Chapter 6 Genomics and Principles of Clinical Genetics
Elementary Understanding of Molecular Genetics
General Organization and Structure of the Human Genome
Ushering in the molecular millennium, the original draft of the Human Genome Project was completed in February 2001 through a multinational effort and has provided the architectural blueprints of essentially every gene in the human genome.1,2 The human genome embodies the total genetic information, or the deoxyribonucleic acid (DNA) content of human cells, and is dispersed among 46 units of tightly packaged linear double-stranded DNA called chromosomes (22 autosomal pairs and the 2 sex chromosomes X and Y).3–5 The 24 unique chromosomes are differentiated visually by chromosome-banding techniques (karyotype analysis) and are classified mainly according to their sizes. Each nucleated cell in a living organism normally has a complete and exact copy of the genome, which is largely made up of single-copy DNA with specific sets of DNA sequences represented only once per genome. The remainder of the genome consists of several classes of either perfectly repetitive or imperfectly repetitive DNA elements. The human genome contains nearly three billion base pairs of genetic information containing the molecular design for approximately 35,000 genes whose highly orchestrated expression renders us human.1,2 Through the mechanism of alternative splicing of the coding sequences within the genes, these approximately 35,000 genes are thought to produce more than 100,000 proteins.6
Basic Structure of DNA and the Gene
In 1953, Watson and Crick described the basic structure of DNA as a polymeric nucleic acid macromolecule comprising deoxyribonucleotides or “building blocks,” of which there are four types: (1) adenine (A), (2) guanine (G), (3) thymine (T), and (4) cytosine (C) (Figure 6-1).3–5 DNA is a double-stranded molecule made up of two anti-parallel complementary strands (sense and antisense strands) that are held together by noncovalent (loosely held) hydrogen bonds between complementary bases, where G and C always form base pairs and T and A always pair (see Figure 6-1). In the literature, typically only the DNA sequence of the sense strand (the strand that transcribes the genetic message in the form of messenger RNA [mRNA]) is provided, and the antisense sequence is inferred through these complementary base pairing rules such that if the sense strand reads AGCCGTA, the antisense strand would be TCGGCAT. DNA natively forms a double helix that resembles a right-handed spiral staircase. DNA elements that store genetic information in the form of a genetic code are called genes (Figure 6-2, A).
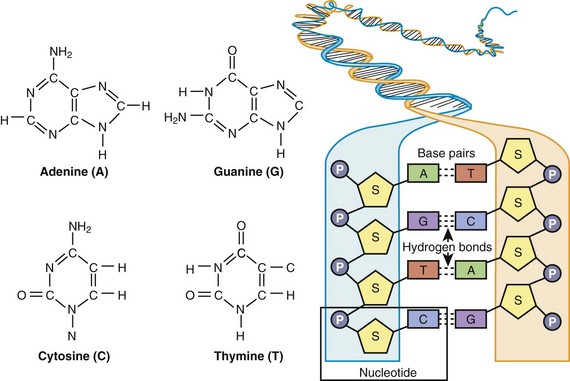
Figure 6-1 The chemical structure of adenine (A), guanine (G), cytosine (C), and thymine (T) and the general organization of DNA, illustrating complimentary base pairing via hydrogen bonds between C and G and between A and T. As defined by the box, a single nucleotide consists of a phosphate (P) group, deoxyribose sugar (S), and a base (A, C, G, or T).
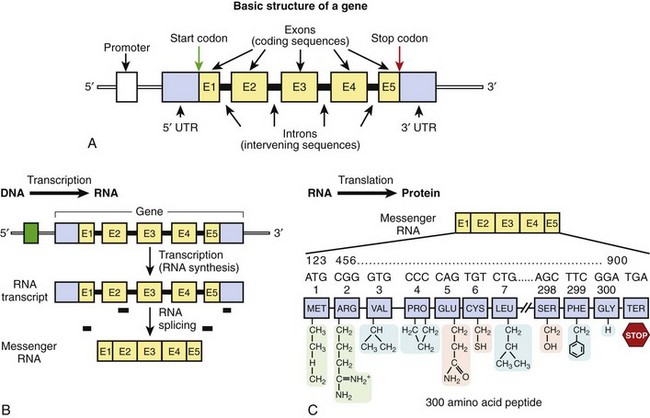
Figure 6-2 A, The basic structure of a gene consisting of DNA segments (exons) that encode for a protein product. Between the exons are intervening sequences called introns. At the 5’ end of the gene is a regulatory element called the promoter, which initiates transcription. At the 5’ and 3’ ends are “untranslated” regions that are considered parts of the first and last exons, respectively. These sequences are not a part of the genetic code but may contain additional regulatory elements. A start codon begins the translation of the genetic message, as encoded by the gene, and a stop codon terminates the message. B, Transcription. C, Translation. B and C depict the two-step process of the transfer of genetic information from DNA to RNA to protein.
Gene sequences account for approximately 30% of the genome; however, less than 2% of the genomic DNA is actually made up of protein-encoding sequences within genes called exons. Between the exons are intervening DNA sequences called introns, which are not a part of the genetic code but may host gene regulatory elements. The approximately 35,000 genes of the genome range in size from one of the smallest of human genes IGF2 (which contains 252 nucleotides and encodes insulin-like growth factor II) to the largest gene DMD (which consists of 2,220,223 nucleotides and encodes dystrophin). The DMD gene consists of over 2 million nucleotides, but only 0.5% of the gene (11,055 nucleotides spanning 79 exons) actually encodes for the dystrophin protein. Typically upstream (20 to 100 bp) from the first exon is a regulatory element called the promoter, which controls transcription of the hereditary message as determined by the gene sequence. Proteins known as transcription factors bind to specific sequences within the promoter region to initiate transcription of the genetic code. The first and last exons of the gene usually consist of an untranslated region (5′ and 3′ UTR, respectively) that is not a part of the genetic code but may host additional sequence elements that regulate gene expression.4
Transfer of the Genetic Code: The Central Dogma of Molecular Biology
DNA sequences in the form of genes contain an encrypted genetic message for the assembly of polypeptides or proteins that serve the biologic function of the cell. This inherited genetic information is transferred to a completed product (protein) through a two-step process.5 First, transcription, which is the process by which the genetic code is transcribed into mRNA, begins with the dissociation of the double-stranded DNA molecule and the formation of a newly synthesized complementary ribonucleic acid (RNA) molecule (Figure 6-2, B). Of note, instead of thymine (T), the nucleotide uracil (U) is in its place on the newly transcribed RNA strand; like thymine, uracil pairs with adenine. The initial mRNA molecule (pre-mRNA) matures into a transferable genetic message by undergoing RNA splicing to expunge the noncoding intronic sequences from the transcript. The vast majority of introns begin with the di-nucleotides GT and end with the di-nucleotides AG. These highly conserved splicing recognition sequences at the beginning and end of the exon-intron and intron-exon boundaries are referred to as the splice donor sites and splice acceptor sites, respectively. These nucleotides allow the RNA splicing apparatus to know precisely where to cleave the sequence in order to excise the noncoding regions (introns) and bring the coding sequences (exons) together. Normal alternative splicing provides the inclusion or exclusion of specific exonic sequences from the mature mRNA transcript to potentially produce several partially unique gene products (proteins) from a single gene that may have unique biologic functions, tissue specificity, or cellular locations. If normal splice recognition sites are disrupted, splicing errors may occur and result in abnormal protein product formation and consequently create a pathogenic substrate for disease. While all cells of the human body, except red blood cells, contain a copy of the genome, not all genes are expressed in all cells. While some genes are ubiquitously expressed, others have exclusive tissue specificity.
The second process, translation, involves the decoding of the mRNA-encrypted message and the assembly of the intended polypeptide (protein) that will serve a biologic role (Figure 6-2, C). Polypeptides are polymers of linear repeating units called amino acids. The assembly of a polypeptide or protein is directed by a triplet genetic code, or codon (three consecutive bases); 64 codons encode for 20 distinct amino acids or the termination of protein assembly. One codon, AUG (ATG on DNA) encodes for the amino acid methionine and is always the first codon (start codon) to start the message and signifies the beginning of the open reading frame (ORF) of the mRNA. Each codon in the linear mRNA is decoded sequentially to give a specific sequence of amino acids that are covalently linked through peptide bonds and ultimately make up a protein. Three codons, UAA, UAG, and UGA, serve as termination codons that stop the linearization of the peptide and signal a release of the finished product. The genetic code is said to be “degenerate” in that specific amino acids may be encoded by more than one codon. For example, when varying the nucleotide in the third position of a codon, often the message does not become altered (the codons GUU, GUC, GUA, and GUG all encode for the amino acid valine).
The accepted nomenclature for naming and numbering nucleotides and codons typically uses the DNA sense strand of the gene and begins with the A of the start codon (ATG) representing nucleotide 1 and ATG as codon 1. Usually, only consecutive nucleotides within the coding region of the gene are numbered. Intronic nucleotides are typically numbered relative to either the first or last nucleotide in the exon preceding or following the intron. For example, the LQT2-associated KCNH2 splice error mutation L799sp (exon 9, nucleotide substitution: 2398 +5 G > T), results from a G-to-T substitution in the intron, five nucleotides from exon 9, where nucleotide 2398 is the last nucleotide in the ninth exon. This substitution results in a splicing error following the last codon of the exon [codon 799 encoding for leucine (L)].7
Non–protein-coding genes are transcribed as well. MicroRNAs (miRNAs) are small ~22 nucleotide-long RNAs that function to inhibit gene expression of targeted genes by binding in a partially complementary fashion to miRNA recognition sequences within the 3′ UTR of target mRNA transcripts and negatively regulate protein-encoding gene mRNA stability or translation into protein.8–10 Each miRNA is thought to regulate the expression of hundreds of target genes at the post-transcriptional mRNA level. To date, hundreds of human miRNAs have been described, three (miR-1, miR-133, and miR-208) of which are abundant in the heart and serve as key regulators of heart development, contraction, and conduction.8
Modes of Inheritance: Genetics of Disease
On average, two unrelated individuals share 99.5% of their approximately three billion nucleotide genomic DNA sequence, and yet their genomic DNA sequences may vary at millions of single nucleotides or small sections of DNA nucleotides dispersed throughout their genomes.2,11 It is this inherited variation in the genome that is the basis of human and medical genetics. Reciprocal forms of genetic information at a specific locus (location) along the genome are called alleles.3 An allele can represent a segment of DNA or even a single nucleotide. The normal form of genetic information is often considered the wild-type or normal allele, and the allele at variance from the normal is often referred to as the mutant allele.
These normal variations at specific loci in the DNA sequence are called polymorphisms. Some polymorphisms are very common, and others represent rare genetic variants. In medical genetics, a disease-causing mutation refers to a DNA sequence variation that embodies an abnormal allele and is not found in the normal healthy population but subsists only in the diseased population and produces a functionally abnormal product. An individual is said to be homozygous when he or she has a pair of identical alleles, one paternal (from father) and one maternal (from mother). When the alleles are different, then that individual is said to be heterozygous for that specific allele. The term genotype refers to a person’s genetic or DNA sequence composition at a particular locus or at a combined body of loci, and the term phenotype refers to a person’s observed clinical expression of disease in terms of a morphologic, biochemical, or molecular trait.5
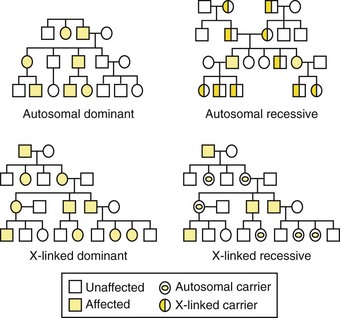
Genetic disorders are described by their patterns of familial transmission (Figure 6-3). The four basic modes of inheritance are (1) autosomal dominant, (2) autosomal recessive, (3) X-linked dominant, and (4) X-linked recessive.3 These modes, or patterns, of inheritance are based mostly on the type of chromosome (autosome or X-chromosome) the gene is located on and whether the phenotype is expressed only when both maternal-derived and paternal-derived chromosomes host an abnormal allele (recessive) or if the phenotype can be expressed even when just one chromosome of the pair (maternal or paternal) harbors the mutant allele (dominant).
In many genetic disorders, the abnormal phenotype can be clearly distinguished from the normal one. However, in certain disorders, the abnormal phenotype is completely absent in some individuals (asymptomatic, with no discerning clinical markers) harboring the disease-causing mutation, while some others show significant variations in the expression of the phenotype in terms of clinical severity, age at onset, and response to therapy. Penetrance is the probability that an abnormal phenotype, as a result of a mutant gene, will have any expression at all. When the frequency of phenotypic expression is less than 100%, the gene is said to show reduced or incomplete penetrance (Figure 6-4). Expressivity refers to the level of expression of the abnormal phenotype, and when the manifestations of the phenotype in individuals who have the same genotype are diverse, the phenotype is said to exhibit variable expressivity (see Figure 6-4). A phenocopy represents an individual who displays the clinical characteristics of a genetically controlled trait but whose observed phenotype is caused by environmental factors rather than determined by his or her genotype (see Figure 6-4). For example, an individual experiencing drug-induced torsades de pointes, a prolonged QT interval on ECG, or both may represent a phenocopy of LQTS. Reduced penetrance, variable expressivity, and observed phenocopies create significant challenges for the appropriate diagnosis, pedigree interpretation, and risk stratification of some genetic disorders, particularly those involving electrical disorders of the heart.
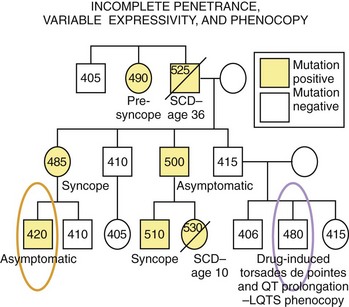
Mutation Types in Human Genetic Disease
The DNA of the human genome is highly stable from generation to generation but not immutable. Instead, it is vulnerable to an array of different types of germline (heritably transmitted) and somatic mutations. In general, mutations can be classified into three categories: (1) genome mutations, (2) chromosome mutations, and (3) gene mutations (Figure 6-5).3,5 Genome mutations are caused by the abnormal segregation of chromosomes during cell division and are illustrated by trisomy 21 (Down syndrome), which results from abnormal cells containing three copies of chromosome 21 instead of the two copies found in a normal cell (see Figure 6-5, A) and Turner’s syndrome (XO), in which the Y chromosome is omitted.
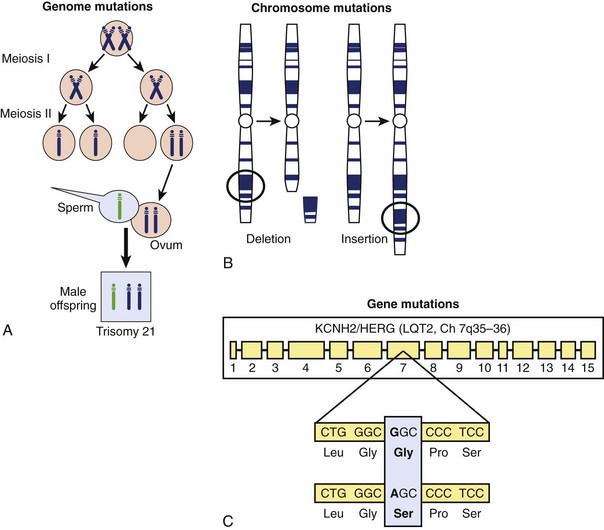
Chromosome mutations involve the structural breakage and rearrangement of chromosomes during cell division, when major portions of a particular chromosome may be missing (deleted) or inserted (Figure 6-5, B). For example, patients with chromosome 22 microdeletion syndrome have variable size deletions involving the long arm of chromosome 22 (22q11.2). Large deletions and duplications of hundreds to thousands of base pairs may lead to copy number variations of genetic material, which may serve as a pathogenic basis for disease. Such gene rearrangements may involve the deletion or duplication of many genes, single genes, or even single exons within specific genes.
Gene mutations involve alterations at the nucleotide level that disrupt the normal function of a single gene product (Figure 6-5, C). Such mutations are classified into three basic categories: (1) nucleotide substitutions, (2) deletions, and (3) insertions. If a single nucleotide substitution, which is the most common, occurs in the coding region (exon), the result may be either a synonymous (silent) mutation in which the new codon still specifies the same amino acid or a nonsynonymous mutation in which the altered codon encodes for a different amino acid or terminates further protein assembly (i.e., introduces a premature stop codon) (Figure 6-6, A). The term missense mutation is also used to indicate a single nucleotide substitution that results in the exchange of a normal amino acid in the protein for a different one (see Figure 6-6, A). Importantly, a missense mutation may or may not result in a functionally perturbed protein that leads to a disease phenotype. The functional consequence of a missense mutation may depend on the differences in biochemical properties between the amino acids that are being exchanged, the location in the protein at which the exchange occurs, or both. A nonsense mutation is a nonsynonymous mutation resulting in a substitution of an amino acid for a stop codon (see Figure 6-6, A). A nonsense mutation results in a truncated (shortened) gene product at the location of the new stop codon. The functional effects could range from no appreciable difference to functional lethality (a nonfunctioning protein), depending on where in the protein a nonsense mutation occurs.
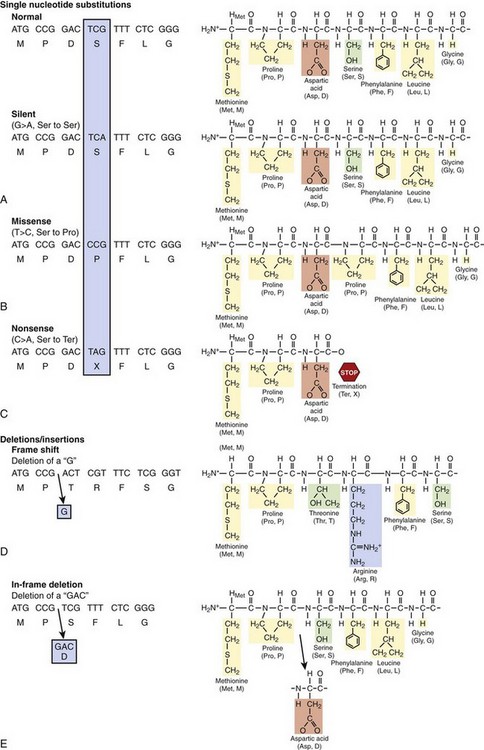
Gene mutations may also involve insertions and deletions of nucleotides that can be as small as a single nucleotide or as large as several hundreds to thousands of nucleotides in length. Most of these insertions and deletions occurring in the exon alter the “reading frame” of translation at the point of the insertion or deletion and produce a new sequence of amino acids in the finished product, the so-called frame-shift mutation (Figure 6-6, B). Many frame-shift mutations often result in a different product length from the normal gene product by creating a new stop codon, which produces a shorter or longer gene product, depending on the location of the new stop codon. In-frame insertions and deletions occur when three nucleotides are affected (see Figure 6-6, B) and result in a single amino acid or multiple amino acids being removed or added without affecting the remainder of the transcript.
Principles of Genetic Testing
Currently, nearly 1500 genetic tests (www.GeneTests.org) are clinically available and are offered by nearly 600 diagnostic laboratories worldwide. In addition, many genetic tests are available on a research basis. Most current genetic tests are performed to identify gene mutations in rare genetic disorders that follow Mendelian inheritance patterns (such as cystic fibrosis, Huntington chorea, sickle cell anemia, and Tay-Sachs disease) or for more complex conditions (such as breast, prostate, and colon cancers). In cardiology, clinical genetic testing is available for most of the cardiac channelopathies and cardiomyopathies.
General Techniques Used in Genetic Testing at the Single Gene Level
Typically, 5 to 15 mL whole blood obtained from venipuncture placed in ethelenediaminetetraacetic acid (EDTA)–containing tubes (“purple top”) is requested as the genomic DNA source for either research-based or clinic-based genetic testing.5 DNA isolated from a buccal (mouth cheek) swab does not provide a sufficient amount of DNA for comprehensive genetic testing, but it may be adequate for mutation-specific confirmatory testing of family relatives. Umbilical cord blood may be acquired at the time of birth for newborn screening. For autopsy-negative cases of sudden unexplained death, a cardiac channel genetic test can be completed on DNA isolated from EDTA blood, a piece of frozen ventricle myocardium tissue, or tissue from any other organ (liver, spleen, thymus) with a high nucleus to cytoplasm ratio.12 DNA from paraffin-embedded tissue, however, remains an unreliable source.13 Both research-based and clinical genetic testing typically require signed and dated informed consent to accompany the samples to be tested.
In genetic testing, the identification of gene mutations usually involves the polymerase chain reaction (PCR) technique. PCR is used to amplify many copies of a specific region of DNA sequence within the gene of interest. Typically, 20 to 25 base pair (bp) forward and reverse single-stranded DNA oligonucleotide primers are designed to be complementary to reciprocal intronic DNA sequences flanking the exon of interest to produce PCR products (200 to 400 bp in length) containing the desired DNA sequence to be analyzed. A well-optimized PCR reaction will yield millions of copies of only the specific sequence of interest.14
PCR amplification is often followed by the use of an intermediate mutation detection platform such as single-stranded conformational polymorphism (SSCP), denaturing gradient gel electrophoresis (DGGE), or denaturing high-performance liquid chromatography (DHPLC). These methods are used to inform the investigator of the presence or absence of a DNA sequence alteration in the samples examined. DHPLC is currently one of the most sensitive and accurate technologies to discover unknown gene mutations.15,16 DHPLC is based on the creation and separation of double-stranded DNA fragments containing a mismatch in the base pairing between the wild-type and mutant DNA strands, known as heteroduplex DNA. This mismatch in base pairing creates a weakness in the double-stranded DNA complex. PCR products are injected onto a solid phase column and eluted by using a linear acetonitrile gradient. Samples containing heteroduplex DNA (as a result of a heterozygote DNA alteration) will elute from the column faster than will normal homoduplex (perfect match) DNA fragments, thus providing an abnormal elution chromatogram profile.14
Together, these techniques provide excellent precision and accuracy to detect (1) single nucleotide substitutions that produce missense, nonsense, and splice site mutations and (2) small insertions and deletions. However, large whole-gene, multiple-exon, or single-exon deletions or duplications elude detection by this approach. Another technique, multiple ligation probe analysis (MLPA), however, will allow for the identification of such large gene rearrangements, which have recently been reported to account for as much as 5% to 10% of patients with LQTS with an otherwise negative genetic test.17–19 In contrast to the traditional PCR/DHPLC/DNA sequencing approach to mutation detection, MLPA relies on specifically engineered probes that are designed to bind to gene sequences (typically exonic sequences) and allows for the detection of copy number changes of the target sequence.19 Large deletion mutations, for example, will result in a loss of copy number of the target (exon), and duplications are represented as an increase in copy number.
Since first described in 1977 by Sanger and colleagues, advances in DNA sequencing methodologies and technical instrumentation have rapidly evolved DNA sequencing capacity and genetic information output. Massively parallel sequencing or next-generation sequencing technology is on the verge of allowing investigators to go well beyond the current capacity of generating DNA sequence reads of 600 to 800 nucleotides for 96 samples per instrument run to the ability to process from hundreds of thousands to tens of millions of base sequence reads in parallel. Through the use of next-generation sequencing and multiplex exon amplification, the possibility exists for molecular interrogation of an individual’s complete library of annotated protein-coding sequences in a single reaction or a few reactions with remarkable cost-effectiveness.20,21
Genetic Testing: Benefits, Limitations, and Family Matters
Genetic testing may have diagnostic value for symptomatic individuals by elucidating the exact molecular basis for the disorder, by establishing a definitive molecular diagnosis or disease prediction when the clinical probability of the disorder is inconclusive, by confirming or excluding the presence of a disease-causing mutation in presymptomatic individuals with a family history of a genetic disorder, and by helping personalize treatment recommendations and management of a patient’s specific disorder by characterization of the precise genotype.22,23 Genetic testing may also prove carrier status in those concerned about recessively inherited disorders such as cystic fibrosis.
While benefits such as certainty of diagnosis, increased psychological well being, and greater awareness of prophylactic treatment and risk stratification may be achieved, genetic testing may also contribute to an increase in risk for depression, anxiety, guilt, stigmatization, discrimination, family conflict, and unnecessary or inappropriate use of risk-reducing strategies.24 Patients therefore need to be well informed on the implications of genetic testing and should not be coerced into providing a DNA sample for analysis. Full disclosure should be given as to the intent of the research or clinical genetic test, the results of the analysis, and who will have access to the results.5
Genetic information must be considered private and personal information that has the potential to be mishandled.25,26 Disclosure of confidential information to third parties such as insurance companies or employers can have negative consequences for the patient in the form of genetics-based discrimination. In May 2009, the Genetic Information Nondiscrimination Act (GINA) was signed into federal law prohibiting employers and health insurers from denying employment or insurance to a healthy individual on the basis of genetic test results.27
Genetic testing is appreciated now as a family as well as an individual experience.24 Even though genetic testing is performed on an individual’s genetic material, the individual’s decision to undergo genetic testing and the test results may have substantial implications for other family members, especially for those with disorders associated with sudden cardiac death. However, under current guidelines, only the individual tested or the legal guardian in the case of a minor may be informed of the genetic test results, and the decision or responsibility to inform unsuspecting relatives of the potential for genetic predisposition for sudden cardiac death rests exclusively on the informed patient.5
Interpretation of Genetic Test Results
The patient and family suspected of having genetic heart disease should be evaluated and managed by a cardiologist with specific expertise in heritable channelopathies or cardiomyopathies.5 Because of issues associated with incomplete penetrance and variable expressivity, the results of the genetic test must be interpreted cautiously and incorporated into the overall diagnostic evaluation for these disorders. The assignment of a specific variant as a true pathogenic disease-causing mutation requires vigilant scrutiny. To illustrate this requirement, recently a comprehensive determination of the spectrum and prevalence of rare nonsynonymous single nucleotide mutations (amino acid–altering variants) in the five LQTS-associated cardiac ion channel genes was performed in approximately 800 ostensibly healthy subjects from four distinct ethnic groups. The study showed that approximately 2% to 5% of healthy individuals are found to host rare amino acid–altering missense variants.28,29 Some of the variants observed in this healthy population may represent subclinical disease modifiers and others simply represent benign background “genetic noise.” This observation of background nonpathogenic missense variants is certainly not confined to the LQTS genes alone but may extend to virtually any gene in the human genome. Therefore, rather than being viewed as a binary yes-or-no test result, genetic testing results more appropriately should be considered as probabilistic in nature. Algorithms based on mutation location, species conservation, and the biophysical nature of the amino acid substitution may assist in distinguishing pathogenic mutations from otherwise rare variants of uncertain significance (VUS) and perhaps allow for the assignment of estimated predictive values to the probability of pathogenicity of each novel mutation identified within a specific gene.30
Future Directions in Cardiovascular Genetics
Genome-Wide Association Studies
Since the completion of the Human Genome Project’s final draft in 2003 and the International HapMap Project in 2005 and with the advent of novel high-throughput genotyping methods, an explosion of genome-wide association studies (GWAS) has taken place. GWAS are large, population-based (involving thousands of individuals), hypothesis-free association studies of common genetic variants and observed phenotypic diversity of complex traits; these studies are conducted through large-scale genotyping of hundreds of thousands of SNPs located across the genome and compare the allelic frequencies of SNPs in cases and controls.31,32 To date, several hundred genetic loci and specific polymorphisms have been found to be associated with a number of complex traits for many diseases categories, including neurodegenerative, neuropsychiatric, metabolic, autoimmune, and musculoskeletal diseases; several forms of cancer; and cardiovascular diseases.31,32
GWAS have been performed recently for electrocardiographic traits that have been associated with risk for ventricular arrhythmias and sudden cardiac death, including the PR interval and QRS duration as measures of cardiac conduction and the QT interval duration as an index of cardiac repolarization.33–35 A GWAS for atrial fibrillation, a disease of irregular rhythm of the heart’s upper chambers and significantly associated with an increased risk for stroke, heart failure, and death, has been completed.36 Cardiovascular GWAS have not only isolated associations between specific genetic variants and complex disease-defining traits but have shed light on novel biologic mechanistic pathways. For example, an initial GWAS and subsequently several replication studies have identified a strong association between variants in the NOS1AP (capon) gene and QT interval duration, thus highlighting the importance of nitric oxide synthase pathway in myocardial function and action potential repolarization that had not been previously known and providing novel physiological information.37 Recently, two large independent GWAS meta-analysis studies identified significant associations with 10 loci that appear to modulate or influence the QT interval duration, including loci mapping near the monogenic LQTS-associated genes KCNQ1, KCNH2, and SCN5A.33,34 Whether or not these novel loci represent the location of additional candidate LQTS-causing or disease-modifying genes remains to be investigated.
Micro-RNAs as Pathogenic Contributors to Electrical Diseases of the Heart
miRNA represent one family of small noncoding RNA molecules, which function as micromanagers of gene expression, as genetic on-off switches to eliminate mRNAs that should not be expressed in a particular cell type or at a particular moment, or as a finetuning mechanism adjusting the physiological levels of gene expression in response to environmental factors. In mammals, miRNAs mediate post-transcriptional gene silencing usually by binding to the 3′ UTR region of mRNAs of their target transcripts and may individually regulate tens to hundreds of gene transcripts.10
Recently, the dysregulation of specific miRNAs has been linked to the development and pathogenesis of numerous disease states, including those of the heart.8,9 For example, miRNA expression array studies have shown upregulation, and/or downregulation of miRNAs in morphologic pathologies of the heart, including aortic stenosis, hypertrophic cardiomyopathy, dilated cardiomyopathy, and ischemic cardiomyopathy, compared with the normal condition of the heart. Additionally, cardiac electrophysiology may be altered by perturbations in miRNA expression profiles, as numerous cardiac action potential repolarizing K+ ion channels, including the LQTS-associated KCNQ1-, KCNH2-, and KCNJ2-encoded channels, are under the finetuning control of miRNAs in maintaining gradients in ion channel density that are critical for the correct chronologic excitation of cardiomyocytes.9 Often, after cardiac infarction, the surviving heart muscle hypertrophies and undergoes electrical remodeling that is associated with continuous alterations in the electrical properties of cardiomyocytes and may prolong the action potential, slow cardiac conduction, and provide a proarrhythmic milieu.
Of the several hundred miRNAs that have been identified, miR-1 and miR-133 are thought to be muscle specific. In a recent study, miR-1 was found to be overexpressed in patients with coronary heart disease; when overexpressed in normal and infarcted rat hearts, miR-1 aggravated cardiac arrythmogenesis through conduction slowing and membrane depolarization by post-transcriptionally repressing the KCNJ2 gene (encoding the K+ channel subunit kir2.1, which is responsible for Andersen-Tawil syndrome) and the GJA1 gene (which encodes for connexin43, which is responsible for intracellular conductance in ventricles).38 Interestingly, the elimination of miR-1 by an antisense inhibitor in infarcted rat hearts was antiarrhythmic, suggesting that therapeutic inhibition of miR-1 following myocardial infarction may reduce the proarrhythmic response and the occurrence of sudden death. Whether or not perturbations to miR-1 or other heart-specific miRNAs in the form of single nucleotide substitutions resulting in mishandling of proper miRNA expression or maturation can lead to a monogenic cardiac electrical disorder or if specific miRNA therapeutic inhibition may be used to repress potentially life-threatening arrhythmias remains to be seen and will certainly be a part of the next decade of cardiovascular genetic and pharmacologic research.
Induced Pluripotent Stem Cell–Derived Cardiomyocytes
In 2006, Takahashi and Yamanaka showed that murine embryonic fibroblast and adult fibroblast acquired embryonic stem cell–like properties following retroviral induction of four transcription factor (Oct3/4, Sox2, Klf4, and c-Myc)–encoding genes.39,40 These newly transformed cells were referred to as induced pluripotent stem cells or iPS cells. In 2007, the application of this groundbreaking technology to human cells was rapidly realized with the generation of iPS cells derived from human fibroblast using either the same cocktail of transcription factors or an independently determined mixture.41,42 The iPS technology allows for the potential to overcome important obstacles that are currently associated with embryonic stem (ES) cells, such as immune rejection of ES-derived tissues after transplantation and the profound ethical concerns associated with destroying human embryos. The biomedical promise of human iPS cells derived from the patient’s own skin biopsy (fibroblast) is enormous and may hugely benefit regenerative medicine, drug or toxicology research, and disease model generation efforts (Figure 6-7). In fact, disease-specific iPS cell lines from patients are now beginning to emerge.
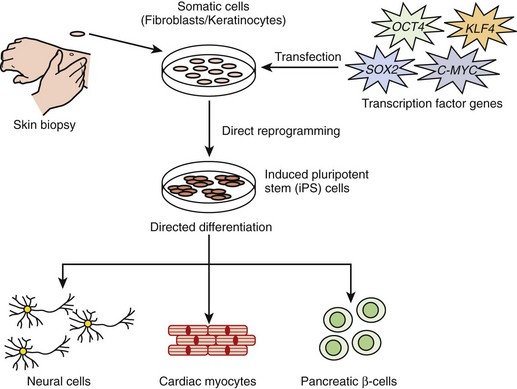
In 2009, Zhang and colleagues showed for the first time that human iPS cells derived from fibroblast could be differentiated into functional cardiomyocytes.43 This holds significant promise for the application of iPS cell–derived cardiomyocytes in cardiac research on disease models, in drug development, and as an autologous source of cells for myocardial repair. In the study of electrical diseases such as LQTS, for example, researchers may be able to take a skin biopsy from a patient with LQTS and subject iPS cell–derived cardiomyocytes to candidate drugs to determine the best therapy for that particular individual. iPS cell–derived cardiomyocytes from mutation-positive patients with LQTS would permit the functional electrophysiological study of that patient’s specific ion channel mutation in its most native environment, rather than using the currently available technology of heterologous overexpression studies that only partially recapitulate the true macromolecular ion channel complex. Such studies may allow for a more precise in vitro characterization of variant channels to assist in deciphering truly pathogenic mutations from benign VUS. This technology may also allow investigators to answer key questions surrounding the observed reduced penetrance and variable expressivity that are common among cardiac electrical disorders; this may be accomplished by generating iPS cell–derived cardiomyocytes from multiple family members who show variable disease expression ranging from an asymptomatic course to a severe symptomatic course. Such studies may further elucidate particular genetic or environmental factors that may contribute to the overall risk of experiencing a cardiac event, including sudden death.
1 Lander ES, Linton LM, Birren B, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860-921.
2 Venter JC, Adams MD, Myers EW, et al. The sequence of the human genome. Science. 2001;291:1304-1351.
3 Nussbaum RLMR, Willard HF. Thompson & Thompson’s genetics in medicine, ed 6. Philadelphia: Saunders; 2001.
4 Strachan TRA. Human molecular genetics, ed 3. New York: Garland Science; 2004.
5 Tester DJ, Ackerman MJ. Genetic testing. In: Gussak I, Antzelevitch C, editors. Electrical diseases of the heart: Genetics, mechanisms, treatment, prevention. London: Springer, 2008.
6 Graveley BR. Alternative splicing: Increasing diversity in the proteomic world. Trends Genet. 2001;17:100-107.
7 Tester DJ, Will ML, Haglund CM, et al. Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Heart Rhythm. 2005;2:507-517.
8 Barringhaus KG, Zamore PD. MicroRNAs: Regulating a change of heart. Circulation. 2009;119:2217-2224.
9 Latronico MVG, Condorelli G. MicroRNAs and cardiac pathology. Nat Rev Cardiol. 2009;6:419-429.
10 Shomron N, Levy C. MicroRNA-biogenesis and pre-mRNA splicing crosstalk. J Biomed Biotechnol. 2009;2009:594678.
11 Guttmacher AE, Collins FS. Genomic medicine—a primer. N Engl J Med. 2002;347:1512-1520.
12 Tester DJ, Ackerman MJ. The role of molecular autopsy in unexplained sudden cardiac death. Curr Opin Cardiol. 2006;21:166-172.
13 Carturan E, Tester DJ, Brost BC, et al. Postmortem genetic testing for conventional autopsy-negative sudden unexplained death: An evaluation of different DNA extraction protocols and the feasibility of mutational analysis from archival paraffin-embedded heart tissue. Am J Clin Pathol. 2008;129:391-397.
14 Tester DJ, Will ML, Ackerman MJ. Mutation detection in congenital long QT syndrome: Cardiac channel gene screen using PCR, dHPLC, and direct DNA sequencing. Methods Mol Med. 2006;128:181-207.
15 Ning L, Moss A, Zareba W, et al. Denaturing high-performance liquid chromatography quickly and reliably detects cardiac ion channel mutations in long QT syndrome. Genet Test. 2003;7:249-253.
16 Spiegelman JI, Mindrinos MN, Oefner PJ. High-accuracy DNA sequence variation screening by DHPLC. Biotechniques. 2000;29:1084-1090. 1092
17 Eddy C-A, MacCormick JM, Chung S-K, et al. Identification of large gene deletions and duplications in KCNQ1 and KCNH2 in patients with long QT syndrome. Heart Rhythm. 2008;5:1275-1281.
18 Koopmann TT, Alders M, Jongbloed RJ, et al. Long QT syndrome caused by a large duplication in the KCNH2 (HERG) gene undetectable by current polymerase chain reaction-based exon-scanning methodologies. Heart Rhythm. 2006;3:52-55.
19 Tester DJ, Ackerman MJ. Novel gene and mutation discovery in congenital long QT syndrome: Let’s keep looking where the street lamp standeth. Heart Rhythm. 2008;5:1282-1284.
20 Mardis ER. Next-generation DNA sequencing methods. Annu Rev Genomics Hum Genet. 2008;9:387-402.
21 Porreca GJ, Zhang K, Li JB, et al. Multiplex amplification of large sets of human exons. Nat Methods. 2007;4:931-936.
22 Tester DJ, Ackerman MJ. Genetic testing for cardiac channelopathies: Ten questions regarding clinical considerations for heart rhythm allied professionals. Heart Rhythm. 2005;2:675-677.
23 Priori SG, Napolitano C. Role of genetic analyses in cardiology: Part I: Mendelian diseases: Cardiac channelopathies. Circulation. 2006;113:1130-1135.
24 Van Riper M. Genetic testing and the family. J Midwifery Women’s Health. 2005;50:227-233.
25 Thomas SM. Society and ethics—the genetics of disease. Curr Opin Genet Dev. 2004;14:287-291.
26 Lea DH, Williams J, Donahue MP. Ethical issues in genetic testing. J Midwifery Women’s Health. 2005;50:234-240.
27 Abiola S. Recent developments in health law. The Genetic Information Nondiscrimination Act of 2008: “First major Civil Rights bill of the century” bars misuse of genetic test results. J Law Med Ethics. 2008;36:856-860.
28 Ackerman MJ, Splawski I, Makielski JC, et al. Spectrum and prevalence of cardiac sodium channel variants among black, white, Asian, and Hispanic individuals: Implications for arrhythmogenic susceptibility and Brugada/long QT syndrome genetic testing. Heart Rhythm. 2004;1:600-607.
29 Ackerman MJ, Tester DJ, Jones GS, et al. Ethnic differences in cardiac potassium channel variants: Implications for genetic susceptibility to sudden cardiac death and genetic testing for congenital long QT syndrome. Mayo Clin Proc. 2003;78:1479-1487.
30 Kapa S, Tester DJ, Salisbury BA, et al. Genetic testing for long QT syndrome: Distinguishing pathogenic mutations from benign variants. Circulation. 2009;120:1752-1760.
31 Frazer KA, Murray SS, Schork NJ, et al. Human genetic variation and its contribution to complex traits. Nat Rev Genet. 2009;10:241-251.
32 McCarthy MI, Abecasis GR, Cardon LR, et al. Genome-wide association studies for complex traits: Consensus, uncertainty and challenges. Nat Rev Genet. 2008;9:356-369.
33 Newton-Cheh C, Eijgelsheim M, Rice KM, et al. Common variants at ten loci influence QT interval duration in the QTGEN Study. Nat Genet. 2009;41:399-406.
34 Pfeufer A, Sanna S, Arking DE, et al. Common variants at ten loci modulate the QT interval duration in the QTSCD Study. Nat Genet. 2009;41:407-414.
35 Smith JG, Lowe JK, Kovvali S, et al. Genome-wide association study of electrocardiographic conduction measures in an isolated founder population: Kosrae. Heart Rhythm. 2009;6:634-641.
36 Gudbjartsson DF, Holm H, Gretarsdottir S, et al. A sequence variant in ZFHX3 on 16q22 associates with atrial fibrillation and ischemic stroke. Nat Genet. 2009;41:876-878.
37 Arking DE, Pfeufer A, Post W, et al. A common genetic variant in the NOS1 regulator NOS1AP modulates cardiac repolarization. Nat Genet. 2006;38:644-651.
38 Yang B, Lin H, Xiao J, et al. The muscle-specific microRNA miR-1 regulates cardiac arrhythmogenic potential by targeting GJA1 and KCNJ2. Nat Med. 2007;13:486-491.
39 Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663-676.
40 Yamanaka S. A fresh look at iPS cells. Cell. 2009;137:13-17.
41 Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861-872.
42 Yu J, Vodyanik MA, Smuga-Otto K, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917-1920.
43 Zhang J, Wilson GF, Soerens AG, et al. Functional cardiomyocytes derived from human induced pluripotent stem cells. Circ Res. 2009;104:e30-e41.