[level-membership-for-hematology-oncology-and-palliative-medicine-category]
Chapter 11 Heme Biosynthesis and Its Disorders
Sideroblastic Anemia
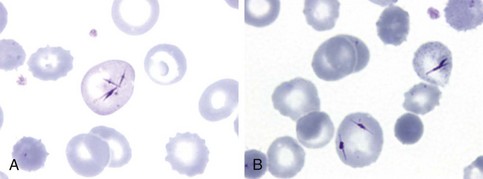
Figure 11-1 NEEDLE-LIKE INCLUSIONS OF PORPHYRIN IN THE CIRCULATING RED CELLS OF A PATIENT WITH CONGENITAL PORPHYRIA AFTER SPLENECTOMY.
(From Merino A, To-Figueras J, Herrero C: Atypical red cell inclusions in congenital erythropoietic porphyria. Br J Haematol 132:124, 2006.)
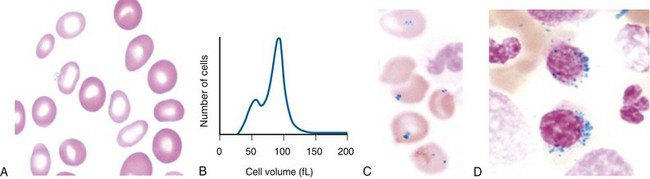
Figure 11-2 A, Peripheral blood smear from a patient with hereditary sideroblastic anemia shows a population of hypochromic and microcytic erythrocytes. B, Erythrocyte volume distribution curve of a patient with hereditary sideroblastic anemia. A dimorphic size distribution is evident. C, Peripheral blood showing Pappenheimer bodies (Prussian blue stain). D, The bone marrow smear stained with Prussian blue shows ring sideroblasts.
Table 11-1 Classification of Sideroblastic Anemias
| HEREDITARY (NONSYNDROMIC) |
| ACQUIRED |
|
Idiopathic acquired* (refractory anemia with ring sideroblasts) Associated with previous chemotherapy, irradiation, or in transition myelodysplasia or myeloproliferative diseases |
| DRUGS |
| RARE CAUSES |
| HEREDITARY (SYNDROMIC) |
*Trial of pyridoxine indicated.
Therapy for Hereditary Sideroblastic Anemia
About 25% to 50% of patients with hereditary sideroblastic anemia show a full or partial response to pyridoxine, and this vitamin should be continued on a lifelong basis in the responders. A lower maintenance dose should be determined for each responding patient by progressive dose reduction, because long-term therapy with pyridoxine at 100 to 200 mg/day has been associated with peripheral neuropathy.1 The adult nutritional requirement for pyridoxine is 1 to 2 mg/day; some patients have been maintained on as little as 4 mg/day as a supplement.2 Folic acid supplements should also be administered because the erythroid hyperplasia increases demand for this vitamin.
There is one report of successful allogeneic peripheral blood stem cell transplantation in a 19-year-old man with transfusion-dependent hereditary sideroblastic anemia.3 Transfusions are the mainstay of treatment for severe anemia unresponsive to pyridoxine. Regular administration of packed red cells using white blood cell filters are given to relieve symptoms and permit normal childhood development. Iron overload and secondary hemosiderosis rapidly progress after transfusions begin; chelation therapy with desferrioxamine or oral deferasirox should be initiated from the onset.
Iron removal may be of great benefit for patients who have mild or moderate anemia and evidence of iron overload.4,5 These patients can often tolerate intermittent phlebotomy, which is preferable to chelation therapy for iron removal, and should be continued to reduce ferritin levels to less than 300 ng/mL. All patients with iron overload should avoid ingestion of ascorbic acid supplements, which enhance iron absorption and increase the tissue toxicity of elemental iron. Alcohol should also be avoided. Splenectomy is contraindicated in this disease.
Therapy for Acquired Sideroblastic Anemia
Transfusions are indicated for relief of symptomatic anemia. A trial of pyridoxine at 100 to 200 mg/day for 3 months is worthwhile in patients who have anemia but who do not display neutropenia or thrombocytopenia. However, few patients with acquired idiopathic sideroblastic anemia respond to this vitamin. If any response is achieved, maintenance therapy with pyridoxine at lower dosage is indicated. Cyclosporin (5 to 6 mg/kg/day) has been reported to benefit the anemia of the closely related myelodysplastic condition of refractory anemia, although the response appeared limited to those with hypoplastic bone marrows.14
Clinical and Laboratory Evaluation of Sideroblastic Anemia
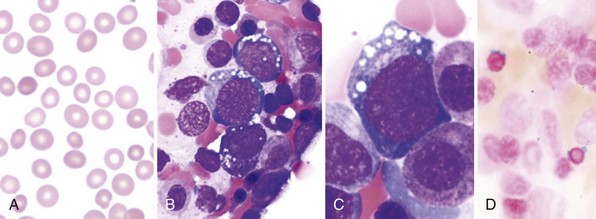
Typically, sideroblastic anemia develops insidiously in a middle-aged or elderly patient with normal or increased mean corpuscular volume (MCV) and a blood smear showing a population of hypochromic red cells. Hepatosplenomegaly may be present. Leukocyte and platelet counts are usually normal, but some patients have thrombocytosis that occasionally exceeds 1000 × 109/L.6 If leukopenia or thrombocytopenia is present, a careful search should be made for myelodysplastic features, which lead to the more descriptive term of refractory cytopenia for the condition.7,8 An iron stain of the bone marrow aspirate shows ring sideroblasts, which should total more than 15% of all erythroblasts to make the diagnosis of acquired sideroblastic anemia.8–10 Iron cannot be assessed in the marrow trephine biopsy core because it may leach out during decalcification.
The bone marrow also shows erythroid hyperplasia. Although mild dyserythropoiesis (e.g., multinuclearity, nuclear budding) and megaloblastoid changes are present, myelopoiesis and megakaryopoiesis are usually normal. When changes are confined to dyserythropoiesis, the condition has been called pure sideroblastic anemia.8,11 However, dysplasia of myelopoietic and megakaryopoietic elements may be present (i.e., trilineage dysplasia) with the following features: Pelger-Huët–like anomaly, hypersegmentation or hypogranularity of neutrophils, micromegakaryocytes, large mononuclear megakaryocytes, and megakaryocytes with multiple small nuclei (see Chapter 24).8 Dysmegakaryopoiesis is more easily detected in trephine biopsies than in marrow smears, although the trephine may also show unsuspected islands of myeloblasts characteristic of myelodysplasia.12 The overall blast count in marrow smears is, by definition, less than 5%, and the peripheral blood monocyte count is less than 1.0 × 109/L. Cytogenetic analysis of marrow aspirates provides important information, because a normal karyotype predicts long survival in any type of acquired sideroblastic anemia.13
1 Parry GJ, Bredesen DE. Sensory neuropathy with low-dose pyridoxine. Neurology. 1985;35:1466.
2 Cox TC, Bottomley SS, Wiley JS, et al. X-linked pyridoxine-responsive sideroblastic anemia due to a THR388-to-SER substitution in erythroid 5-aminolevulinate synthase. N Engl J Med. 1994;330:675.
3 Gonzales MI, Caballero D, Vazquez L, et al. Allogeneic peripheral blood stem cell transplantation in a case of hereditary sideroblastic anaemia. Br J Haematol. 2000;109:658.
4 Peto TEA, Pippard MJ, Weatherall DJ. Iron overload in mild sideroblastic anaemias. Lancet. 1983;1:375.
5 Cazzola M, Barosi G, Bergamaschi G, et al. Iron loading in congenital dyserythropoietic anaemias and congenital sideroblastic anaemias. Br J Haematol. 1983;54:649.
6 Streeter RR, Presant CA, Reinhard E. Prognostic significance of thrombocytosis in idiopathic sideroblastic anemia. Blood. 1977;50:427.
7 Kampmeier P, Anastasi J, Vardiman JW, et al. Issues in the pathology of the myelodysplastic syndromes. Hematol Oncol Clin North Am. 1992;6:501.
8 Gattermann N, Aul C, Schneider W. Two types of acquired idiopathic sideroblastic anaemia (AISA). Br J Haematol. 1990;74:45.
9 Bennett JM, Catovsky D, Daniet MT, et al. Proposals for the classification of the myelodysplastic syndromes. Br J Haematol. 1982;51:189.
10 May SJ, Smith SA, Jacobs A, et al. The myelodysplastic syndrome: Analysis of laboratory characteristics in relation to the FAB classification. Br J Haematol. 1985;59:311.
11 Clatch RJ, Krigman HR, Peters MG, et al. Dysplastic haemopoiesis following orthotopic liver transplantation: Comparison with similar changes in HIV infection and primary myelodysplasia. Br J Haematol. 1994;88:685.
12 Tricot G, De Wolf-Peeters C, Hendricks B, et al. Bone marrow histology in myelodysplastic syndromes. I. Histological findings in myelodysplastic syndromes and comparison with bone marrow smears. Br J Haematol. 1984;57:423.
13 Yunis J, Rydell RE, Oken MM, et al. Refined chromosome analysis as an independent prognostic indicator in de novo myeloplastic synQ dromes. Blood. 1986;67:1721.
14 Jonasova A, Neuwirtova R, Cermak J, et al. Cyclosporin A therapy in hypoplastic MDS patients and certain refractory anemias without hypoplastic bone marrow. Br J Haematol. 1998;100:304.
[/level-membership-for-hematology-oncology-and-palliative-medicine-category][not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
Chapter 11 Heme Biosynthesis and Its Disorders
Sideroblastic Anemia
Figure 11-1 NEEDLE-LIKE INCLUSIONS OF PORPHYRIN IN THE CIRCULATING RED CELLS OF A PATIENT WITH CONGENITAL PORPHYRIA AFTER SPLENECTOMY.
(From Merino A, To-Figueras J, Herrero C: Atypical red cell inclusions in congenital erythropoietic porphyria. Br J Haematol 132:124, 2006.)
Figure 11-2 A, Peripheral blood smear from a patient with hereditary sideroblastic anemia shows a population of hypochromic and microcytic erythrocytes. B, Erythrocyte volume distribution curve of a patient with hereditary sideroblastic anemia. A dimorphic size distribution is evident. C, Peripheral blood showing Pappenheimer bodies (Prussian blue stain). D, The bone marrow smear stained with Prussian blue shows ring sideroblasts.
Table 11-1 Classification of Sideroblastic Anemias
| HEREDITARY (NONSYNDROMIC) |
| ACQUIRED |
|
Idiopathic acquired* (refractory anemia with ring sideroblasts) Associated with previous chemotherapy, irradiation, or in transition myelodysplasia or myeloproliferative diseases |
| DRUGS |
| RARE CAUSES |
| HEREDITARY (SYNDROMIC) |
