Chapter 60 Hemophilia A and B
Table 60-1 Factor VIII Mutant Genotype and Inhibitor Risk in Previously Untreated Hemophilia A Patients
| Multidomain deletions | ≈75% |
| Light chain nonsense mutns | 30%-40% |
| Intron 22 inversion | 20%-25% |
| Single domain deletions | 15%-25% |
| Small non-A run insertions/deltns | 15%-20% |
| Heavy chain nonsense mutns | 10%-20% |
| Factor VIII missense mutns | <10% |
| Small A run insertions/deltns | <5% |
| Splicing mutns | <5% |
Table 60-2 Methods of Factor VIII Measurement
 |
Hemophilia Carrier Detection and Prenatal Diagnosis
Table 60-3 Differential Diagnosis of a Low Factor VIII Level
FV, Factor V; FVIII, factor VIII; vWD, von Willebrand disease.
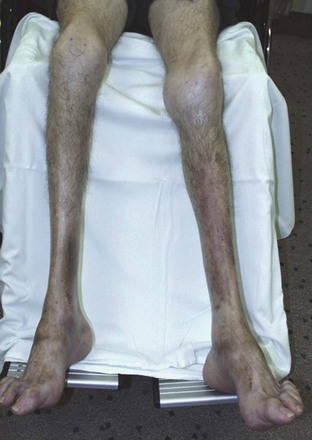
Compartment Syndrome
| Scores | Reference | |
|---|---|---|
| Clinical | World Federation of Hemophilia Joint score (Gilbert score) | Gilbert, Semin Hematol, 1993 |
| The modified WFH joint score Colorado PE-1 and PE-0.5 | Manco-Johnson et al, Haemophilia, 2000 | |
| HJHS (Hemophilia Joint Health Score) | Feldman et al, Arthritis Care Res, 2011 | |
| Plain radiography | Arnold-Hilgartner | Arnold and Hilgartner 1977 |
| Pettersson | Petterson et al, Acta Paediatrica, 1981 | |
| MRI | Progressive Denver scoring system | Nuss et al, Haemophilia, 2000 |
| Additive European scoring system | Lundin et al, Haemophilia, 2004 | |
| Single compatible IPSG MRI scoring system | Doria et al, Haemophilia, 2008 |
IPSG, International Prophylaxis Study Group; MRI, magnetic resonance imaging; WFH, World Federation of Hemophilia.
Home Care
Table 60-5 Recommendations for Clotting Factor Replacement
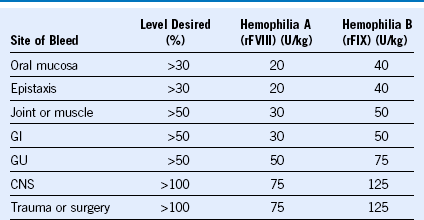
CNS, Central nervous system; GI, gastrointestinal; GU, genitourinary; rFIX, recombinant factor IX; rFVIII, recombinant factor VIII.