Genetics and Genomics in Critical Care
The field of genetics and genomics continues to expand and affect crucial aspects of health care. Genetic testing does not yet have a large role in the critical care unit, but given the exponential growth of knowledge in this field, and the decreasing costs of genomic sequencing, the day when personalized health care will include a genetic screen to tailor treatment to individual biology is on the horizon.1 This chapter includes an overview of the biologic basis of genomics, a description of the different types of genetic and genomic studies, and the growing impact of pharmacogenetics. It also incorporates some examples of genetic diseases and pharmacogenetic syndromes in critical care. A list of genetic and genomic terms is provided at the end of the chapter.
Genetics and Genomics
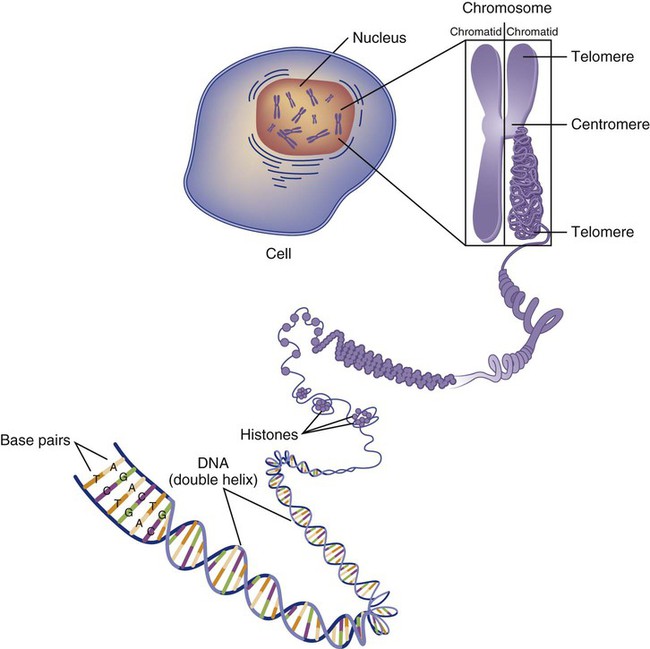
Genetics is the study of heredity, particularly as it relates to the ability of individual genes to transfer heritable physical characteristics. Genes are specific sequences of deoxyribonucleic acid (DNA) located on chromosomes within the nucleus of each cell (Fig. 4-1). Genes contain the blueprint for protein production that results in the physical characteristics of each individual. The pioneering work in human genetics has focused on single-gene variants that are rare in the population but have a large effect for the affected individual and follow classic inheritance patterns. About 6,000 rare disorders have been identified, and while single-gene conditions are relatively rare in the population, cumulatively they affect about 25 million people.1,2 Single-gene disorders include Huntington’s disease, Tourette syndrome, cystic fibrosis, and Duchenne muscular dystrophy.2 In single-gene variant disorders the influence of genetics is strong and the environmental effect is very weak.
Genomics refers to the study of all of the genetic material within the cell and encompasses the environmental interaction and impact on biologic and physical characteristics. Thus, genomics is a much larger and more complex area of study. The genome is the complete set of DNA in an organism. Each human nucleated somatic cell contains a copy of the complete genome. The exceptions are reproductive cells (oocytes and sperm), which contain only one half of the paired chromosomes, and red blood cells, because they do not have a nucleus. The human genome contains between 20,000 and 25,000 protein-coding genes, which represent less than 2% of the total genome.3
Many disease conditions that are commonly seen in the modern world result from a confluence of both environmental and genomic influences. These are described as complex traits or polygenic conditions.4 Furthermore, environmental conditions can alter the expression of different genes and consequently enhance the signs and symptoms associated with a disease. This represents a relatively new field of research known as epigenetics.5 Coronary artery disease is an example of a widespread disease that has both a strong environmental component (diet, obesity, diabetes, smoking) and a genomic effect that alters risk of developing the condition.4 Whole genome sequencing methods and genome wide association studies (GWAS) are employed to examine multiple genes simultaneously. As the cost of genomic sequencing plummets, this will increasingly become a viable clinical option.6 Nowhere is the genomic influence more evident than in pharmacogenomics, or the unique individual response to medications (environmental impact) based upon gene expression (genomic impact).7
Genetic and Genomic Structure and Function
Chromosomes
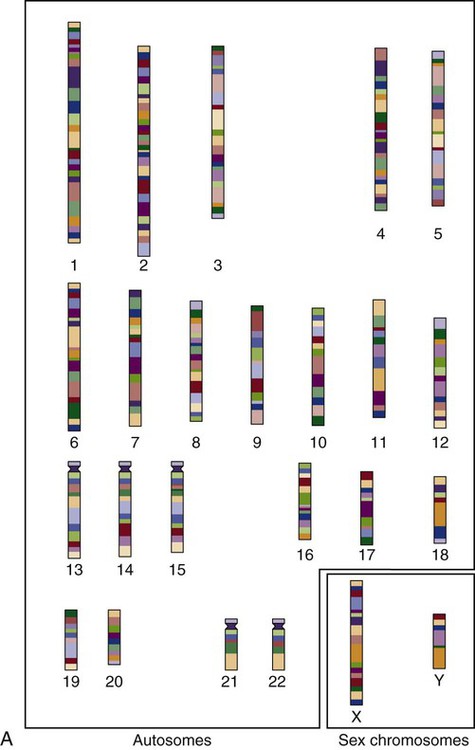
The nucleus inside each human cell contains each person’s full genetic blueprint of 23 pairs of chromosomes—22 pairs of autosomes and 1 pair of sex chromosomes—making the total 46. A number system is used to identify each chromosome. The chromosomes are traditionally arranged in order of size, starting with the largest (chromosome 1) to the smallest (chromosome 22), with the sex chromosomes placed last or to the side. A schematic of this chromosome arrangement to show the variation in size is shown in Figure 4-2A; the chromosomes are obviously not arranged this way inside the cell. A karyotype is the arrangement of human chromosomes from largest to smallest, as shown in the sequence in Figure 4-2B.
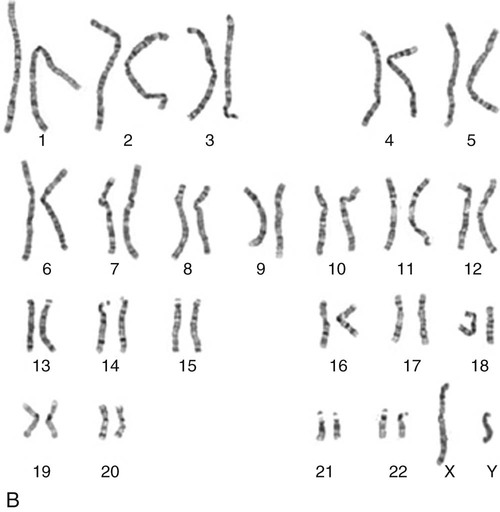
Each chromosome consists of an unbroken strand of DNA. To fit all of this genetic material inside the cell nucleus, the DNA is tightly coiled inside the chromosomes in a hierarchical order of compact structures. A specialized class of proteins called histones organizes the double-stranded DNA into what looks like a tightly coiled telephone cord (see Fig. 4-1).
Each somatic chromosome, also called an autosome, is made of two strands, called chromatids, which are joined near the center (see Fig. 4-1). This central region is called the centromere, and the ends of the chromatids are called telomeres. The segments of the chromosome separated by the centromere are called arms. The shorter arm of each chromosome is called p (for petit, or small), and the longer arm is called q. Differential staining of chromosomes produces alternating dark and light transverse bands. The bands are labeled p1, p2, p3, and so forth on the p arm and q1, q2, q3, and so forth on the q arm, counted from the centromere toward the telomeres.
DNA Base Pairs
The subunits within each DNA strand are called nucleotides or bases, and they are combined in pairs to form the rungs of the DNA ladder. Four nucleotide bases—adenine (A), thymine (T), guanine (G), and cytosine (C)—comprise the “letters” in the genetic DNA “alphabet.” Each nucleotide base is attached to a phosphorylated molecule of the 5-carbon sugar deoxyribose that forms the backbone of the DNA chain and can be visualized as the two sides of a ladder that have been twisted around (Fig. 4-3). The bases in the double helix are paired T with A, and G with C. The nucleotide bases are designed so that only G can pair with C and only T can pair with A to achieve a consistent distance across the width of the DNA strand. The TA and GC combinations are known as base pairs (see Fig. 4-3). There are approximately three billion bases in the human genome, based on findings from the Human Genome Project.3
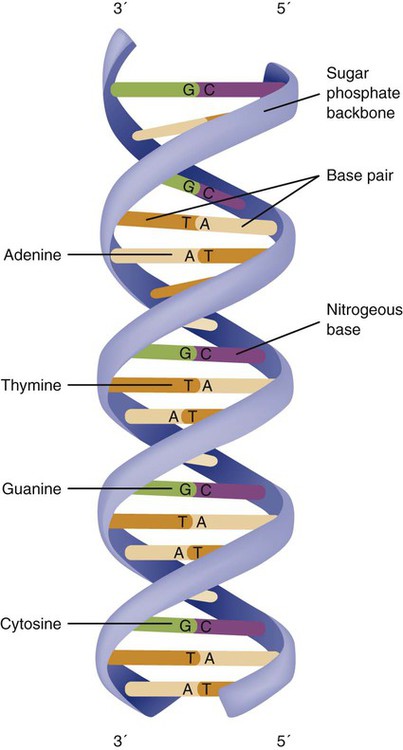
The two DNA strands are orientated in opposite directions. Each DNA strand has a specific direction that is labeled as the 3′ end or the 5′ end (pronounced 3 prime, and 5 prime) (see Fig. 4-3). Because the DNA strands face in opposite directions, the 3′ end of one strand is always matched to the 5′ end of the other strand. This fact becomes important for replication (discussed next). The 3′ end is described as the leading strand because new nucleotides can be added only at the 3′ end.
DNA Replication
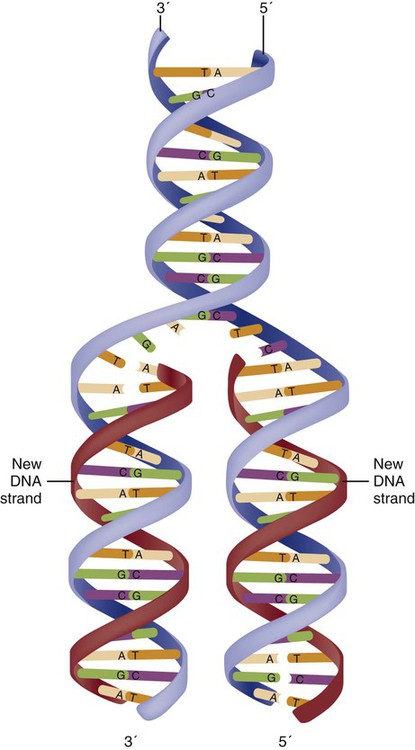
Before a cell divides, it needs to make a second copy of the entire DNA content within the cell. This process is called DNA replication. Sections of the DNA double helix separate longitudinally, creating openings between base pairs, known as replication bubbles, and mirror-image copies are made from the original strands. Stated another way, the original DNA strands provide a template that will be copied. The original is called the parent strands and the mirror-image copies are described as the daughter strands. In Figure 4-4 the parent strand is illustrated by the color blue with the 5′ and 3′ ends of each parent strand labeled. The daughter strands are colored in red. The replication process is facilitated by DNA polymerase, an enzyme that lengthens the DNA strand by the addition of new nucleotide bases at the 3′ end of the daughter strand (Fig. 4-4). After DNA replication is accomplished, the cell uses a sophisticated mechanism for identifying and fixing errors in the replicated strand.8 After this procedure, the cell is ready to divide, and each new cell will contain a copy of the original DNA code.
DNA Alphabet
The nucleotides A, T, C, and G can be thought of as “letters” of a genetic alphabet that are combined into three-letter “words” that are transcribed (written) by the intermediary of ribonucleic acid (RNA). The RNA translates the three-letter words into the amino acids used to make the polypeptide chains that constitute proteins. This process may be written as DNA → RNA → protein.9
Transcription
The process of making an RNA strand from a DNA strand is known as transcription. The strand with the genetic code that is to be transcribed is labeled as the sense strand or sometimes as the coding strand. The other strand, which is the RNA mirror image, is called the antisense or non-coding strand (Fig. 4-5). The reason there are two strands oriented in opposite directions is to facilitate replication of a DNA strand during cell division (see Fig. 4-4), or when proteins are needed (see Fig. 4-5) without compromising the original genetic material. To visualize how the process of transcription works, it may be helpful to study Figure 4-5 and locate the names of the different strands; the DNA strand is colored blue, the RNA strand is colored green to make the distinction clear.
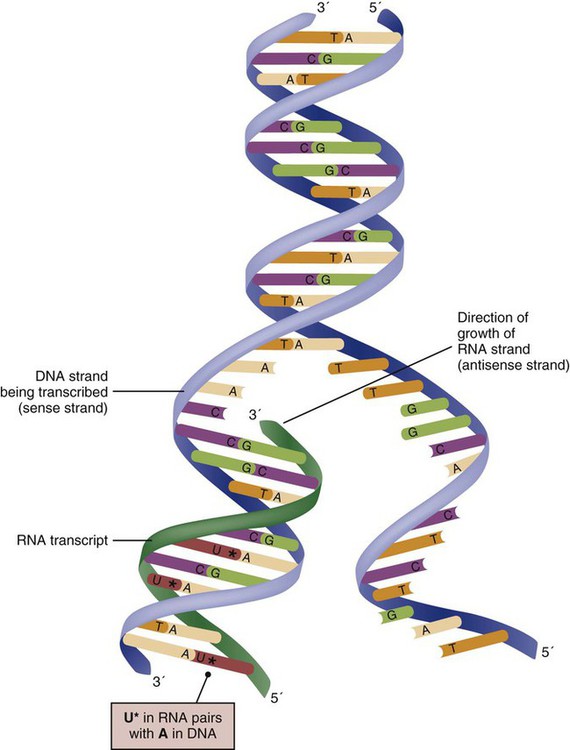
Transcription occurs under the guidance of the enzyme RNA polymerase, sections of the DNA double helix unfold and separate into two single strands within the length of the double helix (see Fig. 4-5). The mirror-image DNA strand (shown in red in Fig. 4-4) serves as a template for the synthesis of a complementary, mirror-image strand of RNA (shown in green in Fig. 4-5). The purpose of transcription is to have the RNA mirror strand replicate the genetic code in the original DNA sense strand.
When RNA transcribes DNA nucleotides, one significant change occurs. The adenosine (A) DNA base is paired with a uracil (U) base in the RNA transcript (see Fig. 4-5). Each RNA strand also has a 3′ end (can be conceptualized as a head) and a 5′ end (conceptualized as a tail), and the growing RNA strand adds bases only at the 3′ leading end. The RNA strand, called messenger RNA, then leaves the nucleus of the cell, and the next action takes place in the cytoplasm.
Translation
The next step is to translate the RNA bases into three-letter words (e.g., AUC, UGA) called codons that can be used to specify an amino acid. The 3-base RNA codons are designed to code for one of 20 amino acids. Some codons signal to stop the sequence; these termination sequences are UAA, UAG, and UGA. The three-letter codons are not unique; for example, both UAU and UUC code for the amino acid phenylalanine. Most amino acids can be made from more than one codon. This can be appreciated by an examination of the list of three-letter codons, the corresponding three-letter amino acid abbreviation, the single-letter abbreviation, and the name of the amino acid in Table 4-1. The process by which proteins are made from instructions encoded in DNA is called gene expression.
TABLE 4-1
THE GENETIC CODE: AMINO ACIDS*
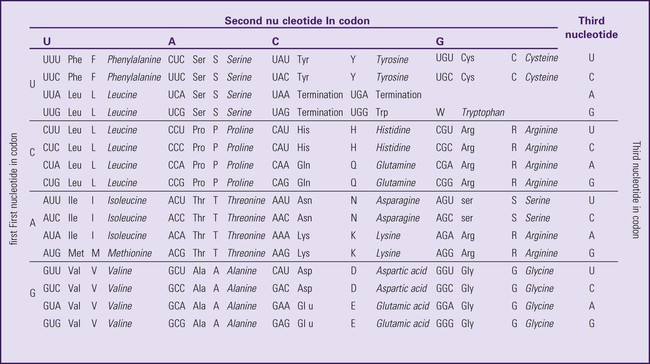 |
*Amino acids are the building blocks of proteins. The 20 amino acids are constructed from information contained in the DNA blueprint that is translated and transcribed by RNA. This transfer of information from DNA to amino acids is called the genetic code. Triple sets of bases (codons) are transcribed into the 20 amino acids. Sixty-four combinations of codons are possible, and several codons code for the same amino acids. The amino acids are connected in long polypeptide chains that form proteins. Three combinations signal the end of a protein chain: UAA, UAG, and UGA. Each three-letter codon also has a single-letter abbreviation, which is shown in the table.
Only a brief review of how DNA contributes to the genetic code is possible in this chapter. The volume of information that underpins genetics and genomics reflects the work of many scientists who performed research to advance this knowledge, and it may take some individual study or additional classes to master the content. An excellent and free online tutorial called “DNA from the Beginning” is available through the Cold Spring Harbor Laboratory website.10 The 41 modules use animation, video interviews, and text to present an interesting and informative introduction to the history and science of genetics.10
Genetic Variation, Mutation and Polymorphism
Variation
Genetic variation is common to all species. It means that individuals do not have the same nucleotides (A, C, T, G) in exactly the same position on the DNA strand. Some nucleotide differences result in the expression of different proteins and physical traits. Many nucleotide changes produce no visible external alteration, although those that have health-related consequences are of great interest to clinicians, patients, and researchers. Genetic variation can result from a variety of changes. It can be a single-letter substitution of one nucleotide base for another that can produce an inappropriate stop codon or produce a codon for a different amino acid. The amino acid codes are shown in Table 4-1. An example of a single-letter switch that results in an increased risk for disease is the G-to-A substitution at nucleotide 1691 in the coagulation factor V gene. This is also an example of a relatively rare single-gene variant that alters the protein product and increases the incidence of deep vein thrombosis (DVT), as described in Box 4-1.
Genetic material in the chromosome can also be deleted (Fig. 4-6A), new information from another chromosome can be inserted (Fig. 4-6B), or a tandem repeat (multiple repeats of the same sequence) may produce duplicate genetic material within the chromosome (Fig. 4-6C).
Translocation of genetic material describes a process in which chromosomes break and genetic material is moved from one chromosome to another. For example, the Philadelphia Chromosome, or Philadelphia translocation, is a specific chromosomal abnormality associated with chronic myelogenous leukemia (CML). It results from a reciprocal translocation between chromosomes 9 and 22, in which parts of these two chromosomes switch places, producing the oncogenic Philadelphia Chromosome and development of dysregulated tyrosine kinase. CML represents 20% of all adult cases of leukemia.11
Single-Nucleotide Polymorphisms
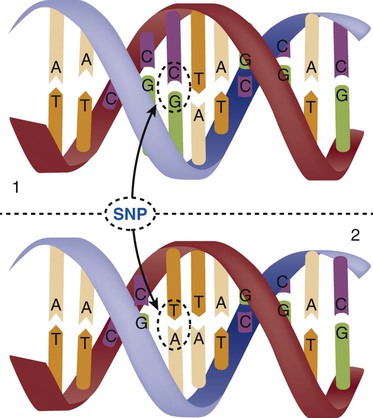
When a genetic variant occurs frequently and is present in 1% or more of the population, it is described as a genetic polymorphism. The most common change is the substitution of one single-nucleotide base. A single-letter switch is known as a single-nucleotide polymorphism (SNP), commonly abbreviated and pronounced “snip” (Fig. 4-7). This is significant when the SNP occurs in a region of DNA that codes for an amino acid. If the SNP change alters the amino acid product that is produced, it is called a nonsynonymous SNP, or missense SNP. If a nonsynonymous SNP occurs in a coding region, it may affect protein structure and lead to alterations in phenotype (disease manifestation). An example is the G-to-A coding SNP at the 1691 site of the factor V gene associated with blood coagulation.12 This polymorphism leads to the substitution of an arginine (A) by glutamine (G) at amino acid position 506, which alters one of the cleavage sites for activated protein C. Factor Va inactivation is delayed because the cleavage site is atypical. This change in one amino acid reduces the anticoagulant activity of factor V, creates a hypercoagulable state, and consequently increases the risk for DVT12 (see Box 4-1).
Alleles
Another name for a variant of a gene that occurs at a single locus is an allele. Allele symbols consist of the gene symbol, an asterisk, and the italicized allele designation. For example, the apolipoprotein E gene (APOE) has three major alleles (APOE*E2, APOE*E3, APOE*E4), and each allele codes for a different isoform of the ApoE protein.13 Apolipoprotein E is involved with cholesterol metabolism, and expression of the APOE*E4 allele is associated with the development of hyperlipidemia. APOE*E4 expression also is associated with late-onset Alzheimer’s disease and with a less favorable outcome after traumatic brain injury and brain hemorrhage.13
Not all changes in the DNA sequence have deleterious effects. Most SNPs have no effect because they are synonymous SNPs, variants that code for the same amino acid (see Table 4-1), or because they are located in non-coding genomic region.
Genetic Inheritance
Genetic Disorders
Complex Gene and Multifactorial Disorders
In some disorders, many genes interact to produce the condition, or there must be an interaction between vulnerable genes and the environment. Cardiovascular atherosclerotic diseases and type 2 diabetes are examples of complex gene disorders that result from an interaction of genetic and environmental factors.4 Pharmacogenetic syndromes are also in this category, resulting from the interactions of genes and medications.
Mitochondrial Disorders
Some diseases are caused by alterations in the DNA of the mitochondria, which are intracellular organelles found in the cytoplasm. Mitochondrial DNA is totally different and separate from the double-helix DNA found in the nucleus. Mitochondrial DNA is transferred to offspring by maternal transmission only, because mitochondria occur in oocytes but not in sperm. Most mitochondrial genetic diseases are associated with disorders of enzyme function that disrupts mitochondrial energy production. Tissues and organs that have high energy requirements such as skeletal muscles, the heart, and the central nervous system are most often affected.14
Genetic History and Family Pedigree
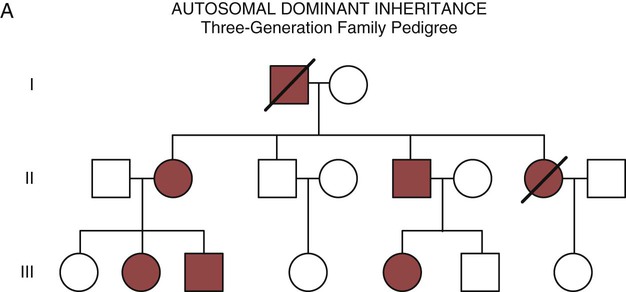
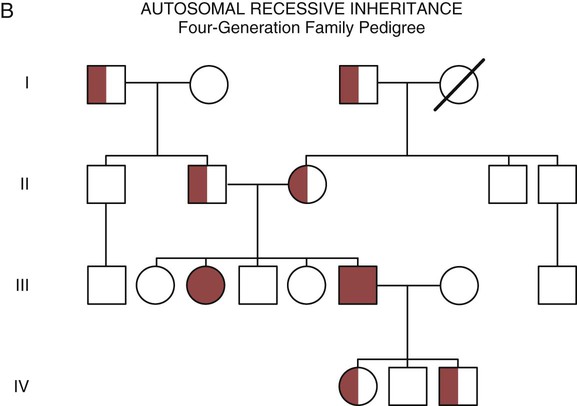
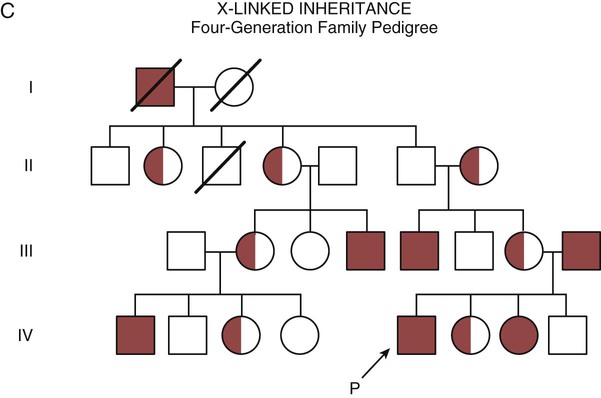
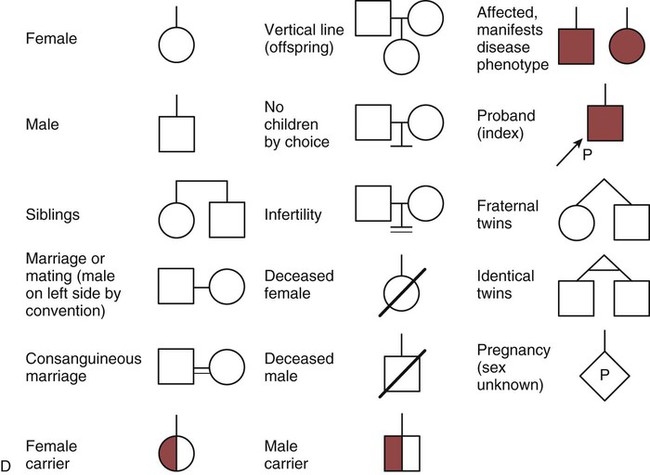
One of the tools used to determine whether a disease has a genetic component is construction of a family pedigree.15 For nurses, it is important to develop the skills to ask questions to elucidate which family members are affected and which are unaffected and then to identify the individuals who may carry the gene in question but do not have symptoms (carriers). Standardized symbols are used in the construction of a pedigree.15 The use of a legend to explain what the symbols mean prevents misinterpretation. The proband is the name given to the first person diagnosed in the family pedigree. Examples of three simple pedigrees that illustrate the single-gene inheritance patterns and the symbols used to construct a pedigree are shown in Figure 4-8 with examples described below.
Autosomal Dominant Inheritance
Autosomal dominant inheritance is described as a dominant pattern because only one copy of an affected gene is required to transmit the condition. The person has inherited one affected gene from one parent and one healthy gene from the other parent, and is described as heterozygous for that gene. Typically, the disease appears in every generation. Each child of an affected parent has a 50% chance of inheriting the condition, depending on whether a parent has transmitted the affected gene or not. Male and female offspring are equally likely to inherit and transmit the condition (see Fig. 4-8A). Examples of conditions with autosomal dominant inheritance patterns include familial hypercholesterolemia (FH) and Marfan syndrome.
Autosomal Recessive Inheritance
With an autosomal recessive pattern of inheritance, the disease or condition manifests only if the person has received an affected gene from both parents. The parents are carriers of the gene but are not themselves affected. The parents are heterozygous, because they have one copy of a normal gene and one copy of an affected gene. The inheritance pattern for this situation is slightly complex. Any of their children has a 25% chance to inherit in the following combinations: one normal and one affected gene (heterozygous carrier), two normal genes (unaffected), or two affected genes (homozygous affected). Persons who are homozygous for the gene associated with the disease always are affected, which means they demonstrate the phenotype of the disease (see Fig. 4-8B). In autosomal recessive inheritance, the phenotype associated with the condition is seen more often in siblings (sibships) than in the parents. Conditions associated with autosomal recessive inheritance include cystic fibrosis and sickle cell disease.
Sex-Linked Inheritance
Hemophilia A and Hemophilia B.
Some diseases are inherited through an X-linked pattern of inheritance. A classic example is the F8 gene, which codes for the protein that makes coagulation factor VIII. The F8 gene is located on the X chromosome.16 Of note, the inheritance of hemophilia B is also sex-linked because the F9 gene, which codes for the protein that makes coagulation factor IX, is also located on the X chromosome.16
In an X-linked disorder, each son has a 50% chance of being hemophiliac, and each daughter has a 50% chance of being a carrier. In a family pedigree, the absence of direct male-to-male transmission makes this condition identifiable as an X-linked disorder. Hemophilia A is an example of a single-gene disorder. Figure 4-8C illustrates an X-linked family pedigree for hemophilia A; a male who is affected (generation I) may pass the X-linked F8 gene only to his daughters (generation II), who may pass the gene to sons who will be affected, or to daughters who will be carriers (generation III). A female will only be affected if she receives a defective X-linked gene from both her father and mother as occurs in generation IV.
Complex Gene-Gene and Gene-Environment Disorders
Obesity and the FTO Gene
The pandemic of obesity in the industrialized world has entrained higher rates of cardiovascular atherosclerotic disease and type 2 diabetes. GWAS have identified polymorphisms in the fat mass and obesity-associated gene (FTO) on chromosome 16 that are linked to obesity.17 A patient who is overweight or obese may blame it on “my bad genes,” and this raises the question of whether the presence of an altered FTO gene automatically means the individual will be obese. From the results of a GWAS analysis of over 38,000 individuals, the 16% who were homozygous for the FTO gene carried about 3 kilograms more weight, with an increased risk of developing obesity.17 Additional genes are now being identified that will further increase our understanding of the shared genetic and environmental contribution to the obesity epidemic.18
How Information about Genetics is Obtained
Genetic Epidemiology
Genetic epidemiology represents the fusion of epidemiologic studies and genetic and genomic research methods. Before a gene can be mapped (genotyped), it is essential to have a reliable phenotype that can be consistently measured.19 One of the challenges in applying genetics to the critical care arena is that the genotype is stable but the phenotype is dynamic. Phenotypes are different at different stages of a disease and are influenced by medications, environmental factors, and gene-gene interactions.19 It is helpful to discuss the methods used outside of the critical care unit as a way of understanding the challenges in the application of genomics in this arena.
Genome-Wide Association Studies
GWAS examine the breadth of the human genome and typically use very large samples. The intent is to use genetic microarray technology and statistical computational power to find SNPs associated with a disease. Some GWAS are conducted with the intent of finding additional genes. However, one of the strengths of the GWAS model is that it does not have to begin with a biologic model in mind as long as the disease of interest is sufficiently common in the population, and by testing thousands of SNPs, the researchers may find associations that have not been detected by other methods. A classic example is a GWAS of 14,000 individuals (cases) with 3000 shared controls genotyped to find SNPs associated with seven common diseases.20 Significant new SNPs were found for five of seven diseases: type 1 diabetes, type 2 diabetes, rheumatoid arthritis, Crohn’s disease, and coronary artery disease.20 New GWAS are published every month, and the National Human Genome Research Institute (NHGRI) maintains an online catalog of published studies that can be searched by disease trait, chromosomal region, gene, or SNP.21
Genome Mapping Projects
The Human Genome Project
The human genome map was created from the DNA of only a few people.22 One of the surprising discoveries was that the number of genes possessed by humans was not as large as expected. The number of protein-coding genes in the chromosome is estimated to be between 20,000 and 25,000.3 Although researchers were initially surprised that the gene number was not higher, it has become apparent than many posttranscriptional alterations occur to actively change the protein product. The ongoing research agenda is to understand all of these additional sequence elements.
ENCODE Project
The ENCyclopedia Of DNA Elements (ENCODE) pilot project involved a detailed analysis of 1% of the human genome using cell lines.23 The goal was to elucidate all of the functional elements that enable relatively few human genes to produce such a wide variety of biologic products (e.g., proteins). The second phase of the ENCODE project examined the entire genome in 147 different cell types. The focus was on the regulatory role of RNAs in modulating cellular functions directly as biologically active molecules or indirectly by encoding other active molecules.23,24 Across coding and non-coding areas, 80% of the human genome is involved in at least one biochemical RNA or chromatin-associated event in the cell-types examined.24 Other areas of research focus on epigenetics histone modification, DNA methylation, and on the transcriptional regulatory elements that control gene expression.24 This database will be a rich resource for future biomedical genomic research.
HapMap Project
The International HapMap Project database25 contains over 4 million SNPs from 270 individuals representing four populations: African (Yoruba, Nigeria), European Americans (Utah), Japanese (Tokyo), and Han Chinese. HapMap displays short linear sections of genetic loci on the same chromosome known as haplotypes or haplotype blocks. Loci are grouped as a haplotype if they are close to each other on a chromosome and are inherited as a linear group. Genetic epidemiologists call this grouped pattern linkage disequilibrium. It means that the loci are linked due to shared ancestry, where genetic information was inherited by offspring in “chunks” or “blocks” rather than as individual genes. The mixing of sections of different chromosomes that are inherited from each parent is called recombination. The resultant linked genetic loci are called haplotypes. The edges of the haplotypes are conceptualized as recombination hotspots, where little ancestral relationship remains.22 More than 32,000 hotspots have been identified. Hotspots account for approximately 60% of recombination in the human genome and represent about 6% of the genomic sequence.26 An example of the expected haplotype change over 150,000 years from African ancestral chromosomes to modern chromosomes is shown in Figure 4-9. The biologic rationale for recombination is to increase genetic diversity.
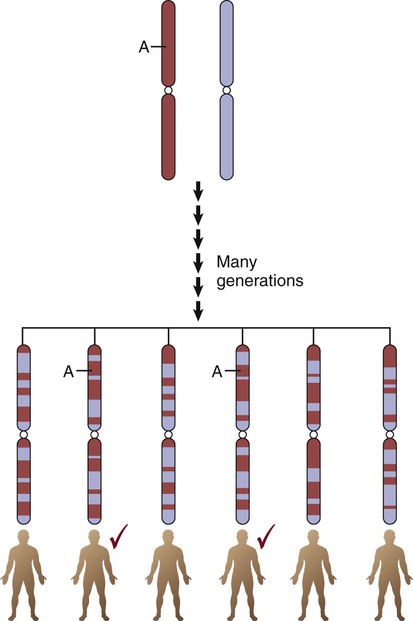
1000 Genomes Project
The 1000 Genomes Project builds on the information gained in the HapMap project because it greatly increases the number of included genomes. Scientific goals of the 1000 Genomes Project were to produce a catalog of human genomes identifying genetic variants that occur at 1% or greater frequency in the human population across most of the genome, and down to 0.5% frequency or lower, within genes.22 The 1000 Genomes Project did not collect any health-related phenotypic information.
Personal Genome Project
The Personal Genome Project (PGP) is in the process of enrolling 100,000 individual volunteers from the general public, with the goal of creating a reference database, to ultimately make individual genome sequencing more affordable.27 The purpose is to create a diverse catalog of the human genome linked to phenotypes such as medical records and medical history. This will provide a reference database against which individual genomes can be compared. As of May 12, 2012, over 1800 participants have enrolled.28 Even with only a small percentage of the intended individuals yet enrolled, the number of new genomic variants being identified with each new genome entered into the database (i.e., not seen in a previously entered genome) has dropped dramatically.26 As the number of participants increases, the researchers plan to examine the genotype-phenotype relationships of specific diseases.26
The Human Microbiome Project
In the healthy human body, microbial cells vastly outnumber human cells sometimes being described as our second genome.29 The human body is host to many microbial communities (human microbiome) located on the skin and in the nose, mouth, gastrointestinal, and urogenital tracts.30 The spectrum of these microbial communities was previously almost entirely unknown. The Human Microbiome Project is designed to identify the core human microbiome, and to determine whether changes in the human microbiome can be correlated with changes in human health.29,31 The human microbiome project is highly relevant to critical care because so many patients are admitted with sepsis or experience disruption of the gastrointestinal tract with diarrhea. The interaction of host bacteria, pathologic bacteria, critical care environment, and pharmacogenetics has the potential to bring novel insights to our current understanding of critical illness.
Genetic Diversity
The oldest human genetic lineages originate from the continent of Africa. The migration of a subset of humans out of Africa resulted in loss of genetic diversity. Numerous studies have shown higher levels of nucleotide and haplotype diversity of nuclear and mitochondrial genomes in Africans than in non-Africans.32,33 Large-scale autosomal studies of African genetic diversity are being conducted.
Copy Number Variation
Copy number variation (CNV) adds another level of variation to the human genome. CNV describes structural changes of DNA, larger than 1 kilobase, that include insertions, deletions, and inversions. The impact of CNVs on frequently seen diseases appears to be negligable,34 but when rare DNA loci associated with a disease state are inserted, the “dose” of affected DNA can alter disease expression. Examples include familial Parkinson’s disease35 and schizophrenia.36 CNVs can be either associated with a Mendelian gene inheritance or appear de novo, as a new variant, in the genome.37
Individual Genome Sequences
It was stratospherically expensive to sequence the genome of an individual. However, the price is dropping dramatically and the goal of a $1000 individual genome may soon be a reality. One of the many challenges is how to use all of the data provided by genome analysis.38 Another issue is personal privacy. These issues will require serious legal and ethical discussion at both individual and societal levels.
Genetics in Critical Care
The next step in the genomics revolution will be to connect the growing amounts of research data to clinical interventions that may help patients. The areas of clinical practice in which this has received most attention are cancer,39 cardiovascular disease,40 and pharmacogenomics.
Cancer Genetics
Somatic Mutations
The Cancer Genome Atlas
There is strong evidence that many forms of cancer are caused by epigenetic alterations; one mechanism is the addition of methyl groups to specific genes to block their effect, a process sometimes referred to as gene silencing. Epigenetic changes can disrupt the normal balance in cell proliferation, cell survival, and cell differentiation. The Cancer Genome Atlas (TCGA) project was established to accelerate understanding of the molecular basis of human cancers. The large-scale genome sequencing techniques, first developed in the Human Genome Project, are used to sequence genes associated with lethal and common cancers.41 The goal is to document all cancer genetic changes from chromosomal rearrangements to DNA mutations to epigenetic changes (chemical modifications of DNA that can turn genes on or off without altering the DNA sequence). TCGA is an international research initiative that has demonstrated it may be more important to identify and target specialized biologic pathways in many lethal cancers than single genes. Biologic pathway disruptions result in multiple gene rearrangements of gene clusters, which at first appear to be unrelated but are connected by cellular signaling systems orchestrated by complex biologic pathways.42–44
Cardiovascular Genetics
Genetic markers are being included in many cardiovascular research studies.45 A classic example is the Framingham Heart Study. This longitudinal, multigenerational study was started in 1948 to identify the common factors that contribute to cardiovascular disease.46 The researchers have phenotypic and pedigree data for several generations, and they have added genetic markers to their investigation of biologic and epidemiologic factors.47 These cardiac genomic research studies will improve understanding of the interactions between genomic and environmental factors that underpin cardiovascular disease.
Long QT Syndrome
Long QT syndrome (LQTS) is a hereditary cardiac channelopathy with several different genotypes (LQTS 1-10), and a phenotype that manifests as a prolongation of the corrected QT interval (QTc) on the electrocardiogram (ECG); affected individuals incur an increased risk of sudden cardiac death (SCD)48 (Box 4-2). Not all patients who have the LQTS genotype are aware of their condition, and not all have a prolonged QTc at rest.48 In the critical care unit these patients are at high risk of gene-environment or gene-medication interactions that can lengthen the QTc interval and entrain polymorphic ventricular tachycardia, also known as torsades de pointes49; see torsades de pointes in Chapter 14.
Cardiomyopathy
There is a very wide spectrum of cardiomyopathy genotypes and phenotypes. Some are single-gene disorders (inherited), and some are complex trait disorders with a strong environmental component where cardiomyopathy has resulted from ischemic or valvular heart disease (see cardiomyopathy in Chapter 15).
Hypertrophic Cardiomyopathy.
Hypertrophic cardiomyopathy is the most frequently encountered inherited cardiomyopathy.50 It is both genetically and phenotypically heterogeneous with documented mutations in more than 11 genes that encode for cardiac sarcomere proteins.50 Most mutations are missense SNPs in which the normal nucleotide is replaced; this alters the resultant protein. The risk of sudden cardiac death (SCD) is high for patients who manifest the phenotype of a hypertrophied ventricle or hypertrophied ventricular septum. To identify other members of the family who may be affected, construction of a three-generation pedigree and genetic testing is recommened.50
Dilated Cardiomyopathy.
Familial dilated cardiomyopathy is associated with mutations in more than 14 genes. The phenotype is left ventricular enlargement with systolic dysfunction with more than two affected family members.51 In patients over 40 years of age a diagnosis of idiopathic dilated cardiomyopathy can be made based upon the phenotype when other structural causes have been ruled out. Genetic testing is rarely performed in older adults, although the falling costs of genomic sequencing may make this feasible in the future.52
Pharmacogenetics
Cytochrome P450 Family and Medication Metabolism
The cytochrome P450 (CYP450) enzymes are a superfamily of heme-containing enzymes that are vital for medication metabolism. The CYP450 family contains 57 genes that code for enzymes involved in medication metabolism.54 The isoenzymes CYP3A4 and CYP3A5 metabolize about 50% of currently marketed medications, and they constitute approximately 60% of the total hepatic CYP450 enzyme content.55 The metabolism of more than 90% of the most clinically important medications can be accounted for by seven CYP isozymes: 3A4, 3A5, 1A2, 2C9, 2C19, 2D6, and 2E1.55
CYP3A4 can be used to understand what the abbreviations represent. CYP represents the symbol for all cytochrome P450 proteins, 3 denotes the gene family, A designates the subfamily, and 4 represents the individual gene.55 The U.S. Food and Drug Administration (FDA) requires that new medications undergo testing for interactions with the CYP450 pathway before release.
Warfarin
Warfarin (Coumadin) is a frequently prescribed anticoagulant for patents with atrial fibrillation, mechanical cardiac valves, or thrombotic disorders. Variants in the cytochrome P450 enzyme CYP2C9 gene and in the vitamin K epoxide reductase complex subunit 1 gene (VKORC1) contribute to the considerable dose variation seen with this anticoagulant.56,57 The vitamin K epoxide reductase (VKOR) enzyme activates the vitamin K–dependent clotting factors (II, VII, IX, X). Warfarin is used to inhibit the VKOR complex to prevent clot formation.
Several prospective studies showed that up to 30% of dose variability during the initiation phase of warfarin anticoagulation could be explained by CYP2C9 and VKORC1 polymorphisms. Genetic testing is not currently recommended for everyone who takes warfarin. Laboratory tests to detect the CYP2C9 and VKORC1 variants are available only at specialized reference laboratories. The FDA has added a warning label on the package to alert clinicians about CYP2C9 and VKORC1 polymorphism interactions and their effects on warfarin dosing.58
Malignant Hyperthermia
Malignant hyperthermia is a rare inherited genetic disorder that negatively affects skeletal muscle upon exposure to volatile anesthetics and depolarizing muscle relaxants. It may be unexpectedly discovered during general anesthesia. In malignant hyperthermia, calcium is released from the muscle sarcoplasmic reticulum and induces life-threatening muscle contracture with skeletal muscle rigidity, acidosis, and elevated temperature. The first gene to be identified was the ryanodine receptor 1 gene (RYR1) associated with calcium release in the sarcoplasmic reticulum.59–61 Malignant hyperthermia is described in more detail in Box 4-3.
Genetics, Genomics, and Nursing
As genetics and genomics exert more influence in clinical practice, it will have implications for the knowledge base of nurses and other professionals in health care. The learning curve may be steep. The addition of genetics also introduces new ethical and legal dilemmas into health care discussions. Genetics and genomics will have a big impact on how health and illness are conceptualized in the future. This is already required content for baccalaureate nursing education, and is now recommended for nurses with graduate degrees.62 Essential competencies for nursing practice have been developed by the American Nurses Association (ANA), and examples are listed in Table 4-2.63 It is always demanding to embrace a new model of health care delivery, and that is the challenge for all health care professionals in the coming decades. The tsunami of genetic information that is now flowing into research journals will rapidly make its way into clinical practice, including into critical care. This flood of new and relevant information will mandate that nurses understand genetic terms, incorporate genetic pedigree information into the history and physical examination, and be knowledgeable about pharmacogenetic interventions. Genetics and genomics constitute a complex area of study, made more so by the rapid evolution of scientific knowledge. One way to begin is to look at some of the free, internet-based, interactive educational tutorials listed in Box 4-4 and to become familiar with the genetic and genomic vocabulary in the key terms list at the end of the chapter.
TABLE 4-2
ESSENTIAL GENETICS AND GENOMICS COMPETENCIES FOR NURSES
| DOMAIN | COMPETENCIES OF THE REGISTERED NURSE |
| Professional Responsibilities | |
| Competent nursing incorporating genetic and genomic knowledge and skills | Recognizes when one’s own attitudes and values related to genetic and genomic science may affect care provided to patients |
| Advocates for patients’ access to desired genetic or genomic services and resources, including support groups | |
| Examines competency of practice on a regular basis, identifying areas of strength and areas in which professional development related to genetics and genomics would be beneficial | |
| Incorporates genetic and genomic technologies and information into registered nurse practice | |
| Demonstrates in practice the importance of tailoring genetic and genomic information and services to patients based on their culture, religion, knowledge level, literacy, and preferred language | |
| Advocates for the rights of all patients for autonomous, informed genetic and genomic related decision making and voluntary action | |
| Professional Practice and Assessment | |
| Application and integration of knowledge | Demonstrates an understanding of the relationship of genetics or genomics to health, prevention, screening, diagnostics, prognostics, selection of treatment, and monitoring of treatment effectiveness |
| Demonstrates an ability to elicit a minimum of three generations of family health history information | |
| Constructs a pedigree from collected family history information using standardized symbols and terminology | |
| Collects personal, health, and developmental histories that consider genetic, environmental, and genomic influences and risks | |
| Conducts comprehensive health and physical assessments that incorporate knowledge about genetic, environmental, and genomic influences and risk factors | |
| Critically analyzes the history and physical assessment findings for genetic, environmental, and genomic influences and risk factors | |
| Assesses patients’ knowledge, perceptions, and responses to genetic and genomic information | |
| Develops plan of care that incorporates genetic and genomic assessment information | |
| Identification of needed information | Identifies patients who may benefit from specific genetic and genomic information or services based on assessment data |
| Identifies credible, accurate, appropriate, and current genetic and genomic information, resources, services, and technologies specific to given patients | |
| Identifies ethical, ethnic or ancestral, cultural, religious, legal, fiscal, and societal issues related to genetic and genomic information and technologies | |
| Defines issues that undermine the rights of all patients for autonomous, informed genetic and genomic-related decision making and voluntary action | |
| Assistance with referrals | Facilitates referrals for specialized genetic and genomic services for patients, as needed |
| Provision of education, care, and support | Provides patients with interpretation of selective genetic and genomic information or services |
| Provides patients with credible, accurate, appropriate, and current genetic and genomic information, resources, services, and technologies that facilitate decision making | |
| Uses health promotion and disease prevention practices that consider genetic and genomic influences on risk with personal and environmental risk factors | |
| Incorporates knowledge of genetic or genomic risk factors (e.g., patient with a genetic predisposition for high cholesterol levels that can benefit from a change in lifestyle to decrease the likelihood that the genotype will be expressed) | |
| Uses genetic and genomic based interventions and information to improve patients’ outcomes | |
| Collaborates with health care providers in providing genetic and genomic health care | |
| Collaborates with insurance providers or payers to facilitate reimbursement for genetic and genomic health care services | |
| Performs interventions and treatments appropriate to patients’ genetic and genomic health care needs | |
| Evaluates impact and effectiveness of genetic and genomic technology, information, interventions, and treatments on patients’ outcome | |
From Consensus Panel on Genetic/Genomic Nursing Competencies. Essentials of Genetic and Genomic Nursing: Competencies, Curricula Guidelines, and Outcome Indicators. 3rd ed. Silver Spring, MD: American Nurses Association; 2009.
Ethical and Legal Issues in Genetics and Genomics
Direct-to-Consumer Tests
A new frontier in genomics testing is the ability of any individual to voluntarily undergo genetic testing. Several private companies advertise this service on the internet. Patients send a sample of their DNA, usually a swab from the inside of the cheek (buccal swab), to check for selected genetic risks. Other websites offer paternity testing from buccal swabs of the child and father. Many countries are now enacting laws to ensure individuals’ genomic information is protected. Genetic counselors caution that the results may be hard to understand or misleading for some individuals but clearly this is of great interest to many people.64
The Genetic Information Nondiscrimination Act
The Genetic Information Nondiscrimination Act (GINA) of 2008 is an essential piece of legislation designed to prevent abuse of genetic information in employment and health insurance decisions in the United States.65 The purpose of GINA is to protect individuals who may have the gene for a disorder—but do not manifest the phenotype—from being penalized. Some people who may be at risk for a disorder disease will not be tested because they fear that a positive result may affect their employability. GINA also mandates that genetic information about individuals and their families has the same protections as health information. It is important that clinicians understand that the GINA legislation does not cover all categories of insurance. It offers no protection against discrimination for life insurance, disability insurance, or long-term care insurance. The GINA legislation does not cover members of the military, the Veteran’s Affairs Service, or Indian Health Service.66,67
Human Genetics Key Terms
Allele: one of several alternative gene variants that can exist at a single locus (location) on a chromosome. The term allele may also be used when referring to SNP variants. The most frequently found allele in a population is called the wild-type allele.
Candidate gene: a gene that is believed to cause or contribute to a disease. This term is typically used when the gene will be included in a research study.
Chromosomes: structures made of DNA and proteins and located in the nuclei of cells. Chromosomes come in pairs, and a normal human cell contains 46 chromosomes: 22 pairs of autosomes and a pair of sex chromosomes. Chromosomes are composed of genes, regulatory sequences, and non-coding DNA segments.
Codon: three-sequence nucleotide bases that code for an amino acid.
DNA: deoxyribonucleic acid is made up of four nucleotide bases: adenine (A), guanine (G), cytosine (C), and thymine (T). DNA is heritable genetic information that resides in chromosomes inside each cell nucleus.
Epigenetic: chemical modifications of DNA that can turn genes on or off without altering the DNA sequence. Epigenetic changes are not heritable.
Gene: a unit of inheritance; a working subunit of DNA. Each of the 20,000 to 25,000 genes in the body contains the code for a specific product, typically a protein such as an enzyme, and other specific tissue cells.
Gene expression: the process by which the coded information of a gene is translated into the structures present and operating in the cell (proteins or ribonucleic acid [RNA]).
Gene map: a description of the relative positions of genes on a chromosome and the distance between them.
Genetic linkage maps: DNA maps that assign relative chromosomal locations to genetic locations—either genes for known traits or distinctive sequences of DNA on the basis of how frequently they are inherited together.
Genetics: the scientific study of heredity, which is how particular qualities or traits are transmitted from parents to offspring. Traditionally, the focus has been on individual genes and their impact on uncommon single-gene disorders. Today the study of genetics also involves multi-gene disorders and gene-environment interactions.
Genomics: the expansive study of all the genes in the human genome, including gene-gene interactions, gene interactions with the environment, and the influence of other psychosocial and cultural factors.
Genotype: the genetic code sequence carried by an individual.
Haplotype: closely linked loci on a chromosome. Haplotype blocks denote chromosomal regions where SNPs are in strong linkage disequilibrium, and they are mapped in the HapMap Project database.
HapMap Project: a map of haplotype blocks that researchers use when searching for candidate genes. Haplotypes are cataloged according to racial and ethnic group in the HapMap database.
Heterozygous: possessing two different sequences (alleles) of a particular gene, with one inherited from each parent.
Homozygous: possessing two identical sequences of a particular gene, with one inherited from each parent.
Linkage: the association of genes or markers that lie very near each other on a chromosome. Linked genes and markers tend to be inherited together.
Linkage analysis: a gene-mapping technique that finds patterns of heredity in large, high-risk families to locate a disease-causing gene mutation by identifying traits that are co-inherited with the gene.
Linkage disequilibrium (LD): the nonrandom association between alleles at different loci. These alleles at loci occur together on the same section of a chromosome more often than would be predicted by chance alone. This technique has been used to determine which genes are linked and therefore inherited together.
Locus and loci: the place on a chromosome where a specific gene is located, similar in concept to a street address for the gene. The singular term is locus, and the plural is loci.
Mendelian diseases: single-gene disorders that appear in families in dominant or recessive inheritance patterns.
Mutation: a change, deletion, or rearrangement in an individual’s DNA sequence that may lead to the synthesis of an altered protein or the inability to produce the protein. The term mutation is used when the variant occurs in less than 1% of the population.
Nucleotide: a building block of DNA or RNA that consists of one nitrogenous base, one phosphate molecule, and one glucose molecule.
Pedigree: a graphic multigenerational family health history that uses standardized symbols.
Pharmacogenomics: the study of genetically determined responses to medications, genetic variation in medication metabolizing enzymes, and consequent alteration in medication effectiveness across the genome.
Phenotype: the observable manifestation of a genetic trait that results from a specific genotype or gene-environment interaction. These are physical characteristics such as the signs and symptoms associated with a disease.
Polymorphism: a common variation in the sequence of DNA that occurs in more than 1% of the population. The most frequent sequence is referred to as the wild type, and less common variants are called polymorphisms.
Proband: the first person diagnosed with a condition in a family pedigree. An arrow in the family pedigree identifies the proband.
Single-nucleotide polymorphism (SNP): a change in the DNA nucleotide sequence caused by replacement of a single-nucleotide base. If the change of nucleotide results in a different protein product, it is called a non-synonymous SNP. If the protein product is not changed, it is called a synonymous SNP.
Somatic cells: all body cells, except the reproductive cells.
Transcription: the process used by DNA to code for messenger RNA.
Summary
• DNA is arranged inside the nucleus of the cell. DNA resembles a ladder with two long strands twisted around each other to form a double-stranded helix. The rungs of the ladder are made up of nucleotide base pairs. There are 20,000 to 25,000 genes in the human genome.
• Several conditions seen in critical care have a genetic component, including factor V Leiden thrombosis, hemophilia A, LQTS, and cardiomyopathy. Pharmacogenetic syndromes represent medication-gene interactions; examples include malignant hyperthermia owing to RYR1 polymorphisms, and warfarin dosage affected by CYP2C9 and VKORC1 polymorphisms.
• The GINA legislation is designed to prevent the use of an individual’s genetic risk profile in employment and insurance decisions in the United States.
• Genetic and genomic competency is recommended for all registered nurses.