[level-membership-for-critical-care-medicine-category]
Hematologic Disorders and Oncologic Emergencies
Overview of Coagulation and Fibrinolysis
Hemostasis, the ability of the body to control bleeding and clotting, is an intricate balancing act between the coagulation mechanism and fibrinolysis. Four major actions are involved in achieving hemostasis: 1) local vasoconstriction to reduce blood flow; 2) platelet aggregation at the injury site and formation of a platelet plug; 3) formation of a fibrin mesh to strengthen the plug; and 4) dissolution of the clot after tissue repair is complete.1 Disruption of the normal hemostatic balance can result in devastating hemorrhagic or thrombotic conditions.
Coagulation Mechanism
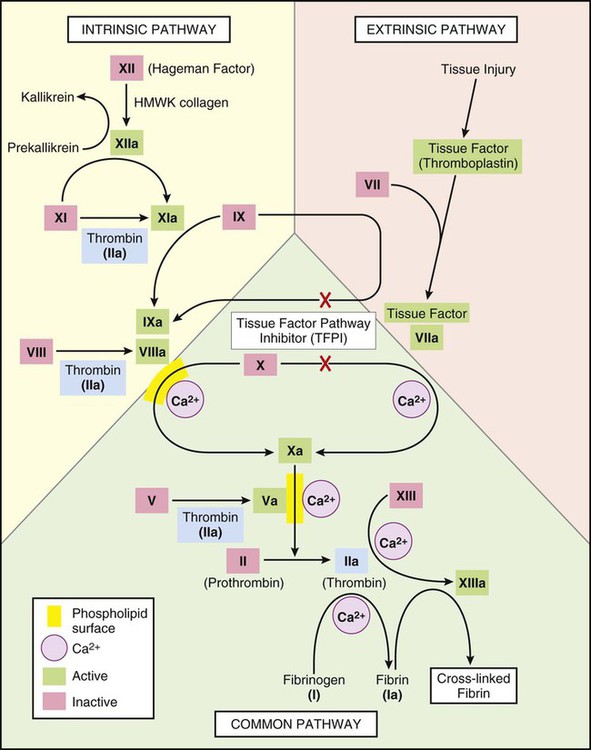
The coagulation mechanism consists of 13 factors that work together through a series of feedback loops to achieve hemostasis (Table 38-1 and Fig. 38-1). Depending on the initial triggering event, the extrinsic or intrinsic coagulation pathway is intiated.1,2 The extrinsic pathway begins when vascular injury occurs, resulting in release of tissue factor and activation of coagulation factor VII. The intrinsic pathway is activated when the damaged subendothelium comes into direct contact with circulating blood. In this contact phase, proteins activate additional coagulation factors (XII, XI, IX, and VIII).1 At this point, the two pathways converge into a common pathway, and prothrombin and fibrinogen are converted to their active forms, resulting in clot formation.3,4
TABLE 38-1
COAGULATION FACTORS AND COMMON NAMES
| FACTOR | COMMON NAME |
| I | Fibrinogen |
| II | Prothrombin |
| III | Tissue factor |
| IV | Calcium |
| V | Proaccelerin |
| VI | Accelerin |
| VII | Proconvertin |
| VIII | Antihemophilic |
| IX | Christmas factor |
| X | Stuart |
| XI | Plasma thromboplastin antecedent |
| XII | Hageman factor |
| XIII | Fibrin stabilizing factor |
Clot Formation
Platelets are activated by the arrival of thrombin at the site of injury (Fig. 38-2). Local platelets change shape, become sticky, and begin to aggregate along the vessel wall. Activated platelets undergo degranulation, releasing several factors to assist in clot formation. Serotonin and histamine, two potent vasoconstrictors, help limit blood loss while the clot is forming. The prostaglandin thromboxane A2 (TXA2) contributes to vasoconstriction and promotes further platelet degranulation. Adenosine diphosphate (ADP) recruits platelets by increasing adherence and degranulation,2,3 and the process continues.
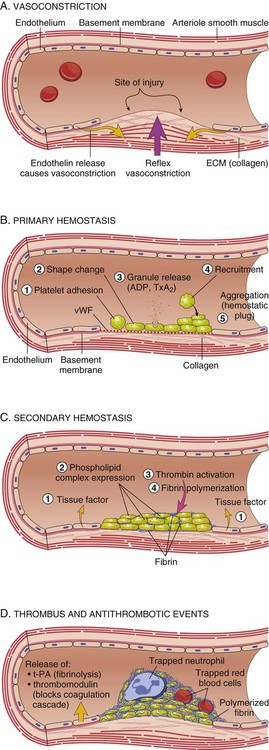
At the convergence of the intrinsic and extrinsic pathways, factor X is converted into its active form, thereby enabling the conversion of prothrombin to thrombin. Thrombin then converts fibrinogen to fibrin. Strands of fibrin form and radiate around the newly formed clot, essentially creating a net in which platelets, red blood cells (RBCs), and white blood cells (WBCs) are trapped.1 The clot is further secured to the site as platelet actomyosin causes it to contract and consolidate.2
Regulatory Mechanisms
Under normal conditions, feedback systems prevent the coagulation process from spinning out of control. Prostacyclin I2 (PGI2) is a prostaglandin released from damaged endothelial cells. It counteracts the effects of TXA2, serotonin, and histamine through vasodilation and inhibition of platelet degranulation.1–3 Another means of regulating clot formation is through inhibition of enzymes necessary for activation of coagulation factors along the intrinsic and extrinsic pathways, preventing the conversion of prothrombin to thrombin. The most important of these regulators is antithrombin III; however, protein C and protein S also play a role in thrombin inhibition.5
Fibrinolysis
The process of fibrinolysis promotes dissolution and remolding of the clot to promote repair of the vessel wall and maintain flow through the vessel lumen (Fig. 38-2D). Fibrinolysis begins as soon as the fibrin clot is formed. Circulating plasminogen, a precursor to the powerful enzyme plasmin, binds to fibrin and is trapped within the newly formed clot. The injured epithelial wall releases tissue-type plasminogen activator (tPA), which converts plasminogen to its active state, plasmin. The plasmin begins to digest the fibrin, rapidly breaking down the clot.2,4 When fibrin is broken down, the fibrin degradation products released act as anticoagulants.
Disseminated Intravascular Coagulation
Description
Disseminated intravascular coagulation (DIC) is a syndrome that arises as a complication of other serious or life-threatening conditions. Although DIC is not seen often, it can seriously hamper diagnostic and treatment efforts for the critically ill patient. An understanding of the etiologic and pathophysiologic mechanisms of DIC can assist in anticipating the syndrome’s occurrence, recognizing its signs and symptoms, and prompting intervention. Also known as consumptive coagulopathy, DIC is characterized by bleeding and thrombosis, both of which result from depletion of clotting factors, platelets, and RBCs. If not treated quickly, DIC will progress to multiple organ failure and death.6
Etiology
Many clinical events can prompt the development of DIC in the critically ill patient, but the exact underlying trigger may not be identifiable (Box 38-1). There are, however, some commonly known conditions associated with the development of DIC.
Sepsis, particularly that caused by gram-negative organisms, can be identified as the culprit in as many as 20% of cases, making it the most common cause of DIC. In this instance, endotoxins serve as a trigger for activation of tissue factor and the extrinsic coagulation pathway. Metabolic acidosis and hypoperfusion associated with shock syndromes can result in increased formation of free radicals and damage to tissues. Tissue factor is activated, resulting in DIC. Massive trauma or burns are frequently associated with DIC. Direct tissue damage activates the extrinsic coagulation pathway, and damage to endothelial surfaces activates the intrinsic pathway.3 Obstetric emergencies, such as abruptio placenta, retained placenta, or incomplete abortion, are also associated with the development of DIC. Tissue factor is concentrated in the placenta, and damage or disruption of this structure can activate coagulation pathways, resulting in coagulopathy.7
Pathophysiology
Regardless of the cause, the common thread in the development of DIC is damage to the endothelium that results in activation of the coagulation mechanism (Fig. 38-3).1 The extrinsic coagulation pathway plays a major role in the development of DIC. Direct damage to the endothelium results in the release of tissue factor and activation of this pathway. The secondary surge of thrombin formation as a result of activation of the intrinsic coagulation pathway leads to the massive disruption of the delicate balance that is hemostasis. Excessive thrombin formation results in rapid consumption of coagulation factors and depletion of regulatory substances—protein C, protein S, and antithrombin.5 With no checks and balances, thrombi continue to form along damaged epithelial walls, resulting in occlusion of the vessels. As occlusion reaches a critical level, tissue ischemia ensues, leading to further tissue damage and perpetuating the process. Eventually, end-organ function is affected by the ischemia, and failure is evident.1
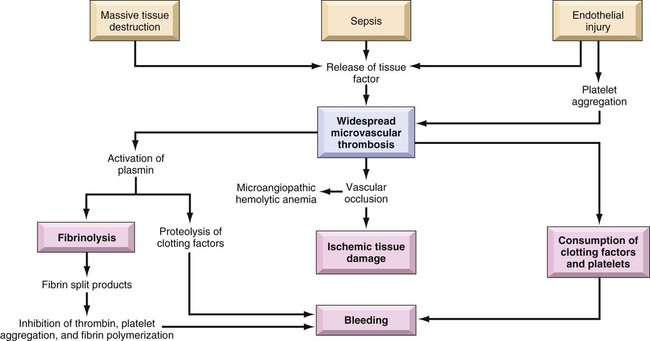
In response to the formation of clots, the fibrinolytic system is activated. As plasmin breaks down the fibrin clots, fibrin split products are released, and they act as anticoagulants.3,7 Coupled with depletion of circulating clotting factors, activation of fibrinolysis results in excessive bleeding. The end result is shock and further tissue ischemia that aggravate end-organ dysfunction and failure. Death is imminent if this destructive cycle is not interrupted.8
Assessment and Diagnosis
Clinical Manifestations
Clinical manifestations are related to the two primary pathophysiologic mechanisms of DIC: the formation of thrombi and bleeding. Thrombi in peripheral capillaries can lead to cyanosis, particularly in the fingers, toes, ears, and nose. In severe, untreated cases, this peripheral ischemia may progress to gangrene.3,6 As the condition progresses, ischemia worsens, and end organs are affected. The result of this more central ischemia can be respiratory insufficiency and failure, acute kidney injury, bowel infarction, and ischemic stroke. The tissue damage that results perpetuates the anomalies of DIC.3
As coagulation factors are depleted, bleeding from intravenous and other puncture sites is observed. Ecchymoses may result from even routine interventions such as the use of a manual blood pressure cuff, bathing, or turning.3,6 Bloody drainage may also occur from surgical sites, drains, and urinary catheters. With progression of DIC, the patient is at risk for severe gastrointestinal or subarachnoid hemorrhage.3,6 Table 38-2 lists many of the common signs and symptoms of DIC.
TABLE 38-2
COMMON SIGNS AND SYMPTOMS OF DISSEMINATED INTRAVASCULAR COAGULATION
| SYSTEM | SIGNS RELATED TO HEMORRHAGE | SIGNS RELATED TO THROMBI |
| Integumentary | Bleeding from gums, venipunctures, and old surgical sites; epistaxis; ecchymoses | Peripheral cyanosis, gangrene |
| Cardiopulmonary | Hemoptysis | Dysrhythmias, chest pain, acute myocardial infarction, pulmonary embolus, respiratory failure |
| Renal | Hematuria | Oliguria, acute kidney injury, renal failure |
| Gastrointestinal | Abdominal distention, hemorrhage | Diarrhea, constipation, bowel infarct |
| Neurologic | Subarachnoid hemorrhage | Altered level of consciousness, ischemic stroke |
Laboratory Findings
Laboratory tests used to diagnose DIC essentially assess the four basic characteristics of this syndrome: 1) increased coagulant activity; 2) increased fibrinolytic activity; 3) impaired regulatory function; and 4) end-organ failure.3
Continuous activation of the coagulation pathways results in consumption of coagulation factors. Because of this, the prothrombin time (PT), the activated partial thromboplastin time (aPTT), and the international normalized ratio (INR) values are elevated. Although the platelet count may fall within normal ranges, serial examination reveals a declining trend in values. An unexpected drop of at least 50% in the platelet count, particularly in the presence of known contributing factors and associated signs and symptoms, strongly indicates DIC.1 Fibrinogen levels drop as more and more clots are formed. Thrombus formation in small vessels narrows the vessel lumen, forcing RBCs to squeeze through. The resulting damage and fragmentation of these cells can be seen on microscopic examination of blood samples. Damaged, fragmented RBCs are called schistocytes.3,6
In response to the excess clotting activity, the fibrinolytic process accelerates, and levels of byproducts increase. This is reflected in markedly elevated levels of fibrin degradation products. Another key laboratory test used to evaluate the degree of clot dissolution—and therefore the severity of the coagulopathy—is the d-dimer level.1 d-dimers exclusively indicate clot degradation because, unlike fibrin degradation products, which also result from the breakdown of free circulating fibrin, d-dimers result only from dissolution of clots.3 With progression of the coagulopathy, normal regulatory mechanisms are disrupted, as reflected in decreasing levels of inhibitory factors such as protein C, factor V, and antithrombin III.3,6,9
Unchecked DIC resulting in occlusion of vessels and tissue ischemia leads to end-organ dysfunction. Respiratory failure, indicated by abnormal arterial blood gas (ABG) levels; liver failure, indicated by increasing liver enzymes; and renal impairment, indicated by rising blood urea nitrogen (BUN) and creatinine levels are common findings in advanced DIC.9
No single laboratory study can confirm the diagnosis of DIC, but several key results are strong indicators of the condition (Table 38-3). The International Society of Thrombosis and Hemostasis emphasizes early detection of DIC through observation of abnormal trends in laboratory values.5
TABLE 38-3
KEY LABORATORY STUDIES IN DISSEMINATED INTRAVASCULAR COAGULATION
| TEST | VALUE |
| Prothrombin time (PT) | >12.5 sec |
| Platelets | <50,000/mm3, or at least 50% drop from baseline |
| Activated partial thromboplastin time (aPTT) | >40 sec |
| d-dimer | >250 ng/mL |
| Fibrin degradation products (FDP) | >40 mg/mL |
| Fibrinogen | <100 mg/dL |
Medical Management
Without question, the primary intervention in DIC is prevention. Being aware of the conditions that commonly contribute to the development of DIC and treating them vigorously and without delay provide the best defense against this devastating condition.3,6,7,9 After DIC is identified, maintaining organ perfusion and slowing consumption of coagulation factors are paramount to achieving a favorable outcome.1,3
Multiple organ dysfunction syndrome (MODS) frequently results from DIC and exacerbates the underlying pathology.8 It is essential to prevent end-organ ischemia and damage by supporting blood pressure and circulating volume. Administration of intravenous fluids and inotropic agents and, if overt hemorrhaging is evident, infusion of packed RBCs are appropriate interventions to replace blood volume and essential, oxygen-carrying RBCs.
In the presence of severe platelet depletion (less than 50,000/mm3) and severe hemorrhage, platelet transfusions are often indicated.5,6 However, caution must be used when administering platelets because antiplatelet antibodies may be formed. These antibodies may become activated during future platelet transfusions and elicit DIC.3
Replacement of clotting factors in the patient with DIC is thought by some authorities to perpetuate the coagulopathy; however, there is little scientific evidence to support this theory.1 Fibrinogen levels less than 100 mg/dL indicate the appropriateness of administering cryoprecipitate. A prolonged PT indicates the need for fresh-frozen plasma.3,5,6
Slowing consumption of coagulation factors by inhibiting the processes involved in clot formation is another strategy used in treating DIC. The use of heparin, particularly low–molecular- weight heparin (LMWH), to prevent formation of future clots is controversial.6 It is contraindicated in patients with DIC associated with recent surgery or with gastrointestinal or central nervous system (CNS) bleeding. However, heparin has been beneficial in obstetric emergencies such as retained placenta or incomplete abortion, severe arterial occlusions, or MODS caused by microemboli.3,6 Inhibitors such as aminocaproic acid may be used in conjunction with heparin.3,6
The use of recombinant activated protein C is gaining popularity in treating DIC, especially in the setting of severe sepsis. Activated protein C acts as an anticoagulant and works to restore normal inhibition of coagulation pathways. However, it has been associated with an increased incidence of intracerebral bleeding and must be used with caution in patients with severely decreased platelets.3,5
Thrombin production in DIC surpasses that of antithrombins and other regulatory factors that would normally be present to inactivate thrombin and its subsequent actions. The use of antithrombin III has recently been approved in the United States. Ongoing research is yielding mixed results in the treatment of DIC.5 One interesting area of research is the use of protease inhibitors. Protease molecules normally inhibit the conversion of fibrinogen to fibrin in the coagulation mechanism, but in DIC, this inhibitory mechanism is impaired. The introduction of protease inhibitors by intravenous infusion may be advantageous in arresting DIC.3
Nursing Management
Nursing management of the patient with DIC incorporates a variety of nursing diagnoses (Box 38-2). Assessment and monitoring are the primary weapons in the critical care nurse’s arsenal against DIC. Knowing the diseases and conditions that are most often associated with DIC and understanding the pathophysiologic mechanisms involved enables the critical care nurse to anticipate its development and intervene quickly. Nursing interventions include supporting the patient’s vital functions, initiating bleeding precautions, providing comfort and emotional support, and maintaining surveillance for complications.
 Box 38-2 Nursing Diagnoses
Box 38-2 Nursing Diagnoses
Disseminated Intravascular Coagulation
Supporting Patient’s Vital Functions
Frequent assessments should include parameters for neurologic status, renal function, cardiopulmonary function, and skin integrity that indicate impaired tissue or organ perfusion. Particular parameters to include are mental status, BUN and creatine levels, urine output, vital signs, hemodynamic values, cardiac rhythm, arterial blood gas and pulse oximetry values, skin breakdown, ecchymoses, or hematomas.6
The critical care nurse must recognize and support the patient’s vital physiologic functions. Administration of intravenous fluids, blood products, and inotropic agents to provide adequate hemodynamic support and tissue oxygenation is essential in preventing or combating end-organ damage. Close monitoring of vital signs, hemodynamic parameters, intake and output, and appropriate laboratory values assists the critical care nurse in administering and titrating appropriate agents.3
Initiating Bleeding Precautions
Awareness of the patient’s bleeding potential necessitates adjustments to normal nursing interventions (Box 38-3). The nurse avoids unnecessary venipunctures that may result in bleeding, bruising, or hematomas by drawing blood from and administering medications through existing arterial or venous lines. The use of manual or automatic blood pressure cuffs is avoided whenever possible. If tracheal or oral suctioning is necessary, the use of low-level suction is recommended.6 Meticulous skin care is advised, keeping the skin moist and using specialty mattresses and beds as appropriate to prevent breakdown. Gentle care should be used when bathing or turning the patient to prevent bruising or hematoma formation.
 Box 38-3
Box 38-3
NIC
Bleeding Precautions
Reduction of stimuli that may induce bleeding or hemorrhage in at-risk patients
Monitor the patient closely for hemorrhage
Note hemoglobin/hematocrit levels before and after blood loss, as indicated
Monitor for signs and symptoms of persistent bleeding (e.g., check all secretions for frank or occult blood)
Monitor coagulation studies, including prothrombin time (PT), partial thromboplastin time (PTT), fibrinogen, fibrin degradation/split products, and platelet counts, as appropriate
Monitor orthostatic vital signs, including blood pressure
Maintain bed rest during active bleeding
Administer blood products (e.g., platelets, fresh-frozen plasma), as appropriate
Protect the patient from trauma, which may cause bleeding
Avoid injections (IV, IM, or SQ), as appropriate
Instruct the ambulating patient to wear shoes
Use soft toothbrush or toothettes for oral care
Use electric razor, instead of straight-edge, for shaving
Tell patient to avoid invasive procedures; if they are necessary, monitor closely for bleeding
Coordinate timing of invasive procedures with platelet or fresh-frozen plasma transfusions, if appropriate
Refrain from inserting objects into a bleeding orifice
Avoid taking rectal temperatures
Administer mediations (e.g., antacids), as appropriate
Instruct patient to avoid aspirin and other anticoagulants
Instruct patient to increase intake of foods rich in vitamin K
Use therapeutic mattress to minimize skin trauma
Avoid constipation (e.g., encourage fluid intake and stool softeners), as appropriate
Instruct the patient and/or family on signs of bleeding and appropriate actions (e.g., notify the nurse), should bleeding occur
From Bulechek GM, et al. Nursing Interventions Classification (NIC). 6th ed. St. Louis: Mosby; 2013.
Thrombocytopenia
Description
Thrombocytopenia is defined as a platelet count less than 140,000/mm3.10,11 Similar to DIC, thrombocytopenia often results from another underlying condition that affects the platelet count. Thrombocytopenia is more common in women than in men, affecting adults most often between the ages of 20 and 50 years.12,13
Etiology
Onset of thrombocytopenia often follows a viral infection, pregnancy, administration of certain medications (e.g., heparin, thiazide diuretics, chemotherapeutic agents), malignancies, splenomegaly, blood transfusions, or alcoholism.12 Regardless of the precipitating condition, the development of thrombocytopenia occurs by means of one of four mechanisms: 1) decreased platelet production; 2) increased platelet destruction; 3) splenic sequestration of platelets; and 4) platelet dilution.10 The most common form of thrombocytopenia seen in the critical care unit is immune thrombocytopenic purpura (ITP).11
Pathophysiology
In ITP, lymphocytes produce antibodies that begin to destroy existing platelets. The cause of this autoimmune response is unknown.13 With insufficient platelets available, the normal coagulation pathways are disrupted. Inadequate hemostasis ensues, and bleeding results. Although life-threatening gastrointestinal or intracerebral bleeding can occur, the bleeding seen in ITP does not result in deep visceral hemorrhage and hematoma.10
Assessment and Diagnosis
ITP is characterized by the gradual onset of signs and symptoms.13 The diagnosis is primarily based on findings in the patient’s history and physical examination.
Clinical Manifestations
Petechial hemorrhages, which manifest as small red spots primarily on legs and oral mucosa, are most indicative of a platelet disorder, unlike larger hematomas, which are most commonly associated with coagulation disorders.11 Bruising unrelated to trauma is another common sign of ITP.12 The signs and symptoms of ITP are listed in Table 38-4.
TABLE 38-4
IDIOPATHIC THROMBOCYTOPENIA PURPURA: SIGNS, SYMPTOMS, AND LABORATORY DATA
| SYSTEM OR STUDY | SIGNS AND SYMPTOMS |
| Integumentary | Petechial hemorrhage of lower extremities, ecchymoses, gingival bleeding, spontaneous epistaxis |
| Neurologic | Sudden, severe headache; nausea and vomiting; seizures; focal neurologic deficits; decreased level of consciousness |
| Renal | Hematuria |
| Gastrointestinal | Hematemesis, melena, hematochezia |
| Other | Heavy menses in women, retinal hemorrhage |
| Laboratory | Decreased platelet count, often <30,000 mm3 |
Unusual bleeding is a hallmark of ITP. Excessive bleeding from the gums after dental work, spontaneous epistaxis, blood in the urine or stool, and in women, unusually heavy menses are typical features of ITP. Rarely, retinal hemorrhage or intracerebral bleeding may be observed.13,14
Medical Management
In most cases, ITP resolves spontaneously, and treatment is not necessary. In mild cases in which diminished platelets counts result in symptoms, administration of oral corticosteroids is appropriate.10,13 Platelet counts will increase to normal levels with 2 to 6 weeks, and dosages can then be tapered.
In patients exhibiting life-threatening hemorrhage, rapid intervention is necessary. Administration of intravenous immunoglobulin suppresses the platelet-destroying antibody response. This therapy is extremely expensive and is reserved for the most severe manifestations of ITP.13 High-dose methylprednisolone can be given intravenously and is very effective in treating ITP.11 Platelet transfusion is recommended after administration of intravenous immunoglobulin or methylprednisolone. When steroid therapy fails to arrest the condition, surgical removal of the spleen is considered.13,14
Nursing Management
Nursing management of the patient with ITP incorporates a variety of nursing diagnoses (Box 38-5). Nursing interventions include supporting the patient’s vital functions, initiating bleeding precautions (see Box 38-3), providing comfort and emotional support, and maintaining surveillance for complications.
 Box 38-5
Box 38-5Collaborative management of the patient with ITP is outlined in Box 38-6.
Heparin-Induced Thrombocytopenia
Description
Another form of thrombocytopenia seen in critical care patients is heparin-induced thrombocytopenia (HIT). There are two distinct types of HIT. The most common form is non–immune-mediated HIT, formally known as type 1 HIT.15 Seen in up to 30% of patients receiving heparin therapy, this nonautoimmune condition manifests within a few days of initiation of therapy. Platelet depletion is moderate, counts are usually less than 100,000/mm3, and the condition is transient, often resolving spontaneously. Discontinuation of heparin is not required. The second form is type 2 HIT, or immune-mediated HIT,15 which is less commonly encountered but has more severe consequences.10,16,17 This discussion is limited to immune-mediated HIT.
Etiology
Immune-mediated HIT is a response to the administration of heparin therapy. It has been observed in 0.5% to 5% of patients treated with unfractionated heparin and has occurred after exposure to LMWH, although to a lesser degree.15 The disorder is characterized by severe thrombocytopenia during heparin therapy. Diagnostically it is identified by a platelet count less than 50,000/mm3 or at least a 50% decrease from the baseline platelet count from the initiation of therapy. Onset usually occurs 5 to 14 days from the first exposure to heparin, but the onset can occur within hours of a re-exposure to heparin.10,16–18 Depending on the source of the disorder, reported mortality rates are as high as 30%.15
Pathophysiology
The thrombocytopenia that occurs with immune-mediated HIT is related to the formation of heparin-antibody complexes. These complexes release a substance known as platelet factor 4 (PF4). PF4 attracts heparin molecules, forming immunogenic complexes that adhere to platelet and endothelial surfaces (Fig. 38-4). Activation of platelets stimulates the release of thrombin and the subsequent formation of platelet clumps.10
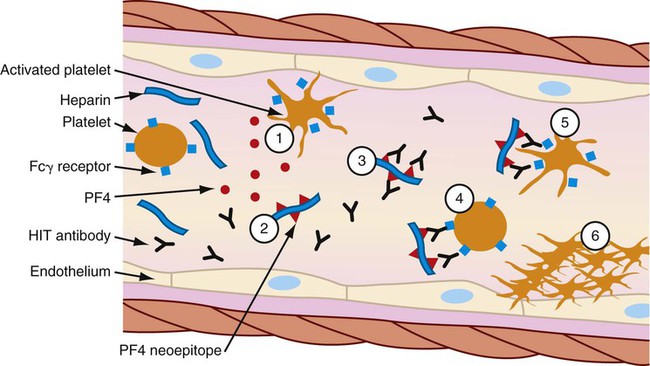
Patients with immune-mediated HIT are at greater risk for thrombosis than bleeding. Vessel occlusion can result in the need for limb amputation, stroke, acute myocardial infarction, and even death.10,16–18 The resultant formation of fibrin-platelet-rich thrombi is the primary characteristic of HIT that distinguishes it from other forms of thrombocytopenia and gives rise to its more descriptive name: white clot syndrome.16
Assessment and Diagnosis
Clinical Manifestations
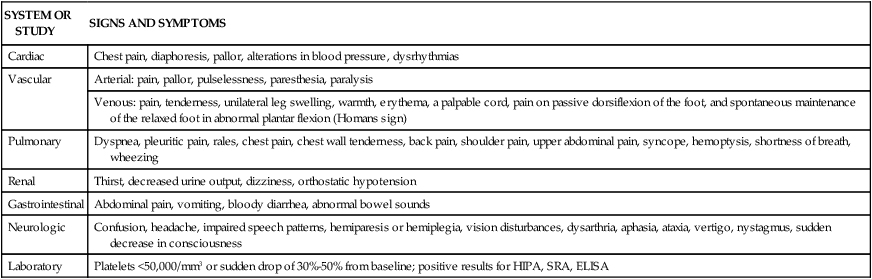
Common signs and symptoms are listed in Table 38-5. The clinical manifestations of HIT are related to the formation of thrombi and subsequent vessel occlusion.15 Most thrombotic events are venous, although venous and arterial thrombosis can occur. Thrombotic events typically include deep vein thrombosis, pulmonary embolism, limb ischemia thrombosis, thrombotic stroke, and myocardial infarction.15,18 The presence of blanching and the loss of peripheral pulses, sensation, or motor function in a limb indicate peripheral vascular thrombi. Neurologic signs and symptoms such as confusion, headache, and impaired speech can signal the onset of cerebral artery occlusion and stroke. Acute myocardial infarction may be heralded by dyspnea, chest pain, pallor, and alterations in blood pressure. Thrombi in the pulmonary vasculature may be evidenced by pleuritic pain, rales, and dyspnea.10,16
TABLE 38-5
HEPARIN-INDUCED THROMBOCYTOPENIA: SIGNS, SYMPTOMS, AND LABORATORY DATA
| SYSTEM OR STUDY | SIGNS AND SYMPTOMS |
| Cardiac | Chest pain, diaphoresis, pallor, alterations in blood pressure, dysrhythmias |
| Vascular | Arterial: pain, pallor, pulselessness, paresthesia, paralysis |
| Venous: pain, tenderness, unilateral leg swelling, warmth, erythema, a palpable cord, pain on passive dorsiflexion of the foot, and spontaneous maintenance of the relaxed foot in abnormal plantar flexion (Homans sign) | |
| Pulmonary | Dyspnea, pleuritic pain, rales, chest pain, chest wall tenderness, back pain, shoulder pain, upper abdominal pain, syncope, hemoptysis, shortness of breath, wheezing |
| Renal | Thirst, decreased urine output, dizziness, orthostatic hypotension |
| Gastrointestinal | Abdominal pain, vomiting, bloody diarrhea, abnormal bowel sounds |
| Neurologic | Confusion, headache, impaired speech patterns, hemiparesis or hemiplegia, vision disturbances, dysarthria, aphasia, ataxia, vertigo, nystagmus, sudden decrease in consciousness |
| Laboratory | Platelets <50,000/mm3 or sudden drop of 30%-50% from baseline; positive results for HIPA, SRA, ELISA |
Laboratory Findings
The key indicator for identifying HIT is the platelet count. General consensus in the literature considers a platelet count of less than 100,000/mm3 or a sudden drop of 50% from the patient’s baseline after initiation of heparin therapy to strongly indicate HIT.16–18
Two types of assays have become available to assist in confirming the diagnosis of HIT: activation assays, based on platelet aggregation or the release of granular contents such as serotonin, and assays that identify the HIT antigen. Activation assays are highly sensitive in detecting the presence of HIT. The most common assay used is heparin-induced platelet aggregation (HIPA). Serotonin release assay (SRA) is used by a few institutions. The enzyme-linked immunosorbent assay (ELISA) identifies the presence of the HIT antigen.16
Medical Management
Early identification is critical to managing the effects of immune-mediated HIT. Current guidelines suggest that for high-risk patients platelet count monitoring be performed every 2 or 3 days from day 4 to day 14.19 When a decrease in the platelet count is detected, heparin therapy should be discontinued immediately, and the patient should be tested for the presence of heparin antibodies.10,17–19 If the original indication for heparin still exists or new thromboses occur, an alternative form of anticoagulation is usually necessary.19
Direct Thrombin Inhibitors
Direct thrombin inhibitors (DTIs) are being used with increasing frequency to treat HIT. DTIs bind directly to the thrombin molecule, thereby inhibiting its action.18 Two such medications for use in the United States are lepirudin and argatroban. Warfarin, although commonly used to treat deep vein thrombosis, is not indicated as a sole agent in treating HIT because of its prolonged onset of action. Studies have shown that the use of warfarin without concomitant use of DTIs can significantly increase the incidence of thrombosis in patients with HIT.15–18 Comparative information on these medications is provided in Table 38-6.
TABLE 38-6
PHARMACOLOGIC MANAGEMENT
Heparin-Induced Thrombocytopenia
| MEDICATION | DOSAGE | ACTION | SPECIAL CONSIDERATIONS |
| Lepirudin (Refludan) | Loading dose: 0.4 mg/kg IV bolus IV infusion: 0.15 mg/kg/hr |
Used to inhibit free and clot-bound thrombin; a recombinant form of leech-derived hirudin | Monitor aPTT; maintain INR 1.5-2.5 times normal Reduce dosage in patients with known or suspected renal insufficiency |
| Side effects include bleeding | |||
| Can develop antilepirudin antibodies that enhance anticoagulant effect | |||
| Argatroban | Loading dose: None | Used to inhibit thrombin | Obtain baseline aPTT 2 hr after therapy started |
| IV infusion: 2 mcg/kg/min not to exceed 10 mcg/kg/min | Monitor aPTT; maintain INR 1.5-3.0 times initial baseline | ||
| Reduce dosage in patients with known or suspected hepatic impairment | |||
| Bivalirudin | Loading dose: 0.75 mg/kg IV bolus |
Used to inhibit thrombin | Monitor aPTT, maintain INR 1.5-2.5 times initial baseline |
| IV infusion: 1.75-2.0 mg/kg/hr | Adjust dose in presence of renal failure |

aPTT, Activated partial thromboplastin time; INR, international normalized ratio; IV, intravenous.
Nursing Management
Nursing management of the patient with HIT incorporates a variety of nursing diagnoses (Box 38-7). Nursing interventions include decreasing the incidence of heparin exposure, maintaining surveillance for complications, providing comfort and emotional support, and educating the patient and family. The critical care nurse plays a pivotal role in prevention and detection of HIT. Initial assessment is crucial to identifying those patients at risk for HIT. Ascertaining a medical history that includes previous heparin therapy, deep vein thrombosis, or cardiovascular surgery that included the use of cardiopulmonary bypass can alert the nurse to potential problems.
 Box 38-7 Nursing Diagnoses
Box 38-7 Nursing Diagnoses
Heparin-Induced Thrombocytopenia
• Risk for Decreased Cardiac Tissue Perfusion, p. 1155
• Risk for Ineffective Peripheral Tissue Perfusion, p. 1158
• Risk for Ineffective Renal Tissue Perfusion, p. 1159
• Risk for Ineffective Gastrointestinal Tissue Perfusion, p. 1157
• Risk for Ineffective Cerebral Tissue Perfusion, p. 1156
• Powerlessness related to lack of control over the current situation or disease progression, p. 1152
• Deficient Knowledge related to lack of previous exposure to information, p. 1134 (see Box 38-8, Patient Education for Heparin-Induced Thrombocytopenia)
Educating the Patient and Family
Prevention of subsequent episodes in patients sensitized to heparin incudes education of the patient and family (Box 38-8). Education should include measures to avoid future exposure to heparin. The use of medical alert bracelets and listing heparin allergies in the medical record are necessary to avoid this serious complication in the future.
Collaborative management of the patient with HIT is outlined in Box 38-9.
Sickle Cell Anemia
Description
Sickle cell anemia (SCA) is a disease passed down through families in which RBCs form an abnormal sickle or crescent shape. RBCs carry oxygen to the body and are normally shaped like a disk.1 The sickle-shaped cells have a shortened life span; are unable to carry adequate oxygen to tissues, and due to their shape become trapped in the vasculature; and can cause severe pain, increased risk of infection, and life-threatening complications.20
Etiology
SCA is an autosomal recessive genetic disorder. An individual with normal hemoglobin has two copies of hemoglobin A (Hb A) gene. An individual with SCA has two copies of hemoglobin S (Hb S) gene. Individuals who have one gene for Hb S and one gene for Hb A are known as “carriers” of the sickle cell trait (Hb AS). When two “carriers” have a child, there is a 25% chance that they will have a child with SCA (Hb SS), a 50% chance of having a child with sickle cell trait (Hb AS), and a 25% chance of having a child with entirely normal hemoglobin (Hb AA).21,22
This genetic trait is primarily found in people of West African descent. The disease has also been linked to persons of sole European or Middle Eastern ancestry; however, this is extremely rare. The disease is not prevalent in persons of Asian or Pacific Islander descent.21
SCA is usually diagnosed during the first few years of life due to manifestation of initial symptoms. Prenatal screening is now available for at-risk couples.22 This consists of DNA analysis from fetal cells. This should be offered as part of their prenatal counseling. There are more than 40 states that undergo universal neonatal screening for hemoglobinopathies.22
Pathophysiology
Sickle cell disease (SCD) is a chronic inflammatory condition characterized by hemolysis and vasoocclusion. The cause of SCA is a mutation in the genetic sequence in the beta chain gene of the hemoglobin. This results in a sequence of the replacement of valine with glutamic acid at the N-terminal amino acid position 6 of the protein chain.21,22 This substitution leads to the production of hemoglobin S.
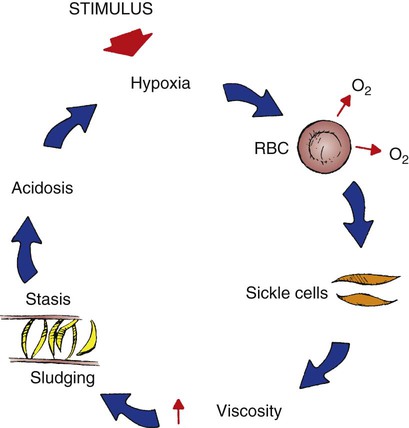
Normal RBCs contain hemoglobin that are flexible, biconcave disks. When deoxygenated, RBCs containing predominantly Hb S distort into a crescent or sickle shape. In this form the hemoglobin becomes is rigid and friable, causing vasoocclusion in the small vessels of the circulatory system. This tends to happen during times of physiologic stress, such as physical overexertion, muscle tissue ischemia, dehydration, infection, or extreme temperatures.22 Even though these conditions have a tendency to exacerbate the condition, the majority of the sickling events have no identifying cause.21
The RBCs become lodged in the vasculature and the microcirculation causing stasis and obstruction of blood flow and damage to the surrounding organs, tissue ischemia, infarction, and if not corrected eventually, necrosis (Fig. 38-5).22 In addition, hemolysis of the RBCs occurs, resulting in anemia.
Assessment and Diagnosis
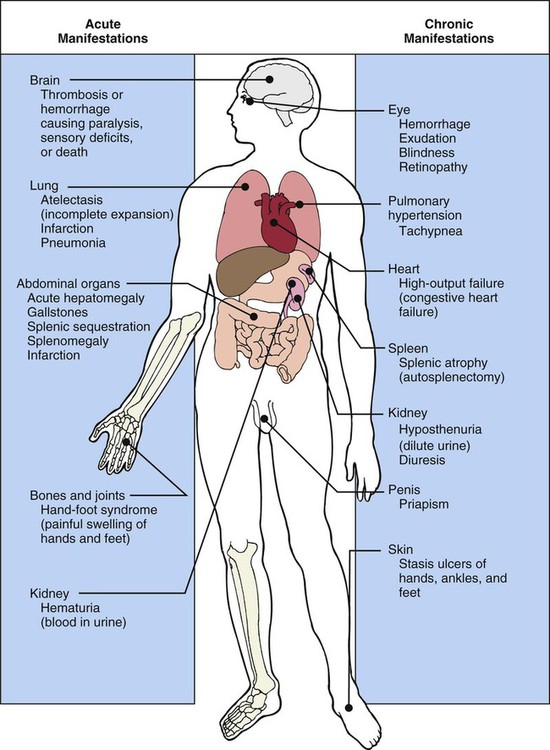
Clinical Manifestations
There are a variety of clinical manifestations associated with SCA (Fig. 38-6). The patient may present with a low-grade fever, bone or joint pain, pinpoint pupils, inability to follow commands, photophobia, tachycardia, tachypnea, decreased respiratory excursion, hepatomegaly, nonpalpable spleen, and pretibial ulcers.23
Laboratory Studies
Initial laboratory studies include a complete blood count (CBC), a peripheral blood smear, and a quantitative hemoglobin electrophoresis. Sickle cells constitute 5% to 10% of the blood smear. The elevated reticulocyte count (greater than 10%) is characteristically accompanied by the presence of Howell-Jolly bodies. Howell-Jolly bodies are small remnants of nuclear material from hemolyzed erythrocytes reflective of hyposplenia or autoinfarction, and target cells (an erythrocyte with a deeply stained core surrounded by a lighter-stained margin; it resembles a target with a bull’s eye).22 Typically an elevated WBC count occurs during and following a crisis. Other tests might include an indirect bilirubin level, which will be elevated following hemolysis. The haptoglobin level will be low or absent because it cannot be replaced quickly enough after severe hemolysis. Haptoglobin, a glycoprotein, exists to bind free hemoglobin that is released from hemolyzed erythrocytes.22
Medical Management
SCA is not a curable disease; however, there are treatment options available for management of symptoms and complications. Bone marrow transplant offers a cure in limited number of cases.20 Medical interventions are aimed at preventing infections, managing pain, transfusing RBCs, and administering hydroxyurea. Patients with chronic disease are more effective managed through a multidisciplinary approach.20 It is also important to look at other issues that may exacerbate the patient disease process such as diet, poor housing, inadequate housing, lack of education, poor access to services, and poor lifestyle choices.
Prevent Infection
Both children and adults with SCA are more prone to infection and have a more difficult time fighting off infection.23 This can result in damage to the spleen from constant sickling of the RBCs. Damage to the spleen can prevent the destruction of bacteria in the blood. Prophylactic administration of oral antibiotics starting at 2 months old can decrease the chances of a pneumococcal infection and early death. Proper vaccinations against pneumococcal infections, meningitis, hepatitis, and influenza are important to prevent future infections.23
Pain Management
Pain associated with SCA can be acute or chronic in nature. The most common type of pain associated with SCA is vasoocclusive pain. It is commonly treated with an anti-inflammatory and opioid or non-opioid analgesics.23,24 The pain associated with SCA can vary enormously; therefore, a number of different approaches may be required. The medication of choice should be influenced by the patient’s history of analgesia use. Some patients may have extremely intricate medication regimes.23 Paracetamol and nonsteroidal anti-inflammatory drugs (NSAIDs) are used for mild to moderate pain relief. If this is not effective, oral or parenteral opiates may be an alternative.23,24
For those who wish to try the nonpharmacologic route, there are other approaches such as psychologic support, massage, acupuncture, and transcutaneous electrical nerve stimulation. Distraction can be another valuable tool to use. The use of television, video games, repeating inspirational phrases and mental calculations can also be a form of distraction. Studies suggested that cognitive behavioral therapy can help teach patients coping strategies for acute and chronic pain.23
Transfusion Therapy
RBC transfusion therapy in SCD is an important lifesaving treatment option but should be done with careful consideration. Transfusion therapy is primarily used for treatment of patients who are experiencing complications due to SCD or as an emergency measure.24 Transfusion therapy should be used with extreme caution due to risks such as iron overload, exposure to hepatitis, HIV and other infectious agents, alloimmunization, induction of hyperviscosity, and limitations on resources. The indication for having a blood transfusion or exchange are recurrent painful vasoocclusive crises with long hospital admissions, acute chest syndrome, stroke, priapism, and leg ulcers. Blood transfusions or exchange can also be performed before major operations, such as hip replacement because of avascular necrosis of the hip bone.23
Administration of Hydroxyurea
Hydroxyurea is an oral agent that is a safe and effective treatment for children and adults that suffer from SCA. It works by increasing the level of fetal hemoglobin in the RBCs, thereby reducing the concentration of sickle hemoglobin and sickling itself.23 The patient is usually started at a dose of 15 mg/kg PO once a day. The dose is increased by 5 mg/kg every 12 weeks until 35 mg/kg is reached, providing the patient’s blood count remains within an acceptable range. Research shows patients receiving hydroxyurea had lower episodes of pain and acute chest syndrome and also had a decreased need for blood transfusion and hospitalizations compared to those that received no treatment. Research states that hydroxyurea can be used as an alternative to regular blood transfusions.24
Nursing Management
Nursing management of the patient with SCA incorporates a variety of nursing diagnoses (Box 38-10). Nursing interventions include supporting the patient’s vital functions, providing comfort and emotional support, maintaining surveillance for complications, and educating the patient and family.
 Box 38-10 Nursing Diagnoses
Box 38-10 Nursing Diagnoses
Sickle Cell Anemia
• Acute Pain related to transmission and perception of cutaneous, visceral, muscular, or ischemic impulses, p. 1123
• Risk for Ineffective Peripheral Tissue Perfusion, p. 1158
• Risk for Ineffective Renal Tissue Perfusion, p. 1159
• Risk for Ineffective Gastrointestinal Tissue Perfusion, p. 1157
• Risk for Ineffective Cerebral Tissue Perfusion, p. 1156
• Powerlessness related to lack of control over the current situation or disease progression, p. 1152
• Deficient Knowledge related to lack of previous exposure to information, p. 1134 (see Box 38-11, Patient Education for Sickle Cell Anemia)
Supporting Patient’s Vital Functions
Frequent assessments should include parameters for neurologic status, renal function, cardiopulmonary function, and skin integrity that indicate impaired tissue or organ perfusion. Particular parameters to include are mental status, BUN and creatine levels, urine output, vital signs, hemodynamic values, cardiac rhythm, arterial blood gas and pulse oximetry values, skin breakdown, ecchymoses, or hematomas.24
The critical care nurse must recognize and support the patient’s vital physiologic functions. Administration of intravenous fluids, blood products, and inotropic agents to provide adequate hemodynamic support and tissue oxygenation is essential in preventing or combating end-organ damage. Close monitoring of vital signs, hemodynamic parameters, intake and output, and appropriate laboratory values assists the critical care nurse in administering and titrating appropriate agents.24
Maintaining Surveillance for Complications
The critical care nurse needs to be vigilant for signs of life-threatening complications such as septicemia, acute myocardial infarction, priapism, ischemic stroke, and shock.20 If there are any significant concerns these must be reported immediately. Other issues that may arise are dehydration, hypoxia, infection, skin and tissue viability, and decreased hemoglobin levels.
Educating the Patient and Family
Even though there is no cure for SCA, the focus of care is prevention. Early in the patient’s hospital stay, the patient and family should be taught about SCA, its etiologies, and its treatment (Box 38-11). Patient and family education focuses on measures to help prevent painful reoccurring episodes. If the patient smokes, he or she should be encouraged to stop smoking and be referred to a smoking cessation program. In addition, the importance of continuous medical follow-up should be stressed. While research continues to try to find a cure, nurses must continue to be sensitive to the effects of the disease on the patient and the family as well as the need to be culturally sensitive.20
Collaborative management of the patient with SCA is outlined in Box 38-12.
Tumor Lysis Syndrome
Description
Tumor lysis syndrome (TLS) refers to a variety of metabolic disturbances that may be seen with the treatment of cancer. A potentially lethal complication of various forms of cancer treatment, TLS occurs when large numbers of neoplastic cells are rapidly killed, resulting in the release of large amounts of potassium, phosphate, and uric acid into the systemic circulation. It is most commonly seen in patients with lymphoma, leukemia, or multiple metastatic conditions.25
Etiology
Although most often associated with the use of chemotherapeutic medications, biologic agents, and irradiation used in the treatment of malignant disorders, TLS can in rare instances occur spontaneously. The development of TLS has been linked to other pathophysiologic conditions such as elevated WBC counts, large tumors, multiple organ involvement by malignancy, and renal insufficiency.25
Pathophysiology
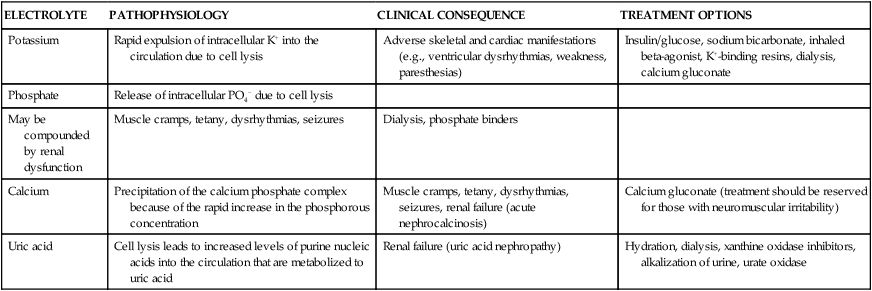
The primary mechanism involved in the development of TLS is the destruction of massive numbers of malignant cells by chemotherapy or radiation therapy (Fig. 38-7). Massive destruction of cells releases large amounts of potassium, phosphorus, and nucleic acids, leading to severe metabolic disturbances such as hyperuricemia, hyperkalemia, hyperphosphatemia, and hypocalcemia (Table 38-7). Vomiting, diarrhea, and other insensible fluid losses from fever or tachypnea also contribute to these electrolyte disturbances.25 Death of patients with TLS is most often caused by complications of renal failure or cardiac arrest.25–27
TABLE 38-7
ELECTROLYTE ABNORMALITIES ENCOUNTERED IN TUMOR LYSIS SYNDROME AND THEIR CLINICAL CONSEQUENCES
| ELECTROLYTE | PATHOPHYSIOLOGY | CLINICAL CONSEQUENCE | TREATMENT OPTIONS |
| Potassium | Rapid expulsion of intracellular K+ into the circulation due to cell lysis | Adverse skeletal and cardiac manifestations (e.g., ventricular dysrhythmias, weakness, paresthesias) | Insulin/glucose, sodium bicarbonate, inhaled beta-agonist, K+-binding resins, dialysis, calcium gluconate |
| Phosphate | Release of intracellular PO4− due to cell lysis | ||
| May be compounded by renal dysfunction | Muscle cramps, tetany, dysrhythmias, seizures | Dialysis, phosphate binders | |
| Calcium | Precipitation of the calcium phosphate complex because of the rapid increase in the phosphorous concentration | Muscle cramps, tetany, dysrhythmias, seizures, renal failure (acute nephrocalcinosis) | Calcium gluconate (treatment should be reserved for those with neuromuscular irritability) |
| Uric acid | Cell lysis leads to increased levels of purine nucleic acids into the circulation that are metabolized to uric acid | Renal failure (uric acid nephropathy) | Hydration, dialysis, xanthine oxidase inhibitors, alkalization of urine, urate oxidase |
From Davidson MB, et al. Pathophysiology, clinical consequences, and treatment of tumor lysis syndrome. Am J Med. 2004;116:546.
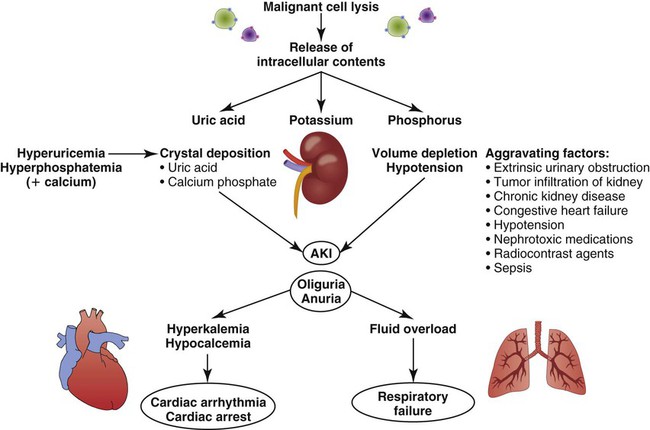
Hyperuricemia
Hyperuricemia occurs 48 to 72 hours after the initiation of anticancer therapy.25 Tumor cells undergo rapid growth and development, and large amounts of nucleic acids are present within them. When therapy is initiated, tumor cell destruction releases nucleic acids, which are metabolized into uric acid. Metabolic acidosis ensues, resulting in crystallization of the uric acid in the distal tubules of the kidney and leading to obstruction of urine flow. Glomerular filtration rates drop as the kidneys are unable to clear the increasing amounts of uric acid. Consequently, acute kidney injury eventually occurs.28 Acute kidney injury is discussed further in Chapter 27.
Hyperuricemia associated with TLS can be potentiated by several other factors, including elevated uric acid levels before the initiation of therapy. Other causes of increased uric acid production are elevated WBC counts, destruction of WBCs, and enlargement of the lymph nodes, spleen, or liver.25
Hyperkalemia
Hyperkalemia occurs within 6 to 72 hours after the initiation of chemotherapy. This is the most deleterious of all the manifestations of TLS.25 In addition to the release of nucleic acids, tumor cell destruction also results in the release of potassium. Renal insufficiency related to hyperuricemia prevents adequate excretion of potassium, and levels rise. The resultant hyperkalemia may have a profound effect on intracellular and extracellular fluid levels.29 Left untreated, hyperkalemia can have devastating consequences, including cardiac arrest and death.25
Hyperphosphatemia and Hypocalcemia
Hyperphosphatemia and hypocalcemia occur 24 to 48 hours after the initiation of therapy.25 Phosphorus levels also rise as a consequence of tumor cell destruction. Calcium ions then bind with the excess phosphorus, creating calcium phosphate salts and bringing about hypocalcemia. These salts precipitate in the kidney tubules, worsening renal insufficiency. Hypocalcemia causes tetany and cardiac dysrhythmias, which can result in cardiac arrest and death.28,29
Assessment and Diagnosis
Detection and recognition of TLS is accomplished through assessment of clinical manifestations, evaluation of laboratory findings, and other diagnostic tests. Table 38-8 summarizes common findings in TLS.25,27
TABLE 38-8
COMMON FINDINGS IN TUMOR LYSIS SYNDROME
| DIAGNOSTIC PARAMETER | FINDINGS |
| Clinical | Weight gain, edema, diarrhea, lethargy, muscle cramps, nausea and vomiting, paresthesia, weakness, oliguria, uremia, seizures |
| Laboratory | ↑ Potassium, phosphorus, uric acid, BUN, Cr |
| ↓ Calcium, creatinine clearance, pH, bicarbonate, Paco2 | |
| Diagnostic | Positive Chvostek and Trousseau signs, hyperactive deep tendon reflexes, dysrhythmias, ECG changes |
Clinical Manifestations
Clinical manifestations are related to the metabolic disturbances associated with TLS.27 The patient’s history reveals an unexplained weight gain after initiation of chemotherapy or radiation therapy. The weight gain is associated with fluid retention due to electrolyte disturbances. Other early signs heralding the onset of TLS include diarrhea, lethargy, muscle cramps, nausea, vomiting, paresthesias, and weakness.26
Laboratory Findings
Laboratory findings demonstrate electrolyte disturbances such as elevated potassium and phosphorus levels and a decreased calcium level. Uric acid levels are increased. Elevated levels of BUN and creatinine and a decreased creatinine clearance also indicate TLS. Metabolic acidosis is confirmed by the presence of decreased pH, bicarbonate levels, and partial pressure of carbon dioxide (Paco2) on arterial blood gas measurements.26
Other Diagnostic Tests
Physical examination reveals positive Chvostek and Trousseau signs related to hypocalcemia. Hyperactive deep tendon reflexes indicate hyperkalemia and hypocalcemia.27 Potassium and calcium disturbances result in changes that can be seen on the electrocardiogram (ECG), such as peaked or inverted T waves, altered QT intervals, widened QRS complexes, and dysrhythmias.25
Medical Management
Medical interventions are aimed at maintaining adequate hydration, treating metabolic imbalances, and preventing life-threatening complications (see Table 38-7).26,29
Adequate Hydration
Administration of intravenous fluids may be necessary early in the course of treatment if inadequate hydration exists. The administration of isotonic saline (0.9% normal saline) reduces serum concentrations of uric acid, phosphate, and potassium.27 The use of nonthiazide diuretics to maintain adequate urine output may be required. If renal failure occurs, hemodialysis should be considered.27
Metabolic Imbalances
Electrolytes and arterial blood gases are closely monitored. Dietary restrictions of potassium and phosphorus may be necessary. The goals in treating hyperuricemia are to inhibit uric acid formation and to increase renal clearance.29 This can be accomplished through the administration of sodium bicarbonate to increase the pH of the urine to above 7.0, which increases the solubility of uric acid, preventing subsequent crystallization. Allopurinol administration can also inhibit uric acid formation.27
Life-Threatening Complications
If potassium levels rise dangerously, Kayexalate (sodium polystyrene sulfonate) may be given orally, or if the patient is unable to tolerate oral medications due to nausea and vomiting, rectal instillation may be used. If the patient is oliguric, glucose and insulin infusions may be given to facilitate lowering the potassium levels. A 10% solution of calcium gluconate may be administered to stabilize cardiac tissue membranes to prevent life-threatening dysrhythmias.30 Phosphorus-binding antacids can be used for treating hyperphosphatemia. Stool softeners may be necessary to treat the constipation often associated with the administration of these antacids. Calcium gluconate may be required to replace calcium, but it should be used judiciously.25
Nursing Management
Nursing management of the patient with TLS incorporates a variety of nursing diagnoses (Box 38-13). Nursing interventions include monitoring fluid and electrolytes, providing comfort and emotional support, maintaining surveillance for complications, and education the patient and family.
 Box 38-13 Nursing Diagnoses
Box 38-13 Nursing Diagnoses
Tumor Lysis Syndrome
• Excess Fluid Volume related to renal dysfunction, p. 1139
• Decreased Cardiac Output related to alterations in contractility, p. 1128
• Anxiety related to threat to biologic, psychologic, and/or social integrity, p. 1125
• Ineffective Coping related to a situational crisis and personal vulnerability, p. 1150
Maintaining Surveillance for Complications
Nursing interventions are aimed at preventing complications. Seizure precautions should be instituted, especially if calcium levels are disrupted. Insertion of a nasogastric tube is appropriate if nausea or vomiting occurs. Dietary adjustments are necessary, such as potassium and phosphorus restrictions in the presence of elevated serum levels and providing additional fiber to combat the constipation associated with the administration of antacids.30
Anemia of Critical Illness
Etiology
Anemia of critical illness (ACI) is a hospital-acquired disorder commonly seen in critically ill patients. It is the result of inflammation, iron deficiency, trauma, surgery, gastrointestinal bleeding, and iatrogenic blood loss from diagnostic testing. Inflammation causes decreased RBC production and increased destruction of RBCs by macrophages. Frequent phlebotomy, coagulopathies, and nutritional deficits are just a few reasons for this increasing problem.31,32 Studies have shown that as many as 50% of transfusions administered in the critical care unit are related to hospital-acquired anemia.33 For these reasons, research has been focused on determining ways to minimize iatrogenic blood losses, improve blood salvage techniques, and develop alternatives to traditional blood transfusion therapy.
Blood Conservation Strategies
As more patients and clinicians opt for the limited use of blood products, strategies for conserving blood and preventing unnecessary loss become an important part of the critical care nurse’s standard of care. These strategies include minimizing blood loss, managing oxygen delivery and consumption, stimulating production of RBCs, and understanding transfusion safety and alternative agents.34,35
Minimizing Blood Loss
Frequent blood draws in the critically ill patient have been associated with the development of anemia. Blood losses correspond to actual volume of samples and discards when drawing from venous access lines. Critical care nurses can be instrumental in significantly decreasing blood loss in this arena. The use of pediatric collection tubes and point-of-care testing are techniques that yield valid diagnostic results but require smaller blood samples. Closed-loop vascular devices that retain the sterility of the potential discard and allow its return to the patient are also being used.36 Noninvasive monitoring devices such as pulse oximetry and capnography can reduce the need for arterial blood gas analysis.
The critical care nurse plays a key role in preventing and managing hemorrhagic blood loss in the critically ill patient. Control of hypertension, which can contribute to significant hemorrhage, can be accomplished through fluid management and the administration of antihypertensive and vasodilatory medications as needed.37 Blood salvage devices can be employed to collect shed blood and return it to the patient.35 Several pharmacologic agents can assist in achieving hemostasis and can prevent further blood loss. Desmopressin is a potent vasoconstrictor that also affects clotting factor VIII.35 Aminocaproic acid (Amicar) inhibits activation of plasminogen.35 All of these agents and devices work to control bleeding.
Managing Oxygen Delivery and Consumption
Illness-related stress, blood loss from surgery, infection, pain, and anxiety contribute to the higher than normal demand for oxygen by the critically ill patient.32 Monitoring pulse oximetry is useful in identifying activities and interventions that can contribute to the imbalance between supply and demand. Supplemental oxygen therapy assists in maintaining available oxygen supplies. Promoting a restful environment through modulation of nursing care activities and providing pain and sedation control can assist in decreasing the demand for oxygen. It is important to monitor cardiac output and other hemodynamic parameters to manage interventions that optimize oxygen delivery. Administration of fluids and inotropic agents optimizes blood pressure and cardiac output, and vasodilators are used to decrease afterload and improve efficiency of cardiovascular function.35
Stimulating Production of Red Blood Cells
Insufficient erythropoiesis can contribute to anemia in the critically ill patient. The administration of epoetin alpha can stimulate the production of RBCs, reducing the need for transfusions. Iron preparations such as ferrous sulfate, iron sucrose, or iron gluconate may be administered to provide necessary iron stores for the increased erythropoiesis.38
Encouraging Safer Transfusions and Alternative Agents
Finding ways to decrease the risks associated with blood transfusions and make the blood supply safer for patients is a high priority. Better, more sensitive screening tests, irradiation, and removal of leukocytes are a few of the current methods in use. Plasma expanders manufactured from nonhuman sources are also available. Autologous transfusions, for which the patient donates his or her own blood before a surgical procedure or other anticipated need, has also been a common practice for many years. Recombinant DNA technology is being used to develop safe alternatives to blood transfusions. The use of blood from other species is being researched in the quest to provide safe and effective products for use in severe anemia.35
Summary
Coagulation and Fibrinolysis
• Hemostasis is the ability of the body to control bleeding and clotting.
• Four actions are involved in achieving hemostasis: local vasoconstriction to reduce blood flow; platelet aggregation at the injury site and formation of a platelet plug; formation of a fibrin mesh to strengthen the plug; and dissolution of the clot after tissue repair.
Disseminated Intravascular Coagulation
• DIC is characterized by bleeding and thrombosis, which result from depletion of clotting factors, platelets, and RBCs and, if left untreated, will result in death.
• Medical management focuses on identification of the underlying cause, provision of hemodynamic support to preserve end-organ function, and administration of blood, blood components, and medications to interrupt the process.
• Nursing actions include initiating bleeding precautions, providing comfort and emotional support, and maintaining surveillance for complications (e.g., hypovolemic shock, ischemia, multiple organ dysfunction syndrome).
Idiopathic Thrombocytopenia Purpura
• Idiopathic thrombocytopenia purpura is caused by an autoimmune response that results in the destruction of existing platelets.
• Medical management focuses on administration of glucocorticoids, immunoglobulin, and platelets.
• Nursing actions include initiating bleeding precautions, providing comfort and emotional support, and maintaining surveillance for complications (e.g., intracranial hemorrhage, severe blood loss).
Heparin-Induced Thrombocytopenia
• There are two forms of heparin-induced thrombocytopenia: type 1 and type 2. Type 1 is more common, milder, and transient. Type 2 is the result of an autoimmune response to the administration of heparin and is more severe than type 1.
• Medical management focuses discontinuation of all heparin, initiation of anticoagulation with an alternative anticoagulant, and treatment of thrombosis.
• Nursing actions include providing comfort and emotional support and maintaining surveillance for complications (e.g., deep vein thrombosis, pulmonary emboli, limb ischemia, ischemic stroke, acute myocardial infarction).
Sickle Cell Anemia
• Sickle cell anemia (SCA) is a disease passed down through families in which red blood cells form an abnormal sickle or crescent shape.
• Medical management focuses on prevention of infection, management of pain, and administration of red blood cells and hydroxyurea.
• Nursing actions include supporting the patient’s vital functions, providing comfort and emotional support, maintaining surveillance for complications (e.g., ischemic stroke, acute myocardial infarction, acute limb ischemia, acute kidney injury), and educating the patient and family.
Tumor Lysis Syndrome
• Tumor lysis syndrome occurs when a large number of neoplastic cells are rapidly killed, resulting in the release of large amounts of potassium, phosphate, and uric acid in the systemic circulation.
• Medical management focuses on preservation of renal function and treatment of electrolyte disorders (hyperkalemia, hyperuricemia, hyperphosphatemia, and hypocalcemia).
• Nursing actions include providing comfort and emotional support and maintaining surveillance for complications (e.g., acute kidney injury, dysrhythmias).
Hospital-Acquired Anemia
• Anemia of critical illness occurs as a result of inflammation, iron deficiency, trauma, surgery, gastrointestinal bleeding, and from iatrogenic blood loss from diagnostic testing.
• Blood conservation strategies include minimizing blood loss, managing oxygen delivery and consumption, and stimulating production of RBCs.
[/level-membership-for-critical-care-medicine-category][not-level-membership-for-critical-care-medicine-category]
Hematologic Disorders and Oncologic Emergencies
Overview of Coagulation and Fibrinolysis
Hemostasis, the ability of the body to control bleeding and clotting, is an intricate balancing act between the coagulation mechanism and fibrinolysis. Four major actions are involved in achieving hemostasis: 1) local vasoconstriction to reduce blood flow; 2) platelet aggregation at the injury site and formation of a platelet plug; 3) formation of a fibrin mesh to strengthen the plug; and 4) dissolution of the clot after tissue repair is complete.1 Disruption of the normal hemostatic balance can result in devastating hemorrhagic or thrombotic conditions.
Coagulation Mechanism
The coagulation mechanism consists of 13 factors that work together through a series of feedback loops to achieve hemostasis (Table 38-1 and Fig. 38-1). Depending on the initial triggering event, the extrinsic or intrinsic coagulation pathway is intiated.1,2 The extrinsic pathway begins when vascular injury occurs, resulting in release of tissue factor and activation of coagulation factor VII. The intrinsic pathway is activated when the damaged subendothelium comes into direct contact with circulating blood. In this contact phase, proteins activate additional coagulation factors (XII, XI, IX, and VIII).1 At this point, the two pathways converge into a common pathway, and prothrombin and fibrinogen are converted to their active forms, resulting in clot formation.3,4
TABLE 38-1
COAGULATION FACTORS AND COMMON NAMES
| FACTOR | COMMON NAME |
| I | Fibrinogen |
| II | Prothrombin |
| III | Tissue factor |
| IV | Calcium |
| V | Proaccelerin |
| VI | Accelerin |
| VII | Proconvertin |
| VIII | Antihemophilic |
| IX | Christmas factor |
| X | Stuart |
| XI | Plasma thromboplastin antecedent |
| XII | Hageman factor |
| XIII | Fibrin stabilizing factor |
Clot Formation
Platelets are activated by the arrival of thrombin at the site of injury (Fig. 38-2). Local platelets change shape, become sticky, and begin to aggregate along the vessel wall. Activated platelets undergo degranulation, releasing several factors to assist in clot formation. Serotonin and histamine, two potent vasoconstrictors, help limit blood loss while the clot is forming. The prostaglandin thromboxane A2 (TXA2) contributes to vasoconstriction and promotes further platelet degranulation. Adenosine diphosphate (ADP) recruits platelets by increasing adherence and degranulation,2,3 and the process continues.
At the convergence of the intrinsic and extrinsic pathways, factor X is converted into its active form, thereby enabling the conversion of prothrombin to thrombin. Thrombin then converts fibrinogen to fibrin. Strands of fibrin form and radiate around the newly formed clot, essentially creating a net in which platelets, red blood cells (RBCs), and white blood cells (WBCs) are trapped.1 The clot is further secured to the site as platelet actomyosin causes it to contract and consolidate.2
Regulatory Mechanisms
Under normal conditions, feedback systems prevent the coagulation process from spinning out of control. Prostacyclin I2 (PGI2) is a prostaglandin released from damaged endothelial cells. It counteracts the effects of TXA2, serotonin, and histamine through vasodilation and inhibition of platelet degranulation.1–3 Another means of regulating clot formation is through inhibition of enzymes necessary for activation of coagulation factors along the intrinsic and extrinsic pathways, preventing the conversion of prothrombin to thrombin. The most important of these regulators is antithrombin III; however, protein C and protein S also play a role in thrombin inhibition.5
Fibrinolysis
The process of fibrinolysis promotes dissolution and remolding of the clot to promote repair of the vessel wall and maintain flow through the vessel lumen (Fig. 38-2D). Fibrinolysis begins as soon as the fibrin clot is formed. Circulating plasminogen, a precursor to the powerful enzyme plasmin, binds to fibrin and is trapped within the newly formed clot. The injured epithelial wall releases tissue-type plasminogen activator (tPA), which converts plasminogen to its active state, plasmin. The plasmin begins to digest the fibrin, rapidly breaking down the clot.2,4 When fibrin is broken down, the fibrin degradation products released act as anticoagulants.
Disseminated Intravascular Coagulation
Description
Disseminated intravascular coagulation (DIC) is a syndrome that arises as a complication of other serious or life-threatening conditions. Although DIC is not seen often, it can seriously hamper diagnostic and treatment efforts for the critically ill patient. An understanding of the etiologic and pathophysiologic mechanisms of DIC can assist in anticipating the syndrome’s occurrence, recognizing its signs and symptoms, and prompting intervention. Also known as consumptive coagulopathy, DIC is characterized by bleeding and thrombosis, both of which result from depletion of clotting factors, platelets, and RBCs. If not treated quickly, DIC will progress to multiple organ failure and death.6
Etiology
Many clinical events can prompt the development of DIC in the critically ill patient, but the exact underlying trigger may not be identifiable (Box 38-1). There are, however, some commonly known conditions associated with the development of DIC.
Sepsis, particularly that caused by gram-negative organisms, can be identified as the culprit in as many as 20% of cases, making it the most common cause of DIC. In this instance, endotoxins serve as a trigger for activation of tissue factor and the extrinsic coagulation pathway. Metabolic acidosis and hypoperfusion associated with shock syndromes can result in increased formation of free radicals and damage to tissues. Tissue factor is activated, resulting in DIC. Massive trauma or burns are frequently associated with DIC. Direct tissue damage activates the extrinsic coagulation pathway, and damage to endothelial surfaces activates the intrinsic pathway.3 Obstetric emergencies, such as abruptio placenta, retained placenta, or incomplete abortion, are also associated with the development of DIC. Tissue factor is concentrated in the placenta, and damage or disruption of this structure can activate coagulation pathways, resulting in coagulopathy.7
Pathophysiology
Regardless of the cause, the common thread in the development of DIC is damage to the endothelium that results in activation of the coagulation mechanism (Fig. 38-3).1 The extrinsic coagulation pathway plays a major role in the development of DIC. Direct damage to the endothelium results in the release of tissue factor and activation of this pathway. The secondary surge of thrombin formation as a result of activation of the intrinsic coagulation pathway leads to the massive disruption of the delicate balance that is hemostasis. Excessive thrombin formation results in rapid consumption of coagulation factors and depletion of regulatory substances—protein C, protein S, and antithrombin.5 With no checks and balances, thrombi continue to form along damaged epithelial walls, resulting in occlusion of the vessels. As occlusion reaches a critical level, tissue ischemia ensues, leading to further tissue damage and perpetuating the process. Eventually, end-organ function is affected by the ischemia, and failure is evident.1
In response to the formation of clots, the fibrinolytic system is activated. As plasmin breaks down the fibrin clots, fibrin split products are released, and they act as anticoagulants.3,7 Coupled with depletion of circulating clotting factors, activation of fibrinolysis results in excessive bleeding. The end result is shock and further tissue ischemia that aggravate end-organ dysfunction and failure. Death is imminent if this destructive cycle is not interrupted.8
Assessment and Diagnosis
Clinical Manifestations
Clinical manifestations are related to the two primary pathophysiologic mechanisms of DIC: the formation of thrombi and bleeding. Thrombi in peripheral capillaries can lead to cyanosis, particularly in the fingers, toes, ears, and nose. In severe, untreated cases, this peripheral ischemia may progress to gangrene.3,6 As the condition progresses, ischemia worsens, and end organs are affected. The result of this more central ischemia can be respiratory insufficiency and failure, acute kidney injury, bowel infarction, and ischemic stroke. The tissue damage that results perpetuates the anomalies of DIC.3
As coagulation factors are depleted, bleeding from intravenous and other puncture sites is observed. Ecchymoses may result from even routine interventions such as the use of a manual blood pressure cuff, bathing, or turning.3,6 Bloody drainage may also occur from surgical sites, drains, and urinary catheters. With progression of DIC, the patient is at risk for severe gastrointestinal or subarachnoid hemorrhage.3,6 Table 38-2 lists many of the common signs and symptoms of DIC.
TABLE 38-2
COMMON SIGNS AND SYMPTOMS OF DISSEMINATED INTRAVASCULAR COAGULATION
| SYSTEM | SIGNS RELATED TO HEMORRHAGE | SIGNS RELATED TO THROMBI |
| Integumentary | Bleeding from gums, venipunctures, and old surgical sites; epistaxis; ecchymoses | Peripheral cyanosis, gangrene |
| Cardiopulmonary | Hemoptysis | Dysrhythmias, chest pain, acute myocardial infarction, pulmonary embolus, respiratory failure |
| Renal | Hematuria | Oliguria, acute kidney injury, renal failure |
| Gastrointestinal | Abdominal distention, hemorrhage | Diarrhea, constipation, bowel infarct |
| Neurologic | Subarachnoid hemorrhage | Altered level of consciousness, ischemic stroke |
Laboratory Findings
Laboratory tests used to diagnose DIC essentially assess the four basic characteristics of this syndrome: 1) increased coagulant activity; 2) increased fibrinolytic activity; 3) impaired regulatory function; and 4) end-organ failure.3
Continuous activation of the coagulation pathways results in consumption of coagulation factors. Because of this, the prothrombin time (PT), the activated partial thromboplastin time (aPTT), and the international normalized ratio (INR) values are elevated. Although the platelet count may fall within normal ranges, serial examination reveals a declining trend in values. An unexpected drop of at least 50% in the platelet count, particularly in the presence of known contributing factors and associated signs and symptoms, strongly indicates DIC.1 Fibrinogen levels drop as more and more clots are formed. Thrombus formation in small vessels narrows the vessel lumen, forcing RBCs to squeeze through. The resulting damage and fragmentation of these cells can be seen on microscopic examination of blood samples. Damaged, fragmented RBCs are called schistocytes.3,6
In response to the excess clotting activity, the fibrinolytic process accelerates, and levels of byproducts increase. This is reflected in markedly elevated levels of fibrin degradation products. Another key laboratory test used to evaluate the degree of clot dissolution—and therefore the severity of the coagulopathy—is the d-dimer level.1 d-dimers exclusively indicate clot degradation because, unlike fibrin degradation products, which also result from the breakdown of free circulating fibrin, d-dimers result only from dissolution of clots.3 With progression of the coagulopathy, normal regulatory mechanisms are disrupted, as reflected in decreasing levels of inhibitory factors such as protein C, factor V, and antithrombin III.3,6,9
Unchecked DIC resulting in occlusion of vessels and tissue ischemia leads to end-organ dysfunction. Respiratory failure, indicated by abnormal arterial blood gas (ABG) levels; liver failure, indicated by increasing liver enzymes; and renal impairment, indicated by rising blood urea nitrogen (BUN) and creatinine levels are common findings in advanced DIC.9
No single laboratory study can confirm the diagnosis of DIC, but several key results are strong indicators of the condition (Table 38-3). The International Society of Thrombosis and Hemostasis emphasizes early detection of DIC through observation of abnormal trends in laboratory values.5
TABLE 38-3
KEY LABORATORY STUDIES IN DISSEMINATED INTRAVASCULAR COAGULATION
| TEST | VALUE |
| Prothrombin time (PT) | >12.5 sec |
| Platelets | <50,000/mm3, or at least 50% drop from baseline |
| Activated partial thromboplastin time (aPTT) | >40 sec |
| d-dimer | >250 ng/mL |
| Fibrin degradation products (FDP) | >40 mg/mL |
| Fibrinogen | <100 mg/dL |
Medical Management
Without question, the primary intervention in DIC is prevention. Being aware of the conditions that commonly contribute to the development of DIC and treating them vigorously and without delay provide the best defense against this devastating condition.3,6,7,9 After DIC is identified, maintaining organ perfusion and slowing consumption of coagulation factors are paramount to achieving a favorable outcome.1,3
Multiple organ dysfunction syndrome (MODS) frequently results from DIC and exacerbates the underlying pathology.8 It is essential to prevent end-organ ischemia and damage by supporting blood pressure and circulating volume. Administration of intravenous fluids and inotropic agents and, if overt hemorrhaging is evident, infusion of packed RBCs are appropriate interventions to replace blood volume and essential, oxygen-carrying RBCs.
In the presence of severe platelet depletion (less than 50,000/mm3) and severe hemorrhage, platelet transfusions are often indicated.5,6 However, caution must be used when administering platelets because antiplatelet antibodies may be formed. These antibodies may become activated during future platelet transfusions and elicit DIC.3
Replacement of clotting factors in the patient with DIC is thought by some authorities to perpetuate the coagulopathy; however, there is little scientific evidence to support this theory.1 Fibrinogen levels less than 100 mg/dL indicate the appropriateness of administering cryoprecipitate. A prolonged PT indicates the need for fresh-frozen plasma.3,5,6
Slowing consumption of coagulation factors by inhibiting the processes involved in clot formation is another strategy used in treating DIC. The use of heparin, particularly low–molecular- weight heparin (LMWH), to prevent formation of future clots is controversial.6 It is contraindicated in patients with DIC associated with recent surgery or with gastrointestinal or central nervous system (CNS) bleeding. However, heparin has been beneficial in obstetric emergencies such as retained placenta or incomplete abortion, severe arterial occlusions, or MODS caused by microemboli.3,6 Inhibitors such as aminocaproic acid may be used in conjunction with heparin.3,6
The use of recombinant activated protein C is gaining popularity in treating DIC, especially in the setting of severe sepsis. Activated protein C acts as an anticoagulant and works to restore normal inhibition of coagulation pathways. However, it has been associated with an increased incidence of intracerebral bleeding and must be used with caution in patients with severely decreased platelets.3,5
Thrombin production in DIC surpasses that of antithrombins and other regulatory factors that would normally be present to inactivate thrombin and its subsequent actions. The use of antithrombin III has recently been approved in the United States. Ongoing research is yielding mixed results in the treatment of DIC.5 One interesting area of research is the use of protease inhibitors. Protease molecules normally inhibit the conversion of fibrinogen to fibrin in the coagulation mechanism, but in DIC, this inhibitory mechanism is impaired. The introduction of protease inhibitors by intravenous infusion may be advantageous in arresting DIC.3
Nursing Management
Nursing management of the patient with DIC incorporates a variety of nursing diagnoses (Box 38-2). Assessment and monitoring are the primary weapons in the critical care nurse’s arsenal against DIC. Knowing the diseases and conditions that are most often associated with DIC and understanding the pathophysiologic mechanisms involved enables the critical care nurse to anticipate its development and intervene quickly. Nursing interventions include supporting the patient’s vital functions, initiating bleeding precautions, providing comfort and emotional support, and maintaining surveillance for complications.
Box 38-2 Nursing Diagnoses
Disseminated Intravascular Coagulation
Supporting Patient’s Vital Functions
Frequent assessments should include parameters for neurologic status, renal function, cardiopulmonary function, and skin integrity that indicate impaired tissue or organ perfusion. Particular parameters to include are mental status, BUN and creatine levels, urine output, vital signs, hemodynamic values, cardiac rhythm, arterial blood gas and pulse oximetry values, skin breakdown, ecchymoses, or hematomas.6
The critical care nurse must recognize and support the patient’s vital physiologic functions. Administration of intravenous fluids, blood products, and inotropic agents to provide adequate hemodynamic support and tissue oxygenation is essential in preventing or combating end-organ damage. Close monitoring of vital signs, hemodynamic parameters, intake and output, and appropriate laboratory values assists the critical care nurse in administering and titrating appropriate agents.3
Initiating Bleeding Precautions
Awareness of the patient’s bleeding potential necessitates adjustments to normal nursing interventions (Box 38-3). The nurse avoids unnecessary venipunctures that may result in bleeding, bruising, or hematomas by drawing blood from and administering medications through existing arterial or venous lines. The use of manual or automatic blood pressure cuffs is avoided whenever possible. If tracheal or oral suctioning is necessary, the use of low-level suction is recommended.6 Meticulous skin care is advised, keeping the skin moist and using specialty mattresses and beds as appropriate to prevent breakdown. Gentle care should be used when bathing or turning the patient to prevent bruising or hematoma formation.
Box 38-3
NIC
Bleeding Precautions
Reduction of stimuli that may induce bleeding or hemorrhage in at-risk patients
Monitor the patient closely for hemorrhage
Note hemoglobin/hematocrit levels before and after blood loss, as indicated
Monitor for signs and symptoms of persistent bleeding (e.g., check all secretions for frank or occult blood)
Monitor coagulation studies, including prothrombin time (PT), partial thromboplastin time (PTT), fibrinogen, fibrin degradation/split products, and platelet counts, as appropriate
Monitor orthostatic vital signs, including blood pressure
Maintain bed rest during active bleeding
Administer blood products (e.g., platelets, fresh-frozen plasma), as appropriate
Protect the patient from trauma, which may cause bleeding
Avoid injections (IV, IM, or SQ), as appropriate
Instruct the ambulating patient to wear shoes
Use soft toothbrush or toothettes for oral care
Use electric razor, instead of straight-edge, for shaving
Tell patient to avoid invasive procedures; if they are necessary, monitor closely for bleeding
Coordinate timing of invasive procedures with platelet or fresh-frozen plasma transfusions, if appropriate
Refrain from inserting objects into a bleeding orifice
Avoid taking rectal temperatures
Administer mediations (e.g., antacids), as appropriate
Instruct patient to avoid aspirin and other anticoagulants
Instruct patient to increase intake of foods rich in vitamin K
Use therapeutic mattress to minimize skin trauma
Avoid constipation (e.g., encourage fluid intake and stool softeners), as appropriate
Instruct the patient and/or family on signs of bleeding and appropriate actions (e.g., notify the nurse), should bleeding occur
From Bulechek GM, et al. Nursing Interventions Classification (NIC). 6th ed. St. Louis: Mosby; 2013.
Thrombocytopenia
Description
Thrombocytopenia is defined as a platelet count less than 140,000/mm3.10,11 Similar to DIC, thrombocytopenia often results from another underlying condition that affects the platelet count. Thrombocytopenia is more common in women than in men, affecting adults most often between the ages of 20 and 50 years.12,13
Etiology
Onset of thrombocytopenia often follows a viral infection, pregnancy, administration of certain medications (e.g., heparin, thiazide diuretics, chemotherapeutic agents), malignancies, splenomegaly, blood transfusions, or alcoholism.12 Regardless of the precipitating condition, the development of thrombocytopenia occurs by means of one of four mechanisms: 1) decreased platelet production; 2) increased platelet destruction; 3) splenic sequestration of platelets; and 4) platelet dilution.10 The most common form of thrombocytopenia seen in the critical care unit is immune thrombocytopenic purpura (ITP).11
Pathophysiology
In ITP, lymphocytes produce antibodies that begin to destroy existing platelets. The cause of this autoimmune response is unknown.13 With insufficient platelets available, the normal coagulation pathways are disrupted. Inadequate hemostasis ensues, and bleeding results. Although life-threatening gastrointestinal or intracerebral bleeding can occur, the bleeding seen in ITP does not result in deep visceral hemorrhage and hematoma.10
Assessment and Diagnosis
ITP is characterized by the gradual onset of signs and symptoms.13 The diagnosis is primarily based on findings in the patient’s history and physical examination.
Clinical Manifestations
Petechial hemorrhages, which manifest as small red spots primarily on legs and oral mucosa, are most indicative of a platelet disorder, unlike larger hematomas, which are most commonly associated with coagulation disorders.11 Bruising unrelated to trauma is another common sign of ITP.12 The signs and symptoms of ITP are listed in Table 38-4.
TABLE 38-4
IDIOPATHIC THROMBOCYTOPENIA PURPURA: SIGNS, SYMPTOMS, AND LABORATORY DATA
| SYSTEM OR STUDY | SIGNS AND SYMPTOMS |
| Integumentary | Petechial hemorrhage of lower extremities, ecchymoses, gingival bleeding, spontaneous epistaxis |
| Neurologic | Sudden, severe headache; nausea and vomiting; seizures; focal neurologic deficits; decreased level of consciousness |
| Renal | Hematuria |
| Gastrointestinal |
