CHAPTER 101 Growth Factors in Glial Tumors
This last part of surgery, namely, operations, is a reflection on the healing art; it is a tacit acknowledgement of the insufficiency of surgery. It is like an armed savage who attempts to get that by force which a civilized man would get by stratagem.1
Progressive loss of cellular control through accumulation of multiple genetic defects leads to tumor formation and malignant progression.2 These changes can involve activation of growth-promoting oncogenes or inactivation of tumor suppressor genes.3 Polypeptide growth factors play a major role in tumor growth and progression.
Growth Factors
Growth factors are defined by Tannock and colleagues as “a polypeptide produced by cells that stimulates or inhibits proliferation by either the same cell or other cells.”4 Most growth factors are secreted soluble polypeptides that activate specific membrane-bound receptors. They usually act via binding to receptors situated on the cell surface. Through complex molecular cascades this signal is transduced and passed on in an alternative format to ultimately bring about changes in growth or function of the cell. Growth factors play a role in regulating diverse cellular processes, including cell growth and division, cell survival and apoptosis, cellular differentiation, and motility. Abnormalities in these functions are seen in virtually all brain tumors. Astrocytomas have been the most intensely studied. The malignant phenotype is associated with genomic instability, immortality, increased cell survival and division, enhanced migration and invasion, neoangiogenesis, and the ability to avoid detection by the immune surveillance system.5
There are three mains ways that growth factors are involved in the genesis of gliomas.6 The first involves concurrent expression of both a growth factor and its corresponding receptor, which allows autocrine stimulation of cellular growth. The second scenario is growth factor expression without receptor expression. Through paracrine actions on tissues such as vascular endothelium, an environment supportive of tumor growth is created. Finally, tumor cells may express receptors for growth-promoting ligands that are produced elsewhere in the body.
Intracellular Signaling
Abnormalities in intracellular signaling are almost universally involved in the transition from normal cells to malignant cells, which results in increased cell division and abnormal survival.7
Phosphorylation of proteins by kinases is a major mechanism by which the function of growth factors is modulated. Cellular signaling will usually involve the following steps, as illustrated in Figure 101-1.7 First, a ligand molecule binds to a receptor situated on the cell surface. This leads to activation of the receptor, which may involve conformational structural changes in the receptor itself or recruitment of other proteins called adaptor proteins, which in turn activate downstream signaling pathways. This process often involves changes in phosphorylation of target proteins and subsequent activation of transcription factors, which alters gene expression in the cell.
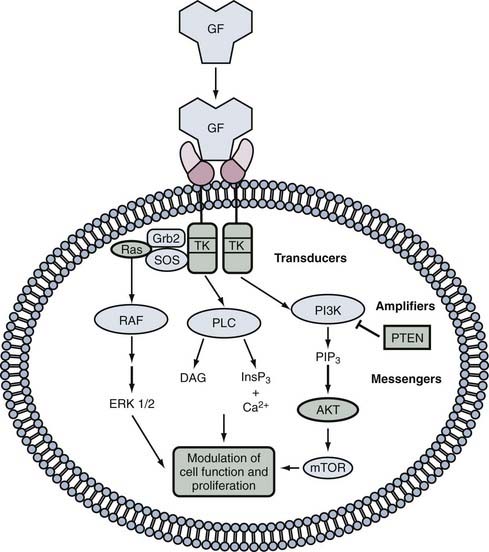
Disruption of a growth factor signal transduction pathway for therapeutic purposes can occur at any level along the signaling pathway. There is considerable overlap and intercommunication among the different pathways. Examples of common systems are given in the subsequent text, but interested readers are referred to the more detailed material cited at the end of this chapter (see, for example, Kapoor and O’Rourke8).
Receptor Tyrosine Kinases
The receptor tyrosine kinases (RTKs) are a very important, evolutionarily conserved family of signaling molecules.9 These receptors are made up of an extracellular ligand-binding domain, a transmembrane region, and a domain contained within the cytoplasm that has the ability to enzymatically phosphorylate target proteins,7 thereby altering their interactions with other molecules. RTKs contain a single hydrophobic transmembrane domain, as opposed to the seven–transmembrane receptor family, which lacks intrinsic protein kinase activity and is coupled to G proteins. There are approximately 60 genes in this family of receptors,10,11 with wide-ranging effects on the cell. They are subdivided into 20 subfamilies.9 Mutations in RTKs are found in approximately 30% of human cancers.9
The enzymatically active intracellular portions are normally locked into an inactive conformation.9 Ligand binding permits oligomerization of the receptor components. This brings the catalytic and cytoplasmic domains into juxtaposition and permits transphosphorylation of a tyrosine residue in a region called the activation loop, which leads to the activation of kinase. Next, phosphorylation of segments in the cytoplasmic segment by the activated enzyme permits interaction with intracellular docking proteins. These docking proteins contain sites, such as SH2 sites,12 that bind specifically to the phosphorylated tyrosine–containing segments of the receptor complex. Downregulation of receptor activity occurs via internalization into endosomes or dephosphorylation.7
Cytokine Receptor Superfamily
Interleukins, prolactin, growth hormone, and interferons are examples of ligands that bind to cytokine receptors.7,13 Ligand binding leads to substrate tyrosine phosphorylation in a process dependent on members of the Janus protein tyrosine kinase (JAK) family.14 There are four members in this protein family: JAK1, JAK2, JAK3, and Tyk2. If a specific JAK molecule is absent, the function of all cytokine receptors with which it normally associates is lost. Protein recruitment to the receptor complex after tyrosine phosphorylation results in activation of mitogen-activated protein kinases (MAPKs) and frequently phosphatidylinositol-3′-kinase (PI3K), which gives rise to further downstream cellular changes, including activation of members of the signal transducer and activator of transcription (STAT) family of transcription factors.7 A further level of complexity is added by the action of the suppressor of cytokine signaling (SOCS) family of proteins, which target the phosphorylated tyrosine–containing proteins for degradation, thereby blocking STAT signaling.13,15
Integrin Signaling
There are at least 24 different integrin receptors, which consist of 1 of 8 core β subunits and 1 of 18 α subunits.16 These receptors are linked to the cellular microfilament system and influence cell adhesion and movement. Intracellular signaling can alter the binding ability of the extracellular domain.15,16 Integrin receptor complexes regulate the activity of the Rho family of guanosine triphosphatases (GTPases) and interact with PI3K, phospholipase C-γ (PLC-γ), Shc, and Grb family adaptor proteins.7
Signal Transducers and Activators of Transcription
The STAT family of proteins acts downstream from the tyrosine phosphorylation step.13 They contain SH2 domains in their carboxyl regions. Phosphorylation of tyrosine residues within this SH2 domain allows STAT dimerization.16 These dimers then undergo translocation to the nucleus, where they activate gene transcription by binding to specific DNA sequences.
Role of Tyrosine Phosphatases
Tyrosine phosphatases contain a catalytic domain that dephosphorylates previously activated tyrosine residues. They can be either cytoplasmic or membrane bound16 and cause both upregulation and downregulation of intracellular signaling processes. SHP-2 is an example of a tyrosine kinase that positively influences cell signaling.16 It seems particularly important in regulation of the MAPK pathway.
Serine/Threonine Phosphorylation Systems
Two major families of receptors, the Toll/interleukin-1 (IL-1) receptor family and the transforming growth factor-β (TGF-β) receptor family, induce their effects via phosphorylation of serine and threonine residues7 rather than tyrosine.
The Toll/IL-1 receptor family includes six Toll-like receptors (TLR1 to TLR6), the IL-1 receptor, and the IL-18 receptor.17 Binding results in activation of the nuclear factor NF-κB and c-Jun N-terminal kinases (JNKs).7
Role of G Proteins in Intracellular Signaling
There are two broad types of G proteins: heterotrimeric G proteins and small G proteins.18 Both types are active when bound to adenosine triphosphate (ATP) and inactive when bound to adenosine diphosphate (ADP). There are more than 1000 G protein–coupled receptors, which represents 2% of the human genome.19 They mediate a wide variety of cellular functions, including hormonal signaling, neurotransmitter signaling, light perception, and chemokine functioning.20 This variety of intracellular effects is mediated in part by molecular differences in the intracellular adaptor molecules created by combinatorial construction of trimers from a diverse pool of subunits, as outlined in the following text.
Structurally, all receptors in this family have seven transmembrane α helices, an extracellular ligand-binding site, and an intracellular cytoplasmic domain that interacts with the so-called heterotrimeric G-protein complex.7,19,20
Various combinations of α, β, and γ subunits form heterotrimers. The guanine nucleotide–binding portion is known as the α subunit.7,20 There are 16 known α subunits.7,20 The α subunit has an inhibitory effect on the function of the combined βγ complex. The β and γ subunits cooperate to activate intracellular protein targets. There are 4 different β and 7 different γ subunits.7,20 In the quiescent nonactivated state, one molecule of guanosine diphosphate (GDP) is bound to the α subunit, which in turn is bound to the βγ complex.21 This trimeric complex is recruited to the receptor upon ligand binding and results in activation of the G-protein complex by exchange of a molecule of guanosine triphosphate (GTP) for the GDP already bound to the α subunit. The activated GTP-α portion then dissociates from the βγ complex, which leaves it free to modulate the function of intracellular targets. Cleavage of a phosphate group from the GTP-α group permits reassociation with the βγ complex and terminates intracellular propagation of the signal.21 Figure 101-2 illustrates the cycle of G-protein activation.
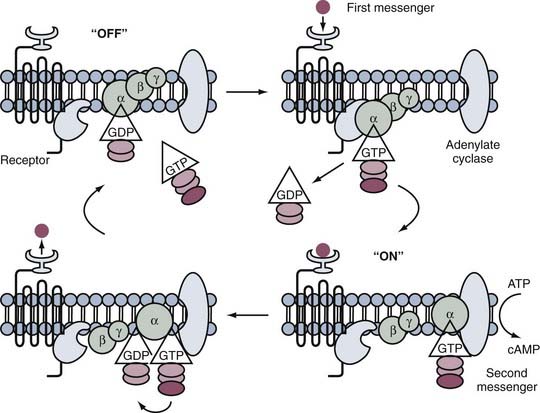
In addition, the GTP-bound α subunit can activate the enzyme adenylyl cyclase, which produces the intracellular second messenger cyclic adenosine monophosphate (cAMP).7 Another common target of G protein–associated receptors is phospholipase C-β (PLC-β), which can interact with either α or βγ complexes. Members of the PLC-β family generate the intracellular messengers inositol 1,4,5-triphosphate (IP3) and diacylglycerol (DAG) via hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2) in the cellular membrane.7 IP3 acts to transiently increase intracellular calcium concentrations, which has a variety of effects, including protein kinase activation.
Finally, ion channel function can be modulated by G proteins via coupling to cyclic guanosine monophosphate (cGMP) phosphodiesterase, as in the case with the light-sensing molecule rhodopsin.7 Free GTP-coupled α subunit lowers cGMP levels by activation of the phosphodiesterase.
Unlike their heterotrimeric counterparts, small G proteins possess intrinsic adenosine triphosphatase (ATPase) activity.19 Two categories of modulatory molecules act on small G proteins. Guanosine nucleotide exchange factors (GEFs) activate small G proteins by promoting the dissociation of GDP, thereby allowing a molecule of GTP to bind and activate the G protein. GTPase-activating proteins (GAPs), in contrast, promote inactivation of the G protein by fostering hydrolysis of GTP to GDP.18 There are five subfamilies of small G proteins, the most well characterized being the p21-ras subfamily.18
G proteins play an important role in tumors of the nervous system. The activated form of the small G protein p21-ras is present at elevated levels in astrocytomas,18 possibly because of overactivation by upstream activators such as the epidermal growth factor receptors (EGFRs). Suppression of p21-ras activity via the farnesyltransferase inhibitor L-744,832 has been shown to suppress astrocytoma growth.22 In the setting of neurofibromatosis type 1, loss of the NF1 gene, which is a downregulator of p21-ras activity, leads to upregulation of ras activity.18
Growth Factors Involved in Brain Tumors
Designing agents to inhibit glioma growth factors requires knowledge not only of the growth factor and its receptor or receptors but also of the downstream signaling chain. Instead of thinking about inhibiting the growth factor in isolation, it is useful to conceptualize intervention to disrupt a signaling cascade (see, for example, Wick and associates23).
Epidermal Growth Factor Receptor
EGFR (HER1, c-erbB1) is a 170-kD glycoprotein encoded by a gene on chromosome 17 (Table 101-1).24 It contains 1186 amino acid residues. Although epidermal growth factor (EGF) is not typically overexpressed in brain tumors, its receptor EGFR is. Microglia found in the vicinity of glioma cells have been shown to secrete EGF, thus suggesting a possible paracrine stimulatory pathway.25 EGFR belongs to the erbB receptor family and is also known as erbB1 receptor. Other members of this receptor family are erbB2 (HER2/Neu), erbB3, and erbB4.81 These transmembrane receptors have a single transmembrane-spanning domain, and ligand binding activates an intracellular tyrosine kinase that normally promotes cell growth and division. TGF-α is overexpressed in malignant gliomas and appears to be a prime ligand for binding to EGFR. Other ligands for EGFR include EGF, amphiregulin, heparin-binding EGF-like growth factor, and epiregulin.26
TABLE 101-1 Receptors and Ligands of the Epidermal Growth Factor Receptor Family
| RECEPTOR | LIGAND |
|---|---|
| EGFR (erbB1, HER1) | EGF, TGF-α, HB-EGF, amphiregulin, decorin, betacellulin, epiregulin |
| HER2 (erbB2, neu) | — |
| HER3 (erbB3) | NRG-1 |
| HER4 (erbB4) | NRG-1 to NRG-4 |
EGF, epidermal growth factor; EGFR, EGF receptor; HB-EGF, heparin-binding EGF; TGF, transforming growth factor.
Adapted from Nicholas MK, Lukas RV, Jafri NF, et al. Epidermal growth factor receptor–mediated signal transduction in the development and therapy of gliomas. Clin Cancer Res. 2006;12:7261.
A constitutively active form of EGFR (EGFRvIII) appears to be involved in the progression of gliomas rather than their initiation. EGFR is overexpressed in 40% to 50% of high-grade gliomas.26 In 50% of grade IV astrocytomas, EGFR gene rearrangement is present and commonly involves deletion of the ligand-binding domains. The resulting EGFRvIII has been shown to interfere with apoptosis by increasing Bcl-xl, which normally decreases apoptosis. In addition, mutant EGFR can increase drug resistance and suppress caspase activation. This EGFR mutation is commonly seen in the primary type of glioblastoma multiforme (GBM), which arises in older individuals and is associated with loss of the p16 tumor suppressor gene and expression of wild-type p53. It seems to be associated with a worse prognosis.27 EGFR-C958, resulting from genomic deletion of exons 23 to 25 of the cytoplasmic domain, is the second most common mutant and is found in 20% of GBMs.28 An autocrine loop involving EGFR and heparin EGF-like growth factor has been demonstrated in glioma cell lines.27
Another member of the EGFR family that is mutated in astrocytomas is neu (HER2/p185).30 The gene for this 185-kD protein is also found on chromosome 17, and it shares 50% sequence homology with EGFR. Its ligand is unknown, but mutations found in high-grade gliomas result in continuous ligand-independent growth signals.30
Therapies have focused mainly on antagonizing EGFR or interfering with tyrosine kinase activity.31 Attempts at therapy with monoclonal antibodies32 and vaccines33 have met with little success to date. A bispecific cytotoxin composed of recombinant IL-13 and EGF moieties bound to the diphtheria toxin is in development and has been shown to inhibit the growth of various cell lines,34 but no success in human trials has been reported.
The small-molecule inhibitors gefitinib (Iressa) and erlotinib (Tarceva)25 both bind to the receptor ATP-binding pocket, thereby preventing ATP binding and inhibiting signal transduction (Table 101-2).33 A phase II trial with gefitinib (Iressa/ZD1839—an EGFR tyrosine kinase inhibitor) in patients with recurrent glioblastoma75 failed to show any objective effects on tumor growth, although it was generally well tolerated.76 Combination with the mTOR (mammalian target of rapamycin) inhibitor sirolimus was more promising, with a 25% 6-month progression-free survival rate in patients with GBM.44 Despite previous studies showing good tolerability,41 a similar phase II study of erlotinib, which also inhibits the EGFRvIII mutant,77 likewise failed to show dramatic results.78 A phase II trial comparing erlotinib with temozolomide and other standard agents as first-line therapy failed to show any responses.79 It appears that cells expressing the constitutively active mutant form of EGFR (EGFRvIII) along with PTEN (phosphatase and tensin homologue from chromosome 10) are more responsive to EGFR kinase inhibitors.80 PTEN is a lipid phosphatase that acts to dephosphorylate the 3′ position of the intracellular signaling molecules inositol-3,4-bisphosphate31 and inositol-3-phosphate,81,82 thereby inhibiting the EGFR/PI3K/Akt/mTOR signaling pathway.33,83
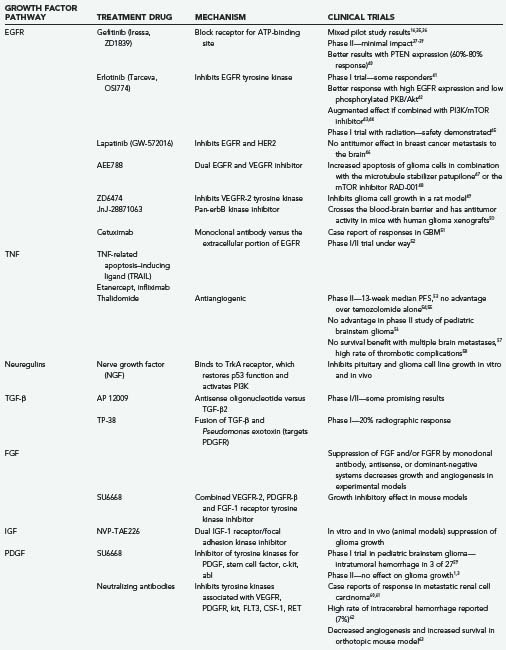
Another approach has been to combine gefitinib with everolimus (RAD-001), an agent that interferes with the downstream mTOR protein kinase. This has resulted in a modest radiographic response, but no increase in survival.84 There is also new interest in combining this agent with radiation therapy,85 and timing of dose administration appears to be important.
Trials of the combined EGFR/erbB2 inhibitor lapatinib (GW-572016) and the combined EGFR/vascular endothelial growth factor receptor (VEGFR) inhibitor AEE788 are under way.86 There have been case reports of successful use of cetuximab87 and other monoclonal antibodies directed against the extracellular portion of EGFR, and clinical trials are also under way.52
Tumor Necrosis Factor
Another receptor family that contains about 25 receptors is the tumor necrosis factor receptor (TNFR) family.88 Common ligands include the tumor necrosis factor (TNF) family, Fas ligand, CD40, CD30, and CD27.89 Ligand binding leads to receptor aggregation and subsequent conformational changes in the receptors. Various receptor-specific adaptor proteins are then recruited to the receptor complex.
TNF-α exists in a membrane-associated and a freely soluble form. The former is a transmembrane protein, whereas the latter is a trimeric complex of 17-kD proteins.90 Expression of TNF-α is upregulated by the hypoxic environment often found in association with high-grade gliomas.91 There are two TNFRs: TNFRI is activated by the soluble form of TNF-α, and TNFRII binds to the membrane-associated form.90
Interestingly, activation of TNFRs, especially TNFRI, can facilitate two diametrically opposite responses. A common motif in this family of receptors is the death domain, which has the ability to induce cellular apoptosis. Examples of receptors with cytoplasmic death domains are TNFRI, Fas, and the nerve growth factor (NGF) receptor p75.7 This motif is also seen in the DR4 receptor for the ligand TRAIL (tumor necrosis factor–related apoptosis-inducing ligand),92 which is also the subject of study as an antitumor agent.93 This cytoplasmic domain interacts with death domains on intracellular effector molecules such as TNFR-associated death domain (TRADD) and Fas-associated death domain (FADD).18 In turn, these molecules activate caspase cascades leading to apoptosis.94
In contrast to this proapoptotic effect, a second effect of TNFRI activation is activation of the transcription factor NF-κB via AP-1, which has a multitude of intracellular effects, including protection of the cell from apoptosis.95 Knockout mice in which the TNF-α gene is lacking exhibit increased resistance to the development of experimental cancers.36 TNFRII has been less well characterized. It is known that activation of TNFRII leads to subsequent activation of several intracellular pathways, including MAPK, extracellular signal–regulated kinase (ERK), p38, NF-κB, and JNK.90,94
Monoclonal antibody–based inhibitors of TNF have seen widespread use in the treatment of chronic inflammatory conditions such as inflammatory bowel disease and arthritis.96 The much maligned drug thalidomide has shown some activity against gliomas and acts to inhibit RNA processing of TNF-α and other cytokines90; clinical trials are ongoing, although to date there have been few clear-cut benefits demonstrated.53–58 Experimental exposure of astrocytes to TNF-α causes dedifferentiation to more primitive radial glia-like phenotypes,97 although a link to the genesis of glioma has not been established.
Transforming Growth Factor-β Receptor Family
Members of the TGF-β superfamily include TGF-β1, TGF-β2, TGF-β3, inhibins, activins, and bone morphogenetic proteins (BMPs).7 These transmembrane receptors possess a catalytic domain with serine/threonine kinase activity. They have variable growth-promoting and grow-inhibiting effects, as well as a central role in glioma cell invasion.23 The Smad transcription factor family is the primary downstream mediator of TGF-β receptors, although effects on the MAPK/JNK/p38 pathway have also been observed.7,98 There are several groups of Smads, which transduce signals from receptors to the nucleus, where they alter genetic transcription. In lower grade tumors, TGF-β acts as a tumor suppressor, whereas in higher grade tumors it promotes cell growth via activation of unmethylated platelet-derived growth factor B (PDGF-B).99
An antisense oligonucleotide TGF-β2 inhibitor named AP 12009 has shown early promise in phase I/II trials in patients with recurrent high-grade gliomas.38 TP38 was created by structurally fusing TGF-β to the Pseudomonas exotoxin. A phase I trial resulted in a 20% radiologic response rate.100
Enzastaurin (LY317615) acts by blocking the PKC-β and PI3K/Akt pathways downstream from certain TGF-β receptors. The result is reduced angiogenesis and increased tumor cell apoptosis.77 A phase III trial (Study Evaluating Enzastaurin in Recurrent Glioblastoma [STEERING]) comparing enzastaurin with lomustine for recurrent gliomas is currently under way.
Fibroblast Growth Factor Family
As a family, the fibroblast growth factors (FGFs) play important roles during human development. To date, a group of more than 20 polypeptides have been assigned to this family based on sequence homology.101 The molecular weights of these molecules are generally between 17 and 34 kD.101
FGF-1 (formerly acidic FGF because of its acidic isoelectric point) and FGF-2 (formerly basic FGF [bFGF]) have clearly been shown to be elevated in astrocytomas.102 Both these polypeptides play a role in cellular proliferation, whereas the role of FGF-9 is less certain. FGF-2 appears to have additional angiogenic effects, probably mediated through upregulation of vascular endothelial growth factor (VEGF) expression.103
High expression of FGF-2 is associated with worse outcomes in children with high-grade gliomas and in adults with astrocytic tumors.81,103 Plasma levels may also be elevated. Cytogenetic studies of astrocytomas do not show any consistent alterations in chromosomes encoding the FGFs or their receptors.104
Four FGF receptors (FGFRs) are known to exist.101 They possess extracellular heparin sulfate proteoglycans that act as low-affinity binding sites for FGFs and appear to be important for eventual binding to the high-affinity receptor and subsequent biologic activity. Binding elevates the activity of the ras pathway.102 FGFR1 is elevated in malignant gliomas, whereas FGFR2 expression is lost.102 FGFR4 expression predicts malignancy in astrocytomas.104
When expressed in normal brain tissue, FGFR1 usually occurs in what is called the alpha form, which has three immunoglobulin domains located in the extracellular binding domain. This is the form expressed in low-grade astrocytic tumors. In contrast, high-grade astrocytomas overexpress a splice variant with two immunoglobulin domains in the ligand-binding region, the so-called beta form.102
Monoclonal antibody–mediated downregulation of FGF-2 activity has tumor growth–suppressing effects,105 thus suggesting an autocrine role for this growth factor in gliomas. Testing has shown that suppression of FGFR1 expression with antisense oligonucleotides can decrease high-grade astrocytoma growth by up to 80%.106,107 Dominant-negative systems have been used to interfere with FGFR1 and FGFR2 in rat C6 glioma models. Tumor invasion, growth, and angiogenesis were affected.108 bFGF has been shown to induce rat C6 glioma cell growth mediated via the Map-erk kinase/extracellular-regulated kinase (MEK/ERK) pathway. Blockade of this pathway or immunoneutralization of TGF-β1, or both, abrogates the growth-promoting effect of FGF.109 Finally, inhibition of FGF by 1,3,6-naphthalenetrisulfonate suppressed tumor cell growth and angiogenesis in a rabbit corneal model.110 Manipulation of this pathway has potential in the treatment of glial tumors.
Insulin-like Growth Factor System
This signaling pathway consists of insulin-like growth factors (IGF-I and IGF-II, or somatomedins) and a group of insulin-like growth factor–binding proteins (IGFBP1 to IGFBP6) that help regulate the system.111,112 The IGFs are 7.5-kD proteins that are similar in structure to proinsulin. IGF-I is also known as somatomedin C, and it mediates many of the growth-promoting effects of growth hormone.111 The cognate receptors IGFR1 and IGFR2 are involved in controlling normal growth and development.112 Type I IGF receptor (IGF-IR) is a heterodimeric protein related to the insulin receptor.111
Elevated levels of IGFs and IGFRs have been documented in both meningiomas and gliomas. The growth of several types of tumors, including gliomas, primitive neuroectodermal tumors (PNETs), and neuroblastomas, is increased by the addition of exogenous IGF. IGFBP2, which is not normally expressed in adult cells, is elevated in high-grade gliomas. An increased ratio of IGF-II to IGFBP2 predicts increasing anaplasia in meningiomas. Expression of IGFR1 has a positive correlation with tumor grade.113
Several experiments have been performed with antisense plasmids and gene therapy to target IGF and its receptors.112 Tumor regression, reduced invasion, and elevated immunogenicity have all been demonstrated. NVP-TAE226, a dual inhibitor of IGFR1 and focal adhesion kinase (FAK), suppresses glioma growth both in vitro and in animal models.113
Neurotrophic Factors
As outlined earlier, there are other receptors in the erbB family. The ligands for these receptors arise from four different genes, alternatively spliced.114 As a group they are called heregulins or neuregulins. The neurotrophins include molecules such as neurotrophic factor-3 (NT-3), neurotrophic factor 4/5 (NT-4/5), NGF, glial cell line–derived neurotrophic factor (GDNF), and brain-derived neurotrophic factor (BDNF).6 The neuregulins NRG1-β, erbB2, and erbB4 are linked to poor clinical outcome in medulloblastoma, whereas TrkC expression is linked to a better prognosis.115
These factors bind to two groups of transmembrane receptors: Trk receptors (TrkRs) and the p75NTR neurotrophin receptor.116 The p75NTR receptor is capable of binding all members of this growth factor family.117 NGF binds to TrkA, BDNF and NT-4/5 bind to TrkB, and NT-3 binds to TrkC. TrkR kinase activity promotes cell survival, whereas p75NTR activates the JNK-p53-Bax pathway leading to apoptosis.116 GDNF may play a role in glioma cell migration.118
In medulloblastomas, activation of TrkC leads to apoptosis,119 and its expression is associated with a more favorable prognosis.120 NGF is involved in an autocrine growth loop in the C6 glioma cell line.121 Likewise, GDNF may be involved in an autocrine loop along with its receptor GDNFRα1 inasmuch as both are overexpressed in rat C6 glioma cells.122 Neuregulin-1 enhances astrocytoma cell survival via activation of Akt.123
In pituitary cell lines, NGF has the ability to activate p53 function via trkA and PI3K activation.124 Likewise, animal studies have shown that NGF has an inhibitory effect on C6 glioma cell growth.125 The true therapeutic potential of this class of growth factors has yet to be realized.
Platelet-Derived Growth Factor
Five distinct homodimeric or heterodimeric ligands (PDGF-AA, PDGF-AB, PDGF-BB, PDGF-CC, and PDGF-DD) are included in this family.103,126 These ligands bind to three tyrosine kinase receptors: PDGF-αα, PDGF-αβ, and PDGF-ββ. They help regulate cell growth, survival, and proliferation via the ras-MAPK, Akt, PI3K, and PLC-γ pathways.126 Autocrine and paracrine signaling involving PDGF-C and PDGF-D may play a role in growth and survival.126,127 PDGF-B is highly homologous to V-sis, which was isolated from simian sarcoma virus and is capable of inducing a variety of tumors, including astrocytomas in primates.128
Overexpression of PDGF and PDGF receptors (PDGFRs) is well documented in gliomas103,129 and tends to be seen in conjunction with TP53 tumor suppressor loss (i.e., in secondary glioblastomas).8,126
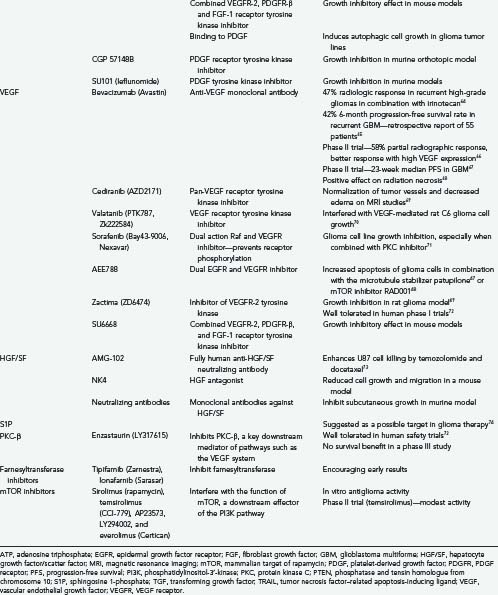
Recent experiments have shown that forced expression of PDGF in retrovirus-infected glial precursor cells can lead to tumor formation in both infected and noninfected progenitor cells,130 thus raising the possibility of paracrine actions. The drug imatinib (Gleevec) interferes with the PDGFR tyrosine kinase and the Bcr-Abl fusion protein.25 Despite clinical success in the treatment of other tumors such as chronic myelogenous leukemia, it has been less successful in the treatment of high-grade gliomas,75,131,132 possibly because of difficulty circumventing the blood-brain barrier and P-glycoprotein–mediated efflux.25,133 Likewise, treatment of oligodendrogliomas and mixed oligoastrocytomas has been disappointing.134 An inhibitor of PDGFR-β called CP-673,451 is also in development.86 Another agent, SU6668, which functions as a combined VEGFR-2, PDGFR-β, and FGF-1 RTK inhibitor, has demonstrated initial growth inhibitory effects in mice.135 In murine models, glioma cell growth could be inhibited with the PDGF RTK inhibitor CGP 57148B.136
The PDGF tyrosine kinase inhibitor SU101 (leflunomide) interferes with glioma growth in mice.137 Neutralizing antibodies directed against PDGF induce nonapoptotic death in glioma cell lines.138 Downregulation of hypoxia-inducible factor-1α (HIF-1α) by small interfering RNA (siRNA) results in downregulation of PDGF-B expression.139
Vascular Endothelial Growth Factor
Initially identified as a factor responsible for peritumoral edema, VEGF (also called vascular permeability factor or vasculotropin) was initially isolated as a 40- to 46-kD protein. VEGF is a dimeric growth factor with 20% homology to the PDGF-A and PDGF-B subunits140 VEGF has multiple actions, the most well studied of which is as a paracrine activator of angiogenesis.25,103 There are several members of the vascular endothelial growth factor (VEGF) gene family, namely, VEGF-A, VEGF-B, VEGF-C, VEGF-D, and PIGF.31 VEGF-A is the best studied and is overexpressed in gliomas.141 VEGF-A is a homodimer of 23-kD monomers40 and has multiple isoforms derived from differential splicing of the gene. They are named according to the number of residues included in the gene product.31,103 VEGF165 (VEGF-A) is the most common and mitogenic isoform of VEGF.31 VEGF-A binds to two main receptors: VEGFR-1 (Flt-1) and VEGFR-2 (KDR, Flk-1).53 There is strong evidence that VEGF plays an important role in angiogenesis and anaplastic progression of astrocytic tumors. VEGF appears to be secreted by some glial tumors, whereas its receptors are found on surrounding endothelial cells.103,142
There has been recent interest in angiogenesis inhibitors such as VEGF-Trap and monoclonal antibodies such as bevacizumab (Avastin). The former, a humanized soluble VEGFR, shows some promise in the experimental treatment of glioma cell lines.143 In combination with the cytotoxic agent irinotecan, the latter has shown some promising results in phase II clinical trials for the treatment of recurrent high-grade gliomas. A 63% radiographic response rate and a 38% 6-month progression-free survival rate was reported with an acceptable side effect profile.67 Based on the pattern of recurrence after bevacizumab therapy, others have suggested that it is more effective against the enhancing vascularized portions of tumor than against the nonenhancing infiltrative portions.65 This makes biologic sense because the tumor angiogenic profile and expression of VEGF predict susceptibility of the tumor to this agent.66 A median progression-free survival of 111 days in patients with recurrent GBM has been reported with the VEGFR inhibitor AZD2171 (cediranib), and a phase III trial in forthcoming.72 Other VEGFR tyrosine kinase inhibitors—vatalanib (PTK787/ZK222584), sorafenib (BAY 43-9006), and AZD2171—are the focus of recent investigation.70,86,144 Studies are also under way with the combined VEGFR/EGFR inhibitor ZD6474 (zactima)72 and the downstream PKC-β inhibitor enzastaurin (LY317615).72 No benefit was seen when using a combination of enzastaurin and standard chemotherapy in phase III trials despite documented radiologic improvements documented in earlier trials.72
Hepatocyte Growth Factor/Scatter Factor
Hepatocyte growth factor (HGF) and scatter factor (SF) are two names for the same molecule. Their different names derive from independent isolation: HGF as a mitogen for hepatocytes and SF for its ability to induce dissociation (scattering) of epithelial cells.145 The HGF/SF gene is found on chromosome 7q21.1. HGF/SF induces a wide spectrum of biologic events, including proliferation, angiogenesis, invasion, branching morphogenesis, and transformation,31 through binding of its receptor Met, located on chromosome 7q21-31.145 This powerful glioma chemokine can form a potential autocrine loop in glioma cells by stimulation of the Met tyrosine kinase receptor.25,146 Binding of c-Met by HGF/SF activates several cell signaling pathways, including the ras-MARK and PI3K pathways. This can protect glioblastoma cells from DNA-damaging agents such as ionizing radiation, cisplatin, paclitaxel, camptothecin, and doxorubicin.147
Killing of U-87 MG cell lines by commonly used chemotherapeutic agents can be augmented by addition of the anti-HGF/SF antibody AMG-102.73 The HGF antagonist NK4 inhibited GBM cell growth, migration, and neoangiogenesis in a mouse model.148 Finally, a combination of monoclonal antibodies against HGF/SF was reported to inhibit the subcutaneous growth of GBMs in a murine model.37
Sphingosine 1-Phosphate
Sphingosine 1-phosphate (S1P) is a lipid that can act as a glioma cell mitogen via activation of one of five G protein–linked receptors.149 Glioma cell motility is induced by binding S1P receptors 1 and 3, whereas S1P receptor 2 inhibits tumor invasion and motility.25 S1P has been suggested as a target in glioma therapy.74
Modulators of Downstream Signaling Elements and Other Therapies
A downstream component of the ras pathway is farnesyltransferase. Two farnesyltransferase inhibitors, tipifarnib (Zarnestra) and lonafarnib (Sarasar) have shown modest encouraging results in several types of cancers.35,150 mTOR is a downstream target of the PI3K pathway. Inhibitors such as sirolimus (rapamycin), temsirolimus (CCI-779), AP23573, LY294002, and everolimus (Certican) have shown antiproliferative activity in vitro39,151,152 and in a phase II trial (CCI-779).151 An Akt inhibitor called perifosine has shown promise in mouse glioma models.153
Antisense approaches directed against such diverse intracellular targets such as vascular endothelial growth factor (VEGF), transforming growth factor-beta (TGB-β), basic fibroblast growth factor (bFGF), endothelial growth factor receptor-1 (EGFR-1), and phosphoenolpyruvate carboxykinase-beta (PCK-β) are also in early stages of development.86,154
Conclusion
We are approaching an exciting time in the treatment of gliomas. As our understanding of the molecular biology of these tumors expands, so do options for rational treatment strategies. Because of the heterogeneity in tumor biology it is unlikely that any single strategy will prove useful for all tumors. In concert with advances in treatment, we need methods of choosing the best therapy for a given tumor. For example, it has become clear that the methylation status of the Ob-omethylguanine-DNA methyltransferase (MGMT) promoter gene has a clear impact on the efficacy of temozolomide treatment155 and that the status of chromosomes 1p and 19q has a striking ability to predict responses to chemotherapy.156,157 The goal of glioma therapy in the coming era will be to identify points of attack based on an individual tumor’s characteristics and to exploit these opportunities with treatments based on detailed knowledge of the molecular biology of tumor growth.
Belda-Iniesta C, de Castro Carpeno J, Sereno M, et al. Epidermal growth factor receptor and glioblastoma multiforme: molecular basis for a new approach. Clin Transl Oncol. 2008;10:73.
Brandsma D, van den Bent MJ. Molecular targeted therapies and chemotherapy in malignant gliomas. Curr Opin Oncol. 2007;19:598.
Dunn IF, Heese O, Black PM. Growth factors in glioma angiogenesis: FGFs, PDGF, EGF, and TGFs. J Neuro Oncol. 2000;50:121.
Guha A, Mukherjee J. Advances in the biology of astrocytomas. Curr Opin Neurol. 2004;17:655.
Hahn WC, Weinberg RA. Modelling the molecular circuitry of cancer. Nat Rev Cancer. 2002;2:331.
Kapoor GS, O’Rourke DM. Mitogenic signaling cascades in glial tumors. Neurosurgery. 2003;52:1425.
Kargiotis O, Rao JS, Kyritsis AP. Mechanisms of angiogenesis in gliomas. J Neuro Oncol. 2006;78:281.
Lefkowitz RJ. G proteins in medicine. N Engl J Med. 1995;332:186.
Neubig RR, Siderovski DP. Regulators of G-protein signalling as new central nervous system drug targets. Nat Rev Drug Discov. 2002;1:187.
Ohgaki H, Kleihues P. Genetic pathways to primary and secondary glioblastoma. Am J Pathol. 2007;170:1445.
Raza SM, Lang FF, Aggarwal BB, et al. Necrosis and glioblastoma: a friend or a foe? A review and a hypothesis. Neurosurgery. 2002;51:2.
Reardon DA, Wen PY. Therapeutic advances in the treatment of glioblastoma: rationale and potential role of targeted agents. Oncologist. 2006;11:152.
Stupp R, Hegi M, van den Bent MJ, et al. Changing paradigms—an update on the multidisciplinary management of malignant glioma. Oncologist. 2006;11:165.
Tayal V, Kalra BS. Cytokines and anti-cytokines as therapeutics—an update. Eur J Pharmacol. 2008;579:1.
Trojan J, Cloix JF, Ardourel MY, et al. Insulin-like growth factor type I biology and targeting in malignant gliomas. Neuroscience. 2007;145:795.
Wick W, Naumann U, Weller M. Transforming growth factor-beta: a molecular target for the future therapy of glioblastoma. Curr Pharm Des. 2006;12:341.
Woods SA, Marmor E, Feldkamp M, et al. Aberrant G protein signaling in nervous system tumors. J Neurosurg. 2002;97:627.
1 Kobler J. The Reluctant Surgeon: A Biography of John Hunter, Medical Genius and Great Inquirer of Johnson’s England. Garden City, NY: Doubleday; 1960.
2 Ohgaki H, Kleihues P. Genetic pathways to primary and secondary glioblastoma. Am J Pathol. 2007;170:1445.
3 Hahn WC, Weinberg RA. Modelling the molecular circuitry of cancer. Nat Rev Cancer. 2002;2:331.
4 Tannock I, Hill R, Bristow R, et al. The Basic Science of Oncology, 4th ed. Toronto: McGraw-Hill; 2005.
5 Hanahan D, Weinberg R. The hallmarks of cancer. Cell. 2000;100:57.
6 Morrison RS, Jarell AD, Schuster JM. Growth factors and brain tumors, 5th ed. Winn H, editor. Youman’s Neurological Surgery, Vol 1. Philidelphia: WB Saunders. 2004:725.
7 Ihle JN. Intracellular signalling. In Abeloff MD, Armitage JO, Niderhuber JE, Kastan MB, editors: Abeloff: Clinical Oncology, 3rd ed, Philadelphia: Churchill Livingstone, 2004.
8 Kapoor GS, O’Rourke DM. Mitogenic signaling cascades in glial tumors. Neurosurgery. 2003;52:1425.
9 Amit I, Wides R, Yarden Y. Evolvable signaling networks of receptor tyrosine kinases: relevance of robustness to malignancy and to cancer therapy. Mol Syst Biol. 2007;3:1.
10 Amit I, Wides R, Yarden Y. Evolvable signaling networks of receptor tyrosine kinases: relevance of robustness to malignancy and cancer therapy. Mol Syst Biol. 2007;3:1.
11 Heldin CH, Ostman A. Ligand-induced dimerization of growth factor receptors: variations on the theme. Cytokine Growth Factor Rev. 1996;7:3.
12 Pawson T. Protein modules and signalling networks. Nature. 1995;373:573.
13 O’Shea JJ, Murray PJ. Cytokine signaling modules in inflammatory responses. Immunity. 2008;28:477.
14 Ihle JN. Janus kinases in cytokine signalling. Philos Trans R Soc Lond B Biol Sci. 1996;351:159.
15 Starr R, Hilton DJ. SOCS: suppressors of cytokine signalling. Int J Biochem Cell Biol. 1998;30:1081.
16 Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673.
17 Medzhitov R. Toll-like receptors and innate immunity. Nat Rev Immunol. 2001;1:135.
18 Woods SA, Marmor E, Feldkamp M, et al. Aberrant G protein signaling in nervous system tumors. J Neurosurg. 2002;97:627.
19 Hollmann MW, Strumper D, Herroeder S, et al. Receptors, G proteins, and their interactions. Anesthesiology. 2005;103:1066.
20 Simon MI, Strathmann MP, Gautam N. Diversity of G proteins in signal transduction. Science. 1991;252:802.
21 Conklin BR, Bourne HR. Structural elements of G alpha subunits that interact with G beta gamma, receptors, and effectors. Cell. 1993;73:631.
22 Feldkamp MM, Lau N, Guha A. The farnesyltransferase inhibitor L-744,832 inhibits the growth of astrocytomas through a combination of antiproliferative, antiangiogenic, and proapoptotic activities. Ann N Y Acad Sci. 1999;886:257.
23 Wick W, Naumann U, Weller M. Transforming growth factor-beta: a molecular target for the future therapy of glioblastoma. Curr Pharm Des. 2006;12:341.
24 Kondo I, Shimizu N. Mapping of the human gene for the epidermal growth factor receptor (EGFR) on the p13-q22 region of chromosome y. Cytogenet Cell Genet. 1983;35:9.
25 Hoelzinger DB, Demuth T, Berens ME. Autocrine factors that sustain glioma invasion and paracrine biology in the brain microenvironment. J Natl Cancer Inst. 2007;99:1583.
26 Belda-Iniesta C, de Castro Carpeno J, Sereno M, et al. Epidermal growth factor receptor and glioblastoma multiforme: molecular basis for a new approach. Clin Transl Oncol. 2008;10:73.
27 Shinojima N, Tada K, Shiraishi S, et al. Prognostic value of epidermal growth factor receptor in patients with glioblastoma multiforme. Cancer Res. 2003;63:6962.
28 Eley G, Frederick L, Wang X, et al. 3′ end structure and rearrangements of EGFR in glioblastomas. Cancer. 1998;23:284.
29 Ramnarain DB, Park S, Lee DY, et al. Differential gene expression analysis reveals generation of an autocrine loop by a mutant epidermal growth factor receptor in glioma cells. Cancer Res. 2006;66:867.
30 Schwechheimer K, Laufle R, Schmahl W, et al. Expression of neu/c-erbB-2 in human brain tumors. Hum Pathol. 1994;25:772.
31 Ho QT, Kuo CJ. Vascular endothelial growth factor: biology and therapeutic applications. Int J Biochem Cell Biol. 2007;39:1349.
32 Stragliotto G, Vega F, Stasiecki P, et al. Multiple infusions of anti–epidermal growth factor receptor (EGFR) monoclonal antibody (EMD 55,900) in patients with recurrent malignant gliomas. Eur J Cancer. 1996;32A:636.
33 Brandes AA, Franceschi E, Tosoni A, et al. Epidermal growth factor receptor inhibitors in neuro-oncology: hopes and disappointments. Clin Cancer Res. 2008;14:957.
34 Stish BJ, Oh S, Vallera DA. Anti-glioblastoma effect of a recombinant bispecific cytotoxin cotargeting human IL-13 and EGF receptors in a mouse xenograft model. J Neuro Oncol. 2008;87:51.
35 Cloughesy TF, Kuhn JG, Wen P. Two phase II trials of R115777 (Zarnestra) in patients with recurrent glioblastoma multiforme: a comparison of patients on enzyme-inducing anti-epileptic drugs (EIAED) and not on EIAED at maximum tolerated dose respectively: a North American Brain Tumor Consortium (NABTC) Report. Neuro Oncol. 2003;5:349.
36 Moore RJ, Owens DM, Stamp G, et al. Mice deficient in tumor necrosis factor-alpha are resistant to skin carcinogenesis. Nat Med. 1999;5:828.
37 Cao B, Su Y, Oskarsson M, et al. Neutralizing monoclonal antibodies to hepatocyte growth factor/scatter factor (HGF/SF) display antitumor activity in animal models. Proc Natl Acad Sci U S A. 2001;98:7443.
38 Hau P, Jachimczak P, Schlingensiepen R, et al. Inhibition of TGF-β2 with AP 12009 in recurrent malignant gliomas: from preclinical to phase I/II studies. Oligonucleotides. 2007;17:201.
39 Kesari S, Ramakrishna N, Sauvageot C. Targeted molecular therapy of malignant gliomas. Curr Neurol Neurosci Rep. 2005;5:186.
40 Ferrara N, Henzel WJ. Pituitary follicular cells secrete a novel heparin-binding growth factor specific for vascular endothelial cells. Biochem Biophys Res Commun. 1989;161:851.
41 Prados MD, Lamborn KR, Chang S, et al. Phase 1 study of erlotinib HCl alone and combined with temozolomide in patients with stable or recurrent malignant glioma. Neuro Oncol. 2006;8:67.
42 Haas-Kogan DA, Prados MD, Tihan T, et al. Epidermal growth factor receptor, protein kinase B/Akt, and glioma response to erlotinib. J Natl Cancer Inst. 2005;97:880.
43 Fan QW, Cheng CK, Nicolaides TP, et al. A dual phosphoinositide-3-kinase alpha/mTOR inhibitor cooperates with blockade of epidermal growth factor receptor in PTEN-mutant glioma. Cancer Res. 2007;67:7960.
44 Doherty L, Gigas DC, Kesari S, et al. Pilot study of the combination of EGFR and mTOR inhibitors in recurrent malignant gliomas. Neurology. 2006;67:156.
45 Krishnan S, Brown PD, Ballman KV, et al. Phase I trial of erlotinib with radiation therapy in patients with glioblastoma multiforme: results of North Central Cancer Treatment Group protocol N0177. Int J Radiat Oncol Biol Phys. 2006;65:1192.
46 Lin NU, Carey LA, Liu MC, et al. Phase II trial of lapatinib for brain metastases in patients with human epidermal growth factor receptor 2-positive breast cancer. J Clin Oncol. 2008;26:1993.
47 Failly M, Korur S, Egler V, et al. Combination of sublethal concentrations of epidermal growth factor receptor inhibitor and microtubule stabilizer induces apoptosis of glioblastoma cells. Mol Cancer Ther. 2007;6:773.
48 Goudar RK, Shi Q, Hjelmeland MD, et al. Combination therapy of inhibitors of epidermal growth factor receptor/vascular endothelial growth factor receptor 2 (AEE788) and the mammalian target of rapamycin (RAD001) offers improved glioblastoma tumor growth inhibition. Mol Cancer Ther. 2005;4:101.
49 Sandstrom M, Johansson M, Andersson U, et al. The tyrosine kinase inhibitor ZD6474 inhibits tumour growth in an intracerebral rat glioma model. Br J Cancer. 2004;91:1174.
50 Emanuel SL, Hughes TV, Adams M, et al. Cellular and in vivo activity of JNJ-28871063, a nonquinazoline pan-ErbB kinase inhibitor that crosses the blood-brain barrier and displays efficacy against intracranial tumors. Mol Pharmacol. 2008;73:338.
51 Belda-Iniesta C, Carpeno Jde C, Saenz EC, et al. Long term responses with cetuximab therapy in glioblastoma multiforme. Cancer Biol Ther. 2006;5:912-914.
52 Combs SE, Heeger S, Haselmann R, et al. Treatment of primary glioblastoma multiforme with cetuximab, radiotherapy and temozolomide (GERT)—phase I/II trial: study protocol. BMC Cancer. 2006;6:133.
53 Puduvalli VK, Giglio P, Groves MD, et al. Phase II trial of irinotecan and thalidomide in adults with recurrent glioblastoma multiforme. Neuro Oncol. 2008;10:216.
54 Riva M, Imbesi F, Beghi E, et al. Temozolomide and thalidomide in the treatment of glioblastoma multiforme. Anticancer Res. 2007;27:1067.
55 Groves MD, Puduvalli VK, Chang SM, et al. A North American Brain Tumor Consortium (NABTC 99-04) phase II trial of temozolomide plus thalidomide for recurrent glioblastoma multiforme. J Neuro Oncol. 2007;81:271.
56 Turner CD, Chi S, Marcus KJ, et al. Phase II study of thalidomide and radiation in children with newly diagnosed brain stem gliomas and glioblastoma multiforme. J Neuro Oncol. 2007;82:95.
57 Knisely JP, Berkey B, Chakravarti A, et al. A phase III study of conventional radiation therapy plus thalidomide versus conventional radiation therapy for multiple brain metastases (RTOG 0118). Int J Radiat Oncol Biol Phys. 2008;71:79.
58 Krown SE, Niedzwiecki D, Hwu WJ, et al. Phase II study of temozolomide and thalidomide in patients with metastatic melanoma in the brain: high rate of thromboembolic events (CALGB 500102). Cancer. 2006;107:1883.
59 Pollack IF, Jakacki RI, Blaney SM, et al. Phase I trial of imatinib in children with newly diagnosed brainstem and recurrent malignant gliomas: a Pediatric Brain Tumor Consortium report. Neuro Oncol. 2007;9:145.
60 Medioni J, Cojocarasu O, Belcaceres JL, et al. Complete cerebral response with sunitinib for metastatic renal cell carcinoma. Ann Oncol. 2007;18:1282.
61 Koutras AK, Krikelis D, Alexandrou N, et al. Brain metastasis in renal cell cancer responding to sunitinib. Anticancer Res. 2007;27:4255.
62 Pouessel D, Culine S. High frequency of intracerebral hemorrhage in metastatic renal carcinoma patients with brain metastases treated with tyrosine kinase inhibitors targeting the vascular endothelial growth factor receptor. Eur Urol. 2008;53:376.
63 de Bouard S, Herlin P, Christensen JG, et al. Antiangiogenic and anti-invasive effects of sunitinib on experimental human glioblastoma. Neuro Oncol. 2007;9:412.
64 Bokstein F, Shpigel S, Blumenthal DT. Treatment with bevacizumab and irinotecan for recurrent high-grade glial tumors. Cancer. 2008;112:2267.
65 Norden AD, Young GS, Setayesh K, et al. Bevacizumab for recurrent malignant gliomas: efficacy, toxicity, and patterns of recurrence. Neurology. 2008;70:779.
66 Sathornsumetee S, Cao Y, Marcello JE, et al. Tumor angiogenic and hypoxic profiles predict radiographic response and survival in malignant astrocytoma patients treated with bevacizumab and irinotecan. J Clin Oncol. 2008;26:271.
67 Vredenburgh JJ, Desjardins A, Herndon JE2nd, et al. Phase II trial of bevacizumab and irinotecan in recurrent malignant glioma. Clin Cancer Res. 2007;13:1253.
68 Gonzalez J, Kumar AJ, Conrad CA, et al. Effect of bevacizumab on radiation necrosis of the brain. Int J Radiat Oncol Biol Phys. 2007;67:323.
69 Batchelor TT, Sorensen AG, di Tomaso E, et al. AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell. 2007;11:83.
70 Goldbrunner RH, Bendszus M, Wood J, et al. PTK787/ZK222584, an inhibitor of vascular endothelial growth factor receptor tyrosine kinases, decreases glioma growth and vascularization. Neurosurgery. 2004;55:426.
71 Jane EP, Premkumar DR, Pollack IF. Coadministration of sorafenib with rottlerin potently inhibits cell proliferation and migration in human malignant glioma cells. J Pharmacol Exp Ther. 2006;319:1070.
72 Fine H. Promising new therapies for malignant gliomas. Cancer J. 2007;13:349.
73 Jun HT, Sun J, Rex K, et al. AMG 102, a fully human anti-hepatocyte growth factor/scatter factor neutralizing antibody, enhances the efficacy of temozolomide or docetaxel in U-87 MG cells and xenografts. Clin Cancer Res. 2007;13:6735.
74 Van Brocklyn JR. Sphingolipid signaling pathways as potential therapeutic targets in gliomas. Mini Rev Med Chem. 2007;7:984.
75 Rich JN, Reardon DA, Peery T, et al. Phase II trial of gefitinib in recurrent glioblastoma. J Clin Oncol. 2004;22:133.
76 Franceschi E, Cavallo G, Lonardi S, et al. Gefitinib in patients with progressive high-grade gliomas: a multicentre phase II study by Gruppo Italiano Cooperativo di Neuro-Oncologia (GICNO). Br J Cancer. 2007;96:1047.
77 Graff JR, McNulty AM, Hanna KR, et al. The protein kinase Cβ-selective inhibitor, Enzastaurin (LY317615.HCl), suppresses signaling through the AKT pathway, induces apoptosis, and suppresses growth of human colon cancer and glioblastoma xenografts. Cancer Res. 2005;65:7462.
78 Vogelbaum MA, Peereboom DM, Stevens G, et al. Phase II study of erlotinib single agent therapy in recurrent glioblastoma multiforme. Eur J Cancer Suppl. 2005;3:135.
79 Van Den Bent M, Brandes A, Rampling R, et al. Randomized phase II trial of erlotinib versus temozolomide (TMZ) or BCNU in recurrent glioblastoma multiforme (GBM): EORTC 26034 [abstract]. J Clin Oncol. 2007;25(Suppl 18):A-2005.
80 Mellinghoff IK, Wang MY, Vivanco I, et al. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med. 2005;353:2012.
81 Fukui S, Nawashiro H, Otani N, et al. Nuclear accumulation of basic fibroblast growth factor in human astrocytic tumors. Cancer. 2003;97:3061.
82 Parsons R, Simpson L. PTEN and cancer. Methods Mol Biol. 2003;222:147.
83 Brandsma D, van den Bent MJ. Molecular targeted therapies and chemotherapy in malignant gliomas. Curr Opin Oncol. 2007;19:598.
84 Nguyen T, Lassmann AB, Lis E, et al. A pilot study to assess the tolerability and efficacy of RAD-001 (everolimus) with gefitinib in patients with recurrent glioblastoma multiforme. J Clin Oncol. 2006;24:1507.
85 Andersson U, Johansson D, Behnam-Motlagh P, et al. Treatment schedule is of importance when gefitinib is combined with irradiation of glioma and endothelial cells in vitro. Acta Oncol. 2007;46:951.
86 Reardon DA, Wen PY. Therapeutic advances in the treatment of glioblastoma: rationale and potential role of targeted agents. Oncologist. 2006;11:152-914.
87 Belda-Iniesta C, Carpeno Jde C, Saenz EC, et al. Long term responses with cetuximab therapy in glioblastoma multiforme. Cancer Biol Ther. 2006;5:912.
88 Kargiotis O, Rao JS, Kyritsis AP. Mechanisms of angiogenesis in gliomas. J Neuro Oncol. 2006;78:281.
89 Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell. 2001;104:487.
90 Balkwill F. TNF-alpha in promotion and progression of cancer. Cancer Metastasis Rev. 2006;25:409.
91 Kaur B, Khwaja FW, Severson EA, et al. Hypoxia and the hypoxia-inducible-factor pathway in glioma growth and angiogenesis. Neuro Oncol. 2005;7:134.
92 Siegel RM. Caspases at the crossroads of immune-cell life and death. Nat Rev Immunol. 2006;6:308.
93 Panner A, Parsa AT, Pieper RO. Use of APO2L/TRAIL with mTOR inhibitors in the treatment of glioblastoma multiforme. Expert Rev Anticancer Ther. 2006;6:1313.
94 Raza SM, Lang FF, Aggarwal BB, et al. Necrosis and glioblastoma: a friend or a foe? A review and a hypothesis. Neurosurgery. 2002;51:2.
95 Ahn KS, Sethi G, Aggarwal BB. Nuclear factor-kappa B: from clone to clinic. Curr Mol Med. 2007;7:619.
96 Tayal V, Kalra BS. Cytokines and anti-cytokines as therapeutics—an update. Eur J Pharmacol. 2008;579:1.
97 Sharif A, Legendre P, Prevot V, et al. Transforming growth factor alpha promotes sequential conversion of mature astrocytes into neural progenitors and stem cells. Oncogene. 2007;26:2695.
98 Massague J. TGFβ signaling: receptors, transducers, and Mad proteins. Cell. 1996;85:947.
99 Bruna A, Darken RS, Rojo F, et al. High TGFbeta-Smad activity confers poor prognosis in glioma patients and promotes cell proliferation depending on the methylation of the PDGF-B gene. Cancer Cell. 2007;11:147.
100 Sampson J, Akabani G, Archer G. Progress report of a phase I study of the intracerebral microinfusion of a recombinant chimeric protein composed of transforming growth factor (TGF)-alpha and a mutated form of the Pseudomonas exotoxin termed PE-38 (TP-38) for the treatment of malignant brain tumors. J Neuro Oncol. 2003;11:329.
101 Mason I. Initiation to end point: the multiple roles of fibroblast growth factors in neural development. Nat Rev Neurosci. 2007;8:583.
102 Morrison RS, Yamaguchi F, Saya H, et al. Basic fibroblast growth factor and fibroblast growth factor receptor I are implicated in the growth of human astrocytomas. J Neuro Oncol. 1994;18:207.
103 Dunn IF, Heese O, Black PM. Growth factors in glioma angiogenesis: FGFs, PDGF, EGF, and TGFs. J Neuro Oncol. 2000;50:121.
104 Yamada SM, Yamada S, Hayashi Y, et al. Fibroblast growth factor receptor (FGFR) 4 correlated with the malignancy of human astrocytomas. Neurol Res. 2002;24:244.
105 Murai N, Ueba T, Takahashi JA, et al. Apoptosis of human glioma cells in vitro and in vivo induced by a neutralizing antibody against human basic fibroblast growth factor. J Neurosurg. 1996;85:1072.
106 Redekop GJ, Naus CC. Transfection with bFGF sense and antisense cDNA resulting in modification of malignant glioma growth. J Neurosurg. 1995;82:83.
107 Yamada SM, Yamaguchi F, Brown R, et al. Suppression of glioblastoma cell growth following antisense oligonucleotide-mediated inhibition of fibroblast growth factor receptor expression. Glia. 1999;28:66.
108 Auguste P, Gursel DB, Lemiere S, et al. Inhibition of fibroblast growth factor/fibroblast growth factor receptor activity in glioma cells impedes tumor growth by both angiogenesis-dependent and -independent mechanisms. Cancer Res. 2001;61:1717.
109 Dhandapani KM, Khan MM, Wade FM, et al. Induction of transforming growth factor-beta1 by basic fibroblast growth factor in rat C6 glioma cells and astrocytes is mediated by MEK/ERK signaling and AP-1 activation. J Neurosci Res. 2007;85:1033.
110 Cuevas P, Carceller F, Reimers D, et al. Inhibition of intra-tumoral angiogenesis and glioma growth by the fibroblast growth factor inhibitor 1,3,6-naphthalenetrisulfonate. Neurol Res. 1999;21:481.
111 Glick R, Unterman T, Van der Woude M, et al. Insulin and insulin-like growth factors in central nervous system tumors; part V. Production of insulin-like growth factors I and II. J Neurosurg. 1992;77:445.
112 Trojan J, Cloix JF, Ardourel MY, et al. Insulin-like growth factor type I biology and targeting in malignant gliomas. Neuroscience. 2007;145:795.
113 Liu TJ, LaFortune T, Honda T, et al. Inhibition of both focal adhesion kinase and insulin-like growth factor-I receptor kinase suppresses glioma proliferation in vitro and in vivo. Mol Cancer Ther. 2007;6:1357.
114 Esper RM, Pankonin MS, Loeb JA. Neuregulins: versatile growth and differentiation factors in nervous system development and human disease. Brain Res Rev. 2006;51:161.
115 Grotzer MA, Janss AJ, Phillips PC, et al. Neurotrophin receptor TrkC predicts good clinical outcome in medulloblastoma and other primitive neuroectodermal brain tumors. Klin Padiatr. 2000;212:196.
116 Assimakopoulou M, Kondyli M, Gatzounis G, et al. Neurotrophin receptors expression and JNK pathway activation in human astrocytomas. BMC Cancer. 2007;7:202.
117 Denkins Y, Reiland J, Roy M, et al. Brain metastases in melanoma: roles of neurotrophins. Neuro Oncol. 2004;6:154.
118 Song H, Moon A. Glial cell-derived neurotrophic factor (GDNF) promotes low-grade Hs683 glioma cell migration through JNK, ERK-1/2 and p38 MAPK signaling pathways. Neurosci Res. 2006;56:29.
119 Kim JY, Sutton ME, Lu DJ, et al. Activation of neurotrophin-3 receptor TrkC induces apoptosis in medulloblastomas. Cancer Res. 1999;59:711.
120 Segal RA, Goumnerova LC, Kwon YK, et al. Expression of the neurotrophin receptor TrkC is linked to a favorable outcome in medulloblastoma. Proc Natl Acad Sci U S A. 1994;91:12867.
121 Watanabe T, Katayama Y, Kimura S, et al. Control of proliferation and survival of C6 glioma cells with modification of the nerve growth factor autocrine system. J Neuro Oncol. 1999;41:121.
122 Wiesenhofer B, Stockhammer G, Kostron H, et al. Glial cell line–derived neurotrophic factor (GDNF) and its receptor (GFR-alpha 1) are strongly expressed in human gliomas. Acta Neuropathol. 2000;99:131.
123 Ritch PS, Carroll SL, Sontheimer H. Neuregulin-1 enhances survival of human astrocytic glioma cells. Glia. 2005;51:217.
124 Facchetti M, Uberti D, Memo M, et al. Nerve growth factor restores p53 function in pituitary tumor cell lines via trkA-mediated activation of phosphatidylinositol 3-kinase. Mol Endocrinol. 2004;18:162.
125 Kimura S, Yoshino A, Katayama Y, et al. Growth control of C6 glioma in vivo by nerve growth factor. J Neuro Oncol. 2002;59:199.
126 Fomchenko E, Holland EC. Platelet-derived growth factor-mediated gliomagenesis and brain tumor recruitment. Neurosurg Clin N Am. 2007;18:39.
127 Lokker NA, Sullivan CM, Hollenbach SJ, et al. Platelet-derived growth factor (PDGF) autocrine signaling regulates survival and mitogenic pathways in glioblastoma cells: evidence that the novel PDGF-C and PDGF-D ligands may play a role in the development of brain tumors. Cancer Res. 2002;62:3729.
128 Deinhardt F. Biology of Primate Retroviruses: Viral Oncology. New York: Raven Press; 1980.
129 Fleming T, Saxena A, Clark W. Amplification and/or overexpression of platelet-derived growth factor receptors and epidermal growth factor receptor in human glial tumors. Cancer Res. 1992;52:4550.
130 Assanah M, Lochhead R, Ogden A, et al. Glial progenitors in adult white matter are driven to form malignant gliomas by platelet-derived growth factor–expressing retroviruses. J Neurosci. 2006;26:6781.
131 Raymond E, Brandes A, Van Oosterom A, et al. Multicenter phase II study of imatinib mesylate in patients with recurrent glioblastoma: an EORTC: NDDG/BTG Intergroup study [abstract]. J Clin Oncol. 2004;22(Suppl 14):A-1501.
132 Wen PY, Yung WK, Lamborn KR, et al. Phase I/II study of imatinib mesylate for recurrent malignant gliomas: North American Brain Tumor Consortium Study 99-08. Clin Cancer Res. 2006;12:4899.
133 Dai H, Marbach P, Lemaire M, et al. Distribution of STI-571 to the brain is limited by P-glycoprotein–mediated efflux. J Pharmacol Exp Ther. 2003;304:1085.
134 van den Bent MJ, Brandes AA, Frenay M. Multicentre phase II study of imatinib mesylate (Glivec) in patients with recurrent anaplastic oligodendroglioma (AOD)/mixed oligoastrocytoma (MOA) and anaplastic astrocytoma (AA)/low grade astrocytoma (LGA): an EORTC New Drug Development Group (NDDG) and Brain Tumor Group (BTG) study. J Clin Oncol. 2005;23:1501a.
135 Farhadi MR, Capelle HH, Erber R, et al. Combined inhibition of vascular endothelial growth factor and platelet-derived growth factor signaling: effects on the angiogenesis, microcirculation, and growth of orthotopic malignant gliomas. J Neurosurg. 2005;102:363.
136 Uhrbom L, Hesselager G, Ostman A, et al. Dependence of autocrine growth factor stimulation in platelet-derived growth factor-B–induced mouse brain tumor cells. Int J Cancer. 2000;85:398.
137 Shawver LK, Schwartz DP, Mann E, et al. Inhibition of platelet-derived growth factor–mediated signal transduction and tumor growth by N-[4-(trifluoromethyl)-phenyl]5-methylisoxazole-4-carboxamide. Clin Cancer Res. 1997;3:1167.
138 Takeuchi H, Kanzawa T, Kondo Y, et al. Inhibition of platelet-derived growth factor signalling induces autophagy in malignant glioma cells. Br J Cancer. 2004;90:1069.
139 Yoshida D, Kim K, Noha M, et al. Hypoxia inducible factor 1-alpha regulates of platelet derived growth factor-B in human glioblastoma cells. J Neuro Oncol. 2006;76:13.
140 Senger DR, Galli SJ, Dvorak AM, et al. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science. 1983;219:983.
141 Chaudhry IH, O’Donovan DG, Brenchley PE, et al. Vascular endothelial growth factor expression correlates with tumour grade and vascularity in gliomas. Histopathology. 2001;39:409.
142 Plate K, Breier G, Millauer B, et al. Upregulation of vascular endothelial growth factor and its cognate receptors in a rat glioma model of tumor angiogenesis. Cancer Res. 1993;53:5822.
143 Wachsberger PR, Burd R, Cardi C, et al. VEGF trap in combination with radiotherapy improves tumor control in u87 glioblastoma. Int J Radiat Oncol Biol Phys. 2007;67:1526.
144 Strumberg D, Richly H, Hilger R. Phase I clinical and pharmaco-kinetic study of the novel Raf kinase and vascular endothelial growth factor receptor inhibitor BAY 43-9006 in patients with advanced refractory solid tumors. J Clin Oncol. 2005;23:965.
145 Maulik G, Shrikhande A, Kijima T, et al. Role of the hepatocyte growth factor receptor, c-Met, in oncogenesis and potential for therapeutic inhibition. Cytokine Growth Factor Rev. 2002;13:41.
146 Abounader R, Laterra J. Scatter factor/hepatocyte growth factor in brain tumor growth and angiogenesis. Neurooncology. 2005;7:436.
147 Bowers D, Fan S, Walter K, et al. Scatter factor/hepatocyte growth factor protects against cytotoxic death in human glioblastoma via phosphatidylinositol 3-kinase– and AKT-dependent pathways. Cancer Res. 2000;60:4277.
148 Brockmann M, Papadimitriou A, Brandt M, et al. Inhibition of intracerebral glioblastoma growth by local treatment with the scatter factor/hepatocyte growth factor-antagonist NK4. Clin Cancer Res. 2003;9:4578.
149 Young N, Van Brocklyn JR. Roles of sphingosine-1-phosphate (S1P) receptors in malignant behavior of glioma cells. Differential effects of S1P2 on cell migration and invasiveness. Exp Cell Res. 2007;313:1615.
150 Caponigro F, Casale M, Bryce J. Farnesyltransferase inhibitors in clinical development. Expert Opin Investig Drugs. 2003;12:943.
151 Chang S, Wen P, Cloughesy TF. Phase II study of CCI-779 in patients with recurrent glioblastoma multiforme. Invest New Drugs. 2005;23:357.
152 Newton H. Molecular neuro-oncology and development of targeted therapeutic strategies for brain tumors. Part 2: PI3K/Akt/PTEN, mTOR, SHH/PTCH and angiogenesis. Expert Rev Anticancer Ther. 2004;4:105.
153 Momota H, Nerio E, Holland EC. Perifosine inhibits multiple signaling pathways in glial progenitors and cooperates with temozolomide to arrest cell proliferation in gliomas in vivo. Cancer Res. 2005;65:7429.
154 Matsuno A, Nagashima T. Specific gene suppression using antisense strategy for growth suppression of glioma. Med Electron Microsc. 2004;37:158.
155 Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997.
156 Cairncross G, Berkey B, Shaw E, et al. Phase III trial of chemotherapy plus radiotherapy compared with radiotherapy alone for pure and mixed anaplastic oligodendroglioma: Intergroup Radiation Therapy Oncology Group Trial 9402. J Clin Oncol. 2006;24:2707.
157 van den Bent MJ, Carpentier AF, Brandes AA, et al. Adjuvant procarbazine, lomustine, and vincristine improves progression-free survival but not overall survival in newly diagnosed anaplastic oligodendrogliomas and oligoastrocytomas: a randomized European Organisation for Research and Treatment of Cancer phase III trial. J Clin Oncol. 2006;24:2715.