CHAPTER 78 Hematologic Arthritis
HEMOPHILIC ARTHROPATHY
The elbow is frequently involved with acute hemarthrosis.14 In addition, the elbow is the second most common site of arthropathy in the hemophilic patient after the knee joint. A 1965 report from Sweden surveyed 114 patients with hemophilia A and 43 patients with hemophilia B.1 Sixty-six of the 95 severely affected and 20 of the 38 moderately affected patients had elbow involvement; 59 patients had bilateral elbow involvement. The severity of the arthropathy in all joints increased with age and disease severity.
PATHOGENESIS
Hemophilia is an X-chromosome–linked disease characterized by a deficiency or functional defect of coagulation factors VIII or IX, which results in an increased bleeding tendency. Hemophilic arthropathy is believed to be the consequence of repeated episodes of intra-articular bleeding, which can occur spontaneously or as a result of trauma.21 Joint destruction is the result of the direct deleterious effect of joint hematoma on the metabolism of the articular cartilage and the secondary hypertrophic synovitis, which leads to cartilage and bone destruction as well as soft tissue contractures (Box 78-1). Increases in intra-articular pressure may also play a role in producing the degenerative changes.39 The growth plates may be affected in children, which may result in associated angular deformity. The pathogenesis and management of hemophilic arthropathy may be further complicated by the presence of a coexistent human immunodeficiency virus (HIV) infection.10
EVALUATION
Patients with hemophilic arthropathy of the elbow may present in various stages of the disease. The first episodes of acute elbow hemarthrosis present with severe pain, effusion, and limited motion. As joint degeneration progresses, patients develop various grades of pain, stiffness, and deformity. Gamble et al18 measured elbow motion in 48 patients with hemophilia followed for an average of 10.8 years. Patients older than 25 years had decreased motion in all planes compared with patients younger than 15 years. Pronation was the first motion to be restricted, and extension was the motion most affected at the end of follow-up. Interestingly, many patients with hemophilic arthropathy are more limited by their lack of pronation and supination than by limited flexion and extension.
Hemophilic arthropathy commonly affects other joints including the knees and ankles. In the upper extremity, the elbow is more commonly affected than the shoulder or the wrist; in a series of 23 moderate and severe hemophiliacs, the elbow joints were the site of recognizable arthropathy in 87% of the cases, whereas the shoulder and wrist were affected in a small proportion of patients.21 When surgery is contemplated, attention should be paid to the order in which the different joints need to be addressed.
Hemophilic pseudotumors are an uncommon but well-known musculoskeletal complication of hemophilia.29 Pseudotumors occur in approximately 1% to 2% of patients with bleeding disorders. They present usually as an enlarging mass secondary to intraosseous, subperiosteal or intramuscular bleeding with various degrees of bone and soft tissue compromise. They can compress adjacent neurovascular structures and be misdiagnosed as true malignancies based on their clinical and radiographic features.
Imaging Studies
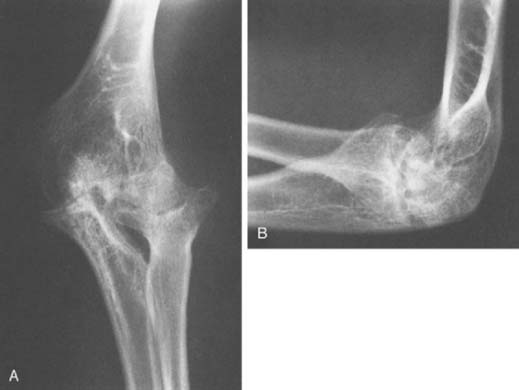
Hemophilic arthropathy usually presents with joint line narrowing, spurs, subperiosteal new bone formation, and occasionally, bone loss or deformity. A number of classification systems have been developed to rate the severity of the radiographic changes in hemophilic arthropathy. The system published by Arnold and Hilgarter3 has been extensively used in the literature and includes five stages as detailed in Box 78-2 (see Figs. 78-1 through 78-4).
BOX 78-2 Radiographic Classification of Hemophilic Arthropathy3
| Stage | Features |
|---|---|
| I | No skeletal abnormalities |
| Soft issue swelling present | |
| II | Osteoporosis, epiphyseal overgrowth |
| Normal joint line | |
| No bone cysts | |
| III | Subchondral joint changes |
| Subchondral cysts visible | |
| Trochlear notch widened | |
| IV | Joint space narrowing |
| V | Loss of joint space |
| Joint contracture | |
| Enlargement of epiphyses or architectural changes |
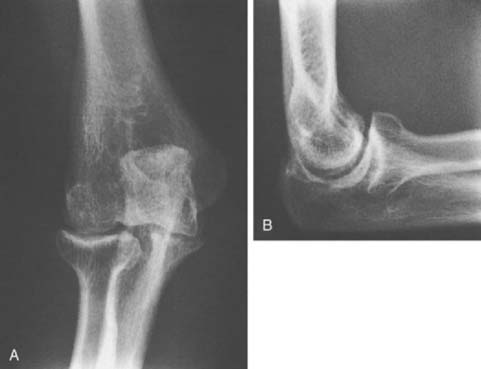
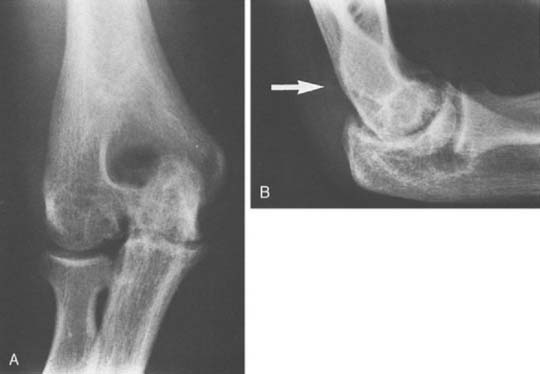
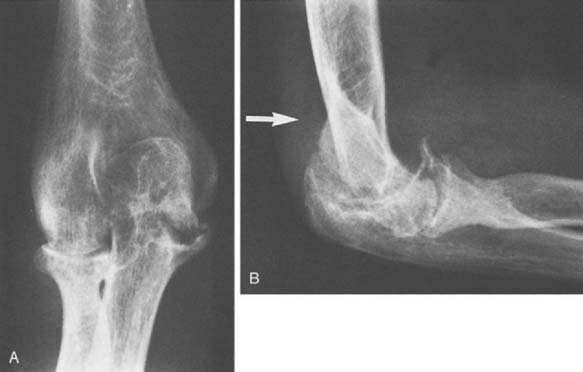
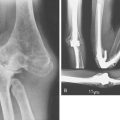
FIGURE 78-3 A, Radiographs of the same patient shown in Figure 78-2 taken 5 years later show progressive changes with loss of the joint space narrowing and loss of cartilage (stage IV). B, Again the arrow shows opacification of the synovium.
More recently, the World Federation of Hemophilia has adopted the radiographic scoring and classification system proposed by Peterson et al32 and based on thefollowing eight aspects: osteoporosis, hypertrophy of the endplate, loss of joint space, irregular subchondral surface, subchondral cysts, erosion of the joint surface, incongruity of the joint surfaces, and deformity of the joint. The score ranges from 0 (no radiographic abnormalities) to 13 (severe arthropathy).
Some authors have identified three main patterns of joint involvement.19,43 Patients with involvement of predominantly the medial side of the joint present with ulnohumeral joint narrowing and medial spurring, which may irritate the ulnar nerve. Involvement of the lateral side of the joint is associated with radial head enlargement, posterolateral elbow pain, and restriction of pronation and supination. Finally, global involvement of the joint is associated usually with more severe stiffness affecting all planes of motion.
As with other inflammatory conditions, magnetic resonance imaging has been used to more precisely evaluate abnormalities of the articular cartilage, soft tissues, and synovium and to monitor the progression of the arthropathy.46 Several scoring systems based on magnetic resonance findings have been developed to identify patients at risk for the development of severe arthropathy and to prevent articular changes by the use of more intensive medical treatment.25,26
TREATMENT
Medical Management
The treatment of hemophilia requires correction of the coagulation defect by replacement of the deficient factor. Patients with mild hemophilia A or Von Willebrand’s disease may benefit from treatment with desmopressin, which releases factor VIII from endothelial storage sites. At present, recombinant concentrates of factors VIII and IX are the products of choice for factor replacement. The regular administration of replacement therapy with the object of reducing spontaneous bleeding is a logical approach. The goal is to maintain the factor VIII or IX level above 1%. This can be achieved by factor concentrate administration two or three times a week. Some physicians recommend factor replacement for all patients, whereas others favor treating only those patients with frequent bleeding episodes. Starting treatment at a younger age seems to protect against later development of hemophilic arthropathy.17
Replacement therapy is also the treatment of choice of bleeding episodes. The desired factor level is a function of the severity and location of the bleeding episode. Joint or muscle bleeding episodes are treated to achieve factor levels of 30% to 50%. Replacement therapy is also critical when surgery is considered.10 Most orthopedic surgery can be managed with initial levels of 80% to 100%, followed in a few days by minimum levels of at least 30%. Joint manipulation under anesthesia requires levels of at least 50%. The number of units required to obtain these titers is calculated based on the body weight and the volume of distribution, which is 1.5 for factor VIII and 1.2 for factor IX. Factor levels should be checked to ensure the effectiveness of the calculated dose. Patients with factor VIII inhibitor require a specific approach; if the inhibitor titer is below 5 BU, recombinant VII factor is administered. If the inhibitor titer is greater than 5 BU, there are three treatment options: porcine factor VIII, factor VIII inhibitor bypassing activity (FEIBA), or recombinant factor VIIa.
Acute Hemarthrosis
The treatment of acute hemarthrosis is mostly medical, and includes factor replacement and analgesia. The goal of replacement therapy is to maintain levels of 50% to 100% (or increase the plasma level between 20% and 50%) until the bleeding stops. Prompt replacement therapy is advised at the earliest sign of bleeding, especially in younger patients, in order to prevent joint degeneration. Well-trained patients and families are able to administer their replacement therapy at home without examination by a physician, which might delay treatment.
Tense painful hemarthrosis may be aspirated under factor replacement coverage. The needle is introduced into the joint usually in the soft spot centered between the radial head, the tip of the olecranon, and the lateral epicondyle. Aspiration should not be performed until the clotting factor has been raised to at least 50%. Uncontrolled massive joint bleeding may be controlled through selective angiographic embolization of the affected blood vessels.30 Acute ulnar nerve compression requiring surgical intervention has been reported as a complication of acute elbow hemarthrosis.27
Established Arthropathy
Synoviorthesis
Synoviorthesis involves injecting a pharmacologic agent into the joint with the aim of stabilizing the synovial membrane. Several agents may be used for this process, including radioactive gold, yttrium-90, phosphorus-32, rifampicin, osmic acid, and hyaluronic acid.9,16,34,37 Phosphorus-32 (chromic phosphate) is the agent approved by the U.S. Food and Drug Administration for synoviorthesis in hemophiliacs.12 When injected into the joint, these agents remove inflamed synovium, which decreases the frequency and severity of intra-articular bleeding episodes.9 Synoviorthesis is considered usually when the synovitis and recurrent bleeding are not controlled with factor replacement, anti-inflammatory medications, and physical therapy used for 3 to 6 months. It is especially recommended in patients with inhibitors to the clotting factor, advanced HIV infection or hepatitis, or multiple joint involvement. The results are somewhat unpredictable: in a series of 29 elbows treated with synoviorthesis using radioactive gold, results were satisfactory in eight cases at an average follow-up of 14 years.34 In this study, synoviorthesis decreased the frequency of bleeding but did not prevent joint degeneration.
Synovectomy, Radial Head Resection, and Contracture Release
Surgical débridement and synovectomy of the elbow joint is indicated when the hemophilic arthropathy fails to respond to replacement therapy and three synoviorthesis attempts or when limited motion impairs elbow function but the joint may still be salvaged.24 The procedure involves synovectomy, resection of any contracted capsule, removal of osteophytes, recreation of the humeral fossae, and occasionally, radial head resection.
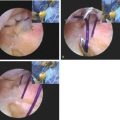
This procedure is most commonly performed through an open lateral approach or arthroscopically. Arthroscopic synovectomy has the potential of decreasing the rate of recurrent hemarthrosis and improving joint function while being associated with lower postoperative morbidity and fewer hospitalizations compared to open surgery (Fig. 78-5).45 Several studies have confirmed these findings in the adult as well as the pediatric population.13,22 In some series, the median bleeding frequency decline approached 85%.13
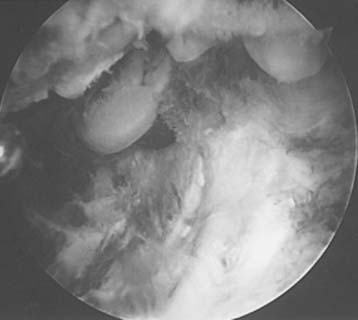
There is some published information on the results of joint débridement for hemophilic arthropathy of the elbow. Kay et al24 reported on 12 elbows treated with synovectomy and followed between 1 and 5 years. The average number of bleeding episodes over a 12-month period was decreased from 24 to 3. Five patients lost an average 28 degrees and six improved an average of 11 degrees of motion. Complications occurred in 25% of the cases. Le Balc’h et al28 reported on 23 elbows followed for 18 to 70 months after synovectomy. Patient age ranged from 8 to 25 years of age. Moderate pain persisted in only three, pronation and supination improved in nine, and flexion-extension improved in 14 elbows. Four elbows presented recurrent bleeding. Interestingly, arthroscopic synovectomy has been shown to be cost effective in a small series of 11 patients who required arthroscopy of the ankle (seven cases), elbow (three cases) or knee (one case). In this group of patients, the average cost per month was $7500 before surgery and decreased to $900 after surgery.42
Arthroplasty
The results of total elbow arthroplasty for hemophilic arthropathy in two separate institutions have been reported recently. Five patients underwent seven elbow arthroplasties at Oxford using several designs, including the Kudo, Souter, and Coonrad-Morrey prostheses.7,23 There were significant improvements in pain and function at an average follow-up of 2 years. Complications included deep infection requiring resection, ulnar nerve palsy, and axillary vein thrombosis.
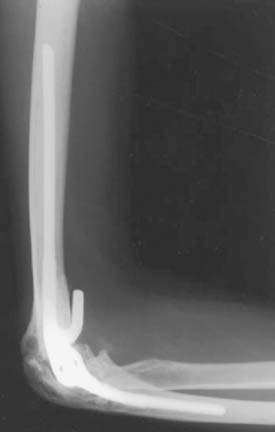
Kamineni et al23 recently reviewed the results of five total elbow arthroplasties performed at the Mayo Clinic for hemophilic arthropathy. The mean age of the patients was 39 years (range, 25 to 58 years). All patients had concurrent HIV infection. At a mean follow-up of 5.8 years (range, 1 to 11 years), one patient developed uncontrollable hemorrhage and was eventually diagnosed with a deep infection requiring implant removal. One additional late hematogenous deep infection could be controlled by joint débridement and polyethylene exchange. At most recent follow-up, pain and motion were improved significantly in all four patients with a surviving implant (Fig. 78-6). Radiographic loosening was evident in one of the four cases.
Pseudotumors
The treatment of pseudotumors is somewhat controversial and depends on the location and severity of the symptoms. Rapidly growing lesions in the distal part of the skeleton in children usually respond better to nonoperative treatment, whereas slowly growing lesions in the proximal part of the skeleton in adults usually leads to bone erosions and fail to respond to nonoperative treatment.2,36
Hemophilic pseudotumors are initially treated with intensive replacement therapy during 6 weeks. If the pseudotumor regresses by 25% to 50%, nonoperative treatment may be continued. Otherwise, consideration should be given to percutaneous drainage or surgicalremoval. Small pseudotumors may be drained and injected with fibrin glue.6 Surgical resection if larger pseudotumors is associated with good results.36
SICKLE CELL DISEASE
The term sickle cell disease is used to include various inherited diseases characterized by the presence of hemoglobin S (sickle hemoglobin). Hemoglobin S imparts a sickle shape to deoxygenated erythrocytes. Sickle cell heterozygosis results in sickle cell trait (hemoglobin AS), whereas sickle cell homozygosity results in sickle cell disease (hemoglobin SS). Bone and joint involvement is exceedingly rare in patients with sickle cell trait. A study of 94 patients with sickle cell trait found no significant difference in the frequency of joint symptoms or objectively demonstrable joint disease when compared with 114 black people with normal hemoglobin electrophoretic patterns.11 In contrast, most if not all patients with homozygous sickle cell anemia experience bone or joint crisis, and bone and joint problems are the most common manifestations (Box 78-3). In a prospective study of 56 adults with SS disease, 31 individuals suffered a total of 61 separate episodes of joint involvement during a 6- to 18-month observation period.15 The elbow was affected at one time or another in 13 patients (42%) usually in association with involvement of other large joints.
PATHOGENESIS
Hemoglobin S results from the inherited substitution of valine for glutamic acid as the sixth amino acid of the beta globin chain. This minor change produces profound alterations in the stability and solubility of the hemoglobin molecule.5 Hemoglobin S molecules polymerize in hypoxic and acidic environments, imparting a sickle shape to erythrocytes.
Sickle erythrocytes show increased adhesion. Interaction of sickle cells with adhesion proteins of the vascular endothelium initiates an inflammatory response, which further increases cellular adhesiveness. Increased adhesion and inflammation decrease blood flow, leading to further sickling. Repeated episodes of decreased blood flow may lead to impaired nourishment of critical structures. Vascular occlusion is responsible for the bone and joint manifestations of sickle cell disease, including avascular necrosis and increased risk for infection. Synovial biopsy specimens generally reveal microvascular thrombosis, with occasional intraluminal sickled cells, peri-vascular fibrosis, and mononuclear inflammatory cell infiltration.35
CLINICAL EVALUATION
Vaso-occlusive Crisis
Repeated painful vaso-occlusive crises are the hallmark of sickle cell disease. However, their prevalence is highly variable. In a prospective observational study of 3578 patients, the average rate of painful crises per patient-year was 0.8; however, almost 40% of all patients had no episodes of pain, whereas 5% of the patients accounted for one third of all episodes.33
Septic Arthritis and Osteomyelitis
The risk of bacterial infection is increased in children with sickle cell disease due to an early decrease in splenic function, genetic polymorphisms of binding proteins, and human leukocyte antigen (HLA) class II alleles.41 Areas of infarcted bone or bone marrow are typical sites for infections.4 Osteomyelitis is usually due to Salmonella or other gram-negative organisms such as Escherichia coli.4 Staphylococcus aureus, the most common cause of osteomyelitis in normal hosts, probably accounts for only one fourth of all cases in sickle cell disease. Septic arthritis is less common than osteomyelitis. The clinical presentation of osteomyelitis and septic arthritis often is similar to a vaso-occlusive crisis, and the exact diagnosis is often difficult to make. In general, infectious episodes are more likely to be associated with fever, longer lasting pain, and decreased range of motion.
Avascular Necrosis and Arthropathy
Joint osteonecrosis is a well-described complication of sickle cell disease.20 The femoral and humeral head are most commonly affected, but the distal humerus may also develop avascular necrosis. It usually presents with elbow pain and stiffness, which usually worsen as secondary degenerative changes ensue. Joint degeneration may also be secondary to microvascular and inflammatory changes in the synovium.35 Finally, inflammatory conditions, such as rheumatoid or gouty arthritis, may coexist with sickle cell disease and contribute to degenerative articular changes.31
Imaging Studies
Magnetic resonance imaging helps diagnose avascular necrosis in the early stages and may play a role in differentiating osteomyelitis from areas of bone infarction or osteonecrosis.35 Bone scans have also been used to identify infection with variable success.38 Microbiologic studies are needed to confirm the diagnosis of osteomyelitis or septic arthritis and guide treatment based on the results of the cultures and sensitivities. When septic arthritis is suspected, joint aspiration should be performed. The diagnosis of osteomyelitis may require culture of surgical biopsies.
MANAGEMENT
Vaso-occlusive Crises
Treatment of vaso-occlusive crises consists of hydration (100% to 150% of normal daily fluids), warm packs to the affected area, analgesics, and anti-inflammatory medication. Narcotics are usually needed to control the pain associated with vaso-occlusive crises. Patients with HbSS or HbS-beta(0) thalassemia and repeated episodes of vaso-occlusive crises should be considered for therapy with hydroxyurea, which may decrease the frequency of painful episodes in children and adults.8 The possible beneficial effects of other drugs such as azatidicine, magnesium pidolate, and clotrimazole derivatives is under investigation.
LEUKEMIA AND OTHER MYELOPROLIFERATIVE DISORDERS
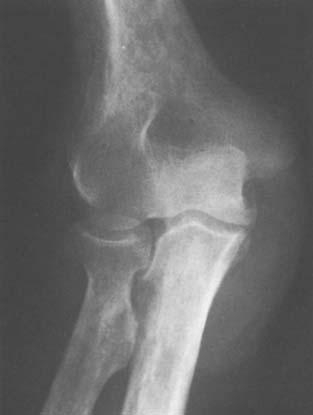
Leukemia and other myeloproliferative disorders may present with symptoms at the elbow secondary to (1) bone involvement with the myeloproliferative disorders or (2) distal humerus osteonecrosis associated with chemotherapy and treatment with steroids.40 These dis-orders are also known to be associated with a higher incidence of fractures,40 but they rarely affect the elbow joint. Joint involvement is more common in acute than chronic leukemia and in children compared with adults (Fig. 78-7).44 Surgical treatment is only required for patients with osteonecrosis.
1 Ahlberg A. Haemophilia in Sweden. VII. Incidence, treatment and prophylaxis of arthropathy and other musculo-skeletal manifestations of haemophilia A and B. Acta Orthop. Scand. Suppl. 1965;77:3.
2 Ahlberg A.K. On the natural history of hemophilic pseudotumor. J. Bone Joint Surg. Am. 1975;57:1133.
3 Arnold W.D., Hilgartner M.W. Hemophilic arthropathy. Current concepts of pathogenesis and management. J. Bone Joint Surg. Am. 1977;59:287.
4 Bennett O.M., Namnyak S.S. Bone and joint manifestations of sickle cell anaemia. J. Bone Joint Surg. Br. 1990;72:494.
5 Bunn H.F. Pathogenesis and treatment of sickle cell disease. N. Engl. J. Med. 1997;337:762.
6 Caviglia H.A., Fernandez-Palazzi F., Gilbert M.S. Haemophilic pseudotumours of the limbs and their percutaneous treatment. Haemophilia. 2002;8:402.
7 Chapman-Sheath P.J., Giangrande P., Carr A.J. Arthroplasty of the elbow in haemophilia. J. Bone Joint Surg. Br. 2003;85:1138.
8 Charache S., Terrin M.L., Moore R.D., Dover G.J., Barton F.B., Eckert S.V., McMahon R.P., Bonds D.R. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. N. Engl. J. Med. 1995;332:1317.
9 Dawson T.M., Ryan P.F., Street A.M., Robertson P.L., Kalff V., Kelly M.J., Cicuttini F.M. Yttrium synovectomy in haemophilic arthropathy. Br. J. Rheumatol. 1994;33:351.
10 DeGnore L.T., Wilson F.C. Surgical management of hemophilic arthropathy. Instr. Course Lect. 1989;38:383.
11 Dorwart B.B., Goldbert M.A., Schumacher H.R., Alavi A. Absence of increased frequency of bone and joint disease with hemoglobin AS and AC. Ann. Intern. Med. 1977;86:66.
12 Dunn A.L., Busch M.T., Wyly J.B., Abshire T.C. Radionuclide synovectomy for hemophilic arthropathy: a comprehensive review of safety and efficacy and recommendation for a standardized treatment protocol. Thromb. Haemost. 2002;87:383.
13 Dunn A.L., Busch M.T., Wyly J.B., Sullivan K.M., Abshire T.C. Arthroscopic synovectomy for hemophilic joint disease in a pediatric population. J. Pediatr. Orthop. 2004;24:414.
14 Dymock I.W., Hamilton E.B., Laws J.W., Williams R. Arthropathy of haemochromatosis. Clinical and radiological analysis of 63 patients with iron overload. Ann. Rheum. Dis. 1970;29:469.
15 Espinoza L.R., Spilberg I., Osterland C.K. Joint manifestations of sickle cell disease. Medicine (Baltimore). 1974;53:295.
16 Fernandez-Palazzi F., Rivas S., Viso R., de Bosch N.B., de Saez A.R., Boadas A. Synovectomy with rifampicine in haemophilic haemarthrosis. Haemophilia. 2000;6:562.
17 Fischer K., van der Bom J.G., Mauser-Bunschoten E.P., Roosendaal G., Prejs R., de Kleijn P., Grobbee D.E., van den Berg M. The effects of postponing prophylactic treatment on long-term outcome in patients with severe hemophilia. Blood. 2002;99:2337.
18 Gamble J.G., Vallier H., Rossi M., Glader B. Loss of elbow and wrist motion in hemophilia. Clin. Orthop. Relat. Res. 1996;328:94.
19 Goddard N.J. Elbow arthropathy in patients with severe haemophilia. In: Rodriguez-Werchan E.C., editor. The Haemophilic Joints, New Perspectives. Oxford UK: Blackwell Publishing Ltd.; 2003:125.
20 Hernigou P., Habibi A., Bachir D., Galacteros F. The natural history of asymptomatic osteonecrosis of the femoral head in adults with sickle cell disease. J. Bone Joint Surg. Am. 2006;88:2565.
21 Hogh J., Ludlam C.A., Macnicol M.F. Hemophilic arthropathy of the upper limb. Clin. Orthop. Relat. Res. 1987;218:225.
22 Journeycake J.M., Miller K.L., Anderson A.M., Buchanan G.R., Finnegan M. Arthroscopic synovectomy in children and adolescents with hemophilia. J. Pediatr. Hematol. Oncol. 2003;25:726.
23 Kamineni S., Adams R.A., O’Driscoll S.W., Morrey B.F. Hemophilic arthropathy of the elbow treated by total elbow replacement. A case series. J. Bone Joint Surg. Am. 2004;86-A(3):584.
24 Kay L., Stainsby D., Buzzard B., Fearns M., Hamilton P.J., Owen P., Jones P. The role of synovectomy in the management of recurrent haemarthroses in haemophilia. Br. J. Haematol. 1981;49:53.
25 Kilcoyne R.F., Lundin B., Pettersson H. Evolution of the imaging tests in hemophilia with emphasis on radiography and magnetic resonance imaging. Acta Radiol. 2006;47:287.
26 Kilcoyne R.F., Nuss R. Radiological evaluation of hemophilic arthropathy. Semin. Thromb. Hemost. 2003;29:43.
27 Lancourt J.E., Gilbert M.S., Posner M.A. Management of bleeding and associated complications of hemophilia in the hand and forearm. J. Bone Joint Surg. Am. 1977;59:451.
28 Le Balc’h T., Ebelin M., Laurian Y., Lambert T., Verroust F., Larrieu M.J. Synovectomy of the elbow in young hemophilic patients. J. Bone Joint Surg. Am. 1987;69:264.
29 Magallon M., Monteagudo J., Altisent C., Ibanez A., Rodriguez-Perez A., Riba J., Tusell J., Martin-Villar J. Hemophilic pseudotumor: multicenter experience over a. 25-year period. Am J Hematol. 1994;45(2):103.
30 Mauser-Bunschoten E.P., Zijl J.A., Mali W., van Rinsum A.C., van den Berg H.M., Roosendaal G. Successful treatment of severe bleeding in hemophilic target joints by selective angiographic embolization. Blood. 2005;105:2654.
31 Nistala K., Murray K.J. Co-existent sickle cell disease and juvenile rheumatoid arthritis. Two cases with delayed diagnosis and severe destructive arthropathy. J. Rheumatol. 2001;28:2125.
32 Petersson H., Ahlberg A., Nilsson I.M. A radiologic classification of haemophilic arthropathy. Clin. Orthop. Relat. Res. 1996;149:153.
33 Platt O.S., Thorington B.D., Brambilla D.J., Milner P.F., Rosse W.F., Vichinsky E., Kinney T.R. Pain in sickle cell disease. Rates and risk factors. N. Engl. J. Med. 1991;325:11.
34 Rodriguez-Merchan E.C., Magallon M., Galindo E., Lopez-Cabarcos C. Hemophilic synovitis of the knee and the elbow. Clin. Orthop. Relat. Res. 1997;343:47.
35 Schumacher H.R., Andrews R., McLaughlin G. Arthropathy in sickle-cell disease. Ann. Intern. Med. 1973;78:203.
36 Shaheen S., Alasha E. Hemophilic pseudotumor of the distal parts of the radius and ulna. A case report. J. Bone Joint Surg. Am. 2005;87:2546.
37 Siegel H.J., Luck J.V.Jr., Siegel M.E., Quines C., Anderson E. Hemarthrosis and synovitis associated with hemophilia: clinical use of P-32 chromic phosphate synoviorthesis for treatment. Radiology. 1994;190:257.
38 Skaggs D.L., Kim S.K., Greene N.W., Harris D., Miller J.H. Differentiation between bone infarction and acute osteomyelitis in children with sickle-cell disease with use of sequential radionuclide bone-marrow and bone scans. J. Bone Joint Surg. Am. 2001;83-A:1810.
39 Sokoloff L. Biochemical and physiological aspects of degenerative joint diseases with special reference to hemophilic arthropathy. Ann. N. Y. Acad. Sci. 1975;240:285.
40 Strauss A.J., Su J.T., Dalton V.M., Gelber R.D., Sallan S.E., Silverman L.B. Bony morbidity in children treated for acute lymphoblastic leukemia. J. Clin. Oncol. 2001;19:3066.
41 Tamouza R., Neonato M.G., Busson M., Marzais F., Girot R., Labie D., Elion J., Charron D. Infectious complications in sickle cell disease are influenced by HLA class II alleles. Hum. Immunol. 2002;63:194.
42 Tamurian R.M., Spencer E.E., Wojtys E.M. The role of arthroscopic synovectomy in the management of hemarthrosis in hemophilia patients: financial perspectives. Arthroscopy. 2002;18:789.
43 Utukuri M.M., Goddard N.J. Haemophilic arthropathy of the elbow. Haemophilia. 2005;11:565.
44 Weinberger A., Schumacher H.R., Schimmer B.M., Myers A.R., Brogadir S.P. Arthritis in acute leukemia. Clinical and histopathological observations. Arch. Intern. Med. 1981;141:1183.
45 Wiedel J.D. Arthroscopic synovectomy: state of the art. Haemophilia. 2002;8:372.
46 Yulish B.S., Lieberman J.M., Strandjord S.E., Bryan P.J., Mulopulos G.P., Modic M.T. Hemophilic arthro-pathy: assessment with MR imaging. Radiology. 1987;164:759.