[level-membership-for-obstetrics-gynecology-category]
CHAPTER 43 Gestational trophoblastic tumours
Introduction
The World Health Organization (WHO) has classified gestational trophoblastic disease (GTD) into two premalignant diseases, termed ‘complete hydatidiform mole’ (CM) and ‘partial hydatidiform mole’ (PM), and three malignant disorders, termed ‘invasive mole’, ‘gestational choriocarcinoma’ and ‘placental-site trophoblastic tumour’ (PSTT) (World Health Organization 1983). These malignant disorders are also frequently referred to as ‘gestational trophoblastic tumours’ (GTTs) or ‘neoplasia’ (GTN). GTTs are important to recognize because they are nearly always curable and fertility can be preserved in most cases. This is mainly because:
Genetics and Pathology
Complete hydatidiform mole
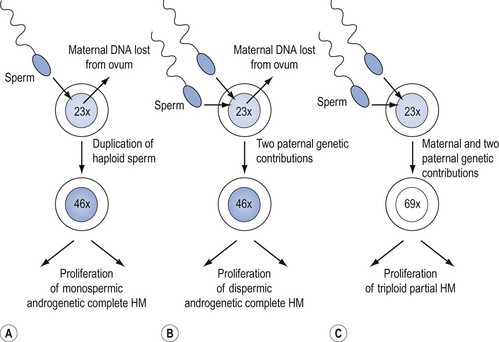
CMs nearly always contain paternal DNA alone and are therefore androgenetic. This occurs in most cases because a single sperm bearing a 23X set of chromosomes fertilizes an ovum lacking maternal genes, and then duplicates to form the homozygote, 46XX (Figure 43.1A). However, in up to 25% of CMs, fertilization can take place with two spermatozoa, resulting in the heterozygous 46XY or 46XX configuration (Figure 43.1B). A 46YY conceptus has not yet been described and is presumably non-viable. Very rarely, a CM can arise from a fertilized ovum which has retained its maternal nuclear DNA and is therefore biparental in origin (Fisher and Newlands 1998). Macroscopically, the classic CM resembles a bunch of grapes due to generalized (complete) swelling of chorionic villi. However, this appearance is only seen in the second trimester and the diagnosis is usually made earlier when the villi are much less hydropic. Indeed, in the first trimester, the villi microscopically contain little fluid, are branching and consist of hyperplastic syncytio- and cytotrophoblast with many vessels. Although it was previously thought that CM produced no fetal tissue, histology from 6–8-week abortions reveals evidence of embryonic elements, including fetal red cells (Paradinas 1998). This has resulted in pathologists incorrectly labelling CMs as PMs. The presence of embryonic tissue from a twin pregnancy comprising a fetus and a CM is another source of error which can lead to the incorrect diagnosis of PM.
Partial hydatidiform mole
Genetically, PMs are nearly all triploid with two paternal and one maternal sets of chromosomes (Figure 43.1C). Rarely, tetraploid PMs are found. Triploidy occurs in 1–3% of all recognized conceptions and in approximately 20% of spontaneous abortions with an abnormal karyotype, but at least two paternal sets of chromosomes are needed for PM development. Triploids due to two sets of maternal chromosome do not become PMs (Lawler et al 1982). Although a variety of reports have suggested that diploid PMs exist, genetic analysis of such lesions has not supported this. In general, a diploid molar gestation is believed to be a CM (Genest et al 2002). Flow cytometry, which can be done in formalin-fixed, paraffin-embedded tissues (Seckl et al 2000), can help in differentiating CM from PM, and PM from diploid non-molar hydropic abortions.
In PMs, villous swelling is less intense and only affects some villi. Both swollen and non-swollen villi can have trophoblastic hyperplasia which is mild and focal. The villi have characteristic indented outlines and round inclusions. An embryo is usually present and can be recognized macroscopically or inferred from the presence of nucleated red cells in villous vasculature. It may survive into the second trimester, but usually dies at approximately 8–9 weeks of gestation and this is followed by loss of vessels and stromal fibrosis. In PMs evacuated early, villous swelling and trophoblastic excess can be so mild and focal that the diagnosis of PM may be missed (Paradinas 1998). Indeed, at uterine evacuation for a ‘miscarriage’, it is likely that many PMs are misclassified as products of conception. Fortunately, the authors only see about one patient per year with persistent GTD related to a previously unrecognized PM. Of the increasing number of PMs which are correctly diagnosed, very few go on to develop persistent GTD. Indeed, in approximately 3000 PMs reviewed and followed at Charing Cross Hospital between 1973 and 1997, only 15 (0.5%) have required chemotherapy (Seckl et al 2000).
Other pregnancies mistaken for hydatidiform mole
Over half of first-trimester non-molar abortions are due to trisomy, monosomy, maternally derived triploidy and translocations. These often develop hydrops but are small (<3 mm), and PM can be excluded if they are diploid on flow cytometry. A significant recent development in the pathological analysis of hydatidiform mole (HM) is the use of p57kip2 immunostaining to make a definitive diagnosis of androgenetic CM as opposed to a hydropic abortion or a PM. p57kip2, a cyclin-dependent kinase inhibitor, is a paternally imprinted gene which is maternally expressed. The absence of maternal genes in androgenetic CM means that the gene cannot be expressed in CM cytotrophoblast. Consequently, p57kip2 staining is negative with CMs, in contrast to PMs, hydropic abortion and normal placenta. This technique is well validated, easy and inexpensive to perform (Sebire et al 2004, Popiolek et al 2006).
Syndromes such as Turner’s, Edward’s and Beckwith-Wiedemann’s can also cause histological confusion with PMs (Paradinas 1998).
Invasive hydatidiform mole
Invasive hydatidiform mole is common and is clinically identified by the combination of an abnormal uterine ultrasound (US) and a persistent or rising hCG level following uterine evacuation of a CM or PM. Pathological confirmation of this condition is rarely required. Moreover, repeat dilatation and curettage (D & C) is often contraindicated because of the risks of uterine perforation, infection, life-threatening haemorrhage and subsequent hysterectomy. In occasional cases where histology is available, invasive mole can be distinguished from choriocarcinoma by the presence of chorionic villi. Identification of cases of CM and PM that will subsequently undergo malignant transformation would be an important development. There is accumulating evidence that such HMs are more likely to show telomerase activity (i.e. have limitless replicative potential) and increased expression of the anti-apoptotic gene Mcl-1 (Wells 2007). These findings need further prospective evaluation.
Choriocarcinoma
Most choriocarcinomas have been shown to have grossly abnormal karyotypes with diverse ploidies and several chromosome rearrangements, none of which are specific for the disease (Arima et al 1994). Studies of the origin of GTTs have confirmed that choriocarcinoma may arise from any type of pregnancy including a normal term pregnancy, or homozygous or heterozygous CM. Until recently, it was thought that PMs could not give rise to choriocarcinoma. However, there is now incontrovertible genetic evidence that PMs can indeed transform into choriocarcinomas (Seckl et al 2000). This is important as some centres wrongly believe that it is safe to discontinue hCG follow-up following the diagnosis of a PM.
Interestingly, choriocarcinoma may not always be due to the antecedent pregnancy. A patient with a history of CM 4 years previously developed choriocarcinoma following the delivery of a twin pregnancy. Using polymerase chain reaction to amplify short tandem repeat polymorphisms in DNA, this tumour was shown to be genetically identical to the previous CM (Fisher et al 1995). As for invasive moles, obtaining tissue to make a formal histological diagnosis of choriocarcinoma is often not appropriate, so doubt frequently exists regarding whether patients have one or the other form of GTT.
Placental-site trophoblastic tumour
PSTTs have been shown to follow term delivery, non-molar abortion or CM. Furthermore, a recent case has shown that PSTTs can develop after a PM (Palmieri et al 2005). Like choriocarcinoma, the causative pregnancy may not be the immediate antecedent pregnancy (Fisher et al 1995). Genetic analysis of some PSTTs has demonstrated that they are mostly diploid, originating from either a normal conceptus and therefore biparental, or androgenetic and arising from a CM (Newlands et al 1998a).
In the normal placenta, placental-site trophoblast is distinct from villous trophoblast and infiltrates the decidua, myometrium and spiral arteries of the uterine wall. PSTTs are rare, slow-growing malignant tumours composed mainly of intermediate trophoblast derived from cytotrophoblast, and so produce little hCG. However, they often stain strongly for human placental lactogen (hPL) and β1-glycoprotein. Elevated Ki-67 levels may help in distinguishing PSTTs from regressing placental nodules (Shih and Kurman 1998). In contrast to other forms of GTT, spread tends to occur late by local infiltration and via the lymphatics, although distant metastases can occur. More than five mitoses per 10 high-power fields may predict tumours with metastasizing potential (Newlands et al 1998a).
Epithelioid trophoblastic tumour
Epithelioid trophoblastic tumours (ETTs) are a recently described neoplastic proliferation of intermediate trophoblast that are thought, by some investigators, to be distinct from PSTTs and choriocarcinoma. It has been proposed that ETTs arise from the intermediate trophoblasts of the chorionic laeve (Shih and Kurman 2001). Histologically, ETTs display a relatively uniform, nodular proliferation of intermediate-sized epithelioid trophoblasts, forming nests and cords. Islands of trophoblast are typically surrounded by areas of hyalinization or eosinophilic debris simulating tumour cell necrosis and resembling keratinous material in a squamous cell carcinoma. ETTs can be associated with focal replacement of the cervical glandular epithelium with stratified neoplastic cells, simulating squamous cervical intraepithelial neoplasia. ETT cells are positive for cytokeratin, epithelial membrane antigen and inhibin-A, whereas trophoblastic markers hPL, hCG and melanoma cell adhesion molecular are only expressed focally (Hui et al 2005).
Epidemiology and Aetiological Factors
Hydatidiform mole
Incidence and ethnic origin
The incidence of HM in the UK is one in 714 pregnancies (Tham et al 2003). Recent results indicate that the previously documented higher rates in the Far East have fallen towards the stable levels found in Europe and North America (Hando et al 1998), possibly because of dietary changes. The incidence of PM has been underestimated in the past and is currently three per 1000 pregnancies (Newlands et al 1998b).
Age
CMs are more common at the extremes of reproductive age. In one study, compared with the lowest rate between 25–29 years, the relative increased risk was six-fold in girls under 15 years, three-fold between 40 and 45 years, 26-fold between 45 and 49 years and more than 400-fold over 50 years of age (Bagshawe and Begent 1983, Newlands et al 1998b). PMs are also more common at the extremes of reproductive age, although the effect is less pronounced (Sebire et al 2002a).
Previous pregnancies
Increasing gravidity does not increase the risk of CM. However, following one CM, the risk of a subsequent pregnancy being a CM rises from one in 1000 to one in 76, and to one in 6.5 with two previous CMs (Bagshawe and Begent 1983). Therefore, patients with a previous CM must be followed-up after each subsequent pregnancy to confirm that their hCG levels return to normal. Similar results have been reported for PMs, with the risk for subsequent molar pregnancies rising to one in 59 for women with one previous PM (Sebire et al 2003).
Choriocarcinoma
The incidence of choriocarcinoma following term delivery without a history of CM is approximately one in 50,000. However, CM is probably the most common antecedent to choriocarcinoma, comprising 29–83% in various studies across the world (World Health Organization 1983). Consequently, the overall incidence of choriocarcinoma after a CM is much higher. Proof of this is frequently difficult to obtain, but when histology was available, these tumours were identified as choriocarcinoma in 3% and invasive mole in 16% of previous CMs (seldom as PSTT). Rarely, PMs can give rise to choriocarcinoma (Seckl et al 2000). Unlike HM, choriocarcinoma does not exhibit any clear geographical trends in incidence, but the effect of age remains important.
Placental-site trophoblastic tumours
First described as a separate disease entity in 1976 (Kurman et al 1976), there are currently approximately 150 recorded cases of this tumour in the literature; therefore, estimates of its true incidence may be quite inaccurate (Newlands et al 1998a, Papadopoulos et al 2002, Hassadia et al 2005, Baergen et al 2006). In the largest series described — 62 patients between 1976 and 2006 — PSTTs represented 0.2% of all trophoblastic tumours (Schmid et al 2008) The relative incidence in the UK has been stable over the past 15 years following an increase in the first years after the introduction of this new disease classification.
Genetic Factors: The Role of Imprinting
Hydatidiform mole
All autosomal genes consist of two alleles (paternal and maternal). However, some alleles are only expressed from one parent and not the other; a phenomenon called ‘genomic imprinting’. Underexpression of an imprinted gene has been demonstrated in cases of both familial and sporadic CM, and abnormal methylation patterns have been described in another family (Fisher et al 2002, Judson et al 2002).
Extensive mapping studies have been performed on the families with CM. These have demonstrated a defective locus at 19q13.4 in five families which has been localized to a single gene — NALP7 (Murdoch et al 2006). This is the first causative single gene defect identified in CM. NALP7 is a member of the CATERPILLER family which is involved in inflammation and apoptosis. It is expressed in oocytes and the endometrium, and is a negative regulator of the proinflammatory cytokine interleukin-1β, which is involved in regulating trophoblast invasion during implantation. It is unclear how defects in NALP7 result in CM formation, but abnormal inflammation during embryogenesis may be involved.
NALP7 does not appear to be involved in the establishment of imprinting; therefore, defects in NALP7 may be a consequence of abnormal imprinting rather than its cause. Further families have been identified that do not map to 19q13.4, proving that familial CM is a genetically heterogeneous condition (Zhao et al 2006). Moreover, to date, abnormal NALP7 has not been demonstrated in sporadic CM or PM cases.
Three other, closely related, genes are imprinted and may be involved in GTT development. These are H19, a putative tumour suppressor gene (Hao et al 1993), and p57kip2 (discussed previously) (Matsuoka et al 1996), both of which are normally expressed by the maternal allele; and the paternally expressed IGF-2, a growth factor commonly implicated in tumour proliferation (Ogawa et al 1993). While p57kip2 showed the expected pattern of expression in CM and choriocarcinoma (Chilosi et al 1998), CM and postmole tumours were unexpectedly found to express H19 (Walsh et al 1995), and some post-term tumours showed biallelic expression of both H19 and IGF-2 (Hashimoto et al 1995). This suggests that loss of the normal imprinting patterns of these genes may be an important factor in the development of GTT.
Choriocarcinoma
Historically, cytogenetic studies of choriocarcinoma have demonstrated a diverse range of abnormalities. Microsatellite studies have shown loss of heterozygosity at specific regions: deletions of 7p12-q11.2, amplification of 7q21-q31 and loss of 8p12-p21 (Matsuda et al 1997) Abnormalities in the latter two are also found in some cases of ovarian and breast cancer, and are postulated to encode novel tumour suppressor genes. Several groups have reproduced these findings in non-molar choriocarcinoma, but results in postmolar choriocarcinoma have been inconsistent (Burke et al 2006). Only a minority of tumours showed loss of heterozygosity for all three regions, suggesting that these defects are acquired late in the development of choriocarcinoma and are not essential for malignant transformation.
Risk of Gestational Trophoblastic Tumours Following Complete or Partial Hydatidiform Mole
Following evacuation of a CM or PM, the risk of developing a GTT is less than 16% and 0.5%, respectively (Bagshawe et al 1990, Seckl et al 2000). Since it is not yet possible to predict which patients with a CM or PM will develop persistent GTD, all patients must be registered for hCG monitoring. Following this strict protocol enables the identification of individuals with persistent trophoblastic growth who could benefit from life-saving chemotherapy.
Human Chorionic Gonadotrophin
Beta-human chorionic gonadotrophin assays
The family of pituitary/placental glycoprotein hormones includes hCG, follicle-stimulating hormone, luteinizing hormone (LH) and thyroid-stimulating hormone (TSH). Each hormone comprises an α-subunit which is common between the family members and a distinct β-subunit. Consequently, assays to measure hCG are directed against the β-subunit. Many different β-hCG assays are available. Some detect intact β-hCG, and others are either selective for individual fragments or detect various combinations of fragments (Cole 1998). In pregnancy, hCG is usually intact and fragments of β-hCG are not produced, although the β chain is hyperglycosylated during the first trimester. However, in cancer, β-hCG may circulate in many different forms which can vary in their glycosylation status. Therefore, assays used in patients with GTD and cancer need to be able to detect all forms of β-hCG. The ideal assay should be able to recognize all forms equally well and be sufficiently sensitive to limit the risk of false-negative results (Mitchell and Seckl 2007). Moreover, the assay should not produce false-positive results as this is well recognized to be associated with unnecessary medical interventions and potentially life-threatening complications (Cole et al 2001). So how good are the existing β-hCG assays?
Commercially available β-hCG tests are based on the sandwich assay principle and rely on two antibodies which generally target different regions of the molecule. These assays are primarily licensed for use in pregnancy detection. However, they are frequently employed for monitoring patients with cancer. The assays can produce both false-positive and false-negative results (Cole et al 2001, Mitchell and Seckl 2007). The false-positive results occur because there is another molecule (often a heterophile antibody) which sticks the capture and detection antibodies together. This can usually be avoided by measuring the hCG in urine, as cross-reacting antibodies are large and cannot pass through the renal glomerulus into the urine. Alternatively, if a false-positive result is suspected, remeasuring the hCG on an alternative assay or serially diluting the serum sample usually resolves the issue. Real hCG will be seen in another assay and will dilute appropriately, whilst a cross-reacting molecule will be negative in another assay and does not serially dilute away. Recent work from the authors’ laboratory shows that commercial assays may be particularly prone to false-negative results, either because they completely fail to detect or as a consequence of poor sensitivity for a particular β-hCG isoform (Mitchell and Seckl 2007). False-negative results can potentially result in failure to diagnose disease or early termination of treatment, and therefore higher relapse rates.
In addition to the commercial assays, there are also several in-house assays in various centres around the world which are usually based on a single antibody to capture the hormone on a competitive basis with labelled (often radioactively) β-hCG. These assays may also produce false-positive and false-negative results. Indeed, all types of assay are only as good as the antibodies used. At Charing Cross Hospital, a rabbit polyclonal antibody is used in a radioimmunoassay (RIA); this recognizes all forms of β-hCG equally well and with sufficient sensitivity that false-negative results appear to be rare (Mitchell and Seckl 2007). Moreover, since this RIA is performed on both serum and urine samples which are serially diluted, the risk of false-positive results is very low (Mitchell and Seckl 2007).
New assays designed to detect specific hCG variants are in development and are leading to further increases in the specificity and sensitivity of hCG monitoring. Elevation of hyperglycosylated hCG (hCG-H) levels may be specific to cases of HM that subsequently require chemotherapy. This rise occurs earlier than in conventional hCG and before clinically apparent GTTs develop (Cole et al 2006a). However, further work is required, and since no commercial hCG-H assays are available, progress in this area is likely to be slow.
Recent data have also shown that proportionately higher levels of free β-hCG fragments are produced in PSTTs, and that this is a highly sensitive test for discriminating PSTTs from choriocarcinoma (Cole et al 2006b, Harvey et al 2008), although β-hCG may also be elevated in some non-trophoblastic malignancies.
Beta-human chorionic gonadotrophin as a tumour marker
hCG has a half-life of 24–36 h and is the most sensitive and specific marker for trophoblastic tissue. However, hCG production is not confined to pregnancy and GTD. Indeed, hCG is produced by any trophoblastic tissue found, for example, in germ cell tumours and in up to 15% of epithelial malignancies (Vaitukaitis 1979). The hCG levels in such cases can be just as high as those seen in GTD or in pregnancy. Therefore, measurements of hCG do not reliably discriminate between pregnancy, GTD or non-gestational trophoblastic tumours.
However, serial measurements of hCG have revolutionized the management of GTD for several reasons. The amount of hCG produced correlates with tumour volume, such that a serum hCG level of 5 IU/l corresponds to approximately 104–105 viable tumour cells. Consequently, these assays are several orders of magnitude more sensitive than the best imaging modalities available today. In addition, hCG levels can be used to determine prognosis (Bagshawe 1976). Serial measurements allow monitoring of disease progression or response to therapy (Figure 43.2). Development of drug resistance can be detected at an early stage, which facilitates appropriate changes in management. Estimates may be made of the time for which chemotherapy should be continued after hCG levels are undetectable in serum in order to reduce the tumour volume to zero. For these reasons, hCG is the best tumour marker known.
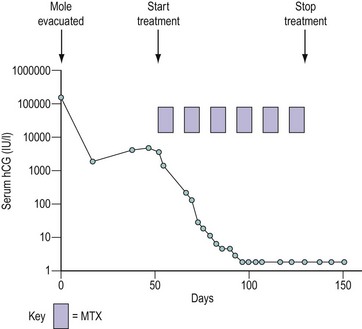
Figure 43.2 Graph demonstrating the use of monitoring the serum human chorionic gonadotrophin (hCG) concentration following evacuation of a hydatidiform mole (HM). In this case, after an initial fall, the hCG level started to rise, indicating the development of invasive HM or choriocarcinoma, so the patient was called up for staging. The prognostic score was low risk (see Table 43.4) and the patient was successfully treated with methotrexate (MTX) and folinic acid (see Table 43.5).
Clinical Features
Complete and partial moles
These most commonly present in the first trimester as a threatened abortion with vaginal bleeding. If the diagnosis is delayed, patients may notice the passing of grape-like structures (vesicles), and occasionally the entire mole may be evacuated spontaneously. The uterus may be any size but is commonly large for gestational age. Patients with marked trophoblastic growth and high hCG levels are particularly prone to hyperemesis, toxaemia and the development of theca lutein cysts which may sometimes be palpable above the pelvis. Toxaemia was diagnosed in 27% of patients with CM (Berkowitz et al 1981), but is seen less frequently today because of early US diagnosis. Convulsions are rare. The high hCG levels may also produce hyperthyroidism because of cross-reactivity between hCG and TSH at the TSH receptor. Although pulmonary, vaginal and cervical metastases can occur, they may disappear spontaneously following removal of the mole. Thus the presence of metastases does not necessarily imply that an invasive mole or choriocarcinoma has developed. Patients may rarely present with acute respiratory distress, not only because of pulmonary metastases or anaemia but occasionally as a result of tumour embolization (Savage et al 1998). The risk of embolization is reduced by avoiding agents which induce uterine contraction before the cervix has been dilated to enable evacuation of the CM.
Patients with PMs do not usually exhibit the dramatic clinical features characteristic of CM (Goldstein and Berkowitz 1994). The uterus is often not enlarged for gestational age, and vaginal bleeding tends to occur later so that patients most often present in the late first or early second trimester with a missed or incomplete abortion. In fact, the diagnosis is often only suspected when the histology of curettings is available. The pre-evacuation hCG level is less than 100,000 IU/l at diagnosis in over 90% of cases.
At present, a proportion of CMs and PMs still go undiagnosed because of miscarriage at home or because termination centres do not carry out histopathological examination of all abortions (Seckl et al 2004a). This can result in late presentation of disease, sometimes with life-threatening complications. Clearly, there is little that can be done about miscarriages at home. However, for women attending termination centres, it may be possible to establish a screening procedure to help prevent subsequent problems from missed diagnosis.
Twin pregnancies
Twin pregnancies comprising a normal fetus and an HM occur in between one in 20,000 and one in 100,000 pregnancies. Some probably abort in the first trimester and so go undiagnosed. However, some are discovered on US examination either routinely or because of complications such as bleeding, excessive uterine size or problems related to a high hCG level. With specialist obstetric care, 40% of such cases have continued into the third trimester and delivered live babies (Sebire et al 2002b).
Invasive moles
Invasive moles are usually diagnosed because serial urine or serum hCG measurements reveal a stable or rising hCG level in the weeks after molar evacuation. Patients may complain of persistent vaginal bleeding and lower abdominal pains and/or swelling. This may occur as a result of haemorrhage from leaking tumour-induced vasculature as the trophoblast invades through the myometrium, or because of vulval, vaginal or intra-abdominal metastases. The tumour may also involve other pelvic structures, including the bladder or rectum, producing haematuria or rectal bleeding, respectively. Enlarging pulmonary metastases or tumour emboli growing in the pulmonary arteries can contribute to life-threatening respiratory complications (Seckl et al 1991). The risk of these complications is clearly higher in patients where the initial diagnosis of a molar pregnancy was missed and who are not, therefore, on hCG follow-up.
Choriocarcinoma
Choriocarcinoma can present after any form of pregnancy, but most commonly occurs after a CM. Histological proof of choriocarcinoma is not usually obtained after a CM because of the risk of fatal haemorrhage caused by biopsy, and so it is impossible to distinguish from an invasive mole. Choriocarcinoma following a normal pregnancy or non-molar abortion usually presents within 1 year of delivery but can occur 17 years later (Tidy et al 1995). The presenting features may be similar to HM with vaginal bleeding, abdominal pain and a pelvic mass.
However, one-third of all choriocarcinomas present without pelvic symptoms but have symptoms from distant metastases. In these cases, lives can be saved by remembering to include choriocarcinoma in the differential diagnosis of metastatic malignancy (particularly in lungs, brain or liver) presenting in a woman of childbearing age. Any site may be involved, including skin (producing a purple lesion), cauda equina and the heart. Pulmonary disease may be parenchymal, pleural or may result from tumour embolism and subsequent growth in the pulmonary arteries (Savage et al 1998). Thus, respiratory symptoms and signs can include dyspnoea, haemoptysis and pulmonary artery hypertension. Cerebral metastases may produce focal neurological signs, convulsions, evidence of raised intracranial pressure, and intracerebral or subarachnoid haemorrhage. Hepatic metastases may cause local pain or referred pain in the right shoulder. Although none of these presentations are specific to choriocarcinoma, performing a simple pregnancy test or quantitative hCG assay can provide a vital clue to the diagnosis.
Infantile choriocarcinoma
Choriocarcinoma in the fetus or newborn is exceptionally rare, with approximately 30 reported cases (Blohm and Gobel 2004, Sebire et al 2005). While a primary choriocarcinoma within the infant is possible, the mother also had the tumour in 17 cases. Interestingly, the diagnosis was often made in the neonate before the mother. In all cases, the infant was anaemic and had a raised hCG level, but the site of metastasis was variable, including brain, liver, lung and skin. Only a few cases have been treated successfully with platinum chemotherapy (Johnson et al 2003), with the rest dying within weeks of the initial diagnosis, which may have been delayed. Consequently, serum or urine hCG levels should be measured in all babies of mothers with choriocarcinoma. As the disease can present up to 6 months after delivery, an argument could be made for serial monitoring of hCG in these infants.
Placental-site trophoblastic tumour
PSTTs grow slowly and can present years after term delivery, non-molar abortion, CM or PM. Unlike choriocarcinoma, PSTTs tend to metastasize late in their natural history, so patients frequently present with gynaecological symptoms alone. In addition to vaginal bleeding, the production of hPL by the cytotrophoblastic cells may cause hyperprolactinaemia which can result in amenorrhoea and galactorrhoea. Rarely, patients can develop nephrotic syndrome, haematuria and disseminated intravascular coagulopathy. Metastases may occur in the vagina, extrauterine pelvic tissues, retroperitoneum, lymph nodes, lungs and brain (Newlands et al 1998a).
Epithelioid trophoblastic tumours
There are currently only a few recorded cases of this recently described tumour in the literature. Although most ETT patients are women of childbearing age, a significant percentage of women are peri- or postmenopausal with a remote history of pregnancy (Coulson et al 2000). Similar to PSTTs, patients present with vaginal bleeding and low levels of serum β-hCG (<2500 IU/l). In half of the reported cases, tumours arise in the lower uterine segment or cervix, and the distinction from a keratinizing squamous cell carcinoma can sometimes be difficult. Extrauterine locations such as broad ligament as the primary site have also been observed (Kuo et al 2004). Similar to PSTTs, ETTs appear to respond less well to conventional chemotherapy compared with choriocarcinoma. Consequently, management of ETT cases is currently identical to that for PSTT cases, with hysterectomy for localized disease and careful follow-up until more experience of this rare lesion becomes available.
Investigations
Human chorionic gonadotrophin, chest X-ray and pelvic Doppler ultrasound
All patients who are suspected of having GTTs should have an hCG, a chest X-ray and pelvic Doppler ultrasound. The most common metastatic appearance on chest X-ray is of multiple, discrete, rounded lesions, but large solitary lesions, a miliary pattern or pleural effusions can occur (Bagshawe and Noble 1965). Furthermore, tumour emboli to the pulmonary arteries can produce an identical picture to venous thromboembolism, with wedge-shaped infarcts and areas of decreased vascular markings. Pulmonary artery hypertension can cause dilatation of the pulmonary arteries. Routine computed tomography (CT) scanning of the chest does not add anything to the management of these cases.
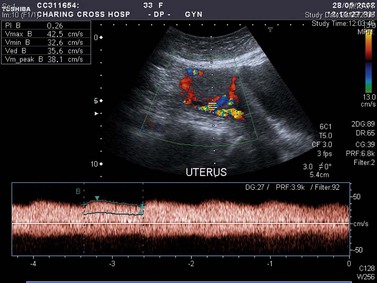
US and colour Doppler imaging are not diagnostic but highly suggestive of persistent GTD when there is a combination of a raised hCG level, no pregnancy and a vascular mass within the uterus (Figure 43.3). The last is seen in more than 75% of patients at the authors’ centre. Detection rates are higher for CM compared with PM, and improve after 14 weeks of gestation (Fowler et al 2006). The uterine volume and uterine artery blood flow correlate with the amount of disease and the degree of abnormal tumour vasculature, respectively. Doppler frequently demonstrates a change in the waveform of the uterine arteries (Figure 43.3). This is attributed to large vascular channels forming in the myometrium, resulting in arteriovenous shunting (Long et al 1992). The uterine artery pulsatility index, an indirect measure of the functional tumour vascularity, has been shown to independently predict the response to chemotherapy in GTTs (Agarwal et al 2002). Furthermore, the increased sensitivity of modern colour Doppler US now reveals abnormal blood vessel encroachment through the myometrium into the endometrium. Although this has not been shown to have prognostic significance, the degree of vascular endometrial encroachment may aid in assessing the risk of major haemorrhage in patients with GTT (Boultebee and Newlands 1995). Interestingly, the vascular abnormalities within the pelvis and uterus can persist long after the disease has been eradicated with chemotherapy. Indeed, patients with repeated vaginal haemorrhage from these vascular malformations may require selective arterial embolization. This is usually successful and does not appear to affect fertility.
Experimental imaging techniques
Radiolabelled anti-hCG antibodies given intravenously can localize tumours producing hCG when the serum hCG level is more than 100 IU/l (Begent et al 1987). However, both false-positive and false-negative results occur, so anti-hCG scanning should be regarded as complementary to other imaging investigations. More recently, positron emission tomography (PET) has provided a novel approach to image many tumour types using a variety of labels. Whole-body PET has already been reported to distinguish GTT emboli from blood clots in two patients with choriocarcinoma (Hebart et al 1996).
Other case reports demonstrate the potential value of PET in the differential diagnosis of undefined pulmonary nodules or in the localization of disease (Numnum et al 2005, Shaw et al 2005). 18Fluorodeoxyglucose (FDG)-PET may be used to locate sites of disease in cases showing serological relapse without visible abnormality on conventional imaging (Dhillon et al 2006). However, the role of PET has yet to be confirmed in larger series of patients with GTTs.
Genetic analysis
On some occasions, it can be helpful to perform a comparative genetic analysis of the patient’s trophoblastic tumour with their normal tissue and, if available, that of their partner. Thus, if the tumour is suspected of being of non-gestational origin, this can be confirmed by the presence of maternal DNA alone and the complete absence of paternal DNA. Genetic studies can also determine which of several antecedent pregnancies is the causal pregnancy of the current GTT. This can have an impact on determining appropriate therapy and prognosis (Fisher and Newlands 1998, Fisher et al 2007). In addition, genetic studies can help to identify whether a woman with repeated CM pregnancies has the rare biparental variant of CM. The latter condition is rarely compatible with a normal pregnancy, and preimplantation diagnosis is not yet possible. In contrast, a woman with repeated androgenetic CM can be offered in-vitro fertilization and preimplantation selection of unaffected concepti.
Management
Molar evacuation
Evacuation of the uterine cavity using suction gives the lowest incidence of sequelae. When the molar trophoblast invades the myometrium, it is relatively easy to perforate the uterus if a metal curette is used. Medical induction involving repeated contraction of the uterus induced by oxytocin or prostaglandin, or other surgical approaches including hysterectomy or hysterotomy increase the risk of requiring chemotherapy two- to three-fold compared with suction evacuation. This is thought to be because tumour is more likely to be disseminated by uterine contraction and manipulation. For similar reasons, the use of prostanoids to ripen a nulliparous cervix is not recommended, even in nulliparous women (Royal College of Obstetricians and Gynaecologists 2004). If bleeding is severe immediately after suction evacuation, a single dose of ergometrine to produce one uterine contraction may stem the haemorrhage, and does not appear to increase the chance of requiring chemotherapy.
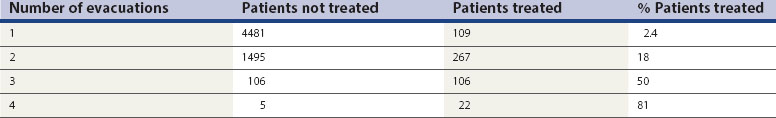
In the past, it has been common practice for gynaecologists to perform a second and sometimes a third evacuation of the uterine cavity in patients with a molar pregnancy. However, the chance of requiring chemotherapy after one evacuation is only 2.4%, but rises markedly to 18% after two evacuations and 81% after four evacuations (Table 43.1). Patients with an hCG plateau or with rising hCG levels are at particularly high risk of this, and 74% require chemotherapy despite undergoing a second evacuation. In addition, high hCG values have been shown to be an indicator for low rates of benefit of a second evacuation, with 80% of patients with a preintervention hCG level of more than 5000 IU/l subsequently requiring chemotherapy (Savage and Seckl 2005). However, second evacuations may be useful for symptom control in selected patients with heavy vaginal bleeding in the presence of persisting molar tissue, or curative if the recurrent molar tissue is confined to the uterine cavity, particularly in those with hCG levels below 1500 IU/l (Pezeshki et al 2004). Such cases should be discussed first with the local GTD centre. The use of US control during this procedure may help to reduce the risk of uterine perforation.
Twin pregnancies
At Charing Cross Hospital, there have been 77 confirmed cases of CM with a separate normal conceptus; 25% of these resulted in a live birth, while the remainder had non-viable pregnancies which mostly ended in spontaneous abortions or suction D & Cs. Interestingly, in both the viable and non-viable pregnancies, only 20% of women subsequently needed chemotherapy to eliminate persistent GTD and none of these women died of resistant disease. Furthermore, although the incidence of pre-eclampsia was 5–10% in those continuing their pregnancies, there were no maternal deaths. Thus, it appears reasonably safe to allow patients with twin pregnancies in which one of the conceptions is a CM to continue to term provided that there are no other complications. This is in line with observations on singleton CMs which suggest that later gestational age at termination does not increase the risk of subsequently requiring chemotherapy (Seckl et al 2004b).
Registration and follow-up after uterine evacuation
In the majority of cases, the molar tissue dies out spontaneously, the hCG concentration returns to normal (= 4 IU/l) and the patient can start a new pregnancy after a further 6 months. If the hCG level has fallen to normal within 8 weeks of evacuation, marker follow-up can be safely reduced to 6 months as 98% of patients requiring chemotherapy presented within this timeframe (Sebire et al 2007). Since patients who have had a previous mole or GTT are at greater risk of having another, all patients should have a further estimation of hCG at 6 and 10 weeks following the completion of each subsequent pregnancy.
Indications for chemotherapy
Factors associated with an increased risk of requiring chemotherapy are summarized in Table 43.2. The hormones in the oral contraceptive pill are probably growth factors for trophoblastic tumours; for this reason, patients are advised not to use the pill until hCG levels have returned to normal.
Table 43.2 Factors increasing the risk of requiring chemotherapy following evacuation of an hydatidiform mole
| Factor |
hCG, human chorionic gonadotrophin.
Sources: Berkowitz RS, Goldstein DP, Marean AR, Bernstein M 1981 Obstetrics and Gynecology 58: 474–477.Curry SL, Hammond CB, Tyrey L, Creasman WT, Parker RT 1975 Obstetrics and Gynecology 45: 1–8.Stone M, Dent J, Kardana A, Bagshawe KD 1976 BJOG: an International Journal of Obstetrics and Gynaecology 86: 913–916.
The indications for intervention with chemotherapy in patients who have had a CM or PM are shown in Box 43.1. An hCG value = 20,000 IU/l 4 weeks after evacuation of a mole or rising values in this range at an earlier stage indicate that the patient is at increased risk of severe haemorrhage or uterine perforation with intraperitoneal bleeding. These complications can be life-threatening and their risk can be reduced by starting chemotherapy. Withholding chemotherapy from women with metastases in the lung, vulva and vagina is only safe if the hCG levels are falling. However, chemotherapy is required if the hCG levels are not dropping or if the patient has metastases at another site, which can indicate the development of choriocarcinoma.
Prognostic factors: scoring vs International Federation of Gynaecology and Obstetrics staging
The principal prognostic variables for GTTs, which were originally identified by Bagshawe in 1976 and since modified by WHO and the authors’ own experience, are summarized in Table 43.3. Anatomical staging systems, such as that of the International Federation of Gynaecology and Obstetrics (FIGO), have been used by several centres managing GTTs. However, surgery is virtually never indicated in the initial management of this disease, so the FIGO system does not appear to add anything in treatment planning to the existing scoring system. Furthermore, the original FIGO staging system did not always predict prognosis correctly, so some patients were under- or overtreated (Smith et al 1992). This problem was overcome by modifying the FIGO system to include some of the WHO variables (Goldstein et al 1998). An international committee was then established, and recommended a new combined WHO/FIGO scoring system in 2000. This was internationally accepted in 2002, so that all centres managing this rare group of diseases can compare their results more easily (Kohorn 2002).
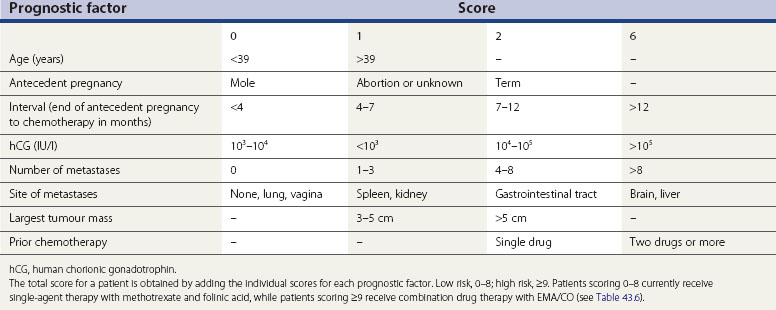
The authors currently use both the Charing Cross system and the new combined system as outlined in Tables 43.3 and 43.4. In both systems, each variable carries a score which, when added together for an individual patient, correlates with the risk of the tumour becoming resistant to single-agent therapy. It is this score, rather than the stage, that determines treatment; thus far, there appears to be excellent concordance for assigning patients to either low- or high-risk groups (see section on chemotherapy below). The most important prognostic variables carry the highest score and include:
Table 43.4 Scoring system for gestational trophoblastic tumours (World Health Organization/ International Federation of Gynaecology and Obstetrics)
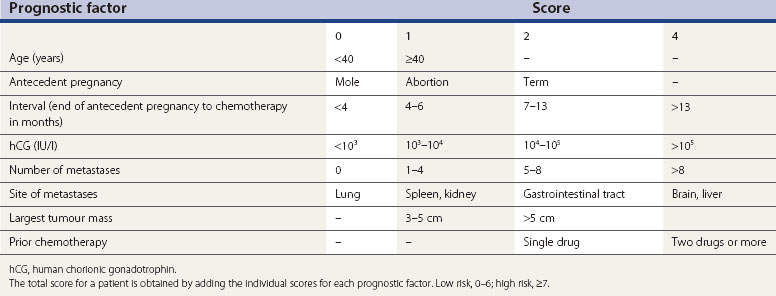
Liver metastases correlate with a worse prognosis than brain metastases (Bower et al 1997), so patients with liver involvement score six points rather than four. Since the ABO blood groups contribute little to the scoring system and it is difficult to have complete data on both patients and relevant partners, this has been dropped from the system.
Chemotherapy
At Charing Cross Hospital, the prognostic scoring system in Table 43.3 has been used to subdivide the patients into three groups (low, medium and high risk) depending on their overall score. Formerly, each risk group corresponded with a separate treatment regimen and so there were three types of therapy (low, medium and high risk). Ten years ago, the medium-risk treatment was discontinued for three reasons:
Low-risk patients
Patients with low-risk disease have a 5-year survival rate of nearly 100% (Bower et al 1997). The regimen used since 1964 at Charing Cross Hospital and widely followed in other centres is shown in Table 43.5. This schedule is well tolerated with no alopecia, and since the folinic acid dose has been increased from 7.5 mg to 15 mg, the incidence of mucosal ulceration has reduced from 20% to just 2%. Methotrexate can induce serositis, resulting in pleuritic chest pain or abdominal pain. Myelosuppression is rare, but a full blood count should be obtained before each course of treatment. Liver and renal function should also be monitored regularly. All patients are advised to avoid sun exposure or use complete sun block for 1 year after chemotherapy because the drugs can induce photosensitivity.
| Methotrexate/folinic acid | |
|---|---|
| Methotrexate | 50 mg by intramuscular injection repeated every 48 h for a total of four doses |
| Calcium folinate | 15 mg orally 30 h after each injection of methotrexate (folinic acid) |
Courses repeated every 2 weeks, i.e. days 1, 15, 29, etc.
Approximately 33% of low-risk patients will need to change therapy: 31% because of drug resistance and 2% due to treatment toxicity (usually mucositis, occasionally severe pleuritic pain or drug-induced hepatitis) (McNeish et al 2002). However, in the authors’ experience, all such patients are cured and the only deaths in this group of patients over the last 30 years were one from concurrent but not therapy-induced non-Hodgkin’s lymphoma and one from hepatitis (Bagshawe et al 1989). Moreover, there is no evidence that methotrexate alone increases the risk of developing a second cancer (Rustin et al 1996).
High-risk patients
These patients are at high risk of developing drug resistance, and since 1979 have been treated with an intensive regimen consisting of etoposide, methotrexate and actinomycin D (EMA) alternating weekly with cyclophosphamide and vincristine, otherwise known as ‘Oncovin’ (CO; see Table 43.6). A recent meta-analysis has failed to demonstrate superiority of any other chemotherapy combination (Xue et al 2006). The EMA/CO regimen requires one overnight stay every 2 weeks and causes reversible alopecia. It is also myelosuppressive, but prolonged gaps in therapy, which may permit tumour regrowth, can usually be avoided by the following measures: continuing to treat unless the white cell count is less than 1.5 × 109/l and/or platelets fall below 60 × 109/l and/or mucosal ulceration develops. The introduction of granulocyte colony-stimulating factor in patients who have a low neutrophil count also helps to maintain treatment intensity and has reduced the number of neutropenic febrile episodes.
| EMA | ||
| Day 1 | Etoposide | 100 mg/m2 by IV infusion over 30 min |
| Actinomycin D | 0.5 mg IV bolus | |
| Methotrexate | 300 mg/m2 by IV infusion over 12 h | |
| Day 2 | Etoposide | 100 mg/m2 by IV infusion over 30 min |
| Actinomycin D | 0.5 mg IV bolus | |
| Folinic acid rescue (starting 24 h after commencing the methotrexate infusion) | 15 mg IM or orally every 12 h for four doses | |
| CO | ||
| Day 8 | Vincristine | 1 mg/m2 IV bolus (max. 2 mg) |
| Cyclophosphamide | 600 mg/m2 IV infusion over 30 min | |
EMA, etoposide, methotrexate and actinomycin D; CO, cyclophosphamide and vincristine.
EMA alternates with CO every week. To avoid extended intervals between courses caused by myelosuppression, it may occasionally be necessary to reduce the EMA by omitting the day 2 doses of etoposide and actinomycin D.
The cumulative 5-year survival of patients treated with this schedule is 86%, with no deaths from GTTs beyond 2 years after the initiation of chemotherapy (Bower et al 1997). While these results were good, the presence of liver or brain metastases correlated with only 30% or 70% long-term survival, respectively. Indeed, long-term survival in patients with both liver and brain metastases is only 10% (Newlands et al 2002). Interestingly, for patients with brain metastasis, if one excludes patients who were too sick to receive chemotherapy and died within a few days of admission, the survival rates appear to be the same as those for patients without brain involvement (Newlands et al 2002). The authors are currently undertaking a new analysis on patients with liver metastasis; of 38 patients treated more recently, the overall survival rate appears to be higher at just under 50% (Seckl et al, unpublished observations). This improvement in outcome may be because of changes in treatment. Other adverse prognostic variables include a longer interval from the antecedent pregnancy, and term delivery in the antecedent pregnancy (Powles et al 2007).
Early deaths accounted for a significant proportion of the overall mortality, with the causes being respiratory failure, cerebral metastases, hepatic failure and pulmonary embolism (Bower et al 1997). Importantly, these women did not have an HM, were not registered for follow-up and consequently presented with extensive disease. Clearly, it will be difficult to improve the survival of this particular subgroup. However, any woman of childbearing age presenting with widespread malignancy should have an hCG measurement as very high levels of this hormone are highly suggestive of choriocarcinoma.
The long-term risk of chemotherapy-induced second tumours in patients treated for GTTs in the authors’ centre has been reviewed previously (Rustin et al 1996), and is discussed below in the section on long-term complications of therapy.
Management of drug-resistant disease
GTN is one of a very small minority of malignant conditions where patients who progress on or after primary chemotherapy still have a good chance of cure [>90% overall 5-year survival in a recent case series (Powles et al 2007)].
Low-risk disease
Frequent (twice-weekly) measurement of serum hCG is a simple way to detect drug resistance at an early stage as the hormone levels will stop falling and may start to rise long before there are other clinical changes. Decisions to alter treatment are not made on the basis of a single hCG result but on a progressive trend over two to three values. For patients receiving methotrexate for low-risk disease, if the hCG is below 100 IU/l when drug resistance occurs, the disease can often be cured simply by substituting actinomycin D (0.5 mg IV total dose daily for 5 days every 2 weeks) (McNeish et al 2002). This drug is more toxic than methotrexate, inducing some hair thinning (occasional complete alopecia), myelosuppression and more oral ulceration. However, it is preferable to EMA/CO. Indeed, more recently, in order to reduce the number of patients proceeding directly to this more toxic regimen, the authors have raised the hCG level for switching from methotrexate to actinomycin D therapy to less than 300 IU/l. The cure rate, even in those rare cases that need three or more lines of treatment, remains nearly 100% (Powles et al 2007).
High-risk disease
Seventy per cent of patients who have failed EMA/CO for high-risk disease can still be salvaged by further chemotherapy and/or surgery (Bower et al 1997). Indeed, the combination of surgical removal of the main site of drug resistance (usually uterus, lung or brain) together with chemotherapy is particularly effective. Preoperative investigations include those outlined above. If all of these investigations are negative, hysterectomy should be considered. Following surgery or when surgery is not appropriate, the authors use the cisplatin-containing regimen, EP (etoposide 150 mg/m2 and cisplatin 75 mg/m2 with hydration) alternating weekly with EMA (omitting day 2 except the folinic acid). Although this regimen is very toxic, the outcome has been impressive with survival rates in excess of 80% (Newlands et al 2000).
Other options that can be considered include use of some of the newer anticancer agents such as the taxanes, topotecan, gemcitabine and temozolomide. Several cases of drug-resistant GTTs that have responded to paclitaxel-based single-agent or combination therapy have been reported (Jones et al 1996, Termrungruanglert et al 1996, Osborne et al 2004, Shorbagi et al 2005). An alternating doublet of paclitaxel/cisplatin and paclitaxel/etoposide (TP/TE) has been found to be well tolerated and effective in patients with relapsed and/or refractory GTN (Wang et al 2008). Overall survival ranged from 44% for patients who failed previous cisplatin-based chemotherapy to 75% for those receiving TP/TE as a result of discontinuing prior chemotherapies due to toxicities.
Another approach in patients with refractory disease involves high-dose chemotherapy with autologous bone marrow or peripheral stem cell transplantation. Patient selection here is important in determining outcome as it has been shown for refractory germ cell tumours that patients with drug-sensitive disease stay in remission (Beyer et al 1996, Lyttelton et al 1998). In the largest series of high-dose chemotherapy for refractory GTT, five out of 11 patients showed a temporary partial or complete response, although all but one patient progressed eventually (El-Helw et al 2005). Since the time of writing, the authors have treated several more cases and now appear to have two long-term survivors. Therefore, high-dose chemotherapy with autologous stem cell support for GTN remains investigational. Further studies are needed to better define the role of high-dose chemotherapy, in particular in patients with high-risk GTN who fail their first salvage treatment for recurrent disease.
Management of acute disease-induced complications
Respiratory failure
Occasionally patients present with respiratory failure due to multiple pulmonary metastases or, more rarely, as a result of massive tumour embolism to the pulmonary circulation (Savage et al 1998). However, in the authors’ experience, these patients can be cured with appropriate management. Oxygen support may be required, including masked continuous positive airway pressure ventilation, but mechanical ventilation is usually contraindicated as it results in trauma to the tumour vasculature, leading to massive intrapulmonary haemorrhage and death.
Management of cerebral metastases
Prophylaxis against possible CNS disease, i.e. in the presence of a normal MRI brain, is given to patients from all risk categories with lung metastases and all high-risk patients regardless of the absence or presence of lung deposits. The prophylaxis consists of 12.5 mg methotrexate administered intrathecally, followed 24 h later by 15 mg folinic acid orally. This is given with every course of low-risk therapy, or with each CO in the high-risk therapy for three doses. Since the introduction of this policy, the development of brain metastases without evidence of drug resistance elsewhere has been much less frequent (Athanassiou et al 1983).
Overt CNS disease requires careful management as therapy can induce haemorrhage into the tumour, leading to a rise in intracranial pressure and subsequent loss of life (Athanassiou et al 1983). Early resection of solitary brain deposits in patients with serious neurological signs can sometimes be life-saving (Ishizuka 1983, Song and Wu 1988, Rustin et al 1989). Cerebral oedema can be reduced with high-dose corticosteroids, so patients are given 24 mg dexamethasone in divided doses before starting chemotherapy. The EMA/CO regimen is modified by increasing the dose of methotrexate to 1 g/m2, given as a 24-h infusion on day 1. The folinic acid rescue is increased to 30 mg given 8-hourly intravenously for 3 days, commencing 32 h after the start of methotrexate infusion. Provided that there is no evidence of raised intracranial pressure, 12.5 mg methotrexate is given intrathecally with each CO until the serum hCG level is normal. Modified EMA/CO is then continued for a further 6–8 weeks. Overall, long-term remission can be achieved in approximately 80% of patients with cerebral metastases (Newlands et al 2002). Patients who survive the first 3 weeks of such treatment have a good prognosis, with an 86–89% chance of cure (Athanassiou et al 1983, Rustin et al 1989, Newlands et al 2002).
Patients who develop cerebral tumours during chemotherapy have a poor prognosis because their disease is almost certainly drug resistant. Nevertheless, a combination of immediate surgery to remove the deposit(s) and modified chemotherapy designed to provide better CNS penetration can be curative in this situation (Athanassiou et al 1983, Rustin et al 1989). Radiotherapy has been advocated as an alternative therapeutic approach. However, it has not been shown to eradicate tumour in its own right, and in combination with chemotherapy has produced less effective results than chemotherapy alone. Nevertheless, stereotactic radiotherapy probably has a role in the treatment of isolated deep lesions that cannot be removed surgically, especially if still present at the end of chemotherapy.
Management of placental-site trophoblastic tumours
PSTTs are relatively resistant to combination chemotherapy regimens, and therefore hysterectomy with pelvic lymph node clearance and ovarian preservation remains the treatment of choice provided that the disease is localized to the uterus (stage I). In these patients, 10-year survival is 90% and there is no clear benefit from pre- and/or postoperative chemotherapy (Schmid et al 2008). However, for patients with disease in the resection margin or lymphovascular invasion, the authors routinely offer adjuvant EP/EMA and, more recently, TE/TP chemotherapy for 8 weeks until more data are available.
When disease has spread beyond the uterus, patients can respond to and be cured by chemotherapy (EP/EMA) either alone or in combination with surgery (Newlands et al 1998b). Ten-year survival rates are 52% for locoregional (stage II) disease and 49% for metastatic (stage III/IV) disease. In the largest series of 62 patients, 9% had refractory disease and died of PSTTs (Schmid et al 2008).
Nineteen per cent of patients treated for PSTTs experience disease recurrence, all within the first 5 years (Schmid et al 2008). In this group, the outlook is poor with a minority achieving long-term survival. Radiotherapy does not appear to add much to the management, and it is unlikely that a high dose will be curative, although the latter should be considered in chemoresponsive patients who have failed first-line therapies. The most important prognostic variable is the interval from the antecedent pregnancy (Newlands et al 2000). A cut-off of 4 years discriminates patients with a positive predictive value of 100% and a negative predictive value of 98% (Schmid et al 2008). PSTTs are so rare that it is unlikely that their treatment will ever be fully optimized.
The major question in the management of PSTTs, which frequently affect women of childbearing age, is whether fertility-sparing surgery is safe in a minority of cases with good prognostic factors and disease localized to a single uterine site. There have been five reported cases of attempted fertility-sparing surgery using a variety of surgical techniques with and without postoperative chemotherapy, with mixed results. One case had a successful pregnancy, two cases achieved long-term remission but no term pregnancy, and two cases went on to need a hysterectomy for relapsed disease (Leiserowitz and Webb 1996, Tsuji et al 2002, Machtinger et al 2005, Pfeffer et al 2007). In one case at relapse, it was found post hysterectomy that disease that appeared localized to a single uterine site on imaging, including 18FDG-PET, was in fact multifocal throughout the uterus. Any attempts at fertility-sparing surgery should be undertaken cautiously with preoperative counselling of the patient and close postoperative follow-up.
Patient Follow-Up after Chemotherapy
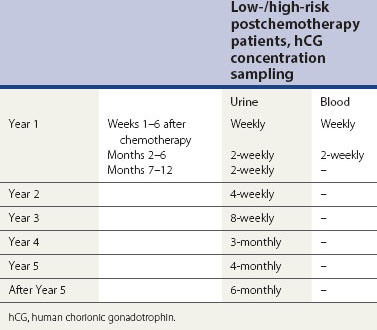
On completion of their chemotherapy, patients are followed-up regularly with hCG estimations (Table 43.7) to confirm that their disease is in remission. The risk of relapse is approximately 3% and is most likely in the first year of follow-up. However, the authors currently continue follow-up for life, until a full set of data are available to more accurately indicate when it may be safe to stop.
Timing of Pregnancy after Treatment
Patients are advised not to become pregnant until 12 months after completing their chemotherapy. This minimizes the potential teratogenicity of treatment and avoids confusion between a new pregnancy and relapsed disease as the cause of rising hCG. Despite this advice, 230 women on follow-up at the authors’ centre between 1973 and 1997 have become pregnant during the first year. Fortunately, this does not appear to be associated with an increased risk of relapse or fetal morbidity, and there were no maternal deaths. Indeed, 72% of women continued their pregnancy to term (Blagden et al 2002). Consequently, although the authors continue to advise women to avoid pregnancy for 1 year after completing chemotherapy, those that become pregnant can be reassured of a likely favourable outcome. When a patient becomes pregnant, it is important to confirm by US and other appropriate means that the pregnancy is normal. Follow-up is then discontinued until 3 weeks after the end of pregnancy, when the hCG due to the pregnancy should have returned to normal.
Patients who do not require chemotherapy following evacuation of their first mole, although not on life-long follow-up, should have their hCG levels measured 6 and 10 weeks after any subsequent pregnancy. This is because they are at increased risk compared with the general population of a further molar pregnancy (Sebire et al 2003). In addition, with a subsequent pregnancy, there is a small risk of reactivation of ‘dormant’ residual molar tissue, even if the pregnancy itself is normal.
Contraceptive Advice
Patients using oral contraceptives before the hCG level is normal following evacuation of an HM have an increased risk of developing persistent GTD (Stone et al 1976). Other centres have not been able to reproduce this finding, so this area is controversial. Nevertheless, the authors’ data set is very large; for this reason, patients in the UK are advised to avoid the oral contraceptive pill until the hCG level has returned to normal after removal of an HM. Patients who have had chemotherapy for their GTT are advised not to use the oral contraceptive pill until their hCG level is normal and chemotherapy is completed.
Long-Term Complications of Therapy
Most patients, including those who have received intensive chemotherapy, return to normal activity within a few months, and the majority of the side-effects are reversible, including alopecia. Late sequelae from chemotherapy have been remarkably rare. In 15,279 patient-years of follow-up, there was no significant increase in the incidence of second tumours (Rustin et al 1996) following methotrexate therapy. In contrast, 26 patients receiving combination chemotherapy for GTTs developed another cancer when the expected rate was only 16.45; a significant difference (Rustin et al 1996).
Fertility is an important issue in the management of patients with GTTs. Although combination chemotherapy induces the menopause 3 years earlier than expected (Bower et al 1998), fertility does not otherwise appear to be affected. In 392 patients receiving single-agent methotrexate, 327 (83.4%) had successful live births, whilst in the 336 patients receiving multi-agent chemotherapy, including EMA/CO, 280 (83.3%) also succeeded in having normal pregnancies (Woolas et al 1998). There is no increase in the incidence of congenital malformations compared with the general population (Rustin et al 1984, Woolas et al 1998).
Prognosis
All patients in the low-risk groups can be expected to be cured of their GTTs (Bagshawe and Begent 1983, Newlands et al 1986). For high-risk patients, survival has improved progressively and is currently 86% (Bower et al 1997). The diagnosis of choriocarcinoma is often not suspected until the disease is advanced. As a result, some deaths occur before chemotherapy has a chance to be effective. The number of such patients can be diminished by a greater awareness of the possibility that multiple metastases in a woman of childbearing age may be due to choriocarcinoma. The simple measurement of the hCG level in such individuals is a very strong indicator of choriocarcinoma, and could help to hasten referrals for life-saving chemotherapy.
Conclusion
KEY POINTS
Agarwal R, Strickland S, McNeish IA, et al. Doppler ultrasonography of the uterine artery and the response to chemotherapy in patients with gestational trophoblastic tumors. Clinical Cancer Reseach. 2002;8:1142-1147.
Arima T, Imamura T, Amada S, Tsuneyoshi M, Wake N. Genetic origin of malignant trophoblastic neoplasms. Cancer Genetics and Cytogenetics. 1994;73:95-102.
Athanassiou A, Begent RH, Newlands ES, Parker D, Rustin GJ, Bagshawe KD. Central nervous system metastases of choriocarcinoma: 23 years’ experience at Charing Cross Hospital. Cancer. 1983;52:1728-1735.
Baergen RN, Rutgers JL, Young RH, Osann K, Scully RE. Placental site trophoblastic tumor: a study of 55 cases and review of the literature emphasizing factors of prognostic significance. Gynecologic Oncology. 2006;100:511-520.
Bagshawe KD. Risk and prognostic factors in trophoblastic neoplasia. Cancer. 1976;38:1373-1385.
Bagshawe KD, Noble MI. Cardiorespiratory effects of trophoblastic tumours. Quarterly Journal of Medicine. 1965;137:39-54.
Bagshawe KD, Begent RH. Staging markers and prognostic factors in germ cell tumours. In: Bagshawe KD, Newlands EH, Begent RH, editors. Clinics in Oncology 2. Germ Cell Tumours; 1983:159-181.
Bagshawe KD, Dent J, Newlands ES, Begent RH, Rustin GJ. The role of low dose methotrexate and folinic acid in gestational trophoblastic tumours (GTT). BJOG: an International Journal of Obstetrics and Gynaecology. 1989;96:795-802.
Bagshawe KD, Lawler SD, Paradinas FJ, Dent J, Brown P, Boxer GM. Gestational trophoblastic tumours following initial diagnosis of partial hydatidiform mole. The Lancet. 1990;334:1074-1076.
Begent RH, Bagshawe KD, Green AJ, Searle F. The clinical value of imaging with antibody to human chorionic gonadotrophin in the detection of residual choriocarcinoma. British Journal of Cancer. 1987;55:657-660.
Berkowitz RS, Goldstein DP. Pathogenesis of gestational trophoblastic neoplasms. Pathology Annual. 1981;11:391.
Berkowitz RS, Goldstein DP, Marean AR, Bernstein M. Oral contraceptives and postmolar trophoblastic disease. Obstetrics and Gynecology. 1981;58:474-477.
Beyer J, Kramar A, Mandanas R, et al. High-dose chemotherapy as salvage treatment in germ cell tumors: a multivariate analysis of prognostic variables. Journal of Clinical Oncology. 1996;14:2638-2645.
Blagden SP, Foskett MA, Fisher RA, et al. The effect of early pregnancy following chemotherapy on disease relapse and foetal outcome in women treated for gestational trophoblastic tumours. British Journal of Cancer. 2002;86:26-30.
Blohm ME, Gobel U. Unexplained anaemia and failure to thrive as initial symptoms of infantile choriocarcinoma: a review. European Journal of Pediatrics. 2004;163:1-6.
Boultebee JE, Newlands ES. New diagnostic and therapeutic approaches to gestational trophoblastic tumours. In: Bourne TH, Jauniaux E, Jurkovic D, editors. Transvaginal Colour Doppler. The Scientific Basis and Practical Application of Colour Doppler in Gynaecology. Springer; 1995:57-65.
Bower M, Newlands ES, Holden L, et al. EMA/CO for high-risk gestational trophoblastic tumours: results from a cohort of 272 patients. Journal of Clinical Oncology. 1997;15:2636-2643.
Bower M, Rustin GJ, Newlands ES, et al. Chemotherapy for gestational trophoblastic tumours hastens menapause by 3 years. European Journal of Cancer. 1998;34:1204-1207.
Burke B, Sebire NJ, Moss J, et al. Evaluation of deletions in 7q11.2 and 8p12-p21 as prognostic indicators of tumour development following molar pregnancy. Gynecologic Oncology. 2006;103:642-648.
Chilosi M, Piazzola E, Lestani M, et al. Differential expression of p57kip2, a maternally imprinted cdk inhibitor, in normal human placenta and gestational trophoblastic disease. Laboratory Investigation. 1998;78:269-276.
Cole LA. hCG, its free subunits and its metabolites. Roles in pregnancy and trophoblastic disease. Journal of Reproductive Medicine. 1998;43:3-10.
Cole LA, Shahabi S, Butler SA, et al. Utility of commonly used commercial human chorionic gonadotropin immunoassays in the diagnosis and management of trophoblastic diseases. Clinical Chemistry. 2001;47:308-315.
Cole LA, Butler SA, Khanlian SA, et al. Gestational trophoblastic diseases: 2. Hyperglycosylated hCG as a reliable marker of active neoplasia. Gynecologic Oncology. 2006;102:151-159.
Cole LA, Khanlian SA, Muller CY, Giddings A, Kohorn E, Berkowitz R. Gestational trophoblastic diseases: 3. Human chorionic gonadotropin-free beta-subunit, a reliable marker of placental site trophoblastic tumors. Gynecologic Oncology. 2006;102:160-164.
Coulson LE, Kong CS, Zaloudek C. Epithelioid trophoblastic tumor of the uterus in a postmenopausal woman: a case report and review of the literature. American Journal of Surgical Pathology. 2000;24:1558-1562.
Curry SL, Hammond CB, Tyrey L, Creasman WT, Parker RT. Hydatidiform moles; diagnosis, management and long term follow-up of 347 patients. Obstetrics and Gynecology. 1975;45:1-8.
Dhillon T, Palmieri C, Sebire NJ, et al. Value of whole body 18FDG-PET to identify the active site of gestational trophoblastic neoplasia. Journal of Reproductive Medicine. 2006;51:879-887.
El-Helw LM, Seckl MJ, Haynes R, et al. High-dose chemotherapy and peripheral blood stem cell support in refractory gestational trophoblastic neoplasia. British Journal of Cancer. 2005;93:620-621.
Fisher RA, Soteriou BA, Meredith L, Paradinas FJ, Newlands ES. Previous hydatidiform mole identified as the causative pregnancy of choriocarcinoma following birth of normal twins. International Journal of Cancer. 1995;5:64-70.
Fisher RA, Newlands ES. Gestational trophoblastic disease: molecular and genetic studies. Journal of Reproductive Medicine. 1998;43:81-97.
Fisher RA, Hodges MD, Rees HC, et al. The maternally transcribed gene p57(KIP2) (CDNK1C) is abnormally expressed in both androgenetic and biparental complete hydatidiform moles. Human Molecular Genetics. 2002;11:3267-3272.
Fisher RA, Savage PM, MacDermott C, et al. The impact of molecular genetic diagnosis on the management of women with hCG-producing malignancies. Gynecologic Oncology. 2007;107:413-419.
Fowler DJ, Lindsay I, Seckl MJ, Sebire NJ. Routine pre-evacuation ultrasound diagnosis of hydatidiform mole: experience of more than 1000 cases from a regional referral center. Ultrasound in Obstetrics and Gynecology. 2006;27:56-60.
Genest DR, Ruiz RE, Weremowicz S, Berkowitz RS, Goldstein DP, Dorfman DM. Do nontriploid partial hydatidiform moles exist? A histologic and flow cytometric reevaluation of nontriploid specimens. Journal of Reproductive Medicine. 2002;47:363-368.
Goldstein DP, Berkowitz RS. Current management of complete and partial molar pregnancy. Journal of Reproductive Medicine. 1994;39:139-146.
Goldstein DP, Zanten-Przybysz IV, Bernstein MR, Berkowitz RS. Revised FIGO staging system for gestational trophoblastic tumors. Journal of Reproductive Medicine. 1998;43:37-43.
Hando T, Ohno M, Kurose T. Recent aspects of gestational trophoblastic disease in Japan. International Journal of Gynaecology and Obstetrics. 1998;60(Suppl 1):S71-S76.
Hao Y, Crenshaw T, Moulton T, Newcomb E, Tycko B. Tumor suppressor activity of H19 RNA. Nature. 1993;365:764-767.
Harvey RA, Pursglove HD, Schmid P, Savage PM, Mitchell HD, Seckl MJ. Human chorionic gonadotrophin free beta-subunit measurement as a marker of placental site trophoblastic tumours. Journal of Reproductive Medicine. 2008;53:643-648.
Hashimoto K, Azuma C, Koyama M, et al. Loss of imprinting in choriocarcinoma. Nature Genetics. 1995;9:109-110.
Hassadia A, Gillespie A, Tidy J, et al. Placental site trophoblastic tumour: clinical features and management. Gynecologic Oncology. 2005;99:603-607.
Hebart H, Erley C, Kaskas B, et al. Positron emission tomography helps to diagnose tumor emboli and residual disease in choriocarcinoma. Annals of Oncology. 1996;7:416-418.
Hui P, Martel M, Parkash V. Gestational trophoblastic diseases: recent advances in histopathologic diagnosis and related genetic aspects. Advances in Anatomic Pathology. 2005;12:116-125.
Ishizuka T. Intracranial metastases of choriocarcinoma: a clinicopathologic study. Cancer. 1983;52:1896-1903.
Johnson EJ, Crofton PM, O’Neill JM, et al. Infantile choriocarcinoma treated with chemotherapy alone. Medical and Pediatric Oncology. 2003;41:550-557.
Jones WB, Schneider J, Shapiro F, Lewis JLJr. Treatment of resistant gestational choriocarcinoma with taxol: a report of two cases. Gynecologic Oncology. 1996;61:126-130.
Judson H, Hayward BE, Sheridan E, Bonthron DT. A global disorder of imprinting in the human female germ line. Nature. 2002;416:539-542.
Kohorn EI. Negotiating a staging and risk factor scoring system for gestational trophoblastic neoplasia. A progress report. Journal of Reproductive Medicine. 2002;47:445-450.
Kuo KT, Chen MJ, Lin MC. Epithelioid trophoblastic tumor of the broad ligament: a case report and review of the literature. American Journal of Surgical Pathology. 2004;28:405-409.
Kurman RJ, Scully RE, Norris HJ. Trophoblastic pseudotumor of the uterus: an exaggerated form of syncytial endometritis simulating a malignant tumor. Cancer. 1976;38:1214-1226.
Lawler SD, Fisher RA, Pickthall VJ, Povey S, Evans MW. Genetic studies on hydatidiform moles I: the origin of partial moles. Cancer Genetics and Cytogenetics. 1982;4:309-320.
Leiserowitz GS, Webb MJ. Treatment of placental site trophoblastic tumor with hysterotomy and uterine reconstruction. Obstetrics and Gynecology. 1996;88:696-699.
Long MG, Boultbee JE, Langley R, Newlands ES, Begent RH, Bagshawe KD. Doppler assessment of the uterine circulation and the clinical behaviour of gestational trophoblastic tumours requiring chemotherapy. British Journal of Cancer. 1992;66:883-887.
Lyttelton MP, Newlands ES, Giles C, et al. High-dose therapy including carboplatin adjusted for renal function in patients with relapsed germ cell tumor: outcome and prognostic factors. British Journal of Cancer. 1998;77:1672-1676.
Machtinger R, Gotlieb WH, Korach J, et al. Placental site trophoblastic tumor: outcome of five cases including fertility preserving management. Gynecologic Oncology. 2005;96:56-61.
Matsuda T, Sasaki M, Kato H, et al. Human chromosome 7 carries a putative tumor suppressor gene(s) involved in choriocarcinoma. Oncogene. 1997;15:2773-2781.
Matsuoka S, Thompson JS, Edwards MC, et al. Imprinting of the gene encoding a human cyclin-dependent kinase inhibitor, p57kip2, on chromosome 11p15. Proceedings of the National Academy of Sciences USA. 1996;93:3026-3030.
McNeish IA, Strickland S, Holden L, et al. Low risk persistent gestational trophoblastic disease: outcome following initial treatment with low-dose methotrexate and folinic acid, 1992–2000. Journal of Clinical Oncology. 2002;20:1838-1844.
Mitchell H, Seckl MJ. Discrepancies between commercially available immunoassays in the detection of tumour-derived hCG. Molecular and Cellular Endocrinology. 2007;260–262:310-313.
Murdoch S, Djuric U, Mazhar B, et al. Mutations in NALP7 cause recurrent hydatidiform moles and reproductive wastage in humans. Nature Genetics. 2006;38:300-302.
Newlands ES, Bagshawe KD, Begent RH, Rustin GJ, Holden L, Dent J. Development of chemotherapy for medium- and high-risk patients with gestational trophoblastic tumours 1979–1984. BJOG: an International Journal of Obstetrics and Gynaecology. 1986;93:63-69.
Newlands ES, Bower M, Fisher RA, Paradinas FJ. Management of placental site trophoblastic tumours. Journal of Reproductive Medicine. 1998;43:53-59.
Newlands ES, Paradinas FJ, Fisher RA. Recent advances in gestational trophoblastic disease. Hematology/Oncology Clinics of North America. 1998;13:225-244.
Newlands ES, Mulholland PJ, Holden L, Seckl MJ, Rustin GJ. Etoposide and cisplatin/etoposide, methotrexate, and actinomycin D (EMA) chemotherapy for patients with high-risk gestational trophoblastic tumors refractory to EMA/cyclophosphamide and vincristine chemotherapy and patients presenting with metastatic placental site trophoblastic tumors. Journal of Clinical Oncology. 2000;18:854-859.
Newlands ES, Holden L, Seckl MJ, McNeish I, Strickland S, Rustin GJ. Management of brain metastases in patients with high-risk gestational trophoblastic tumors. Journal of Reproductive Medicine. 2002;47:465-471.
Numnum TM, Leath CA3rd, Straughn JMJr, Conner MG, Barnes MN3rd. Occult choriocarcinoma discovered by positron emission tomography/computed tomography imaging following a successful pregnancy. Gynecologic Oncology. 2005;97:713-715.
Ogawa O, Eccles MR, Szeto J, et al. Relaxation in insulin-like growth factor II gene imprinting implicated in Wilm’s tumour. Nature. 1993;362:749-751.
Osborne R, Covens A, Mirchandani D, Gerulath A. Successful salvage of relapsed high-risk gestational trophoblastic neoplasia patients using a novel paclitaxel-containing doublet. Journal of Reproductive Medicine. 2004;49:655-661.
Palmieri C, Fisher RA, Sebire NJ, et al. Placental site trophoblastic tumour arising from a partial hydatidiform mole. The Lancet. 2005;366:688.
Papadopoulos AJ, Foskett M, Seckl MJ, et al. Twenty-five years’ clinical experience of placental site trophoblastic tumors. Journal of Reproductive Medicine. 2002;47:460-464.
Paradinas FJ. The diagnosis and prognosis of molar pregnancy. The experience of the National Referral Centre in London. International Journal of Gynaecology and Obstetrics. 1998;60(Suppl 1):S57-S64.
Pezeshki M, Hancock BW, Silcocks P, et al. The role of repeat uterine evacuation in the management of persistent gestational trophoblastic disease. Gynecologic Oncology. 2004;95:423-429.
Pfeffer PE, Sebire N, Lindsay I, McIndoe A, Lim A, Seckl MJ. Fertility-sparing partial hysterectomy for placental-site trophoblastic tumour. Lancet Oncology. 2007;8:744-746.
Popiolek DA, Yee H, Mittal K, et al. Multiplex short tandem repeat DNA analysis confirms the accuracy of p57(KIP2) immunostaining in the diagnosis of complete hydatidiform mole. Human Pathology. 2006;37:1426-1434.
Powles T, Savage PM, Stebbing J, et al. A comparison of patients with relapsed and chemo-refractory gestational trophoblastic neoplasia. British Journal of Cancer. 2007;96:732-737.
Royal College of Obstetricians and Gynaecologists 2004 The Management of Gestational Trophoblastic Neoplasia. Guideline No. 38. RCOG, London.
Rustin GJ, Booth M, Dent J, Salt S, Rustin F, Bagshawe KD. Pregnancy after cytotoxic chemotherapy for gestational trophoblastic tumours. BMJ (Clinical Research Ed.). 1984;288:103-106.
Rustin GJ, Newlands ES, Begent RH, Dent J, Bagshawe KD. Weekly alternating chemotherapy (EMA/CO) for treatment of central nervous systems of choriocarcinoma. Journal of Clinical Oncology. 1989;7:900-903.
Rustin GJ, Newlands ES, Lutz JM, et al. Combination but not single agent methotrexate chemotherapy for gestational trophoblastic tumours (GTT) increases the incidence of second tumours. Journal of Clinical Oncology. 1996;14:2769-2773.
Savage P, Roddie M, Seckl MJ. A 28-year-old woman with a pulmonary embolus. The Lancet. 1998;352:30.
Savage P, Seckl MJ. The role of repeat uterine evacuation in trophoblast disease. Gynecologic Oncology. 2005;99:251-252. author reply 252–253
Schmid P, Nagai Y et al 2008 Prognostic markers and long-term outcome of placental-site trophoblastic tumours: 30 years of UK experience. Submitted to The Lancet.
Sebire NJ, Foskett M, Fisher RA, Rees H, Seckl M, Newlands ES. Risk of partial and complete hydatidiform molar pregnancy in relation to maternal age. BJOG: an International Journal of Obstetrics and Gynaecology. 2002;109:99-102.
Sebire NJ, Foskett M, Paradinas FJ, et al. Outcome of twin pregnancies with complete hydatidiform mpole and healthy co-twin. The Lancet. 2002;359:2165-2166.
Sebire NJ, Fisher RA, Foskett M, Rees H, Seckl MJ, Newlands ES. Risk of recurrent hydatidiform mole and subsequent pregnancy outcome following complete or partial hydatidiform molar pregnancy. BJOG: an International Journal of Obstetrics and Gynaecology. 2003;110:22-26.
Sebire NJ, Rees HC, Peston D, Seckl MJ, Newlands ES, Fisher RA. p57(KIP2) immunohistochemical staining of gestational trophoblastic tumours does not identify the type of the causative pregnancy. Histopathology. 2004;45:135-141.
Sebire NJ, Lindsay I, Fisher RA, Seckl MJ. Intraplacental choriocarcinoma: experience from a tertiary referral center and relationship with infantile choriocarcinoma. Fetal and Pediatric Pathology. 2005;24:21-29.
Sebire NJ, Foskett M, Short D, et al. Shortened duration of human chorionic gonadotrophin surveillance following complete or partial hydatidiform mole: evidence for revised protocol of a UK regional trophoblastic disease unit. BJOG: an International Journal of Obstetrics and Gynaecology. 2007;114:760-762.
Seckl MJ, Rustin GJ, Newlands ES, Gwyther SJ, Bomanji J. Pulmonary embolism, pulmonary hypertension, and choriocarcinoma. The Lancet. 1991;338:1313-1315.
Seckl MJ, Fisher RA, Salerno G, et al. Choriocarcinoma and partial hydatidiform moles. The Lancet. 2000;356:36-39.
Seckl MJ, Gillmore R, Foskett M, Sebire NJ, Rees H, Newlands ES. Routine terminations of pregnancy — should we screen for gestational trophoblastic neoplasia. The Lancet. 2004;364:705-707.
Seckl MJ, Dhillon T, Dancey G, et al. Increased gestational age at evacuation of a complete hydatidiform mole: does it correlate with increased risk of requiring chemotherapy? Journal of Reproductive Medicine. 2004;49:527-530.
Shaw SW, Wang CW, Ma SY, Ng KK, Chang TC. Exclusion of lung metastases in placental site trophoblastic tumor using [18F]fluorodeoxyglucose positron emission tomography: a case report. Gynecologic Oncology. 2005;99:239-242.
Shih IM, Kurman RJ. Ki-67 labeling index in the differential diagnosis of exaggerated placental site, placental site trophoblastic tumour, and choriocarcinoma: a double staining technique using Ki-67 and Mel-CAM antibodies. Human Pathology. 1998;29:27-33.
Shih IM, Kurman RJ. The pathology of intermediate trophoblastic tumors and tumor-like lesions. International Journal of Gynecological Pathology. 2001;20:31-47.
Shorbagi A, Aksoy S, Kilickap S, Güler N. Successful salvage therapy of resistant gestational trophoblastic disease with ifosfamide and paclitaxel. Gynecologic Oncology. 2005;97:722-723.
Smith DB, Holden L, Newlands ES, Bagshawe KD. Correlation between clinical staging (FIGO) and prognostic groups in gestational trophoblastic disease. BJOG: an International Journal of Obstetrics and Gynaecology. 1992;100:157-160.
Song HZ, Wu PC. Treatment of brain metastases in choriocarcinoma and invasive mole. In: Song HZ, Wu PC, editors. Studies in Trophoblastic Disease in China. Oxford: Pergamon; 1988:231-237.
Stone M, Dent J, Kardana A, Bagshawe KD. Relationship of oral contraceptive to development of trophoblastic tumour after evacuation of hydatidiform mole. BJOG: an International Journal of Obstetrics and Gynaecology. 1976;86:913-916.
Termrungruanglert W, Kudelka AP, Piamsomboon S, et al. Remission of refractory gestational trophoblastic disease with high-dose paclitaxel. Anti-Cancer Drugs. 1996;7:503-506.
Tham BW, Everard JE, Tidy JA, Drew D, Hancock BW. Gestational trophoblastic disease in the Asian population of Northern England and North Wales. BJOG: an International Journal of Obstetrics and Gynaecology. 2003;110:555-559.
Tidy JA, Rustin GJ, Newlands ES, et al. Presentation and management of choriocarcinoma after nonmolar pregnancy. BJOG: an International Journal of Obstetrics and Gynaecology. 1995;102:715-719.
Tsuji Y, Tsubamoto H, Hori M, Ogasawara T, Koyama K. Case of PSTT treated with chemotherapy followed by open uterine tumor resection to preserve fertility. Gynecologic Oncology. 2002;87:303-307.
Vaitukaitis JL. Human chorionic gonadotrophin — a hormone secreted for many reasons. New England Journal of Medicine. 1979;301:324-326.
Walsh C, Miller SJ, Flam F, Fisher RA, Ohlsson R. Paternally derived H19 is differentially expressed in malignant and non-malignant trophoblast. Cancer Research. 1995;55:1111-1116.
Wang J, Short D, Sebire NJ, et al. Salvage chemotherapy of relapsed or high-risk gestational trophoblastic neoplasia (GTN) with paclitaxel/cisplatin alternating with paclitaxel/etoposide (TP/TE). Annals of Oncology. 2008;19:1578-1583.
Wells M. The pathology of gestational trophoblastic disease: recent advances. Pathology. 2007;39:88-96.
World Health Organization. Gestational Trophoblastic Diseases. Technical Report Series 692. Geneva: WHO; 1983. pp 7–81
Woolas RP, Bower M, Newlands ES, Seckl M, Short D, Holden L. Influence of chemotherapy for gestational trophoblastic disease on subsequent pregnancy outcome. BJOG: an International Journal of Obstetrics and Gynaecology. 1998;105:1032-1035.
Xue Y, Zhang J, Wu TX, An RF 2006 Combination chemotherapy for high-risk gestational trophoblastic tumour. Cochrane Database Systematic Reviews 3: CD005196.
Zhao J, Moss J, Sebire NJ, et al. Analysis of the chromosomal region 19q13.4 in two Chinese families with recurrent hydatidiform mole. Human Reproduction. 2006;21:536-541.
[/level-membership-for-obstetrics-gynecology-category][not-level-membership-for-obstetrics-gynecology-category]
CHAPTER 43 Gestational trophoblastic tumours
Introduction
The World Health Organization (WHO) has classified gestational trophoblastic disease (GTD) into two premalignant diseases, termed ‘complete hydatidiform mole’ (CM) and ‘partial hydatidiform mole’ (PM), and three malignant disorders, termed ‘invasive mole’, ‘gestational choriocarcinoma’ and ‘placental-site trophoblastic tumour’ (PSTT) (World Health Organization 1983). These malignant disorders are also frequently referred to as ‘gestational trophoblastic tumours’ (GTTs) or ‘neoplasia’ (GTN). GTTs are important to recognize because they are nearly always curable and fertility can be preserved in most cases. This is mainly because:
Genetics and Pathology
Complete hydatidiform mole
CMs nearly always contain paternal DNA alone and are therefore androgenetic. This occurs in most cases because a single sperm bearing a 23X set of chromosomes fertilizes an ovum lacking maternal genes, and then duplicates to form the homozygote, 46XX (Figure 43.1A). However, in up to 25% of CMs, fertilization can take place with two spermatozoa, resulting in the heterozygous 46XY or 46XX configuration (Figure 43.1B). A 46YY conceptus has not yet been described and is presumably non-viable. Very rarely, a CM can arise from a fertilized ovum which has retained its maternal nuclear DNA and is therefore biparental in origin (Fisher and Newlands 1998). Macroscopically, the classic CM resembles a bunch of grapes due to generalized (complete) swelling of chorionic villi. However, this appearance is only seen in the second trimester and the diagnosis is usually made earlier when the villi are much less hydropic. Indeed, in the first trimester, the villi microscopically contain little fluid, are branching and consist of hyperplastic syncytio- and cytotrophoblast with many vessels. Although it was previously thought that CM produced no fetal tissue, histology from 6–8-week abortions reveals evidence of embryonic elements, including fetal red cells (Paradinas 1998). This has resulted in pathologists incorrectly labelling CMs as PMs. The presence of embryonic tissue from a twin pregnancy comprising a fetus and a CM is another source of error which can lead to the incorrect diagnosis of PM.
Partial hydatidiform mole
Genetically, PMs are nearly all triploid with two paternal and one maternal sets of chromosomes (Figure 43.1C). Rarely, tetraploid PMs are found. Triploidy occurs in 1–3% of all recognized conceptions and in approximately 20% of spontaneous abortions with an abnormal karyotype, but at least two paternal sets of chromosomes are needed for PM development. Triploids due to two sets of maternal chromosome do not become PMs (Lawler et al 1982). Although a variety of reports have suggested that diploid PMs exist, genetic analysis of such lesions has not supported this. In general, a diploid molar gestation is believed to be a CM (Genest et al 2002). Flow cytometry, which can be done in formalin-fixed, paraffin-embedded tissues (Seckl et al 2000), can help in differentiating CM from PM, and PM from diploid non-molar hydropic abortions.
In PMs, villous swelling is less intense and only affects some villi. Both swollen and non-swollen villi can have trophoblastic hyperplasia which is mild and focal. The villi have characteristic indented outlines and round inclusions. An embryo is usually present and can be recognized macroscopically or inferred from the presence of nucleated red cells in villous vasculature. It may survive into the second trimester, but usually dies at approximately 8–9 weeks of gestation and this is followed by loss of vessels and stromal fibrosis. In PMs evacuated early, villous swelling and trophoblastic excess can be so mild and focal that the diagnosis of PM may be missed (Paradinas 1998). Indeed, at uterine evacuation for a ‘miscarriage’, it is likely that many PMs are misclassified as products of conception. Fortunately, the authors only see about one patient per year with persistent GTD related to a previously unrecognized PM. Of the increasing number of PMs which are correctly diagnosed, very few go on to develop persistent GTD. Indeed, in approximately 3000 PMs reviewed and followed at Charing Cross Hospital between 1973 and 1997, only 15 (0.5%) have required chemotherapy (Seckl et al 2000).
Other pregnancies mistaken for hydatidiform mole
Over half of first-trimester non-molar abortions are due to trisomy, monosomy, maternally derived triploidy and translocations. These often develop hydrops but are small (<3 mm), and PM can be excluded if they are diploid on flow cytometry. A significant recent development in the pathological analysis of hydatidiform mole (HM) is the use of p57kip2 immunostaining to make a definitive diagnosis of androgenetic CM as opposed to a hydropic abortion or a PM. p57kip2, a cyclin-dependent kinase inhibitor, is a paternally imprinted gene which is maternally expressed. The absence of maternal genes in androgenetic CM means that the gene cannot be expressed in CM cytotrophoblast. Consequently, p57kip2 staining is negative with CMs, in contrast to PMs, hydropic abortion and normal placenta. This technique is well validated, easy and inexpensive to perform (Sebire et al 2004, Popiolek et al 2006).
Syndromes such as Turner’s, Edward’s and Beckwith-Wiedemann’s can also cause histological confusion with PMs (Paradinas 1998).
Invasive hydatidiform mole
Invasive hydatidiform mole is common and is clinically identified by the combination of an abnormal uterine ultrasound (US) and a persistent or rising hCG level following uterine evacuation of a CM or PM. Pathological confirmation of this condition is rarely required. Moreover, repeat dilatation and curettage (D & C) is often contraindicated because of the risks of uterine perforation, infection, life-threatening haemorrhage and subsequent hysterectomy. In occasional cases where histology is available, invasive mole can be distinguished from choriocarcinoma by the presence of chorionic villi. Identification of cases of CM and PM that will subsequently undergo malignant transformation would be an important development. There is accumulating evidence that such HMs are more likely to show telomerase activity (i.e. have limitless replicative potential) and increased expression of the anti-apoptotic gene Mcl-1 (Wells 2007). These findings need further prospective evaluation.
Choriocarcinoma
Most choriocarcinomas have been shown to have grossly abnormal karyotypes with diverse ploidies and several chromosome rearrangements, none of which are specific for the disease (Arima et al 1994). Studies of the origin of GTTs have confirmed that choriocarcinoma may arise from any type of pregnancy including a normal term pregnancy, or homozygous or heterozygous CM. Until recently, it was thought that PMs could not give rise to choriocarcinoma. However, there is now incontrovertible genetic evidence that PMs can indeed transform into choriocarcinomas (Seckl et al 2000). This is important as some centres wrongly believe that it is safe to discontinue hCG follow-up following the diagnosis of a PM.
Interestingly, choriocarcinoma may not always be due to the antecedent pregnancy. A patient with a history of CM 4 years previously developed choriocarcinoma following the delivery of a twin pregnancy. Using polymerase chain reaction to amplify short tandem repeat polymorphisms in DNA, this tumour was shown to be genetically identical to the previous CM (Fisher et al 1995). As for invasive moles, obtaining tissue to make a formal histological diagnosis of choriocarcinoma is often not appropriate, so doubt frequently exists regarding whether patients have one or the other form of GTT.
Placental-site trophoblastic tumour
PSTTs have been shown to follow term delivery, non-molar abortion or CM. Furthermore, a recent case has shown that PSTTs can develop after a PM (Palmieri et al 2005). Like choriocarcinoma, the causative pregnancy may not be the immediate antecedent pregnancy (Fisher et al 1995). Genetic analysis of some PSTTs has demonstrated that they are mostly diploid, originating from either a normal conceptus and therefore biparental, or androgenetic and arising from a CM (Newlands et al 1998a).
In the normal placenta, placental-site trophoblast is distinct from villous trophoblast and infiltrates the decidua, myometrium and spiral arteries of the uterine wall. PSTTs are rare, slow-growing malignant tumours composed mainly of intermediate trophoblast derived from cytotrophoblast, and so produce little hCG. However, they often stain strongly for human placental lactogen (hPL) and β1-glycoprotein. Elevated Ki-67 levels may help in distinguishing PSTTs from regressing placental nodules (Shih and Kurman 1998). In contrast to other forms of GTT, spread tends to occur late by local infiltration and via the lymphatics, although distant metastases can occur. More than five mitoses per 10 high-power fields may predict tumours with metastasizing potential (Newlands et al 1998a).
Epithelioid trophoblastic tumour
Epithelioid trophoblastic tumours (ETTs) are a recently described neoplastic proliferation of intermediate trophoblast that are thought, by some investigators, to be distinct from PSTTs and choriocarcinoma. It has been proposed that ETTs arise from the intermediate trophoblasts of the chorionic laeve (Shih and Kurman 2001). Histologically, ETTs display a relatively uniform, nodular proliferation of intermediate-sized epithelioid trophoblasts, forming nests and cords. Islands of trophoblast are typically surrounded by areas of hyalinization or eosinophilic debris simulating tumour cell necrosis and resembling keratinous material in a squamous cell carcinoma. ETTs can be associated with focal replacement of the cervical glandular epithelium with stratified neoplastic cells, simulating squamous cervical intraepithelial neoplasia. ETT cells are positive for cytokeratin, epithelial membrane antigen and inhibin-A, whereas trophoblastic markers hPL, hCG and melanoma cell adhesion molecular are only expressed focally (Hui et al 2005).
Epidemiology and Aetiological Factors
Hydatidiform mole
Incidence and ethnic origin
The incidence of HM in the UK is one in 714 pregnancies (Tham et al 2003). Recent results indicate that the previously documented higher rates in the Far East have fallen towards the stable levels found in Europe and North America (Hando et al 1998), possibly because of dietary changes. The incidence of PM has been underestimated in the past and is currently three per 1000 pregnancies (Newlands et al 1998b).
Age
CMs are more common at the extremes of reproductive age. In one study, compared with the lowest rate between 25–29 years, the relative increased risk was six-fold in girls under 15 years, three-fold between 40 and 45 years, 26-fold between 45 and 49 years and more than 400-fold over 50 years of age (Bagshawe and Begent 1983, Newlands et al 1998b). PMs are also more common at the extremes of reproductive age, although the effect is less pronounced (Sebire et al 2002a).
Previous pregnancies
Increasing gravidity does not increase the risk of CM. However, following one CM, the risk of a subsequent pregnancy being a CM rises from one in 1000 to one in 76, and to one in 6.5 with two previous CMs (Bagshawe and Begent 1983). Therefore, patients with a previous CM must be followed-up after each subsequent pregnancy to confirm that their hCG levels return to normal. Similar results have been reported for PMs, with the risk for subsequent molar pregnancies rising to one in 59 for women with one previous PM (Sebire et al 2003).
Choriocarcinoma
The incidence of choriocarcinoma following term delivery without a history of CM is approximately one in 50,000. However, CM is probably the most common antecedent to choriocarcinoma, comprising 29–83% in various studies across the world (World Health Organization 1983). Consequently, the overall incidence of choriocarcinoma after a CM is much higher. Proof of this is frequently difficult to obtain, but when histology was available, these tumours were identified as choriocarcinoma in 3% and invasive mole in 16% of previous CMs (seldom as PSTT). Rarely, PMs can give rise to choriocarcinoma (Seckl et al 2000). Unlike HM, choriocarcinoma does not exhibit any clear geographical trends in incidence, but the effect of age remains important.
Placental-site trophoblastic tumours
First described as a separate disease entity in 1976 (Kurman et al 1976), there are currently approximately 150 recorded cases of this tumour in the literature; therefore, estimates of its true incidence may be quite inaccurate (Newlands et al 1998a, Papadopoulos et al 2002, Hassadia et al 2005, Baergen et al 2006). In the largest series described — 62 patients between 1976 and 2006 — PSTTs represented 0.2% of all trophoblastic tumours (Schmid et al 2008) The relative incidence in the UK has been stable over the past 15 years following an increase in the first years after the introduction of this new disease classification.
Genetic Factors: The Role of Imprinting
Hydatidiform mole
All autosomal genes consist of two alleles (paternal and maternal). However, some alleles are only expressed from one parent and not the other; a phenomenon called ‘genomic imprinting’. Underexpression of an imprinted gene has been demonstrated in cases of both familial and sporadic CM, and abnormal methylation patterns have been described in another family (Fisher et al 2002, Judson et al 2002).
Extensive mapping studies have been performed on the families with CM. These have demonstrated a defective locus at 19q13.4 in five families which has been localized to a single gene — NALP7 (Murdoch et al 2006). This is the first causative single gene defect identified in CM. NALP7 is a member of the CATERPILLER family which is involved in inflammation and apoptosis. It is expressed in oocytes and the endometrium, and is a negative regulator of the proinflammatory cytokine interleukin-1β, which is involved in regulating trophoblast invasion during implantation. It is unclear how defects in NALP7 result in CM formation, but abnormal inflammation during embryogenesis may be involved.
NALP7 does not appear to be involved in the establishment of imprinting; therefore, defects in NALP7 may be a consequence of abnormal imprinting rather than its cause. Further families have been identified that do not map to 19q13.4, proving that familial CM is a genetically heterogeneous condition (Zhao et al 2006). Moreover, to date, abnormal NALP7 has not been demonstrated in sporadic CM or PM cases.
Three other, closely related, genes are imprinted and may be involved in GTT development. These are H19, a putative tumour suppressor gene (Hao et al 1993), and p57kip2 (discussed previously) (Matsuoka et al 1996), both of which are normally expressed by the maternal allele; and the paternally expressed IGF-2, a growth factor commonly implicated in tumour proliferation (Ogawa et al 1993). While p57kip2 showed the expected pattern of expression in CM and choriocarcinoma (Chilosi et al 1998), CM and postmole tumours were unexpectedly found to express H19 (Walsh et al 1995), and some post-term tumours showed biallelic expression of both H19 and IGF-2 (Hashimoto et al 1995). This suggests that loss of the normal imprinting patterns of these genes may be an important factor in the development of GTT.
Choriocarcinoma
Historically, cytogenetic studies of choriocarcinoma have demonstrated a diverse range of abnormalities. Microsatellite studies have shown loss of heterozygosity at specific regions: deletions of 7p12-q11.2, amplification of 7q21-q31 and loss of 8p12-p21 (Matsuda et al 1997) Abnormalities in the latter two are also found in some cases of ovarian and breast cancer, and are postulated to encode novel tumour suppressor genes. Several groups have reproduced these findings in non-molar choriocarcinoma, but results in postmolar choriocarcinoma have been inconsistent (Burke et al 2006). Only a minority of tumours showed loss of heterozygosity for all three regions, suggesting that these defects are acquired late in the development of choriocarcinoma and are not essential for malignant transformation.
Risk of Gestational Trophoblastic Tumours Following Complete or Partial Hydatidiform Mole
Following evacuation of a CM or PM, the risk of developing a GTT is less than 16% and 0.5%, respectively (Bagshawe et al 1990, Seckl et al 2000). Since it is not yet possible to predict which patients with a CM or PM will develop persistent GTD, all patients must be registered for hCG monitoring. Following this strict protocol enables the identification of individuals with persistent trophoblastic growth who could benefit from life-saving chemotherapy.
Human Chorionic Gonadotrophin
Beta-human chorionic gonadotrophin assays
The family of pituitary/placental glycoprotein hormones includes hCG, follicle-stimulating hormone, luteinizing hormone (LH) and thyroid-stimulating hormone (TSH). Each hormone comprises an α-subunit which is common between the family members and a distinct β-subunit. Consequently, assays to measure hCG are directed against the β-subunit. Many different β-hCG assays are available. Some detect intact β-hCG, and others are either selective for individual fragments or detect various combinations of fragments (Cole 1998). In pregnancy, hCG is usually intact and fragments of β-hCG are not produced, although the β chain is hyperglycosylated during the first trimester. However, in cancer, β-hCG may circulate in many different forms which can vary in their glycosylation status. Therefore, assays used in patients with GTD and cancer need to be able to detect all forms of β-hCG. The ideal assay should be able to recognize all forms equally well and be sufficiently sensitive to limit the risk of false-negative results (Mitchell and Seckl 2007). Moreover, the assay should not produce false-positive results as this is well recognized to be associated with unnecessary medical interventions and potentially life-threatening complications (Cole et al 2001). So how good are the existing β-hCG assays?
Commercially available β-hCG tests are based on the sandwich assay principle and rely on two antibodies which generally target different regions of the molecule. These assays are primarily licensed for use in pregnancy detection. However, they are frequently employed for monitoring patients with cancer. The assays can produce both false-positive and false-negative results (Cole et al 2001, Mitchell and Seckl 2007). The false-positive results occur because there is another molecule (often a heterophile antibody) which sticks the capture and detection antibodies together. This can usually be avoided by measuring the hCG in urine, as cross-reacting antibodies are large and cannot pass through the renal glomerulus into the urine. Alternatively, if a false-positive result is suspected, remeasuring the hCG on an alternative assay or serially diluting the serum sample usually resolves the issue. Real hCG will be seen in another assay and will dilute appropriately, whilst a cross-reacting molecule will be negative in another assay and does not serially dilute away. Recent work from the authors’ laboratory shows that commercial assays may be particularly prone to false-negative results, either because they completely fail to detect or as a consequence of poor sensitivity for a particular β-hCG isoform (Mitchell and Seckl 2007). False-negative results can potentially result in failure to diagnose disease or early termination of treatment, and therefore higher relapse rates.
In addition to the commercial assays, there are also several in-house assays in various centres around the world which are usually based on a single antibody to capture the hormone on a competitive basis with labelled (often radioactively) β-hCG. These assays may also produce false-positive and false-negative results. Indeed, all types of assay are only as good as the antibodies used. At Charing Cross Hospital, a rabbit polyclonal antibody is used in a radioimmunoassay (RIA); this recognizes all forms of β-hCG equally well and with sufficient sensitivity that false-negative results appear to be rare (Mitchell and Seckl 2007). Moreover, since this RIA is performed on both serum and urine samples which are serially diluted, the risk of false-positive results is very low (Mitchell and Seckl 2007).
New assays designed to detect specific hCG variants are in development and are leading to further increases in the specificity and sensitivity of hCG monitoring. Elevation of hyperglycosylated hCG (hCG-H) levels may be specific to cases of HM that subsequently require chemotherapy. This rise occurs earlier than in conventional hCG and before clinically apparent GTTs develop (Cole et al 2006a). However, further work is required, and since no commercial hCG-H assays are available, progress in this area is likely to be slow.
Recent data have also shown that proportionately higher levels of free β-hCG fragments are produced in PSTTs, and that this is a highly sensitive test for discriminating PSTTs from choriocarcinoma (Cole et al 2006b, Harvey et al 2008), although β-hCG may also be elevated in some non-trophoblastic malignancies.
Beta-human chorionic gonadotrophin as a tumour marker
hCG has a half-life of 24–36 h and is the most sensitive and specific marker for trophoblastic tissue. However, hCG production is not confined to pregnancy and GTD. Indeed, hCG is produced by any trophoblastic tissue found, for example, in germ cell tumours and in up to 15% of epithelial malignancies (Vaitukaitis 1979). The hCG levels in such cases can be just as high as those seen in GTD or in pregnancy. Therefore, measurements of hCG do not reliably discriminate between pregnancy, GTD or non-gestational trophoblastic tumours.
However, serial measurements of hCG have revolutionized the management of GTD for several reasons. The amount of hCG produced correlates with tumour volume, such that a serum hCG level of 5 IU/l corresponds to approximately 104–105 viable tumour cells. Consequently, these assays are several orders of magnitude more sensitive than the best imaging modalities available today. In addition, hCG levels can be used to determine prognosis (Bagshawe 1976). Serial measurements allow monitoring of disease progression or response to therapy (Figure 43.2). Development of drug resistance can be detected at an early stage, which facilitates appropriate changes in management. Estimates may be made of the time for which chemotherapy should be continued after hCG levels are undetectable in serum in order to reduce the tumour volume to zero. For these reasons, hCG is the best tumour marker known.
Figure 43.2 Graph demonstrating the use of monitoring the serum human chorionic gonadotrophin (hCG) concentration following evacuation of a hydatidiform mole (HM). In this case, after an initial fall, the hCG level started to rise, indicating the development of invasive HM or choriocarcinoma, so the patient was called up for staging. The prognostic score was low risk (see Table 43.4) and the patient was successfully treated with methotrexate (MTX) and folinic acid (see Table 43.5).
Clinical Features
Complete and partial moles
These most commonly present in the first trimester as a threatened abortion with vaginal bleeding. If the diagnosis is delayed, patients may notice the passing of grape-like structures (vesicles), and occasionally the entire mole may be evacuated spontaneously. The uterus may be any size but is commonly large for gestational age. Patients with marked trophoblastic growth and high hCG levels are particularly prone to hyperemesis, toxaemia and the development of theca lutein cysts which may sometimes be palpable above the pelvis. Toxaemia was diagnosed in 27% of patients with CM (Berkowitz et al 1981), but is seen less frequently today because of early US diagnosis. Convulsions are rare. The high hCG levels may also produce hyperthyroidism because of cross-reactivity between hCG and TSH at the TSH receptor. Although pulmonary, vaginal and cervical metastases can occur, they may disappear spontaneously following removal of the mole. Thus the presence of metastases does not necessarily imply that an invasive mole or choriocarcinoma has developed. Patients may rarely present with acute respiratory distress, not only because of pulmonary metastases or anaemia but occasionally as a result of tumour embolization (Savage et al 1998). The risk of embolization is reduced by avoiding agents which induce uterine contraction before the cervix has been dilated to enable evacuation of the CM.
Patients with PMs do not usually exhibit the dramatic clinical features characteristic of CM (Goldstein and Berkowitz 1994). The uterus is often not enlarged for gestational age, and vaginal bleeding tends to occur later so that patients most often present in the late first or early second trimester with a missed or incomplete abortion. In fact, the diagnosis is often only suspected when the histology of curettings is available. The pre-evacuation hCG level is less than 100,000 IU/l at diagnosis in over 90% of cases.
At present, a proportion of CMs and PMs still go undiagnosed because of miscarriage at home or because termination centres do not carry out histopathological examination of all abortions (Seckl et al 2004a). This can result in late presentation of disease, sometimes with life-threatening complications. Clearly, there is little that can be done about miscarriages at home. However, for women attending termination centres, it may be possible to establish a screening procedure to help prevent subsequent problems from missed diagnosis.
Twin pregnancies
Twin pregnancies comprising a normal fetus and an HM occur in between one in 20,000 and one in 100,000 pregnancies. Some probably abort in the first trimester and so go undiagnosed. However, some are discovered on US examination either routinely or because of complications such as bleeding, excessive uterine size or problems related to a high hCG level. With specialist obstetric care, 40% of such cases have continued into the third trimester and delivered live babies (Sebire et al 2002b).
Invasive moles
Invasive moles are usually diagnosed because serial urine or serum hCG measurements reveal a stable or rising hCG level in the weeks after molar evacuation. Patients may complain of persistent vaginal bleeding and lower abdominal pains and/or swelling. This may occur as a result of haemorrhage from leaking tumour-induced vasculature as the trophoblast invades through the myometrium, or because of vulval, vaginal or intra-abdominal metastases. The tumour may also involve other pelvic structures, including the bladder or rectum, producing haematuria or rectal bleeding, respectively. Enlarging pulmonary metastases or tumour emboli growing in the pulmonary arteries can contribute to life-threatening respiratory complications (Seckl et al 1991). The risk of these complications is clearly higher in patients where the initial diagnosis of a molar pregnancy was missed and who are not, therefore, on hCG follow-up.
Choriocarcinoma
Choriocarcinoma can present after any form of pregnancy, but most commonly occurs after a CM. Histological proof of choriocarcinoma is not usually obtained after a CM because of the risk of fatal haemorrhage caused by biopsy, and so it is impossible to distinguish from an invasive mole. Choriocarcinoma following a normal pregnancy or non-molar abortion usually presents within 1 year of delivery but can occur 17 years later (Tidy et al 1995). The presenting features may be similar to HM with vaginal bleeding, abdominal pain and a pelvic mass.
However, one-third of all choriocarcinomas present without pelvic symptoms but have symptoms from distant metastases. In these cases, lives can be saved by remembering to include choriocarcinoma in the differential diagnosis of metastatic malignancy (particularly in lungs, brain or liver) presenting in a woman of childbearing age. Any site may be involved, including skin (producing a purple lesion), cauda equina and the heart. Pulmonary disease may be parenchymal, pleural or may result from tumour embolism and subsequent growth in the pulmonary arteries (Savage et al 1998). Thus, respiratory symptoms and signs can include dyspnoea, haemoptysis and pulmonary artery hypertension. Cerebral metastases may produce focal neurological signs, convulsions, evidence of raised intracranial pressure, and intracerebral or subarachnoid haemorrhage. Hepatic metastases may cause local pain or referred pain in the right shoulder. Although none of these presentations are specific to choriocarcinoma, performing a simple pregnancy test or quantitative hCG assay can provide a vital clue to the diagnosis.
Infantile choriocarcinoma
Choriocarcinoma in the fetus or newborn is exceptionally rare, with approximately 30 reported cases (Blohm and Gobel 2004, Sebire et al 2005). While a primary choriocarcinoma within the infant is possible, the mother also had the tumour in 17 cases. Interestingly, the diagnosis was often made in the neonate before the mother. In all cases, the infant was anaemic and had a raised hCG level, but the site of metastasis was variable, including brain, liver, lung and skin. Only a few cases have been treated successfully with platinum chemotherapy (Johnson et al 2003
[/not-level-membership-for-obstetrics-gynecology-category]
