[level-membership-for-opthalmology-category]
Chapter 57 Hemoglobinopathies
In a landmark 1910 Archives of Internal Medicine article,1 Dr James Herrick noted “peculiar, elongated, sickle-shaped red blood corpuscles” from a peripheral blood smear of a Grenadian dental student with recurring medical illnesses and, thus, penned the first written description of sickle-cell disease (SCD).1–3 Linus Pauling and colleagues in 1949 were the first to implicate a defective hemoglobin molecule within the erythrocyte as the cause of the disease.4,5
The term “sickle-cell disease” encompasses the group of hemoglobinopathies characterized by intravascular hemolysis and by defective oxygen transport. In a normal red blood cell, two α-globin subunits, two β-globin subunits, and a central heme molecule combine to form adult hemoglobin (Hb A). The β-globin gene, an oxygen transport gene, is found on the short arm of chromosome 11.6,7 Hemoglobin S (Hb S) results from a single amino acid point mutation in which a valine substitutes for a glutamic acid at the sixth position within the β-globin chain. Hemoglobin C (Hb C) is caused by a glutamic acid to lysine mutation in the β-globin molecule.
SCD is transmitted through the autosomal recessive mode of inheritance. Two copies of Hb S may combine with one another (SS disease), or one copy of Hb S and another β-globin variant, such as Hb C, may combine (double heterozygous SC disease).7 Individuals with one copy of Hb A and one copy of Hb S are described as having the sickle-cell trait, the carrier state for SCD. β-thalassemia occurs when a reduced amount of β-globin is present. This condition is beta-plus (β+) thalassemia, whereas the absence of β-globin is called beta-zero (β0). Either of these conditions may combine with Hb S, leading to a compound heterozygous state.7
Prevalence
To date, SCD remains the most common inherited blood disorder, and affects nearly 80 000 people of African American and Hispanic descent.8 SCD occurs in 1/500 African American births and every 1/36 000 Hispanic-American births.8–10 SCD results in 5% of deaths in children under 5 years in Africa, more than 9% of such deaths in West Africa, and up to 16% of under-5 deaths in individual West African countries.11 Approximately 8% (or one in 12) of black Americans possess sickle-cell trait, which is not associated with increased mortality or morbidity (except in hyphema), and is thought to confer a genetic protection against malarial infection.10 Their hemoglobin concentration is typically 35–40% Hb S and 55–60% Hb A.7 Although sickle-cell trait subjects are generally asymptomatic, they may encounter systemic complications of SCD under conditions of extreme hypoxia.12 Populations of African descent possess the highest frequency of Hb S. At-risk genotypes for SCD are also observed in people of Mediterranean, Caribbean, South and Central American, Arab, and East Indian descent.7 SS disease is displayed in approximately 0.15% of African descendants in North America.7,13 Those with SS disease do not typically become symptomatic until after 6 months of age, when the fetal hemoglobin (Hb F) is replaced with Hb S. These individuals have a decreased life expectancy, as they are prone to developing severe anemia and are highly susceptible to recurrent infections.14 Individuals with Hb SC disease, the double heterozygotes, typically exhibit 50% Hb S and 50% Hb C hemoglobin composition and typically display less morbidity from the systemic complications of SCD, and have a normal life expectancy.7 With a hemoglobin composition of 60–90% Hb S and 10–30% Hb F, sickle β-thalassemia patients are typically encountered in central African and Mediterranean countries. Those with other forms of sickle β-thalassemia may have Hb A as 10–30% of their hemoglobin composition.7,15
Genetic modifiers
The degree of phenotypic expression of SCD is quite variable, even among those with the same genotype. Environmental effects and multiple gene interactions are thought to be at play. Further, the ability of an individual to generate Hb F may lead to reduced disease severity. Different β-globin cluster haplotypes may result in different levels of Hb F.7,16 Levels of Hb F have been shown to correlate with the clinical manifestations of SCD.17
Pathophysiology
The interplay among abnormal erythrocytes and hypoxic, hyperosmolar, or acidotic conditions leads to the abnormal rheology and hemolysis characteristic of SCD. In Hb S, a strongly hydrophobic polar valine takes the place of a nonpolar strongly hydrophilic glutamic acid residue.5 Upon deoxygenation in the microcirculation, hydrophobic residues within Hb S are exposed and associate with hydrophobic regions of adjacent molecules. This polymerization results in the generation of rigid fibers of Hb S which damage the red blood cell membrane and cytoskeleton and cause the cell to assume a sickle shape. This polymerization process is reversible as oxygenation increases, and the cell may resume its native discoid shape. However, the repeated cycle of sickling and unsickling of the red blood cell membrane may lead to permanent damage to the erythrocyte membrane, irreversible sickling, and hemolysis. Mean corpuscular hemoglobin concentration (MCHC) may be the most important factor contributing to the rate of Hb S polymerization.5 The higher the MCHC, the more hemoglobin molecules that are available to participate in polymerization, and the closer these molecules are to one another, further promoting a favorable environment for Hb S polymerization.18,19
The erythrocyte’s original state of oxygenation also impacts the extent and rate of polymerization.15,16 An intrinsic property of the normal erythrocyte is the ability to deform easily in order to pass through capillaries with smaller diameters than its own. Decreased erythrocyte deformability and increased rigidity can cause increased capillary transit time.19 Deoxygenation and sickling promote increased permeability of the cell membrane to potassium, sodium, and calcium cations, which leads to water efflux from the cell, cellular volume contraction, and resultant increase in Hb S concentration.5,19,20,21
In addition to mechanical obstruction of blood vessels by dense, sickled erythrocytes, these sickled erythrocytes display increased adhesion to vascular endothelium matrix proteins, such as laminin,22,23 and thus cause direct damage to the endothelium. Integrin α4β1, integrin-associated protein, sulfated glycolipid, lutheran protein, phosphatidylserine, band 3 protein, and CD36 are adhesion molecules expressed in sickled red blood cells.24–26 Immature red blood cells called reticulocytes increase in SCD following intravascular hemolysis. These cells also have increased adhesion molecules, such as integrin α4β1,27–29 which promote pathologic adhesion to the vascular endothelium, specifically to vascular cell adhesion molecule-1 (VCAM-1). Direct activation of endothelial cells occurs in response to elevated circulating cytokines such as tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β),30,31 which upregulate expression of endothelial adhesion molecules like intercellular adhesion molecule-1 (ICAM-1), VCAM-1, E-selectin, and P-selectin.32,33
Inflammation likely plays a role in the vaso-occlusive process in SCD. Lutty and colleagues demonstrated retention of SS red cells and adherence of red cells in reticulocyte-rich fractions in retina and choroid of rat eyes in hypoxic conditions or following TNF-α stimulation.34–36 TNF-α and IL-1 may contribute to vaso-occlusion by accelerating the production of adhesion molecules on the vascular endothelium and by activating polymorphonuclear leukocytes.30 TNF-α and IL-1 have been shown to be upregulated in the sera of individuals with SCD at baseline.31,37,38
Nitric oxide (NO) is a potent vasodilator and regulator of vascular tone; it is derived from the vascular endothelial NO synthase. SCD has been associated with elevated reactive oxygen species, which scavenge NO and metabolize arginine, its precursor.7 l-arginine as an oral supplement has been given to induce NO production in transgenic sickle-cell mice.38,39
Vascular endothelial growth factor (VEGF), which is upregulated by hypoxia, is present in the serum of SCD patients at baseline, and is elevated during vaso-occlusive crises.40 Elevated VEGF has been demonstrated in eyes with sickle-cell retinopathy.41,42 VEGF has also been shown to increase levels of cell adhesion molecules, ICAM-1 and VCAM-1.43,44 Angiopoietin-1 (Ang-1) and angiopoietin-2 (Ang-2) interact with the Tie-2 receptor on endothelial cells, regulating angiogenesis. Ang-1 is responsible for the maintenance and stabilization of mature blood vessels, while Ang-2 leads to vessel destabilization and dissociation of pericytes and is upregulated by hypoxia and VEGF.45 Interaction among these proteins may be important in the pathogenesis of SCD.
Systemic manifestations
Over recent years, as the life expectancy of those affected by SCD has increased, so have the number of complications these individuals experience.12,46 Sickling of the red blood cells, with resultant intravascular hemolysis, thrombosis, tissue necrosis, and ischemia, causes a myriad of systemic complications, including cerebrovascular accident, acute chest syndrome, pulmonary hypertension, splenic sequestration, priapism, osteonecrosis, cholelithiasis, pneumonia, leg ulcers, aplastic crisis, renal disease, need for recurrent transfusions, episodic, painful vaso-occlusive crises, and death.7
Severe visual impairment and blindness from complications of proliferative sickle-cell retinopathy (PSR) may also occur. For reasons that are as yet unclear, subjects with Hb SC disease typically display less systemic morbidity from their hemoglobinopathy, but have a higher likelihood of experiencing the ophthalmic complications, than their homozygous Hb SS counterparts.47 In one study of patients with SCD, retinopathy was detected in 33% of patients with Hb SC disease compared to 3% of patients with Hb SS disease.37
Several theories have been proposed to explain this difference. One explanation could be that Hb SS patients typically have a shorter life expectancy than Hb SC patients, and may not live long enough to manifest the ophthalmic disease. Alternatively, the difference in retinopathy among sickle-cell genotypes may be due to the higher hematocrit and cell density, and the lower Hb F, of individuals with Hb SC as compared to Hb SS individuals.7 The overall lower hematocrit in Hb SS patients, and the resultant lowered viscosity of blood, may confer a relative protection from vaso-occlusion in the retinal circulation.48 Another theory is that the retinal vaso-occlusions in SS disease may be so complete that no viable tissue remains to produce the growth factors necessary to mount a proliferative response. In Hb SC disease, the vascular occlusions may be less complete, and they occur in the setting of a chronic hypoxic rather than anoxic state. Thus, a steady-state release of angiogenic growth factors from the remaining viable tissue may occur.49 Finally, Lutty and colleagues showed that irreversibly sickled Hb SS erythrocytes are easily trapped in retinal capillaries and precapillary arterioles in hypoxic conditions in a rat model.16,35 Hb SC cells, however, regardless of oxygen levels, were less easily trapped in the retinal microvasculature. When the vascular endothelium was stimulated with cytokines, retention of Hb SC cells did occur. The authors suggested that vaso-occlusion in Hb SC disease may be the result of the complex interaction among cytokines, the fibrinolytic cascade, leukocyte interactions, and activation of the vascular endothelium with induction of adhesion molecules.13,35 This data suggests that retention of sickled cells in the retina is not mechanical (rigid cells retained in a small lumen) but rather due to hypoxia-initiated cytokines and receptors.
Ophthalmic clinical features
Retrobulbar and orbital involvement
Orbital involvement of SCD is uncommon, but has been reported. One study from Oman reported periorbital swelling during vaso-occlusive crises in five patients.50 The patients ranged in age from 6 to 15 years old. Four of these patients had SS disease, while one had sickle-beta thalassemia. Infarction of orbital bones and orbital hematomas occur, and may lead to the orbital compression syndrome. These patients typically display lid edema, fever, facial pain, proptosis, restriction of motility, and resultant diplopia. The authors concluded that these syndromes usually resolve with conservative management with treatment of the underlying crisis, antibiotics, and steroids. Orbital involvement in SCD may cause sudden permanent unilateral loss of vision without detectable retinal arterial changes due to retrobulbar ischemic optic neuropathy.7,51 Urgent decompression of the orbit may be required. Recurrent bilateral lacrimal gland swelling has also been reported.52
Anterior-segment involvement
Conjunctival and iris findings may be present in SCD. An early reported finding is the saccular and sausage-like dilatations of the tiniest conjunctival vessels.53 They are most easily seen with the highest power of the slit-lamp biomicroscope. Paton described these comma-shaped capillary segments in the inferior bulbar conjunctival vessels, and coined the term “conjunctival sign.”54 This sign was uncommon in those with high Hb F levels. Local heat from the slit lamp induces vasodilation, and causes reversal of the comma-shaped vessels,55,56 whereas topical vasoconstricting drops increase the abnormality.57,58 These conjunctival vascular changes vary with oxygenation status, and are more prevalent in Hb SS disease than in Hb SC disease.54 Pathophysiology studies show constriction of these conjunctival vessels during painful crises, with normal blood flow returning on recovery.58 On histopathologic examination, these vessels exhibit endothelial proliferation, dilatation, and thinning of the proximal blood vessel segments, and aggregation of red blood cells in the distal portions of capillaries.58
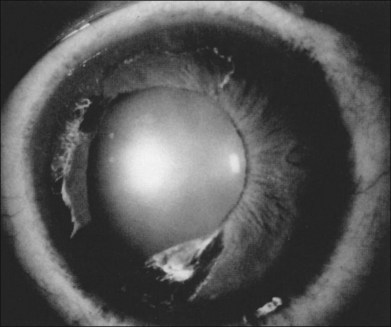
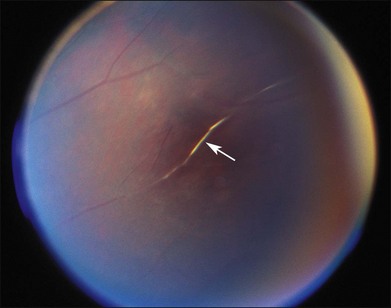
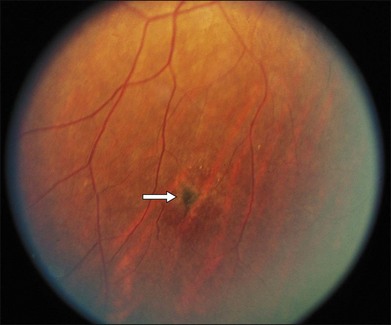
Segmental iris atrophy and pupil abnormalities can be seen in individuals with SCD (Fig. 57.1).59,60 Iris atrophy is thought to result from sectoral ischemic necrosis involving radial iris vessels. Secondary neovascularization of the iris stroma has also been described and documented with intravenous fluorescein angiography, and in one instance this neovascular frond resembled that classically seen as the “sea fan” in PSR.13,61 Neovascular glaucoma rarely occurs as a consequence of PSR.7,62
Hyphema in a patient with SCD and in those with sickle-cell trait represents a sight-threatening emergency, as even modest elevations of intraocular pressure (IOP) have resulted in vision loss from central retinal artery occlusion or macular branch retinal artery occlusion.62–65 This is likely a mechanical phenomenon, as sickled red blood cells clog the trabecular meshwork,61 causing elevation in IOP. Although intensive medical management to lower IOP in SCD patients with hyphema is recommended, IOP-lowering medications must be used cautiously. Repetitive use of carbonic anhydrase inhibitors, for example, is contraindicated in SCD patients with hyphema as most of these medications cause intracameral acidotic conditions that may worsen erythrocyte sickling. Early surgical intervention for IOP control should be considered, typically by paracentesis of the anterior chamber.66 Intracameral tissue plasminogen activator may be a potentially useful agent in the treatment of posttraumatic hyphema in some cases.67 Hyperoxygenation of the patient may also be helpful.
Posterior-segment involvement
Vitreoretinal interface
The pathologic retinal neovascularization caused by retinal ischemia in SCD can result in vision loss from vitreous hemorrhage.68,69 Other abnormalities of the vitreoretinal interface have been described, such as peripheral retinal whitening, which may represent abnormal or particularly strong adhesion at the vitreoretinal interface. This has been described in 93% of Hb SS patients,70 83% of Hb SC patients,71 and 82% of Hb S Thal patients.72 There is no known pathologic significance to this finding. This peripheral whitening is noted in the peripheral retina in the normal population as “white without pressure,” and may not be truly unique to those with sickle-cell retinopathy.13,57
Correspondingly, flat, brown, ovoid lesions in the retinal periphery have been noted in SCD, and termed “dark without pressure.” These lesions can be transient, and may disappear without sequelae. The underlying choroid appears normal on ophthalmoscopy, and the corresponding fluorescein angiogram is also normal.13,73
Optic nerve
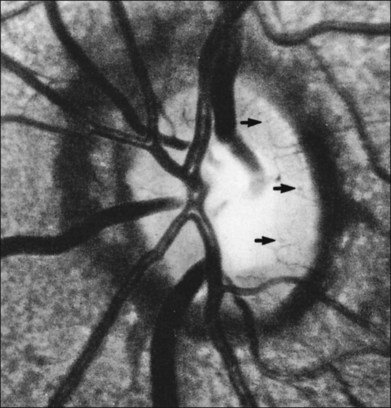
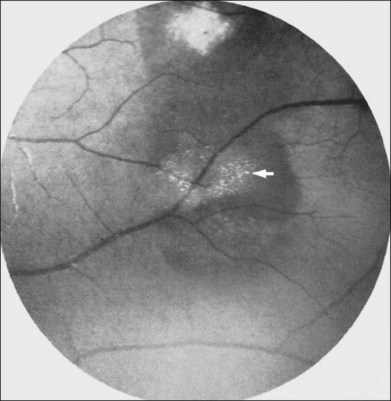
The optic nerve in patients with SCD may exhibit vascular changes. Dark, dilated capillaries at the optic nerve head appear as small red dots (Fig. 57.2), and represent precapillary arterioles plugged with sickled erythrocytes. These abnormal vessels have a linear or Y-shaped configuration on high-magnification ophthalmoscopy.13,57,70,74 These changes are not visually significant as the vascular occlusions are transient. Optic disc neovascularization is rare but has been reported in four Hb SC patients13,75–78 and one Hb SS patient.13,70
Macula
Though the hallmark of ocular SCD has been the peripheral retinopathy, the macula is also susceptible to infarction from vaso-occlusive disease. The “macular depression sign” has been described as an oval depression of the bright foveal or parafoveal reflex as a result of macular thinning due to ischemic atrophy.79 This finding is best detected using red-free illumination. The extent of macular vascular changes has not been shown to correlate with visual acuity. In a study by Sanders and colleagues, an enlarged foveal avascular zone (FAZ) was identified in patients with SCD on fluorescein angiography as compared to healthy, age-matched controls.80 Within the sickle-cell group, visual acuity did not correlate with FAZ size. The authors reported an enlarged FAZ in patients with SCD irrespective of the type of hemoglobinopathy or degree of retinopathy.
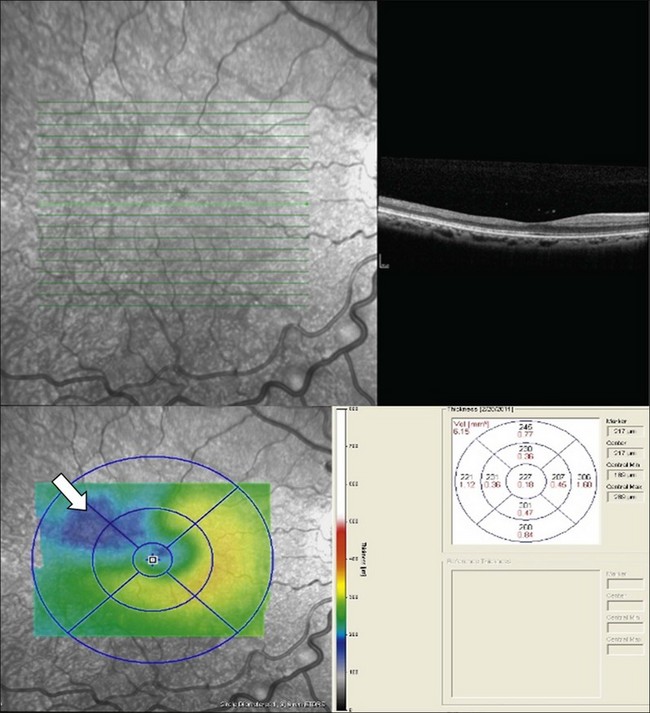
Macular infarcts can now be confirmed using optical coherence tomography.81 In a recent study utilizing spectral-domain optical coherence tomography (SD-OCT), Hoang and coworkers82 described retinal thinning of the central macula compared to healthy controls, most notably in the outer retinal layers in the central macula and parafoveal retina. Although several previous studies have reported atrophy of inner retinal layers after macular infarction, Hoang et al. hypothesize that the outer retinal layers may be thinned due to ischemia and resultant atrophy of the choriocapillaris, as these larger-caliber vessels may possibly be more prone to occlusion than the retinal vessels. Additionally, this study reported “splaying,” or blunting of the foveal contour on SD-OCT in asymptomatic patients with SCD with areas of focal parafoveal thinning (Fig. 57.3). A case series by Murthy et al. also describes thinning of the temporal macula in patients with PSR on SD-OCT, and suggests that this finding should lead clinicians to perform peripheral wide-field angiography to evaluate the retinal periphery for ischemia.83 Macular hole,84 epiretinal membranes,85 macular schisis,86 and posterior pole neovascularization87 have all been reported as rare complications of sickle-cell retinopathy.
Angioid streaks
Angioid streaks, breaks in Bruch’s membrane, have a well-documented association with SCD, occurring in 1–2% of patients.13,70,88 These irregular, reddish subretinal bands are most commonly found in patients with the Hb SS genotype.70,89 The incidence of angioid streaks in these eyes increases with age, and one Jamaican study found that 22% of 60 patients with Hb SS disease over age 40 had angioid streaks, compared to 2% of 150 patients under 40.90 Why angioid streaks occur in SCD is unclear. One hypothesis is that elastic tissue injury may occur as the result of oxidative damage due to hypoxia, and inflammatory cells may release cytokines that cause tissue damage.7,91 Angioid streaks in SCD usually have a benign course and resultant choroidal neovascularization (CNV) is rare.13
Retinal vasculature
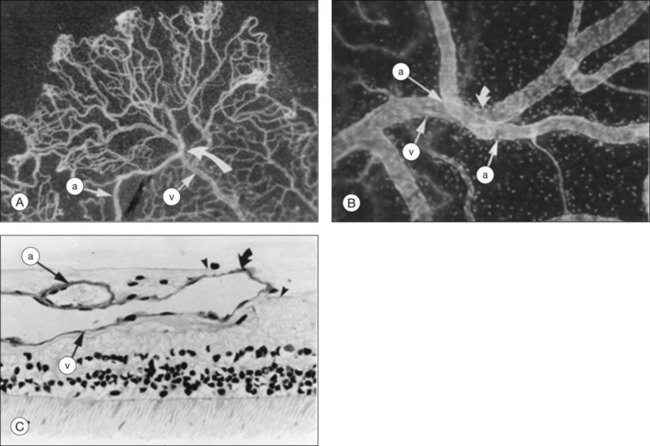
Vascular tortuosity caused by arteriovenous anastomoses may be more commonly observed in Hb SS patients (Fig. 57.4).7 One study reported increased retinal vascular tortuosity in 47% of Hb SS patients and 32% of Hb SC patients,92 while another reported 11% vascular tortuosity in both Hb SS and Hb SC patients.70,71 This discrepancy could be due to an imprecise definition of vascular tortuosity.7
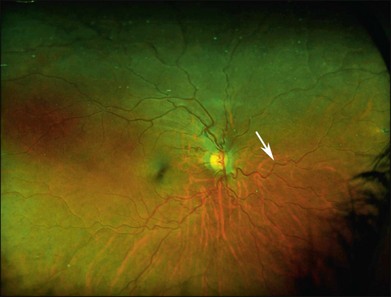
Retinal vascular occlusions frequently occur in the peripheral retina in patients with SCD, and peripheral retinal nonperfusion is a common finding in these eyes. Arteriovenous anastomoses may occur in this setting (Fig. 57.5). Retinal arteriole “silver-wiring” represents permanently occluded arterioles (Fig. 57.6).
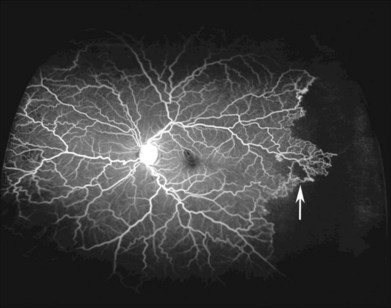
Depending on its location, vascular occlusion may cause temporary or permanent vision loss. The precapillary arterioles in eyes with SCD are particularly susceptible to obstruction. The cause of these occlusions may be multifactorial, potentially involving sickled red blood cells attaching to vascular endothelium, or the activation of the coagulation cascade, with secondary intimal injury.2,93 In the peripheral temporal retina, where vessels become markedly narrow, arterial occlusions are common.
Vaso-occlusion arising from the posterior ciliary arterial circulation may potentially cause choroidal infarction, an event that has been described in numerous reports in the ophthalmic literature in SCD. Histopathologic studies show impacted red blood cells, increased fibrin, and platelet fibrin thrombi in cases of choroidal occlusion.94,95 As in the retinal circulation, the interaction among adhesion molecules, facilitating adhesion of dense reticulocytes to the endothelium, may play a role.34 Choroidal neovascularization may rarely occur spontaneously96 or result after high-energy laser burns.97
Nonproliferative sickle retinopathy
Salmon patch hemorrhages
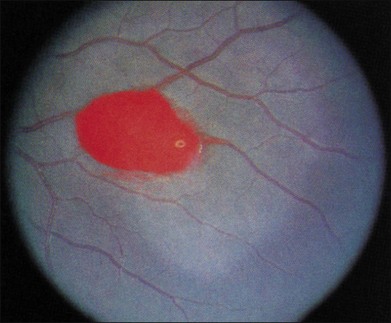
Salmon patch lesions are pinkish orange hemorrhages located between the retina and its internal limiting membrane (Fig. 57.7). These may arise from areas of bleeding into the retina adjacent to areas of nonperfusion, and have been described as a “blowout” of an occluded arteriole.98,99 Although the hemorrhage is initially red, it may turn a red-orange or salmon color over time because of progressive hemolysis. The localized collection of blood may remain under the internal limiting membrane, travel into the subretinal space, or spread into the vitreous cavity.13,98,99
Iridescent spots
If arteriolar occlusion causes retinal hemorrhage, a small schisis cavity may develop after the intraretinal portion of the hemorrhage resolves.13 The cavity may contain hemosiderin-laden macrophages, which may appear as multiple glistening, refractile, or iridescent spots (Fig. 57.8).
Black sunburst
Black sunbursts are flat, stellate, or round areas of hyperpigmentation, and result when intraretinal hemorrhage tracks into the subretinal space (Fig. 57.9).98,100 On histopathologic study, black sunbursts appear as focal hypertrophy of the retinal pigment epithelium (RPE).98 This “sunburst sign” may also represent localized choroidal ischemic damage to the overlying RPE.101 Another hypothesis is that the black sunburst may be the RPE response to an area of underlying CNV.96,102
Proliferative sickle retinopathy
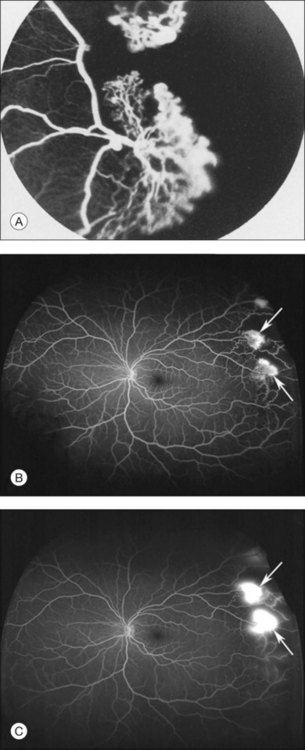
A critical event in the development of PSR is the formation of the sea fan, the peripheral vascular lesion so named for its close resemblance to the marine invertebrate, Gorgonia flabellum (Fig. 57.10). Peripheral retinal arteriolar occlusions are the inciting events in sea-fan formations. Occlusion of the vasculature causes release of growth factors, resulting in formation of these neovascular fronds. PSR complications are the major contributor to vision loss. Sea fans are predisposed to hemorrhage into the vitreous, and to cause vitreous membrane formation, tractional retinoschisis, and tractional or combined rhegmatogenous–tractional retinal detachment.48 Sea fans often form at arteriovenous crossings and may have multiple feeding arterioles and draining venules (Fig. 57.11).103
In 1971, Goldberg devised the widely utilized classification system for PSR (Table 57.1).104
Table 57.1 Goldberg classification of proliferative sickle-cell retinopathy
| Stage I | Peripheral arterial occlusions |
| Stage II | Peripheral arteriovenous anastomoses |
| Stage III | Neovascular and fibrous proliferations |
| Stage IV | Vitreous hemorrhage |
| Stage V | Retinal detachment |
Goldberg stages
Stage I
Stage I retinopathy is defined by peripheral vascular occlusion.57 The peripheral retina may show multiple simultaneous arteriolar occlusions, and silver-wiring of the arterioles may be present. Vaso-occlusion occurs primarily in the peripheral temporal retina due to longer arteriovenous transit times (with deoxygenation), an increased number of occludable bifurcation sites, and decreased perfusion.
Stage II
In this stage, vascular remodeling at the border of perfused and nonperfused retina occurs. Arteriovenous anastomoses form connections between occluded arterioles and adjacent terminal venules by way of pre-existing capillaries (Fig. 57.5). The anastomoses do not leak, confirming that these early vascular lesions are derived from pre-existing blood vessels with an intact blood–retinal barrier, as opposed to being true neovascular complexes.
Stage III
Sea-fan fronds are the hallmark of stage III PSR, and are perhaps the most recognizable trademark lesion of sickle retinopathy (Fig. 57.10). These lesions are most commonly found in the superotemporal retina, followed by the inferotemporal, superonasal, and inferonasal quadrants. Sea fans represent true neovascularization, and thus, display diffuse leakage on fluorescein angiography (Fig. 57.10). The blood–retinal barrier is not intact. Therefore, sea fans cause chronic transudation into the vitreous from the neovascular lesions, which in turn leads to vitreous degeneration and retinal traction. These may be precursors to vitreous hemorrhage and retinal detachment.13,48,57
Most sea fans are found at the border of perfused and nonperfused retina, and they grow toward the ora serrata.48,78 Sea fan neovascular complexes arise from the venous aspect of the arteriovenous anastomoses, and develop approximately 18 months following the formation of the arteriovenous connections13,48 (Fig. 57.11). Chronic ischemia within the peripheral retina leads to an increase in angiogenic factors, such as VEGF and basic fibroblastic growth factor.13,41,94 Pigment epithelial growth factor/VEGF balance may play a role in the angiogenesis of these lesions as well as the subsequent, spontaneous regression of some neovascular complexes.42
Another potential mechanism for sea-fan development has been proposed.95 Occlusive events may create hydrostatic back-pressure, causing extrusion of the upstream segment of the blocked vessel into the preretinal space. The rise in intraluminal pressure may cause expansion of the extruded vessel, with resultant stretching of the pericytes and endothelial cells. This process may stimulate endothelial cells, leading to endothelial cell proliferation105 and subsequent neovascularization.95,106
Sea fans typically possess at least one feeding arteriole and one draining venule, and are most commonly found at the sites of arteriovenous anastomoses and arteriovenous crossings.103,104 A network of these lesions with multiple anastomoses may develop with multiple feeding and draining vessels, and they may form tractional vitreous bands.
Stage IV
Vitreous hemorrhage represents Goldberg stage IV. Sea fans grow or are pulled into the vitreous chamber, and vitreous traction on the delicate neovascular fronds may cause bleeding into the vitreous. Vitreous hemorrhage may be localized over the sea fan, and an individual may remain asymptomatic. However, dramatic, sudden vision loss may occur as the hemorrhage disseminates into the vitreous gel. Vitreous hemorrhage occurs more commonly in the Hb SC than the Hb SS genotype (23% versus 3%).70,88,92 The risk of recurrent vitreous hemorrhage also increases if an eye has more than 60° of circumferential retinal neovascularization, or if a patient initially presents with vitreous hemorrhage.107 Chronic vitreous hemorrhage may give rise to fibroglial membranes and vitreous strands, which may produce traction and resultant retinal detachment.48,57
Stage V
Presence of traction retinal detachment (TRD) defines Goldberg stage V. TRD develops as the result of chronic vitreous hemorrhage or chronic transudation from neovascular tissue and resultant vitreous membrane formation. Round or horseshoe retinal breaks may also occur, and may be due to localized retinal atrophy and thinning from chronic vaso-occlusion and ischemia.48 In contrast to the TRD commonly seen in proliferative diabetic retinopathy, the TRD in SCD most commonly involves the peripheral retina as opposed to the posterior pole. Combined TRD and rhegmatogenous retinal detachment may also occur.
Alternative classification schemes
Another classification scheme for PSR was proposed by Penman and colleagues108 and applies a prognostic significance to varying degrees of PSR. In this angiographic study of eyes of children in a Jamaican sickle cell cohort, two groups were assigned. The type I group showed a normal vascular pattern, and the type 2 group displayed an abnormal vascular pattern. An abnormal vascular pattern was most commonly seen in individuals with Hb SC disease versus individuals with Hb SS disease, and was more likely to develop with age. Over time, the type 2 eyes were more likely to develop proliferative disease. Yet another grading system for PSR has been proposed by Sayag and coworkers109 to compare clinical outcome of peripheral sea fans in Goldberg stage III lesions treated by laser photocoagulation as compared to the natural course of the disease. The following grades for PSR were described: (A) flat sea fan with leakage <1 Macular Photocoagulation Study (MPS) disc area; (B) elevated sea fan with hemorrhage; (C) elevated sea fan with partial fibrosis; (D) a sea fan with complete fibrosis and without well-demarcated vessels; and (E) a sea fan with complete fibrosis and well-demarcated vessels. In this study, scatter laser photocoagulation is suggested for grade B, grade D, or grade E lesions; however, the authors suggest that grade A or grade C should be observed without laser treatment due to the low rate of progression.109
Incidence/prevalence
Among the hemoglobinopathies, the incidence of PSR is higher in those individuals with Hb SC disease and S-β thalassemia than in individuals with Hb SS disease. In the Hb SC genotype, PSR has an earlier onset, with peak prevalence at 15–24 years of age in men and 20–39 years in women. In the SS genotype, peak prevalence of PSR onset falls in the age range of 25–39 years in men and women.7 In one natural history study of SCD patients followed longitudinally over 20 years, prevalence of PSR was greater in those with Hb SC disease, and by the ages of 24–26 years, PSR had occurred in 43% subjects with Hb SC disease and in 14% subjects with Hb SS disease.110
Risk factors
Fox and colleagues described other risk factors that increase an individual’s likelihood of developing PSR in addition to genotype, age, and sex.111 The development of an unstable type IIa border (hairpin loop) conferred an increased risk for PSR. In the Hb SS genotype, high total hemoglobin in males and a low Hb F in both males and females were associated with the development of PSR. In the Hb SC genotype, increased mean cell volume and low Hb F increased the risk in men and women. High total hemoglobin and high MCHC incurred a higher PSR risk in men with Hb SC genotype.7 In this same study, 70% of patients with Hb SC disease developed bilateral PSR, while 49% of patients with Hb SS disease developed bilateral PSR.111
Natural history
Autoinfarction of the proliferative sickle lesions occurs frequently. Spontaneous regression of PSR may occur in 32% of eyes.110 The mechanism of autoinfarction is not completely understood. This process may be due to chronic and repetitive vaso-occlusion within the vascular channels of the delicate sea-fan neovascular complexes. In a cohort of 120 patients with Hb SS disease and 222 patients with Hb SC disease followed for 10 years, visual acuity loss occurred in 10% of untreated eyes.112 Vision loss most commonly resulted from PSR, specifically vitreous hemorrhage, TRD, and epiretinal membranes. Patients with nonproliferative disease had a lower incidence of vision loss over the 10-year period than did their counterparts with PSR.112 Downes and colleagues reported that patients with unilateral PSR had a 16% (11% Hb SS, 17% Hb SC) probability of regressing to no PSR and a 14% (16% Hb SS, 13% Hb SC) probability of progressing to bilateral PSR. Those with bilateral PSR had an 8% (both Hb SC and Hb SS genotypes) probability of regressing to unilateral PSR and a 1% (0 Hb SS, 2% Hb SC) probability of regressing to a PSR-free state.110
Ophthalmic treatments
No intervention is indicated for small, asymptomatic peripheral lesions, given the relatively high probability of autoinfarction.113,114 However, treatment of PSR may be considered in the eye of an individual with significant visual loss from PSR or vision impairment in the contralateral eye. Treatment may also be required in cases of rapid growth of a sea fan, presence of large, elevated sea fans, spontaneous hemorrhage, or bilateral proliferative disease.13 The goal of intervention in these cases is to prevent the progression of stage III PSR to stages IV or V.
Historically, a goal in PSR treatment had been to occlude the feeder arterioles of the sea-fan lesions.113 This was accomplished through laser photocoagulation, diathermy, or cryotherapy.114 This technique was shown to reduce the incidence of vitreous hemorrhage in a small study.113 Feeder vessel photocoagulation is no longer widely used because of its high rate of complications, which include CNV from breaks in Bruchs membrane and retinal break formation.
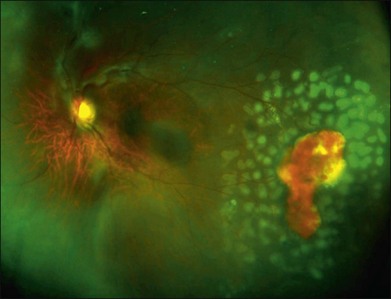
Scatter laser photocoagulation is the current mainstay of treatment in PSR. The objective of scatter laser treatment is similar to that employed in the management of proliferative diabetic retinopathy, in which ischemic retina is ablated by laser photocoagulation, thus decreasing the stimulus for pathologic growth factor secretion. Laser photocoagulation of ischemic retina may also help to decrease the overall oxygen requirement of the retina. Through laser destruction of ischemic, damaged peripheral retina, oxygen may be shunted to healthier, more viable retinal tissue (Fig. 57.12).
Anti-VEGF agents have been successful in the treatment of neovascular age-related macular degeneration, retinal venous occlusion, diabetic macular edema, and proliferative diabetic retinopathy. Intravitreal bevacizumab has been reported to cause complete regression of retinal neovascularization and resolution of vitreous hemorrhage following intravitreal injection in eyes with PSR.115,116 Future study is warranted to assess the role of intravitreal anti-VEGF therapy in PSR.
Vitrectomy may be considered for nonclearing vitreous hemorrhage. Early intervention with vitrectomy may be considered if an individual has an associated retinal detachment or visual impairment in the contralateral eye, or may be indicated to improve visualization of retinal pathology to facilitate treatment. A possible therapeutic approach is combination vitrectomy and scatter laser photocoagulation, with consideration of an anti-VEGF agent in an eye with active PSR. Exchange transfusion, erythropheresis, and hyperbaric oxygen have been attempted to minimize complications from surgically induced anterior-segment ischemia. These methods were without clear benefit and were associated with systemic complications.117 Vitrectomy in an eye with PSR can be safe with today’s modern vitrectomy techniques without these extra measures. The vitreoretinal surgeon should take special care to maximize perfusion in the eye, and work closely with the anesthesiologist to insure the patient is well oxygenated throughout and after the procedure.
Given the propensity for peripheral retinal breaks and other peripheral vitreoretinal pathology to develop, scleral buckling may be considered in SCD to relieve traction from anterior lesions. Though anterior-segment ischemia is a feared complication of scleral buckling in sickle-cell retinopathy eyes, scleral buckles have safely been utilized in sickle-cell retinal detachment surgery.118 Williamson and colleagues reported their experience with management of vitreoretinal complications of sickle-cell retinopathy.119 These authors observed flattening of the retina in one patient with rhegmatogenous retinal detachment and one patient with TRD without surgical intervention, though such events are unusual. A high rate of iatrogenic retinal tears was noted by vitreoretinal surgeons while peeling membranes from the atrophic, ischemic peripheral retina with delamination (tangential peeling of preretinal scar tissue from the retinal surface) techniques. Therefore, these authors recommend segmentation (removal of anterior–posterior traction) techniques instead.
Imaging
Selected imaging modalities facilitate diagnosis, monitoring, and assessment of treatment response in sickle-cell retinopathy. As impaired choroidal circulation may be present in eyes with SCD, indocyanine green (ICG) angiography has been studied as a way to assess choroidal perfusion, but the utility of ICG in sickle retinopathy is as yet undetermined.120 The use of ultrawide-field imaging is particularly helpful to monitor peripheral lesions and to assess treatment response (Fig. 57.12). Fluorescein angiography remains the gold-standard imaging tool for assessment of retinal perfusion status. A potential limitation of conventional fluorescein angiography is the inability to image the pathology of the far peripheral retina in some eyes with sickle retinopathy.108 Accordingly, ultrawide-field fundus photography and angiography have become useful in the evaluation of the retinal periphery in eyes with sickle-cell retinopathy. As previously mentioned, OCT and especially SD-OCT, with its high resolution, may provide important details about foveal anatomy, which may aid in the diagnosis and management of sickle retinopathy,79,81–83 even in asymptomatic patients.
Potential therapeutic options for the future
Treatment strategies that decrease Hb S and increase Hb F have been shown to decrease the systemic morbidity of SCD. Higher Hb F may interfere with polymerization of Hb S. Agents to increase Hb F include hydroxyurea, omega-3 fatty acids, and erythropoietin. Hydroxyurea may also possess anti-inflammatory properties, and may reduce the load of inflammatory mediators in SCD. The burden of Hb S cells may be reduced through transfusion or hemapheresis. Warfarin, heparin, and ticlopidine all work as antithrombotic agents. Vasodilation of occluded small blood vessels may be improved with NO, or its precursor, arginine. Additional future areas of investigation include hematopoietic stem cell transplantation and gene therapy.7
1 Herrick JB. Peculiar elongated and sickle-shaped red blood corpuscles in a case of severe anemia. Arch Intern Med. 1910;6:517–521.
2 Savitt TL. Tracking down the first recorded sickle cell patient in western medicine. J Natl Med Assoc. 2010;102:981–992.
3 Savitt TL, Goldberg MF. Herrick’s 1910 case report of sickle cell anemia. The rest of the story. JAMA. 1989;261:266–271.
4 Pauling L, Itano HA, Singer SJ, et al. Sickle cell anemia, a molecular disease. Science. 1949;110:543–548.
5 Barabino GA, Platt NO, Kaul DK. Sickle cell biomechanics. Annu Rev Biomed Eng. 2010;12:345–367.
6 Ashley-Koch A, Yang Q, Olney RS. Sickle hemoglobin (HbS) allele and sickle cell disease: a HuGE review. Am J Epidemiol. 2000;151:839–845.
7 Elagouz M, Jyothi S, Gupta B, et al. Sickle cell disease and the eye: old and new concepts. Surv Ophthalmol. 2010:359–377.
8 Neumayr L, Pringle S, Giles S, et al. Chart card: feasibility of a tool for improving emergency department care in sickle cell disease. J Natl Med Assoc. 2010;102:1017–1023.
9 Rosenberg JB, Hutcheson K. Pediatric sickle retinopathy: correlation with clinical factors. J AAPOS. 2011;15:49–53.
10 National Heart, Lung and Blood Institute. Who is at risk for sickle cell anemia? Available online at http://www.nhlbi.nih.gov/health/dci/Diseases/Sca/SCA_WhoIsAtRisk.html, 2008. [accessed June 10, 2011]
11 World Health Organization. Sickle-cell anaemia: report by the Secretariat. Available online at http://www.who.int/gb/ebwha/pdf_files/WHA59/A59_9-en.pdf, 2006. [accessed May 26, 2011]
12 Serjeant G. Sickle cell disease. Oxford: Oxford University Press; 1985.
13 Emerson GG, Harlan JB, Fekrat S, et al. Hemoglobinopathies. Retina. 4th ed. Ryan SJ, ed. Retina. Edinburgh: Elsevier; 2006;vol. II:1429–1445.
14 Gill FM, Sleeper LA, Weiner SJ, et al. Clinical events in the first decade in a cohort of infants with sickle cell disease: Cooperative Study of Sickle Cell Disease. Blood. 1995;86:776–783.
15 Bunn HF. Disorders of hemoglobin. In: Braunwald E, Isselbacher KJ, Petersdorf RG, et al. Harrison’s principles of internal medicine. 11th ed. New York: McGraw-Hill; 1987:1518–1523.
16 Powara D, Hiti A. Sickle cell anemia: beta s gene cluster haplotypes as genetic markers for severe disease expression. Am J Dis Child. 1993;147:1197–1202.
17 Thomas PW, Higgs DR, Serjeant GR. Benign clinical course in homozygous sickle cell disease: a search for predictors. J Clin Epidemiol. 1997;50:121–126.
18 Ferrone FA. Polymerization and sickle cell disease: a molecular review. Microcirculation. 2004;11:115–128.
19 Bunn HF. Pathogenesis and treatment of sickle cell disease. N Engl J Med. 1997;337:762–769.
20 Mohandas N, Rossi ME, Clark MR. Association between morphologic distortion of sickle cells and deoxygenation-induced cation permeability increase. Blood. 1986;68:450–454.
21 Brugnara C, Bunn HF, Tosteson DC. Regulation of erythrocyte cation and water content in sickle cell anemia. Science. 1986;232:388–390.
22 Fabry ME, Kaul DK. Sickle cell vasocclusion. Hematol/Oncol Clin North Am. 1991;5:375–398.
23 Hebbel RP. Adhesive interactions of sickle erythrocytes with endothelium. J Clin Invest. 1997;100:S83–S86.
24 Sugihara K, Sugihara T, Mohandas N, et al. Thrombospondin mediates adherence of CD36+ sickle erythrocytes to endothelial cells. Blood. 1992;80:2634–2642.
25 Swerlick RA, Eckman JR, Kumar A, et al. Alpha 4 beta 1 expression on sickle reticulocytes; vascular cell adhesion molecule 1 dependent binding to the endothelium. Blood. 1993;82:1891–1899.
26 Wun T, Paglieroni T, Field CL, et al. Platelet-erythrocyte adhesion in sickle cell disease. J Invest Med. 1999;47:121–127.
27 Kaul DK, Fabry ME, Nagel RL. The pathophysiology of vascular obstruction in the sickle syndromes. Blood Rev. 1996;10:29–44.
28 Joneckis CC, Ackley RL, Orringer EP, et al. Integrin alpha 4 beta 1 and glycoprotein IV (CD36) are expressed on circulating reticulocytes in sickle cell anemia. Blood. 1993;82:3548–3555.
29 Setty BN, Stuart MJ. Vascular cell adhesion molecule 1 is involved in mediating hypoxia-induced sickle red blood cell adherence to endothelium; potential role in sickle cell disease. Blood. 1996;88:2311–2320.
30 Francis RB, Jr., Haywood LJ. Elevated immunoreactive tumor necrosis factor and interleukin-1 in sickle cell disease. J Natl Med Assoc. 1992;84:611–615.
31 Malave I, Perdomo Y, Escalona E, et al. Levels of tumor necrosis factor α/cachectin (TNFα) in sera from patients with sickle cell disease. Acta Haematol. 1993;90:172–176.
32 Solovey A, Lin Y, Browne P, et al. Circulating activated endothelial cells in sickle cell anemia. N Engl J Med. 1997;337:1584–1590.
33 Kunz Mathews M, McLeod DS, Merges C, et al. Neutrophils and leukocyte adhesion molecules in sickle cell retinopathy. Br J Ophthalmol. 2002;86:684–690.
34 Lutty GA, Otsuji T, Taomoto M, et al. Mechanisms for sickle red blood cell retention in choroid. Curr Eye Res. 2002;25:163–171.
35 Lutty GA, Phelan A, Mcleod DS, et al. A rat model for sickle-cell mediated vaso-occlusion in retina. Microvasc Res. 1996;52:270–280.
36 Lutty GA, Taomoto M, Cao J, et al. Inhibition of TNF-alpha-induced sickle RBC retention in retina with a VLA-4 antagonist. Invest Ophthalmol Vis Sci. 2001;42:1349–1355.
37 Lutty GA, Goldberg MF. Ophthalmological complications. In: Embury SH, Hebbel RP, Mohandas N, et al. Sickle cell disease: basic principles and clinical practice. New York: Raven Press; 1992:703–724.
38 Vichinsky E. New therapies in sickle cell disease. Lancet. 2002;360:629–631.
39 Wood KC, Hsu LL, Gladwin MT. Sickle cell disease vasculopathy; a state of nitric oxide resistance. Free Radic Biol Med. 2008;44:1506–1528.
40 Gurkan E, Tanriverdi K, Baslamish F. Clinical relevance of vascular endothelial growth factor levels in sickle cell disease. Ann Hematol. 2005;84:71–75.
41 Cao J, Kunz Mathews MK, McLeod DS, et al. Angiogenic factors in human proliferative sickle cell retinopathy. Br J Ophthalmol. 1999;83:838–846.
42 Kim DY, Mocanu V, McLeod DS, et al. Expression of pigment epithelium-derived factor (PEDF) and vascular endothelial growth factor (VEGF) in sickle cell retina and choroid. Exp Eye Res. 2003;7:433–445.
43 Lu M, Perez VL, Ma N. VEGF increases retinal vascular ICAM-1 expression in vivo. Invest Ophthalmol Vis Sci. 1999;40:1808–1812.
44 Perlman N, Selvaraj SK, Batra S, et al. Placenta growth factor activates monocytes and correlates with sickle cell disease severity. Blood. 2003;102:1506–1514.
45 Oh H, Takagi H, Suzuma K, et al. Hypoxia and vascular endothelial growth factor selectively upregulate angiopoietin-2 in bovine microvascular endothelial cells. J Biol Chem. 1999;274:15732–15739.
46 Fadugbagbe AO, Gurgel RQ, Mendonca CQ, et al. Ocular manifestations of sickle cell disease. Ann Trop Paediatr. 2010;30:19–26.
47 Ballas SK, Lewis CN, Noone AM, et al. Clinical, hematological, and biochemical features of Hb SC disease. Am J Hematol. 1982;13:37–51.
48 Goldberg MF. Retinal neovascularization in sickle cell retinopathy. Trans Am Acad Ophthalmol Otolaryngol. 1977;83:Op409–Op431.
49 Gagliano DA, Jampol L, Rabb M. Sickle cell disease. Duane’s clinical ophthalmology. Tasman WS, Jaeger E, eds. Duane’s clinical ophthalmology. Philadelphia: Lippincott Raven; 1996;vol. 3:1–40.
50 Ganesh A, William RR, Mitra S, et al. Orbital involvement in sickle cell disease: a report of 5 cases and review literature. Eye. 2001;15:774–780.
51 Perlman JI, Forman S, Gonzalez ER. Retrobulbar ischemic optic neuropathy associated with sickle cell disease. J Neuro-Ophthalmol. 1994;14:45–48.
52 Adewoye AH, Ramsey J, McMahon L, et al. Lacrimal gland enlargement in sickle cell disease. Am J Hematol. 2006;81:888–889.
53 Condon PI, Sergeant GR. Ocular findings in elderly cases of homozygous sickle cell disease in Jamaica. Br J Ophthalmol. 1976;60:361–364.
54 Paton D. The conjunctival sign of sickle cell disease. Arch Ophthalmol. 1961;66:90–94.
55 Fink AI, Funahashi T, Robinson M, et al. Conjunctival blood flow in sickle cell disease. Preliminary report. Arch Ophthalmol. 1961;66:824–829.
56 Paton D. The conjunctival sign of sickle-cell disease. Further observations. Arch Ophthalmol. 1962;68:627–632.
57 Nagpal KC, Goldberg MF, Rabb MF. Ocular manifestations of sickle hemoglobinopathies. Surv Ophthalmol. 1977;21:391–411.
58 Funahashi T, Fink A, Robinson M, et al. Pathology of conjunctival vessels in sickle-cell disease. A preliminary report. Am J Ophthalmol. 1964;57:713–718.
59 Chambers J, Puglisi J, Kernitsky R, et al. Iris atrophy in hemoglobin SC disease. Am J Ophthalmol. 1974;77:247–249.
60 Galinos S, Rabb MF, Goldberg MF, et al. Hemoglobin SC disease and iris atrophy. Am J Ophthalmol. 1973;75:421–425.
61 Bergren RL, Brown GC. Neovascular glaucoma secondary to sickle cell retinopathy. Am J Ophthalmol. 1992;113:718–719.
62 Goldberg MF. The diagnosis and treatment of secondary glaucoma after hyphema in sickle cell patients. Am J Ophthalmol. 1979;87:43–49.
63 Goldberg MF. Sickled erythrocytes, hyphema,and secondary glaucoma IV. The rate and percentage of sickling of erythrocytes in rabbit aqueous humor, in vitro and in vivo. Ophthalmic Surg. 1979;10:62–69.
64 Goldberg MF. Sickled erythrocytes, hyphema, and secondary glaucoma I. The diagnosis and treatment of sickled erythrocytes in human hyphemas. Ophthalmic Surg. 1979;10:17–31.
65 Goldberg MF, Dizon R, Raichand M. Sickled erythrocytes, hyphema, and secondary glaucoma II. Injected sickle cell erythrocytes into human, monkey, and guinea pig anterior chambers: the introduction of sickling and secondary glaucoma. Ophthalmic Surg. 1979;10:32–51.
66 Deutsch TA, Weinreb RN, Goldberg MF. Indications for surgical management of hyphema in patients with sickle cell trait. Arch Ophthalmol. 1984;102:566–569.
67 Karaman K, Culic S, Erceg I, et al. Treatment of post-traumatic trabecular meshwork thrombosis and secondary glaucoma with intracameral tissue plasminogen activator in previously unrecognized sickle cell anemia. Coll Antropol. 2005;29(Suppl 1):123–126.
68 Henry MD, Chapman AZ. Vitreous hemorrhage and retinopathy associated with sickle cell disease. Am J Ophthalmol. 1954;38:204–209.
69 Hannon JF. Vitreous hemorrhages associated with sickle cell-hemoglobin C disease. Am J Ophthalmol. 1956;42:707–712.
70 Condon PI, Serjeant GR. Ocular findings in homozygous sickle cell anemia in Jamaica. Am J Ophthalmol. 1972;73:533–543.
71 Condon PI, Serjeant GR. Ocular findings in hemoglobin SC disease in Jamaica. Am J Ophthalmol. 1972;74:921–931.
72 Condon PI, Serjeant GR. Ocular findings in sickle cell thalassemia in Jamaica. Am J Ophthalmol. 1972;74:1105–1109.
73 Nagpal KC, Goldberg MF, Asdourian G, et al. Dark-without-pressure fundus lesions. Br J Ophthalmol. 1975;59:476–479.
74 Goldberg MF. Retinal vaso-occlusion in sickling hemoglobinopathies. Birth Defects Orig Artic Ser. 1976;12:475–515.
75 Condon PI, Serjeant GR. Behaviour of untreated proliferative sickle retinopathy. Br J Ophthalmol. 1980;64:404–411.
76 Kimmel AS, Magargal LE, Tasman WS. Proliferative sickle retinopathy and neovascularization at the disc: regression following treatment with peripheral scatter laser photocoagulation. Ophthalm Surg. 1986;17:20–22.
77 Ober RR, Michels RG. Optic disk neovascularization in hemoglobin SC disease. Am J Ophthalmol. 1978;85:711–714.
78 Raichand M, Goldberg MF, Nagpal KC, et al. Evolution of neovascularization in sickle cell retinopathy. A prospective fluorescein angiographic study. Trans Am Ophthalmol Soc. 1992;90:481–504.
79 Goldbaum MH. Retinal depression sign indicating a small retinal infarct. Am J Ophthalmol. 1978;86:45–55.
80 Sanders RJ, Brown GC, Rosenstein RB, et al. Foveal avascular zone diameter and sickle cell disease. Arch Ophthalmol. 1991;109:812–815.
81 Witkin AJ, Rogers AH, Ko TH, et al. Optical coherence tomography demonstration of macular infarction in sickle cell retinopathy. Arch Ophthalmol. 2006;124:747.
82 Hoang QV, Chau FY, Shahidi M, et al. Central macular splaying and outer retinal thinning in asymptomatic sickle cell patients by spectral-domain optical coherence tomography. Am J Ophthalmol. 2011;151:990–994.
83 Murthy RK, Grover S, Chalam K. Temporal macular thinning on spectral-domain optical coherence tomography in proliferative sickle cell retinopathy. Arch Ophthalmol. 2011;129:247–249.
84 Raichland M, Dizon RV, Nagpal KC, et al. Macular holes associated with proliferative sickle cell retinopathy. Arch Ophthalmol. 1987;96:1592–1596.
85 Moriarty BJ, Acheson RW, Serjeant GR. Epiretinal membranes in sickle cell disease. Br J Ophthalmol. 1987;71:466–469.
86 Schubert HD. Schisis in sickle cell retinopathy. Arch Ophthalmol. 2005;123:1607–1609.
87 Frank RN, Cronin MA. Posterior pole neovascularization in a patient with hemoglobin SC disease. Am J Ophthalmol. 1979;88:680–682.
88 Clarkson JG. The ocular manifestations of sickle cell disease: a prevalence and natural history study. Trans Am Ophthalmol Soc. 1992;90:481–504.
89 Clarkson JG, Altman RD. Angioid streaks. Surv Ophthalmol. 1982;26:235–246.
90 Condon PI, Serjeant GR. Ocular findings in elderly cases of homozygous sickle cell disease in Jamaica. Br J Ophthalmol. 1976;60:361–364.
91 Aessopos A, Farmakis D, Loukopoulos D. Elastic tissue abnormalities resembling pseudoxanthoma elasticum in beta thalassemia and the sickling syndromes. Blood. 2002;99:30–35.
92 Welch RB, Goldberg MF. Sickle-cell hemoglobin and its relation to fundus abnormality. Arch Ophthalmol. 1966;75:353–362.
93 Fine LC, Petrovic V, Irvine AR, et al. Spontaneous central retinal artery occlusion in hemoglobin SC disease. Am J Ophthalmol. 2000;130:680–681.
94 Lutty GA, Merges C, Crone S, et al. Immunohistochemical insights into sickle cell retinopathy. Curr Eye Res. 1994;13:125–138.
95 McLeod DS, Goldberg MF, Lutty GA. Dual-perspective analysis of vascular formations in sickle cell retinopathy. Arch Ophthalmol. 1993;111:1234–1245.
96 Liang JC, Jampol LM. Spontaneous peripheral chorioretinal neovascularization in association with sickle cell anemia. Br J Ophthalmol. 1983;67:107–110.
97 Condon PI, Jampol LM, Ford SM, et al. Choroidal neovascularization induced by photocoagulation in sickle cell disease. Br J Ophthalmol. 1981;65:192–197.
98 Romayananda N, Goldberg MF, Green WR. Histopathology of sickle cell retinopathy. Trans Am Acad Opthalmol Otol. 1973;77:652–676.
99 Gagliano DA, Goldberg MF. The evolution of salmon-patch hemorrhages in sickle cell retinopathy. Arch Ophthalmol. 1989;107:1814–1815.
100 Serjeant GR, Serjeant BE. The eyes. In: Serjeant GR, Serjeant BE. Sickle cell disease. 3rd ed. Oxford: Oxford University Press; 2001:366–392.
101 Emerson GG, Lutty GA. Effects of sickle cell disease on the eye: clinical features and treatment. Hematol Oncol Clin North Am. 2005;19:957–963.
102 Lutty GA, McLeod DS, Pachinis A, et al. Retinal and choroidal neovascularization in a transgenic mouse model of sickle cell disease. Am J Pathol. 1994;145:490–497.
103 McLeod DS, Merges C, Fukushima A, et al. Histopathologic features of neovascularization in sickle cell retinopathy. Am J Ophthalmol. 1997;124:455–472.
104 Goldberg MF. Natural history of untreated proliferative sickle retinopathy. Arch Ophthalmol. 1971;85:428–437.
105 Curtis AS, Seehar GM. The control of cell division by tension or diffusion. Nature. 1978;274:52–53.
106 van Meurs JC. Evolution of a retinal hemorrhage in a patient with sickle-cell hemoglobin C disease. Arch Ophthalmol. 1995;113:1074–1075.
107 Condon PI, Whitelocke RA, Bird AC, et al. Recurrent visual loss in homozygous sickle cell disease. Br J Ophthalmol. 1985;69:700–706.
108 Penman AD, Talbot JF, Chuang EL, et al. New classification of peripheral retinal vascular changes in sickle cell disease. Br J Ophthalmol. 1994:681–689.
109 Sayag D, Binaghi M, Souied EH, et al. Retinal photocoagulation for proliferative sickle cell retinopathy: A prospective clinical trial with new sea fan classification. Eur J Ophthalmol. 2008;18:248–254.
110 Downes SM, Hambleton IR, Chuang EL, et al. Incidence and natural history of proliferative sickle cell retinopathy: observations from a cohort study. Ophthalmol. 2005:1869–1875.
111 Fox PD, Dunn DT, Morris JS, et al. Risk factors for proliferative sickle retinopathy. Br J Ophthalmol. 1990;74:172–176.
112 Moriarty BJ, Acheson RW, Condon PI, et al. Patterns of visual loss in untreated sickle cell retinopathy. Eye. 1988;2:330–335.
113 Condon P, Jampol LM, Farber MD, et al. A randomized clinical trial of feeder vessel photocoagulation of proliferative sickle cell retinopathy.II. Update and analysis of risk factors. Ophthalmology. 1984;91:1496–1498.
114 Rednam KR, Jampol LM, Goldberg MF. Scatter retinal photocoagulation for proliferative sickle cell retinopathy. Am J Ophthalmol. 1982;93:594–599.
115 Shaikh S. Intravitreal bevacizumab (Avastin) for the treatment of proliferative sickle retinopathy. Indian J Ophthalmol. 2008;56:259.
116 Siquiera RC, Costa RA, Scott IU, et al. Intravitreal bevacizumab (Avastin) injection associated with regression of retinal neovascularization caused by sickle cell retinopathy. Acta Ophthalmol Scand. 2006;84:834–835.
117 Bove JR. Transfusion-transmitted diseases: current problems and challenges. Prog Hematol. 1986;14:123–147.
118 Pulido JS, Flynn HW, Clarkson JG, et al. Pars plana vitrectomy in the management of the complications of proliferative sickle retinopathy. Arch Opthalmol. 1988;106:1553–1557.
119 Williamson TH, Rajput R, Laidlaw DAH, et al. Vitreoretinal management of the complications of sickle cell retinopathy by observation or pars plana vitrectomy. Eye. 2009;23:1314–1320.
120 Diallo JW, Kuhn D, Hayman-Gawrilow P, et al. Contribution of indocyanine green angiography in sickle cell retinopathy. J Fr Ophthalmol. 2009;32:430–435.
[/level-membership-for-opthalmology-category][not-level-membership-for-opthalmology-category]
Chapter 57 Hemoglobinopathies
In a landmark 1910 Archives of Internal Medicine article,1 Dr James Herrick noted “peculiar, elongated, sickle-shaped red blood corpuscles” from a peripheral blood smear of a Grenadian dental student with recurring medical illnesses and, thus, penned the first written description of sickle-cell disease (SCD).1–3 Linus Pauling and colleagues in 1949 were the first to implicate a defective hemoglobin molecule within the erythrocyte as the cause of the disease.4,5
The term “sickle-cell disease” encompasses the group of hemoglobinopathies characterized by intravascular hemolysis and by defective oxygen transport. In a normal red blood cell, two α-globin subunits, two β-globin subunits, and a central heme molecule combine to form adult hemoglobin (Hb A). The β-globin gene, an oxygen transport gene, is found on the short arm of chromosome 11.6,7 Hemoglobin S (Hb S) results from a single amino acid point mutation in which a valine substitutes for a glutamic acid at the sixth position within the β-globin chain. Hemoglobin C (Hb C) is caused by a glutamic acid to lysine mutation in the β-globin molecule.
SCD is transmitted through the autosomal recessive mode of inheritance. Two copies of Hb S may combine with one another (SS disease), or one copy of Hb S and another β-globin variant, such as Hb C, may combine (double heterozygous SC disease).7 Individuals with one copy of Hb A and one copy of Hb S are described as having the sickle-cell trait, the carrier state for SCD. β-thalassemia occurs when a reduced amount of β-globin is present. This condition is beta-plus (β+) thalassemia, whereas the absence of β-globin is called beta-zero (β0). Either of these conditions may combine with Hb S, leading to a compound heterozygous state.7
Prevalence
To date, SCD remains the most common inherited blood disorder, and affects nearly 80 000 people of African American and Hispanic descent.8 SCD occurs in 1/500 African American births and every 1/36 000 Hispanic-American births.8–10 SCD results in 5% of deaths in children under 5 years in Africa, more than 9% of such deaths in West Africa, and up to 16% of under-5 deaths in individual West African countries.11 Approximately 8% (or one in 12) of black Americans possess sickle-cell trait, which is not associated with increased mortality or morbidity (except in hyphema), and is thought to confer a genetic protection against malarial infection.10 Their hemoglobin concentration is typically 35–40% Hb S and 55–60% Hb A.7 Although sickle-cell trait subjects are generally asymptomatic, they may encounter systemic complications of SCD under conditions of extreme hypoxia.12 Populations of African descent possess the highest frequency of Hb S. At-risk genotypes for SCD are also observed in people of Mediterranean, Caribbean, South and Central American, Arab, and East Indian descent.7 SS disease is displayed in approximately 0.15% of African descendants in North America.7,13 Those with SS disease do not typically become symptomatic until after 6 months of age, when the fetal hemoglobin (Hb F) is replaced with Hb S. These individuals have a decreased life expectancy, as they are prone to developing severe anemia and are highly susceptible to recurrent infections.14 Individuals with Hb SC disease, the double heterozygotes, typically exhibit 50% Hb S and 50% Hb C hemoglobin composition and typically display less morbidity from the systemic complications of SCD, and have a normal life expectancy.7 With a hemoglobin composition of 60–90% Hb S and 10–30% Hb F, sickle β-thalassemia patients are typically encountered in central African and Mediterranean countries. Those with other forms of sickle β-thalassemia may have Hb A as 10–30% of their hemoglobin composition.7,15
Genetic modifiers
The degree of phenotypic expression of SCD is quite variable, even among those with the same genotype. Environmental effects and multiple gene interactions are thought to be at play. Further, the ability of an individual to generate Hb F may lead to reduced disease severity. Different β-globin cluster haplotypes may result in different levels of Hb F.7,16 Levels of Hb F have been shown to correlate with the clinical manifestations of SCD.17
Pathophysiology
The interplay among abnormal erythrocytes and hypoxic, hyperosmolar, or acidotic conditions leads to the abnormal rheology and hemolysis characteristic of SCD. In Hb S, a strongly hydrophobic polar valine takes the place of a nonpolar strongly hydrophilic glutamic acid residue.5 Upon deoxygenation in the microcirculation, hydrophobic residues within Hb S are exposed and associate with hydrophobic regions of adjacent molecules. This polymerization results in the generation of rigid fibers of Hb S which damage the red blood cell membrane and cytoskeleton and cause the cell to assume a sickle shape. This polymerization process is reversible as oxygenation increases, and the cell may resume its native discoid shape. However, the repeated cycle of sickling and unsickling of the red blood cell membrane may lead to permanent damage to the erythrocyte membrane, irreversible sickling, and hemolysis. Mean corpuscular hemoglobin concentration (MCHC) may be the most important factor contributing to the rate of Hb S polymerization.5 The higher the MCHC, the more hemoglobin molecules that are available to participate in polymerization, and the closer these molecules are to one another, further promoting a favorable environment for Hb S polymerization.18,19
The erythrocyte’s original state of oxygenation also impacts the extent and rate of polymerization.15,16 An intrinsic property of the normal erythrocyte is the ability to deform easily in order to pass through capillaries with smaller diameters than its own. Decreased erythrocyte deformability and increased rigidity can cause increased capillary transit time.19 Deoxygenation and sickling promote increased permeability of the cell membrane to potassium, sodium, and calcium cations, which leads to water efflux from the cell, cellular volume contraction, and resultant increase in Hb S concentration.5,19,20,21
In addition to mechanical obstruction of blood vessels by dense, sickled erythrocytes, these sickled erythrocytes display increased adhesion to vascular endothelium matrix proteins, such as laminin,22,23 and thus cause direct damage to the endothelium. Integrin α4β1, integrin-associated protein, sulfated glycolipid, lutheran protein, phosphatidylserine, band 3 protein, and CD36 are adhesion molecules expressed in sickled red blood cells.24–26 Immature red blood cells called reticulocytes increase in SCD following intravascular hemolysis. These cells also have increased adhesion molecules, such as integrin α4β1,27–29 which promote pathologic adhesion to the vascular endothelium, specifically to vascular cell adhesion molecule-1 (VCAM-1). Direct activation of endothelial cells occurs in response to elevated circulating cytokines such as tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β),30,31 which upregulate expression of endothelial adhesion molecules like intercellular adhesion molecule-1 (ICAM-1), VCAM-1, E-selectin, and P-selectin.32,33
Inflammation likely plays a role in the vaso-occlusive process in SCD. Lutty and colleagues demonstrated retention of SS red cells and adherence of red cells in reticulocyte-rich fractions in retina and choroid of rat eyes in hypoxic conditions or following TNF-α stimulation.34–36 TNF-α and IL-1 may contribute to vaso-occlusion by accelerating the production of adhesion molecules on the vascular endothelium and by activating polymorphonuclear leukocytes.30 TNF-α and IL-1 have been shown to be upregulated in the sera of individuals with SCD at baseline.31,37,38
Nitric oxide (NO) is a potent vasodilator and regulator of vascular tone; it is derived from the vascular endothelial NO synthase. SCD has been associated with elevated reactive oxygen species, which scavenge NO and metabolize arginine, its precursor.7 l-arginine as an oral supplement has been given to induce NO production in transgenic sickle-cell mice.38,39
Vascular endothelial growth factor (VEGF), which is upregulated by hypoxia, is present in the serum of SCD patients at baseline, and is elevated during vaso-occlusive crises.40 Elevated VEGF has been demonstrated in eyes with sickle-cell retinopathy.41,42 VEGF has also been shown to increase levels of cell adhesion molecules, ICAM-1 and VCAM-1.43,44 Angiopoietin-1 (Ang-1) and angiopoietin-2 (Ang-2) interact with the Tie-2 receptor on endothelial cells, regulating angiogenesis. Ang-1 is responsible for the maintenance and stabilization of mature blood vessels, while Ang-2 leads to vessel destabilization and dissociation of pericytes and is upregulated by hypoxia and VEGF.45 Interaction among these proteins may be important in the pathogenesis of SCD.
Systemic manifestations
Over recent years, as the life expectancy of those affected by SCD has increased, so have the number of complications these individuals experience.12,46 Sickling of the red blood cells, with resultant intravascular hemolysis, thrombosis, tissue necrosis, and ischemia, causes a myriad of systemic complications, including cerebrovascular accident, acute chest syndrome, pulmonary hypertension, splenic sequestration, priapism, osteonecrosis, cholelithiasis, pneumonia, leg ulcers, aplastic crisis, renal disease, need for recurrent transfusions, episodic, painful vaso-occlusive crises, and death.7
Severe visual impairment and blindness from complications of proliferative sickle-cell retinopathy (PSR) may also occur. For reasons that are as yet unclear, subjects with Hb SC disease typically display less systemic morbidity from their hemoglobinopathy, but have a higher likelihood of experiencing the ophthalmic complications, than their homozygous Hb SS counterparts.47 In one study of patients with SCD, retinopathy was detected in 33% of patients with Hb SC disease compared to 3% of patients with Hb SS disease.37
Several theories have been proposed to explain this difference. One explanation could be that Hb SS patients typically have a shorter life expectancy than Hb SC patients, and may not live long enough to manifest the ophthalmic disease. Alternatively, the difference in retinopathy among sickle-cell genotypes may be due to the higher hematocrit and cell density, and the lower Hb F, of individuals with Hb SC as compared to Hb SS individuals.7 The overall lower hematocrit in Hb SS patients, and the resultant lowered viscosity of blood, may confer a relative protection from vaso-occlusion in the retinal circulation.48 Another theory is that the retinal vaso-occlusions in SS disease may be so complete that no viable tissue remains to produce the growth factors necessary to mount a proliferative response. In Hb SC disease, the vascular occlusions may be less complete, and they occur in the setting of a chronic hypoxic rather than anoxic state. Thus, a steady-state release of angiogenic growth factors from the remaining viable tissue may occur.49 Finally, Lutty and colleagues showed that irreversibly sickled Hb SS erythrocytes are easily trapped in retinal capillaries and precapillary arterioles in hypoxic conditions in a rat model.16,35 Hb SC cells, however, regardless of oxygen levels, were less easily trapped in the retinal microvasculature. When the vascular endothelium was stimulated with cytokines, retention of Hb SC cells did occur. The authors suggested that vaso-occlusion in Hb SC disease may be the result of the complex interaction among cytokines, the fibrinolytic cascade, leukocyte interactions, and activation of the vascular endothelium with induction of adhesion molecules.13,35 This data suggests that retention of sickled cells in the retina is not mechanical (rigid cells retained in a small lumen) but rather due to hypoxia-initiated cytokines and receptors.
Ophthalmic clinical features
Retrobulbar and orbital involvement
Orbital involvement of SCD is uncommon, but has been reported. One study from Oman reported periorbital swelling during vaso-occlusive crises in five patients.50 The patients ranged in age from 6 to 15 years old. Four of these patients had SS disease, while one had sickle-beta thalassemia. Infarction of orbital bones and orbital hematomas occur, and may lead to the orbital compression syndrome. These patients typically display lid edema, fever, facial pain, proptosis, restriction of motility, and resultant diplopia. The authors concluded that these syndromes usually resolve with conservative management with treatment of the underlying crisis, antibiotics, and steroids. Orbital involvement in SCD may cause sudden permanent unilateral loss of vision without detectable retinal arterial changes due to retrobulbar ischemic optic neuropathy.7,51 Urgent decompression of the orbit may be required. Recurrent bilateral lacrimal gland swelling has also been reported.52
Anterior-segment involvement
Conjunctival and iris findings may be present in SCD. An early reported finding is the saccular and sausage-like dilatations of the tiniest conjunctival vessels.53 They are most easily seen with the highest power of the slit-lamp biomicroscope. Paton described these comma-shaped capillary segments in the inferior bulbar conjunctival vessels, and coined the term “conjunctival sign.”54 This sign was uncommon in those with high Hb F levels. Local heat from the slit lamp induces vasodilation, and causes reversal of the comma-shaped vessels,55,56 whereas topical vasoconstricting drops increase the abnormality.57,58 These conjunctival vascular changes vary with oxygenation status, and are more prevalent in Hb SS disease than in Hb SC disease.54 Pathophysiology studies show constriction of these conjunctival vessels during painful crises, with normal blood flow returning on recovery.58 On histopathologic examination, these vessels exhibit endothelial proliferation, dilatation, and thinning of the proximal blood vessel segments, and aggregation of red blood cells in the distal portions of capillaries.58
Segmental iris atrophy and pupil abnormalities can be seen in individuals with SCD (Fig. 57.1).59,60 Iris atrophy is thought to result from sectoral ischemic necrosis involving radial iris vessels. Secondary neovascularization of the iris stroma has also been described and documented with intravenous fluorescein angiography, and in one instance this neovascular frond resembled that classically seen as the “sea fan” in PSR.13,61 Neovascular glaucoma rarely occurs as a consequence of PSR.7,62
Hyphema in a patient with SCD and in those with sickle-cell trait represents a sight-threatening emergency, as even modest elevations of intraocular pressure (IOP) have resulted in vision loss from central retinal artery occlusion or macular branch retinal artery occlusion.62–65 This is likely a mechanical phenomenon, as sickled red blood cells clog the trabecular meshwork,61 causing elevation in IOP. Although intensive medical management to lower IOP in SCD patients with hyphema is recommended, IOP-lowering medications must be used cautiously. Repetitive use of carbonic anhydrase inhibitors, for example, is contraindicated in SCD patients with hyphema as most of these medications cause intracameral acidotic conditions that may worsen erythrocyte sickling. Early surgical intervention for IOP control should be considered, typically by paracentesis of the anterior chamber.66 Intracameral tissue plasminogen activator may be a potentially useful agent in the treatment of posttraumatic hyphema in some cases.67 Hyperoxygenation of the patient may also be helpful.
Posterior-segment involvement
Vitreoretinal interface
The pathologic retinal neovascularization caused by retinal ischemia in SCD can result in vision loss from vitreous hemorrhage.68,69 Other abnormalities of the vitreoretinal interface have been described, such as peripheral retinal whitening, which may represent abnormal or particularly strong adhesion at the vitreoretinal interface. This has been described in 93% of Hb SS patients,70 83% of Hb SC patients,71 and 82% of Hb S Thal patients.72 There is no known pathologic significance to this finding. This peripheral whitening is noted in the peripheral retina in the normal population as “white without pressure,” and may not be truly unique to those with sickle-cell retinopathy.13,57
Correspondingly, flat, brown, ovoid lesions in the retinal periphery have been noted in SCD, and termed “dark without pressure.” These lesions can be transient, and may disappear without sequelae. The underlying choroid appears normal on ophthalmoscopy, and the corresponding fluorescein angiogram is also normal.13,73
Optic nerve
The optic nerve in patients with SCD may exhibit vascular changes. Dark, dilated capillaries at the optic nerve head appear as small red dots (Fig. 57.2), and represent precapillary arterioles plugged with sickled erythrocytes. These abnormal vessels have a linear or Y-shaped configuration on high-magnification ophthalmoscopy.13,57,70,74 These changes are not visually significant as the vascular occlusions are transient. Optic disc neovascularization is rare but has been reported in four Hb SC patients13,75–78 and one Hb SS patient.13,70
Macula
Though the hallmark of ocular SCD has been the peripheral retinopathy, the macula is also susceptible to infarction from vaso-occlusive disease. The “macular depression sign” has been described as an oval depression of the bright foveal or parafoveal reflex as a result of macular thinning due to ischemic atrophy.79 This finding is best detected using red-free illumination. The extent of macular vascular changes has not been shown to correlate with visual acuity. In a study by Sanders and colleagues, an enlarged foveal avascular zone (FAZ) was identified in patients with SCD on fluorescein angiography as compared to healthy, age-matched controls.80 Within the sickle-cell group, visual acuity did not correlate with FAZ size. The authors reported an enlarged FAZ in patients with SCD irrespective of the type of hemoglobinopathy or degree of retinopathy.
Macular infarcts can now be confirmed using optical coherence tomography.81 In a recent study utilizing spectral-domain optical coherence tomography (SD-OCT), Hoang and coworkers82 described retinal thinning of the central macula compared to healthy controls, most notably in the outer retinal layers in the central macula and parafoveal retina. Although several previous studies have reported atrophy of inner retinal layers after macular infarction, Hoang et al. hypothesize that the outer retinal layers may be thinned due to ischemia and resultant atrophy of the choriocapillaris, as these larger-caliber vessels may possibly be more prone to occlusion than the retinal vessels. Additionally, this study reported “splaying,” or blunting of the foveal contour on SD-OCT in asymptomatic patients with SCD with areas of focal parafoveal thinning (Fig. 57.3). A case series by Murthy et al. also describes thinning of the temporal macula in patients with PSR on SD-OCT, and suggests that this finding should lead clinicians to perform peripheral wide-field angiography to evaluate the retinal periphery for ischemia.83 Macular hole,84 epiretinal membranes,85 macular schisis,86 and posterior pole neovascularization87 have all been reported as rare complications of sickle-cell retinopathy.
Angioid streaks
Angioid streaks, breaks in Bruch’s membrane, have a well-documented association with SCD, occurring in 1–2% of patients.13,70,88 These irregular, reddish subretinal bands are most commonly found in patients with the Hb SS genotype.70,89
[/not-level-membership-for-opthalmology-category]
