[level-membership-for-neurosurgery-category]
Chapter 18 Growth Hormone–Secreting Tumors
Phenotypic features of acromegaly include acral enlargement; coarse facial features with frontal bossing, prognathism, diastema and macroglossia; skin thickening, hypertrichosis, malodorous hyperhidrosis, and acanthosis nigricans; and deepening of the voice due to laryngeal hypertrophy. Other manifestations include headaches, lethargy, obstructive sleep apnea, peripheral neuropathies such as carpal tunnel syndrome, and bony deformation including bone thickening and vertebral osteophyte formation. Other consequences include abnormal carbohydrate metabolism and diabetes mellitus; cardiovascular diseases including hypertension, atherosclerosis, and cardiomyopathy; and an increased risk of other neoplasms including colon cancer.1–6 Patients with acromegaly have a two- to three-fold increase in mortality due to cardiovascular and cerebrovascular diseases. Normalization of GH levels may decrease this risk substantially to levels comparable to the general population.7,8 Other clinical manifestations are related to mass effect of the pituitary tumor, including headaches, visual loss (classically bitemporal hemianopia), and hormonal deficiencies (hypogonadism, hypothyroidism, and hypoadrenalism).
The disease is insidious, which leads to a delayed diagnosis, often seven to ten years after onset of symptoms.9 Recently, this lag has shortened significantly, likely due to the increase in magnetic resonance imaging.10 Patients with acromegaly generally present in the third to fifth decade, and both sexes are affected equally.11
The average annual incidence of acromegaly is three to four per million, and the prevalence is 40 to 70 cases per million people.7,10 The vast majority of cases, 95% to 98%, are due to a GH-secreting pituitary adenoma.9,10,12 Pituitary adenomas from somatotroph cells may lead to excessive secretion of GH, while adenomas from acidophil stem cells or mammosomatotrophs often secrete both GH and prolactin (PRL).9,12 Most GH-secreting tumors (75%–80%) are macroadenomas (>1 cm in diameter),7 and 20% to 50% co-secrete PRL or other pituitary hormones.13 Rare causes of acromegaly include ectopic GH-secreting tumors such as bronchial carcinoid or pancreatic islet cell tumors; hypothalamic GH-releasing hormone (GHRH)-secreting tumors; exogenous administration of GH; or familial syndromes such as McCune-Albright syndrome, multiple endocrine neoplasia I (MEN-I) or Carney complex.9
Growth Hormone
There is some heterogeneity in GH due to differential splicing and post-translational modification.14–16 Two main forms of GH are found in the circulation: the 22K form comprises 90% of serum GH, and the 20K form makes up 5%.16 This heterogeneity may explain differences between levels measured by radioimmunoassays and actual biological activity in some patients.
Diagnosis
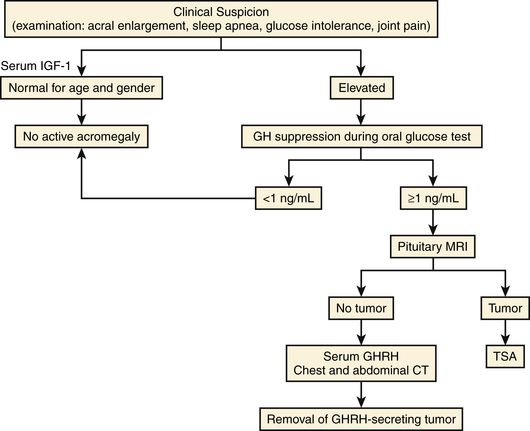
The suspicion of acromegaly is usually based on physical examination, while incidental radiologic detection in patients without typical manifestations is rare. Laboratory testing is needed to prove GH excess, while radiology techniques are used to visualize the tumor (Fig. 18-1). Screening laboratory tests include measurement of basal GH and IGF-1, and laboratory confirmation occurs when GH fails to suppress with oral glucose tolerance testing.
Laboratory Diagnosis
Random Serum GH Measurement
In healthy subjects, random GH levels are less than 5 ng/ml while most acromegalic patients have levels greater than 10 ng/ml. In active acromegaly, the normal episodic GH pattern is replaced by a constantly elevated GH level throughout the day.1 However, GH levels fluctuate widely and GH has a short half-life, so some acromegalic patients have normal GH levels on initial testing. Serum GH levels may be elevated in other conditions including uncontrolled diabetes mellitus, renal failure, malnutrition and during physical or emotional stress, even in the absence of acromegaly.13 Therefore, random GH measurement is not the preferred screening test for acromegaly.
IGF-1 Measurement
Serum IGF-1 measurement is the best single test for screening. The measurement of IGF-1 is used to provide an indicator of the body’s overall exposure to GH. Normal ranges for IGF-1 vary between different assays and are age- and gender-dependent.13 IGF-1 is increased in nearly all acromegalic patients, even those in whom random single GH levels are within the normal range. IGF-1 is also a reliable indicator for post-treatment hormonal remission, as it reflects GH secretion over the prior 24 hours.4 One of the IGF-1 binding proteins (IGFBPs) can be measured to assist in the diagnosis of acromegaly. The level of IGFBP-3 correlates directly with GH, but the overlap with normal persons limits its use.
GH Suppression to Hyperglycemia
The oral glucose suppression test (75 g) is used to confirm a diagnosis of acromegaly. In a normal control, GH decreases to below 1 ng/ml after a glucose load, whereas in an acromegalic patient, this suppression fails to occur.17 This test is useful to document biochemical remission after surgical removal of the pituitary tumor,18 but does not appear to be useful to assess control in patients receiving therapy with somatostatin analogues.17
Other Dynamic Tests
Other dynamic tests are rarely needed to diagnose acromegaly. Thyrotropin-releasing hormone (TRH) stimulation leads to a significant increase in GH in untreated acromegalics, although it does not cause a significant change in GH levels in normal subjects. This response may also occur in the setting of liver disease, renal failure, or depression. TRH-stimulation may identify patients who, despite a normal postsurgical GH level, have residual GH-secreting tumor.19 Administration of oral L-Dopa or bromocriptine, a dopamine agonist, to a fasting normal subject increases GH secretion, though it paradoxically decreases GH levels in a fasting acromegalic patient.20
GHRH
GHRH levels should not be routinely used in diagnosis of acromegaly. However, they are useful in patients with confirmed acromegaly who do not harbor a pituitary tumor. Ectopic acromegaly, due to non-central nervous system tumors such as a pancreatic islet cell tumor or a bronchial carcinoid, results in a significantly elevated serum GHRH, whereas GHRH is usually low with a GH-secreting adenoma.21
Other Hormones
Hormonal assessments are needed to measure prolactin co-secretion, although moderately elevated prolactin can be due to stalk effect. Pituitary hormone deficiencies may be measured by ACTH, cortisol, TSH, free T4, FSH, LH levels in both genders, estradiol in women, and testosterone in men.
Treatment
Surgery
Surgical adenomectomy by an experienced neurosurgeon remains the first-line treatment for most patients with acromegaly.22,23 The goals of surgical resection are to eliminate mass effect, preserve or restore pituitary and visual function, and obtain tissue for histopathologic analysis.24
Most of these tumors are sellar or suprasellar lesions that may be removed trans-sphenoidally using a direct endonasal, sublabial, or trans-septal approach with an endoscope or microscope. The first trans-sphenoidal resection of a pituitary lesion was performed by Hermann Schloffer in 1907; the procedure was popularized by Harvey Cushing in the two decades that followed.25,26 Neurosurgeons have been improving the trans-sphenoidal adenomectomy (TSA) since. A craniotomy may rarely be necessary when a tumor has extensive suprasellar or parasellar extension.
Endoscopy
The lesion is removed in the same manner as described with the microscopic approach. Duraform is then placed over the sella, and the sphenoid sinus is packed using Gelfoam. NasoPore is laid over the posterior septectomy site and the sphenoethmoid recesses bilaterally. A mucosal septal flap may be used if a CSF leak is present. A speculum is not needed with this approach.27,28
The straight surgical endoscope provides a wide field of view, while angled scopes permit enhanced visualization of the sellar wall, suprasellar, retrosellar, or parasellar regions. Three-dimensional endoscopes have been recently introduced and provide a stereoscopic, nondistorted view of the regional anatomy in contrast to older two-dimensional endoscopes (Figure 18-2).
Sublabial Trans-Sphenoidal Approach
Sublabial may be the appropriate approach for patients with large tumors or pediatric patients with small nares. The upper lip is retracted, an incision is made horizontally in the gingival mucosa, and the maxilla and nasal cavity floor are accessed. A vertical incision is made to separate the nasal mucosa from the septum, and the anterior septum is subluxed and deviated. The speculum is inserted and the microscope or endoscope is brought into the field. The operation continues in the same manner as described above.29
Neuronavigation
Frameless stereotactic neuronavigation permits the surgeon to confirm his position at any point during the TSA to assess the proximity of surrounding structures or determine when he is approaching the limits of the tumor (Figure 18-3). Navigation may be used to assist with large lesions that involve the carotid arteries or recurrent lesions where the normal anatomy has been altered by a prior operation.30,31
Outcomes of Trans-Sphenoidal Surgery
Clinical Outcomes
Following operative decompression, visual field defects improve in 70% to 89% of patients,32,33 remain unchanged in 7% percent, and rarely worsen (<4%).34 Headaches usually improve in a few days, while soft-tissue swelling and glucose and blood pressure control improve progressively in the next few weeks postoperatively. Bony abnormalities persist, while joint symptoms often improve, although complete resolution is unlikely.
Postoperative Biochemical Outcome
Plasma GH levels rapidly decrease within hours after surgery,35 while IGF-1 levels improve over the next several weeks to months.22
The success rate in achieving biochemical control of GH levels varies widely due to the diverse criteria used by different authors and changes in hormonal assays.36 Recent studies have established more stringent criteria for “biochemical cure” or remission, requiring a random GH less than 2.5 μg/l, GH nadir less than 0.4 μg/l after an oral glucose tolerance test, as well as a normal IGF-1.36,37 Until 2010, GH nadir used as the threshold during oral glucose tolerance testing was 1.0 μg/l. Using these criteria, a 2003 study of 59 patients followed for an average of more than 13 years after TSA showed that 52% achieved long-term biochemical remission after surgery alone.38 Overall, the postoperative remission rate is 80% for patients with GH-secreting microadenomas and less than 50% in macroadenomas, while long-term recurrence rate is 3% to 10%.
The factors predictive of surgical outcome include: tumor size, extrasellar growth, dural invasion, and preoperative GH level.18,22,23,35,39,40 More than 40% of acromegalic patients treated with surgery initially will require additional treatment.41–43 Reoperation for acromegaly has a lower success rate and higher complication rate than does the initial surgery. Reoperation is generally reserved for patients unresponsive to other forms of therapy or who have progressive visual impairment despite medical therapy.43
Complications
Potential complications from trans-sphenoidal surgery include hemorrhage, carotid artery injury, ischemic stroke, visual impairment, cerebrospinal fluid leak, nasal septal perforation, and epistaxis. The risk of stroke or death is less than 1%, while the risk of CSF rhinorrhea is 5%. Postoperative hypopituitarism may occur in 3% to 15% patients. In the first few days postoperatively, morning serum cortisol levels should be measured. Patients with symptoms of hypocortisolemia and a morning serum cortisol less than 10 μg/dl usually receive glucocorticoid replacement upon discharge. A full evaluation of pituitary hormones should be repeated at six to 12 weeks postoperatively to determine need for replacement.
Mortality for patients undergoing trans-sphenoidal surgery is very low, approximately 0.5%. Among patients with giant macroadenomas, the mortality is approximately 1%. Following resection of a giant macroadenoma, when some residual tumor may remain, a rare but potentially fatal postoperative complication is apoplexy. In one study of 134 surgically resected giant adenomas, four patients suffered fatal postoperative pituitary apoplexy.44 These patients must be closely watched.
Trans-sphenoidal pituitary exploration for patients with acromegaly with no pituitary tumor seen on MRI and a negative workup for an ectopic GHRH-secreting lesion is controversial. Surgical exploration may rarely reveal a small lesion hidden by the wall of the cavernous sinus or along the sellar floor.45
Pharmacologic Therapy
There are three main categories of pharmacologic treatment of acromegaly: Somatostatin analogues, growth hormone-receptor antagonists, and dopamine agonists.46
Somatostatin Analogues
Somatostatin analogues (octreotide and lanreotide) should be considered in patients with abnormal GH or IGF-1 levels postoperatively, either alone or while awaiting the effects of radiation. They are also recommended for patients who are medically unstable for surgery (e.g., those with uncontrolled hypertension, diabetes mellitus, or sleep apnea), or patients in whom tumors are clearly invasive and complete surgical removal is unlikely.6,47 Poor-risk patients may be reconsidered for surgery if their medical condition improves after treatment with somatostatin analogues.48
Most acromegalic patients treated with somatostatin analogues achieve a significant decrease in serum GH and IGF-1 levels.49,50 A double-blind, randomized, multi-center study of 115 acromegalic patients showed that octreotide, at a dose of 100 μg three times per day for six months, reduced mean GH levels by 70% and IGF-1 levels by 60%.51 Tumor size shrinks by 25% to 50% in 20% to 47% of patients with acromegaly on chronic octreotide therapy.47,48,51–53 Eight to 12 weeks of preoperative somatostatin analogue therapy shrinks GH-secreting macroadenomas by about 40%,54 and short-term preoperative octreotide administration decreases surgical risk in patients who have cardiac and metabolic complications of acromegaly.48,55 It is debatable, however, whether this affects surgical outcome. Two prospective, randomized studies demonstrated no benefit of preoperative octreotide on rate of hormone normalization postoperatively or on duration of hospital stay.56,57
With somatostatin analogues, relief of clinical symptoms is often seen immediately after starting the injections and before serum GH levels have declined. Many clinicians discontinue somatostatin analogues in patients who are undergoing radiation therapy, as there is a theoretical concern that it may be radioprotective, based on the experience with dopamine agonists in prolactinomas.58 Side effects associated with the long-term administration of somatostatin analogues are relatively minor and include pain at the injection site, nausea, abdominal cramps, mild steatorrhea, hyperglycemia, and cholesterol gallstones.51,52,59 Discontinuation of medical therapy may result in rebound of pretreatment hormone levels and tumor size.54,60
Growth Hormone–Receptor Antagonist
Pegvisomant is an alternative for patients in whom surgery and medical therapy with somatostatin analogues are ineffective or poorly tolerated.61 Pegvisomant, a mutated derivative of GH, is a highly effective antagonist of GH action in patients with acromegaly, and normalizes IGF-1 in more than 90% of patients.6,46 The inhibition of GH action, rather than pituitary GH secretion, represents a paradigm shift in the medical management of acromegaly.46,62–64 Pegvisomant does not act directly on the adenoma, and there is a potential risk of increase in GH secretion and tumor enlargement due to a loss of negative feedback from serum IGF-1. Early results, however, suggest that while there is some increase in GH level with pegvisomant therapy, it is not progressive and does not lead to a significant increase in tumor growth in up to two years of follow-up.46,62–65 Nevertheless, serial MRI scans to monitor tumor size are recommended. IGF-1 and not GH levels should be followed to monitor treatment with pegvisomant. Combined therapy with somatostatin analogues and pegvisomant normalized IGF-1 in most patients.66
Side effects from pegvisomant are generally minor. Some patients have asymptomatic hepatic enzyme elevation after initiation of pegvisomant, which normalizes after the stopping the drug,46,67 but few serious cases of hepatitis were reported.6
Dopamine Agonists
Therapy with dopamine agonists may be attempted for tumors that co-secrete prolactin. Bromocriptine is effective in lowering GH and IGF-1 levels in only 20% of patients, with only 10% achieving normalization.6,68,69 Side effects include nausea, vertigo, hypotension, and nasal stuffiness. The newer generation of long-acting D2-receptor agonists, including cabergoline and quinagolide, appear to be more effective and better tolerated than bromocriptine.48,70,71 Long-term administration of cabergoline to 64 patients normalized plasma IGF-1 levels in 39% of patients and decreased IGF-1 modestly in another 28%.71 It is most effective for patients with less active disease (IGF-1 < 750 ng/ml) and those with mixed GH and prolactin-secreting tumors.68
Radiation Therapy
Radiotherapy is indicated for patients who are unsuitable for or have failed surgery, and in whom medical therapy with somatostatin analogues and dopamine agonists has failed to achieve biochemical remission. In the first two years after radiation, a significant decrease in GH levels is usually achieved, followed by a slow decline for years thereafter.72–77 GH concentrations of approximately 5 μg/l are achieved in 80% of patients, though this may take 10 to 15 years to achieve. Few patients are actually “cured” if one applies strict criteria.61,78
Risks of conventional radiotherapy include visual changes from radiation damage to the optic nerve or chiasm, cognitive and neurologic deficits from radiation necrosis in adjacent brain, and a small risk for the development of secondary brain tumors such as gliomas.72–74,77,79 Hypopituitarism occurs in 30% to 70% of patients within 10 years.72,80,81 Stereotactic radiosurgery, such as the gamma knife or linear accelerator, is replacing conventional radiotherapy at most medical centers.77,80,82,83 In one study, gamma knife radiosurgery (GKS) was used as the primary procedure in 68 of 79 patients with GH-secreting tumors. All patients saw a decline in GH levels within six months.83 In a small series using CyberKnife, 44% of patients achieved biochemical remission after an average 25 months of follow-up.84
The major risk from SRS is radiation damage to the visual pathways, but this can be decreased by limiting the radiation dose to the optic chiasm to less than 10 Gy. Structures in the cavernous sinus are less radiosensitive, so an ablative dose may be administered to tumors with lateral invasion or impingement on the cranial nerves. This allows SRS to function as an adjuvant to surgical resection in patients with tumors that have invaded the cavernous sinus. Stereotactic radiosurgery has lower long-term risks of developing cognitive changes or a second neoplasm than does conventional radiation, and SRS appears to induce remission more rapidly than does fractionated radiotherapy.80,83 As with conventional radiotherapy, approximately 30% of patients develop new hormone deficiencies after SRS.80,84 In patients who cannot safely undergo SRS due to the proximity of the visual pathways to the tumor margin, fractionated stereotactic radiotherapy may be a safer and more effective alternative to conventional radiotherapy.80,83
Pediatric
GH-secreting tumors in children and adolescents with gigantism are more often aggressive and invasive than in adults.85 Trans-sphenoidal resection of GH-secreting tumors is safe in pediatric patients. For patients with small nares, a sublabial approach may be used. The risk of recurrence after surgical resection is higher in pediatric than in adult patients, however. The rate of postoperative complications including DI is higher in a series of pediatric patients than in adult series, and the likelihood of achieving biochemical remission is lower, 60% versus 78%.85
Although there are less data than for adults, medical therapy with somatostatin analogues decreases serum IGF-1 levels in children. A small study of children treated with pegvisomant decreased IGF-1 levels, decreased somatic growth, and improved acromegalic features in all three patients.86 Radiotherapy has special risks in the pediatric population because of the danger of panhypopituitarism and its effects on growth and puberty. The years-long delay in effectiveness with radiation may preclude any effect on the rapid growth seen in children with gigantism. Generally, the risks of pituitary radiotherapy outweigh the potential benefits in children.85
Abe T., Tara L.A., Lüdecke D.K. Growth hormone-secreting pituitary adenomas in childhood and adolescence: features and results of transnasal surgery. Neurosurgery. 1999;45(1):1-10.
Abosch A., Tyrrell J.B., Lamborn K.R., et al. Transsphenoidal microsurgery for growth hormone-secreting pituitary adenomas: initial outcome and long-term results. J Clin Endocrinol Metab. 1998;83(10):3411-3418.
Beauregard C., Truong U., Hardy J., Serri O. Long-term outcome and mortality after transsphenoidal adenomectomy for acromegaly. Clin Endocrinol. 2003;58(1):86-91.
Ben-Shlomo A., Melmed S. Acromegaly. Endocrinol Metab Clin North Am. 2008;37(1):565-583.
Biermasz N.R., Dulken H.V., Roelfsema F. Postoperative radiotherapy in acromegaly is effective in reducing GH concentration to safe levels. Clin Endocrinol (Oxf). 2000;53(3):321-327.
Buhk J.H., Jung S., Psychogios M.K., et al. Tumor volume of growth hormone–secreting pituitary adenomas during treatment with pegvisomant: a prospective multicenter study. J Clin Endocrinol Metab. 2010;95(2):552-558.
Chanson P., Salenave S., Kamenicky P., et al. Acromegaly. Best Pract Res Clin Endocrinol Metab. 2009;23:555-574.
Cohen-Gadol A.A., Liu J.K., Laws E.R.Jr. Cushing’s first case of transsphenoidal surgery: the launch of the pituitary surgery era. J Neurosurg. 2005;103:570-574.
Colao A., Ferone D., Marzullo P., et al. Long-term effects of depot long-acting somatostatin analog octreotide on hormone levels and tumor mass in acromegaly. J Clin Endocrinol Metab. 2001;86(6):2779-2786.
Feenststra J., de Herder W.W., ten Have S.M., et al. Combined therapy with somatostatin analogues and weekly pegvisomant in active acromegaly. Lancet. 2005;365(9471):1644-1646.
Freda P.U., Katznelson L., van der Lely A.J., et al. Long-acting somatostatin analog therapy of acromegaly: a meta-analysis. J Clin Endocrinol Metab. 2005;90(8):4465-4473.
Giustina A., Chanson P., Bronstein M.D., et al. A consensus on criteria for cure of acromegaly. J Clin Endocrinol Metab. 2010;95(7):3141-3148.
Gutt B., Hatzack C., Morrison K., et al. Conventional pituitary irradiation is effective in normalising plasma IGF-I in patients with acromegaly. Eur J Endocrinol. 2001;144(2):109-116.
Jho H.D., Alfieri A. Endoscopic endonasal pituitary surgery: E: evolution of surgical technique and equipment in 150 operations. Minim Invasive Neurosurg. 2001;44:1-12.
Laws E.R., Vance M.L., Thapar K. Pituitary surgery for the management of acromegaly. Horm Res. 2000;53(Suppl 3):71-75.
Melmed S. Medical progress: Acromegaly. N Engl J Med. 2006;355(24):2558-2573.
Melmed S., Casanueva F., Cavagnini F., et al. Consensus statement: medical management of acromegaly. Eur J Endocrinol. 2005;153(6):737-740.
Nachtigall L., Delgado A., Swearingen B., et al. Changing patterns in diagnosis and therapy of acromegaly over two decades. J Clin Endocrinol Metab. 2008;93(6):2035-2041.
Parkinson C., Trainer P.J. Pegvisomant: a growth hormone receptor antagonist for the treatment of acromegaly. Growth Horm IGF Res. 2000;10:S119-S123.
Powell J.S., Wardlaw S.L., Post K.D., Freda P.U. Outcome of radiotherapy for acromegaly using normalization of insulin-like growth factor I to define cure. J Clin Endocrinol Metab. 2000;85(5):2068-2071.
Racine M.S., Barkan A.L. Medical management of growth hormone-secreting pituitary adenomas. Pituitary. 2002;5:67-76.
Roberts B.K., Ouyang D.L., Lad S.P. Efficacy and safety of CyberKnife radiosurgery for acromegaly. Pituitary. 2007;10(1):19-25.
Swearingen B., Barker F.G., Kaznelson L., et al. Long-term mortality after transsphenoidal surgery and adjunctive therapy for acromegaly. J Clin Endocrinol Metab. 1998;83(10):3419-3426.
Zada G., Kelly D.F., Cohan P., et al. Endonasal transsphenoidal approach for pituitary adenomas and other sellar lesions: an assessment of efficacy, safety, and patient impressions. J Neurosurg. 2003;98:350-358.
Zhang N., Pan L., Wang E.M., et al. Radiosurgery for growth hormone-producing pituitary adenomas. J Neurosurg. 2000;93(suppl 3):6-9.
1. Barkan A. Acromegaly. Endocrinol Metab Clin North Am. 1989;18:277-310.
2. Molitch M.E. Pathologic hyperprolactinemia. Endocrinol Metab Clin North Am. 1992;21:877-901.
3. Ezzat S., Wilkins G.E., Patel Y., et al. The diagnosis and management of acromegaly: a Canadian consensus report. Clin Invest Med. 1996;19(4):259-270.
4. Melmed S. Acromegaly. N Engl J Med. 1990;322:966-977.
5. Melmed S., Ho K., Klibanski A., et al. Clinical review 75: recent advances in pathogenesis, diagnosis, and management of acromegaly. J Clin Endocrinol Metab. 1995;80(12):3395-3402.
6. Chanson P., Salenave S., Kamenicky P., et al. Acromegaly. Best Pract Res Clin Endocrinol Metab. 2009;23:555-574.
7. Melmed S., Jackson I., Kleinberg D., Klibanski A. Current treatment guidelines for acromegaly. J Clin Endocrinol Metab. 1998;83(8):2646-2652.
8. Swearingen B., Barker F.G., Kaznelson L., et al. Long-term mortality after transsphenoidal surgery and adjunctive therapy for acromegaly. J Clin Endocrinol Metab. 1998;83(10):3419-3426.
9. Ben-Shlomo A., Melmed S., Acromegaly. Endocrinol Metab Clin North Am, 2008;1(37).
10. Nachtigall L., Delgado A., Swearingen B., et al. Changing patterns in diagnosis and therapy of acromegaly over two decades. J Clin Endocrinol Metab. 2008;93(6):2035-2041.
11. Bengtsson B.A., Edén S., Ernest I., et al. Epidemiology and long-term survival in acromegaly. A study of 166 cases diagnosed between 1955 and 1984. Acta Med Scand. 1988;223(4):327-335.
12. Melmed S. Medical progress: acromegaly. N Engl J Med. 2006;355(24):2558-2573.
13. Cohen K., Nissley S. The serum half-life of somatomedin activity: evidence for growth hormone dependence. Acta Endocrinol. 1991;124:74-85.
14. Baumann G. Growth hormone heterogeneity: genes, isohormones, variants and binding proteins. Endocrinol Rev. 1991;12:424-449.
15. Denoto F., Moore D., Goodman H. Human growth hormone DNA sequence and mRNA structures: Possible alternative splicing. Nucleic Acids Res. 1981;9:3719.
16. McCarter J., Shaw M., Winer L., et al. The 20,000-dalton variant of human growth hormone growth hormone receptors in human liver. Mol Cell Endocrinol. 1990;64:596.
17. Carmichael J.D., Bonert V.S., Mirocha J.M., Melmed S. The utility of oral glucose tolerance testing for diagnosis and assessment of treatment outcomes in 166 patients with acromegaly. J Clin Endocrinol Metab. 2009;94(2):523-527.
18. Serri O., Somma M., Comtois R., et al. Acromegaly: biochemical assessment of cure after long term follow-up of transsphenoidal selective adenomectomy. J Clin Endocrinol Metab. 1985;61:1185-1189.
19. De Marinis L., Mancini A., Zuppi P., et al. Paradoxical growth hormone response to thyrotropin releasing hormone in active acromegaly. Acta Endocrinol (Copenh). 1990;122:433-449.
20. Boyd A., Lebovitz H., Pfeiffer J. Stimulation of human growth hormone secretion by L-dopa. N Engl J Med. 1970;283:1425-1429.
21. Barkan A., Shenker Y., Grekin R. Acromegaly due to ectopic growth hormone (GH)-releasing hormone production: dynamic studies of GH and ectopic GHRH secretion. J Clin Endocrinol Metab. 1986;63:1057-1064.
22. Abosch A., Tyrrell J.B., Lamborn K.R., et al. Transsphenoidal microsurgery for growth hormone-secreting pituitary adenomas: Initial outcome and long-term results. J Clin Endocrinol Metab. 1998;83(10):3411-3418.
23. Tindall G.T., Oyesiku N.M., Watts N.B., et al. Transsphenoidal adenomectomy for growth hormone-secreting pituitary adenomas in acromegaly: outcome analysis and determinants of failure. J Neurosurg. 1993;78(2):205-215.
24. Colao A., Ferone D., Cappabianca P., et al. Effect of octreotide pretreatment on surgical outcome in acromegaly. J Clin Endocrinol Metab. 1997;82(10):3308-3314.
25. Cohen-Gadol A.A., Liu J.K., Laws E.R.Jr. Cushing’s first case of transsphenoidal surgery: the launch of the pituitary surgery era. J Neurosurg. 2005;103:570-574.
26. Kante A.S., Dumont A.S., Asthagiri A.R., et al. The transsphenoidal approach: a historical perspective. Neurosurg Focus. 18(4), 2005.
27. Jho H.D., Alfieri A. Endoscopic endonasal pituitary surgery: E: evolution of surgical technique and equipment in 150 operations. Minim Invasive Neurosurg. 2001;44:1-12.
28. Jho H.D. Endoscopic transsphenoidal surgery. J Neuro-Oncol. 2001;54:187-195.
29. Zada G., Kelly D.F., Cohan P., et al. Endonasal transsphenoidal approach for pituitary adenomas and other sellar lesions: An assessment of efficacy, safety, and patient impressions. J Neurosurg. 2003;98:350-358.
30. Jane J.A.Jr., Thapar K., Alden T.D., Laws E.R.Jr. Fluoroscopic frameless stereotaxy for transsphenoidal surgery. Neurosurgery. 2001;48:1302-1308.
31. Yano S., Kawano T., Kudo M., et al. Endoscopic endonasal transsphenoidal approach through the bilateral nostrils for pituitary adenomas. Neurol Med Chir (Tokyo). 2009;49(1):1-7.
32. Shimon I., Melmed S. Management of pituitary tumors. Ann Intern Med. 1998;129:472-483.
33. Orrego J.J., Barkan A.L. Pituitary disorders. Drug treatment options. Drugs. 2000;59:93-106.
34. Powell M. Recovery of vision following transsphenoidal surgery for pituitary adenomas. Br J Neurosurg. 1995;9(3):367-373.
35. Fahlbusch R., Honegger J., Buchfelder M. Evidence supporting surgery as treatment of choice for acromegaly. J Endocrinol. 1997;155(suppl 1):S53-S55.
36. Giustina A., Barkan A., Casanueva F.F., et al. Criteria for cure of acromegaly: a consensus statement. J Clin Endocrinol Metab. 2000;85(2):526-529.
37. Giustina A., Chanson P., Bronstein M.D., et al. A consensus on criteria for cure of acromegaly. J Clin Endocrinol Metab. 2010;95(7):3141-3148.
38. Beauregard C., Truong U., Hardy J., Serri O. Long-term outcome and mortality after transsphenoidal adenomectomy for acromegaly. Clin Endocrinol. 2003;58(1):86-91.
39. Freda P.U., Wardlaw S.L., Post K.D. Long-term endocrinological follow-up evaluation in 115 patients who underwent transsphenoidal surgery for acromegaly. J Neurosurg. 1998;89(3):353-358.
40. Laws E.R., Vance M.L., Thapar K. Pituitary surgery for the management of acromegaly. Horm Res. 2000;53(suppl 3):71-75.
41. Buchfelder M., Brockmeier S., Fahlbusch R., et al. Recurrence following transsphenoidal surgery for acromegaly. Horm Res. 1991;35(3-4):113-118.
42. Buchfelder M., Fahlbusch R., Schott W., Honegger J. Long-term follow-up results in hormonally active pituitary adenomas after primary successful transsphenoidal surgery. Acta Neurochir Suppl. 1991;53:72-76.
43. Long H., Beauregard H., Somma M., et al. Surgical outcome after repeated transsphenoidal surgery in acromegaly. J Neurosurg. 1996;85(2):239-247.
44. Ahmad F.U., Pandey P., Mahapatra A.K. Post operative ‘pituitary apoplexy’ in giant pituitary adenomas: a series of cases. Neurol India. 53(3), 2005. 326–238
45. Daud S., Hamrahian A.H., Weil R.J., et al. Acromegaly with negative pituitary MRI and no evidence of ectopic source: the role of transsphenoidal pituitary exploration? Pituitary. 2009;Nov 11. [Epub ahead of print]
46. Trainer P.J., Drake W.M., Katznelson L., et al. Treatment of acromegaly with the growth hormone-receptor antagonist pegvisomant. N Engl J Med. 2000;342(16):1171-1177.
47. Newman C.B., Melmed S., George A., et al. Octreotide as primary therapy for acromegaly. J Clin Endocrinol Metab. 1998;83(9):3034-3040.
48. Colao A., Ferone D., Cappabianca P., et al. Effect of octreotide pretreatment on surgical outcome in acromegaly. J Clin Endocrinol Metab. 1997;82(10):3308-3314.
49. Freda P.U., Katznelson L., van der Lely A.J., et al. Long-acting somatostatin analog therapy of acromegaly: a meta-analysis. J Clin Endocrinol Metab. 2005;90(8):4465-4473.
50. Colao A., Ferone D., Marzullo P., et al. Long-term effects of depot long-acting somatostatin analog octreotide on hormone levels and tumor mass in acromegaly. J Clin Endocrinol Metab. 2001;86(6):2779-2786.
51. Ezzat S., Snyder P.J., Young W.F., et al. Octreotide treatment of acromegaly. A randomized, multicenter study. Annals of Internal Medicine. 1992;117(9):711-718.
52. Sassolas G., Harris A.G., James-Deidier A. Long term effect of incremental doses of the somatostatin analog SMS 201-995 in 58 acromegalic patients. French SMS 201-995 approximately equal to Acromegaly Study Group. J Clin Endocrinol Metab. 1990;71(2):391-397.
53. Plockinger U., Reichel M., Fett U., et al. Preoperative octreotide treatment of growth hormone-secreting and clinically nonfunctioning pituitary macroadenomas: E: effect on tumor volume and lack of correlation with immunohistochemistry and somatostatin receptor scintigraphy. J Clin Endocrinol Metab. 1994;79:1416-1423.
54. Barkan A.L., Lloyd R.V., Chandler W.F., et al. Preoperative treatment of acromegaly with long-acting somatostatin analog SMS 201-995: shrinkage of invasive pituitary macroadenomas and improved surgical remission rate. J Clin Endocrinol Metab. 1988;67(5):1040-1048.
55. Racine M.S., Barkan A.L. Medical management of growth hormone-secreting pituitary adenomas. Pituitary. 2002;5:67-76.
56. Biermasz N.R., van Dulken H., Roelfsema F. Direct postoperative and follow-up results of transsphenoidal surgery in 19 acromegalic patients pretreated with octreotide compared to those in untreated matched controls. J Clin Endocrinol Metab. 1999;84(10):3551-3555.
57. Kristof R.A., Stoffel-Wagner B., Klingmüller D., Schramm J. Does octreotide treatment improve the surgical results of macro-adenomas in acromegaly? A randomized study. Acta Neurochir (Wien). 1999;141(4):399-405.
58. Landolt A.M., Lomax N. Gamma knife radiosurgery for prolactinomas. J Neurosurg. 2000;93(suppl 3):14-18.
59. Lamberts S.W., van der Lely A.J., de Herder W.W., Hofland L.J. Octreotide. N Engl J Med. 1996;334(4):246-254.
60. Barakat S., Melmed S. Reversible shrinkage of a growth hormone-secreting pituitary adenoma by a long-acting somatostatin analogue, octreotide. Arch Intern Med. 1989;149(6):1443-1445.
61. Melmed S., Casanueva F., Cavagnini F., et al. Consensus statement: medical management of acromegaly. Eur J Endocrinol. 2005;153(6):737-740.
62. Herman-Bonert V.S., Zib K., Scarlett J.A., Melmed S. Growth hormone receptor antagonist therapy in acromegalic patients resistant to somatostatin analogs. J Clin Endocrinol Metab. 2000;85:2958-2961.
63. Parkinson C., Trainer P.J. Pegvisomant: a growth hormone receptor antagonist for the treatment of acromegaly. Growth Horm IGF Res. 2000;10:S119-S123.
64. Ross R.J., Leung K.C., Maamra M., et al. Binding and functional studies with the growth hormone receptor antagonist, B2036-PEG (pegvisomant), reveal effects of pegylation and evidence that it binds to a receptor dimer. J Clin Endocrinol Metab. 2001;86:1716-1723.
65. Buhk J.H., Jung S., Psychogios M.K., et al. Tumor volume of growth hormone-secreting pituitary adenomas during treatment with pegvisomant: a prospective multicenter study. J Clin Endocrinol Metab. 2010;95(2):552-558.
66. Feenststra J., de Herder W.W., ten Have S.M., et al. Combined therapy with somatostatin analogues and weekly pegvisomant in active acromegaly. Lancet. 2005;365(9471):1644-1646.
67. van der Lely A.J., Hutson R.K., Trainer P.J., et al. Long-term treatment of acromegaly with pegvisomant, a growth hormone receptor antagonist. Lancet. 2001;358(9295):1754-1759.
68. Colao A., Ferone D., Marzullo P., et al. Effect of different dopaminergic agents in the treatment of acromegaly. J Clin Endocrinol Metab. 1997;82(2):518-523.
69. Jaffe C.A., Barkan A.L. Treatment of acromegaly with dopamine agonists. Endocrinol Metab Clin North Am. 1992;21(3):713-735.
70. Cozzi R., Attanasio R., Barausse M., et al. Cabergoline in acromegaly: a renewed role for dopamine agonist treatment? Eur J Endocrinol. 1998;139(5):516-521.
71. Abs R., Verhelst J., Maiter D., et al. Cabergoline in the treatment of acromegaly: a study in 64 patients. J Clin Endocrinol Metab. 1998;83(2):374-378.
72. Eastman R.C., Gorden P., Glatstein E., Roth J. Radiation therapy of acromegaly. Endocrinol Metab Clin North Am. 1992;21(3):693-712.
73. Goffman T.E., Dewan R., Arakaki R., et al. Persistent or recurrent acromegaly. Long-term endocrinologic efficacy and neurologic safety of postsurgical radiation therapy. Cancer. 1992;69(1):271-275.
74. Feek C.M., McLelland J., Seth J., et al. How effective is external pituitary irradiation for growth hormone-secreting pituitary tumors? Clin Endocrinol (Oxf). 1984;20(4):401-408.
75. Biermasz N.R., Dulken H.V., Roelfsema F. Postoperative radiotherapy in acromegaly is effective in reducing GH concentration to safe levels. Clin Endocrinol (Oxf). 2000;53(3):321-327.
76. Biermasz N.R., van Dulken H., Roelfsema F. Long-term follow-up results of postoperative radiotherapy in 36 patients with acromegaly. J Clin Endocrinol Metab. 2000;85(7):2476-2482.
77. Jaffe C.A. Reevaluation of conventional pituitary irradiation in the therapy of acromegaly. Pituitary. 1999;2:55-62.
78. Gutt B., Hatzack C., Morrison K., et al. Conventional pituitary irradiation is effective in normalising plasma IGF-I in patients with acromegaly. Eur J Endocrinol. 2001;144(2):109-116.
79. Critides S.D. Glioblastoma after radiotherapy. Neurosurgery. 1988;22(6 Pt 1):1115.
80. Powell J.S., Wardlaw S.L., Post K.D., Freda P.U. Outcome of radiotherapy for acromegaly using normalization of insulin-like growth factor I to define cure. J Clin Endocrinol Metab. 2000;85(5):2068-2071.
81. Snyder P.J., Fowble B.F., Schatz N.J., et al. Hypopituitarism following radiation therapy of pituitary adenomas. Am J Med. 1986;81(3):457-462.
82. Jackson I.M., Noren G. Role of gamma knife radiosurgery in acromegaly. Pituitary. 1999;2:71-77.
83. Zhang N., Pan L., Wang E.M., et al. Radiosurgery for growth hormone-producing pituitary adenomas. J Neurosurg. 2000;93(suppl 3):6-9.
84. Roberts B.K., Ouyang D.L., Lad S.P. Efficacy and safety of cyberknife radiosurgery for acromegaly. Pituitary. 2007;10(1):19-25.
85. Abe T., Tara L.A., Lüdecke D.K. Growth hormone-secreting pituitary adenomas in childhood and adolescence: features and results of transnasal surgery. Neurosurgery. 1999;45(1):1-10.
86. Goldenberg N., Racine M.S., Thomas P., et al. Treatment of pituitary gigantism with the growth hormone receptor antagonist pegvisomant. J Clin Endocrinol Metab. 2008;93:2953-2956.
[/level-membership-for-neurosurgery-category][not-level-membership-for-neurosurgery-category]
Chapter 18 Growth Hormone–Secreting Tumors
Phenotypic features of acromegaly include acral enlargement; coarse facial features with frontal bossing, prognathism, diastema and macroglossia; skin thickening, hypertrichosis, malodorous hyperhidrosis, and acanthosis nigricans; and deepening of the voice due to laryngeal hypertrophy. Other manifestations include headaches, lethargy, obstructive sleep apnea, peripheral neuropathies such as carpal tunnel syndrome, and bony deformation including bone thickening and vertebral osteophyte formation. Other consequences include abnormal carbohydrate metabolism and diabetes mellitus; cardiovascular diseases including hypertension, atherosclerosis, and cardiomyopathy; and an increased risk of other neoplasms including colon cancer.1–6 Patients with acromegaly have a two- to three-fold increase in mortality due to cardiovascular and cerebrovascular diseases. Normalization of GH levels may decrease this risk substantially to levels comparable to the general population.7,8 Other clinical manifestations are related to mass effect of the pituitary tumor, including headaches, visual loss (classically bitemporal hemianopia), and hormonal deficiencies (hypogonadism, hypothyroidism, and hypoadrenalism).
The disease is insidious, which leads to a delayed diagnosis, often seven to ten years after onset of symptoms.9 Recently, this lag has shortened significantly, likely due to the increase in magnetic resonance imaging.10 Patients with acromegaly generally present in the third to fifth decade, and both sexes are affected equally.11
The average annual incidence of acromegaly is three to four per million, and the prevalence is 40 to 70 cases per million people.7,10 The vast majority of cases, 95% to 98%, are due to a GH-secreting pituitary adenoma.9,10,12 Pituitary adenomas from somatotroph cells may lead to excessive secretion of GH, while adenomas from acidophil stem cells or mammosomatotrophs often secrete both GH and prolactin (PRL).9,12 Most GH-secreting tumors (75%–80%) are macroadenomas (>1 cm in diameter),7 and 20% to 50% co-secrete PRL or other pituitary hormones.13 Rare causes of acromegaly include ectopic GH-secreting tumors such as bronchial carcinoid or pancreatic islet cell tumors; hypothalamic GH-releasing hormone (GHRH)-secreting tumors; exogenous administration of GH; or familial syndromes such as McCune-Albright syndrome, multiple endocrine neoplasia I (MEN-I) or Carney complex.9
Growth Hormone
There is some heterogeneity in GH due to differential splicing and post-translational modification.14–16 Two main forms of GH are found in the circulation: the 22K form comprises 90% of serum GH, and the 20K form makes up 5%.16 This heterogeneity may explain differences between levels measured by radioimmunoassays and actual biological activity in some patients.
Diagnosis
The suspicion of acromegaly is usually based on physical examination, while incidental radiologic detection in patients without typical manifestations is rare. Laboratory testing is needed to prove GH excess, while radiology techniques are used to visualize the tumor (Fig. 18-1). Screening laboratory tests include measurement of basal GH and IGF-1, and laboratory confirmation occurs when GH fails to suppress with oral glucose tolerance testing.
Laboratory Diagnosis
Random Serum GH Measurement
In healthy subjects, random GH levels are less than 5 ng/ml while most acromegalic patients have levels greater than 10 ng/ml. In active acromegaly, the normal episodic GH pattern is replaced by a constantly elevated GH level throughout the day.1 However, GH levels fluctuate widely and GH has a short half-life, so some acromegalic patients have normal GH levels on initial testing. Serum GH levels may be elevated in other conditions including uncontrolled diabetes mellitus, renal failure, malnutrition and during physical or emotional stress, even in the absence of acromegaly.13 Therefore, random GH measurement is not the preferred screening test for acromegaly.
IGF-1 Measurement
Serum IGF-1 measurement is the best single test for screening. The measurement of IGF-1 is used to provide an indicator of the body’s overall exposure to GH. Normal ranges for IGF-1 vary between different assays and are age- and gender-dependent.13 IGF-1 is increased in nearly all acromegalic patients, even those in whom random single GH levels are within the normal range. IGF-1 is also a reliable indicator for post-treatment hormonal remission, as it reflects GH secretion over the prior 24 hours.4 One of the IGF-1 binding proteins (IGFBPs) can be measured to assist in the diagnosis of acromegaly. The level of IGFBP-3 correlates directly with GH, but the overlap with normal persons limits its use.
GH Suppression to Hyperglycemia
The oral glucose suppression test (75 g) is used to confirm a diagnosis of acromegaly. In a normal control, GH decreases to below 1 ng/ml after a glucose load, whereas in an acromegalic patient, this suppression fails to occur.17 This test is useful to document biochemical remission after surgical removal of the pituitary tumor,18 but does not appear to be useful to assess control in patients receiving therapy with somatostatin analogues.17
Other Dynamic Tests
Other dynamic tests are rarely needed to diagnose acromegaly. Thyrotropin-releasing hormone (TRH) stimulation leads to a significant increase in GH in untreated acromegalics, although it does not cause a significant change in GH levels in normal subjects. This response may also occur in the setting of liver disease, renal failure, or depression. TRH-stimulation may identify patients who, despite a normal postsurgical GH level, have residual GH-secreting tumor.19 Administration of oral L-Dopa or bromocriptine, a dopamine agonist, to a fasting normal subject increases GH secretion, though it paradoxically decreases GH levels in a fasting acromegalic patient.20
GHRH
GHRH levels should not be routinely used in diagnosis of acromegaly. However, they are useful in patients with confirmed acromegaly who do not harbor a pituitary tumor. Ectopic acromegaly, due to non-central nervous system tumors such as a pancreatic islet cell tumor or a bronchial carcinoid, results in a significantly elevated serum GHRH, whereas GHRH is usually low with a GH-secreting adenoma.21
Other Hormones
Hormonal assessments are needed to measure prolactin co-secretion, although moderately elevated prolactin can be due to stalk effect. Pituitary hormone deficiencies may be measured by ACTH, cortisol, TSH, free T4, FSH, LH levels in both genders, estradiol in women, and testosterone in men.
Treatment
Surgery
Surgical adenomectomy by an experienced neurosurgeon remains the first-line treatment for most patients with acromegaly.22,23 The goals of surgical resection are to eliminate mass effect, preserve or restore pituitary and visual function, and obtain tissue for histopathologic analysis.24
Most of these tumors are sellar or suprasellar lesions that may be removed trans-sphenoidally using a direct endonasal, sublabial, or trans-septal approach with an endoscope or microscope. The first trans-sphenoidal resection of a pituitary lesion was performed by Hermann Schloffer in 1907; the procedure was popularized by Harvey Cushing in the two decades that followed.25,26 Neurosurgeons have been improving the trans-sphenoidal adenomectomy (TSA) since. A craniotomy may rarely be necessary when a tumor has extensive suprasellar or parasellar extension.
Endoscopy
The lesion is removed in the same manner as described with the microscopic approach. Duraform is then placed over the sella, and the sphenoid sinus is packed using Gelfoam. NasoPore is laid over the posterior septectomy site and the sphenoethmoid recesses bilaterally. A mucosal septal flap may be used if a CSF leak is present. A speculum is not needed with this approach.27,28
The straight surgical endoscope provides a wide field of view, while angled scopes permit enhanced visualization of the sellar wall, suprasellar, retrosellar, or parasellar regions. Three-dimensional endoscopes have been recently introduced and provide a stereoscopic, nondistorted view of the regional anatomy in contrast to older two-dimensional endoscopes (Figure 18-2).
Sublabial Trans-Sphenoidal Approach
Sublabial may be the appropriate approach for patients with large tumors or pediatric patients with small nares. The upper lip is retracted, an incision is made horizontally in the gingival mucosa, and the maxilla and nasal cavity floor are accessed. A vertical incision is made to separate the nasal mucosa from the septum, and the anterior septum is subluxed and deviated. The speculum is inserted and the microscope or endoscope is brought into the field. The operation continues in the same manner as described above.29
Neuronavigation
Frameless stereotactic neuronavigation permits the surgeon to confirm his position at any point during the TSA to assess the proximity of surrounding structures or determine when he is approaching the limits of the tumor (Figure 18-3). Navigation may be used to assist with large lesions that involve the carotid arteries or recurrent lesions where the normal anatomy has been altered by a prior operation.30,31
Outcomes of Trans-Sphenoidal Surgery
Clinical Outcomes
Following operative decompression, visual field defects improve in 70% to 89% of patients,32,33 remain unchanged in 7% percent, and rarely worsen (<4%).34
[/not-level-membership-for-neurosurgery-category]
