 12 hours not to exceed 200 units/kg in 24 hours. The dose-response relationship of FEIBA is variable, and because FEIBA contains activated coagulation factors it raises the risk of disseminated intravascular coagulation (DIC). NovoSeven has been cleared by the Food and Drug Administration (FDA) for use in treating hemophilia A or B when factor VIII or factor IX inhibitors are present, respectively; its application in the treatment of ACOTS is off label. A NovoSeven dosage of 30 mcg/kg is instantly effective in stopping microvascular hemorrhage in nonhemophilic trauma victims, and NovoSeven does not cause DIC.17,18 However, one study found a possible link between off-label NovoSeven use and arterial and venous thrombosis in patients with existing thrombotic risk factors.19 Cyklokapron may be used at a loading dose of 1 g administered over 10 minutes followed by infusion of 1 g over 8 hours. First approved in 1986 to prevent bleeding in hemophilic patients who are about to undergo invasive procedures, Cyklokapron is effective for ACOTS, but this is an off-label use.
Liver Disease
Procoagulant Deficiency in Liver Disease
Liver disease predominantly alters the production of the vitamin K–dependent factors II (prothrombin), VII, IX, and X and control proteins C, S, and Z. In liver disease, these seven factors are produced in their des-γ-carboxyl forms, which cannot participate in coagulation (see Chapter 40). At the onset of liver disease, factor VII, which has the shortest plasma half-life at 6 hours, is the first coagulation factor to exhibit decreased activity. Because the PT is particularly sensitive to factor VII activity, it is characteristically prolonged in mild liver disease, serving as a sensitive early marker.20 Vitamin K deficiency caused by dietary insufficiency independent of liver disease produces a similar effect on the PT (see the case study in this chapter).
Declining coagulation factor V activity is a more specific marker of liver disease than deficient factor II, VII, IX or X because factor V is non–vitamin K dependent and is not affected by dietary vitamin K deficiency. The factor V activity assay, performed in conjunction with the factor VII assay, may be used to distinguish liver disease from vitamin K deficiency.21
Fibrinogen is an acute phase reactant that is frequently elevated in early or mild liver disease. Moderately and severely diseased liver produces fibrinogen that is coated with excessive sialic acid, a condition called dysfibrinogenemia in which the fibrinogen functions poorly. Dysfibrinogenemia causes generalized soft tissue bleeding associated with a prolonged thrombin time and an exceptionally prolonged reptilase clotting time.22 In end-stage liver disease, the fibrinogen level may fall to less than 100 mg/dL, which is a mark of liver failure.23
VWF and factors VIII and XIII are acute phase reactants that may be unaffected or elevated in mild to moderate liver disease.24,25 In contrast to the other coagulation factors, VWF is produced from endothelial cells and megakaryocytes and is stored in endothelial cells and platelets.
Platelet Abnormalities in Liver Disease
Moderate thrombocytopenia occurs in a third of patients with liver disease. Platelet counts of less than 150,000/mcL may result from sequestration and shortened platelet survival associated with portal hypertension and resultant hepatosplenomegaly. In alcoholism-related hepatic cirrhosis, acute alcohol toxicity also suppresses platelet production. Platelet aggregation and secretion properties are often suppressed; this is reflected in reduced platelet aggregometry and lumiaggregometry results. Occasionally platelets are hyperreactive. Aggregometry may be used to predict bleeding and thrombosis risk.26
Disseminated Intravascular Coagulation in Liver Disease
Chronic or compensated DIC (see Chapter 42) is a significant complication of liver disease that is caused by decreased liver production of regulatory antithrombin, protein C, or protein S and by the release of activated procoagulants from degenerating liver cells. In addition, the failing liver cannot clear activated coagulation factors that are regularly produced by abdominal organs and transported through the portal circulation. In primary or metastatic liver cancer, hepatocytes also may produce procoagulant substances that trigger chronic DIC, leading to ischemic complications.
In acute, uncompensated DIC, the PT, PTT, and thrombin time are prolonged; the fibrinogen level is reduced to less than 100 mg/dL; and fibrin degradation products, including D-dimers, are significantly increased.27 If the DIC is chronic and compensated, the only elevated test result may be the D-dimer assay value, a hallmark of unregulated coagulation and fibrinolysis.28 Although DIC can be resolved only by removing its underlying cause, its hemostatic deficiencies may be temporarily corrected by administering RBCs, FP, platelet concentrates, activated protein C, or antithrombin concentrates.29,30
Hemostasis Laboratory Tests in Liver Disease
The PT, PTT, thrombin time, fibrinogen concentration, platelet count, and D-dimer concentration are useful in characterizing the hemostatic abnormalities in liver disease (Table 41-2). Factor V and VII assays may be used in combination to differentiate liver disease from vitamin K deficiency. Both factors are decreased in liver disease, but only factor VII is decreased in vitamin K deficiency.
TABLE 41-2
Hemostasis Laboratory Tests in Liver Disease
| Assay | Interpretation |
| Fibrinogen | >400 mg/dL in early, mild liver disease; <200 mg/dL in moderate to severe liver disease causing hypofibrinogenemia or dysfibrinogenemia |
| Thrombin time | Prolonged in dysfibrinogenemia, hypofibrinogenemia, elevation of fibrin degradation products, and therapy with unfractionated heparin |
| Reptilase time | Prolonged in hypofibrinogenemia, significantly prolonged in dysfibrinogenemia; assay rarely used |
| PT | Prolonged even in mild liver disease due to presence of des-γ-carboxyl factors in place of normal factors II (prothrombin), VII, IX, and X. Report PT in seconds, not INR, when testing for liver disease. |
| PTT | Mildly prolonged in severe liver disease due to DIC or des-γ-carboxyl factors II (prothrombin), IX, and X |
| Platelet count | Mild thrombocytopenia, platelet count <150,000/mcL |
| Platelet aggregometry | Mild suppression of platelet aggregation and secretion to most agonists |
| D-dimer | >240 ng/mL by quantitative assay |
| Euglobulin clot lysis time | Lysis in <2 hr in primary or secondary systemic fibrinolysis; assay rarely used |
Renal Failure and Hemorrhage
Acute renal failure may be associated with acute gastrointestinal bleeding caused by specific anatomic lesions, but this does not necessarily indicate that a coagulopathy or platelet dysfunction is present.31 Chronic renal failure of any cause is commonly associated with platelet dysfunction and mild to moderate mucocutaneous bleeding.32 Platelet adhesion to blood vessels and platelet aggregation are suppressed, perhaps because the platelets are coated by guanidinosuccinic acid or dialyzable phenolic compounds.33 Decreased RBC mass (anemia) and thrombocytopenia contribute to the bleeding and may be corrected with dialysis, erythropoietin or RBC transfusions, and interleukin-11 therapy.34,35
Hemostasis activation syndromes that deposit fibrin in the renal microvasculature reduce the function of the glomeruli. Examples of such disorders are DIC, hemolytic uremic syndrome, and thrombotic thrombocytopenic purpura. Although these are by definition thrombotic disorders, they invariably cause thrombocytopenia, which may lead to mucocutaneous bleeding. Fibrin also may be deposited in renal transplant rejection and in the glomerulonephritis syndrome of systemic lupus erythematosus. This may be associated with a rise in quantitative plasma D-dimer, thrombin-antithrombin complexes, or prothrombin fragment 1 + 2, which are markers of coagulation activation (see Chapter 45).36
Laboratory tests for bleeding in renal disease provide only modest information with little predictive or management value. The bleeding time may be prolonged, but is too unreliable to provide an accurate diagnosis or to assist in monitoring treatment.37 Platelet aggregometry test results may predict bleeding. The PT and PTT are expected to be normal.
Management of renal failure–related bleeding typically focuses on the severity of the hemorrhage without reliance on laboratory test results. Renal dialysis improves platelet function, particularly when anemia is well controlled. Desmopressin acetate may be administered intravenously (DDAVP) or intranasally (Stimate) to increase the plasma concentration of high-molecular-weight multimers of VWF, which also aids platelet adhesion and aggregation.38 Patients with renal failure should not take aspirin, clopidogrel, or other platelet inhibitors, because these drugs increase the risk of hemorrhage.
Nephrotic Syndrome and Hemorrhage
Nephrotic syndrome is a state of increased glomerular permeability associated with a variety of conditions, such as chronic glomerulonephritis, diabetic glomerulosclerosis, systemic lupus erythematosus, amyloidosis, and renal vein thrombosis.39 In nephrotic syndrome, low-molecular-weight proteins are lost through the glomerulus into the glomerular filtrate and the urine. Coagulation factors II (prothrombin), VII, IX, X and XII have been detected in the urine, as have the coagulation regulatory proteins antithrombin and protein C. In 25% of cases, loss of regulatory proteins takes precedence over loss of procoagulants and leads to a tendency toward venous thrombosis.40
Vitamin K Deficiency
Hemorrhagic Disease of the Newborn Caused by Vitamin K Deficiency
Because of their sterile intestines and the minimal concentration of vitamin K in human milk, newborns are constitutionally vitamin K deficient.41 Hemorrhagic disease of the newborn was common in the United States before routine administration of vitamin K to infants was legislated in the 1960s, and it still occurs in developing countries. The activity levels of factors II (prothrombin), VII, IX, and X are lower in normal newborns than in adults, and premature infants have even lower concentrations of these factors.42 Breastfeeding prolongs the deficiency, because passively acquired maternal antibodies delay the establishment of gut flora.
Vitamin K Antagonists
The γ-carboxylation cycle of coagulation factors is interrupted by coumarin-type oral anticoagulants such as warfarin that disrupt the vitamin K epoxide reductase and vitamin K quinone reductase reactions (Figure 41-1). In deficient γ-carboxylation, the liver releases dysfunctional des-γ-carboxyl factors II (prothrombin), VII, IX, and X, and proteins C, S, and Z; these inactive forms are called proteins in vitamin K antagonism (PIVKA). Therapeutic overdose or the accidental or felonious administration of warfarin-containing rat poisons may result in moderate to severe hemorrhage because of the lack of functional factors. The effect of brodifacoum or “superwarfarin,“ often used as a rodenticide, lasts for weeks to months, and treatment of poisoning with this substance requires repeated administration of vitamin K with follow-up PT monitoring.43 Warfarin overdose is the single most common reason for hemorrhage-associated emergency department visits.
Autoanti-VIII Inhibitor and Acquired Hemophilia
Acquired autoantibodies that specifically inhibit factors II (prothrombin), V, VIII, IX, and XIII and VWF have been described in nonhemophilic patients.44 Anti-VIII is the most common. Patients who develop an autoantibody to factor VIII, which is diagnostic of acquired hemophilia, are frequently older than age 60 and have no apparent underlying disease. Acquired hemophilia is occasionally associated with rheumatoid arthritis, inflammatory bowel disease, systemic lupus erythematosus, or lymphoproliferative disease. It also may occur in women of childbearing age 2 to 5 months after delivery. Patients with inhibitor autoantibodies are given immunosuppressive therapy, although autoantibodies that develop after pregnancy typically disappear spontaneously. Acquired hemophilia has an overall incidence of 1 per 1 million people per year. Patients experience sudden and severe bleeding in soft tissues or bleeding in the gastrointestinal or genitourinary tract. Acquired hemophilia, even when treated, remains fatal in at least 20% of cases. Autoantibodies to other procoagulants are less frequent, but create similar symptoms.45
Clot-Based Studies in Acquired Hemophilia
The in vitro kinetics of factor VIII neutralization by inhibitor are nonlinear. Although there is early rapid loss of factor VIII activity, residual activity remains, which indicates that the reaction has reached equilibrium. This is called type II kinetics (Figure 41-2). In contrast, alloantibodies to factor VIII, which develop in 20% to 25% of patients with severe hemophilia in response to factor VIII therapy, exhibit type I kinetics. In the latter, there is linear in vitro neutralization of factor VIII activity over 1 to 2 hours, which results in complete inactivation. In type I kinetics in vitro measurement is relatively accurate, whereas in type II kinetics the titration of inhibitor activity is semiquantitative.46
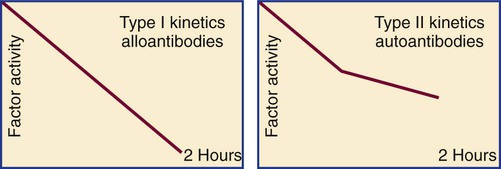
[/not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
