[level-membership-for-endocrinology-diabetes-and-metabolism-category]Chapter 9
Glucagon and the Glucagon-Like Peptides
Biosynthesis of Pancreatic Glucagon
Proglucagon is encoded by a single gene in mammals that gives rise to a proglucagon mRNA with major sites of expression in pancreatic islets, brain, and enteroendocrine cells of the small and large intestine. Proglucagon also may be expressed in subsets of salivary glands and taste buds.1 The proglucagon gene contains six exons, several of which encode distinct functional peptide domains (Fig. 9-1). Glucagon, a 29 amino acid peptide, is synthesized in the A cells of the pancreatic islets of Langerhans. Islet A cells are distinguishable from insulin-producing β cells in part by the morphology of their respective secretory granules and by their anatomic distribution, predominantly at the periphery of the islet. The peripheral location of islet A cells, together with functional studies demonstrating central (from a core of β cells) to peripheral (A cells) islet blood flow, raises the possibility of a tightly regulated islet microenvironment. Nevertheless, as the distribution of islet α and β cells may vary from species to species, the functional importance of islet cell distribution remains unclear.

FIGURE 9-1 Structure of proglucagon and the proglucagon-derived peptides. The amino acid sequences of glucagon, glucagon-like peptide (GLP)-1, and GLP-2 are shown, as well as the cleavage site for dipeptidyl peptidase-4 (DPP-4). IP-1 and IP-2, Intervening peptides-1 and -2, respectively.
Islet Transcription Factors and the α Cell
Studies of targeted gene disruption in mice have provided new insights into the organization of islet endocrine cells in the pancreas. Neurogenin-3 is critical for pancreatic islet cell development and differentiation and controls the expression of many genes important for α cell differentiation and glucagon gene transcription. Ngn3 interacts in a regulatory network with the zinc-finger protein Myt1 to promote glucagon expression in α cells.2 Mice with a null allele in the basic helix-loop-helix transcription factor p48 fail to develop exocrine pancreatic tissue; however, hormone-secreting islet cells, including glucagon-producing α cells, are found within the mesentery during embryonic development, and later in the spleen.3 A key role for cell adhesion molecules in the control of spatial organization of islet cells is illustrated by analysis of the endocrine pancreas in mice expressing a dominant negative E-cadherin receptor in islet β cells. These mice exhibit abnormal clustering of β cells, yet glucagon-producing α cells are still capable of aggregating into islet-like clusters.4 In contrast, the normal peripheral distribution of islet α cells is markedly perturbed in neural cell adhesion molecule (NCAM)-/- mice; however, the number of α cells and the glucagon content of the pancreas remain unaffected.5 It is intriguing that mice with genetic disruption of the ciliogenic transcription factor regulatory factor X (RFX)-3 exhibit small disorganized islets with markedly reduced insulin and glucagon content, implying a role for cilia in optimal islet organization and cell development.6
Considerable insight into the developmental biology of the endocrine pancreas and islet α cells has been derived from studies of islet transcription factors in cell lines and mice. Gene knockout studies demonstrate that the LIM domain protein isl-1 is necessary for the formation of differentiated islet cells, including α cells, in the developing pancreas.7 Targeted deletion of the homeobox transcription factor Arx results in mice that exhibit complete failure of α cell development, leading to glucagon deficiency and neonatal hypoglycemia.8 In contrast, although mice deficient in the homeobox transcription factor Ipf/Pdx-1 fail to develop a pancreas, a few islet cells immunopositive for insulin or glucagon are observed in Ipf/Pdx-1-/- mice at E11.9 These observations suggest that Ipf/Pdx-1 is not essential for the formation of islet α cells, whereas loss of Pdx-1 expression appears to be required for acquisition of a differentiated α cell phenotype.
The homeobox transcription factor Nkx2.2 is expressed in adult α, β, and PP islet cells; mice with targeted disruption of NKx2.2 lack β cells, develop diabetes, and exhibit a marked reduction in the numbers of islet α cells.10 A related phenotype is observed in NeuroD/BETA2-/- mice, which exhibit marked reductions in the numbers of islet β and α cells and develop diabetes shortly after birth,11 a phenotype dependent on the specific genetic background. Similarly, disruption of pax6 function in mice results in poorly formed islets with disorganized islet architecture and markedly reduced but detectable numbers of both β and islet α cells.12,13 It is intriguing that mice that harbor mutations in islet transcription factors also may exhibit paradoxically increased numbers of islet α cells. For example, targeted deletion of the Pax4 gene results in poorly formed islets, with a marked reduction in islet β cells and comparatively increased numbers of islet α cells.14 A related phenotype is observed in mice with a homozygous deletion in the glucose transporter 2 (GLUT-2) gene, with a marked increase in the ratio of α to β cells observed in GLUT2-/-islets.15 Whether these findings are due to a block in the normal islet differentiation program, or to loss of inhibitors that restrain α cell proliferation, is not known. Evidence in support of glucagon itself regulating the numbers of islet α cells derives from analysis of mice with targeted disruption of the genes that encode prohormone convertase-2, or the glucagon receptor. These mice represent models for loss of bioactive glucagon16 or glucagon action,17 respectively, and they exhibit marked α cell hyperplasia, which is corrected in the former instance by glucagon replacement therapy.
Proglucagon Gene Transcription Factors
A combination of gene transfection experiments using cell lines in vitro and transgenic studies of promoter function in vivo has yielded insight into the molecular control of glucagon gene transcription. Differential gene expression analysis has revealed a remarkably complex pattern of transcription factors preferentially expressed in α versus β cell lines18 and in single α cells isolated from the developing endocrine pancreas.19 The rat proglucagon gene promoter directs transcriptional initiation from a single transcription start site that maps to an identical location in brain, pancreas, and intestine.20 Cell transfection experiments utilizing islet cell lines have identified five distinct regions, designated G1 through G5, within the proximal proglucagon promoter that exhibit functional importance for activation of islet cell–specific proglucagon gene transcription. The proximal G1 region is AT rich, interacts with both widely expressed and islet cell–specific proteins, and is functionally important for specifying expression in islet A cells. G1 interacts with the homeobox transcripton factors Isl-1, Cdx-2/3, Brn4, and Pax6.13,21–24 These transcription factors bind G1 and activate reporter genes containing G1-derived sequences in transfection assays. Pax6 and c-Maf exhibit cooperativity in their ability to increase G1-dependent transcriptional activation,25 whereas Nkx6.1 inhibits Pax6-dependent G1 activation. Reduction of isl-1 expression leads to reduced G1-dependent proglucagon promoter activity and decreased levels of proglucagon mRNA transcripts.21 Similarly, increased expression of Cdx-3 in islet cells is associated with induction of both transfected G1-dependent reporter genes and endogenous proglucagon gene expression.22,26 The G4 element, located just upstream of G1, binds a complex resembling insulin-enhancer factor 1 (IEF1), and recent experiments suggest that IEF1 represents a heterodimer of the helix-loop-helix (HLH) proteins E47 and BETA2.27
The more distal G3 region functions as an islet enhancer–like element and has been divided into two distinct functional domains. Subdomain A contains a sequence element similar to sequences found within the insulin and somatostatin promoters, leading to the designation of this composite sequence as a pancreatic islet cell–specific enhancer sequence, or PISCES element.28,29 Sequences within domain A of G3 also appear to mediate the inhibition of glucagon gene transcription by insulin.30–32 The pax6 protein has been identified as a positive activator of proglucagon gene transcription that exerts its function in part through interaction with domain A of the G3 element.13 Pax6 also binds the G1 element, either as a monomer or via heterodimer formation with cdx-2/3.33 Disruption of Pax6 expression in gene-targeted mice results in loss of islet A cells.12,13 The levels of pancreatic and intestinal proglucagon mRNA transcripts are also markedly reduced in SEYNEU mice that harbor a dominant negative mutation in the pax6 gene. Thus Pax6, a PISCES binding protein, is functionally important for both islet and enteroendocrine cell development and activation of proglucagon gene transcription via the G3 enhancer and G1 promoter elements.12,13,33,34
The G2 element mediates the positive and negative actions of HNF-3 (Foxa) proteins, with isoforms of HNF-3β and HNF-3α competing for binding to the G2 element and serving as repressors and activators, respectively, of proglucagon gene transcription.35,36 HNF-3γ also binds to and transactivates the proglucagon promoter G2 element but does not appear to be essential for islet cell formation or glucagon gene transcription.37–39 Whether HNF-3β plays an essential role in the development of islet α cell formation or glucagon gene transcription in vivo remains unknown, as targeted inactivation of the HNF-3β gene results in embryonic lethality before formation of the endocrine pancreas.40,41 Although the numbers of α cells and the morphology of islets appear histologically normal in HNF3α-/- mice, the development of neonatal hypoglycemia in the face of inappropriately reduced levels of circulating glucagon and proglucagon mRNA transcripts demonstrates the essential role of HNF-3α in pancreatic proglucagon gene transcription.37
Activation of the cyclic adenosine monophosphate (cAMP)-dependent pathway leads to transcriptional induction of proglucagon gene expression in both islet and intestinal cells through a cAMP response element (CRE) present upstream of the G3 element in the proximal promoter region of the rat proglucagon gene.42,43 The CRE also mediates transcriptional activation by ATF3 family members44 and is the target for pharmacologic depolarization–dependent induction of rat proglucagon gene transcription.45 A second calcium response element has been localized to the G2 element, and calcium responsiveness may be mediated via interaction of nuclear factor of activated T cells (NFAT)-like proteins with members of the HNF-3 family.46 The sequence of the CRE element is less well conserved in the human proglucagon promoter, and its functional relevance for proglucagon gene expression in human islets has not yet been elucidated.
Transgenic experiments in mice have identified distinct DNA sequences essential for tissue-specific proglucagon gene transcription in vivo. The first 1253 nucleotides of the rat proglucagon promoter direct heterologous transgene expression to the islets and brain but not the intestine in transgenic mice.47 In contrast, targeting of transgene expression to enteroendocrine cells in vivo requires the presence of additional rat proglucagon gene 5′-flanking sequences between −1253 and −2252.48 Foxo1 binds to an upstream site at −1798 and appears to be important for both basal and insulin-regulated proglucagon gene transcription in α cells.49 DNA sequences constituting the human proglucagon gene promoter exhibit different functional properties than homologous rat sequences, as the first 1600 bp of the human proglucagon gene 5′-flanking region target transgene expression to appropriate cell types in the brain and intestine, but not the pancreatic islets in transgenic mice.50
Islet Proglucagon Biosynthesis
Consistent with the increased circulating glucagon levels in patients with diabetes, experimental diabetes in rodents is associated with increased levels of pancreatic proglucagon mRNA; correction of the insulin deficiency, but not the hyperglycemia, normalizes the levels of proglucagon mRNA.51 Paradoxically, insulin may increase proglucagon gene expression in enteroendocrine cells.52 Orexin-A also inhibits glucagon secretion and proglucagon gene expression via the phosphatidylinositol-3-kinase–dependent pathway, requiring Foxo1.53 In contrast, few hormones or metabolites have been identified that increase pancreatic proglucagon biosynthesis in vivo. Although experiments using in situ hybridization suggested that 4 days of fasting in the rat leads to a doubling of pancreatic proglucagon mRNA transcripts,54 hypoglycemia induced by insulin failed to demonstrate upregulation of proglucagon mRNA.51,55 Available data suggest that regulation of glucagon secretion, and not biosynthesis, may be a more relevant locus of control in vivo.
Following transcription of proglucagon mRNA and translation of the proglucagon precursor, 29 amino acid mature glucagon is liberated via posttranslational processing by specific prohormone convertases differentially expressed in the islet α cell. In contrast to processing in the enteroendocrine cell (see Fig. 9-1), the amino acid sequences carboxyterminal to glucagon remain unprocessed and are secreted as part of a larger polypeptide designated the major proglucagon fragment (MPGF). PC2 is essential for processing of proglucagon to glucagon in α cells as PC2-/- mice exhibit hypoglycemia, α cell hyperplasia, and glucagon deficiency with an accumulation of incompletely processed PGDPs in the pancreas.16 Whether PC2 directly liberates glucagon from proglucagon, or cleaves proglucagon to glicentin, which is subsequently processed to glucagon, remains unclear. The enzyme PC1 is responsible for cleavage of proglucagon to yield an intestinal profile of PGDPs,56,57 and PC1 null mice exhibit impaired processing of proglucagon to the glucagon-like peptides.58 Moreover, human subjects with an inactivating mutation in the PC1 gene exhibit incompletely processed proglucagon in gut endocrine cells and deficiency of both GLP-1 and GLP-2.59 The convertases important for proglucagon processing in the brain have not been identified definitively.
Glucagon Secretion
The secretion of glucagon is regulated, both positively and negatively, by neuropeptides, hormones, metabolites, and the autonomic nervous system. The islet α cell plays a central role in the defense of blood glucose, with hypoglycemia stimulating and hyperglycemia suppressing glucagon secretion in vivo. A cells express voltage-dependent Na+, K+, and Ca2+ channels that interact in the regulation of membrane potential and, ultimately, depolarization. Amino acids such as glutamine, alanine, pyruvate, and arginine stimulate both insulin and glucagon secretion, and this observation provides the basis for the use of arginine in the assessment of islet and α cell function in rodent and human physiology.60 Both α- and β-adrenergic receptors modulate glucagon secretion. Epinephrine stimulates glucagon secretion via protein kinase A (PKA)-dependent enhancement of Ca2+ influx through L-type calcium channels, leading to granule exocytosis. Peptidergic activators of glucagon secretion include cholecystokinin (CCK), pituitary adenylate cyclase–activating polypeptide (PACAP), gastrin, urocortin III, vasopressin, and glucose-dependent insulinotropic peptide (GIP).
In contrast, inhibitors of glucagon secretion include glucose, somatostatin-14, and γ-aminobutyric acid (GABA).61 Somatostatin tonically inhibits glucagon release, and mice with disruption of the somatostatin receptor 2 (SSTR2) gene exhibit mild hyperglycemia and elevated levels of nonfasting glucagon.62 Glucose induces GABA release from β cells, providing an indirect mechanism for glucose-mediated suppression of glucagon secretion.63–65 Glucose also reduces electrical activity and exocytosis via depolarization-induced inactivation of ion channels, specifically KATP, Na(+) (TTX) and N-type Ca(2+) channels, as a direct mechanism for inhibition of glucagon secretion. Insulin also may directly inhibit glucagon release, possibly via insulin receptors expressed on α cells. The anatomic arrangement of peripheral islet α cells surrounding a core of β cells, together with the functional results of immunoneutralization studies, provides additional evidence for intra-islet insulin inhibiting downstream of α cells in some species.66 Although the mechanisms underlying the paracrine inhibition of α cell activity remain unclear, experimental evidence supports an important role for β cell secretory products such as zinc as negative regulators of glucagon secretion.65,67
The effects of glucose on glucagon secretion are integrated with the inhibitory effects of insulin on α cell secretion, as hyperglycemia stimulates insulin secretion, whereas a drop in blood glucose suppresses insulin release from the β cell, thereby relieving the α cell from the tonic inhibitory actions of insulin. These actions are exemplified by meal ingestion, which is associated with increased circulating nutrients and insulin secretion, and reduced levels of circulating glucagon. In contrast, blood glucose is maintained in the fasting state by hepatic glucose production due in part to increased levels of glucagon secretion and suppression of insulin release from the β cell.68,69 Consistent with the effects of individual nutrients on α cell function, infusion of arginine stimulates glucagon secretion in both normal subjects and individuals with diabetes mellitus.69 The response to arginine infusion in normal human subjects, namely, stimulation of insulin and glucagon release, is reproducible, and as expected, the insulin response is significantly greater and the glucagon response attenuated as the glucose concentration increases.70
In normal subjects, glucagon secretion rises as glucose falls in the fasting state and may be further stimulated by exercise, consistent with the physiologic role for glucagon in regulating glucose production. The levels of both glucagon and plasma catecholamines increase with graded exercise in normal human subjects, with the greatest increments in plasma glucagon observed in subjects undergoing prolonged exhaustive exercise.71 The exercise-induced increment in plasma glucagon appears dependent on the degree and duration of exercise, with some studies demonstrating no significant changes in plasma glucagon during exercise under normoglycemic conditions.72 Trained healthy male subjects exhibit increased hepatic glucose production following glucagon infusion, implying that exercise may be associated with the development of increased glucagon sensitivity.73 Although prevention of exercise-induced rises in plasma glucagon by concomitant somatostatin infusion may result in mild hypoglycemia,74 suppression of the rise in plasma glucagon by somatostatin infusion does not always prevent increased hepatic glucose production, likely because of the redundant compensatory mechanisms for maintaining normoglycemia.74,75
The control of hepatic glucose production (HGP) is highly sensitive to the glucagon/insulin ratio, and secretion of these two hormones generally is regulated in a reciprocal manner. The effect of insulin to suppress HGP actually may be determined in part by the levels of circulating glucagon.76 Following meal ingestion, nutrient absorption is associated with energy assimilation and suppression of glucagon secretion. Nevertheless, the integrated response of islet α cells is dependent in part on the nutrient composition of the meal and reflects positive and negative enteric-derived regulators of glucagon secretion. For example, ingestion of carbohydrate, especially glucose, is associated with a decline in plasma glucagon.69 Nutrients also promote release of gut-derived peptides such as CCK and GIP, which stimulate glucagon release,77,78 and GLP-1, which inhibits glucagon release.79,80
Glucagon secretion is increased during times of stress, and “stress-induced hormones” such as cortisol, vasopressin, and β-endorphin increase glucagon secretion from the α cell. The classic stress hormones epinephrine and norepinephrine also stimulate glucagon secretion, and several mechanisms link the autonomic nervous system to increased secretion from the α cell. Epinephrine secreted from the adrenal medulla increases glucagon secretion in normoglycemic subjects. Furthermore, the pancreas receives innervation from both sympathetic and parasympathetic nerves, and stimulation of these autonomic inputs, all of which are activated by hypoglycemia, increases glucagon secretion.81 It is intriguing that mice with targeted inactivation of the pro-opiomelanocortin gene (and hence adrenocorticotropic hormone [ACTH]) exhibit a profound defect in the counterregulatory response to insulin-induced hypoglycemia, largely as the result of defective glucagon secretion. Although specific genetic defects in glucagon secretion have not been described, nondiabetic subjects with mutations in the MODY1/HNF-4α gene exhibit decreased arginine-stimulated glucagon secretion and reduced glucose suppression of plasma glucagon,82 raising the possibility that the HNF-4α transcription factor influences islet α cell function through a direct or indirect mechanism.
Hypoglycemia
Increasing evidence suggests that multiple complementary mechanisms activate α cell secretion during hypoglycemia. Analysis of the glycemic threshold for activation of counterregulatory mechanisms demonstrates that increased epinephrine and glucagon secretion constitute the initial hormonal responses to decreasing blood sugar. Furthermore, the glucose threshold for activation of counterregulatory responses is clearly higher than the threshold for triggering hypoglycemic symptoms in normal subjects.83 Whether hypoglycemia itself directly stimulates glucagon secretion independent of autonomic input remains unclear. Nevertheless, hypoglycemia suppresses insulin (and β cell) secretion, which removes an important inhibitory influence on glucagon secretion. The finding that α cells express the GLUT1 transporter and glucokinase suggests that glucose transport does not appear to be a critical rate-limiting step for glucose metabolism in α cells and provides important insight into how α cells directly sense ambient changes in glucose concentration.84,85 Hypoglycemia stimulates glutamate release from α cells, which, in turn, activates glucagon secretion in an autocrine manner.86 Glutamate release from ventromedial hypothalamic neurons is also a key component of the counterregulatory response, as mice lacking the synaptic vesicular transporters (VGLUT2) exhibit fasting hypoglycemia and a defective glucagon response to hypoglycemia.87 A role for gastrin in the stimulation of glucagon secretion has been proposed, and gastrin-/- mice exhibit defective glucagon secretion in response to insulin-induced hypoglycemia.88
The role of circulating epinephrine in the autonomic response to hypoglycemia is well established. Epinephrine also directly stimulates glucagon secretion in normal human subjects, and stimulation of autonomic sympathetic nervous system innervation to the pancreas elicits an increase in α cell secretion.89,90 Furthermore, parasympathetic nerve stimulation or the neurotransmitter acetylcholine and the neuropeptide VIP all stimulate glucagon secretion. Studies using nerve transection or pharmacologic blockade have illustrated that these pathways exhibit some degree of functional redundancy, providing multiple backup mechanisms to ensure that the α cell responds appropriately to hypoglycemia. Nevertheless, the ganglionic blocker trimethaphan, which impairs autonomic transmission both in ganglia and in the adrenal, markedly attenuated the glucagon response to hypoglycemia in normal subjects,91 emphasizing the important link between autonomic activation and the glucagon counterregulatory response in vivo. In contrast however, pancreas transplantation restores the glucagon response to hypoglycemia, even in patients with severe autonomic neuropathy,92 illustrating the functional redundancy of mechanisms essential for the α cell response to hypoglycemia.
The identity of the specific glucose sensors that trigger the appropriate counterregulatory response to hypoglycemia remains under active investigation. Mice with genetic disruption of the Kir6.2 channel exhibit normal functional α cells, yet display a marked defect in the glucagon response to hypoglycemia.93 GLUT2 expression in glial cells is essential for the glucagon response to hypoglycemia and the central nervous system, principally the ventromedial hypothalamus (VMH),87 and the splanchnic region, specifically consisting of the portal system and liver, contains multiple glucose sensing systems. Selective perfusion of the portal venous system in rats suggests that the portal vein glucose concentration is a key determinant of the sympathoadrenal response to hypoglycemia; however, whether portal glucose sensors are directly or indirectly linked to control of islet glucagon secretion is not clear.94 It is intriguing that injection of glucose into the portal vein activates glucose-sensitive neurons in the lateral hypothalamus and brain stem, suggesting the possibility of a portal-CNS glucoregulatory axis. The detection of a markedly defective counterregulatory response to hypoglycemia, including absent glucagon and catecholamine secretion, in a patient with hypothalamic sarcoidosis further emphasizes the importance of the hypothalamus in sensing glucose and triggering the release of counterregulatory hormones.95 In contrast, analysis of patients after removal of craniopharyngiomas revealed selective impairment of counterregulatory sympathoadrenal activation but normal glucagon responses to hypoglycemia. Furthermore, human subjects with liver transplants and denervated livers exhibit increased levels of circulating glucagon and defective insulin suppression of glucagon, and human islet transplantation is associated with a defective glucagon response to hypoglycemia. These findings emphasize the importance of the autonomic nervous system and the CNS for the regulation of basal and hypoglycemia-stimulated glucagon secretion.
Glucagon Secretion and Diabetes
Glucagon levels are elevated in many patients with poorly controlled diabetes, and excess glucagon secretion in response to meal ingestion is an early event that is detectable within months of the onset of type 1 diabetes.96 The correction of hyperglucagonemia, using somatostatin or insulin, reverses most metabolic derangements associated with insulin-deficient diabetes.97 Hence there is considerable interest in development of glucagon antagonists for the treatment of diabetes. Moreover, genetic attenuation of glucagon receptor expression in diabetic rodents results in significant amelioration of experimental diabetes.98,99 Despite the central importance of glucagon for control of glucose production, hyperglucagonemia alone, without insulin deficiency, does not significantly increase plasma glucose.100 In the presence of adequate amounts of insulin, glucose production is suppressible despite glucagon excess, emphasizing the insulin:glucagon ratio, and not just the absolute level of glucagon, as a key determinant of glucose homeostasis.101 Nevertheless, hyperglucagonemia is associated with increased leucine oxidation and resting metabolic rate in subjects with type 1 diabetes, reemphasizing the importance of both insulin and glucagon in the catabolism associated with suboptimally treated diabetes.102
The application of intensive insulin therapy to the management of patients with type 1 and type 2 diabetes is associated with an increased incidence of hypoglycemia and heightened awareness of the importance of counterregulatory mechanisms for maintaining normoglycemia. Several factors, including intensive insulin administration, hyperglycemia, and diminished autonomic stimulation of the α cell, inhibit glucagon release in insulin-treated patients with type 1 diabetes.81 Although the glucagon response to hypoglycemia is initially normal in patients with type 1 diabetes, this response frequently becomes impaired, increasing the susceptibility of patients to hypoglycemia.103 Impaired counterregulation and hypoglycemia have been observed in some patients with a very short duration of diabetes, suggesting that frequent episodes of hypoglycemia represent an independent risk for development of an abnormal α cell response. The α cell dysfunction in type 1 diabetes is often selective, as the response to hypoglycemia may be absent, yet glucagon secretion may respond normally to arginine stimulation.104 Further evidence emphasizing the importance of intra-islet hyperinsulinemia or other β cell–derived products in the suppression of glucagon secretion derives from observations that tolbutamide-infused subjects exhibit profound defects in the glucagon response to hypoglycemia.105
It is important to note that restoration of normoglycemia for several months may decrease hypoglycemia unawareness in association with improvement in the glucagon response to hypoglycemia in some studies.106 However, a dissociation between improvement in hypoglycemia unawareness and persistence of defective counterregulatory responses also has been observed.107 Although not completely understood, antecedent hypoglycemia in type 1 diabetes can produce counterregulatory failure during subsequent episodes of prolonged moderate intensity exercise,108 emphasizing the complex interrelationship between insulin-induced and exercise-associated counterregulation.
Multiple defects likely contribute to α cell dysfunction in patients with diabetes. Patients with some residual β cell function, as assessed by C-peptide stimulation, appear to be at decreased risk for hypoglycemia, in part because of preserved counterregulatory glucagon responses. Additional contributing factors may include subtle impairment of autonomic stimulation following repeated hypoglycemic episodes. Nevertheless, impaired epinephrine and glucagon responses to insulin-induced hypoglycemia have been observed after a single antecedent hypoglycemic episode in nondiabetic subjects,109 although increased levels of cortisol alone do not acutely induce defects in hypoglycemic counterregulation. It is intriguing that oral administration of amino acids improves cognitive function and the glucagon response to hypoglycemia in both normal subjects and individuals with type 1 diabetes. Taken together, these observations emphasize the susceptibility of the normal α cell to episodes of hypoglycemia, ultimately leading to sustained defects in the glucagon response to hypoglycemia.
Glucagon Action
The importance of glucagon in the control of hepatic glucose metabolism provides a useful model for analysis of hormone action in metabolic pathways. Glucagon stimulates glucose production via activation of hepatic glycogenolysis and gluconeogenesis, and by inhibition of glycolysis. Following activation of the glucagon receptor, adenylyl cyclase activity is increased, leading to activation of protein kinase A and phosphorylase kinase, and increased rates of glycogenolysis via glycogen phosphorylase and inactivation of glycogen synthase.110 Glucagon action serves to modify the activity of enzymes important for glucose production via effects on specific kinases and phosphatases. Glucagon also modulates the expression of genes encoding enzymes of the glycolytic or gluconeogenic pathways111 and regulates fatty acid metabolism via reduction of malonyl CoA and stimulation of fatty acid oxidation. The cAMP-dependent transcription factor is an important downstream mediator for glucagon action, acting in part through activation of the nuclear receptor coactivator PGC-1, and in part through suppression of peroxisome-proliferator–activated receptor-γ (PPAR-γ) agonist activity, which induces a metabolic program that results in the activation of hepatic gluconeogenesis.112
In adipocytes, glucagon increases cAMP and stimulates lipolysis, thereby providing free fatty acids as substrate for fat-burning tissues. Glucagon inhibits insulin-stimulated glucose transport in adipocytes through effects on insulin binding and via postreceptor mechanisms,113 and short-term glucagon administration reduces glucose uptake in hepatic and nonhepatic tissues.114 In the peripheral vascular system, glucagon functions as a vasodilator via effects on local vascular tone, and glucagon increases both cardiac output and heart rate, possibly via direct effects on the heart.115 Pharmacologic doses of glucagon increase renal blood flow, glomerular filtration rate, and urinary electrolyte excretion116; the kidney also exhibits significant gluconeogenic capacity and may account for up to 25% of systemic glucose production in humans.117 Although renal glucose output is markedly increased in subjects with diabetes, and hypoglycemia increases renal glucose output in association with increased release of counterregulatory hormones, the available evidence does not support an important role for glucagon in the control of renal glucose output.118
The actions of glucagon are transduced via activation of the glucagon receptor (Gcgr), a seven-transmembrane–spanning G protein–coupled receptor. The cloned receptor responds to glucagon with an increase in both intracellular cAMP and intracellular calcium.119 The human glucagon receptor gene has been localized to chromosome 17q25. Although activating mutations of the Gcgr have been generated by mutagenesis in vitro,120 no constitutively active Gcgr mutations have been reported in human subjects. Although several population studies have described a Gly40Ser mutation linked to increased development of diabetes and/or hypertension, whether this mutation itself contributes to a diabetes predisposition or is associated with other genes that increase diabetes susceptibility in certain populations remains unclear.
The tissue distribution of Gcgr expression correlates well with studies localizing high-affinity glucagon-binding sites with Gcgr mRNA transcripts detected in liver, brain, adipocytes, heart, kidney, and islet β cells. Rat Gcgr mRNA transcripts also have been detected in spleen, thymus, adrenal gland, ovary, and testis—nonclassical target tissues where glucagon action remains poorly defined.121 Although glucagon action has been studied extensively in the liver and adipocytes, the precise biological importance of glucagon in the brain remains unclear. The brain stem is the principal site of central nervous system (CNS) proglucagon gene expression122; however, PGDPs are transported from the brain stem along nerve fibers to multiple brain regions.123 Consistent with these findings, glucagon binding sites are detected in multiple brain regions, and the glucagon receptor is expressed in cortex, cerebellum, hypothalamus, and brain stem; however, the specific biological action of glucagon in each of these CNS regions remains unclear. Although intracerebroventricular injection of glucagon in rodents causes hyperglycemia and increases sympathetic nervous system discharge, whether these findings are relevant to physiologic control of glucose homeostasis requires further analysis.
The β cell expresses receptors for glucagon, GIP, and GLP-1 that all are coupled to cAMP and stimulation of insulin secretion. The threshold for glucagon-stimulated cAMP accumulation in isolated β cells is ≈1 nM glucagon—higher than the concentrations required for cAMP stimulation by GLP-1 or GIP.124 The physiologic importance of endogenous glucagon for β cell physiology in vivo remains unclear, given the direction of islet blood flow, the peripheral location of islet α cells in rodents, and the high concentrations of glucagon required to stimulate the β cell in vitro.
Insights into glucagon action derive from characterization of glucagon receptor knockout mice.17,125 Gcgr-/- mice exhibit marked elevations in levels of plasma glucagon, mild fasting hypoglycemia, increased pancreatic weight, α cell hyperplasia, and increased circulating levels of the PGDPs. The compensatory mechanisms sufficient for maintaining glucose production despite the complete absence of hepatic glucagon receptor signaling remain unknown. Similarly, the glucagon receptor–dependent signals regulating the number and secretory function of islet α cells have not yet been elucidated.
Both the liver and the kidney contribute to glucagon clearance from the circulation; however, these sites account for less than 50% of glucagon clearance, implicating additional tissues as sites for glucagon clearance or degradation.126 Glucagon action is terminated via both extracellular and intracellular degradation pathways. Glucagon-degrading activity within hepatic endosomes has been attributed to cathepsins B and D in studies using cathepsin inhibitors,127 and both glucagon and GLP-1 are substrates for the widely expressed membrane-bound neutral ectopeptidase (NEP) 24.11.128 An endopeptidase activity has been described that cleaves 29 amino acid glucagon to “mini-glucagon,” which is also known as glucagon (19-29) in various tissues. The physiologic importance of mini-glucagon remains uncertain; whether glucagon (19-29) exerts physiologically relevant effects in the heart or β cell via a separate unique receptor remains unclear.
Pharmaceutical Use of Glucagon in Human Patients
Glucagon is employed as both a diagnostic and a therapeutic agent. Although historically useful for the diagnosis of pheochromocytoma, stimulation of catecholamine secretion in such patients may be dangerous, and hence the glucagon stimulation test is not widely used. Glucagon may also be employed as part of a diagnostic test in patients with hypoglycemia of unknown origin. Perhaps the most common therapeutic clinical application of glucagon is seen in the adjunctive management of severe hypoglycemia. Diabetic patients with hypoglycemia generally respond quickly with a rapid increase in blood glucose to intranasal, intramuscular, or subcutaneous glucagon.129,130 Glucagon is also used to inhibit gastrointestinal motility during radiologic investigations, and several studies have reported the efficacy of glucagon administration in small numbers of patients with bronchospasm, or symptomatic bradycardia.131,132
Glucagon Excess and Deficiency
Glucagonomas presenting as solitary lesions or as part of a multiple endocrine neoplasia syndrome are most commonly detected in the pancreas and often are associated with significant elevations in the levels of circulating glucagon and PGDPs.133 Rarely, extrapancreatic glucagon-producing tumors have been reported in sites including the kidney and ovary. Although many gut carcinoid tumors contain PGDP immunoreactivity, they are not generally associated with the development of a “glucagonoma syndrome.” Patients with glucagonoma generally present with a pathognomonic skin rash termed necrolytic migratory erythema, a detectable pancreatic mass, weight loss, glossitis, anemia, and some degree of glucose intolerance.134 The clinical presentation can be variable, likely reflecting adaptation to the metabolic effects of tumor-secreted PGDPS and tumor-specific differences in posttranslational processing of proglucagon. Rarely, patients may present with glucagonomas and associated manifestations of intestinal hyperplasia, presumably due to release of GLP-2.135 Treatment for benign glucagonomas usually involves surgical resection, whereas chemotherapy, attempted suppression of PGDP secretion and tumor growth with somatostatin analogues, or adjunctive radiotherapy may be indicated in patients with malignant disease. Experimental glucagonomas have been studied in rodents, and several intriguing phenotypes, including severe anorexia and reduction in islet size, remain poorly understood.136,137 Several reports have described isolated cases of glucagon deficiency in infants with hypoglycemia; however, these cases are extremely rare. The molecular basis for the putative glucagon deficiency and whether congenital glucagon deficiency is compatible with survival remain unknown.
The Glucagon-like Peptides: GLP-1 and GLP-2
The proglucagon gene is expressed in the gastrointestinal tract in the stomach and both small and large intestine. The intestinal, brain, and pancreatic mammalian proglucagon mRNA transcripts are identical in structure; hence tissue-specific posttranslational processing underlies the liberation of the glucagon-like peptides in the brain and gastrointestinal tract. In contrast to the pancreas, much less is known about the control of intestinal GLP-1 and GLP-2 biosynthesis. The proglucagon gene islet transcription factors cdx-2/3, pax6 and members of the HNF3 (Foxa) family are also expressed in enteroendocrine cells and presumably regulate proglucagon gene transcription in both cell types. Neurogenin-3 has been identified as an essential upstream determinant of global enteroendocrine cell development. SEYNEU mice with a dominant negative pax6 mutation exhibit a marked reduction in the levels of proglucagon mRNA transcripts in both small and large intestine. Hence, the pax6 gene is essential for both islet and enteroendocrine proglucagon gene transcription. Proglucagon biosynthesis in the gut is also regulated by nutrient intake,138 with feeding and fiber-enriched diets increasing proglucagon gene expression in the proximal and distal intestine, respectively.
Glucagon-Like Peptide 1: Preclinical Studies
GLP-1, in both (7-37) and (7-36amide) molecular forms, is liberated from proglucagon via posttranslational processing and is secreted from intestinal endocrine cells in a nutrient-dependent manner.139 Although enteroendocrine L cells are distributed along the length of the entire gastrointestinal tract from the stomach to the rectum, the largest numbers of L cells are found in the terminal ileum and proximal colon. The rapid increase in plasma GLP-1 following food ingestion has fostered interest in the existence of a proximal-distal loop, whereby nutrients entering the duodenum and the proximal jejunum promote one or more endocrine and/or neural signals that activate GLP-1 secretion from the distal small bowel.140 Studies in rats have identified GIP as one putative component of such a signaling system. The specific signaling mechanisms utilized by nutrients for stimulation of GLP-1 secretion in humans are not completely understood.
The regulation of GLP-1 bioactivity is dependent to a large extent on the rate of GLP-1 degradation and clearance in vivo. Both GLP-1 and GLP-2 contain an alanine residue at position 2, rendering these molecules substrates for enzymatic inactivation by dipeptidyl peptidase-4 (DPP-4). DPP-4 expressed locally in the intestine proximal to sites of GLP-1 synthesis cleaves circulating GLP-1 to yield GLP-1 (9-37/9-36amide).141 Although GLP-1 (9-37/9-36amide) displays weak binding affinity for the GLP-1 receptor and is theoretically a circulating antagonist of GLP-1 action, infusion of GLP-1 (9-36amide) has modest insulin-independent effects on glucose tolerance and insulin secretion in human subjects.142,143 Nevertheless, a substantial amount of total GLP-1 immunoreactivity circulates as GLP-1 (9-37/9-36amide), and assays that do not distinguish between intact GLP-1 and cleaved GLP-1 (9-37/9-36amide) will overestimate the circulating concentrations of bioactive GLP-1. More recent studies indicate that GLP-1 (9-36) amide may enhance cardiac function in preclinical studies144,145 and may inhibit hepatic glucose production in obese human subjects.146
Largely as a result of rapid DPP-4–mediated inactivation of GLP-1, the 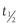 of circulating intact bioactive GLP-1 is very short, generally less than 1 minute. These findings have encouraged efforts at developing more potent long-acting GLP-1 analogues resistant to DPP-4–mediated inactivation. Alternatively, inhibitors of the DPP-4 enzyme have been identified that lower blood sugar in subjects with type 2 diabetes.147–149
of circulating intact bioactive GLP-1 is very short, generally less than 1 minute. These findings have encouraged efforts at developing more potent long-acting GLP-1 analogues resistant to DPP-4–mediated inactivation. Alternatively, inhibitors of the DPP-4 enzyme have been identified that lower blood sugar in subjects with type 2 diabetes.147–149
GLP-1 exerts several complementary actions that control postprandial glycemic excursion, including stimulation of glucose-dependent insulin secretion and inhibition of glucagon secretion and gastric emptying (Fig. 9-2). GLP-1R agonists also regulate growth and development of the endocrine pancreas.150 GLP-1 receptor activation promotes cell survival in β cells via increased levels of cAMP, leading to CREB activation, enhanced IRS-2 activity, and, ultimately, activation of Akt. These actions of GLP-1 are reflected by expansion of β cell mass and enhanced resistance to β cell injury demonstrated in multiple experimental models of diabetes in vivo. Moreover, Glp1r-/- mice exhibit modest defects in the formation of large islets and enhanced susceptibility to apoptotic injury.151 Of potential relevance to the therapeutic use of GLP-1 is the demonstration that primary human islet cultures exhibit improved glucose-dependent insulin secretion and enhanced survival following short-term exposure to GLP-1 in vitro.152 Similarly, use of the GLP-1R agonist exenatide is being explored in human subjects with type 1 diabetes following islet transplantation.153
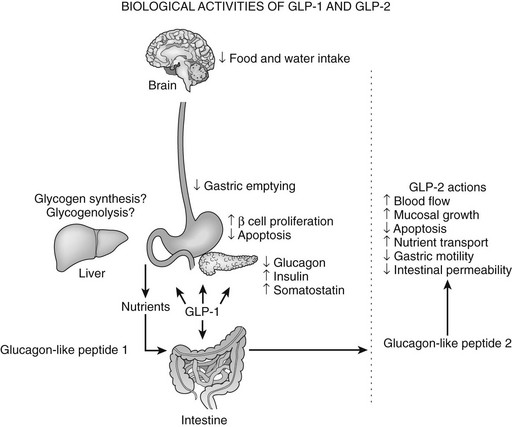
FIGURE 9-2 Biological activities of glucagon-like peptide (GLP)-1 and GLP-2. The various actions of GLP-1 (left panel) and GLP-2 (right panel) are shown.
Inhibition of gastric emptying accounts for a significant portion of the glucose-lowering actions of GLP-1, especially in patients with type 1 diabetes. Sensations of increased satiety and decreased appetite are common following GLP-1 infusion in human subjects,154 and GLP-1 likely decreases appetite via central effects on the hypothalamic nuclei involved in feeding and via peripheral effects on gastric emptying. Although administration of GLP-1 and its analogues to rodents also results in decreased water intake, these actions do not appear to be clinically significant in human studies.
GLP-1 exerts its actions through activation of a G protein–coupled receptor that is structurally related to the glucagon/secretin receptor superfamily.155 GLP-1 transduces its signal through both cAMP and calcium-dependent pathways. Although a potential candidate diabetes gene, the GLP-1 receptor (GLP-1R) localized to human 6p21 has not been associated with linkage to families with type 2 diabetes. Similarly, no GLP-1R mutations have been identified in individuals with diabetes, nor have activating mutations of GLP-1R been described in human subjects.
Evidence for the physiologic importance of GLP-1 in glucose control derives from studies using GLP-1 antagonists to inhibit GLP-1 action in vivo. Infusion of the GLP-1 antagonist exendin (9-39) into rats or humans increased blood glucose in association with decreased levels of glucose-stimulated insulin.156,157 Similarly, immunoneutralization of GLP-1 activity using GLP-1 antiserum increased both fasting- and meal-related glycemic excursions in baboons.158 Furthermore, Glp1r-/- mice exhibit mild diabetes, with abnormalities in both fasting and postprandial glycemia and subnormal levels of glucose-stimulated insulin.159
Therapeutic Use of GLP-1R Agonists for the Treatment of Patients with Type 2 Diabetes
Studies in human subjects have demonstrated the safety and efficacy of using GLP-1 to achieve glucose control in patients with type 2 diabetes. Native GLP-1 was administered via continuous subcutaneous infusion for 6 weeks to obese subjects with type 2 diabetes, resulting in highly significant improvement in glucose control, reduced glycosylated hemoglobin (HbA1c), improved insulin sensitivity, and a small but significant mean 1.9 kg weight loss.160
Because the glucose-lowering effects of native GLP-1 administered by single subcutaneous administration are transient and are no longer evident 1 to 2 hours following peptide injection,161 therapeutic efforts were refocused on stable degradation-resistant GLP-1R agonists, including the lizard peptide exendin-4 (exenatide) and the human GLP-1 analogue liraglutide.162 As continuous enhancement of GLP-1 action for 24 hours a day appears superior for glucose control compared with shorter durations of peptide administration, current dogma supports the elevation of circulating levels of GLP-1 over a 24-hour period.
Exendin-4 (exenatide = synthetic exendin-4) is a 39 amino acid naturally occurring lizard GLP-1R agonist that exhibits 53% amino acid identity to mammalian GLP-1,163,164 yet binds to and activates the GLP-1 receptor. Furthermore, exendin-4 is highly resistant to the proteolytic activity of DPP-4 and exhibits a longer duration of action in vivo. Exenatide was evaluated in phase 3 trials, in combination with metformin, sulfonylurea agents, or both, for the treatment of patients with type 2 diabetes (T2DM), and its use led to significant lowering of HbA1c in a substantial percentage of treated subjects.165–167 Subsequent studies confirmed the efficacy of adding exenatide to patients suboptimally controlled on a thiazolidinedione,168 or as an alternative to insulin therapy.169,170 Dose-limiting side effects of exenatide include nausea and vomiting. Most exenatide-treated subjects experience modest weight loss of 2 to 4 pounds over 6 months; however, about 20% of patients experience more substantial weight loss. Pancreatitis has been reported as a rare complication of therapy with GLP-1R agonists. A once-weekly form of exenatide is also in late-stage clinical development for the treatment of patients with type 2 diabetes.
Liraglutide is a fatty acid DPP-4–resistant human GLP-1 analogue administered as a once-daily therapeutic agent.171 Liraglutide reduces fasting and postprandial glycemia, inhibits gastric emptying, and reduces levels of circulating glucagon.172 Liraglutide has completed phase 3 clinical trials and is pending approval. Additional agents being evaluated in the clinic include a long-acting GLP-1 analogue suitable for once-weekly administration (taspoglutide) and a recombinant human albumin–GLP-1 protein (Albiglutide).
Oxyntomodulin is a 37 amino acid peptide that contains the sequence of glucagon together with an 8 amino acid carboxyterminal extension (see Fig. 9-1). Oxyntomodulin is capable of engaging both GLP-1 and glucagon receptors173; however, a separate oxyntomodulin receptor has not yet been identified. Exogenous oxyntomodulin stimulates insulin secretion, lowers blood glucose, reduces food intake, and promotes weight loss in preclinical studies. Moreover, short-term oxyntomodulin administration is associated with enhanced satiety, weight loss, and increased energy expenditure in human subjects.174,175 Hence there is active interest in evaluation of oxyntomodulin as a glucoregulatory and anorectic agent for the treatment of patients with T2DM and/or obesity.
Dipeptidyl Peptidase-4 Inhibitors
The challenges inherent in overcoming the rapid degradation of native GLP-1, together with the disadvantages and limitations of injectable peptides, have fostered complementary efforts at enhancing endogenous levels of native GLP-1 through the use of dipeptidyl peptidase-4 inhibitors.147,149 DPP-4 inhibitors preserve circulating levels of bioactive GLP-1 and GIP and exert significant glucose-lowering actions in rodents and human subjects. The DPP-4 gene is essential for incretin degradation and glucose homeostasis in mice, as genetic elimination of DPP-4 results in increased levels of intact plasma incretins, enhanced insulin secretion, and reduced glycemic excursion.176 The observation that DPP-4 inhibitors produced significant glucose-lowering effects in four 12-week human studies177 has fostered development of several highly selective orally available DPP-4 inhibitors suitable for once-daily administration. Sitagliptin, generally administered as a 100 mg once-daily tablet, is the first widely approved DPP-4 inhibitor148; it lowers blood glucose when used as monotherapy or as add-on therapy to other oral antidiabetic agents in a broad spectrum of patients with T2DM.178–180 The most compelling use of DPP-4 inhibitors in subjects with T2DM is seen in early initial combination therapy with metformin,181 as metformin appears to independently increase the circulating levels of GLP-1. DPP-4 inhibitors are well tolerated, are weight neutral, and are associated with a low risk for hypoglycemia. A second highly selective DPP-4 inhibitor, alogliptin, has also completed phase 3 clinical trials in subjects with T2DM. As DPP-4 regulates immune function and controls the degradation of a broad spectrum of endocrine and regulatory peptides, the long-term safety of DPP-4 inhibitors, which specifically target the kinase activity of the molecule, remains under scrutiny. Studies of the mechanism of action of DPP-4 inhibitors in vivo reveal that the glucoregulatory actions of DPP-4 inhibitors are abolished in mice with combined genetic disruption of both GIP and GLP-1 receptors.182 The overlapping and contrasting actions of GLP-1R agonists versus DPP-4 inhibitors are illustrated in Table 9-1.
Table 9-1
GLP-1 Receptor Agonists Versus DPP-4 Inhibitors
| GLP-1R Agonists | DPP-4 Inhibitors | |
| Administration | Injection | Orally available |
| GLP-1 concentrations | Pharmacological | Physiological |
| Mechanisms of action | GLP-1 | GLP-1 + GIP |
| Activation of portal glucose | ||
| Sensor | No | Yes |
| ↑ Insulin secretion | +++ | + |
| ↓ Glucagon secretion | ++ | ++ |
| Gastric emptying | Inhibited | +/− |
| Weight loss | Yes | No |
| Expansion of β-cell mass | ||
| In preclinical studies | Yes | Yes |
| Nausea and vomiting | Yes | No |
| Potential immunogenicity | Yes | No |
Adapted from Cell Metab 3(3):153-165, 2006.
Glucagon-Like Peptide-2
GLP-2 is co-secreted with GLP-1 from intestinal endocrine cells and is trophic to the mucosal epithelium in both the small and large intestine (see Fig. 9-2).183 The presence of an alanine residue at position 2 (see Fig. 9-1) predicted that GLP-2, like GLP-1, would be a substrate for inactivation by DPP-4, and significant amounts of biologically inactive GLP-2 (3-33) have been demonstrated in both rodent and human plasma.184 Similarly, analogues of GLP-2 designed to resist DPP-4–mediated cleavage are biologically more potent in vivo. Intravenous administration of GLP-2 stimulates intestinal glucose transport within 30 minutes, and a single injection of GLP-2 rapidly enhances barrier function in the mouse gut, demonstrating that GLP-2 exerts acute actions independent of bowel growth in the gastrointestinal tract.
The importance of GLP-2 for maintenance of the intestinal villous epithelium is illustrated by studies of rats receiving total parenteral nutrition (TPN), as co-infusion of GLP-2 and TPN prevented mucosal villous hypoplasia in the small intestine.185 The physiologic importance of GLP-2 for the physiology or growth of the normal mucosal epithelium has been examined in mice administered GLP-2 (3-33), a partial GLP-2 antagonist. Refeeding of fasted mice results in enhanced mucosal growth; however, coadministration of GLP-2 (3-33) significantly blunted the adaptive response to enteral nutrients.186 Similarly, treatment of diabetic rats with immunoneutralizing GLP-2 antisera significantly reduces adaptive mucosal growth in the small bowel.187
A G protein–linked GLP-2 receptor, related in sequence to the glucagon and GLP-1 receptors, has been isolated188 and has been localized to human enteroendocrine cells, myofibroblasts, and enteric neurons. GLP-2 promotes regeneration and prevents apoptosis in experimental models of intestinal resection and inflammation, including small bowel enteritis, chemotherapy-induced mucosal injury, and ischemic damage.189–191 Furthermore, the antiapoptotic actions of GLP-2 have been demonstrated in primary rat and mouse hippocampal cell cultures and in heterologous cells expressing a transfected GLP-2 receptor. Administration of native GLP-2 twice daily to human subjects with short bowel syndrome resulted in improved energy absorption, decreased fluid loss, and significant weight gain, in association with increased crypt plus villus height in mucosal biopsy specimens.192,193 A degradation-resistant stable human GLP-2 analogue, teduglutide, is currently being evaluated in clinical trials of patients with short bowel syndrome and inflammatory bowel disease.
References
1. Shin, YK, Martin, B, Golden, E, et al. Modulation of taste sensitivity by GLP-1 signaling. J Neurochem. 2008;106(1):455–463.
2. Wang, S, Hecksher-Sorensen, J, Xu, Y, et al. Myt1 and Ngn3 form a feed-forward expression loop to promote endocrine islet cell differentiation. Dev Biol. 2008;317(2):531–540.
3. Krapp, A, Knofler, M, Ledermann, B, et al. The bHLH protein PTF1-p48 is essential for the formation of the exocrine and the correct spatial organization of the endocrine pancreas. Genes Dev. 1998;12(23):3752–3763.
4. Dahl, U, Sjodin, A, Semb, H. Cadherins regulate aggregation of pancreatic beta-cells in vivo. Development. 1996;122(9):2895–2902.
5. Esni, F, Taljedal, IB, Perl, AK, et al. Neural cell adhesion molecule (N-CAM) is required for cell type segregation and normal ultrastructure in pancreatic islets. J Cell Biol. 1999;144(2):325–337.
6. Ait-Lounis, A, Baas, D, Barras, E, et al. Novel function of the ciliogenic transcription factor RFX3 in development of the endocrine pancreas. Diabetes. 2007;56(4):950–959.
7. Ahlgren, U, Pfaff, SL, Jessell, TM, et al. Independent requirement for ISL1 in formation of pancreatic mesenchyme and islet cells. Nature. 1997;385:257–260.
8. Collombat, P, Mansouri, A, Hecksher-Sorensen, J, et al. Opposing actions of Arx and Pax4 in endocrine pancreas development. Genes Dev. 2003;17(20):2591–2603.
9. Jonsson, J, Carlsson, L, Edlund, T, et al. Insulin-promoter-factor 1 is required for pancreas development in mice. Nature. 1994;371:606–609.
10. Sussel, L, Kalamaras, J, Hartigan-O’Connor, DJ, et al. Mice lacking the homeodomain transcription factor Nkx2.2 have diabetes due to arrested differentiation of pancreatic beta cells. Development. 1998;125:2213–2221.
11. Naya, FJ, Huang, H, Qiu, Y, et al. Diabetes, defective pancreatic morphogenesis, and abnormal enteroendocrine differentiation in BETA2/NeuroD-deficient mice. Genes Dev. 1997;11:2323–2334.
12. St-Onge, L, Sosa-Pineda, B, Chowdhury, K, et al. Pax6 is required for differentiation of glucagon-producing α-cells in mouse pancreas. Nature. 1997;387:406–409.
13. Sander, M, Neubuser, A, Kalamaras, J, et al. Genetic analysis reveals that PAX6 is required for normal transcription of pancreatic hormone genes and islet development. Genes Dev. 1997;11:1662–1673.
14. Sosa-Pineda, B, Chowdhury, K, Torres, M, et al. The Pax4 gene is essential for differentiation of insulin-producing β cells in the mammalian pancreas. Nature. 1997;386:399–402.
15. Guillam, MT, Hummler, E, Schaerer, E, et al. Early diabetes and abnormal postnatal pancreatic islet development in mice lacking Glut-2 [see comments] [published errata appear in Nat Genet 17(4):503, 1997 Dec, and 18(1):88, 1998 Jan]. Nat Genet. 1997;17(3):327–330.
16. Furuta, M, Yano, H, Zhou, A, et al. Defective prohormone processing and altered pancreatic islet morphology in mice lacking active SPC2. Proc Natl Acad Sci USA. 1999;94:6646–6651.
17. Gelling, RW, Du, XQ, Dichmann, DS, et al. Lower blood glucose, hyperglucagonemia, and pancreatic α cell hyperplasia in glucagon receptor knockout mice. Proc Natl Acad Sci U S A. 2003;100:1438–1443.
18. Wang, J, Webb, G, Cao, Y, et al. Contrasting patterns of expression of transcription factors in pancreatic alpha and beta cells. Proc Natl Acad Sci U S A. 2003;100(22):12660–12665.
19. Chiang, MK, Melton, DA. Single-cell transcript analysis of pancreas development. Dev Cell. 2003;4(3):383–393.
20. Lee, YC, Brubaker, PL, Drucker, DJ. Developmental and tissue-specific regulation of proglucagon gene expression. Endocrinology. 1990;127:2217–2222.
21. Wang, M, Drucker, DJ. The LIM domain homeobox gene isl-1 is a positive regulator of islet cell-specific proglucagon gene transcription. J Biol Chem. 1995;270:12646–12652.
22. Jin, T, Drucker, DJ. Activation of proglucagon gene transcription through a novel promoter element by the caudal-related homeodomain protein cdx-2/3. Mol Cell Biol. 1996;16:19–28.
23. Laser, B, Meda, P, Constant, I, et al. The caudal-related homeodomain protein Cdx-2/3 regulates glucagon gene expression in islet cells. J Biol Chem. 1996;271:28984–28994.
24. Hussain, MA, Lee, J, Miller, CP, et al. POU domain transcription factor brain 4 confers pancreatic a-cell-specific expression of the proglucagon gene through interaction with a novel proximal promoter G1 element. Mol Cell Biol. 1997;17:7186–7194.
25. Gosmain, Y, Avril, I, Mamin, A, et al. Pax-6 and c-Maf functionally interact with the alpha-cell-specific DNA element G1 in vivo to promote glucagon gene expression. J Biol Chem. 2007;282(48):35024–35034.
26. Jin, T, Trinh, DKY, Wang, F, et al. The caudal homeobox protein cdx-2/3 activates endogenous proglucagon gene expression in InR1-G9 islet cells. Mol Endocrinol. 1997;11:203–209.
27. Dumonteil, E, Laser, B, Constant, I, et al. Differential regulation of the glucagon and insulin I gene promoters by the basic helix-loop-helix transcription factors E47 and BETA2. J Biol Chem. 1998;273:19945–19954.
28. Knepel, W, Vallejo, M, Chafitz, JA, et al. The pancreatic islet-specific glucagon G3 transcription factors recognize control elements in the rat somatostatin and insulin-I genes. Mol Endocrinol. 1991;5:1457–1466.
29. Wrege, A, Diedrich, T, Hochhuth, C, et al. Transcriptional activity of domain A of the rat glucagon G3 element conferred by an islet-specific nuclear protein that also binds to similar pancreatic islet cell-specific enhancer sequences (PISCES). Gene Expression. 1995;4:205–216.
30. Philippe, J. Glucagon gene transcription is negatively regulated by insulin in a hamster islet cell line. J Clin Invest. 1989;84:672–677.
31. Philippe, J, Morel, C, Cordier-Bussat, M. Islet-specific proteins interact with the insulin-response element of the glucagon gene. J Biol Chem. 1995;270:3039–3045.
32. Philippe, J. Insulin regulation of the glucagon gene is mediated by an insulin-responsive DNA element. Proc Natl Acad Sci USA. 1991;88:7224–7227.
33. Ritz-Laser, B, Estreicher, A, Klages, N, et al. Pax-6 and Cdx-2/3 interact to activate glucagon gene expression on the G1 control element. J Biol Chem. 1999;274:4124–4132.
34. Hill, ME, Asa, SL, Drucker, DJ. Essential requirement for Pax6 in control of enteroendocrine proglucagon gene transcription. Mol Endocrinol. 1999;13:1474–1486.
35. Philippe, J, Morel, C, Prezioso, VR. Glucagon gene expression is negatively regulated by hepatocyte nuclear factor 3β. Mol Cell Biol. 1994;14:3514–3523.
36. Philippe, J. Hepatocyte-nuclear factor 3β gene transcripts generate protein isoforms with different transactivation properties on the glucagon gene. Mol Endocrinol. 1995;9:368–374.
37. Kaestner, KH, Katz, J, Liu, Y, et al. Inactivation of the winged helix transcription factor HNF3α affects glucose homeostasis and islet glucagon gene expression in vivo. Genes Dev. 1999;13:495–504.
38. Kaestner, KH, Hiemisch, H, Schutz, G. Targeted disruption of the gene encoding hepatocyte nuclear factor 3 gamma results in reduced transcription of hepatocyte-specific genes. Mol Cell Biol. 1998;18(7):4245–4251.
39. Liu, Y, Shen, W, Brubaker, PL, et al. Foxa3 (HNF-3gamma) binds to and activates the rat proglucagon gene promoter but is not essential for proglucagon gene expression. Biochem J. 2002;366(Pt 2):633–641.
40. Ang, SL, Rossant, J. HNF-3 beta is essential for node and notochord formation in mouse development. Cell. 1994;78:561–574.
41. Weinstein, DC, Ruiz, I, Altaba, A, et al. The winged-helix transcription factor HNF-3 beta is required for notochord development in the mouse embryo. Cell. 1994;78:575–588.
42. Drucker, DJ, Jin, T, Asa, SL, et al. Activation of proglucagon gene transcription by protein kinase A in a novel mouse enteroendocrine cell line. Mol Endocrinol. 1994;8:1646–1655.
43. Knepel, W, Chafitz, J, Habener, JF. Transcriptional activation of the rat glucagon gene by the cyclic AMP-responsive element in pancreatic islet cells. Mol Cell Biol. 1990;10:6799–6804.
44. Wang, J, Cao, Y, Steiner, DF. Regulation of proglucagon transcription by activated transcription factor (ATF) 3 and a novel isoform, ATF3b, through the cAMP-response element/ATF site of the proglucagon gene promoter. J Biol Chem. 2003;278(35):32899–32904.
45. Schwaninger, M, Lux, G, Blume, R, et al. Membrane depolarization and calcium influx induce glucagon gene transcription in pancreatic islet cells through the cyclic AMP-responsive element. J Biol Chem. 1993;268:5168–5177.
46. Furstenau, U, Schwaninger, M, Blumes, R, et al. Characterization of a novel calcium response element in the glucagon gene. J Biol Chem. 1999;274:5851–5860.
47. Efrat, S, Teitelman, G, Anwar, M, et al. Glucagon gene regulatory region directs oncoprotein expression to neurons and pancreatic alpha cells. Neuron. 1988;1:605–613.
48. Lee, YC, Asa, SL, Drucker, DJ. Glucagon gene 5′-flanking sequences direct expression of SV40 large T antigen to the intestine producing carcinoma of the large bowel in transgenic mice. J Biol Chem. 1992;267:10705–10708.
49. McKinnon, CM, Ravier, MA, Rutter, GA. FoxO1 is required for the regulation of preproglucagon gene expression by insulin in pancreatic alphaTC1-9 cells. J Biol Chem. 2006;281(51):39358–39369.
50. Nian, M, Drucker, DJ, Irwin, D. Divergent regulation of human and rat proglucagon gene promoters in vivo. Am J Physiol. 1999;277:G829–G837.
51. Dumonteil, E, Magnan, C, Ritz-Laser, B, et al. Insulin, but not glucose lowering corrects the hyperglucagonemia and increased proglucagon messenger ribonucleic acid levels observed in insulinopenic diabetes. Endocrinology. 1998;139:4540–4546.
52. Yi, F, Sun, J, Lim, GE, et al. Cross talk between the insulin and Wnt signaling pathways: evidence from intestinal endocrine L cells. Endocrinology. 2008;149(5):2341–2351.
53. Goncz, E, Strowski, MZ, Grotzinger, C, et al. Orexin-A inhibits glucagon secretion and gene expression through a Foxo1-dependent pathway. Endocrinology. 2008;149(4):1618–1626.
54. Chen, L, Komiyo, I, Inman, L, et al. Effects of hypoglycemia and prolonged fasting on insulin and glucagon gene expression. J Clin Invest. 1989;84:711–714.
55. Shi, ZQ, Rastogi, KS, Lekas, M, et al. Glucagon response to hypoglycemia is improved by insulin-independent restoration of normoglycemia in diabetic rats. Endocrinology. 1996;137:3193–3199.
56. Dhanvantari, S, Seidah, NG, Brubaker, PL. Role of prohormone convertases in the tissue-specific processing of proglucagon. Mol Endocrinol. 1996;10:342–355.
57. Rothenberg, ME, Eilertson, CD, Klein, K, et al. Processing of mouse proglucagon by recombinant prohormone convertase 1 and immunopurified prohormone convertase 2 in vitro. J Biol Chem. 1995;270:10136–10146.
58. Zhu, X, Zhou, A, Dey, A, et al. Disruption of PC1/3 expression in mice causes dwarfism and multiple neuroendocrine peptide processing defects. Proc Natl Acad Sci U S A. 2002;99:10293–10298.
59. Jackson, RS, Creemers, JW, Farooqi, IS, et al. Small-intestinal dysfunction accompanies the complex endocrinopathy of human proprotein convertase 1 deficiency. J Clin Invest. 2003;112(10):1550–1560.
60. Ward, WK, Bolgiano, DC, McKnight, B, et al. Diminished B cell secretory capacity in patients with noninsulin-dependent diabetes mellitus. J Clin Invest. 1984;74:1318–1328.
61. Rorsman, P, Ashcroft, FM, Berggren, P-O. Regulation of glucagon release from pancreatic A-cells. Biochem Pharmacol. 1991;41:1783–1790.
62. Singh, V, Grotzinger, C, Nowak, KW, et al. Somatostatin receptor subtype-2-deficient mice with diet-induced obesity have hyperglycemia, nonfasting hyperglucagonemia, and decreased hepatic glycogen deposition. Endocrinology. 2007;148(8):3887–3899.
63. Smismans, A, Schuit, F, Pipeleers, D. Nutrient regulation of gamma-aminobutyric acid release from islet beta cells. Diabetologia. 1997;40:1411–1415.
64. Gaskins, HR, Baldeon, ME, Selassie, L, et al. Glucose modulates gamma-aminobutyric acid release from the pancreatic beta TC6 cell line. J Biol Chem. 1995;270:30286–30289.
65. Ishihara, H, Maechler, P, Gjinovci, A, et al. Islet beta-cell secretion determines glucagon release from neighbouring alpha-cells. Nat Cell Biol. 2003;5(4):330–335.
66. Maruyama, H, Hisatomi, A, Orci, L, et al. Insulin within islets is a physiologic glucagon release inhibitor. J Clin Invest. 1984;74:2296–2299.
67. Zhou, H, Zhang, T, Harmon, JS, et al. Zinc, not insulin, regulates the rat alpha-cell response to hypoglycemia in vivo. Diabetes. 2007;56(4):1107–1112.
68. Marliss, EB, Aoki, TT, Unger, RH, et al. Glucagon levels and metabolic effects in fasting man. J Clin Invest. 1970;49(12):2256–2270.
69. Unger, RH, Aguilar-Parada, E, Muller, WA, et al. Studies of pancreatic alpha cell function in normal and diabetic subjects. J Clin Invest. 1970;49(4):837–848.
70. Larsson, H, Ahren, B. Glucose-dependent arginine stimulation test for characterization of islet function: studies on reproducibility and priming effect of arginine. Diabetologia. 1998;41(7):772–777.
71. Galbo, H, Holst, JJ, Christensen, NJ. Glucagon and plasma catecholamine responses to graded and prolonged exercise in man. J Appl Physiol. 1975;38(1):70–76.
72. Sotsky, MJ, Shilo, S, Shamoon, H. Regulation of counterregulatory hormone secretion in man during exercise and hypoglycemia. J Clin Endocrinol Metab. 1989;68(1):9–16.
73. Drouin, R, Lavoie, C, Bourque, J, et al. Increased hepatic glucose production response to glucagon in trained subjects. Am J Physiol. 1998;274(1 Pt 1):E23–E28.
74. Hirsch, IB, Marker, JC, Smith, LJ, et al. Insulin and glucagon in prevention of hypoglycemia during exercise in humans. Am J Physiol. 1991;260(5 Pt 1):E695–E704.
75. Coggan, AR, Raguso, CA, Gastaldelli, A, et al. Regulation of glucose production during exercise at 80% of VO2peak in untrained humans. Am J Physiol. 1997;273(2 Pt 1):E348–E354.
76. Lewis, GF, Vranic, M, Giacca, A. Glucagon enhances the direct suppressive effect of insulin on hepatic glucose production in humans. Am J Physiol. 1997;272(3 Pt 1):E371–E378.
77. Taminato, T, Seino, Y, Goto, Y, et al. Synthetic gastric inhibitory polypeptide. Stimulatory effect on insulin and glucagon secretion in the rat. Diabetes. 1977;26(5):480–484.
78. Rossetti, L, Shulman, GI, Zawalich, WS. Physiological role of cholecystokinin in meal-induced insulin secretion in conscious rats. Studies with L 364718, a specific inhibitor of CCK- receptor binding. Diabetes. 1987;36(10):1212–1215.
79. Wettergren, A, Schjoldager, B, Mortensen, PE, et al. Truncated GLP-1 (proglucagon 78-107-amide) inhibits gastric and pancreatic functions in man. Dig Dis Sci. 1993;38:665–673.
80. Komatsu, R, Matsuyama, T, Namba, M, et al. Glucagonostatic and insulinotropic action of glucagonlike peptide I-(7-36)-amide. Diabetes. 1989;38:902–905.
81. Taborsky, GJ, Jr., Ahren, B, Havel, PJ. Autonomic mediation of glucagon secretion during hypoglycemia. Diabetes. 1998;47:995–1005.
82. Herman, WH, Fajans, SS, Smith, MJ, et al. Diminished insulin and glucagon secretory responses to arginine in nondiabetic subjects with a mutation in the hepatocyte nuclear factor-4a/MODY1 gene. Diabetes. 1997;46:1749–1754.
83. Schwartz, NS, Clutter, WE, Shah, SD, et al. Glycemic thresholds for activation of glucose counterregulatory systems are higher than the threshold for symptoms. J Clin Invest. 1987;79(3):777–781.
84. Heimberg, H, De Vos, A, Pipeleers, D, et al. Differences in glucose transporter gene expression between rat pancreatic alpha- and beta-cells are correlated to differences in glucose transport but not in glucose utilization. J Biol Chem. 1995;270(15):8971–8975.
85. Heimberg, H, De Vos, A, Moens, K, et al. The glucose sensor protein glucokinase is expressed in glucagon- producing alpha-cells. Proc Natl Acad Sci U S A. 1996;93(14):7036–7041.
86. Cabrera, O, Jacques-Silva, MC, Speier, S, et al. Glutamate is a positive autocrine signal for glucagon release. Cell Metab. 2008;7(6):545–554.
87. Tong, Q, Ye, C, McCrimmon, RJ, et al. Synaptic glutamate release by ventromedial hypothalamic neurons is part of the neurocircuitry that prevents hypoglycemia. Cell Metab. 2007;5(5):383–393.
88. Boushey, RP, Abadir, A, Flamez, D, et al. Hypoglycemia, defective islet glucagon secretion, but normal islet mass in mice with a disruption of the gastrin gene. Gastroenterology. 2003;125(4):1164–1174.
89. Marliss, EB, Girardier, L, Seydoux, J, et al. Glucagon release induced by pancreatic nerve stimulation in the dog. J Clin Invest. 1973;52(5):1246–1259.
90. Ahren, B, Veith, RC, Paquette, TL, et al. Sympathetic nerve stimulation versus pancreatic norepinephrine infusion in the dog: 2). Effects on basal release of somatostatin and pancreatic polypeptide. Endocrinology. 1987;121(1):332–339.
91. Havel, PJ, Ahren, B. Activation of autonomic nerves and the adrenal medulla contributes to increased glucagon secretion during moderate insulin-induced hypoglycemia in women. Diabetes. 1997;46(5):801–807.
92. Kendall, DM, Rooney, DP, Smets, YF, et al. Pancreas transplantation restores epinephrine response and symptom recognition during hypoglycemia in patients with long-standing type I diabetes and autonomic neuropathy. Diabetes. 1997;46(2):249–257.
93. Miki, T, Liss, B, Minami, K, et al. ATP-sensitive K+ channels in the hypothalamus are essential for the maintenance of glucose homeostasis. Nat Neurosci. 2001;4(5):507–512.
94. Hevener, AL, Bergman, RN, Donovan, CM. Novel glucosensor for hypoglycemic detection localized to the portal vein. Diabetes. 1997;46(9):1521–1525.
95. Fery, F, Plat, L, van de Borne, P, et al. Impaired counterregulation of glucose in a patient with hypothalamic sarcoidosis. N Engl J Med. 1999;340(11):852–856.
96. Brown, RJ, Sinaii, N, Rother, KI. Too much glucagon, too little insulin: time course of pancreatic islet dysfunction in new-onset type 1 diabetes. Diabetes Care. 2008;31(7):1403–1404.
97. Raskin, P, Unger, RH. Hyperglucagonemia and its suppression. Importance in the metabolic control of diabetes. N Engl J Med. 1978;299(9):433–436.
98. Liang, Y, Osborne, MC, Monia, BP, et al. Reduction in glucagon receptor expression by an antisense oligonucleotide ameliorates diabetic syndrome in db/db mice. Diabetes. 2004;53(2):410–417.
99. Sloop, KW, Cao, JX, Siesky, AM, et al. Hepatic and glucagon-like peptide-1-mediated reversal of diabetes by glucagon receptor antisense oligonucleotide inhibitors. J Clin Invest. 2004;113(11):1571–1581.
100. Sherwin, RS, Fisher, M, Hendler, R, et al. Hyperglucagonemia and blood glucose regulation in normal, obese and diabetic subjects. N Engl J Med. 1976;294(9):455–461.
101. Mittelman, SD, Fu, YY, Rebrin, K, et al. Indirect effect of insulin to suppress endogenous glucose production is dominant, even with hyperglucagonemia. J Clin Invest. 1997;100(12):3121–3130.
102. Charlton, MR, Nair, KS. Role of hyperglucagonemia in catabolism associated with type 1 diabetes: effects on leucine metabolism and the resting metabolic rate. Diabetes. 1998;47(11):1748–1756.
103. Gerich, JE. Lilly lecture 1988. Glucose counterregulation and its impact on diabetes mellitus. Diabetes. 1988;37(12):1608–1617.
104. Gerich, J, Langlois, M, Noacco, C, et al. Lack of glucagon response to hypoglycemia in diabetes: evidence for an intrinsic pancreatic alpha cell defect. Science. 1973;182:171–173.
105. Banarer, S, McGregor, VP, Cryer, PE. Intraislet hyperinsulinemia prevents the glucagon response to hypoglycemia despite an intact autonomic response. Diabetes. 2002;51(4):958–965.
106. Fanelli, CG, Epifano, L, Rambotti, AM, et al. Meticulous prevention of hypoglycemia normalizes the glycemic thresholds and magnitude of most of neuroendocrine responses to, symptoms of, and cognitive function during hypoglycemia in intensively treated patients with short-term IDDM. Diabetes. 1993;42(11):1683–1689.
107. Dagogo-Jack, S, Rattarasarn, C, Cryer, PE. Reversal of hypoglycemia unawareness, but not defective glucose counterregulation, in IDDM. Diabetes. 1994;43(12):1426–1434.
108. Galassetti, P, Tate, D, Neill, RA, et al. Effect of antecedent hypoglycemia on counterregulatory responses to subsequent euglycemic exercise in type 1 diabetes. Diabetes. 2003;52(7):1761–1769.
109. Heller, SR, Cryer, PE. Reduced neuroendocrine and symptomatic responses to subsequent hypoglycemia after 1 episode of hypoglycemia in nondiabetic humans. Diabetes. 1991;40(2):223–226.
110. Bollen, M, Keppens, S, Stalmans, W. Specific features of glycogen metabolism in the liver. Biochem J. 1998;336(Pt 1):19–31.
111. Burcelin, R, Katz, EB, Charron, MJ. Molecular and cellular aspects of the glucagon receptor:role in diabetes and metabolism. Diabetes & Metabolism. 1996;22:373–396.
112. Herzig, S, Long, F, Jhala, US, et al. CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature. 2001;413(6852):179–183.
113. Sato, N, Irie, M, Kajinuma, H, et al. Glucagon inhibits insulin activation of glucose transport in rat adipocytes mainly through a postbinding process. Endocrinology. 1990;127(3):1072–1077.
114. Chen, SS, Zhang, Y, Santomango, TS, et al. Glucagon chronically impairs hepatic and muscle glucose disposal. Am J Physiol Endocrinol Metab. 2007;292(3):E928–935.
115. Gonzalez-Munoz, C, Nieto-Ceron, S, Cabezas-Herrera, J, et al. Glucagon increases contractility in ventricle but not in atrium of the rat heart. Eur J Pharmacol. 2008;587(1–3):243–247.
116. Briffeuil, P, Thu, TH, Kolanowski, J. A lack of direct action of glucagon on kidney metabolism, hemodynamics, and renal sodium handling in the dog. Metabolism. 1996;45(3):383–388.
117. Stumvoll, M, Chintalapudi, U, Perriello, G, et al. Uptake and release of glucose by the human kidney. Postabsorptive rates and responses to epinephrine. J Clin Invest. 1995;96(5):2528–2533.
118. Stumvoll, M, Meyer, C, Kreider, M, et al. Effects of glucagon on renal and hepatic glutamine gluconeogenesis in normal postabsorptive humans. Metabolism. 1998;47(10):1227–1232.
119. Jelinek, LJ, Lok, S, Rosenberg, GB, et al. Expression cloning and signaling properties of the rat glucagon receptor. Science. 1993;259(5101):1614–1616.
120. Hjorth, SA, Orskov, C, Schwartz, TW. Constitutive activity of glucagon receptor mutants. Mol Endocrinol. 1998;12:78–86.
121. Hansen, LH, Abrahamsen, N, Nishimura, E. Glucagon receptor mRNA distribution in rat tissues. Peptides. 1995;16:1163–1166.
122. Drucker, DJ, Asa, S. Glucagon gene expression in vertebrate brain. J Biol Chem. 1988;263:13475–13478.
123. Larsen, PJ, Tang-Christensen, M, Holst, JJ, et al. Distribution of glucagon-like peptide-1 and other preproglucagon-derived peptides in the rat hypothalamus and brainstem. Neuroscience. 1997;77:257–270.
124. Moens, K, Heimberg, H, Flamez, D, et al. Expression and functional activity of glucagon, glucagon-like peptide 1 and glucose-dependent insulinotropic peptide receptors in rat pancreatic islet cells. Diabetes. 1996;45:257–261.
125. Parker, JC, Andrews, KM, Allen, MR, et al. Glycemic control in mice with targeted disruption of the glucagon receptor gene. Biochem Biophys Res Commun. 2002;290(2):839–843.
126. Dobbins, RL, Davis, SN, Neal, DW, et al. Compartmental modeling of glucagon kinetics in the conscious dog. Metabolism. 1995;44:452–459.
127. Authier, F, Mort, JS, Bell, AW, et al. Proteolysis of glucagon within hepatic endosomes by membrane-associated cathepsins B and D. J Biol Chem. 1995;270:15798–15807.
128. Hupe-Sodmann, K, McGregor, GP, Bridenbaugh, R, et al. Characterisation of the processing by human neutral endopeptidase 24.11 of GLP-1(7-36) amide and comparison of the substrate specificity of the enzyme for other glucagon-like peptides. Regul Pept. 1995;58:149–156.
129. Hvidberg, A, Djurup, R, Hilsted, J. Glucose recovery after intranasal glucagon during hypoglycaemia in man. Eur J Clin Pharmacol. 1994;46(1):15–17.
130. Muhlhauser, I, Koch, J, Berger, M. Pharmacokinetics and bioavailability of injected glucagon: differences between intramuscular, subcutaneous, and intravenous administration. Diabetes Care. 1985;8(1):39–42.
131. Melanson, SW, Bonfante, G, Heller, MB. Nebulized glucagon in the treatment of bronchospasm in asthmatic patients. Am J Emerg Med. 1998;16(3):272–275.
132. Love, JN, Sachdeva, DK, Bessman, ES, et al. A potential role for glucagon in the treatment of drug-induced symptomatic bradycardia. Chest. 1998;114(1):323–326.
133. Wermers, RA, Fatourechi, V, Kvols, LK. Clinical spectrum of hyperglucagonemia associated with malignant neuroendocrine tumors. Mayo Clin Proc. 1996;71:1030–1038.
134. Mallinson, CN, Bloom, SR, Warin, AP, et al. A glucagonoma syndrome. The Lancet. 1974:1–5. [Saturday, 6 July].
135. Drucker, DJ. Intestinal growth factors. Am J Physiol. 1997;273:G3–G6.
136. Blume, N, Skouv, J, Larsson, LI, et al. Potent inhibitory effects of transplantable rat glucagonomas and insulinomas on the respective endogenous islet cells are associated with pancreatic apoptosis. J Clin Invest. 1995;96:2227–2235.
137. Ehrlich, P, Tucker, D, Asa, SL, et al. Inhibition of pancreatic proglucagon gene expression in mice bearing subcutaneous endocrine tumors. Am J Physiol Endocrinol Metab. 1994;267:E662–E671.
138. Hoyt, EC, Lund, PK, Winesett, DE, et al. Effects of fasting, refeeding and intraluminal triglyceride on proglucagon expression in jejunum and ileum. Diabetes. 1996;45:434–439.
139. Roberge, JN, Brubaker, PL. Secretion of proglucagon-derived peptides in response to intestinal luminal nutrients. Endocrinology. 1991;128:3169–3174.
140. Roberge, JN, Brubaker, PL. Regulation of intestinal proglucagon-derived peptide secretion by glucose-dependent insulinotropic peptide in a novel enteroendocrine loop. Endocrinology. 1993;133:233–240.
141. Kieffer, TJ, McIntosh, CH, Pederson, RA. Degradation of glucose-dependent insulinotropic polypeptide and truncated glucagon-like peptide 1 in vitro and in vivo by dipeptidyl peptidase IV. Endocrinology. 1995;136(8):3585–3596.
142. Vahl, TP, Paty, BW, Fuller, BD, et al. Effects of GLP-1-(7-36)NH(2), GLP-1-(7-37), and GLP-1- (9-36)NH(2) on intravenous glucose tolerance and glucose-induced insulin secretion in healthy humans. J Clin Endocrinol Metab. 2003;88(4):1772–1779.
143. Meier, JJ, Gethmann, A, Nauck, MA, et al. The glucagon-like peptide 1 metabolite GLP-1 (9-36) amide reduces postprandial glycemia independently of gastric emptying and insulin secretion in humans. Am J Physiol Endocrinol Metab. 2006;290(6):E1118–E1123.
144. Nikolaidis, LA, Elahi, D, Shen, YT, et al. Active metabolite of GLP-1 mediates myocardial glucose uptake and improves left ventricular performance in conscious dogs with dilated cardiomyopathy. Am J Physiol Heart Circ Physiol. 2005;289(6):H2401–2408.
145. Ban, K, Noyan-Ashraf, MH, Hoefer, J, et al. Cardioprotective and vasodilatory actions of glucagon-like peptide 1 receptor are mediated through both glucagon-like peptide 1 receptor-dependent and -independent pathways. Circulation. 2008;117(18):2340–2350.
146. Elahi, D, Egan, JM, Shannon, RP, et al. GLP-1 (9-36) Amide, Cleavage Product of GLP-1 (7-36) amide, is a glucoregulatory peptide. Obesity (Silver Spring). 2008;16(7):1501–1509.
147. Drucker, DJ, Nauck, MA. The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet. 2006;368(9548):1696–1705.
148. Drucker, D, Easley, C, Kirkpatrick, P. Sitagliptin. Nature Reviews Drug Discovery. 2007;6(2):109–110.
149. Drucker, DJ. Dipeptidyl peptidase-4 inhibition and the treatment of type 2 diabetes: preclinical biology and mechanisms of action. Diabetes Care. 2007;30(6):1335–1343.
150. Drucker, DJ. Glucagon-like peptides: regulators of cell proliferation, differentiation, and apoptosis. Mol Endocrinol. 2003;17(2):161–171.
151. Li, Y, Hansotia, T, Yusta, B, et al. Glucagon-like peptide-1 receptor signaling modulates beta cell apoptosis. J Biol Chem. 2003;278:471–478.
152. Farilla, L, Bulotta, A, Hirshberg, B, et al. Glucagon-like peptide 1 inhibits cell apoptosis and improves glucose responsiveness of freshly isolated human islets. Endocrinology. 2003;144(12):5149–5158.
153. Froud, T, Faradji, RN, Pileggi, A, et al. The use of exenatide in islet transplant recipients with chronic allograft dysfunction: safety, efficacy, and metabolic effects. Transplantation. 2008;86(1):36–45.
154. Flint, A, Raben, A, Astrup, A, et al. Glucagon-like peptide 1 promotes satiety and suppresses energy intake in humans. J Clin Invest. 1998;101:515–520.
155. Thorens, B, Porret, A, Bÿhler, L, et al. Cloning and functional expression of the human islet GLP-1 receptor: demonstration that exendin-4 is an agonist and exendin-(9-39) an antagonist of the receptor. Diabetes. 1993;42:1678–1682.
156. Wang, Z, Wang, RM, Owji, AA, et al. Glucagon-like peptide 1 is a physiological incretin in rat. J Clin Invest. 1995;95:417–421.
157. Edwards, CM, Todd, JF, Mahmoudi, M, et al. Glucagon-like peptide 1 has a physiological role in the control of postprandial glucose in humans: studies with the antagonist exendin 9-39. Diabetes. 1999;48(1):86–93.
158. D’alessio, DA, Vogel, R, Prigeon, R, et al. Elimination of the action of glucagon-like peptide 1 causes an impairment of glucose tolerance after nutrient ingestion by healthy baboons. J Clin Invest. 1996;97:133–138.
159. Scrocchi, LA, Brown, TJ, MacLusky, N, et al. Glucose intolerance but normal satiety in mice with a null mutation in the glucagon-like peptide receptor gene. Nature Med. 1996;2:1254–1258.
160. Zander, M, Madsbad, S, Madsen, JL, et al. Effect of 6-week course of glucagon-like peptide 1 on glycaemic control, insulin sensitivity, and beta-cell function in type 2 diabetes: a parallel-group study. Lancet. 2002;359(9309):824–830.
161. Deacon, CF, Johnsen, AH, Holst, JJ. Degradation of glucagon-like peptide-1 by human plasma in vitro yields an N-terminally truncated peptide that is a major endogenous metabolite in vivo. J Clin Endocrinol Metab. 1995;80:952–957.
162. Drucker, DJ. Enhancing incretin action for the treatment of type 2 diabetes. Diabetes Care. 2003;26(10):2929–2940.
163. Eng, J, Kleinman, WA, Singh, L, et al. Isolation and characterization of exendin-4, an exendin-3 analogue, from Heloderma suspectum venom. Further evidence for an exendin receptor on dispersed acini from guinea pig pancreas. J Biol Chem. 1992;267:7402–7405.
164. Chen, YE, Drucker, DJ. Tissue-specific expression of unique mRNAs that encode proglucagon-derived peptides or exendin 4 in the lizard. J Biol Chem. 1997;272:4108–4115.
165. Buse, JB, Henry, RR, Han, J, et al. Effects of exenatide (exendin-4) on glycemic control over 30 weeks in sulfonylurea-treated patients with type 2 diabetes. Diabetes Care. 2004;27(11):2628–2635.
166. DeFronzo, RA, Ratner, RE, Han, J, et al. Effects of exenatide (exendin-4) on glycemic control and weight over 30 weeks in metformin-treated patients with type 2 diabetes. Diabetes Care. 2005;28(5):1092–1100.
167. Kendall, DM, Riddle, MC, Rosenstock, J, et al. Effects of exenatide (exendin-4) on glycemic control over 30 weeks in patients with type 2 diabetes treated with metformin and a sulfonylurea. Diabetes Care. 2005;28(5):1083–1091.
168. Zinman, B, Hoogwerf, BJ, Duran Garcia, S, et al. The effect of adding exenatide to a thiazolidinedione in suboptimally controlled type 2 diabetes: a randomized trial. Ann Intern Med. 2007;146(7):477–485.
169. Heine, RJ, Van Gaal, LF, Johns, D, et al. Exenatide versus insulin glargine in patients with suboptimally controlled type 2 diabetes: a randomized trial. Ann Intern Med. 2005;143(8):559–569.
170. Nauck, MA, Duran, S, Kim, D, et al. A comparison of twice-daily exenatide and biphasic insulin aspart in patients with type 2 diabetes who were suboptimally controlled with sulfonylurea and metformin: a non-inferiority study. Diabetologia. 2007;50(2):259–267.
171. Vilsboll, T, Zdravkovic, M, Le-Thi, T, et al. Liraglutide, a long-acting human GLP-1 Analog, given as monotherapy significantly improves glycemic control and lowers body weight without risk of hypoglycemia in patients with type 2 diabetes mellitus. Diabetes Care. 2007;6:1608–1610.
172. Juhl, CB, Hollingdal, M, Sturis, J, et al. Bedtime administration of NN2211, a long-acting GLP-1 derivative, substantially reduces fasting and postprandial glycemia in type 2 diabetes. Diabetes. 2002;51(2):424–429.
173. Baggio, LL, Huang, Q, Brown, TJ, et al. Oxyntomodulin and glucagon-like peptide-1 differentially regulate murine food intake and energy expenditure. Gastroenterology. 2004;127(2):546–558.
174. Wynne, K, Park, AJ, Small, CJ, et al. Subcutaneous oxyntomodulin reduces body weight in overweight and obese subjects: a double-blind, randomized, controlled trial. Diabetes. 2005;54(8):2390–2395.
175. Wynne, K, Park, AJ, Small, CJ, et al. Oxyntomodulin increases energy expenditure in addition to decreasing energy intake in overweight and obese humans: a randomised controlled trial. Int J Obes (Lond). 2006;30:68–72.
176. Marguet, D, Baggio, L, Kobayashi, T, et al. Enhanced insulin secretion and improved glucose tolerance in mice lacking CD26. Proc Natl Acad Sci U S A. 2000;97(12):6874–6879.
177. Ahren, B, Simonsson, E, Larsson, H, et al. Inhibition of dipeptidyl peptidase IV improves metabolic control over a 4-week study period in type 2 diabetes. Diabetes Care. 2002;25(5):869–875.
178. Aschner, P, Kipnes, MS, Lunceford, JK, et al. Effect of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy on glycemic control in patients with type 2 diabetes. Diabetes Care. 2006;29(12):2632–2637.
179. Charbonnel, B, Karasik, A, Liu, J, et al. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin added to ongoing metformin therapy in patients with type 2 diabetes inadequately controlled with metformin alone. Diabetes Care. 2006;29(12):2638–2643.
180. Rosenstock, J, Brazg, R, Andryuk, PJ, et al. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin added to ongoing pioglitazone therapy in patients with type 2 diabetes: a 24-week, multicenter, randomized, double-blind, placebo-controlled, parallel-group study. Clin Ther. 2006;28(10):1556–1568.
181. Goldstein, BJ, Feinglos, MN, Lunceford, JK, et al. Effect of initial combination therapy with sitagliptin, a dipeptidyl peptidase-4 inhibitor, and metformin on glycemic control in patients with type 2 diabetes. Diabetes Care. 2008;31(8):1713.
182. Flock, G, Baggio, LL, Longuet, C, et al. Incretin receptors for glucagon-like peptide 1 and glucose-dependent insulinotropic polypeptide are essential for the sustained metabolic actions of vildagliptin in mice. Diabetes. 2007;56(12):3006–3013.
183. Drucker, DJ, Ehrlich, P, Asa, SL, et al. Induction of intestinal epithelial proliferation by glucagon-like peptide 2. Proc Natl Acad Sci USA. 1996;93:7911–7916.
184. Drucker, DJ, Shi, Q, Crivici, A, et al. Regulation of the biological activity of glucagon-like peptide 2 in vivo by dipeptidyl peptidase IV. Nat Biotechnol. 1997;15(7):673–677.
185. Chance, WT, Foley-Nelson, T, Thomas, I, et al. Prevention of parenteral nutrition-induced gut hypoplasia by coinfusion of glucagon-like peptide-2. Am J Physiol. 1997;273:G559–G563.
186. Shin, ED, Estall, JL, Izzo, A, et al. Mucosal adaptation to enteral nutrients is dependent on the physiologic actions of glucagon-like peptide-2 in mice. Gastroenterology. 2005;128(5):1340–1353.
187. Hartmann, B, Thulesen, J, Hare, KJ, et al. Immunoneutralization of endogenous glucagon-like peptide-2 reduces adaptive intestinal growth in diabetic rats. Regul Pept. 2002;105(3):173–179.
188. Munroe, DG, Gupta, AK, Kooshesh, F, et al. Prototypic G protein-coupled receptor for the intestinotrophic factor glucagon-like peptide 2. Proc Natl Acad Sci U S A. 1999;96(4):1569–1573.
189. Boushey, RP, Yusta, B, Drucker, DJ. Glucagon-like peptide 2 decreases mortality and reduces the severity of indomethacin-induced murine enteritis. Am J Physiol. 1999;277:E937–E947.
190. Boushey, RP, Yusta, B, Drucker, DJ. Glucagon-like peptide (GLP)-2 reduces chemotherapy-associated mortality and enhances cell survival in cells expressing a transfected GLP-2 receptor. Cancer Res. 2001;61(2):687–693.
191. Prasad, R, Alavi, K, Schwartz, MZ. GLP-2alpha accelerates recovery of mucosal absorptive function after intestinal ischemia/reperfusion. J Pediatr Surg. 2001;36(4):570–572.
192. Jeppesen, PB, Hartmann, B, Thulesen, J, et al. Glucagon-like peptide 2 improves nutrient absorption and nutritional status in short-bowel patients with no colon. Gastroenterology. 2001;120(4):806–815.
193. Jeppesen, PB, Sanguinetti, EL, Buchman, A, et al. Teduglutide (ALX-0600), a dipeptidyl peptidase IV resistant glucagon-like peptide 2 analogue, improves intestinal function in short bowel syndrome patients. Gut. 2005;54(9):1224–1231.
[/level-membership-for-endocrinology-diabetes-and-metabolism-category][not-level-membership-for-endocrinology-diabetes-and-metabolism-category]Chapter 9
Glucagon and the Glucagon-Like Peptides
Biosynthesis of Pancreatic Glucagon
Proglucagon is encoded by a single gene in mammals that gives rise to a proglucagon mRNA with major sites of expression in pancreatic islets, brain, and enteroendocrine cells of the small and large intestine. Proglucagon also may be expressed in subsets of salivary glands and taste buds.1 The proglucagon gene contains six exons, several of which encode distinct functional peptide domains (Fig. 9-1). Glucagon, a 29 amino acid peptide, is synthesized in the A cells of the pancreatic islets of Langerhans. Islet A cells are distinguishable from insulin-producing β cells in part by the morphology of their respective secretory granules and by their anatomic distribution, predominantly at the periphery of the islet. The peripheral location of islet A cells, together with functional studies demonstrating central (from a core of β cells) to peripheral (A cells) islet blood flow, raises the possibility of a tightly regulated islet microenvironment. Nevertheless, as the distribution of islet α and β cells may vary from species to species, the functional importance of islet cell distribution remains unclear.
FIGURE 9-1 Structure of proglucagon and the proglucagon-derived peptides. The amino acid sequences of glucagon, glucagon-like peptide (GLP)-1, and GLP-2 are shown, as well as the cleavage site for dipeptidyl peptidase-4 (DPP-4). IP-1 and IP-2, Intervening peptides-1 and -2, respectively.
Islet Transcription Factors and the α Cell
Studies of targeted gene disruption in mice have provided new insights into the organization of islet endocrine cells in the pancreas. Neurogenin-3 is critical for pancreatic islet cell development and differentiation and controls the expression of many genes important for α cell differentiation and glucagon gene transcription. Ngn3 interacts in a regulatory network with the zinc-finger protein Myt1 to promote glucagon expression in α cells.2 Mice with a null allele in the basic helix-loop-helix transcription factor p48 fail to develop exocrine pancreatic tissue; however, hormone-secreting islet cells, including glucagon-producing α cells, are found within the mesentery during embryonic development, and later in the spleen.3 A key role for cell adhesion molecules in the control of spatial organization of islet cells is illustrated by analysis of the endocrine pancreas in mice expressing a dominant negative E-cadherin receptor in islet β cells. These mice exhibit abnormal clustering of β cells, yet glucagon-producing α cells are still capable of aggregating into islet-like clusters.4 In contrast, the normal peripheral distribution of islet α cells is markedly perturbed in neural cell adhesion molecule (NCAM)-/- mice; however, the number of α cells and the glucagon content of the pancreas remain unaffected.5 It is intriguing that mice with genetic disruption of the ciliogenic transcription factor regulatory factor X (RFX)-3 exhibit small disorganized islets with markedly reduced insulin and glucagon content, implying a role for cilia in optimal islet organization and cell development.6
Considerable insight into the developmental biology of the endocrine pancreas and islet α cells has been derived from studies of islet transcription factors in cell lines and mice. Gene knockout studies demonstrate that the LIM domain protein isl-1 is necessary for the formation of differentiated islet cells, including α cells, in the developing pancreas.7 Targeted deletion of the homeobox transcription factor Arx results in mice that exhibit complete failure of α cell development, leading to glucagon deficiency and neonatal hypoglycemia.8 In contrast, although mice deficient in the homeobox transcription factor Ipf/Pdx-1 fail to develop a pancreas, a few islet cells immunopositive for insulin or glucagon are observed in Ipf/Pdx-1-/- mice at E11.9 These observations suggest that Ipf/Pdx-1 is not essential for the formation of islet α cells, whereas loss of Pdx-1 expression appears to be required for acquisition of a differentiated α cell phenotype.
The homeobox transcription factor Nkx2.2 is expressed in adult α, β, and PP islet cells; mice with targeted disruption of NKx2.2 lack β cells, develop diabetes, and exhibit a marked reduction in the numbers of islet α cells.10 A related phenotype is observed in NeuroD/BETA2-/- mice, which exhibit marked reductions in the numbers of islet β and α cells and develop diabetes shortly after birth,11 a phenotype dependent on the specific genetic background. Similarly, disruption of pax6 function in mice results in poorly formed islets with disorganized islet architecture and markedly reduced but detectable numbers of both β and islet α cells.12,13 It is intriguing that mice that harbor mutations in islet transcription factors also may exhibit paradoxically increased numbers of islet α cells. For example, targeted deletion of the Pax4 gene results in poorly formed islets, with a marked reduction in islet β cells and comparatively increased numbers of islet α cells.14 A related phenotype is observed in mice with a homozygous deletion in the glucose transporter 2 (GLUT-2) gene, with a marked increase in the ratio of α to β cells observed in GLUT2-/-islets.15 Whether these findings are due to a block in the normal islet differentiation program, or to loss of inhibitors that restrain α cell proliferation, is not known. Evidence in support of glucagon itself regulating the numbers of islet α cells derives from analysis of mice with targeted disruption of the genes that encode prohormone convertase-2, or the glucagon receptor. These mice represent models for loss of bioactive glucagon16 or glucagon action,17 respectively, and they exhibit marked α cell hyperplasia, which is corrected in the former instance by glucagon replacement therapy.
Proglucagon Gene Transcription Factors
A combination of gene transfection experiments using cell lines in vitro and transgenic studies of promoter function in vivo has yielded insight into the molecular control of glucagon gene transcription. Differential gene expression analysis has revealed a remarkably complex pattern of transcription factors preferentially expressed in α versus β cell lines18 and in single α cells isolated from the developing endocrine pancreas.19 The rat proglucagon gene promoter directs transcriptional initiation from a single transcription start site that maps to an identical location in brain, pancreas, and intestine.20 Cell transfection experiments utilizing islet cell lines have identified five distinct regions, designated G1 through G5, within the proximal proglucagon promoter that exhibit functional importance for activation of islet cell–specific proglucagon gene transcription. The proximal G1 region is AT rich, interacts with both widely expressed and islet cell–specific proteins, and is functionally important for specifying expression in islet A cells. G1 interacts with the homeobox transcripton factors Isl-1, Cdx-2/3, Brn4, and Pax6.13,21–24 These transcription factors bind G1 and activate reporter genes containing G1-derived sequences in transfection assays. Pax6 and c-Maf exhibit cooperativity in their ability to increase G1-dependent transcriptional activation,25 whereas Nkx6.1 inhibits Pax6-dependent G1 activation. Reduction of isl-1 expression leads to reduced G1-dependent proglucagon promoter activity and decreased levels of proglucagon mRNA transcripts.21 Similarly, increased expression of Cdx-3 in islet cells is associated with induction of both transfected G1-dependent reporter genes and endogenous proglucagon gene expression.22,26 The G4 element, located just upstream of G1, binds a complex resembling insulin-enhancer factor 1 (IEF1), and recent experiments suggest that IEF1 represents a heterodimer of the helix-loop-helix (HLH) proteins E47 and BETA2.27
The more distal G3 region functions as an islet enhancer–like element and has been divided into two distinct functional domains. Subdomain A contains a sequence element similar to sequences found within the insulin and somatostatin promoters, leading to the designation of this composite sequence as a pancreatic islet cell–specific enhancer sequence, or PISCES element.28,29 Sequences within domain A of G3 also appear to mediate the inhibition of glucagon gene transcription by insulin.30–32 The pax6 protein has been identified as a positive activator of proglucagon gene transcription that exerts its function in part through interaction with domain A of the G3 element.13 Pax6 also binds the G1 element, either as a monomer or via heterodimer formation with cdx-2/3.33 Disruption of Pax6 expression in gene-targeted mice results in loss of islet A cells.12,13 The levels of pancreatic and intestinal proglucagon mRNA transcripts are also markedly reduced in SEYNEU mice that harbor a dominant negative mutation in the pax6 gene. Thus Pax6, a PISCES binding protein, is functionally important for both islet and enteroendocrine cell development and activation of proglucagon gene transcription via the G3 enhancer and G1 promoter elements.12,13,33,34
The G2 element mediates the positive and negative actions of HNF-3 (Foxa) proteins, with isoforms of HNF-3β and HNF-3α competing for binding to the G2 element and serving as repressors and activators, respectively, of proglucagon gene transcription.35,36 HNF-3γ also binds to and transactivates the proglucagon promoter G2 element but does not appear to be essential for islet cell formation or glucagon gene transcription.37–39 Whether HNF-3β plays an essential role in the development of islet α cell formation or glucagon gene transcription in vivo remains unknown, as targeted inactivation of the HNF-3β gene results in embryonic lethality before formation of the endocrine pancreas.40,41 Although the numbers of α cells and the morphology of islets appear histologically normal in HNF3α-/- mice, the development of neonatal hypoglycemia in the face of inappropriately reduced levels of circulating glucagon and proglucagon mRNA transcripts demonstrates the essential role of HNF-3α in pancreatic proglucagon gene transcription.37
Activation of the cyclic adenosine monophosphate (cAMP)-dependent pathway leads to transcriptional induction of proglucagon gene expression in both islet and intestinal cells through a cAMP response element (CRE) present upstream of the G3 element in the proximal promoter region of the rat proglucagon gene.42,43 The CRE also mediates transcriptional activation by ATF3 family members44 and is the target for pharmacologic depolarization–dependent induction of rat proglucagon gene transcription.45 A second calcium response element has been localized to the G2 element, and calcium responsiveness may be mediated via interaction of nuclear factor of activated T cells (NFAT)-like proteins with members of the HNF-3 family.46 The sequence of the CRE element is less well conserved in the human proglucagon promoter, and its functional relevance for proglucagon gene expression in human islets has not yet been elucidated.
Transgenic experiments in mice have identified distinct DNA sequences essential for tissue-specific proglucagon gene transcription in vivo. The first 1253 nucleotides of the rat proglucagon promoter direct heterologous transgene expression to the islets and brain but not the intestine in transgenic mice.47 In contrast, targeting of transgene expression to enteroendocrine cells in vivo requires the presence of additional rat proglucagon gene 5′-flanking sequences between −1253 and −2252.48 Foxo1 binds to an upstream site at −1798 and appears to be important for both basal and insulin-regulated proglucagon gene transcription in α cells.49 DNA sequences constituting the human proglucagon gene promoter exhibit different functional properties than homologous rat sequences, as the first 1600 bp of the human proglucagon gene 5′-flanking region target transgene expression to appropriate cell types in the brain and intestine, but not the pancreatic islets in transgenic mice.50
Islet Proglucagon Biosynthesis
Consistent with the increased circulating glucagon levels in patients with diabetes, experimental diabetes in rodents is associated with increased levels of pancreatic proglucagon mRNA; correction of the insulin deficiency, but not the hyperglycemia, normalizes the levels of proglucagon mRNA.51 Paradoxically, insulin may increase proglucagon gene expression in enteroendocrine cells.52 Orexin-A also inhibits glucagon secretion and proglucagon gene expression via the phosphatidylinositol-3-kinase–dependent pathway, requiring Foxo1.53 In contrast, few hormones or metabolites have been identified that increase pancreatic proglucagon biosynthesis in vivo. Although experiments using in situ hybridization suggested that 4 days of fasting in the rat leads to a doubling of pancreatic proglucagon mRNA transcripts,54 hypoglycemia induced by insulin failed to demonstrate upregulation of proglucagon mRNA.51,55 Available data suggest that regulation of glucagon secretion, and not biosynthesis, may be a more relevant locus of control in vivo.
Following transcription of proglucagon mRNA and translation of the proglucagon precursor, 29 amino acid mature glucagon is liberated via posttranslational processing by specific prohormone convertases differentially expressed in the islet α cell. In contrast to processing in the enteroendocrine cell (see Fig. 9-1), the amino acid sequences carboxyterminal to glucagon remain unprocessed and are secreted as part of a larger polypeptide designated the major proglucagon fragment (MPGF). PC2 is essential for processing of proglucagon to glucagon in α cells as PC2-/- mice exhibit hypoglycemia, α cell hyperplasia, and glucagon deficiency with an accumulation of incompletely processed PGDPs in the pancreas.16 Whether PC2 directly liberates glucagon from proglucagon, or cleaves proglucagon to glicentin, which is subsequently processed to glucagon, remains unclear. The enzyme PC1 is responsible for cleavage of proglucagon to yield an intestinal profile of PGDPs,56,57 and PC1 null mice exhibit impaired processing of proglucagon to the glucagon-like peptides.58 Moreover, human subjects with an inactivating mutation in the PC1 gene exhibit incompletely processed proglucagon in gut endocrine cells and deficiency of both GLP-1 and GLP-2.59 The convertases important for proglucagon processing in the brain have not been identified definitively.
Glucagon Secretion
The secretion of glucagon is regulated, both positively and negatively, by neuropeptides, hormones, metabolites, and the autonomic nervous system. The islet α cell plays a central role in the defense of blood glucose, with hypoglycemia stimulating and hyperglycemia suppressing glucagon secretion in vivo. A cells express voltage-dependent Na+, K+, and Ca2+ channels that interact in the regulation of membrane potential and, ultimately, depolarization. Amino acids such as glutamine, alanine, pyruvate, and arginine stimulate both insulin and glucagon secretion, and this observation provides the basis for the use of arginine in the assessment of islet and α cell function in rodent and human physiology.60 Both α- and β-adrenergic receptors modulate glucagon secretion. Epinephrine stimulates glucagon secretion via protein kinase A (PKA)-dependent enhancement of Ca2+ influx through L-type calcium channels, leading to granule exocytosis. Peptidergic activators of glucagon secretion include cholecystokinin (CCK), pituitary adenylate cyclase–activating polypeptide (PACAP), gastrin, urocortin III, vasopressin, and glucose-dependent insulinotropic peptide (GIP).
In contrast, inhibitors of glucagon secretion include glucose, somatostatin-14, and γ-aminobutyric acid (GABA).61 Somatostatin tonically inhibits glucagon release, and mice with disruption of the somatostatin receptor 2 (SSTR2) gene exhibit mild hyperglycemia and elevated levels of nonfasting glucagon.62 Glucose induces GABA release from β cells, providing an indirect mechanism for glucose-mediated suppression of glucagon secretion.63–65 Glucose also reduces electrical activity and exocytosis via depolarization-induced inactivation of ion channels, specifically KATP, Na(+) (TTX) and N-type Ca(2+) channels, as a direct mechanism for inhibition of glucagon secretion. Insulin also may directly inhibit glucagon release, possibly via insulin receptors expressed on α cells. The anatomic arrangement of peripheral islet α cells surrounding a core of β cells, together with the functional results of immunoneutralization studies, provides additional evidence for intra-islet insulin inhibiting downstream of α cells in some species.66 Although the mechanisms underlying the paracrine inhibition of α cell activity remain unclear, experimental evidence supports an important role for β cell secretory products such as zinc as negative regulators of glucagon secretion.65,67
The effects of glucose on glucagon secretion are integrated with the inhibitory effects of insulin on α cell secretion, as hyperglycemia stimulates insulin secretion, whereas a drop in blood glucose suppresses insulin release from the β cell, thereby relieving the α cell from the tonic inhibitory actions of insulin. These actions are exemplified by meal ingestion, which is associated with increased circulating nutrients and insulin secretion, and reduced levels of circulating glucagon. In contrast, blood glucose is maintained in the fasting state by hepatic glucose production due in part to increased levels of glucagon secretion and suppression of insulin release from the β cell.68,69 Consistent with the effects of individual nutrients on α cell function, infusion of arginine stimulates glucagon secretion in both normal subjects and individuals with diabetes mellitus.69 The response to arginine infusion in normal human subjects, namely, stimulation of insulin and glucagon release, is reproducible, and as expected, the insulin response is significantly greater and the glucagon response attenuated as the glucose concentration increases.70
In normal subjects, glucagon secretion rises as glucose falls in the fasting state and may be further stimulated by exercise, consistent with the physiologic role for glucagon in regulating glucose production. The levels of both glucagon and plasma catecholamines increase with graded exercise in normal human subjects, with the greatest increments in plasma glucagon observed in subjects undergoing prolonged exhaustive exercise.71 The exercise-induced increment in plasma glucagon appears dependent on the degree and duration of exercise, with some studies demonstrating no significant changes in plasma glucagon during exercise under normoglycemic conditions.72 Trained healthy male subjects exhibit increased hepatic glucose production following glucagon infusion, implying that exercise may be associated with the development of increased glucagon sensitivity.73 Although prevention of exercise-induced rises in plasma glucagon by concomitant somatostatin infusion may result in mild hypoglycemia,74 suppression of the rise in plasma glucagon by somatostatin infusion does not always prevent increased hepatic glucose production, likely because of the redundant compensatory mechanisms for maintaining normoglycemia.74,75
The control of hepatic glucose production (HGP) is highly sensitive to the glucagon/insulin ratio, and secretion of these two hormones generally is regulated in a reciprocal manner. The effect of insulin to suppress HGP actually may be determined in part by the levels of circulating glucagon.76 Following meal ingestion, nutrient absorption is associated with energy assimilation and suppression of glucagon secretion. Nevertheless, the integrated response of islet α cells is dependent in part on the nutrient composition of the meal and reflects positive and negative enteric-derived regulators of glucagon secretion. For example, ingestion of carbohydrate, especially glucose, is associated with a decline in plasma glucagon.69 Nutrients also promote release of gut-derived peptides such as CCK and GIP, which stimulate glucagon release,77,78 and GLP-1, which inhibits glucagon release.79,80
Glucagon secretion is increased during times of stress, and “stress-induced hormones” such as cortisol, vasopressin, and β-endorphin increase glucagon secretion from the α cell. The classic stress hormones epinephrine and norepinephrine also stimulate glucagon secretion, and several mechanisms link the autonomic nervous system to increased secretion from the α cell. Epinephrine secreted from the adrenal medulla increases glucagon secretion in normoglycemic subjects. Furthermore, the pancreas receives innervation from both sympathetic and parasympathetic nerves, and stimulation of these autonomic inputs, all of which are activated by hypoglycemia, increases glucagon secretion.81 It is intriguing that mice with targeted inactivation of the pro-opiomelanocortin gene (and hence adrenocorticotropic hormone [ACTH]) exhibit a profound defect in the counterregulatory response to insulin-induced hypoglycemia, largely as the result of defective glucagon secretion. Although specific genetic defects in glucagon secretion have not been described, nondiabetic subjects with mutations in the MODY1/HNF-4α gene exhibit decreased arginine-stimulated glucagon secretion and reduced glucose suppression of plasma glucagon,82 raising the possibility that the HNF-4α transcription factor influences islet α cell function through a direct or indirect mechanism.
Hypoglycemia
Increasing evidence suggests that multiple complementary mechanisms activate α cell secretion during hypoglycemia. Analysis of the glycemic threshold for activation of counterregulatory mechanisms demonstrates that increased epinephrine and glucagon secretion constitute the initial hormonal responses to decreasing blood sugar. Furthermore, the glucose threshold for activation of counterregulatory responses is clearly higher than the threshold for triggering hypoglycemic symptoms in normal subjects.83 Whether hypoglycemia itself directly stimulates glucagon secretion independent of autonomic input remains unclear. Nevertheless, hypoglycemia suppresses insulin (and β cell) secretion, which removes an important inhibitory influence on glucagon secretion. The finding that α cells express the GLUT1 transporter and glucokinase suggests that glucose transport does not appear to be a critical rate-limiting step for glucose metabolism in α cells and provides important insight into how α cells directly sense ambient changes in glucose concentration.84,85 Hypoglycemia stimulates glutamate release from α cells, which, in turn, activates glucagon secretion in an autocrine manner.86 Glutamate release from ventromedial hypothalamic neurons is also a key component of the counterregulatory response, as mice lacking the synaptic vesicular transporters (VGLUT2) exhibit fasting hypoglycemia and a defective glucagon response to hypoglycemia.87 A role for gastrin in the stimulation of glucagon secretion has been proposed, and gastrin-/- mice exhibit defective glucagon secretion in response to insulin-induced hypoglycemia.88
The role of circulating epinephrine in the autonomic response to hypoglycemia is well established. Epinephrine also directly stimulates glucagon secretion in normal human subjects, and stimulation of autonomic sympathetic nervous system innervation to the pancreas elicits an increase in α cell secretion.89,90 Furthermore, parasympathetic nerve stimulation or the neurotransmitter acetylcholine and the neuropeptide VIP all stimulate glucagon secretion. Studies using nerve transection or pharmacologic blockade have illustrated that these pathways exhibit some degree of functional redundancy, providing multiple backup mechanisms to ensure that the α cell responds appropriately to hypoglycemia. Nevertheless, the ganglionic blocker trimethaphan, which impairs autonomic transmission both in ganglia and in the adrenal, markedly attenuated the glucagon response to hypoglycemia in normal subjects,91 emphasizing the important link between autonomic activation and the glucagon counterregulatory response in vivo. In contrast however, pancreas transplantation restores the glucagon response to hypoglycemia, even in patients with severe autonomic neuropathy,92 illustrating the functional redundancy of mechanisms essential for the α cell response to hypoglycemia.
The identity of the specific glucose sensors that trigger the appropriate counterregulatory response to hypoglycemia remains under active investigation. Mice with genetic disruption of the Kir6.2 channel exhibit normal functional α cells, yet display a marked defect in the glucagon response to hypoglycemia.93 GLUT2 expression in glial cells is essential for the glucagon response to hypoglycemia and the central nervous system, principally the ventromedial hypothalamus (VMH),87 and the splanchnic region, specifically consisting of the portal system and liver, contains multiple glucose sensing systems. Selective perfusion of the portal venous system in rats suggests that the portal vein glucose concentration is a key determinant of the sympathoadrenal response to hypoglycemia; however, whether portal glucose sensors are directly or indirectly linked to control of islet glucagon secretion is not clear.94 It is intriguing that injection of glucose into the portal vein activates glucose-sensitive neurons in the lateral hypothalamus and brain stem, suggesting the possibility of a portal-CNS glucoregulatory axis. The detection of a markedly defective counterregulatory response to hypoglycemia, including absent glucagon and catecholamine secretion, in a patient with hypothalamic sarcoidosis further emphasizes the importance of the hypothalamus in sensing glucose and triggering the release of counterregulatory hormones.95 In contrast, analysis of patients after removal of craniopharyngiomas revealed selective impairment of counterregulatory sympathoadrenal activation but normal glucagon responses to hypoglycemia. Furthermore, human subjects with liver transplants and denervated livers exhibit increased levels of circulating glucagon and defective insulin suppression of glucagon, and human islet transplantation is associated with a defective glucagon response to hypoglycemia. These findings emphasize the importance of the autonomic nervous system and the CNS for the regulation of basal and hypoglycemia-stimulated glucagon secretion.
Glucagon Secretion and Diabetes
Glucagon levels are elevated in many patients with poorly controlled diabetes, and excess glucagon secretion in response to meal ingestion is an early event that is detectable within months of the onset of type 1 diabetes.96 The correction of hyperglucagonemia, using somatostatin or insulin, reverses most metabolic derangements associated with insulin-deficient diabetes.97 Hence there is considerable interest in development of glucagon antagonists for the treatment of diabetes. Moreover, genetic attenuation of glucagon receptor expression in diabetic rodents results in significant amelioration of experimental diabetes.98,99 Despite the central importance of glucagon for control of glucose production, hyperglucagonemia alone, without insulin deficiency, does not significantly increase plasma glucose.100 In the presence of adequate amounts of insulin, glucose production is suppressible despite glucagon excess, emphasizing the insulin:glucagon ratio, and not just the absolute level of glucagon, as a key determinant of glucose homeostasis.101 Nevertheless, hyperglucagonemia is associated with increased leucine oxidation and resting metabolic rate in subjects with type 1 diabetes, reemphasizing the importance of both insulin and glucagon in the catabolism associated with suboptimally treated diabetes.102
The application of intensive insulin therapy to the management of patients with type 1 and type 2 diabetes is associated with an increased incidence of hypoglycemia and heightened awareness of the importance of counterregulatory mechanisms for maintaining normoglycemia. Several factors, including intensive insulin administration, hyperglycemia, and diminished autonomic stimulation of the α cell, inhibit glucagon release in insulin-treated patients with type 1 diabetes.81 Although the glucagon response to hypoglycemia is initially normal in patients with type 1 diabetes, this response frequently becomes impaired, increasing the susceptibility of patients to hypoglycemia.103 Impaired counterregulation and hypoglycemia have been observed in some patients with a very short duration of diabetes, suggesting that frequent episodes of hypoglycemia represent an independent risk for development of an abnormal α cell response. The α cell dysfunction in type 1 diabetes is often selective, as the response to hypoglycemia may be absent, yet glucagon secretion may respond normally to arginine stimulation.104 Further evidence emphasizing the importance of intra-islet hyperinsulinemia or other β cell–derived products in the suppression of glucagon secretion derives from observations that tolbutamide-infused subjects exhibit profound defects in the glucagon response to hypoglycemia.105
It is important to note that restoration of normoglycemia for several months may decrease hypoglycemia unawareness in association with improvement in the glucagon response to hypoglycemia in some studies.106 However, a dissociation between improvement in hypoglycemia unawareness and persistence of defective counterregulatory responses also has been observed.107 Although not completely understood, antecedent hypoglycemia in type 1 diabetes can produce counterregulatory failure during subsequent episodes of prolonged moderate intensity exercise,108 emphasizing the complex interrelationship between insulin-induced and exercise-associated counterregulation.
Multiple defects likely contribute to α cell dysfunction in patients with diabetes. Patients with some residual β cell function, as assessed by C-peptide stimulation, appear to be at decreased risk for hypoglycemia, in part because of preserved counterregulatory glucagon responses. Additional contributing factors may include subtle impairment of autonomic stimulation following repeated hypoglycemic episodes. Nevertheless, impaired epinephrine and glucagon responses to insulin-induced hypoglycemia have been observed after a single antecedent hypoglycemic episode in nondiabetic subjects,109 although increased levels of cortisol alone do not acutely induce defects in hypoglycemic counterregulation. It is intriguing that oral administration of amino acids improves cognitive function and the glucagon response to hypoglycemia in both normal subjects and individuals with type 1 diabetes. Taken together, these observations emphasize the susceptibility of the normal α cell to episodes of hypoglycemia, ultimately leading to sustained defects in the glucagon response to hypoglycemia.
