Genetics
Anencephaly and spina bifida, occurring with a prevalence of about 0.5 to 2 of 1000 live births, and congenital heart disease, with a prevalence of approximately 1%, are the most common. 1
2. Should an asymptomatic infant with a single umbilical artery have a screening ultrasound scan done for renal anomalies?
This point has been argued for years. A single umbilical artery is a rare phenomenon. In one study of nearly 35,000 infants, examination of the placenta showed that only 112 (0.32%) had a single umbilical artery. All 112 underwent renal ultrasonography, and 17% had abnormalities (45% of which persisted). A more recent study demonstrated that left umbilical arteries tend to be absent more often than right umbilical arteries when only a single artery is present. In addition, there was a high incidence of associated congenital malformations in nearly 25% of the infants diagnosed prenatally with a single umbilical artery. Because of the rarity of the condition and the increased association of abnormalities, patients with single umbilical arteries probably should receive a screening renal ultrasound. 2
3. Excluding chromosomal analysis, what laboratory test results suggest that a woman is carrying a fetus with trisomy 21 syndrome?
Recently a new test has been developed that analyzes circulating cell-free DNA extracted from a maternal blood sample. The test detects an increased representation of chromosome 21 material, which is associated with trisomy 21. The Down syndrome detection rate was 98.6% with a false-positive rate of 0.20% This test is not recommended for population-based screening but may be used for women who screen positive on serum screening before proceeding to an invasive diagnostic test. 34
4. Which other conditions may be detected by a raised amniotic fluid AFP level?










5. A woman had a positive serum screen in the second trimester and normal fetal ultrasound results but declined amniocentesis. She delivers a healthy-appearing infant. What further studies are indicated given the triple screen?
6. How would you evaluate a newborn with Down syndrome to ensure you are discharging a healthy infant? What serious abnormalities are likely?
 Order a chromosome study on peripheral blood (G-banding) to rule out translocation or mosaicism.
Order a chromosome study on peripheral blood (G-banding) to rule out translocation or mosaicism.




 Perform a thyroid screen (included in all newborn screens automatically).
Perform a thyroid screen (included in all newborn screens automatically).

 KEY POINTS: COMMON CAUSES OF BIRTH DEFECTS
KEY POINTS: COMMON CAUSES OF BIRTH DEFECTS
7. A macrosomic infant is born with an omphalocele and large tongue. What would you anticipate monitoring closely in this baby, and why?
This baby may have Beckwith–Wiedemann syndrome and may be at risk for hypoglycemia. Other signs of Beckwith–Wiedemann syndrome include grooves or pits on the ear lobes, hemihypertrophy, and visceromegaly ( Fig. 11-1). These children are at risk for Wilms tumor and hepatoblastoma and should be monitored with an abdominal ultrasound and AFP testing every 4 months for the first 6 years of life.

Figure 11-1 Beckwith–Wiedemann Syndrome. Macroglossia can be significant in babies with Beckwith–Wiedemann Syndrome
8. Are there any known factors that increase the risk of having a child with Beckwith–Wiedemann syndrome?
 This midline anterior abdominal well defect is covered by a transparent sac consisting of amnion and peritoneum with Wharton jelly between them.
This midline anterior abdominal well defect is covered by a transparent sac consisting of amnion and peritoneum with Wharton jelly between them.
 Intraabdominal viscera is herniated through the umbilical ring. The size varies with contents.
Intraabdominal viscera is herniated through the umbilical ring. The size varies with contents.
 The umbilical cord inserts into the sac.
The umbilical cord inserts into the sac.
 Other malformations are present in 67% of cases.
Other malformations are present in 67% of cases.
 This defect can be associated with chromosome abnormalities, especially trisomy 13.
This defect can be associated with chromosome abnormalities, especially trisomy 13.
 The umbilical cord is attached to the abdominal wall to the left of the defect. There is a normal umbilical cord insertion.
The umbilical cord is attached to the abdominal wall to the left of the defect. There is a normal umbilical cord insertion.

 In 15% of cases, gastroschisis is associated with other major malformations.
In 15% of cases, gastroschisis is associated with other major malformations.
 It is not commonly found in fetuses with chromosome abnormalities.
It is not commonly found in fetuses with chromosome abnormalities.
Congenital diaphragmatic hernia is an associated inherited condition in the following syndromes:




 Simpson–Golabi–Behmel syndrome
Simpson–Golabi–Behmel syndrome

 Pallister–Killian syndrome (mosaic tetrasomy 12p)
Pallister–Killian syndrome (mosaic tetrasomy 12p)

 Aneuploidy and chromosomal disorders, including numerous microdeletions and microduplication
Aneuploidy and chromosomal disorders, including numerous microdeletions and microduplication
11. A female infant is born with the following features: puffiness of the dorsum of the hands and feet, excessive skin at the nape of the neck with a low posterior hairline, and a broad chest and widely spaced nipples. What is the differential diagnosis?

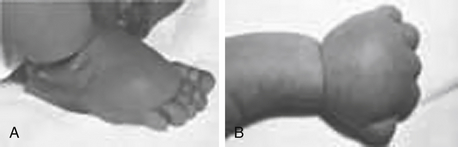

12. How would you work up the baby in Question 11?
 Chromosome study on peripheral blood (G-banding)
Chromosome study on peripheral blood (G-banding)
 Genetic testing for Noonan syndrome if chromosomes are normal
Genetic testing for Noonan syndrome if chromosomes are normal
 Cardiac evaluation, including echocardiogram
Cardiac evaluation, including echocardiogram

 Referral for genetic counseling and early intervention
Referral for genetic counseling and early intervention
 KEY POINTS: PRINCIPLES OF BIRTH DEFECTS
KEY POINTS: PRINCIPLES OF BIRTH DEFECTS
1. Single minor anomalies such as an isolated simian crease are common and not necessarily associated with an underlying genetic problem. However, as the number of minor anomalies increases, the likelihood of an underlying genetic diagnosis also increases.
2. Susceptibility for some birth defects such as cleft lip and palate are multifactorial and caused by a combination of genetic and environmental factors.
3. The findings of intrauterine growth restriction or birth defects (or both) should warrant investigation into genetic, infectious, and teratogenic etiologies.
14. Can Noonan syndrome be diagnosed prenatally, and if so, what are the most common prenatal features?
16. Intrauterine growth restriction (IUGR) has many causes. What approach would you use to evaluate a newborn with IUGR?
 Establish whether the growth restriction is proportionate or disproportionate.
Establish whether the growth restriction is proportionate or disproportionate.
 Perform a detailed physical examination for anomalies or dysmorphic features.
Perform a detailed physical examination for anomalies or dysmorphic features.
 If dysmorphic or multiple anomalies are present, chromosome studies are indicated.
If dysmorphic or multiple anomalies are present, chromosome studies are indicated.

 Viral studies and antibody titers should be ordered as indicated.
Viral studies and antibody titers should be ordered as indicated.
 Uncontrolled maternal phenylketonuria can be associated with IUGR and microcephaly.
Uncontrolled maternal phenylketonuria can be associated with IUGR and microcephaly.



19. What conditions are associated with UPD?
 Maternal UPD 15 is associated with Prader–Willi syndrome, whereas paternal UPD 15 is associated with Angelman syndrome.
Maternal UPD 15 is associated with Prader–Willi syndrome, whereas paternal UPD 15 is associated with Angelman syndrome.
 Paternal UPD 11 is associated with Beckwith–Wiedemann syndrome.
Paternal UPD 11 is associated with Beckwith–Wiedemann syndrome.
 Maternal UPD 7 has been seen in some cases of Russell–Silver syndrome.
Maternal UPD 7 has been seen in some cases of Russell–Silver syndrome.
 Paternal UPD 6 causes growth retardation (sometimes severe) and transient neonatal diabetes.
Paternal UPD 6 causes growth retardation (sometimes severe) and transient neonatal diabetes.

20. What conditions are commonly diagnosed by fluorescent in situ hybridization (FISH)?








21. Can microdeletion syndromes such as DiGeorge syndrome be detected by a routine karyotype?
 KEY POINTS: CHROMOSOMAL DEFECTS
KEY POINTS: CHROMOSOMAL DEFECTS
1. The risk of aneuploidy increases with advanced maternal age; however, most aneuploid births are to mothers who are not of advanced maternal age. The majority of trisomies are a result of maternal meiotic errors; however, almost half of Klinefelter cases are caused by errors in paternal meiosis.
2. Screening for Down syndrome with a combination of noninvasive ultrasound markers of nuchal translucency and maternal serum markers allows for greater than 95% sensitivity in detecting Down syndrome.
3. Microdeletions are too small to be resolved by a karyotype and require fluorescence in situ hybridization. The majority are de novo and not inherited. They are associated with a variety of clinical syndromes such as DiGeorge syndrome, Williams syndrome, Angelman syndrome, Miller–Dieker syndrome, Smith–Magenis syndrome, and Kallmann syndrome.
4. Balanced translocations have the total correct amount of DNA. If the balanced translocation is inherited, the phenotype in the child is predicted to be that of the balanced translocation carrier parent. If the translocation is de novo, there is an approximately 15% chance it will be associated with a phenotypic consequence such as a birth defect, cognitive impairment, or medical problem.
5. UPD means that for one set of chromosomes, both chromosomes were inherited from the same parent. If genes on that chromosome are imprinted, this can produce specific clinical symptoms, such as Prader–Willi syndrome. UPD cannot be detected by a standard karyotype but can be detected with other molecular genetic techniques.
24. For a newborn baby with congenital heart disease and a diaphragmatic hernia, which test should you order first, a karyotype or a chromosome microarray?
Chromosome microarray is now considered the first line test to evaluate chromosomes. The only anomalies missed by a chromosome microarray are balanced translocations and low levels of mosaicism, but the prevalence of these types of chromosomal disorders is low. 5
25. The geneticist cannot be reached, and you have to evaluate an intrauterine fetal demise. What do you do?







Anophthalmia is the medical term used to describe the absence of the globe and ocular tissue from the orbit. Anophthalmia and microphthalmia are often used interchangeably because, in most cases, the magnetic resonance imaging (MRI) or computed tomography (CT) scan shows some remnants of either the globe or surrounding tissue. Anophthalmia may be unilateral or bilateral and is often associated with other anomalies. There are many causes of anophthalmia including single gene mutations, syndromes, chromosome abnormalities, and teratogenic exposures. Anophthalmia is rare, with an incidence of about 1 in 10,000.






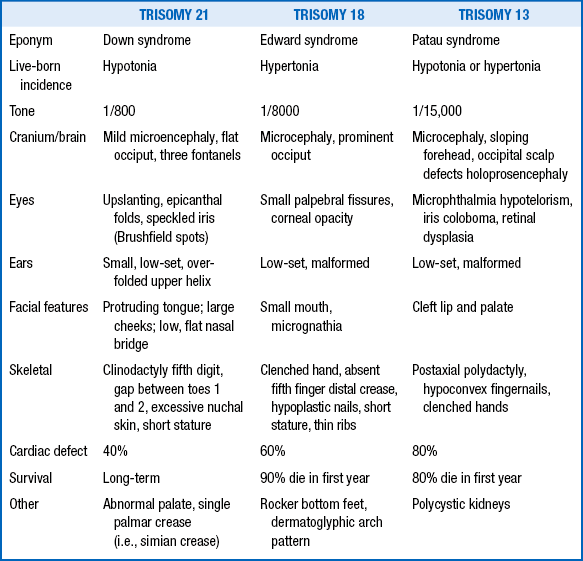
29. What are the major characteristics of the three major chromosomal malformations: trisomy 21, trisomy 18, and trisomy 13?
31. Why was the maternal age of 35 years at delivery chosen as the cutoff for recommending amniocentesis for chromosome analysis, and is this still relevant?
There is a well-known association between advanced maternal age and trisomies (including XXY; XXX; and trisomies 13, 18, and 21). Most cases of Down syndrome involve nondisjunction at meiosis I in the mother. This may be related to the lengthy stage of meiotic arrest between oocyte development in the fetus and ovulation, which may occur as many as 40 years later ( Table 11-2). With the advent of multiple noninvasive methods to screen for aneuploidies, invasive testing is driven more by results on the screening tests rather than maternal age alone.
TABLE 11-2
ASSOCIATION BETWEEN MATERNAL AGE AND RISK OF TRISOMY 21 SYNDROME
| MATERNAL AGE (YEARS) | APPROXIMATE RISK |
| 30 | 1/1000 |
| 35 | 1/365 |
| 40 | 1/100 |
| 45 | 1/50 |
Only 34% of babies are born to mothers older than 35 years of age. Although their individual risk is higher, women in this age bracket account for only 5% of all pregnancies in the United States. 6
Advanced paternal age is associated with de novo point mutations in all genes.
 KEY POINTS: MONOGENIC CONDITIONS
KEY POINTS: MONOGENIC CONDITIONS
1. Genetic susceptibility to some neurologic conditions that are dominantly inherited is due to expansion of triplet repeats. The likelihood of repeat expansions depends on the sex of the transmitting parent and is specific to each disease. For instance, triplet repeats in the gene for myotonic dystrophy are more likely to expand when transmitted maternally, whereas the triplet repeats in the gene for Huntington disease are more likely to expand when transmitted paternally.
2. Most monogenic conditions affecting children are autosomal recessively inherited.
3. Genes encoding mitochondrial proteins can be found either in the mitochondrial genome and be maternally inherited or in the nuclear genome and autosomal recessively inherited.
4. Genetic mosaicism may be detectable by differences in skin pigmentation that appear as swirls.
5. Genetic testing is currently clinically available for more than 2500 disorders.
36. What genetically inherited disease has the highest known mutation rate per gamete per generation?
37. Which disorders with ethnic and racial predilections most commonly warrant maternal screening for carrier status?
TABLE 11-3
SCREENING FOR GENETIC DISORDERS
| DISORDER | ETHNIC GROUP | TEST |
| Tay–Sachs disease | Ashkenazi Jewish, French Canadian | Hexosaminidase A enzymatic testing and genetic testing |
| Sickle cell disease | African, Black, Hispanic, Arab, Indian, Mediterranean | Hemoglobin electrophoresis |
| Alpha- and beta-thalassemia | Mediterranean, southern southeastern Asian, Chinese | MCV <80 |
| Cystic fibrosis | All ethnicities; more common among Caucasians | Genetic testing for the common mutations |
38. Why are mitochondrially encoded disorders transmitted from generation to generation by the mother and not the father?
Mitochondrial DNA abnormalities (e.g., many cases of ragged red fiber myopathies) are passed on from the mother because mitochondria are present in the cytoplasm of the egg and not the sperm. Transmission to male or female offspring is equally likely; however, expression can be variable within a family because of heteroplasmy with normal and abnormal mitochondria in differing proportions in different family members. 7
Advanced paternal age is associated with new dominant mutations. The assumption is that the increased mutation rate is caused by accumulation of new mutations from many cell divisions in the spermatid as men age. The more cell divisions, the more likely that an error (mutation) will occur. The mutation rate in fathers older than 50 years is five times higher than the mutation rate in fathers younger than 20 years of age. New, common autosomal dominant mutations that are often the result of de novo mutations are achondroplasia, craniosynostosis, neurofibromatosis, and Marfan syndrome. 8
41. What is the most common genetic disease that is lethal without intervention within the first year of life?
Hyperphagia, hypotonia, hypogonadism, and obesity. Up to 75% of patients have a paternal microdeletion on the long arm of chromosome 15. The gene or genes responsible for Prader–Willi syndrome are subject to parental imprinting. Imprinting is the process by which expression of a gene depends on whether it has been inherited from the mother or the father. The gene or genes associated with Prader–Willi syndrome are maternally imprinted or maternally silenced, meaning that loss of the paternal copy will result in the phenotype of Prader–Willi ( Fig. 11-3) because only the father’s copy is active. A closely related area of the long arm of chromosome 15 is maternally imprinted, and loss of the maternal copy leads to Angelman syndrome. Angelman syndrome is characterized by severe developmental delay; abnormal ataxic gait; seizures; inappropriate laughter; and jerking movements, especially of the arms. 91011

Figure 11-3 Children with Prader–Willi syndrome often have fair hair and are hypotonic, as demonstrated in the oral hypotonia in this child.
Achondroplasia is the most common viable skeletal dysplasia, occurring 1 in 26,000 live births. Its features are short stature (mean adult height, 4 feet 2 inches), macrocephaly, depressed nasal bridge, lordosis, and a trident hand. Some patients develop hydrocephalus because of a small foramen magnum. Radiographic findings include narrowing of the interpedicular distance as one proceeds caudally. Both achondroplasia and thanatophoric dysplasia are due to mutations in fibroblast growth factor receptor 3. In achondroplasia the mutation is in the transmembrane domain, whereas the mutation in thanatophoric dysplasia is either in the intracellular domain (type 2) or in the extracellular domain (type 1). 12
44. What chromosomal abnormality is found in cri du chat syndrome and what are the clinical features?
Cri du chat syndrome is due to a deletion of material from the short arm of chromosome 5 (i.e., 5p-) that causes many problems, including growth retardation, microcephaly, and severe mental retardation. Patients have a characteristic catlike cry during infancy from which the syndrome derives its name. In 85% of cases the deletion is a de novo event. In 15% it is caused by malsegregation resulting from a balanced parental translocation.






 KEY POINTS: GENETIC CARE IN PREGNANCY
KEY POINTS: GENETIC CARE IN PREGNANCY
1. Standard genetic care in pregnancy should include evaluating the family history; screening for hemoglobinopathies; offering carrier testing for cystic fibrosis; and evaluating for aneuploidy, including noninvasive testing such as nuchal translucency and serum screening or invasive testing such as chorionic villus sampling or amniocentesis. Certain populations or individuals should be offered specific genetic testing for monogenic disorders depending on their personal or family history or their ethnicity.
2. When taking a family history, the clinician should gather information about the number and relationships of family members with birth defects, growth problems, mental retardation, serious medical problems (especially those at a young age), auditory or visual impairment, ethnicity, and consanguinity.
3. Prenatal genetic testing can be performed by chorionic villus sampling or amniocentesis. There is a chance that a genetic abnormality could be identified by chorionic villus sampling that is confined to the placenta (confined placental mosaicism) that will have little bearing on the fetus if the placenta develops normally.
4. Carrier screening for cystic fibrosis is currently offered to all couples contemplating pregnancy or currently pregnant, regardless of ethnicity.
5. The sensitivity of carrier screening for diseases such as cystic fibrosis depends critically on the ethnicity of the patient.
47. In the evaluation of a stillborn infant, how does the general appearance of the fetus suggest a likely etiology?






In addition to an autopsy, other studies that should be considered include chromosomal analysis; skeletal radiographs; placental and cord histologic studies; titers for congenital infection; and, if hydropic, evaluation for a hemoglobinopathy (e.g., alpha-thalassemia) or possible metabolic storage disease. 13
 Cytogenic analysis of both parents to rule out mosaicism or a balanced translocation
Cytogenic analysis of both parents to rule out mosaicism or a balanced translocation

 Infectious work-up for Mycoplasma species, Chlamydia species, and other pathogens
Infectious work-up for Mycoplasma species, Chlamydia species, and other pathogens

 Hormonal-endometrial biopsy or progesterone level analysis to rule out a luteal phase defect
Hormonal-endometrial biopsy or progesterone level analysis to rule out a luteal phase defect



50. How are structural dysmorphisms categorized?
 Malformation: a problem of poor formation (likely genetically based) in which the abnormality is present at the onset of development (e.g., hypoplastic thumbs of Fanconi anemia)
Malformation: a problem of poor formation (likely genetically based) in which the abnormality is present at the onset of development (e.g., hypoplastic thumbs of Fanconi anemia)



51. What is the difference between a major and a minor malformation?
52. What are the features of most common associations, CHARGE, VATER, and VACTERL?
 CHARGE: coloboma of the eye, heart defects, atresia of the choanae, retardation (mental and growth), genital anomalies (in males), ear anomalies. Some cases of CHARGE association are due to mutations in the gene chromodomain helicase DNA-binding protein 7 (CHD7).
CHARGE: coloboma of the eye, heart defects, atresia of the choanae, retardation (mental and growth), genital anomalies (in males), ear anomalies. Some cases of CHARGE association are due to mutations in the gene chromodomain helicase DNA-binding protein 7 (CHD7).
 VATER: vertebral, anal, tracheoesophageal, renal or radial anomalies
VATER: vertebral, anal, tracheoesophageal, renal or radial anomalies

53. What malformations and conditions are associated with oligohydramnios?
In early pregnancy (before 4 months), the majority of amniotic fluid is produced by transudation through the placental membranes and fetal skin. Later in pregnancy the bulk of amniotic fluid arises from fetal urination and fetal lung fluid production. At term the fetus swallows approximately 500 mL of amniotic fluid per day and urinates an equivalent amount. Fetal urine production increases rapidly from 3.5 mL/h at 25 weeks to 25 mL/h at term. Any malformation that leads to impaired urine production will cause oligohydramnios, including renal dysplasia, renal agenesis, and bladder outlet obstruction. When uteroplacental insufficiency occurs, the fetus is often faced with poor nutritive and volume support. The fetus becomes intravascularly depleted, leading to increased fluid conservation and decreased urine output, causing oligohydramnios. Oligohydramnios is often associated with IUGR.
Potter syndrome has come to be synonymous with fetal malformations caused by extreme oligohydramnios. A lack of amniotic fluid leads to fetal compression; a squashed, flat face; clubbing of the feet; pulmonary hypoplasia; and, commonly, breech presentation ( Fig. 11-4). Normal fetal lung development depends on in utero “breathing” and production of fetal lung fluid. In the absence of amniotic fluid, pulmonary hypoplasia occurs and is the cause of death for most fetuses with Potter syndrome. The underlying mechanism in Potter syndrome was initially reported to be renal agenesis or renal dysplasia. However, bladder outlet obstruction and prolonged premature rupture of the membranes may also cause this sequence. Some prefer that Potter syndrome be defined solely as renal agenesis.
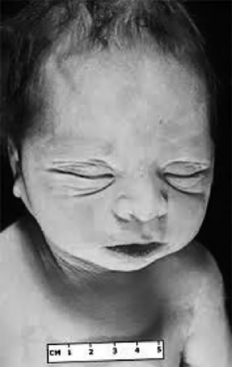
Figure 11-4 Potter syndrome is associated with oliguria and low levels of amniotic fluid, which lead to compression of the face during development and dysmorphic facial features that can include hypertelorism.
57. How do clinodactyly, syndactyly, and camptodactyly differ?
 Clinodactyly: Curvature of a toe or finger (usually the fifth) due to hypoplasia of the middle phalanx, which is the last fetal bone to develop in the hands and feet. Normal curvature can consist of up to 8 degrees of in-turning. Curvature beyond this is considered a minor anomaly.
Clinodactyly: Curvature of a toe or finger (usually the fifth) due to hypoplasia of the middle phalanx, which is the last fetal bone to develop in the hands and feet. Normal curvature can consist of up to 8 degrees of in-turning. Curvature beyond this is considered a minor anomaly.
 Syndactyly: An incomplete separation of fingers (usually third and fourth) or toes (usually second or third) ( Fig. 11-5).
Syndactyly: An incomplete separation of fingers (usually third and fourth) or toes (usually second or third) ( Fig. 11-5).
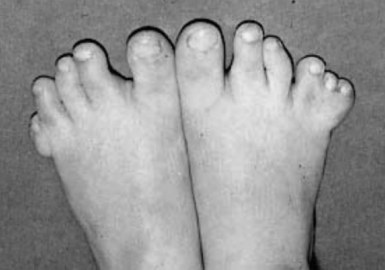
Figure 11-5 Syndactyly and postaxial polydactyly can be observed with genetic conditions such as Bardet–Biedl syndrome.
 Camptodactyly: Abnormal persistent flexion of fingers or toes.
Camptodactyly: Abnormal persistent flexion of fingers or toes.
Preauricular pits and tags are minor anomalies that occur in about 0.3–1.0% of persons, with a wide variance in frequency among racial groups ( Fig. 11-6). They are twice as common in females as in males and can be inherited as an autosomal dominant trait. They are believed to represent remnants of early embryonic bronchial cleft or arch structures. As isolated findings, they do not warrant additional evaluations.
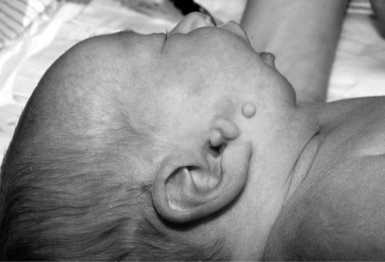
This designation is made when the upper portion of the ear (i.e., helix) meets the head at a level below a horizontal line drawn from the lateral aspect of the palpebral fissure. The best way to measure is to align a straight edge between the two inner canthi and determine whether the ears lie completely below this plane. Normally, approximately 10% of the ear is above this plane. 14
Most cases of cleft lip and palate are inherited in a polygenic or multifactorial pattern. The male-to-female ratio is 3:2, and the incidence in the general population is approximately 1 in 1000. The recurrence risk after one affected child is 3% to 4%; after two affected children, it rises to 8% to 9%.
Colobomas of the iris result from abnormal ocular development and embryogenesis. They are frequently associated with chromosomal syndromes, most commonly trisomy 13, 4p-, 13q-, and triploidy. In addition, they may be commonly found in the CHARGE association, Goltz syndrome, and Axenfeld-Rieger syndrome ( Fig. 11-7). Whenever iris colobomas are noted, chromosome microarray is recommended.
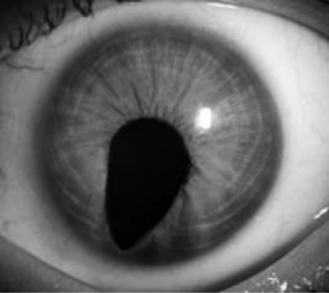
Figure 11-7 Coloboma of the iris can be observed with many heritable eye conditions, including CHARGE.
In the fetus hair follicles on the skin surface grow downward during weeks 10 through 16. During this time the brain and scalp expand outward in a domelike fashion, pulling the follicles in different directions, and at 18 weeks when the hair erupts, patterns are set. The “crown,” or parietal hair whorl, is the focal point of this outgrowth. At birth it is usually a few centimeters anterior to the posterior fontanel. Approximately 55% of single parietal scalp whorls are left of midline (presumably secondary to the larger size of the left brain), 30% are right-sided, and 15% are midline. Bilateral hair whorls are present in 5% of normal persons. Abnormal positioning of the hair whorl (particularly a posterior location) can be seen in microcephaly.
 KEY POINTS: RISK OF RECURRENCE
KEY POINTS: RISK OF RECURRENCE
1. The risk of having an affected child with an autosomal recessive condition if both parents are carriers is 25%.
2. The risk of having an affected child with an autosomal recessive condition after having three previously affected children (if both parents are carriers) is still 25%. Each pregnancy is an independent event.
3. The risk of having a child with Duchenne muscular dystrophy if the mother is a carrier is 25%. None of the girls will be affected for this X-linked disorder, but 50% of the boys will be affected.
4. A karyotype should be determined for every baby with Down syndrome to determine whether the condition was caused by an inherited translocation of chromosome 21, because this would significantly increase the risk of recurrence for the parents.
69. What is the risk of having a child with a recessive disorder when the parents are first or second cousins?
A chromosome translocation is a transfer of chromosomal material between two (or more) nonhomologous chromosomes. The exchange is usually reciprocal (the two segments trading places). The genetic content of the person is therefore complete but rearranged. A Robertsonian translocation represents a special variety of chromosome translocation in which the long arms of two acrocentric chromosomes (13, 14, 15, 21, or 22) fuse at their centromeres. The breaks may occur within, above, or below the centromeres. The short arms are usually lost, but this does not produce an abnormality because the genetic material on the short arms of acrocentric chromosomes occurs in multiple copies throughout the genome. A phenotypically normal person with a Robertsonian translocation has only 45 chromosomes inasmuch as the long arms of two acrocentric chromosomes are fused into one.




Only infants with Turner syndrome have physical features easily identifiable at birth.
77. Describe the similarities and differences between Turner syndrome and Noonan syndrome.
 Similarities include short stature, web neck, cardiac defects, low posterior hairline, broad chest, wide-spaced nipples, edema of the dorsum of the hands and feet, and cubitus valgus.
Similarities include short stature, web neck, cardiac defects, low posterior hairline, broad chest, wide-spaced nipples, edema of the dorsum of the hands and feet, and cubitus valgus.
 Differences are summarized in Table 11-5.
Differences are summarized in Table 11-5.
TABLE 11-5
DIFFERENCES BETWEEN TURNER AND NOONAN SYNDROMES
| TURNER SYNDROME | NOONAN SYNDROME |
| Females only | Both males and females |
| Chromosomal disorder (45,X) | Normal chromosomes (autosomal dominant) Near-normal IQ, mild to moderate intellectual disability |
| Coarctation the most common | Pulmonary stenosis the most common, cardiac defect |
| Amenorrhea and sterility | Normal menstrual cycle in females |
78. What is the most common inherited form of mental retardation?
Fragile X syndrome is most common.
When the lymphocytes of an affected male are grown in a folate-deficient medium and the chromosomes examined, a substantial number of X chromosomes demonstrate a break near the distal end of the long arm. This site, the fragile X mental retardation 1 gene (FMR1), was identified and sequenced in 1991. At the center of the gene is a repeating trinucleotide sequence (CGG) that in normal persons repeats between 6 and 45 times. However, in carriers the sequence expands to between 50 and 200 times (called a premutation), and in fully affected persons it expands to between 200 and 600 copies. These longer sequences cause malfunctioning of the gene. The repeat expansion is most sensitively and accurately determined by Southern blot analysis. Male as well as female subjects can be affected, although it is an X-linked disorder.
Premature ovarian failure is one potential manifestation.
83. What disorder should be considered in a neonate with a maternal history of acute fatty liver of pregnancy or Hemolysis, Elevated Liver enzymes, and Low Platelet count (HELLP) syndrome?
Long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD), a fatty acid oxidation disorder.
89. If you suspect a baby has congenital myotonic dystrophy, which parent would you examine for symptoms, and what would you look for?
You would examine the mother for evidence of a myopathic face, difficulty with speech or swallowing, myotonia and inability to release her grip, or cataracts. The AGC/CTG repeat size is unstable and much more likely to expand when transmitted through a female patient than through a male patient. As the repeat increases in size, the severity increases, and age of onset decreases.
95. A fetus is found to have a cardiac rhabdomyoma on prenatal ultrasound, and both parents are healthy. What is the most likely diagnosis, how would you confirm this, and how would you counsel the parents?
TSC involves other organ systems, including abnormalities of the skin later in life (facial angiofibromas, shagreen patches, ungual fibromas); cortical tubers in the brain; seizures; intellectual disability or developmental delay; kidney problems (angiomyolipomas, cysts, renal cell carcinomas); and, rarely, lymphangioleiomyomatosis in the lungs.15
96. A fetus is found to have polycystic kidneys on prenatal ultrasound. What is the most likely diagnosis, what would you do to confirm the diagnosis, and how would you counsel the parents?
1Hoffman JI, Kaplan S. The incidence of congenital heart disease. J Am Coll Cardiol 2002;39:1890–1900.
2Murphy-Kaulbeck L, Dodds L, Joseph KS, et al. Single umbilical artery risk factors and pregnancy outcomes. Obstet Gynecol 2010;116(4):843–50.
3Palomaki GE, Kloza EM, Lambert-Messerlian GM, et al. DNA sequencing of maternal plasma to detect Down syndrome: an international clinical validation. Genet Med 2011;13(11):913–20.
4Cuckle HS, Malone FD, Wright D, et al. Contingent screening for Down syndrome—results from the FaSTER trial. Prenat Diagn 2008;28(2):89–94.
5Miller DT, Adam MP, Aradhya S, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet 2010;86(5):749–64.
6Olsen CL, Cross PK, Gensburg LJ, et al. The effects of prenatal diagnosis, population ageing, and changing fertility rates on the live birth prevalence of Down syndrome in New York State, 1983-1992. Prenat Diagn;16(11):991–1002.
7DiMauro S, Schon EA. Mitochondrial respiratory-chain diseases. N Engl J Med 2003;348(26):2656–68.
8Goriely A, Wilkie AO. Paternal age effect mutations and selfish spermatogonial selection: causes and consequences for human disease. Am J Hum Genet 2012;90(2):175–200.
9Cassidy SB, Schwartz S, Miller JL, et al. Prader-Willi syndrome. Genet Med 2012;14(1):10–26.
10Buiting K. Prader-Willi syndrome and Angelman syndrome. Am J Med Genet C Semin Med Genet 2010;154C(3):365–76.
11Williams CA, Driscoll DJ, Dagli AI. Clinical and genetic aspects of Angelman syndrome. Genet Med 2010;12(7):385–95.
12Tavormina PL, Shiang R, Thompson LM, et al. Thanatophoric dysplasia (types 1 and 2) caused by mutations in fibroblast growth factor receptor 3. Nature Gen 1995;9:321–8.
13Bove KE. Practice guidelines for autopsy pathology: the perinatal and pediatric autopsy. Autopsy Committee of the College of American Pathologists. Arch Pathol Lab Med 1997;121(4):368–76.
14Feingold M, Bossert VM. Normal values for selected physical parameters: an aid to syndrome delineation. In: Bergsma D, editor. The National Foundation-March of Dimes Birth Defects Series 10:9. White Plains, NY: March of Dimes Birth Defects Foundation; 1974. p. 1–16.
15Northrup H, Koenig MK. Tuberous Sclerosis Complex, <http://www.ncbi.nlm.nih.gov/books/NBK1220/>; 2011 [accessed 11.05.13]

